

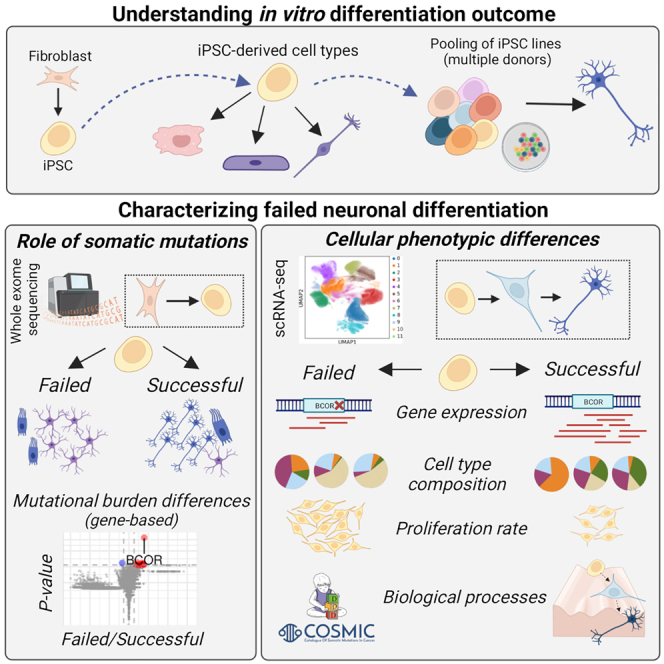
Summary
The use of induced pluripotent stem cells (iPSC) as models for development and human disease has enabled the study of otherwise inaccessible tissues. A remaining challenge in developing reliable models is our limited understanding of the factors driving irregular differentiation of iPSCs, particularly the impact of acquired somatic mutations. We leveraged data from a pooled dopaminergic neuron differentiation experiment of 238 iPSC lines profiled with single-cell RNA and whole-exome sequencing to study how somatic mutations affect differentiation outcomes. We found that deleterious somatic mutations in key developmental genes, notably the BCOR gene, are strongly associated with failure in dopaminergic neuron differentiation and a larger proliferation rate in culture. We further identified broad differences in cell type composition between incorrectly and successfully differentiating lines, as well as significant changes in gene expression contributing to the inhibition of neurogenesis. Our work calls for caution in interpreting differentiation-related phenotypes in disease-modeling experiments.
Keywords: stem cells, human iPSC, cellular differentiation, somatic mutations, single-cell RNA-sequencing, whole-exome sequencing, dopaminergic neurons, neurodevelopment, developmental disorders
Graphical abstract
Highlights
-
•
Differentiation ability of individual iPSC lines is highly variable
-
•
Somatic mutations’ effects on differentiation outcome were evaluated in 238 iPSC lines
-
•
Loss-of-function mutations in BCOR compromise the production of dopaminergic neurons
-
•
Differentiation failure due to BCOR mutations is linked to increased proliferation
A remaining challenge in the use of iPSCs to study neurodevelopment is the variability of differentiation outcomes. Puigdevall et al. analyzed 238 iPSC lines and showed that those with deleterious somatic mutations in BCOR produce fewer neurons, proliferate faster, and undergo major early changes in cell type composition and gene expression.
Introduction
Induced pluripotent stem cells (iPSC) are widely used to model human diseases, as they can differentiate to cell types and tissues that are otherwise not accessible. However, in vitro differentiation is subject to substantial technical and biological confounders that often lead to variable differentiation outcomes, a major challenge to scaling up and interpreting results from disease-modeling studies.1 The underlying reasons for this variability are not well understood, but different factors have been proposed: protocol optimization,2 culture maintenance,3 passage number,4 molecular determinants,5 inter-laboratory variation,6 cell line intrinsic properties,7 or the loss of iPSC heterogeneity in culture.8 Controlling differentiation variability of iPSCs is an essential step in achieving reliable disease models, in particular in the field of developmental biology where substantial efforts are under way to model the cell-level consequences of genetic findings in developmental and neuropsychiatric disorders.
The genetic background of an individual has been shown to account for 8%–23% of phenotypic variation in iPSCs.7 While this donor effect was driven primarily by common variants, rare variants and somatic mutations acquired either in the parental tissue (in vivo) or during the iPSC reprogramming process and culture maintenance (in vitro)9 are also likely to contribute to the observed variation. For example, it has been shown that sub-clonal cancer-associated mutations in P53 may provide growth advantage to stem cells in culture, given their increased frequency in embryonic stem cell lines.10 Also, the reprogramming of parental cells, such as skin-derived fibroblasts, can act as a bottleneck, leading to variants increasing or decreasing in frequency in the resulting population of iPSCs.11 This can be particularly pronounced if the parental cells contain a higher-than-average number of mutations, as can be the case with skin-derived, UV-exposed cells. Although such acquired mutations might not cause a phenotype in iPSCs, they have the potential to affect specific differentiated cell types and lineages,12 altering both their functionality and the overall cell type composition. Still, the contribution of somatic mutations to cellular differentiation has not been systematically explored.
In this study, we hypothesized that somatic mutations and rare germline variants in individual iPSC lines can affect their ability to differentiate. To test this hypothesis, we analyzed differentiation outcomes from four independent experiments that produced different target cell types from iPSC lines of the HipSci project7: dopaminergic neurons (DA),8 macrophages,13 sensory neurons,14 and definitive endoderm tissue15 (Figure 1A). We compared the exome-wide burden of acquired mutations and rare germline variants to the differentiation outcomes of each line (Figure 1B), and used single-cell transcriptomes of the dopaminergic neurons8 to delineate in detail how deleterious variants influence the differentiation process of iPSC-derived neurons. We found that deleterious mutations in developmentally important genes can compromise the success of iPSC differentiation by increasing the growth rate of cell lines, leading to large differences in cell type composition, gene expression, and general outlier behavior of individual lines. Our work highlights somatic mutations as an important source of variation in iPSC-based models and calls for caution when interpreting differentiation-related phenotypes to understand disease. Our results support the notion that mutational processes that affect iPSCs and their differentiation to neurons reflect those that lead to somatic mosaicism during corticogenesis.
Figure 1.

Study overview—cellular basis of variable differentiation outcomes in human iPSCs
(A) Human iPSC lines from the HipSci project were differentiated into macrophages, sensory neurons, endoderm tissue, and dopaminergic neurons, the latter being profiled with scRNA-seq in pools. iPSC lines were classified as having a failed or a successful differentiation outcome in each dataset. n, Number of iPSC lines per differentiation with available whole-exome sequencing (WES) also from the parental fibroblasts.
(B) We evaluated the genetic determinants of iPSC differentiation outcomes using WES data from 832 iPSC lines. For 384 of the 832 lines, WES data were also available for the parental fibroblasts, which allowed the identification of somatic mutations acquired in vitro.
Results
The exome-wide burden of acquired mutations does not explain the differentiation outcome
To test whether the overall burden of somatic mutations acquired by iPSC lines in vitro influenced their differentiation ability, we studied 384 cell lines (251 individual donors) from the HipSci project. Exomes were sequenced for both the parental fibroblast of donors and their derived iPSC lines, allowing us to distinguish between variants present already in the donors from those acquired, or positively selected for, in the subsequent reprogramming process (hereon “in vitro-acquired mutations”)11 (STAR Methods). After excluding 17 hypermutated lines (>240 mutations), a median of 35 mutations (37 including copy number variants) was observed per exome in the iPSCs, of which 14 were annotated as deleterious. The mutational burden was mainly donor-specific, as multiple lines from the same donor showed good correlation of mutation sites (R2 = 0.774). In line with previous publications,16 half (50.5%) of the mutations (single nucleotide variants and dinucleotides) were predicted to be missense or loss-of-function (LoF). Also, 30% of the CNVs acquired by iPSCs overlapped with known CNVs annotated as pathogenic in ClinVar dbVar.17
We considered differentiation outcomes from four different tissues and cell types derived from the same cohort of HipSci donors: dopaminergic neurons8 (DA; n = 126 observed differentiations and n = 349 predicted differentiations, see STAR Methods, Table S1), sensory neurons14 (n = 85), macrophages13 (n = 102) and definitive endoderm15 (n = 86). For each of the cell types, we split the cell lines to those that differentiated successfully and to those that failed or had impaired capacity to give rise to the desired cell type (from hereon “successful” and “failed” lines) (STAR Methods). We then tested for association between the exome-wide mutational burden and the differentiation outcomes, and found no difference between successful and failed lines (all mutations, deleterious mutations (Figure 2A), and other variant classes (Figures S1A and S1B), pAdj>0.05). Similarly, no association was found between mutational burden and endoderm differentiation efficiency, which was defined as a continuous trait (Figure 2B). The identified somatic variants were located in longer genes than unmutated ones (t-test, p = 1.02 · 10−149) and were closer to repetitive elements (t-test, pAdj = 1.16 · 10−23), as previously described for somatic variants acquired in postmitotic neurons (Figures S1C and S1D).18,19 The mutated genes showed reduced transcription (p < 0.05, t-test), but no differences in replication timing (p = 0.49, t-test, Roadmap Epigenomics Project) or promoter states (p = 0.34, chi-squared test, ENCODE BG02ES line) when compared with unmutated genes, in contrast to mutated genes in neurons (Figures S1E and S1F).20,21 Still, mutated genes in our study were enriched in published sets of somatic mutations originating early in brain development, mainly in the postzygotic stage22 (pAdj = 9.46 · 10−4, hypergeometric test) or in the stem cell and neural progenitor stage18 (pAdj = 9.02 · 10−5). This overlap indicates that mutational processes driving in vivo somatic mosaicism in human brain development may be partially mirrored by in vitro somatic mosaicism in iPSCs.
Figure 2.

Effect of the mutational burden on iPSC differentiations
(A) Burden of somatic mutations in each cell line, either total (left) or deleterious (right), was not associated (p > 0.05, Wilcoxon rank-sum test) with the differentiation outcome in macrophages, sensory neurons, or dopaminergic neurons. Boxplot whiskers are within the 1.5 IQR value. See also Figures S1A and S1B and Table S1.
(B) Burden of somatic mutations in each cell line, either total (left) or deleterious (right), was not associated with the differentiation efficiency in the endoderm (pAdj<0.05, Pearson correlation).
(C) Only the BCOR gene was consistently more mutated in failed lines than in successful lines from the dopaminergic differentiation, when considering damaging variation (FC > 2.5 (red), Wilcoxon rank-sum test, pAdj<0.05). See also Figures S1G and S1H and Table S2.
(D) Gene ontology (GO) enrichment analysis revealed the impact of deleterious mutations on brain-related biological processes (hypergeometric test for GO term association, pAdj<0.05, red-colored tiles) contributing to either failed or successful DA differentiation in lines whose outcome was either observed or predicted. The number in each tile corresponds to the rank of the significant GO term, ordered by decreasing odds ratio within each analysis (only top-30 positions are illustrated). See also Table S3.
The burden of deleterious variants in BCOR is linked to differentiation failure in dopaminergic neurons
Mutations that impair the function of active genes in development might also have a critical role in altering cell line differentiation efficiency, even when they do not compromise cell survival in culture. We analyzed how burden differences in individual genes were linked to the differentiation outcome in the DA dataset. We focused on the total burden of deleterious mutations carried by each iPSC line, which includes somatic mutations acquired in vitro and in vivo, as well as germline variants (STAR Methods). We found that only one gene, BCOR, was significantly more mutated in failed lines compared with the successful lines (Wilcoxon rank-sum test, pAdj <0.05, log2FC >2.5) (Figure 2C). This effect was consistently observed with all deleterious variants as well as LoF variants alone, and with both observed (n = 183 cell lines, one line per donor; 48 failed, 135 successful) and predicted DA differentiation outcomes8 (n = 793 cell lines from 529 donors; 99 failed, 694 successful). Importantly, none of the lines that differentiated successfully in culture (i.e., with observed outcomes) carried an LoF mutation in the BCOR gene, while 22 of the 48 failed lines carried at least one (Figure S1G, Table S2). Beyond the binary outcome classification, we also observed the association between the deleterious mutational burden in BCOR with the continuous differentiation efficiency (pAdj = 1.06 · 10−8, n = 183 lines), defined as the fraction of dopaminergic and serotonergic neurons at day 52, or with the predicted model scores8 (pAdj = 7.22 · 10−57, n = 793 lines) (STAR Methods). We confirmed a significant reduction of BCOR expression between mutated (BCOR LoF) and unmutated lines across all time points (p < 0.05, Wilcoxon rank-sum test) (Figure S1H).
Although the mechanism for this association is unknown, the BCOR gene (a BCL6 repressor) is a known epigenetic regulator23 that is both an oncogenic driver gene24 and a developmental disorder-causing gene,25 highlighting its key role in development. Previously, pathogenic mutations in the BCOR gene have been found to be recurrently mutated in blood-derived iPSC lines and positively selected for under iPSC culture conditions.11 The gene is under strong mutational constraint, with only two predicted LoF SNVs (pLoF) observed in gnomAD26 (version 2.1.1: 44.6 expected; LOEUF mutational constraint score 0.141). In addition, we did not find any BCOR LoF variants among the parental fibroblasts of the iPSC lines (n = 253), although they could still be present at very low frequencies as subclones. In our study, while the BCOR mutations driving impaired DA differentiation clearly increased in frequency during the reprogramming process, we cannot determine for certain whether they originated in vivo or in vitro.
A fraction of the lines that were classified as failed (26 of 48, differentiation efficiency <0.2) do not carry any deleterious BCOR mutations, which likely indicates that other genes also contribute to DA differentiation failure but are not identified in our analysis due to limited sample size. To overcome this, we focused on the biological processes that control cellular differentiation and performed a gene ontology (GO) enrichment analysis on those genes that presented the largest mutational burden differences between failed and successful cell lines (top 10% and bottom 10% in fold change [FC], corresponding to 1,865 genes for each outcome, STAR Methods). We found that the genes mostly mutated in failed lines are involved in key neurodevelopmental functions, such as neuron fate commitment (GO:0048663, pAdj = 0.03, OR = 1.99), response to axon injury (GO:0048678, pAdj = 0.015, OR = 2.08) and midbrain development (GO:0030901, pAdj = 0.03, OR = 1.8), consistent with the failed differentiation phenotype (Figure 2D, Table S3). Among the genes contributing to this enrichment in failed lines, we found examples of disease-associated genes27: CDC42 in intellectual disability,28 BMP4 in syndromic microphthalmia,29 and PMP22 in hereditary neuropathy.30 On the contrary, mutations in successful lines disrupted genes involved in the positive regulation of neuron apoptotic process (GO:0043525, pAdj = 0.006, OR = 2.59) and neuron fate specification (GO:0048665, pAdj = 0.001, OR = 3.8), potentially contributing to a higher production of neurons throughout the differentiation.
Differentiation failure is driven by increased proliferation rate and BCOR LoF mutations
One of the unique features of the DA dataset is that the cell lines were differentiated as pools.8 When cell lines are cultured and differentiated together, individual lines may acquire mutations that give them growth advantage, such as those in cancer-associated genes, leading to an imbalanced representation of lines in the pool. In order to understand the potential mechanism by which the observed BCOR mutations impair DA differentiation, we studied the growth dynamics in the pooled DA dataset. We reanalyzed RNA-sequencing data from 846,841 single cells from 238 cell lines (230 donors) at three differentiation time points (days 11, 30, and 52, corresponding to progenitors, young neurons, and mature neurons, respectively) (STAR Methods, Figure S2), leading to a dataset consisting of 19 different pooled differentiations, including 7 to 24 cell lines per pool (Table S4).
We first evaluated how the proportion of cell lines sampled at the three time points (n = 164) changed over the course of DA differentiation, assuming initial equal amounts of each iPSC line (Figure 3A, STAR Methods). Specifically, we calculated an in silico proliferation rate for each line in a given pool by contrasting cell line proportions at specific time points to day 0. Although the Day52/Day0 proportion remained constant for most of the lines (mean = 1.0, 95% confidence interval 0.8–1.2) (Figure S3A), almost every pool contained at least one overrepresented line (up to 3–10x, depending on the pool), with a small fraction of lines being underrepresented (down to 7–700x). Interestingly, lines that failed to differentiate into neurons (n = 50) showed on average larger proliferation rates than successful lines (n = 114), with the difference being most significant at the last time point (p = 2.8 · 10−3, Wilcoxon rank-sum test) (Figure S3B). When we correlated this behavior with the mutational burden, we found that increased proliferation rates were driven by BCOR LoF mutations (Figure 3B). Specifically, failed lines with BCOR LoF mutations (n = 20) had a significantly higher proliferation rate than successful lines (n = 114) across the time points, a difference that was not observed with failed lines without BCOR LoF mutations (n = 30). Consistent with this, we found that the BCOR gene had the highest ratio (5:0) of annotated cancer driver mutations (LoF pathogenic Cosmic-Tier1 mutations) in failed lines compared with successful ones (STAR Methods). Taken together, these results suggest that proliferation advantage, caused by recurrent somatic mutations in key developmental genes like BCOR, has a negative effect on differentiation efficiency. Of note, when we compared cell line abundances in different types of replicate samples (Figure S3C), we found that the correlation of the fractions was substantially lower when replicates were differentiated as part of different pools (R2 = 0.136, n = 41 lines) compared with replicates with the same pool background (R2 = 0.693, n = 31 lines) (Figure 3C, STAR Methods). Still, pool replicates were mostly concordant in their differentiation outcome (33 of 36 lines). This suggests that other cell lines of a given pool can influence the growth dynamics of individual lines, likely due to the presence of non-cell-autonomous effects, but they are not sufficient to alter the differentiation outcome.
Figure 3.

The proliferation rate of cell lines is influenced by the pool environment, but the highest rates were observed among failed lines with BCOR LoF mutations
(A) Cell line proportion throughout dopaminergic neuron differentiation (days 11, 30, and 52). We observed a consistent outlier behavior featured by 1–2 lines that proliferate significantly faster than other lines during pooled differentiation.
(B) Failed cell lines carrying at least one BCOR LoF mutation showed, on average, a higher proliferation rate than neurons that can differentiate successfully (D52/D0, p = 0.0015, Wilcoxon rank-sum test). Boxplot whiskers are within the 1.5 IQR value. See also Figures S3A and S3B.
(C) Cell line proportion could not be replicated between pool replicates, but it remained similar between biological replicates and showed minimal differences between technical and 10X replicates, as expected (linear regression, p<0.05). The shaded area indicates 95% confidence interval on the proportion values. See also Figure S3C.
Poor differentiation outcomes manifest as shifts in cell type composition already at the progenitor stage
Next, we studied how early in the differentiation process cell type composition differences between failed (n = 58) and successful (n = 163) lines start to appear (STAR Methods). In order to better characterize cell type composition changes of cell lines across the three time points, we processed and clustered all cells in the DA dataset together (119 10x samples, Tables S5 and S6 and STAR Methods), contrary to the original study where time points were analyzed separately.8
We then used a negative binomial regression model to evaluate cell type composition changes between failed and successful lines (STAR Methods). The analysis revealed significant shifts in abundance for all major cell types (>2% fraction) as early as day 11, except for some floor-plate progenitors (FPP-1) and ependymal cells (Epend-1) (Figure 4A). Interestingly, cell lines that failed to generate mature neurons at day 52 showed an earlier commitment to either the dopaminergic (DA, pAdj = 9.9 · 10−47) or serotonergic (Sert-like, pAdj = 6,1 · 10−14) fate at day 11, with their neuroblasts clustering with young neurons, but failing to express the same neuronal markers. Similar evidence of accelerated neuronal maturation in vivo has been observed in an iPSC model of Kabuki syndrome caused by a heterozygous nonsense mutation in KMT2D,31 and in iPSC-derived brain organoids modeling schizophrenia.32 Also, different risk genes linked to autism spectrum disorders were recently shown to converge on an accelerated differentiation phenotype of GABAergic and deep-layer projection neurons.33
Figure 4.

Failed and successful lines show major differences in cell type composition, gene expression, and functional enrichment
(A) Major cell types showed significant differences in composition as early as day 11 (negative binomial regression, STAR Methods), except for floor-plate progenitors type 1 (FPP-1) and ependymal-like cells (Epend-1). Neuroblasts from failed lines showed an earlier commitment to dopaminergic (DA) or serotonergic-like (Sert-like) fate. Minor cell types (<2% abundance) are shown in red. Boxplot whiskers are within the 1.5 IQR value. See STAR Methods for cell type abbreviations.
(B) High proliferation rates were associated with significant changes on cell type composition at day 52 linked to a depletion (in orange) of dopaminergic (DA, p < 0.05(∗), linear regression) neurons and an excess (in blue) of astrocytes (Astro) and proliferative floor-plate progenitors type 1 (proFPP-1, ∗∗p < 0.01). See STAR Methods for cell type abbreviations.
(C) Most of the differentially expressed genes at day 11 are enriched in developmental disorder genes (DDD, cyan), especially when considering those that act in a dominant fashion (DDD-dominant, blue) (chi-squared test, STAR Methods). Also, some of the cell types show an enrichment in cancer-associated genes (Cosmic Tier 1, magenta). See also Figures S4A and S4B.
The significance level in (A–C) was indicated as follows: pAdj >0.05 (ns), pAdj <0.05 (∗), pAdj <0.01 (∗∗), pAdj <0.001 (∗∗∗).
(D) Functional enrichment of biological processes among differentially expressed (DE) genes from all (allDE), several (signifDDD), or unique cell types (hypergeometric test for GO term association, pAdj <0.05 and FC >1.5). Overrepresented processes in failed lines include neuron development and neuron maturation as previously observed with genes showing differential deleterious burden between failed and successful lines. The number in each tile corresponds to the ranked position of the significant GO term, ordered by decreasing odds ratio within each analysis. See also Table S7.
At day 52, the overall lower fraction of neurons in failed lines was accompanied by a significantly larger proportion of astrocytes (pAdj = 1.5 · 10−36), ependymal-like cells (pAdj = 7.7 · 10−14), and of an unknown cell type (Unk-1, pAdj = 1.1 · 10−19). In agreement with these observations, iPSC lines with deleterious BCOR mutations also presented an altered cell type composition compared with lines without BCOR mutations, with a significant depletion of neuronal cell types (DA, p.Adj = 5,3 · 10−6; Sert-like, p.Adj = 8.9 · 10−8, Wilcoxon rank-sum test) accompanied by a significant excess of astrocytes (p.Adj = 1.3 · 10−5), proliferative floor-plate progenitors (FPP-1, p.Adj = 1.9 · 10−3), and one unknown cell type (Unk-1, p.Adj = 6.9 · 10−3) (Figure S3D, STAR Methods). Importantly, we also found an association between the cell line proliferation rate and the abundance of those cell types, with faster-proliferating lines showing a depletion of DA neurons (p.Adj = 0.01, Pearson correlation) and an excess of astrocytes (p.Adj = 0.004) and proliferating progenitors (pro-FPP1, p.Adj = 0.004) at day 52. (Figure 4B, STAR Methods). This suggests that differentiation failure is a consequence of a global, early shift in cell type composition caused by abnormally fast proliferation of cell lines that carry damaging mutations in key developmental genes such as BCOR.
Biological processes linked to failed differentiation manifest as differential gene expression
We tested whether differences in cell type composition between successful and failed lines also manifest as gene expression changes. For this, we performed a differential gene expression (DE) analysis between failed (n = 58) and successful (n = 163) cell lines within cell types and time points (STAR Methods). We identified between 50 and 500 DE genes per test, which, importantly, were not correlated with the number of cells observed per outcome (Figure S4A). While the number of DE genes was relatively constant throughout differentiation in certain lineages (dopaminergic, astrocytes, floor-plate progenitors type 1), others showed time-point-specific differences. Remarkably, in most of the cell types at the progenitor stage, gene sets known to cause developmental disorders34 (DDD) were overrepresented among the DE list (adj.p < 0.05, chi-squared test), in particular when only dominant DDD genes were considered. Likewise, cancer-associated genes35 (Cosmic-Tier1) known to influence cellular proliferation were overrepresented in five cell types at day 11, one at day 30, and another at day 52 (Figure 4C).
We then performed a GO enrichment analysis (STAR Methods) on the 10 cell types with a significant proportion of differentially expressed DDD genes. We found several biological processes related to neurodevelopment among the top-25 enriched terms (adj.p < 0.05, ordered by odds ratio) (Figure 4D, Table S7), including the regulation of glial cell differentiation (GO:0045685, day 11) and cerebral cortex development in progenitors (GO:0021987, day 11), as well as the neuron projection extension in ependymal-like cells (GO:1990138, day 11). When aggregating all the detected DE genes in the analysis (any cell type), we found processes strongly linked to failed differentiation, such as the positive regulation of neuron apoptotic process (GO:0043525) and the negative regulation of neuron differentiation (GO:0045665). We also analyzed the changes in pathway regulation on the seven cell types with an excess of cancer-associated DE genes (STAR Methods, Figure S4B), and consistently identified hallmarks of cellular proliferation: upregulation of the tumor suppressor P53, activation of MYC targets, and exacerbated oxidative phosphorylation. This functional enrichment of DE genes is consistent with that of mutated genes in failed lines (Figure 2D).
Proliferation rate predicts cell type outlier status of cell lines
Cellular differentiation is a dynamic process with global changes in the composition of cell populations over time. Although differentiation success is usually defined by the final yield of the desired cell type, this can give an incomplete picture of the variability in the differentiation process. To characterize such variability, we analyzed all cell lines with reliable cell fraction estimates (day 11, n = 172; day 30, n = 187; day 52, n = 209, STAR Methods) to identify those that were outliers in terms of their cell type composition. For this purpose, we computed a Z score per line for each cell type and time point combination and assigned as outliers those cell lines with |Z score|>2 (Figure 5A, STAR Methods). Under this classification, we identified 156 cell lines (175 considering pool replicates as independent lines) that were an outlier in at least one of the time points (day 11, n = 55; day 30, n = 78; day 52, n = 104), most of which had abnormally large cell type fractions. Only on day 11, 16 cell lines showed abnormally low fractions of progenitors and astrocytes, which were compensated by abnormally large fractions of other cell types. We also observed an overall increase in cell type fraction variability at later stages of differentiation.
Figure 5.

Cell lines in the pooled differentiation displayed common outlier behavior in cell type composition
(A) Most of the outlier cell lines showed an excess of a particular cell type throughout the differentiation, except for some lines with a reduced population of astrocytes and progenitors at day 11.
(B) Upper: On average, each outlier line showed 2.21 outlier events per differentiation, but less than 1 event per time point (bars, 95% CI). Also, most of the lines displayed the outlier behavior either simultaneously at days 30 and 52 (young and mature stage), or just at day 11 (progenitor stage); lower: Lines with at least one abnormal cell type abundance throughout the differentiation (outlier event) showed a significant reduction in the somatic burden of acquired mutation in vitro than non-outliers (total burden, p = 3.6 · 10−3; deleterious, p = 2.7 · 10−3; Wilcoxon rank-sum test). Boxplot whiskers are within the 1.5 IQR value. See also Figure S5A.
(C) The mean proliferation rate per line (day 52/day 0) was higher in outlier lines than in non-outlier lines (p = 9.68 · 10−4, logistic regression). Boxplot whiskers are within the 1.5 IQR value.
(D) Upper: Z score correlation of gene expression (i.e., KMT2D) and cell type proportions of floor-plate progenitors type 1 (FPP-1) and neuroblasts committing to dopaminergic neurons (DA) at day 11. Each dot is an iPSC line; lower: The distribution of Pearson correlation coefficients for all genes expressed in previous cell types at day 11. See also Figure S5B.
(E) The abundance of progenitor populations was associated with the expression of genes in developmental disorder (DD) and cancer-associated (Cosmic-Tier1) genes, and to a lesser extent in adult-onset neurodegenerative disorder genes. In schizophrenia, the association is only observed with genes expressed in neurons and no enrichment is observed for non-brain disorders (hypergeometric test for overrepresentation, Benjamini-Hochberg multiple-test correction). Significance levels: pAdj >0.05 (ns, yellow), pAdj <0.05 (∗, golden yellow), pAdj <0.01 (∗∗, orange), pAdj <0.001 (∗∗∗, red).
To further characterize the outlier behavior, we calculated the number of times an outlier line shows an abnormal cell type fraction across the differentiation (outlier event, STAR Methods). On average, we observed 2.21 outlier events per line (2.03–2.39, 95% CI) with even contributions per time point (Figure 5B, upper). When focusing only on the cell lines profiled at the three time points (n = 112 lines), we observed that they tend to show outlier events most frequently at the two consecutive latest time points (day 30 and day 52) or just initially (day 11), likely due to the final plating of neurons occurring between the first and later time points. Only 12 cell lines showed outlier behavior in all time points. Unexpectedly, when we explored the correlation of somatic mutational burden acquired in vitro with outlier behavior (n = 148 lines), we observed a significant reduction of burden (p < 0.01, Wilcoxon rank-sum test) in the outlier group (Figure 5B, lower). This difference was observed for both total and deleterious mutations, but when detaching the outlier status per time point, the difference remained significant only at day 30 (Figure S5A, STAR Methods).
Although observing uneven cell type fractions is common among wild-type iPSC lines in pooled experiments, we found that larger proliferation rates at day 52 were associated with the outlier behavior (p = 9.68 · 10−4, logistic regression, n = 159 lines) (Figure 5C). To identify which genes might be driving this behavior, we correlated cell-type-specific gene expression with changes in cell type composition (Figure 5D, STAR Methods). Among the significant associations, including positively and negatively correlated genes, we observed a strong enrichment of DDD genes (p.Adj<0.001, hypergeometric test) in most of the cell types at the progenitor stage (Figure 5E, STAR Methods), including BCOR (Figure S5B). Similarly, we observed that seven of the nine cell type associations enriched in cancer-associated genes were also enriched among DDD genes, as expected from the significant overlap between the two gene sets (p < 2.2 · 10−16, chi-squared test). A more limited enrichment in adult-onset neurodegenerative disorder genes36 was also observed for floor-plate progenitors (FPP). However, for schizophrenia genes,37 weaker but still significant associations were observed only for dopaminergic (day 30) and serotonergic neurons (day 52). As expected, no enrichment was observed in gene panels for non-brain disorders.36 Overall, this suggests that the regulation of developmental genes during early neural induction is critical for determining progenitor abundance, and as shown for the BCOR gene, for influencing proliferation rate and differentiation success in vitro.
Discussion
One of the biggest limitations of iPSC-based disease modeling is our poor understanding, and control, of factors that influence the capacity of cell lines to differentiate successfully and reproducibly. Recently, it was proposed that the large variability associated with differentiation is primarily explained by cell-intrinsic factors,8 rather than experimental or other technical factors. Genetic variation has previously been shown to drive molecular heterogeneity in iPSCs7,38,39,40 and in hESC,41 but large-scale studies looking at the role of genetic variation in differentiation are only beginning to emerge. In particular, somatic mutations acquired prior to the differentiation, either in vivo during parental tissue clonal evolution or in vitro during iPSC reprogramming and culture maintenance, are potential contributors to variation in differentiation efficiency. Interpreting such variation correctly is critical for developing better models of development and diseases, particularly in the field of neurodevelopment, where disease-modeling efforts are currently rapidly increasing, due to advances in identifying the genetic underpinnings of psychiatric and developmental disorders.42,43
Here, we present an attempt to link differentiation outcomes to somatic mutations in human iPSC lines from the HipSci resource, which offers a unique opportunity to study multiple cell types independently derived from the same iPSC lines. Further, with exome sequencing available for both iPSCs and their parental fibroblasts, we were able to focus also on the subset of damaging mutations acquired in vitro, which are particularly relevant for abnormal differentiation outcomes.
One of the key insights from our work is that although the total burden of acquired mutations in iPSC lines is not predictive of their differentiation outcome, deleterious mutations in the core genes of a given differentiation system can cause unwanted effects on differentiation. This effect is likely not limited to mutations acquired in vitro, as mutations and rare variants in the genetic background of the parental cells selected for reprogramming may account for a considerable fraction of differentiation variability, even if not affecting reprogramming directly. In support of this, we found that somatic deleterious mutations in the BCOR gene are strongly associated with differentiation failure in human dopaminergic neurons. The effect was seen with 183 observed differentiations as well as 793 predicted differentiation outcomes of iPSC to dopaminergic neurons.8 The sensitivity to BCOR deleterious mutations is supported by the strong selection against predicted LoF variants in the Genome Aggregation Database.44 Further, a high prevalence of acquired BCOR mutations was previously found in blood-derived iPSC lines, and it was shown that they likely arose after reprogramming through positive selection for BCOR dysfunction.11 In our dataset, damaging BCOR variants were only observed in the iPSC lines, although they could have originated in vivo and be present in parental fibroblasts as subclones at very low frequencies that later underwent positive selection in vitro.
BCOR is a key transcriptional regulator during embryogenesis. It is part of a specific type of polycomb repressive complex that mediates transcriptional repression through epigenetic modifications of histones24 and has been shown to have a key role in regulating the pluripotent state and differentiation. Like many other chromatin-related genes, BCOR is annotated both as a developmental disorder gene and a cancer driver gene.35,45,46 In line with this dual role, we observed that failed lines with deleterious BCOR mutations showed significantly larger proliferation rates than lines that differentiated successfully, suggesting that monitoring cell line proliferation rates prior to differentiation may be an effective way to screen out lines that will not differentiate correctly.47 Further, abnormal proliferation has been reported as a phenotype in multiple iPSC-based disease models, such as Kabuki syndrome31 or in tuberous sclerosis, where an hyperproliferative population of interneuron progenitors in brain organoids was identified as the underlying cause for the distinctive phenotype of brain tumors and cortical malformations.48
Although the BCOR gene was not part of the expression signature of failed differentiation in specific iPSC populations (n = 184 lines),8 we observed that BCOR expression in dopaminergic neurons was negatively associated with final neuron abundance, and was also linked to the abundance of progenitor populations earlier in the differentiation. As an epigenetic modulator of stemness and differentiation, it remains unknown whether the expressivity of BCOR LoF mutations manifests already in iPSCs, at precursor stage or at both, diverting neuron differentiation toward astrocytes and ependymal-like cells.
Despite the strong association of BCOR mutations with differentiation failure, not all of the failed lines carried damaging variants in that gene, suggesting that other genes are involved as well. Therefore, we also analyzed the most differentially mutated genes in each differentiation outcome to pinpoint the biological processes that were disrupted by deleterious mutations. Interestingly, genes that were mutated mostly in failed lines were enriched in neurodevelopmental processes linked to neuron fate commitment, response to axon injury, or midbrain development. In successful lines, we observed an enrichment of brain-related processes that are likely to boost neuron production by disrupting the positive regulation of neuron apoptotic processes. While our study is limited to identifying acquired mutations only during iPSC reprogramming, it is possible that somatic mosaicism in neurons also plays a role in determining the final differentiation outcome. To this end, mutated genes from our study significantly overlapped with published lists of somatic mutations that originated during early cortical development,18,22 highlighting that mutation-selection processes driving in vitro somatic mosaicism could mirror those observed in vivo during brain formation. If confirmed, this would have major implications for disease modeling, as somatic mosaicism observed in vitro could be used to understand the clinical impact of this process in vivo. Future efforts in profiling exomes of iPSC-derived neurons will provide further insights on the role of somatic mutations in differentiation systems and on new potential applications for developmental studies.
To better characterize the cell type composition dynamics throughout the DA differentiation process, we introduced a critical modification to the analysis in the original study.8 Specifically, we clustered all cells at once, rather than per time point. While we lose some granularity in the definition of cell types, this approach allowed us to observe cell type composition changes per line across cellular lineages, tracing the commitment of neuroblasts to young and mature neurons. We identified a larger fraction of neuroblasts committing to dopaminergic and serotonergic neurons in failed lines, suggesting an accelerated maturation of progenitors potentially linked to the proliferative phenotype. In this scenario, failed lines could progress faster to differentiation initiation after neural induction, promoting an early production of neuroblasts with defects in neuronal commitment.
Finally, we compared the extent of DE in each cell type across time points and conditions between failed and successful lines. With these comparisons, we sought to identify the key regulator genes across the different stages of neurodevelopment and across different biological processes. Many cell types at the progenitor stage (day 11) showed an enrichment of DE genes corresponding to key developmental genes, either DDD or cancer-associated. In those cell types, the differentially regulated neurodevelopmental processes clearly overlap with those affected by deleterious mutations in failed lines. We hypothesize that among the DE genes, there is a potential list of new developmental disorder (DD) candidates, whose clinical significance should be evaluated.
Any differentiation process involves a dynamic evolution of cell types, which does not necessarily fit into a failed or successful outcome based on an arbitrary threshold. To avoid overlooking other relevant changes, we analyzed general behavior in cell type composition. We found that 64.3% of lines in pooled experiments occasionally display abnormal cell type fractions during the differentiation process. This outlier behavior reflects the large variability in cell type composition during in vitro pooled differentiations, likely resulting from the combination of donor effects, non-cell-autonomous effects between lines and stochasticity. Although such effects are indeed a limitation of the pooled study design that can affect cell line abundance, they rarely compromise the ultimate differentiation outcome in our dataset. Also, the periodicity of outlier events suggests that they tend to happen either consecutively in the last two time points or only at the first one, which can be explained by the experimental design, as cells were only passaged at day 20. Even more importantly, we found that outlier behavior was strongly associated with larger proliferation rates in cell lines, possibly implying that acquired mutations in other genes that also increase proliferation activity could be behind the abnormal cell type composition. However, since the mutational recurrence in all other genes was substantially lower than in BCOR, this study was not sufficiently powered to detect population-level evidence for this possibility. Finally, we did not observe a higher burden of acquired mutations in outlier lines when compared to non-outlier lines, but rather the opposite. These observations suggest that while individual deleterious mutations can define differentiation outcomes, the determinants of outlier behavior during neuronal differentiation are likely more varied.
In summary, our study demonstrates that although iPSC models are an excellent tool for studying neurodevelopment and developmental disorders, results from differentiated cell types should be interpreted with caution. We studied a large number of iPSC lines derived from healthy individuals and observed that deleterious mutations in genes known to cause developmental disorders cause differentiation defects via transcriptional and cell type composition changes during neuronal differentiation. Our work highlights somatic mutations as a significant source of variation in iPSC-based disease models and further emphasizes the importance of comprehensively assaying the genomes of iPSC lines prior to their experimental use to achieve reproducible research.
Limitations of the study
This study considers only mutations that originated or were positively selected during iPSC reprogramming from parental fibroblasts, or that were present at iPSC culture. Consequently, we lack mutational information from differentiated cell types, i.e., somatic mutations that may have occurred during the differentiation process and might also contribute to variable differentiation outcomes. As for the DA dataset, the pooling strategy reduces the unwanted batch effect between lines, but introduces non-cell-autonomous effects that might alter the growth dynamics of individual lines. Finally, we did not functionally validate the impact of BCOR LoF mutations on differentiation failure and the gene expression reduction that we observed with single-cell transcriptomics data.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Pooled dopaminergic differentiation | Jerber et al.8 | HipSci Managed access (EGA: EGAS00001002885) HipSci Open Access (ENA: ERP121676) |
| Whole exome sequencing (WES) of HipSci lines | HipSci Project https://www.hipsci.org |
HipSci Managed access: EGA HipSci Open Access (ENA: ERP006946) |
| HipSci genotype array data | HipSci Project https://www.hipsci.org Kilpinen et al.7 |
HipSci Managed access (EGA: EGAS00001000866, EGA: EGAS00001001272) HipSci Open Access (ENA: PRJEB11750) |
| Metadata for the dopaminergic differentiation single-cell dataset | This paper |
https://zenodo.org/record/6079122#.YlSkxNPMJhE (Open Access) |
| Processed single-cell count (dopaminergic differentiation) for days 11, 30 and 52. | This paper |
https://zenodo.org/record/6079122#.YlSkxNPMJhE (Open Access) |
| Macrophage differentiation | Alasoo et al.13 | Table S1 |
| Sensory neuron differentiation | Schwartzentruber et al.14 | Table S1 |
| Endoderm differentiation | Cuomo et al.15 | EGA:EGAS00001002278 EGA: EGAD0001005741 ENA:ERP016000 |
| Somatic mutations (acquired in vitro) | Rouhani et al.11 | https://doi.org/10.1101/2021.02.04.429731 |
| Software and algorithms | ||
| Variant effect predictor (VEP, release 99) | McLaren et al.49 | https://www.ensembl.org/info/docs/tools/vep/index.html |
| BCFtools (version 1.4.25) | Danecek et al.50 | https://samtools.github.io/bcftools/bcftools.html |
| Scanpy toolkit | Wolf et al.51 | https://scanpy.readthedocs.io/en/stable/ |
| Cellranger v3.1.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| R (4.0.4) / Bioconductor (3.12) | R Core Team, 2021 Bioconductor Project, 2021 |
https://www.r-project.org/ https://www.bioconductor.org/ |
| Seurat (v4.0.1) | Butler et al.52 | https://satijalab.org/seurat/ |
| GOstats (v2.56) | Falcon and Gentleman53 | https://bioconductor.org/packages/release/bioc/html/GOstats.html |
| Harmony | Korsunsky et al.54 | https://portals.broadinstitute.org/harmony/articles/quickstart.html |
| Demuxlet | Kang et al.51 | https://github.com/statgen/demuxlet |
| Other | ||
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Developmental disorder genes (DDG2P, version 2.2) | Wright et al.34 | https://nhsgms-panelapp.genomicsengland.co.uk/panels/484/v2.2 |
| Cosmic database (cancer-associated genes, version 90 – GRCh37) | Tate et al.35 | https://cancer.sanger.ac.uk/cosmic |
| MSigDB hallmark gene set signatures | Liberzon et al.55 | https://www.gsea-msigdb.org/gsea/msigdb/ |
| 1000 Genomes Project | The 1000 Genomes Project Consortium et al.56 | https://www.internationalgenome.org/ |
| Exome Aggregation Consortium 0.3.1 | Lek et al.57 | https://gnomad.broadinstitute.org/downloads |
| CADD Phred scores (version 1.6) | Rentzsch et al.58 | https://cadd.gs.washington.edu/score |
| Original code (Zenodo) |
This paper |
https://doi.org/10.5281/zenodo.7641259 (Open Access, frozen repository) |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Helena Kilpinen (helena.kilpinen@helsinki.fi).
Materials availability
This study did not generate new unique reagents or new human iPSC lines.
Method details
Somatic mutations acquired in vitro (SMAV)
A joint variant calling (BCFtools/mpileup and BCFtools/call, version 1.4.25, human genome assembly GRCh37d5) between 384 pairs of parental fibroblasts and their corresponding iPSC lines (251 donors) was performed to identify 18,999 somatic mutations that were acquired or positively selected throughout iPSC reprogramming as described in Rouhani et al.11 This calling was performed for single-nucleotide variants (SNVs), dinucleotides, indels and copy number variants (CNVs) for both autosomal chromosomes and chromosome X. Only in the case of indels, chromosome X was not included.
We filtered the initial call set11 following these steps: the variants out of the exome sequencing baits were excluded, germline variants were filtered-out assuming they show a minor allele frequency MAF>0.1% in 1000 Genomes Phase356 or in ExAC 0.3.1,57 or be carried by the parental fibroblast of more than one donor. Only high-quality variants were filtered-in (PASS filter). Variants with an allelic fraction larger than 0.6 in iPSC or in fibroblasts were removed to filter potential spurious mutation calls. We classified variants either as acquired in vitro or positively selected only when a significant rise in allele frequency was observed between the iPSC and the parental fibroblast (Fisher’s exact test p<1.6·10-4, equivalent to FDR 5% using the Benjamini-Hochberg multiple test correction procedure). 17 out of the 384 iPSC lines were found to be hypermutated in vitro (>240 mutations corresponding to a Z-score>2) and were discarded for further analysis downstream. Finally, we defined our gene universe as the 19,653 genes that were protein-coding genes in the Ensembl gene annotation (GRCh37, version 87) and were covered by the exome sequencing baits. In line with that, we discarded all those variants that could not be annotated to the gene universe, finally releasing a call set of 18,999 mutations, including 460 CNVs, 642 indels, 2,445 dinucleotides and 15,452 SNVs (Table S1).
Functional annotation of SMAV
We annotated the somatic mutations acquired in vivo (SNVs, dinucleotides and indels from the 384 pairs of iPSC lines and their corresponding parental fibroblasts, n=251 donors) by predicting their functional consequence using the variant effect predictor49 (VEP, release 99) and the haplotype-aware BCFtools/csq tool50 (version 1.9). We used Ensembl gene annotations (GRCh37, version 87) and recorded only the most impactful consequence for each mutation, as determined by the following decreasing order of severity: https://www.ensembl.org/info/genome/variation/prediction/predicted_data.html.
We defined mutations as loss-of-function (LoF) when they were annotated as frameshift, stop-gain, splice acceptor or splice donor variants; and as missense pathogenic (or damaging missense) when annotated as missense or start loss with a CADD58 Phred score cutoff > 15 (version 1.6). The definition of deleterious mutations included the union of LoF and missense pathogenic mutations. The remaining mutations were annotated as synonymous or as “others” (either as coding, non-coding or unannotated). Overall, we annotated 1,002 LoF mutations, 5,722 missense pathogenic, 2,631 missense non-pathogenic, 3,158 synonymous and 6,026 other mutations (Table S1).
Differentiation outcome of iPSC-derived cell types
Sensory neurons
We processed the Table S1 of the original publication14 (“IPSDSNs”) that contained the metadata for all cell lines differentiated to sensory neurons. We annotated the differentiation outcome by re-labelling the neuron quality status per line from “Poor” to failed and from “Good” to successful. We excluded those cell lines with undefined neuron quality status and renamed the non-neuronal differentiation outcome to failed differentiation. We identified 13 iPSC lines with differentiation replicates, all of which had a concordant outcome (2 failed, 10 successful), except for one line ("HPSI0613i-eojr_3") that was discarded. In total, we annotated the outcome for 105 cell lines (5 failed and 100 successful), of which 85 lines (3 failed, 82 successful) had also WES data for the parental fibroblasts (N=83 donors) (Table S1).
Macrophages
We processed the Table S1 of the original publication13 that contained the status of the differentiation outcome per line. We removed those cell lines that showed a low macrophage purity (“FC_QC_fail”) or presented degraded RNA (“RNA_QC_fail”). We identified 11 iPSC lines with differentiation replicates, all of which had a concordant outcome (7 failed, 4 successful). In total, we annotated the outcome for 123 cell lines (23 failed and 90 successful), of which 102 lines (22 failed, 80 successful) had also WES data for the parental fibroblasts (N=102 donors) (Table S1).
Dopaminergic neurons
We reprocessed the whole scRNA-seq dataset (excluding cells treated with rotenone), re-clustering all cells at once and annotating the resulting cell types using the same markers from the original publication8 (see also Reanalysis of pooled single-cell data). We then computed the total number of cells and the cell type proportion per line in each pool. For each time point, we removed those cell lines with the lowest number of cells (first twentile) to increase the confidence level of the cell type proportion estimates. We then defined the neuronal differentiation efficiency per cell line in each pool as the proportion of dopaminergic and serotonergic neurons observed at day 52 of the differentiation. We annotated this efficiency for 209 cell lines distributed in 18 pools, and classified lines accordingly either as failed lines (<0.2, this includes lines with poor/impaired outcome) or successful lines (≥0.2). Only 3 out of the 36 iPSC lines placed in more than one pool (pool replicates) were discordant. The DA outcomes for the remaining 206 lines corresponded to 56 failed and 150 successful lines, of which a subset of 126 lines (35 failed, 91 successful) had also WES data for the parental fibroblasts (N=126 donors). Overall, we reached a 98.8% agreement with the neuronal differentiation outcome classification of the original paper.
Additionally, we processed the Table S5 from Jerber et al.8 which contained the model scores from the predicted efficiency. Those scores were obtained from a logistic regression trained with a binary outcome definition per line (either successful lines with >20% measured efficiency or failed lines with <20%) and an independent dataset of bulk RNA-seq that uses all expressed genes from 184 iPSC lines. The model scores classified 812 HipSci iPSC lines as failed (N=103) and successful (N=709) differentiators (precision=0.9 and recall=0.35 for threshold=0.02231), of which 349 lines (33 failed, 316 successful) had also WES data for the parental fibroblasts (N=231 donors) (see Figure 1A, Table S1).
Endoderm
We processed a table obtained from the authors of the original publication15 with the differentiation efficiency for 108 lines, of which 86 also had WES data for the parental fibroblasts. Here, differentiation efficiency is computed as the average pseudotime on day 3, having a continuous distribution of efficiencies rather than a binary outcome (“failed”, “successful”).
Gene burden differences upon differentiation outcome
We leveraged the 832 iPSC lines (from 547 donors) profiled with WES available from the HipSci project and annotated the most severe consequence for each variant using the variant effect predictor49 (VEP, release 99) and the Ensembl gene annotation from the release (version 75). We summarise the results by building a matrix of mutation counts per variant category either for LoF or deleterious mutations representing gene damage categories, or synonymous variants as mutational burden control. The inclusion criteria in each of the variant categories was the same as provided with the annotation of somatic acquired mutations in vivo (see Functional annotation of SMAV). Each matrix contained the 19,653 genes (of the gene universe) as rows and the 832 lines as columns.
We then combined the mutational burden data per line for each variant category (LoF, deleterious and synonymous) with the corresponding binary differentiation outcome (DA predicted (N=793 lines from 529 donors), DA observed (N=183 lines, one line per donor), macrophages (N=118 lines, one line per donor)). We excluded the sensory neurons from the analysis due to the low number of failed lines in that dataset (N=3). For each variant category and outcome combination, we performed a Wilcoxon Rank Sum Test per gene (N=19,653 tests) to identify those that presented a differential burden between failed and successful lines. For each combination, we performed a multiple test correction using the Benjamini & Hochberg approach. The level of statistical significance was set at FDR=5%. To compute a fold change of the mutational differences per gene between failed and successful lines, we initially normalised the mutation ratio by the gene length (as in Ensembl Annotation release 87) and the number of lines per outcome and divided the ratios using pseudocounts. The pseudocounts used for each combination corresponded to the minimum non-zero normalised mutation rate observed across all genes for any of the outcomes. We used a threshold of FC>2.5 or FC<1/2.5 to classify genes as disproportionately mutated in failed lines or in successful lines, respectively. Alternatively in the case of the DA dataset, we also combined the burden data with the continuous distribution of differentiation efficiencies (N=183 lines) and predicted scores (N=793 lines) to check the robustness of the BCOR association. In this case, we performed a Pearson’s correlation per gene for each combination.
GO enrichment analysis (mutational burden)
For the gene ontology enrichment analysis on biological processes, we only focused on the deleterious burden of iPSC lines. Given the number of lines for the DA differentiation (183 lines for the observed outcomes, one line per donor: 48 failed vs 135 successful; and 793 lines for the predicted ones from 529 donors: 99 failed vs 694 successful), it is the variant category linked to gene damage with the best power to detect enrichment. We proceeded with the matrix of counts previously generated for the identification of gene burden differences linked to the differentiation outcome. Likewise, we computed the fold change of mutational differences per gene between failed and successful lines using the ratio of the mutation burden (mutations per Kb) normalised by gene length, number of lines per outcome and adding 0.001 as pseudocounts. Prior to the GO analysis, we annotated all genes with their corresponding Entrez gene identifiers and selected the most differentially mutated genes in successful lines (top-10% in log2FC) and the most differentially mutated in failed lines (bottom-10% in log2FC). We then run the hypergeometric test for GO term overrepresentation of biological processes conditional to the hierarchical GO structure (package GOstats53 from R). For each of the four tests, we provided the selected genes in each DA outcome combination: failed/observed, failed/predicted, successful/observed and successful/predicted. We used the following thresholds: the cutoff for significance was set at p<0.05, we considered only those gene sets defined with more than 20 genes and each gene set had to account for at least 10 counts in each analysis. All gene sets found to be significantly enriched are shown in Table S3. Finally, we highlighted only those significant GO terms related to neurodevelopment or chromatin modification, so we highlighted any gene set with the following words in Figure 2D: “Axon”, “neuron”, “glial”, “brain”, “hindbrain”, “forebrain”, “midbrain”, “synapse”, “chromatin”, “cerebellum”, “neural”, “cortex”, “neurogenesis”, “axonogenesis”, “nervous”, “hippocampus”, “neurotransmitter”, “dopaminergic”, “axenome”, “action potential” and “synaptic”.
Reanalysis of pooled single-cell data (DA)
Sample selection and data pre-processing
The dopaminergic neuron differentiation was profiled by droplet-based scRNA-seq (10x Genomics). We processed a subset of the dopaminergic neuron differentiation dataset8 that consisted of 119 10x samples out of the total 166 (Table S5). Here, a 10x sample is defined as the cells sequenced from one inlet of a 10x chip. For the sake of this study, we did not include those samples from the original experiment profiled under rotenone treatment at day 52 or containing iPSC-derived cerebral organoids (day 119). We also did not process samples for pool 10 (day 11) with reported problems on library preparation. We processed the 119 10x samples using CellRanger software (version 3.1.0) and aligned them to the GRCh37/hg19 reference genome. Gene counts were quantified by the “count” option of the software, using the Ensembl 87 reference gene annotation (N=32,738 genes). After pre-processing, we excluded 4 additional 10x samples due to quality control issues, mainly due to low percentages of cell singletons in deconvolution (≤50%) and low cell viability: two technical replicates from pool 12 on day 52, one sample from pool 8 on day 30 and a sample from pool 1 on day 30 (Figure S2B). The final 115 10x samples covered all pooled experiments (N=19, pools 1-17 and pools 20-21), including 238 different cell lines (230 donors, 7-24 lines per pool). Only one cell line (“HPSI0913i-gedo_33”) was previously removed due to an abnormally high cell line proportion (>90%) in pool 14.
Quality control and deconvolution of cell donor identity
Each 10x sample went through a quality control step in which we removed dying cells or those with broken membranes, displaying a low number of genes per cell (<200) and an excess of mitochondrial count fraction (>5%). Also, we discarded those cells with an abnormal percentage of reads consumed by the top-100 mostly expressed genes (<75%), which indicate technical artefacts compromising the coverage of the full transcriptome of the cells. On the other hand, we filtered out those genes that were not expressed in at least 0.1% of the total cells.
For each of the 19 pools, we performed cell deconvolution using demuxlet51 using existing genetic variation (genotypes of common biallelic exonic variants, MAF>5%) available from the HipSci Project7 as in the original paper. In those cases when iPSC cell lines had not been genotyped (intended cell lines from Table S4), we used the genotypes from the primary fibroblast instead when available. Demuxlet was run using a default prior doublet rate of 0.5. We only retained those (singletons) cells that could unambiguously be linked to a donor and discarded those 10x samples for which the overall singleton outcome was low (<50%) (Figure S2B). After deconvolution, 236 (n=228 donors) out of the 238 lines of the experiment had at least one cell detected. We performed the quality control and the integration of the 10x samples using the Scanpy Python-based toolkit59 (version 1.4.5.1).
Normalisation, dimensionality reduction, batch correction and clustering
We performed a combined analysis of all the three time points (day 11, day 30 and day 52) to have a shared embedding for all 236 lines. Initially, genes that were not expressed in at least 0.1% of total cells were removed. Then, gene counts were normalised to the total number of counts per cell and log-transformed (log1p). After adjusting for mean-variance dependence, we selected the 2,928 highly variable genes and scaled gene counts to unit variance and zero mean. We then calculated the first 50 principal components (PCs) and batch-corrected them with Harmony,54 treating each 10x sample as a different batch (parameters: theta=2, max.iter.harmony=25, max.iter.cluster=500). We then used the batch-transformed PCs to compute a neighbourhood graph (n_neighbors=10), visualise it using UMAP and perform the cell clustering using the Leiden algorithm (resolutioN=0.3) identifying 12 different clusters (Figures S2H–S2J). We also used the Scanpy toolkit (version 1.4.5.1) for all the steps, except for Harmony that was run in R version 4.0.3.
Cell type annotation
Cell type annotation was performed using the same set of literature-curated markers as in Jerber et al.8 (Figures S2E–S2G and Table S6). We confidently annotated 10 out of the 12 identified clusters. Interestingly, we could also identify neuroblasts at day 11 with a commitment to dopaminergic neurons, as they clustered together, but at the same time did not exhibit the neuron marker expression.
At day 11, we characterised four populations of floor-plate midbrain progenitors, either proliferating (proFPP-1, proFPP-2) or non-proliferating (FPP-1, FPP-3). We could also link the population of neuroblasts to their early dopaminergic or serotonergic commitment. At day 30 and 52, we identified six additional cell types, including another cell type of non-proliferating progenitors (FPP-2), two neuronal populations (dopaminergic-like (DA) and serotonergic-like (Sert-like) neurons) and three non-neuronal ones (astrocytes (Astro), ependymal-like cells (Epend-1) and an unknown population (Unk-1) potentially linked to Cajal-Retzius transient neurons. At day 30, two additional rare cell types (<2%) were identified, either belonging to a subgroup of Sert-like neurons associated with proliferation markers (pro.Sert-like), or to an unknown population (Unk-2) only detected in a single 10x sample of pool 12.
Pool reproducibility of cell line abundance
We started processing the metadata object containing annotations from all cells (available online, see Data and code availability). To define different types of replicates, we used the information provided by the metadata of the 115 10x samples (Table S5). Four replicate types were defined per cell line (Figure S3C).
-
-
Pool replicates (41 cell lines, N=90 replicate comparisons): The same cell line was placed in different pooled experiments (different cell lines in the background).
-
-
Biological replicates (31 cell lines, N=31 replicate comparisons): One cell line underwent independent differentiations (different time and plate), but within the same pool (same background).
-
-
Technical replicates (200 cell lines, N=644 replicate comparisons): One cell line underwent differentiation within the same pool, same time, but different wells of the same plate.
-
-
10x replicates (109 cell lines, N=699 replicate comparisons): One cell line underwent differentiation within the same pool, same time and same well of the plate.
Initially, we calculated the cell line proportion within each 10X sample. Then, for each cell line, the corresponding replicate group and a given time-point, we calculated the averaged cell line proportion per replicate taking into account the contributing 10X samples. For instance, to compare the cell line proportion of the two biological replicates for “HPSI1014i-tuju_1” at day 11, we averaged the cell line proportion of the four 10X samples contributing to replicate 1 on one side, and the four 10x samples contributing to replicate 2, on the other. To evaluate the reproducibility between replicate proportions, we fitted a linear regression and computed the adjusted R-squared and the p-value of the association. Data points on Figure 3C correspond to matched replicates per line/time-point combination. Note here that the designation of replicate 1 or replicate 2 before the regression is random.
Cell line proliferation in DA differentiation
The cell suspension for each pool was prepared with an equal amount of each iPSC line.8 For this analysis, we only considered those cell lines within a given pool that had been sampled in the three time points of the differentiation (N=164 cell lines used in 187 pool/line combinations, one cell line per donor). For each pool and time-point, we computed the log-transformed (log1p) cell line proportion. Then, we calculated the proliferation rate at day 11, day 30 and day 52, dividing the cell line proportions observed at each respective time point by the equal proportions from day 0. We then annotated each cell line with the observed outcome in the DA dataset, either successful or failed, based on the neuron differentiation efficiency threshold of 0.2. Additionally, we annotated failed cell lines with either BCOR+ (N=20) or BCOR- (N=30) based on the presence of LOF BCOR variants in each line. Note here that none of the successful lines (N=114) carried any LoF mutation in our iPSC exome-sequencing data.
Annotation of cancer-driver mutations
For each of the 832 iPSC cell lines with exome sequencing data, we annotated the most severe consequence for each variant using the variant effect predictor (VEP, release 99) as described earlier (see Functional annotation of SMAV). We then overlapped the predicted LoF variants from each line with those listed in the database for the cancer-associated genes (Cosmic Tier 1, version 94) under the strongest evidence for oncogenic activity (Tier 1). We restricted the overlap search to those Cosmic Tier 1 variants that have a defined genomic position and a FATHMM11 score >= 0.7. We identified 726 potential driver mutations with 606 iPSC lines carrying at least one driver mutation. The most mutated driver genes were PDE4DIP (564), CCND3 (35), TCF3 (29) and the BCOR (24), also after normalising the burden by the CDS length. We then annotated each cell line with both the observed (183 lines, 48 failed and 135 successful) and predicted DA differentiation (793 lines from 529 donors: 99 failed and 694 successful) and computed the driver mutational ratio (log2-transformed) given the outcome. In both cases, BCOR ranked as the gene with the highest ratio of driver mutations in failed lines versus successful ones (5:0 with the observed DA outcome, 17:6 with the predicted DA outcome).
Differential abundance analysis
After annotating the cell type, we used the metadata information for each cell to compute the cell type proportions per line (available online, see Data and code availability). We used the outcome annotation for the 206 lines classified either as failed or successful, as described for dopaminergic neurons. For 8 pool replicates missing day 52 time-point in one of the pools, we imputed the same outcome as observed in the other replicate. We also included 15 out of the 18 lines that were not profiled at day 52, but with data from previous time-points (either from day 11 or 30, or from both). To take advantage of those lines in the cell type composition analysis, we classified them either as successful or failed using the predicted model scores from Jerber et al.8 Following those steps, we end up annotating the differentiation outcome for 221 lines (58 failed, 163 successful) from 214 donors.
For each time-point and cell type, we used a negative binomial regression model to evaluate the composition changes between failed and successful lines. We modelled the total number of cells per line as an offset variable, given that the accuracy of cell type proportion estimates increases with their magnitude.
We finally performed a multiple-test correction (N=36 tests) using the Benjamini & Hochberg approach (FDR<5%). The significance level of the mean cell type proportion difference between mutated and unmutated groups is indicated by different levels: pAdj<0.05 (∗), pAdj<0.01 (∗∗), pAdj<0.001 (∗∗∗).
Impact of deleterious burden in BCOR
We considered 141 iPSC lines (one line per donor) with these available information layers: cell type proportion estimates on day 52 (see Differentiation outcomes of iPSC-derived cell types), in vitro proliferation rates (see Cell line proliferation in DA differentiation) and deleterious burden (see Gene burden differences upon the differentiation outcome). For each gene (N=19,653), we compared the cell type composition differences at the end of the differentiation between those lines carrying at least one deleterious mutation and those lines unmutated (Wilcoxon Rank Sum Test). For each cell type, we applied multiple-test correction (Benjamini & Hochberg, FDR<5%) to the raw p-values obtained from each gene and indicated whether they are more abundant in mutated (blue) or unmutated lines (orange). Note here that only those cell types that show >2% of abundance at day 52 were considered, discarding FPP-1, FPP-3, proliferative serotonergic-like neurons, proliferative FPP-2 and the unknown cell type 2.
Alternatively, for each major cell type at day 52 (>2% abundance), we tested for the association between cell type proportions per line and their corresponding proliferation rates between day 52 and day 0, using Pearson’s correlation (N=154 lines, one line per donor). We corrected for multiple-test correction using the Benjamini & Hochberg method (N=7 major cell types, FDR<5%). We also indicated if the cell type proportions correlate (blue) or anti-correlate (orange) in Figure 4B. The significance level of all comparisons was indicated as follows: pAdj<0.05 (∗), pAdj<0.01 (∗∗), pAdj<0.001 (∗∗∗).
DE analysis between failed and successful lines
We leveraged the gene expression data from 221 lines (214 donors) annotated with the DA outcome (58 failed, 163 successful), see Differential abundance analysis. Overall, we processed 273,804, 266,226 and 306,811 cells from day 11, day 30 and day 52, respectively. For each time-point, we load the “AnnData/H5AD” object with the unscaled log-transformed gene expression per cell (available online, see Data and code availability) and filtered out those genes expressed in less than 1% of the cells, finally processing 12,912, 14,149 and 14,737 genes, respectively. We performed differential gene expression analysis between failed and successful lines for each cell type and time point combination using the Wilcoxon Rank Sum test (as implemented in Seurat52). We required more than 10 cells to be represented in each of the outcomes to run the DE test for each combination. DE genes were selected based on an adjusted P-value < 0.05 and a FC >1.5.
For each cell type and time point, we also tested the overrepresentation of three gene sets (Cosmic-Tier1, DDD and a subset of dominant DDD genes) among the list of DE genes (Chi-squared test using p-values computed by Monte Carlo simulation using 100,000 replicates). The employed gene universe consists of the union of pass-filtered genes across all time points (N=15,367). We corrected for multiple-test correction using the Benjamini & Hochberg method (N=102, FDR<5%). The significance level of each test was indicated as follows: pAdj<0.05 (∗), pAdj<0.01 (∗∗), pAdj<0.001 (∗∗∗).
GO enrichment analysis (gene expression)
We performed 12 gene ontology enrichment tests on biological processes based on.
-
-
The union of genes found to be differentially expressed in any given time point and cell type combination (labeled allDE).
-
-
The union of genes found to be differentially expressed in any given time point and cell type combination with an overrepresentation of DDD DE genes (labeled signifDDD).
-
-
The lists of DE genes for each of the 10 time point and cell type combinations with an overrepresentation of DDD DE genes (cyan circles with p<0.05, Figure 4C).
Previous to the GO analysis, we annotated each feature of the gene universe (N=15,367) with their corresponding Entrez gene identifiers using the package org.Hs.eg.db from R/Bioconductor. We discarded from the analysis those genes without correspondence or showing duplicate identifiers. We then run the hypergeometric test for GO term overrepresentation of biological processes conditional to the hierarchical GO structure (package GOstats53 from R). Given the different magnitude of DE genes between allDE (N=1,884) or signifDDD (N=972) and each of the combination tests (N=131-372), we used different thresholds for significance in each case: allDE/signifDDD: {minimum gene set size = 30, maximum gene set size=200, pAdj<0.001, minimum number of counts per gene set = 20}; combinations: {minimum gene set size = 10, maximum gene set size=200, pAdj<0.001, minimum number of counts per gene set = 7}.
All gene sets found to be significantly enriched are shown in Table S7. Finally, we highlighted only those significant GO terms related to neurodevelopment or chromatin modification, so we highlighted any gene set with the following words in Figure 4D: “Axon”, “neuron”, “glial”, “brain”, “hindbrain”, “forebrain”, “midbrain”, “synapse”, “chromatin”, “cerebellum”, “neural”, “cortex”, “neurogenesis”, “axonogenesis”, “nervous”, “hippocampus”, “neurotransmitter”, “dopaminergic”, “axenome”, “action potential” and “synaptic”.
Gene set enrichment analysis (cancer genes)
We performed a gene set enrichment analysis (GSEA) on those time point and cell type combinations in which the list of differentially expressed genes were enriched in cancer-associated genes. For this purpose, we used the curated MSigDB hallmark gene set signatures (version 7.4 for symbol identifiers).55 We considered only those gene sets with a larger size than 10 genes. For each gene set, we then ran the preranked gene set enrichment analysis with a maximum gene set size of 500 genes, an eps parameter of 0 and used 10,000 permutations for preliminary estimation of p-values. We highlighted significantly enriched gene sets as those with a BH-adjusted p-value < 0.05 and an enrichment score normalised to mean enrichment of random samples of the same size (NES): NES≥ 1.5 for upregulated pathways and NES ≤ -1.5 for downregulated ones.
Outlier lines in cell type composition
We calculated the cell type fraction per line within each pool and time point, removing those lines with the lowest number of cells (first twentile, 227 lines from 219 donors). Those cell lines pooled in more than one experiment (pool replicates) were treated as independent lines (N=272 combinations). We then computed the z-score associated with the calculated fractions and marked as outliers those lines showing a cell type fraction with a |Z-score|>2, either showing a deficiency or an excess of a given cell type.
We characterised the outlier behaviour focusing on those cell lines represented in the three time points of the differentiation (112 lines, 121 pool-line combinations). Based on that, we computed the number of times each line shows an abnormal cell type fraction (outlier event) throughout the entire differentiation or specifically per time point (bars represent 95% confidence interval in Figure 5B, top-left). For each cell line, we explored when outlier events occurred and identified the most common time point combinations.
For those lines profiled with WES for the iPSC and the corresponding parental fibroblasts, we annotated the burden of somatic acquired mutations in vivo (Table S1) considering either total or deleterious variants. We then evaluated whether cell lines defined as outliers of cell type composition (91 lines, 103 pool-line combinations) showed mean differences on the mutational burden (Wilcoxon Rank Sum Test) to the non-outliers (N=57 lines, 60 pool-line combinations). Alternatively, we also evaluated the outlier lines within each specific time point.
Finally, we also annotated each cell line with their corresponding proliferation rate at day 52 (see Cell line proliferation in DA differentiation) and fitted a logistic regression to predict the outlierness of cell type composition (N=159 lines, 182 pool-line combinations).
Gene expression and cell type abundance links
We analysed the correlation between the cell type abundance and the cell-type-specific expression for all genes using the existing cell line variability throughout the differentiation (N=236 lines from 228 donors). We computed the z-scores for cell type composition as in Outlier lines in cell type composition. As for the gene expression (log1p normalised counts, not-scaled), we computed the z-score per cell type using the average gene expression per line at each time point. The average gene expression per line was calculated considering all the cells of that given line in one specific pool experiment (N=281 combinations), including those from different 10x samples when available. We required a minimum of 10 cells per line (in any time point - cell type combination) to calculate the average gene expression. We discarded all those combinations in which less than 10 lines matched this threshold (proliferative Sert-like neurons at day 11 and the glial cells (Unk-2) at days 11 and 52).
We then correlated the expression z-scores with the cell type fraction z-scores, as shown in the example for KMT2D gene for DA and FPP-1 in day 11 (Figure 5D). To identify the key genes driving the outliereness in cell type composition, we performed the z-score correlations for all the detected genes per time point (day 11, N=12,912; day30, N=14,149; day 52, N=14,737). Those genes with no detectable expression in at least 10 lines of a given combination were not considered. We then sampled the genes with either positive or negative significant associations (p.Adj<0.05) from the resulting distribution of Pearson correlation coefficients per cell type (Figure 5D, lower).
From the list of genes with significant correlation (or anti-correlation) per cell type and time point, we tested whether there was a gene set enrichment on several panels of genetic disease associations: developmental disorder genes34 (DDD), cancer-associated genes35 (Cosmic-Tier1), schizophrenia37 (Open Targets Platform), and three panels from Genomics England:36 adult onset neurodegenerative disorder (Panel App v2.178), bleeding and platelet disorders (v1.2) and family pulmonary fibrosis (v1.29). We performed an hypergeometric test for overrepresentation considering the list of significantly and non-significantly correlated genes and the overlap with each gene set and cell type separately. We then performed multiple-test correction using the Benjamini & Hochberg method (N=198 tests, FDR<5%). The significance level of each test was indicated as follows: pAdj>0.05 (ns), pAdj<0.05 (∗), pAdj<0.01 (∗∗), pAdj<0.001 (∗∗∗).
Acknowledgments
This work was funded by the UK Medical Research Council (MR/L016311/1; MRC eMedLab Medical Bioinformatics career development award to H.K.), Helsinki Institute of Life Science (H.K.), the ICH CIO (S.C. and P.P.) and the NIHR GOSH BRC (S.C., P.P., and H.K.). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health. The authors acknowledge Zenodo, the data repository developed under the European Open Science Infrastructure (OpenAIRE) to facilitate the open access to our metadata and normalized gene count matrices. The authors gratefully acknowledge Drs. Daniel Gaffney, Oliver Stegle, and Florian Merkle for early access to the dopaminergic neuron single-cell dataset, and Drs. Daniel Seaton and Anna Cuomo for assistance with the DA data. The authors also acknowledge Drs. Matthew Hurles, Sebastian Gerety, and Osama Arshad for their feedback on the project. Illustration in Figures 1 and S4C and the graphical abstract were created with BioRender.com.
Author contributions
The main analysis (single-cell data processing, data preparation, and data analysis) was performed by P.P. under the supervision of S.C. and H.K. J.J. performed the cell type annotation and provided guidance with the data preparation. P.D. produced the mutation callset for the somatic acquired variation. P.P., H.K., and S.C. wrote and edited the manuscript. P.P. and H.K. conceived the study.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: March 23, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100280.
Contributor Information
Pau Puigdevall, Email: pau.puigdevall@helsinki.fi.
Helena Kilpinen, Email: helena.kilpinen@helsinki.fi.
Supplemental information
Data and code availability
Exome sequencing from the HipSci project (www.hipsci.org) is available for both the parental fibroblasts and the iPSC lines (raw data). Data for the open access samples is deposited in the European Nucleotide Archive under study accession number (ENA: ERP006946). It includes 325 iPSC lines, of which 260 are also available for the parental fibroblasts. WES data for the managed access samples is deposited in the European Genotype-Phenotype Archive (EGA, https://ega-archive.org), with normal and specific disease cohort datasets available upon request and data access agreement. The variant call sets of acquired mutations in vitro and the code used to generate them are available at Rouhani et al.11
As for the single-cell data from the iPSC dopaminergic differentiation, managed access data is available in the European Genome–phenome Archive (EGA: EGAS00001002885), as part of the EGAD00001006157 dataset. Open access samples are available in the European Nucleotide Archive (ENA: ERP121676) from the project PREJB38269. Metadata information for 828,937 processed cells from the DA dataset and AnnData/H5AD files containing the single-cell expression for days 11, 30 and 52 that are related to the STAR Methods are publicly available at Zenodo, https://zenodo.org/record/6079122#.YlSkxNPMJhE. Chip genotypes for HipSci lines are available from the EGA (EGAS00001000866, EGAS00001001272) and the ENA (PRJEB11750) portals.
All original code is publicly available in a frozen Zenodo repository (https://doi.org/10.5281/zenodo.7641259). The repository includes the single-cell processing scripts, the downstream analysis, and the code to reproduce the main figures and the supplemental ones.
References
- 1.Rowe R.G., Daley G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019;20:377–388. doi: 10.1038/s41576-019-0100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flippe L., Gaignerie A., Sérazin C., Baron O., Saulquin X., Themeli M., Guillonneau C., David L. Rapid and reproducible differentiation of hematopoietic and T cell progenitors from pluripotent stem cells. Front. Cell Dev. Biol. 2020;8:577464. doi: 10.3389/fcell.2020.577464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen K.G., Mallon B.S., McKay R.D.G., Robey P.G. Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell. 2014;14:13–26. doi: 10.1016/j.stem.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koehler K.R., Tropel P., Theile J.W., Kondo T., Cummins T.R., Viville S., Hashino E. Extended passaging increases the efficiency of neural differentiation from induced pluripotent stem cells. BMC Neurosci. 2011;12:82. doi: 10.1186/1471-2202-12-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cacchiarelli D., Qiu X., Srivatsan S., Manfredi A., Ziller M., Overbey E., Grimaldi A., Grimsby J., Pokharel P., Livak K.J., et al. Aligning single-cell developmental and reprogramming trajectories identifies molecular determinants of myogenic reprogramming outcome. Cell Syst. 2018;7:258–268.e3. doi: 10.1016/j.cels.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Volpato V., Smith J., Sandor C., Ried J.S., Baud A., Handel A., Newey S.E., Wessely F., Attar M., Whiteley E., et al. Reproducibility of molecular phenotypes after long-term differentiation to human iPSC-derived neurons: a multi-site omics study. Stem Cell Rep. 2018;11:897–911. doi: 10.1016/j.stemcr.2018.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kilpinen H., Goncalves A., Leha A., Afzal V., Alasoo K., Ashford S., Bala S., Bensaddek D., Casale F.P., Culley O.J., et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature. 2017;546:370–375. doi: 10.1038/nature22403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jerber J., Seaton D.D., Cuomo A.S.E., Kumasaka N., Haldane J., Steer J., Patel M., Pearce D., Andersson M., Bonder M.J., et al. Population-scale single-cell RNA-seq profiling across dopaminergic neuron differentiation. Nat. Genet. 2021;53:304–312. doi: 10.1038/s41588-021-00801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D’Antonio M., Benaglio P., Jakubosky D., Greenwald W.W., Matsui H., Donovan M.K.R., Li H., Smith E.N., D’Antonio-Chronowska A., Frazer K.A. Insights into the mutational burden of human induced pluripotent stem cells from an integrative multi-omics approach. Cell Rep. 2018;24:883–894. doi: 10.1016/j.celrep.2018.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merkle F.T., Ghosh S., Kamitaki N., Mitchell J., Avior Y., Mello C., Kashin S., Mekhoubad S., Ilic D., Charlton M., et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature. 2017;545:229–233. doi: 10.1038/nature22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rouhani F.J., Zou X., Danecek P., Badja C., Amarante T.D., Koh G., Wu Q., Memari Y., Durbin R., Martincorena I., et al. Substantial somatic genomic variation and selection for BCOR mutations in human induced pluripotent stem cells. Nat. Genet. 2022;54:1406–1416. doi: 10.1038/s41588-022-01147-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Volpato V., Webber C. Addressing variability in iPSC-derived models of human disease: guidelines to promote reproducibility. Dis. Model. Mech. 2020;13:dmm042317. doi: 10.1242/dmm.042317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alasoo K., Rodrigues J., Mukhopadhyay S., Knights A.J., Mann A.L., Kundu K., HIPSCI Consortium. Hale C., Dougan G., Gaffney D.J. Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat. Genet. 2018;50:424–431. doi: 10.1038/s41588-018-0046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartzentruber J., Foskolou S., Kilpinen H., Rodrigues J., Alasoo K., Knights A.J., Patel M., Goncalves A., Ferreira R., Benn C.L., et al. Molecular and functional variation in iPSC-derived sensory neurons. Nat. Genet. 2018;50:54–61. doi: 10.1038/s41588-017-0005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuomo A.S.E., Seaton D.D., McCarthy D.J., Martinez I., Bonder M.J., Garcia-Bernardo J., Amatya S., Madrigal P., Isaacson A., Buettner F., et al. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat. Commun. 2020;11:810. doi: 10.1038/s41467-020-14457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yizhak K., Aguet F., Kim J., Hess J.M., Kübler K., Grimsby J., Frazer R., Zhang H., Haradhvala N.J., Rosebrock D., et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 2019;364:eaaw0726. doi: 10.1126/science.aaw0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W., et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei P.-C., Chang A.N., Kao J., Du Z., Meyers R.M., Alt F.W., Schwer B. Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell. 2016;164:644–655. doi: 10.1016/j.cell.2015.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coufal N.G., Garcia-Perez J.L., Peng G.E., Yeo G.W., Mu Y., Lovci M.T., Morell M., O’Shea K.S., Moran J.V., Gage F.H. L1 retrotransposition in human neural progenitor cells. Nature. 2009;460:1127–1131. doi: 10.1038/nature08248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lodato M.A., Woodworth M.B., Lee S., Evrony G.D., Mehta B.K., Karger A., Lee S., Chittenden T.W., D’Gama A.M., Cai X., et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 2015;350:94–98. doi: 10.1126/science.aab1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reid D.A., Reed P.J., Schlachetzki J.C.M., Nitulescu I.I., Chou G., Tsui E.C., Jones J.R., Chandran S., Lu A.T., McClain C.A., et al. Incorporation of a nucleoside analog maps genome repair sites in postmitotic human neurons. Science. 2021;372:91–94. doi: 10.1126/science.abb9032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodin R.E., Dou Y., Kwon M., Sherman M.A., D’Gama A.M., Doan R.N., Rento L.M., Girskis K.M., Bohrson C.L., Kim S.N., et al. The landscape of somatic mutation in cerebral cortex of autistic and neurotypical individuals revealed by ultra-deep whole-genome sequencing. Nat. Neurosci. 2021;24:176–185. doi: 10.1038/s41593-020-00765-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wamstad J.A., Corcoran C.M., Keating A.M., Bardwell V.J. Role of the transcriptional corepressor Bcor in embryonic stem cell differentiation and early embryonic development. PLoS One. 2008;3:e2814. doi: 10.1371/journal.pone.0002814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Astolfi A., Fiore M., Melchionda F., Indio V., Bertuccio S.N., Pession A. BCOR involvement in cancer. Epigenomics. 2019;11:835–855. doi: 10.2217/epi-2018-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ragge N., Isidor B., Bitoun P., Odent S., Giurgea I., Cogné B., Deb W., Vincent M., Le Gall J., Morton J., et al. Expanding the phenotype of the X-linked BCOR microphthalmia syndromes. Hum. Genet. 2019;138:1051–1069. doi: 10.1007/s00439-018-1896-x. [DOI] [PubMed] [Google Scholar]
- 26.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. Author Correction: the mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2021;590:E53. doi: 10.1038/s41586-020-03174-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stark Z., Foulger R.E., Williams E., Thompson B.A., Patel C., Lunke S., Snow C., Leong I.U.S., Puzriakova A., Daugherty L.C., et al. Scaling national and international improvement in virtual gene panel curation via a collaborative approach to discordance resolution. Am. J. Hum. Genet. 2021;108:1551–1557. doi: 10.1016/j.ajhg.2021.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinelli S., Krumbach O.H.F., Pantaleoni F., Coppola S., Amin E., Pannone L., Nouri K., Farina L., Dvorsky R., Lepri F., et al. Functional dysregulation of CDC42 causes diverse developmental phenotypes. Am. J. Hum. Genet. 2018;102:309–320. doi: 10.1016/j.ajhg.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reis L.M., Tyler R.C., Schilter K.F., Abdul-Rahman O., Innis J.W., Kozel B.A., Schneider A.S., Bardakjian T.M., Lose E.J., Martin D.M., et al. BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum. Genet. 2011;130:495–504. doi: 10.1007/s00439-011-0968-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Paassen B.W., van der Kooi A.J., van Spaendonck-Zwarts K.Y., Verhamme C., Baas F., de Visser M. PMP22 related neuropathies: charcot-Marie-Tooth disease type 1A and Hereditary Neuropathy with liability to Pressure Palsies. Orphanet J. Rare Dis. 2014;9:38. doi: 10.1186/1750-1172-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carosso G.A., Boukas L., Augustin J.J., Nguyen H.N., Winer B.L., Cannon G.H., Robertson J.D., Zhang L., Hansen K.D., Goff L.A., Bjornsson H.T. Precocious neuronal differentiation and disrupted oxygen responses in Kabuki syndrome. JCI Insight. 2019;4:e129375. doi: 10.1172/jci.insight.129375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Räsänen N., Tiihonen J., Koskuvi M., Lehtonen Š., Koistinaho J. The iPSC perspective on schizophrenia. Trends Neurosci. 2022;45:8–26. doi: 10.1016/j.tins.2021.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Paulsen B., Velasco S., Kedaigle A.J., Pigoni M., Quadrato G., Deo A.J., Adiconis X., Uzquiano A., Sartore R., Yang S.M., et al. Autism genes converge on asynchronous development of shared neuron classes. Nature. 2022;602:268–273. doi: 10.1038/s41586-021-04358-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wright C.F., Fitzgerald T.W., Jones W.D., Clayton S., McRae J.F., van Kogelenberg M., King D.A., Ambridge K., Barrett D.M., Bayzetinova T., et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385:1305–1314. doi: 10.1016/S0140-6736(14)61705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E., et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin A.R., Williams E., Foulger R.E., Leigh S., Daugherty L.C., Niblock O., Leong I.U.S., Smith K.R., Gerasimenko O., Haraldsdottir E., et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 2019;51:1560–1565. doi: 10.1038/s41588-019-0528-2. [DOI] [PubMed] [Google Scholar]
- 37.Ochoa D., Hercules A., Carmona M., Suveges D., Gonzalez-Uriarte A., Malangone C., Miranda A., Fumis L., Carvalho-Silva D., Spitzer M., et al. Open Targets Platform: supporting systematic drug–target identification and prioritisation. Nucleic Acids Res. 2021;49:D1302–D1310. doi: 10.1093/nar/gkaa1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merkle F.T., Ghosh S., Genovese G., Handsaker R.E., Kashin S., Karczewski K., O’Dushlaine C., Pato C., Pato M., MacArthur D.G., et al. Biological insights from the whole genome analysis of human embryonic stem cells. bioRxiv. 2020 doi: 10.1101/2020.10.26.337352. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amir H., Touboul T., Sabatini K., Chhabra D., Garitaonandia I., Loring J.F., Morey R., Laurent L.C. Spontaneous single-copy loss of TP53 in human embryonic stem cells markedly increases cell proliferation and survival. Stem Cell. 2017;35:872–885. doi: 10.1002/stem.2550. [DOI] [PubMed] [Google Scholar]
- 40.Rouhani F., Kumasaka N., de Brito M.C., Bradley A., Vallier L., Gaffney D. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 2014;10:e1004432. doi: 10.1371/journal.pgen.1004432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merkle F.T., Ghosh S., Genovese G., Handsaker R.E., Kashin S., Meyer D., Karczewski K.J., O’Dushlaine C., Pato C., Pato M., et al. Whole-genome analysis of human embryonic stem cells enables rational line selection based on genetic variation. Cell Stem Cell. 2022;29:472–486.e7. doi: 10.1016/j.stem.2022.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deciphering Developmental Disorders Study Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–438. doi: 10.1038/nature21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh T., Poterba T., Curtis D., Akil H., Al Eissa M., Barchas J.D., Bass N., Bigdeli T.B., Breen G., Bromet E.J., et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature. 2022;604:509–516. doi: 10.1038/s41586-022-04556-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gudmundsson S., Singer-Berk M., Watts N.A., Phu W., Goodrich J.K., Solomonson M., Genome Aggregation Database Consortium. Rehm H.L., MacArthur D.G., ODonnell-Luria A. Variant interpretation using population databases: lessons from gnomAD. arXiv. 2021 doi: 10.48550/arXiv.2107.11458. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruijtenberg S., van den Heuvel S. Coordinating cell proliferation and differentiation: antagonism between cell cycle regulators and cell type-specific gene expression. Cell Cycle. 2016;15:196–212. doi: 10.1080/15384101.2015.1120925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu L., Michowski W., Kolodziejczyk A., Sicinski P. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 2019;21:1060–1067. doi: 10.1038/s41556-019-0384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitchell J.M., Nemesh J., Ghosh S., Handsaker R.E. Mapping genetic effects on cellular phenotypes with “cell villages”. bioRxiv. 2020 doi: 10.1101/2020.06.29.174383. Preprint at. [DOI] [Google Scholar]
- 48.Eichmüller O.L., Corsini N.S., Vértesy Á., Morassut I., Scholl T., Gruber V.-E., Peer A.M., Chu J., Novatchkova M., Hainfellner J.A., et al. Amplification of human interneuron progenitors promotes brain tumors and neurological defects. Science. 2022;375:eabf5546. doi: 10.1126/science.abf5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The Ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Danecek P., McCarthy S.A. BCFtools/csq: haplotype-aware variant consequences. Bioinformatics. 2017;33:2037–2039. doi: 10.1093/bioinformatics/btx100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kang H.M., Subramaniam M., Targ S., Nguyen M., Maliskova L., McCarthy E., Wan E., Wong S., Byrnes L., Lanata C.M., et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018;36:89–94. doi: 10.1038/nbt.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Falcon S., Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- 54.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.-R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.1000 Genomes Project Consortium. Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rentzsch P., Witten D., Cooper G.M., Shendure J., Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Exome sequencing from the HipSci project (www.hipsci.org) is available for both the parental fibroblasts and the iPSC lines (raw data). Data for the open access samples is deposited in the European Nucleotide Archive under study accession number (ENA: ERP006946). It includes 325 iPSC lines, of which 260 are also available for the parental fibroblasts. WES data for the managed access samples is deposited in the European Genotype-Phenotype Archive (EGA, https://ega-archive.org), with normal and specific disease cohort datasets available upon request and data access agreement. The variant call sets of acquired mutations in vitro and the code used to generate them are available at Rouhani et al.11
As for the single-cell data from the iPSC dopaminergic differentiation, managed access data is available in the European Genome–phenome Archive (EGA: EGAS00001002885), as part of the EGAD00001006157 dataset. Open access samples are available in the European Nucleotide Archive (ENA: ERP121676) from the project PREJB38269. Metadata information for 828,937 processed cells from the DA dataset and AnnData/H5AD files containing the single-cell expression for days 11, 30 and 52 that are related to the STAR Methods are publicly available at Zenodo, https://zenodo.org/record/6079122#.YlSkxNPMJhE. Chip genotypes for HipSci lines are available from the EGA (EGAS00001000866, EGAS00001001272) and the ENA (PRJEB11750) portals.
All original code is publicly available in a frozen Zenodo repository (https://doi.org/10.5281/zenodo.7641259). The repository includes the single-cell processing scripts, the downstream analysis, and the code to reproduce the main figures and the supplemental ones.