

Abstract
The objective response rate of immune checkpoint blockade (ICB) in hepatocellular carcinoma (HCC) with anti PD-L1/PD-1 therapy is low. Discovering the signaling pathways regulating PD-L1 might help to improve ICB response rates. Here, we investigate transcription factors IRF-1 and IRF-2 signaling pathways regulating PD-L1 in HCC cells. In vivo studies show that IRF-1 and PD-L1 mRNA expression in human HCC tumors are significantly repressed compared with noncancerous background liver. IRF-1, IRF-2, and PD-L1 mRNA expression correlated positively in HCC tumors. Increased IRF-1 mRNA expression was observed in patients with well-differentiated or early stage HCC tumors. In vitro studies show that IFN-γ induces PD-L1 mRNA and protein expression through upregulation of IRF-1 in mouse and human HCC cells. IRF-1, IRF-2, and PD-L1 mRNA expression is upregulated in murine HCC by co-culture with effector T cells from spleen cells incubated with anti-CD3/CD28 antibodies. IRF-2 over-expression down-regulates IFN-γ induced PD-L1 promoter activity and protein levels in a dose-dependent manner. We identify two IRF-1 response elements (IRE1/IRE2) in the upstream 5′-flanking region of the CD274 (PD-L1) gene promoter. Site-directed mutagenesis shows both IRE1 and IRE2 are functional in transfection promoter assays. IRF-1 traditionally functions as tumor suppressor gene. However, these novel findings show a complex role for IRF-1 which upregulates PD-L1 in the inflammatory tumor microenvironment. IRF-1 antagonizes IRF-2 for binding to the IRE promoter element in PD-L1 which gives new insight to the regulation of PD-L1/PD-1 pathways in HCC ICB therapy.
Electronic supplementary material
The online version of this article (10.1007/s00262-020-02586-9) contains supplementary material, which is available to authorized users.
Keywords: IRF-1, IRF-2, PD-L1, IFN-γ, HCC
Introduction
Hepatocellular carcinoma (HCC) is the fifth most frequently diagnosed cancer worldwide, and the second leading reason of cancer death in men worldwide [1]. HCC carcinogenesis involves mutations in p53 tumor suppressor gene, WNT/β-catenin signaling, chronic viral hepatitis infection [2, 3], mTOR signaling, and/or STAT3/CD133 pathways [4, 5]. Immune checkpoint blockade (ICB) therapy has been regarded as a breakthrough in cancer immunotherapy; however, the objective response rate of ICB treatment (anti PD-L1/PD-1 antibody therapy) in HCC is about 10–20% [6, 7]. Tumor cell intrinsic factors that contribute to ICB therapy resistance include lack of tumor antigen expression, antigenic mutation, loss of T cell infiltration or functionality, as well as alterations of several signaling pathways (IFN, WNT, MARK, PI3K) [8, 9]. Additionally, tumor cell extrinsic mechanisms that cause inhibition of anti-tumor immunity involve immune suppressive cell populations including regulatory T cell, myeloid-derived suppressor cell, type II macrophage, as well as cytokine and chemokine released within tumor microenvironment [6, 8].
The IFN-γ pathway has a key role in ICB therapy. IFN-γ produced by tumor-specific T cells can induce an effective antitumor immune response through enhanced tumor antigen presentation, recruitment of other immune cells, as well as direct anti-proliferative and pro-apoptotic effects on tumor cells [10]. However, continuous IFN- γ exposure can cause expression of PD-L1 by cancer cells, thereby allowing cancer cells to inactivate antitumor T cells [11]. Identifying the mechanisms whereby IFN-γ regulates PD-L1 pathway may be beneficial for improving response of ICB treatment in HCC.
It is known that type I and II IFN regulate PD-L1 through cooperation of multiple distinct signaling cascades, which involves in ATR–CHEK1–STAT1/3–IRF-1 pathway [12, 13], cGAS–STING–IFNα/β–IRF3–PD-L1 pathway [14], as well as JAK1/2–STAT1/2/3–IRF-1 pathway [15, 16]. IRF-1 is a central transcription factor regulating IFN signaling. IRF-2 is a constitutively expressed transcription factor that is known to antagonize IRF-1 signaling; however, the role of IRF-2/IRF-1 interaction and potential regulation of PD-L1 in HCC has not been studied.
IRF-2 and IRF-1 are structurally similar but functionally different transcription factors. IRF-2 can compete with IRF-1 for binding to the same regulatory elements of IFN inducible genes [17]. Previously, we showed that IRF-2 overexpression protected against ischemia–reperfusion injury by decreasing IRF-1 dependent injury [18]. In this study, we investigate IRF-1 and PD-L1 mRNA expression in HCC tumor, and show that they are significantly repressed compared with noncancerous background. IRF-1, IRF-2 and PD-L1 mRNA expression are significantly positively correlated in HCC tumors. IRF-1 mRNA expression in HCC serves as a biomarker for predicting prognosis of patients with HCC. Recombinant IFN-γ or secreted by effector T cells can induce PD-L1 mRNA and protein expression through IRF-1 in HCC cells. Importantly, IRF-2 over-expression down-regulates IFN-γ/IRF-1-induced PD-L1 protein expression in a dose-dependent manner.
Materials and methods
Patient samples
Thirty-one paired HCC and adjacent liver tissues were acquired from patients who had a hepatectomy at the Liver Cancer Center of the University of Pittsburgh School of Medicine (Pittsburgh, PA, USA). Clinical characteristics of the included patients are summarized in Table 1. All human tissues were obtained in accordance with a University of Pittsburgh Institutional Review Board (IRB) approved protocol.
Table 1.
IRF-1, IRF-2, and PD-L1 mRNA in relationship with clinical characteristics of 31 HCC patients included in this study
| Variable | IRF-1 | IRF-2 | PD-L1 | ||||
|---|---|---|---|---|---|---|---|
| N | Mean ± SEM | p | Mean ± SEM | p | Mean ± SEM | p | |
| Age | |||||||
| ≤ 71 | 17 | 1.48 ± 0.41 | 0.106 | 1.52 ± 0.31 | 0.105 | 0.59 ± 0.16 | 0.329 |
| > 71 | 14 | 0.67 ± 0.14 | 0.90 ± 0.15 | 0.40 ± 0.07 | |||
| Sex | |||||||
| Male | 23 | 1.23 ± 0.32 | 0.403 | 1.32 ± 0.24 | 0.481 | 0.52 ± 0.13 | 0.702 |
| Female | 8 | 0.76 ± 0.24 | 1.01 ± 0.23 | 0.44 ± 0.10 | |||
| HBs Ag | |||||||
| + | 4 | 0.70 ± 0.33 | 0.531 | 1.42 ± 0.53 | 0.719 | 0.61 ± 0.35 | 0.667 |
| − | 27 | 1.17 ± 0.27 | 1.21 ± 0.20 | 0.48 ± 0.10 | |||
| HCV Ab | |||||||
| + | 7 | 1.45 ± 0.49 | 0.462 | 1.06 ± 0.13 | 0.617 | 0.50 ± 0.25 | 0.9886 |
| − | 24 | 1.01 ± 0.28 | 1.29 ± 0.24 | 0.50 ± 0.10 | |||
| Differentiation | |||||||
| Well | 6 | 2.31 ± 0.95 | 0.037* | 1.91 ± 0.83 | 0.218 | 0.76 ± 0.31 | 0.269 |
| Moderate | 22 | 0.84 ± 0.18 | 1.09 ± 0.13 | 0.46 ± 0.10 | |||
| Poor | 3 | 0.55 ± 0.03 | 1.07 ± 0.13 | 0.18 ± 0.07 | |||
| TNM stage | |||||||
| I | 8 | 2.49 ± 0.72 | 0.004** | 1.73 ± 0.62 | 0.514 | 0.75 ± 0.21 | 0.159 |
| II | 13 | 0.54 ± 0.10 | 1.10 ± 0.18 | 0.38 ± 0.12 | |||
| IIIA | 6 | 0.58 ± 0.23 | 1.01 ± 0.30 | 0.37 ± 0.11 | |||
| IIIB | 4 | 1.01 ± 0.30 | 1.07 ± 0.27 | 0.22 ± 0.07 | |||
Bold indicates *p < 0.05, **p < 0.01
HBs Ag, hepatitis; B, surface antigen; HCV Ab, hepatitis C virus antibody; TNM, Tumor Node Metastasis
Cell culture
The mouse Hepa 1-6 and human Huh-7, HepG2, and Hep3B hepatic cancer cells were purchased from ATCC (Manassas, VA, USA). The cell lines were authenticated by STR profiling (most recently in 2018/19) and regularly tested for mycoplasma contamination (most recently in 2019) throughout the study. The cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Lonza, Alpharetta, GA, USA), containing 10% heat-inactivated fetal bovine serum (FBS) (Clontech, Mountain View, CA, USA), 100 µ/ml penicillin, 100 µg/ml streptomycin, 15 mmol/l HEPES and 200 mmol/l l-glutamine. The mouse splenocytes and human peripheral blood mononuclear cells (PBMC) were cultured in RPMI medium 1640 (Gibco, UK). All cells were incubated at 37 °C in a humidified incubator containing 5% CO2.
Reagents and antibodies
Recombinant mouse and human IFN-γ were purchased from R&D (Minneapolis, MN, USA). Antibodies used for Western blot are as follows: antibodies against IRF-1, IRF-2, human PD-L1, and Lamin A/C (Cell signaling technology, MA, USA); anti β-actin and mouse PD-L1 antibodies (Abcam, MA, USA); IRDye 800CW anti-mouse and 680RD anti-rabbit secondary antibodies (LI-COR, Lincoln, USA). Antibodies used for immunofluorescent staining are as follows: antibody against PD-L1 (Proteintech, Rosemont, IL, USA), Alexa Fluor 546 anti-rabbit secondary antibody and anti F-actin antibody (Abcam).
Real-time RT-PCR
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and reversely transcribed to single-stranded cDNA with RNA to cDNA EcoDry™ Premix Kit (Takara, Kusatsu, Shiga Prefecture, Japan). Then, real-time polymerase chain reaction (PCR) was performed using SYBR Premix Kit (Takara) on the ABI Stepone PCRSystem (Applied Biosystems, Foster City, CA, USA). Relative expression of each gene was normalized to β-actin mRNA or GAPDH mRNA for mouse and human studies, respectively. Primer sequences used in this study were provided in Supplementary Table 1.
Western blot
Cytoplasmic and nuclear proteins were extracted as previously described [19]. A total of 30 µg or 40 µg nuclear protein or 60ug cytoplasmic protein was electrophoresed on 10% SDS–polyacrylamide gels and transferred to polyvinylidene difluoride membranes. The membranes were incubated with a 1:1000 dilution of primary antibodies overnight. After washed with Tris-buffered saline and Tween-20 (TBST) for three times, and incubated with a 1:5000 dilution of anti-rabbit secondary antibody for 30 m, the membrane was scanned by Li-Cor odyssey. β-actin or Lamin A/C protein was detected as standardization of cytoplasmic protein or nuclear protein, respectively.
Adenovirus infection
An E1- and E3- deficient adenoviral vector carrying the mouse or human AdIRF-1, AdIRF-2, or AdLacZ cDNA was constructed as previously described [20, 21]. HCC cells were infected with adenoviral concentration of 50 MOI for 48 h. After 48 h of infection, the cells were harvested, and then total RNA and cytoplasmic protein and nuclear protein were extracted to determine the expression of IRF-1 and PD-L1.
Immunofluorescent staining
Immunofluorescent staining was performed as previously described [22]. Hepa1-6 or Huh-7 cells were cultured on coverslips, fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min, permeabilized with 0.1% Triton X-100 and 10% FBS in PBS for 30 min at room temperature, and incubated with the primary PD-L1 antibodies for 1 h, which was diluted in a 1:300 ratio. Next, Alexa Fluor 546 anti-mouse secondary antibody was applied with F-actin counterstain. After washing with PBS, the slides were stained with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) and mounted, and then observed with a Olympus Fluoview FV1000 III microscope (Olympus, Tokyo, Japan).
Preparation of effector T cells
Splenocytes were isolated from 8–10 weeks old female C57BL/6 mice purchased from Jackson Labs (Bar Harbor, ME, USA). Red blood cells were lysed with red blood cell lysing buffer (Sigma). The splenocytes were incubated with plate-bound anti-mouse CD3 (clone 145-2C11, 2 µg/ml, BioXcell, NH, USA) and CD28 antibodies (clone PV-1, 2 µg/ml) for 72 h to generate activated effector T cells.
PBMCs were isolated from peripheral blood of healthy donors according to standard criteria by Ficoll-Paque (Cedar Lane, Burlington, ON, Canada), and incubated with plate-bound anti-human CD3 (clone OKT-3, 2 µg/ml, BioXcell) and CD28 antibodies (clone 9.3, 2 µg/ml) for 72 h to acquire activated effector T cells.
Co-culture effector T cells with HCC cells
A total of 8 × 104 Hepa1-6 or Huh-7 cells were cultured in 6-well plate. The following day, a total of 1 × 106 activated T cells were co-cultured with HCC cells (ratio of 10:1) according to our previous study [23]. The T cells and tumor cells were separated by transwell coated with 0.4 um pore membrane insert (Sigma, St. Louis, MO, USA) and T cells were seeded in the insert. INF-γ was neutralized in the effector T cell culture with anti-mouse IFN-γ (10 µg/ml, clone XMG1.2, eBioscience, San Diego, CA, USA) or anti-human IFN-γ (10 µg/ml, clone NIB42). IgG1 isotype (10 µg/ml, clone eBRG1) was used as control. The tumor cells were harvested after 24 h or 48 h.
Plasmid construct
The plasmid pGL3 2 kb prom CD274 (PD-L1) was a gift from Julian Downward (Addgene plasmid # 107,003) [24]. Mutations of the IRF-1 respond elements were generated from the pGL3 prom CD274 according to the manufacturer recommendations by QuickChange II Site-Directed Mutagenesis Kit (Agilent Technologies). Primer sequences used were provided in supplementary Table 1. Mutations were confirmed by DNA sequence analysis by the University of Pittsburgh Health Sciences Core Research Facilities.
Transfection
The human IRF-1 siRNA (sc-35706) and negative control (NC, sc-37007) were purchased from Santa Cruz Biotech (CA, USA). The cells were seeded in a 6-well plate and the following day transfected siRNA (80 pmols) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Serum-free medium was replaced with growth medium after 8 h. 48 h after transfection, the protein level assay was performed.
Luciferase assay
The β-gal reporter control plasmid was used to normalize the transfection efficiency. Huh-7 and Hepa1-6 were cultured in 6-well plate and co-transfected with β-gal and either pGL3 empty vector Basic (Sigma) or pGL3 PD-L1 promoter luciferase reporter using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). 24 h after transfection, the cells were induced by IFN-γ for 6 h or infected with AdLacZ, AdIRF-1, or AdIRF-2 for 24 h before relative luciferase and β-galactosidase activities were measured with the reporter lysis buffer and luciferase substrate (Promega, Madison, WI, USA). Serum-free medium was replaced with growth medium after 6 h. The relative luciferase unit (RLU) was measured using the Dual-Luciferase Report Assay (BioTek, Winooski, VT, USA).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8 software (GraphPad Software Inc, San Diego, CA). To test for statistical significance, the t test, one-way ANOVA and correlation analyses were used to compare between different groups. For survival curves, log-rank test was used to compare between individual groups. For all analyses, p < 0.05 was considered significant. Results are collective data from 2 to 4 repeat experiments.
Results
IRF-1 and PD-L1 mRNA expression were repressed in HCC tumor compared to noncancerous tissue, and IRF-1, IRF-2 & PD-L1 mRNA expression were significantly positively correlative in tumor tissue
To investigate the interaction of IRF-1, IRF-2, and PD-L1 gene expression in patients diagnosed with HCC, we analyzed tumor and background tissue from patients that underwent liver resection for HCC. A total 31 patients were included in this study. Patients’ clinical characteristics are in Table 1. Comparing HCC to the non-cancerous liver in 31 patients, IRF-1 and PD-L1 mRNA expression were significantly repressed in the HCC tumor compared to adjacent background liver (Fig. 1a, b, respectively). IRF-2 mRNA expression showed no significant difference between tumor and background liver (Fig. 1c). There was a significant positive correlation between IRF-1 & IRF-2, IRF-1 & PD-L1, and IRF-2 and PD-L1 mRNA expression (Fig. 1d–f). Furthermore, we analyzed the interaction of IRF-1, IRF-2 & PD-L1 mRNA expression in HCC tumor from The Cancer Genome Atlas (TCGA) data through cBioPortal [25, 26]. Correlative analyses showed that expressions of these three genes were significantly positively correlated with each other, respectively (Supplementary Table 2). Likewise, correlative analyses confirmed that IFN-γ gene (IFNg) mRNA expression correlated positively with IRF-1, IRF-2 & PD-L1 mRNA expression in HCC tumors according to TCGA data (Supplementary Table 3).
Fig. 1.

IRF-1, IRF-2 & PD-L1 mRNA expression were decreased in HCC tumor compared to noncancerous tissue and their mRNA expression were significantly positively correlated with each other in HCC tissue, respectively. a, b IRF-1 and PD-L1 mRNA expression as determined by qPCR were significantly repressed in 31 cases of HCC tumor tissues compared to adjacent background liver (*p < 0.05, **p < 0.01). c IRF-2 mRNA expression was similar to background liver. d–f IRF-1, IRF-2, & PD-L1 mRNA expression are significantly positively correlated with each other in tumor tissues (p < 0.0001, p = 0.0008, p < 0.0001, respectively)
IRF-1 predicted outcomes in HCC
To determine if IRF-1, IRF-2 and PD-L1 gene expression correlated with clinical characteristics of the HCC patients, we analyzed gene expression in patients with different background liver (HBV/HCV), tumor differentiation, and TNM stage. As described in Table 1, significantly increased IRF-1 mRNA expression in HCC tumor was observed in those patients with well-differentiated or in early TNM stage HCC tumors.
To analyze whether the IRF-1 gene served as a biomarker for HCC prognosis, we analyzed the data from the TCGA database through cBioPortal. The log-rank test showed that IRF-1 gene alterations including gene mutation would predict worse overall survival in patients with HCC (Supplementary Fig. 1). A caveat was the small sample size (n = 7) in the number of patients with IRF-1 alterations, compared to the wild-type (n = 600), which suggested alterations might be rare in HCC tumors.
IFN-γ upregulated PD-L1 expression through IRF-1 in HCC cell lines
To explore the role of IRF-1 in regulating PD-L1, we stimulated HCC cell lines with recombinant cytokine IFN-γ. In vitro studies showed that IRF-1 mRNA expression was induced by IFN-γ in mouse Hepa1-6 (Fig. 2a) and human Huh-7 (Fig. 2d) HCC cell lines in a dose-dependent manner. IFN-γ also induced PD-L1 expression in the mouse and human HCC cell lines, respectively (Fig. 2b, e). Since IRF-1 was one of the dominant IFN-γ activated transcription factors, we transduced these mouse and human HCC cell lines with adenoviral IRF-1 (AdIRF-1) and found a marked increase in PD-L1 expression, respectively (Fig. 2c, f). The results showed that IRF-1 upregulated PD-L1 gene expression directly.
Fig. 2.

IFN-γ upregulated PD-L1 expression through IRF-1 in HCC cell lines. a, b IRF-1 and PD-L1 mRNA expression in Hepa1-6 cells determined by qPCR was induced by mouse IFN-γ (25-100µ/ml for 24 h) in a dose-dependent manner (*p < 0.05, **p < 0.01, respectively). c PD-L1 mRNA expression in Hepa1-6 cell was upregulated by transduction with AdIRF-1 at 50 MOI for 48 h (*p < 0.05). d, e IRF-1 and PD-L1 mRNA expression in Huh-7 cells were induced by human IFN-γ at 50-100µ/ml for 24 h, (*p < 0.05, **p < 0.01, respectively). f PD-L1 mRNA expression in Huh-7 cells was upregulated by transduction with AdIRF-1 at 50 MOI for 48 h (*p < 0.05). g Nuclear IRF-1 and cytoplasmic PD-L1 protein level measured by Western Blot in Hepa1-6 cells were increased by mouse IFN-γ (25-50µ/ml for 24 h) in a dose-dependent manner. AdIRF-1 (but not AdLacZ) transduction at 50 MOI for 48 h induce IRF-1 and PD-L1 protein expression. h Nuclear IRF-1 and cytoplasmic PD-L1 protein in human Huh-7 cells were increased by human IFN-γ (50–100 µ/ml for 24 h). AdIRF-1 (but not AdLacZ) transduction at 50 MOI for 48 h induce IRF-1 and to a lesser extent, PD-L1 protein expression. i Nuclear IRF-1 and cytoplasmic PD-L1 protein in human HepG2 and Hep3B cells were increased by human IFN-γ (100µ/ml for 24 h). AdIRF-1 (but not AdLacZ) transduction at 50 MOI for 48 h induce IRF-1 and PD-L1 protein level. j Nuclear IRF-1 and cytoplasmic PD-L1 protein levels in Huh-7 and Hep3B cells were decreased by human IRF-1 siRNA transfection
Next, we examined the effect of IFN-γ or IRF-1 expression on IRF-1 and PD-L1 protein levels. Nuclear IRF-1 and cytoplasmic PD-L1 were increased by IFN-γ in a dose-dependent manner (Fig. 2g, h). Likewise, AdIRF-1 (but not AdLacZ) upregulated PD-L1 protein in mouse Hepa1-6 and human Huh-7 cell lines (Fig. 2g, h).
To determine whether IRF-1 regulating PD-L1 is a general phenomenon rather than specific Huh-7 cell line only, we examined the effect of IFN-γ or IRF-1 expression on IRF-1 and PD-L1 protein level in two additional human HepG2 and Hep3B HCC cell lines. IRF-1 and PD-L1 proteins in HepG2 and Hep3B cells increased by IFN-γ or AdIRF-1 (Fig. 2i). In contrast, silencing of endogenous IRF-1 expression in human Huh-7 and Hep3B cells decreased basal PD-L1 protein levels by IRF-1 siRNA (Fig. 2j). These results support the notion that this regulatory inflammatory pathway occurs in many human HCC cells as well as rodent murine HCC cells.
IFN-γ upregulated IRF-2 expression in HCC cell lines
Since IFN-γ induced IRF-1, and since IRF-2 positively correlated with IRF-1 and PD-L1 expression in human HCC tumors (Fig. 1, Supp. Table 2), we next sought to determine if IFN-γ also induced IRF-2 expression in HCC tumor cell lines. IFN-γ did induce a modest, but significant increase in IRF-2 expression in the murine Hepa1-6 and human Huh-7 HCC cell lines, respectively (Fig. 3a, b).
Fig. 3.

Expression of PD-L1 in Hepa1-6 was upregulated by co-culture with activated mouse effector T cells. IRF-2 mRNA expression was increased in Hepa1-6 cells (a) and Huh-7 cells (b) by recombinant IFN-γ at dose of 50 µ/ml and 100 µ/ml (*p < 0.05, **p < 0.01, respectively). c IRF-1, IRF-2, and PD-L1 mRNA expression were increased in Hepa1-6 after 48 h co-culture with effector T cells separated from mouse splenocytes incubated with anti-mouse CD3 and anti-mouse CD28 antibodies for 72 h (*p < 0.05, **p < 0.01, respectively). d Immunofluorescent staining of PD-L1 protein in Hepa1-6 cells increased by co-culture with effector T cells for 24 h or 48 h compared with Hepa1-6 cell without co-culture. In contrast, PD-L1 level in Hepa1-6 cells decreased when IFN-γ in the effector T cells culture was neutralized by anti-mouse IFN-γ antibody (× 400 magnification). e IRF-1, IRF-2 and PD-L1 mRNA expression decreased in Hepa1-6 after 48 h co-culture with effector T cells when IFN-γ were neutralized by anti-mouse IFN-γ antibody (*p < 0.05, **p < 0.01, respectively)
PD-L1 expression in HCC cell lines was upregulated by IFN-γ from effector T cells
To investigate whether the tumor cell intrinsic PD-L1 expression was induced by IFN-γ produced by effector T cells in the tumor microenvironment, we co-cultured mouse HCC cell line Hepa1-6 with mouse effector T cells. The effector T cells were isolated from mouse spleen and incubated with anti-mouse CD3 and CD28 antibodies to activate them for IFN-γ secretion. IRF-1 and PD-L1 mRNA expression significantly increased in the Hepa1-6 HCC cells after 48 h of co-culture with the T cells (Fig. 3c). IRF-2 mRNA expression was modestly increased by co-culture (Fig. 3c). Immunofluorescent staining for PD-L1 protein expression (red staining) in the Hepa1-6 cells increased in a time-dependent manner with T cell co-culture. In contrast, PD-L1 protein expression in Hepa1-6 cells decreased when IFN-γ secreted by effector T cells was removed with anti-mouse IFN-γ antibody (Fig. 3d). Likewise, IRF-1, IRF-2 and PD-L1 mRNA expression significantly decreased in the Hepa1-6 cells after 48 h of co-culture with the effector T cells when INF-γ was neutralized by anti-mouse INF-γ antibody (Fig. 3e).
Next, we co-cultured human HCC cell line Huh-7 with effector T cells. The effector T cells were acquired from PBMCs activated by anti-human CD3 and CD28 antibodies. Immunofluorescent staining for PD-L1 protein expression (red staining) in the Huh-7 cells increased after 48 h of co-culture with the T cells. In contrast, PD-L1 protein expression decreased when IFN-γ in the T cells culture was neutralized by anti-human IFN-γ antibody (Supplementary Fig. 2).
IRF-2 repressed PD-L1 expression by competing with IRF-1 for binding to the IRF response element in the CD274 (PD-L1) promoter
IRF-1 and IRF-2 are nuclear transcription factors that are known to compete for binding to the same IRF response element (IRE) in the promoter region of IFN-responsive genes. Resting Hepa1-6 murine (Fig. 4a) or Huh-7 human (Fig. 4b) HCC cells exhibited low-level basal IRF-1 and PD-L1 protein expression. IRF-1 overexpression with AdIRF-1 increased IRF-1 protein levels as expected, and also increased PD-L1 protein levels. AdIRF-2 alone had no significant effect on IRF-1 or PD-L1 levels. IFN-γ increased both IRF-1 and PD-L1 proteins levels. Importantly, IRF-2 overexpression inhibited IFN-γ-mediated PD-L1 induction, but did not have any effect on IRF-1 protein levels (Fig. 4a, b).
Fig. 4.

IRF-2 repressed PD-L1 expression by competing with IRF-1 for binding to the same IRF response element (IRE) in the CD274 (PD-L1) gene promoter. PD-L1, IRF-1, and IRF-2 protein expression determined by Western blot in Hepa1-6 cells (a, c) and Huh-7 cells (b, d). IRF-1 over-expression enhanced PD-L1 protein expression. IRF-2 over-expression attenuated IFN-γ or IRF-1 expression-induced PD-L1 protein level. eCD274 promoter luciferase reporter activity was significantly increased by IFN-γ (100 µ/ml for 24 h) in human Huh-7 cells (**p < 0.01). AdIRF-1 (but not AdLacZ or AdIRF-2) transduction also increased CD274 promoter activity (*p < 0.05). Transduction with AdIRF-2 significantly decreased IFN-γ induced CD274 promoter activity (*p < 0.05). f Wild type (WT) CD274 promoter luciferase reporter activity was significantly increased by AdIRF-1 infected for 24 h compared to AdLacZ (*p < 0.05). In contrast, mutation of either IRE1 or IRE2 significantly decreased IRF-1 driven CD274 promoter activity in HCC cells. The double mutation of both IRE1 and IRE2 (D-mutant) had even lower promoter activity (*p < 0.05 vs AdIRF-1)
To investigate whether combining IRF-1 and IRF-2 overexpression had same effect on PD-L1 protein level as IRF-2 overexpression inhibiting IFN-γ mediated PD-L1 induction, we used AdIRF-1 and AdIRF-2 infection together. As expected, IRF-2 overexpression also suppressed IRF-1 specific mediated PD-L1 induction (Fig. 4c, d).
Next, we analyzed two kilobases in the 5′-upstream flanking region of the human CD274 (PD-L1) gene using PROMO bioinformatics software. We identified two putative IRF-1 response elements (IRE1 and IRE2) conserved in both human and murine sequence in the CD274 (PD-L1) promoter at − 1490 and − 1057 nucleotides in the 5′-flaning region (Table 2). The CD274 promoter was previously cloned [24], and we confirmed this promoter plasmid contained the two putative IRF-1 response elements aligned by the Nucleotide BLAST program. Transfection in human Huh7 and murine Hepa1-6 HCC cells showed basal luciferase activity which was significantly increased greater than 3 fold by IFN-γ stimulation (Fig. 4e). AdIRF-1 transduction (but not AdLacZ or AdIRF-2) also increased promoter reporter activity. When IRF-2 was over-expressed, this significantly decreased IFN-γ stimulated CD274 promoter activity (Fig. 4e).
Table 2.
IRE1 and IRE2 in the CD274 promoter sequence and site-directed mutations
| CD274 5′ upstream DNA | Position | Sequence (5′–3′) |
|---|---|---|
| IRE1 | ||
| WT | − 1057 to – 1049 | AGAAGGAAA |
| Mutation | − 1057 to – 1049 | AGCCGGCCA |
| IRE2 | ||
| WT | − 1490 to − 1482 | TTTCCTATT |
| Mutation | − 1490 to − 1482 | TGGCCTGGT |
Binding sites are shown in bold type
To determine the role of IRE in the PD-L1 promoter region, we generated site-directed mutants of IRE1, IRE2, or double mutant of both IRE1 and IRE2 in the CD274 promoter plasmid which were verified by sequencing (Table 2). AdIRF-1 transduction significantly increased wild type (WT) CD274 promoter activity (Fig. 4f). Mutation of either IRE1 or IRE2 significantly decreased IRF-1 driven CD274 promoter activity in HCC cells. The double mutation of both IRE1 and IRE2 (D-mutant) had even lower promoter activity compared to WT CD274 promoter activity driven by AdIRF-1 transduction.
A summary illustration in resting HCC tumor cells portrayed that IRF-1 was down-regulated compared to background liver. Constitutively expressed IRF-2 binded to the cis-acting IRE motif and basally represses PD-L1 gene expression (Fig. 5a). In contrast, in the setting of an inflammatory tumor microenvironment, IFN-γ and other cytokines were released by infiltrating T cells or other immune cells which significantly upregulates trans-acting IRF-1 expression which displaced IRF-2 at the IRE sites and derived PD-L1 gene expression (Fig. 5b).
Fig. 5.
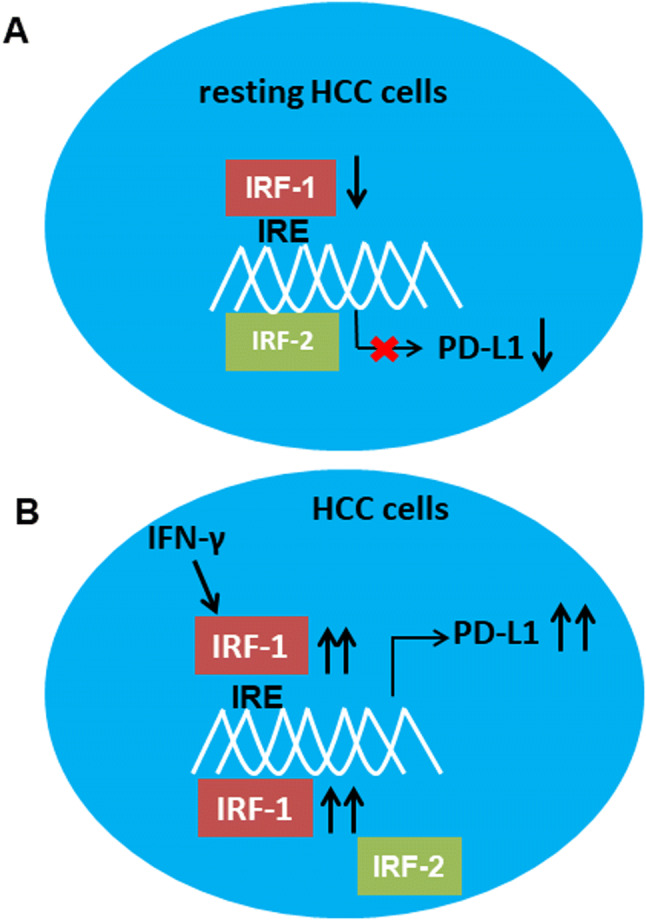
Regulation of PD-L1 expression in HCC by binding of IRF-1 and IRF-2 to IRF response elements (IRE) in the CD274 promoter. a In resting human HCC cells, IRF-1 was relatively down-regulated, and constitutive binding of IRF-2 to the IRE trans-represses PD-L1 gene expression. b Under inflammatory conditions, IFN-γ induced IRF-1 which competed with IRF-2 for binding to the IRE, thereby driving PD-L1 gene expression
Discussion
IRF-1 has been regarded as a tumor suppressor gene functioning to promote apoptosis or autophagy, as well as inhibiting proliferation of tumor cells [20, 22, 27–29]. Our data show that increased IRF-1 mRNA expression in HCC tumors is observed in patients with well-differentiated or early stage HCC, which supports that IRF-1 is considered as tumor suppressor gene. However, it has been shown that IRF-1 functions as a driver of PD-L1 transcription that allows tumor cells to evade antitumor immunity by T cell interaction and potential T cell exhaustion. From this vantage point, IRF-1 can also function as an oncogene to promote cancer cell survival. One recent study showed that IRF-1 inhibits antitumor immunity through upregulation of PD-L1 in melanoma tumor cells, and IRF-1 knockout cells showed reduced tumor growth [30]. Hence, IRF-1 can be regarded as a double-edged sword similar to IFN-γ with capacity for not only promoting antitumor effects but also suppressing antitumor immunity.
The major and novel findings of this study are: (1) IFN-γ induces PD-L1 mRNA and protein expression through upregulation of IRF-1 in mouse and human HCC cells. (2) IRF-2 over-expression down-regulates IFN-γ induced PD-L1 promoter activity and protein levels in human and murine HCC tumor cells. (3) The CD274 (PD-L1) gene promoter has two functionally active IRF-1 response elements (IRE1 & IRE2) that induce PD-L1 gene transcription in response to IFN-γ regulated by IRF-1 and IRF-2.
In addition to IFN-γ regulating PD-L1 through IRF-1 in HCC cells, a novel finding in our study is that the PD-L1 upregulation induced by IRF-1 can be attenuated by IRF-2. IRF-1 and IRF-2 are similar in structure but typically play opposite roles in regulating expression of IFN-responsive genes [17]. A recent study shows that IRF-2 can promote antitumor immunity through inhibiting PD-L1 expression in tumors [31], or reduce chemokines that attract immune cells mediating suppressing antitumor immunity [32]. Although we had HCC tumors with higher IRF-2 levels compared to background liver, we did not find a statistical significance in our 31 patients. This likely relates to a small sample size. Therefore, we analyzed the larger TCGA data through UALCAN and found that IRF-2 is overexpressed in HCC tumor compared to normal liver (50 normal vs 371 HCC, p = 0.0001) [33]. In a resting or non-inflammatory state, HCC cells have lower IRF-1 and relatively higher IRF-2 which inhibits PD-L1 expression in the HCC cells. However, during inflammation drive by IFN-γ, IRF-1 is increased which displaces IRF-2 to upregulate PD-L1 expression in the inflammatory environment.
To identify the molecular basis for IRF-1/IRF-2 transcriptional regulation of PD-L1, we identified two specific IRF-1 response elements in the upstream 5′-flanking region of the CD274 gene promoter. Site-directed mutagenesis shows both IRE1 and IRE2 are functional in transfection promoter assays. Hence, the molecular mechanism is dependent on IRF-1 competing with IRF-2 for binding to the IRE1 and IRE2 promoter elements in the PD-L1 gene. These findings provide new insight into PD-L1 signaling pathway in HCC and may be beneficial for understanding PD-1/PD-L1 blockade therapy in the inflammatory tumor setting.
In this study, we also show that PD-L1 expression is repressed in the majority of human HCC tumors with 28 of 31 patients showing less PD-L1 mRNA expression than background liver. Thus, 10% of our human HCC tumors have increased PD-L1 expression. This down-regulation of PD-L1 in human HCC tumors is similar to prior studies where expression of PD-L1 was only observed in 17% and 19% of HCC tumors, respectively [34, 35]. A predictive biomarker in patients with ICB therapy is a T cell inflammatory tumor microenvironment, manifested as increased number of CD8 + cytotoxic T cells infiltrating in proximity to PD-L1 positive tumor cells [11, 36, 37]. It is controversial whether PD-L1 expression in HCC can be applied as predictive biomarker for ICB therapy [38]; however, greater expression of PD-L1 in tumor cells results in effector T cells exhaustion and predicts significant association with tumor aggressiveness and postoperative recurrence in HCC [39].
As a primary driver of PD-L1 transcription, IRF-1 has been identified as predictive biomarker for PD-1/PD-L1 blockade therapy in the patients with melanoma [40, 41]. IRF-1 expression may serve as a more valuable predictive biomarker for ICB therapy than PD-L1 itself. Our data and evidence from TCGA have shown that the expression of IRF-1, IRF-2 and PD-L1 in HCC cells are significantly correlative. Furthermore, IRF-1 gene serves a biomarker for predicting HCC prognosis. However, additional caveat in our data is the lack of in vivo experiment to investigate IRF-1, IRF-2 and PD-L1 expression induced by IFN-γ in the tumor microenvironment. Therefore, whether IRF-1 and IRF-2 can be used to predict ICB therapy response in HCC will need additional studies.
In summary, our results demonstrate a correlation of IRF-1, IRF-2, and PD-L1 expression in HCC cells. IRF-1 alterations are a negative prognostic biomarker in HCC. IFN-γ regulates PD-L1 through IRF-1 binding to two specific IREs in the CD274 promoter, and this upregulation can be attenuated by IRF-2. These findings provide new insight into the molecular mechanisms regulating the PD-L1 signaling pathway in HCC and have implications for overcoming resistance to anti PD-L1/PD-1 therapy in HCC.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Abbreviations
- ATR
Ataxia telangiectasia and Rad3-related protein
- cGAS
Cyclic guanosine monophosphate (GMP)–adenosine monophosphate (AMP) synthase
- CHEK-1
Check point kinase 1
- CTLA-4
Cytotoxic T-lymphocyte-associated protein 4
- HCC
Hepatocellular carcinoma
- ICB
Immune checkpoint blockade
- IFN-γ
Interferon-γ
- IRE
IRF response element
- IRF-1
Interferon regulatory factor 1
- IRF-2
Interferon regulatory factor 2
- JAK
Janus kinase
- MARK
Mitogen-activated protein kinase
- PD-L1
Programmed death-ligand 1
- PD-1
Programmed death protein 1
- PI3K
Phosphatidylinositol 3-kinase
- RLU
Relative luciferase unit
- STAT
Signal transducer and activator of transcription
- STING
Stimulator of interferon gene
- TCGA
The Cancer Genome Atlas
- TNM
Tumor node metastasis classification
Author contributions
YY and DAG designed the research. YY, LZ, QD, BY, and DAG performed research and also analyzed the data. YY and DAG wrote the paper. All authors edited and approved the submission of this work.
Funding
DAG was supported by funding from National Institute of Health (HHSN276201200017C and P30DK120531-01). YY was supported by funding from Guangxi Natural Science Foundation (2017GXNSFAA198014) and Guangxi High Education Institute Science Foundation (2017KY0099).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval and ethical standards
All individuals provided written informed consent and human tissues were obtained in accordance with the University of Pittsburgh Institutional Review Board (IRB) approved protocol (No. MOD08010372/PRO08010372).
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yihe Yan, Email: yany5@upmc.edu.
David A. Geller, Email: gellerda@upmc.edu
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene. 2007;26:2166–2176. doi: 10.1038/sj.onc.1210279. [DOI] [PubMed] [Google Scholar]
- 3.Wang XW, Hussain SP, Huo TI, Wu CG, Forgues M, Hofseth LJ, Brechot C, Harris CC. Molecular pathogenesis of human hepatocellular carcinoma. Toxicology. 2002;181–182:43–47. doi: 10.1016/s0300-483x(02)00253-6. [DOI] [PubMed] [Google Scholar]
- 4.Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol. 2014;60:855–865. doi: 10.1016/j.jhep.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Won C, Kim BH, Yi EH, et al. Signal transducer and activator of transcription 3-mediated CD133 up-regulation contributes to promotion of hepatocellular carcinoma. Hepatology. 2015;62:1160–1173. doi: 10.1002/hep.27968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun C, Mezzadra R, Schumacher TN. Regulation and Function of the PD-L1 Checkpoint. Immunity. 2018;48:434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prestipino A, Zeiser R. Clinical implications of tumor-intrinsic mechanisms regulating PD-L1. Sci Transl Med. 2019 doi: 10.1126/scitranslmed.aav4810. [DOI] [PubMed] [Google Scholar]
- 10.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 11.Shin DS, Zaretsky JM, Escuin-Ordinas H, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7:188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Permata TBM, Hagiwara Y, Sato H, Yasuhara T, Oike T, Gondhowiardjo S, Held KD, Nakano T, Shibata A. Base excision repair regulates PD-L1 expression in cancer cells. Oncogene. 2019;38:4452–4466. doi: 10.1038/s41388-019-0733-6. [DOI] [PubMed] [Google Scholar]
- 13.Sato H, Niimi A, Yasuhara T, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. 2017;8:1751. doi: 10.1038/s41467-017-01883-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sen T, Rodriguez BL, Chen L, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9:646–661. doi: 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Diaz A, Shin DS, Moreno BH, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–1201. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shevtsov M, Sato H, Multhoff G, Shibata A. Novel approaches to improve the efficacy of immuno-radiotherapy. Front Oncol. 2019;9:156. doi: 10.3389/fonc.2019.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harada H, Fujita T, Miyamoto M, Kimura Y, Maruyama M, Furia A, Miyata T, Taniguchi T. Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell. 1989;58:729–739. doi: 10.1016/0092-8674(89)90107-4. [DOI] [PubMed] [Google Scholar]
- 18.Klune JR, Dhupar R, Kimura S, et al. Interferon regulatory factor-2 is protective against hepatic ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2012;303:G666–G673. doi: 10.1152/ajpgi.00050.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ueki S, Dhupar R, Cardinal J, Tsung A, Yoshida J, Ozaki KS, Klune JR, Murase N, Geller DA. Critical role of interferon regulatory factor-1 in murine liver transplant ischemia reperfusion injury. Hepatology. 2010;51:1692–1701. doi: 10.1002/hep.23501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim PK, Armstrong M, Liu Y, Yan P, Bucher B, Zuckerbraun BS, Gambotto A, Billiar TR, Yim JH. IRF-1 expression induces apoptosis and inhibits tumor growth in mouse mammary cancer cells in vitro and in vivo. Oncogene. 2004;23:1125–1135. doi: 10.1038/sj.onc.1207023. [DOI] [PubMed] [Google Scholar]
- 21.Yokota S, Yoshida O, Dou L, et al. IRF-1 promotes liver transplant ischemia/reperfusion injury via hepatocyte IL-15/IL-15Ralpha production. J Immunol. 2015;194:6045–6056. doi: 10.4049/jimmunol.1402505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li P, Du Q, Cao Z, et al. Interferon-gamma induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon-regulatory factor-1 (IRF-1) Cancer Lett. 2012;314:213–222. doi: 10.1016/j.canlet.2011.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang MQ, Du Q, Varley PR, Goswami J, Liang Z, Wang R, Li H, Stolz DB, Geller DA. Interferon regulatory factor 1 priming of tumour-derived exosomes enhances the antitumour immune response. Br J Cancer. 2018;118:62–71. doi: 10.1038/bjc.2017.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coelho MA, de Carne TS, Rana S, et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity. 2017;47(1083–99):e6. doi: 10.1016/j.immuni.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013 doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kroger A, Ortmann D, Krohne TU, Mohr L, Blum HE, Hauser H, Geissler M. Growth suppression of the hepatocellular carcinoma cell line Hepa1-6 by an activatable interferon regulatory factor-1 in mice. Cancer Res. 2001;61:2609–2617. [PubMed] [Google Scholar]
- 28.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz-Roberts JL, Cook KL, Chen C, et al. Interferon regulatory factor-1 signaling regulates the switch between autophagy and apoptosis to determine breast cancer cell fate. Cancer Res. 2015;75:1046–1055. doi: 10.1158/0008-5472.CAN-14-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shao L, Hou W, Scharping NE, et al. IRF1 Inhibits Antitumor Immunity through the Upregulation of PD-L1 in the Tumor Cell. Cancer Immunol Res. 2019;7:1258–1266. doi: 10.1158/2326-6066.CIR-18-0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dorand RD, Nthale J, Myers JT, et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science. 2016;353:399–403. doi: 10.1126/science.aae0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liao W, Overman MJ, Boutin AT, et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell. 2019;35(559–72):e7. doi: 10.1016/j.ccell.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, Varambally S. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19:649–658. doi: 10.1016/j.neo.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calderaro J, Rousseau B, Amaddeo G, et al. Programmed death ligand 1 expression in hepatocellular carcinoma: relationship With clinical and pathological features. Hepatology. 2016;64:2038–2046. doi: 10.1002/hep.28710. [DOI] [PubMed] [Google Scholar]
- 35.Huang CY, Wang Y, Luo GY, Han F, Li YQ, Zhou ZG, Xu GL. Relationship between PD-L1 expression and CD8+ T-cell immune responses in hepatocellular carcinoma. J Immunother. 2017;40:323–333. doi: 10.1097/CJI.0000000000000187. [DOI] [PubMed] [Google Scholar]
- 36.Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064–5074. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solinas A, Calvisi DF. Programmed death ligand 1 expression in hepatocellular carcinoma: a prognostic marker and therapeutic target for liver cancer? Hepatology. 2016;64:1847–1849. doi: 10.1002/hep.28803. [DOI] [PubMed] [Google Scholar]
- 39.Gao Q, Wang XY, Qiu SJ, et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009;15:971–979. doi: 10.1158/1078-0432.CCR-08-1608. [DOI] [PubMed] [Google Scholar]
- 40.Smithy JW, Moore LM, Pelekanou V, et al. Nuclear IRF-1 expression as a mechanism to assess "Capability" to express PD-L1 and response to PD-1 therapy in metastatic melanoma. J Immunother Cancer. 2017;5:25. doi: 10.1186/s40425-017-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gupta S, McCann L, Chan YGY, et al. Closed system RT-qPCR as a potential companion diagnostic test for immunotherapy outcome in metastatic melanoma. J Immunother Cancer. 2019;7:254. doi: 10.1186/s40425-019-0731-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.