

Abstract
The identification and role of endothelial progenitor cells in pulmonary arterial hypertension (PAH) remain controversial. Single-cell omics analysis can shed light on endothelial progenitor cells and their potential contribution to PAH pathobiology. We aim to identify endothelial cells that may have stem/progenitor potential in rat lungs and assess their relevance to PAH. Differential expression, gene set enrichment, cell–cell communication, and trajectory reconstruction analyses were performed on lung endothelial cells from single-cell RNA sequencing of Sugen–hypoxia, monocrotaline, and control rats. Relevance to human PAH was assessed in multiple independent blood and lung transcriptomic data sets. Rat lung endothelial cells were visualized by immunofluorescence in situ, analyzed by flow cytometry, and assessed for tubulogenesis in vitro. A subpopulation of endothelial cells (endothelial arterial type 2 [EA2]) marked by Tm4sf1 (transmembrane 4 L six family member 1), a gene strongly implicated in cancer, harbored a distinct transcriptomic signature enriched for angiogenesis and CXCL12 signaling. Trajectory analysis predicted that EA2 has a less differentiated state compared with other endothelial subpopulations. Analysis of independent data sets revealed that TM4SF1 is downregulated in lungs and endothelial cells from patients and PAH models, is a marker for hematopoietic stem cells, and is upregulated in PAH circulation. TM4SF1+CD31+ rat lung endothelial cells were visualized in distal pulmonary arteries, expressed hematopoietic marker CD45, and formed tubules in coculture with lung fibroblasts. Our study uncovered a novel Tm4sf1-marked subpopulation of rat lung endothelial cells that may have stem/progenitor potential and demonstrated its relevance to PAH. Future studies are warranted to further elucidate the role of EA2 and Tm4sf1 in PAH.
Keywords: pulmonary hypertension, single-cell RNA sequencing, endothelial progenitor cells, monocrotaline, Sugen–hypoxia
Clinical Relevance
Our study uncovered a novel Tm4sf1 (transmembrane 4 L six family member 1)–marked subpopulation of rat lung endothelial cells that may have stem/progenitor potential and demonstrated its relevance to pulmonary arterial hypertension (PAH). This study will generate renewed interest in stem/progenitor endothelial cells in PAH and motivate future studies with the ultimate goal of developing more effective PAH therapies.
Despite advances in our understanding of pulmonary arterial hypertension (PAH), it remains an incurable disease characterized by pathological pulmonary arterial remodeling and endothelial dysfunction. Endothelial progenitor cells (EPCs) are involved in endothelial homeostasis and angiogenesis and have been extensively studied in PAH animal models and patients using heterogeneous methods for EPC isolation by flow cytometry and/or culture (1). However, results have been conflicting and potentially confounding as to whether EPCs in PAH are protective or harmful (2), a reflection of the broader controversy in the stem cell field as to the precise definition and reliable isolation of EPCs (3). For example, conventional markers used to identify putative EPCs in numerous studies, including PAH studies, are now believed to be unreliable in isolating pure EPCs, and a specific marker for EPCs has yet to be identified (3).
The advent of single-cell omics can shed light on EPCs and their potential role in disease in a data-driven and unbiased approach. However, although single-cell studies in PAH have recently been published, including our recent work using lungs from PAH rat models (4–7), stem/progenitor endothelial cells have yet to be identified. In this study, we used advanced computational, flow cytometry, and in vitro methods to uncover a Tm4sf1-marked subpopulation of rat lung endothelial cells from single-cell RNA sequencing (scRNA-seq) that may have stem/progenitor potential. Further integrative analysis using independent blood and lung transcriptomic data demonstrates its dysregulation and relevance to PAH.
Some of the results of these studies have been previously reported in the form of an abstract (8).
Methods
Main methods are below, with additional details provided in the data supplement.
Animals
Adult male Sprague-Dawley rats (250–350 g) were used for animal experiments, which were approved by the University of California, Los Angeles, Animal Research Committee. As previously described (7), Sugen–hypoxia (SuHx) rats were injected subcutaneously with Sugen 5416 (20 mg/kg) followed by hypoxia at 10% O2 for 21 days and then normoxia for 14 days. Monocrotaline (MCT) rats were injected subcutaneously with MCT (60 mg/kg) followed by normoxia for 28 days. Age-matched control rats were kept in normoxia for 28 days. Lungs were then harvested and enzymatically dissociated into single-cell suspensions followed by scRNA-seq (7) (n = 6/group) or snap frozen for RNA isolation and bulk RNA sequencing (RNA-seq) (n = 4/group). Quality control results and clustering of cells from our overall lung scRNA-seq data set can be found in our prior publication (7).
Differential Expression Analysis
Expression data were normalized, filtered, and clustered using the R (https://www.r-project.org/) package Seurat (https://satijalab.org/seurat/) (9). Cell types were identified on the basis of known cell type marker genes such as Cdh5 (cadherin 5) for endothelial cells. Endothelial clusters were then used for all downstream analyses. Differentially expressed genes (DEGs) of individual endothelial subclusters were determined using MAST (https://github.com/RGLab/MAST) (10). To functionally annotate DEGs, gene set enrichment analysis (GSEA) was performed using R (https://www.r-project.org/) package fgsea (https://github.com/ctlab/fgsea) version 1.18.0.
Cell–Cell Communication Analysis
CellPhoneDB, a repository of ligands and receptors, was used to infer cell–cell communication on the basis of mean expression of multisubunit ligand–receptor complexes in our scRNA-seq data (11).
Trajectory Reconstruction
To predict differentiation states of the endothelial subclusters, we used cellular trajectory reconstruction analysis using gene counts and expression (CytoTRACE), a computational framework that leverages single-cell gene counts as a determinant of developmental potential, covariant gene expression, and local neighborhoods of transcriptionally similar cells to predict ordered differentiation states from scRNA-seq data (12).
Independent Data Sets for Validation
Select genes were queried in independent bulk and single-cell transcriptomic data sets of human lung, blood, and in vitro endothelial cell samples from Gene Expression Omnibus or hosted on laboratory web servers.
Immunofluorescence
Distal pulmonary arteries (external diameter <500 μm in human, <200 μm inrat) were visualized by staining of lung sections from control, MCT, and SuHx rats and donor and PAH subjects from the Pulmonary Hypertension Breakthrough Initiative using fluorescently labeled antibodies (see Table E2 in the data supplement). Images were acquired using a confocal microscope and analyzed in ImageJ (https://imagej.nih.gov/).
FACS
Lung single-cell suspensions from control rats were stained with a mixture of fluorochrome-labeled antibodies (see Table E2) and sorted at the University of California, Los Angeles, Flow Cytometry Core Laboratory on a FACSAria III (BD Biosciences) using a 100-μm nozzle and 23 psi pressure to isolate TM4SF1+CD31+ cells.
Endothelial-Specific Tubulogenesis Assay
Adapted from a previously described EPC angiogenesis assay (13), TM4SF1+CD31+ cells sorted from control rats were cocultured with donor human lung fibroblasts obtained from the Pulmonary Hypertension Breakthrough Initiative and fixed with paraformaldehyde after 6–72 hours. Cells were then stained with endothelial markers Dil-acetylated low-density lipoprotein (Kalen) and fluorescently labeled isolectin GS-IB4 (Invitrogen) and imaged using a confocal microscope.
Results
Tm4sf1-marked Subpopulation of Endothelial Cells Expresses a Distinct Transcriptomic Signature Relevant to PAH
The transcriptomes of 758 endothelial cells generated by scRNA-seq of 18 lungs from MCT, SuHx, and control rats (n = 6/group) (7) were clustered, yielding three distinct subclusters: 454 endothelial arterial type 1 (EA1) cells, 255 endothelial arterial type 2 (EA2) cells, and 49 endothelial capillary cells (Figure 1A). Each of the three subpopulations was represented in MCT, SuHx, and control lungs (Figure 1B). Subcluster-specific markers were identified such as Nostrin (nitric oxide synthase trafficking) for EA1, Tm4sf1 for EA2, and Car4 (carbonic anhydrase 4), a known marker for endothelial capillary cells (Figures 1C–1E). Arterial markers such as Sox17 (SRY-box transcription factor 17) (Figure 1F) and Efnb2 (ephrin B2) (see Figure E1) were also identified, confirming the arterial origin of EA1 and EA2 (14).
Figure 1.
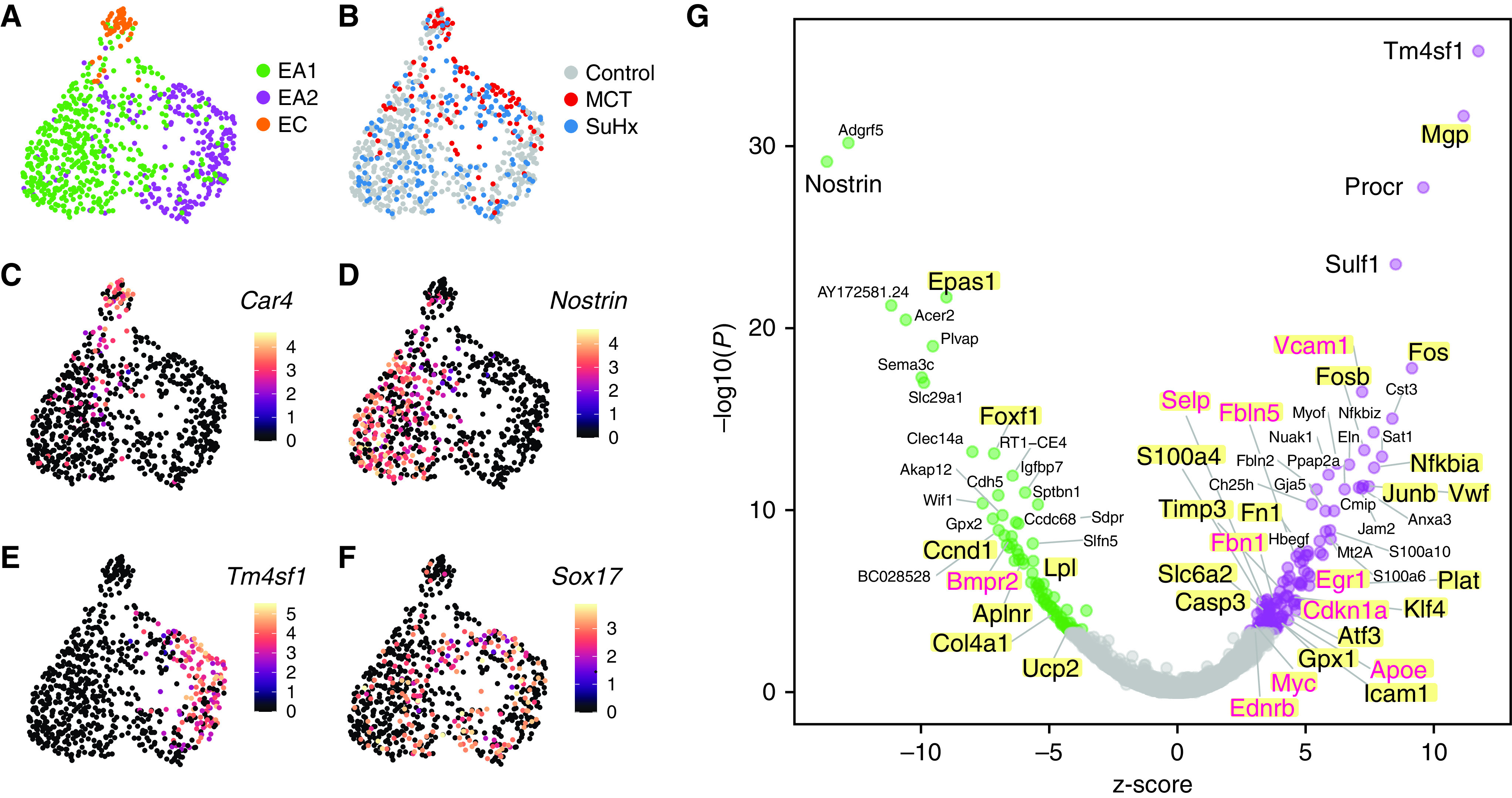
Tm4sf1 (transmembrane 4 L six family member 1)–marked subpopulation of endothelial cells expresses a distinct transcriptomic signature relevant to PAH. (A–F) Uniform manifold approximation and projection (UMAP) plots showing clustering of 758 endothelial cell transcriptomes from 18 rat lungs with individual cells colored by (A) subpopulation, (B) disease condition (n = 6/group), and expression amounts of the following marker genes: (C) Car4 for EC, (D) Nostrin for endothelial arterial type 1 (EA1), (E) Tm4sf1 for endothelial arterial type 2 (EA2), and (F) Sox17 as a known arterial marker expressed in both EA1 and EA2. Expression amounts are represented as log normalized counts. (G) Volcano plot showing DEGs in 255 EA2 cells compared with 454 EA1 cells, where x-axis represents MAST z-scores and y-axis indicates −log10(P). Significant upregulated (z > 0) or downregulated (z < 0) genes with FDR < 0.05 are shown as purple and green dots, respectively. Select top DEGs are labeled with their gene names. DEGs highlighted in yellow represent human PAH-associated genes from either (black text) or both (red text) CTD and DisGeNET database. (H–K) Dot plots showing gene set enrichment analysis of the EA2 DEG signature using (H) GO, (I) hallmark, (J) HuBMAP, and (K) DisGeNET gene sets, where x-axis represents normalized enrichment scores (NESs) in which NES greater than or less than zero and FDR < 0.05 represent gene sets significantly enriched in up- or downregulated genes, respectively, and are colored in purple or green, respectively. The y-axis represents gene sets ordered by their NESs. Select gene sets are labeled and numbered by their ordering as top gene sets enriched in up- or downregulated genes. Dots larger in size represent higher −log10(FDR) values. Acer2 = alkaline ceramidase 2; Adgrf5 = adhesion G protein-coupled receptor F5; Akap12 = A-kinase anchoring protein 12; Anxa3 = annexin A3; Aplnr = apelin receptor; Apoe = apolipoprotein E; Atf3 = activating transcription factor 3; BC028528 = cDNA sequence BC028528; Bmpr2 = bone morphogenetic protein receptor type 2; Cap = cyclase associated actin cytoskeleton regulatory protein; Car4 = carbonic anhydrase 4; Casp3 = caspase 3; Ccdc68 = coiled-coil domain containing 68; Ccnd1 = cyclin D1; Cdh5 = cadherin 5; Cdkn1a = cyclin-dependent kinase inhibitor 1A; Ch25h = cholesterol 25-hydroxylase; Clec14a = C-type lectin domain containing 14A; Cmip = c-Maf-inducing protein; Col4a1 = collagen type IV alpha 1 chain; Cst3 = cystatin C; CTD = Comparative Toxicogenomics Database; DEG = differentially expressed gene; EC = endothelial capillary; Ednrb = endothelin receptor type B; Egr1 = early growth response 1; Eln = elastin; Epas1 = endothelial PAS domain protein 1; ER = endoplasmic reticulum; Fbln = fibulin; Fbn1 = fibrillin 1; FDR = false discovery rate; Fezf2 = Fez family zinc finger 2; Fn1 = fibronectin 1; Fos = Fos protooncogene, AP-1 transcription factor subunit; Fosb = FosB protooncogene, AP-1 transcription factor subunit; Foxf1 = forkhead box F1; Gja5 = gap junction protein, alpha 5; GO = Gene Ontology; Gpx = glutathione peroxidase; Hbegf = heparin binding EGF like growth factor; Hs3st3A1 = heparan sulfate-glucosamine 3-sulfotransferase 3A1; HuBMAP = Human BioMolecular Atlas Program; Icam1 = intercellular adhesion molecule 1; Igfbp7 = insulin-like growth factor binding protein 7; Jam2 = junctional adhesion molecule 2; Junb = JunB protooncogene, AP-1 transcription factor subunit; Klf4 = KLF transcription factor 4; Linc01107 = long intergenic non–protein-coding RNA 1107; Lpl = lipoprotein lipase; MCT = monocrotaline; Mgp = matrix Gla protein; Mt2a = metallothionein 2A; Myc = MYC protooncogene, bHLH transcription factor; Myof = myoferlin; Nfkbia = NFKB inhibitor alpha; Nfkbiz = NFKB inhibitor zeta; Nostrin = nitric oxide synthase trafficking; Nuak1 = NUAK family kinase 1; PAH = pulmonary arterial hypertension; Plat = plasminogen activator, tissue type; Plvap = plasmalemma vesicle associated protein; Ppap2a = phosphatidic acid phosphatase type 2A; Procr = protein C receptor; RT1-CE4 = RT1 class I, locus CE4; S100 = S100 calcium-binding protein; Sat1 = spermidine/spermine N1-acetyl transferase 1; Sdpr = caveolae associated protein 2; Selp = selectin P; Sema3c = semaphorin 3C; Slc29a1 = solute carrier family 29 member 1; Slc6a2 = solute carrier family 6 member 2; Slfn5 = schlafen family member 5; Sox17 = SRY-box transcription factor 17; Sptbn1 = spectrin, beta, nonerythrocytic 1; Srgn = serglycin; SuHx = Sugen–hypoxia; Sulf1 = sulfatase 1; TGF = transforming growth factor; Timp3 = TIMP metallopeptidase inhibitor 3; Ucp2 = uncoupling protein 2; Vcam1 = vascular cell adhesion molecule 1; Vwf = von Willebrand factor; Wif1 = Wnt inhibitory factor 1.
Given that the pathological hallmark of vascular remodeling in PAH occurs predominantly in the pulmonary arteries, we then evaluated the differences in gene expression between the two arterial subclusters EA1 and EA2, having previously shown that many known PAH genes are dysregulated within EA1 and EA2 cells of SuHx and MCT compared with control rats (7). We found a total of 196 DEGs (false discovery rate < 0.05), with 117 genes upregulated and 79 genes downregulated in EA2 compared with EA1 (Figure 1G; see Table E1). Tm4sf1 was the top upregulated DEG in EA2 versus EA1, and its expression was relatively specific to EA2 compared with other lung cell types recovered in our data set (see Figure E2). Tm4sf1 encodes the cell-surface protein transmembrane 4 L six family member 1, which plays a role in the regulation of cell development, growth, and motility and has been strongly implicated in various cancers (15) but has never before been studied in PAH. Given the importance of cancer-related processes and their overlap with PAH, we focused on the Tm4sf1-marked subpopulation of endothelial cells, EA2. Nostrin was the top downregulated gene in EA2 versus EA1; it encodes nitric oxide synthase trafficking, an adaptor protein that binds to endothelial nitric oxide synthase and attenuates NO production.
We then queried a single-cell atlas of human lung endothelial cells integrated from six independent scRNA-seq data sets spanning 73 individuals (14). A uniform manifold approximation and projection plot showed that focal areas protruding from the perimeter of the major clusters, including the arterial cluster, had particularly high expression of TM4SF1 and low expression of NOSTRIN (see Figure E3). This finding supports the presence of an EA2-like subpopulation in human lungs.
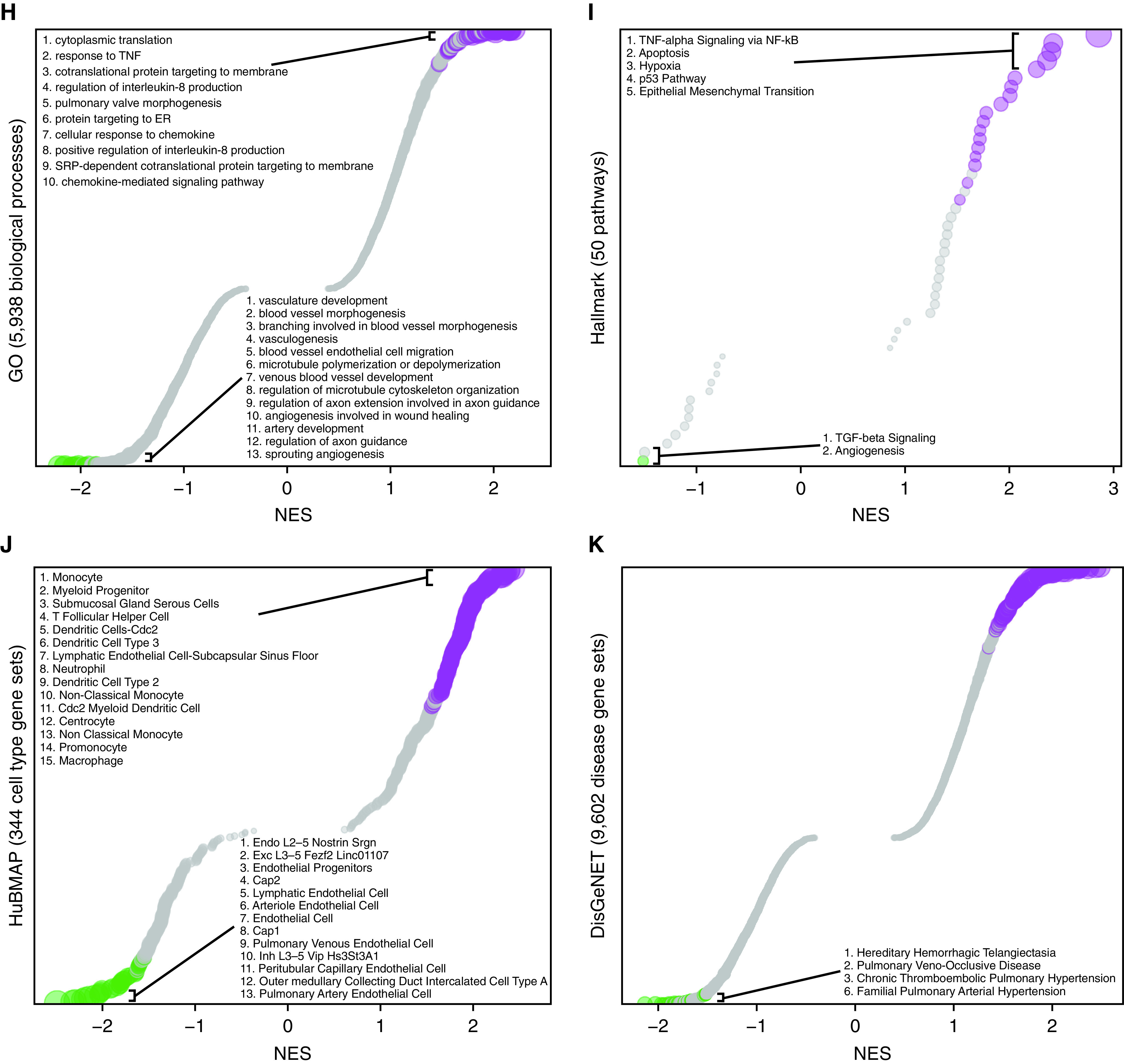
When intersecting EA2 DEGs with known PAH genes curated from DisGeNET and the Comparative Toxicogenomics Database, we found that a number of DEGs are also known PAH genes, such as Bmpr2 (bone morphogenetic protein receptor type 2), which was decreased in EA2 compared with EA1 (Figure 1G). We then used GSEA to determine what biological processes, pathways, cell types, and diseases are enriched in the gene signature of EA2 (vs. EA1) cells. Among 5,938 biological processes from Gene Ontology, cytoplasmic translation and response to TNF were most enriched in upregulated EA2 genes. Furthermore, angiogenesis-related processes were most enriched in downregulated EA2 genes (Figure 1H). Of the 50 hallmark pathways (16), many known to be involved in PAH were enriched in genes upregulated in EA2, such as TNF-α/NF-κB signaling, apoptosis, hypoxia, p53 pathway, and epithelial-to-mesenchymal transition (EMT) (Figure 1I).
To determine what cell type signatures may be enriched in EA2 versus EA1 DEGs, we tested 344 cell type–specific gene sets from the Human BioMolecular Atlas Program (17) and found that immune cell signatures, particularly from monocytes, were most enriched in genes differentially upregulated in EA2, whereas endothelial signatures were most enriched in genes upregulated in EA1 (Figure 1J).
In addition, we found that across 9,602 disease-specific gene sets from DisGeNET, genes downregulated in EA2 versus EA1 were most enriched for diseases associated with primary or secondary forms of pulmonary hypertension: hereditary hemorrhagic telangiectasia, pulmonary venoocclusive disease, chronic thromboembolic pulmonary hypertension, and familial PAH (Figure 1K). Thus, not only was Bmpr2 downregulated in EA2, as it is in human PAH lungs (18), but the downregulated EA2 signature was also highly associated with PAH.
EA2-Specific Cell-to-Cell Signaling Networks Are Activated in PAH Rat Models
Having identified a transcriptionally distinct subpopulation of lung endothelial cells relevant to PAH, we next hypothesized that communication between EA2 and other cell types occurs in a disease-specific manner, given that many other cell types besides endothelial cells have also been implicated in the complex pathobiology of PAH. Using CellPhoneDB to predict ligand–receptor interactions between cell types, we found overall increased intercellular communication among various immune, mesenchymal, and epithelial cells in the lungs of PAH rat models compared with control (Figure 2A). When quantifying the number of statistically significant ligand–receptor interactions between endothelial cells and other cell types, EA2 exhibited stronger cell-to-cell signaling in MCT and SuHx lungs compared with control that was not seen in EA1 nor endothelial capillary cells (Figure 2B).
Figure 2.
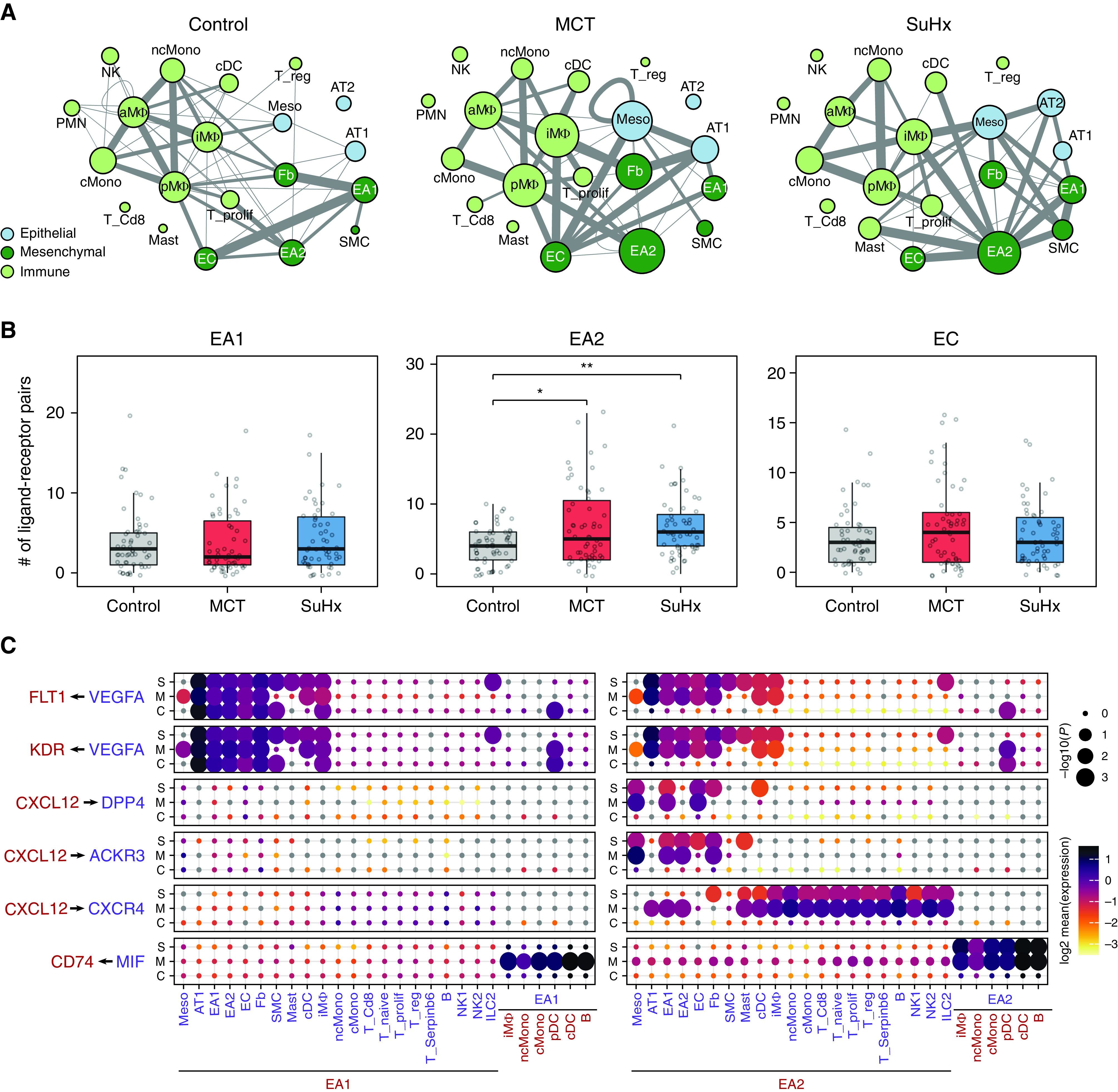
EA2-specific cell-to-cell signaling networks are activated in PAH. (A) Network visualizations of control, MCT, and SuHx lungs, where nodes are represented as cell types and edge weight is proportional to the number of distinct and statistically significant ligand–receptor interaction pairs between two cell types as determined by CellPhoneDB. Thicker edges correspond to a larger number of ligand–receptor pairs. Node size is proportional to the sum of edge weights representing a given cell type’s interactions with all other cell types. The top 10% of edges by weight are shown for each condition. (B) Box plots comparing cell–cell interactions, where each dot represents the number of statistically significant ligand–receptor interaction pairs between endothelial subpopulations (EA1, EA2, and EC) and all other cell types in control, MCT, and SuHx lungs. *P < 0.05 and **P < 0.01 (Wilcoxon rank sum test). (C) Dot plots showing the log2 mean expression of select ligand–receptor pairs in control, MCT, and SuHx between select cell types and EA1 or EA2. Sizes of dots are proportional to strength of P value. ACKR3 = atypical chemokine receptor 3; aMΦ = alveolar macrophages; AT1 = alveolar type 1 cells; AT2 = alveolar type 2 cells; C = control; cDC = conventional dendritic cells; cMono = classical monocytes; DPP4 = dipeptidyl peptidase 4; Fb = fibroblasts; FLT1 = fms-related receptor tyrosine kinase 1; ILC2 = type 2 innate lymphoid cells; iMΦ = interstitial macrophages; KDR = kinase insert domain receptor; M = monocrotaline; meso = mesothelial; MIF = macrophage migration inhibitory factor; ncMono = nonclassical monocytes; NK1 = natural killer cells 1; NK2 = natural killer cells 2; pDC = plasmacytoid dendritic; PMN = neutrophils; pMΦ = proliferating macrophages; S = Sugen–hypoxia; Serpinb6 = serpin family B member 6; SMC = smooth muscle cells; T_Cd8 = CD8-positive T cells; T_naive = naive T cells; T_prolif = proliferating T cells; T_reg = regulatory T cells; VEGFA = vascular endothelial growth factor A.
We further examined specific ligand–receptor interactions and found that signaling molecules known to play a role in PAH, such as VEGFA (vascular endothelial growth factor A) (19), CXCL12 (20, 21), and MIF (macrophage migration inhibitory factor) (22) were not only differentially active in PAH models compared with control but also specific to cell–cell communication involving EA2 but not EA1 (Figure 2C) nor endothelial capillary cells (see Figure E4). For instance, VEGFA from cell types such as fibroblasts and interstitial macrophages was predicted to interact in a PAH- and EA2-specific manner with its cognate receptors FLT1 (fms-related receptor tyrosine kinase 1) (also known as VEGFR-1) and KDR (kinase insert domain receptor) (also known as VEGFR-2). CXCL12 signaling was highly specific to EA2, with distinct cell–cell communication profiles specific to the particular receptor expressed on target cell types. For example, EA2’s CXCL12 interaction with CXCR4 was observed in various PAH immune cell types of both lymphoid and myeloid lineage, whereas its interaction with ACKR3 (atypical chemokine receptor 3) (also known as CXCR7) and DPP4 (dipeptidyl peptidase 4) was noted in PAH fibroblasts, endothelial cells, and mesothelial cells (Figure 2C). MIF is a key proinflammatory immune regulator whose interaction with its receptor CD74 on myeloid and B cells was statistically significant for both MCT and SuHx EA2 compared with control, a pattern that was unique to EA2 (Figure 2C). Overall, the ligand–receptor analysis demonstrated that immune signaling across cell types involving EA2 was particularly activated in PAH rat models.
Trajectory Analysis Suggests That EA2 May Have a Stem/Progenitor Cell Phenotype
Given the important role of CXCL12 and its chemokine receptor CXCR4 in the homing and function of EPCs (23) and the specificity of this signaling axis for EA2 communication in our rat lung scRNA-seq analysis, we asked whether EA2 might have a stem/progenitor cell phenotype. We used CytoTRACE (12), an unbiased computational method that leverages the number of expressed genes per cell as a determinant of developmental potential, to predict the differentiation state of single cells from our endothelial scRNA-seq data (Figure 3A). Compared with EA1 and endothelial capillary cells, we found that EA2 cells had significantly higher CytoTRACE scores corresponding to a less differentiated state (Figure 3B). Comparing CytoTRACE scores by condition, endothelial cells from MCT and SuHx lungs were predicted to be less differentiated compared with control.
Figure 3.

Trajectory analysis reveals that EA2 has a stem/progenitor cell phenotype. (A) UMAP plots showing clustering of endothelial cells colored by cellular trajectory reconstruction analysis using gene counts and expression (CytoTRACE) score (left) and cell type (right). Higher CytoTRACE scores represent a less differentiated state. (B and C) Box plots comparing cell CytoTRACE scores grouped by (B) endothelial subpopulation and (C) disease condition. ****P < 0.0001 (Wilcoxon rank-sum test). (D) Heatmap showing normalized expression of top genes most positively or negatively correlated with cell CytoTRACE scores. Each column is a cell ordered from left to right by decreasing CytoTRACE scores. Cell annotations for cell type and condition are shown at the bottom. Amigo2 = adhesion molecule with Ig-like domain 2; CytoTRACE = cellular trajectory reconstruction analysis using gene counts and expression; Gata2 = GATA binding protein 2; Gpihbp1 = glycosylphosphatidylinositol anchored high density lipoprotein binding protein 1; Lyve1 = lymphatic vessel endothelial hyaluronan receptor 1; Mapt = microtubule-associated protein tau; Mettl7a = methyltransferase like 7A; Rpl18a = ribosomal protein L18A; Rplp = ribosomal protein lateral stalk; Rps = ribosomal protein S; Thbd = thrombomodulin; Tmem100 = transmembrane protein 100; Tmsb4x = thymosin beta 4, X-linked.
To identify markers of the less differentiated phenotype, all 12,116 genes with detectable expression in our data set were rank ordered on the basis of their correlation with CytoTRACE scores across all 758 endothelial cells analyzed. Ribosomal genes correlated most strongly with a less differentiated state (Figure 3D) and thus were downregulated with a more differentiated state, as has been reported for stem cell differentiation (24, 25). GSEA revealed many Gene Ontology biological processes significantly enriched in genes correlated with a less differentiated state compared with a more differentiated state (219 vs. 0 of 5,994 total gene sets) (see Figure E5). Top enriched biological processes included cotranslational protein targeting to membrane and cytoplasmic translation, which were also top enriched Gene Ontology biological processes of the EA2 gene signature (Figure 1H).
Tm4sf1 Is a Marker for Hematopoietic Stem Cells and Is Upregulated in the Peripheral Blood of Patients with PAH
Given prior studies implicating circulating EPCs in patients with PAH (23, 26–28), we asked whether Tm4sf1, the most highly specific marker for rat lung EA2, could also be a marker for circulating stem/progenitor cells in human PAH. Using a data set of 7,551 human blood cell transcriptomes representing 43 cell type clusters derived from 21 healthy donors (29), we found that TM4SF1 was specifically expressed in and 1 of 19 marker genes for hematopoietic stem cell (HSC)/multipotent progenitor 1 cells (false discovery rate = 2.0 × 10−15) (Figure 4A), further suggesting that Tm4sf1-marked EA2 cells may be a stem/progenitor subpopulation of endothelial cells.
Figure 4.
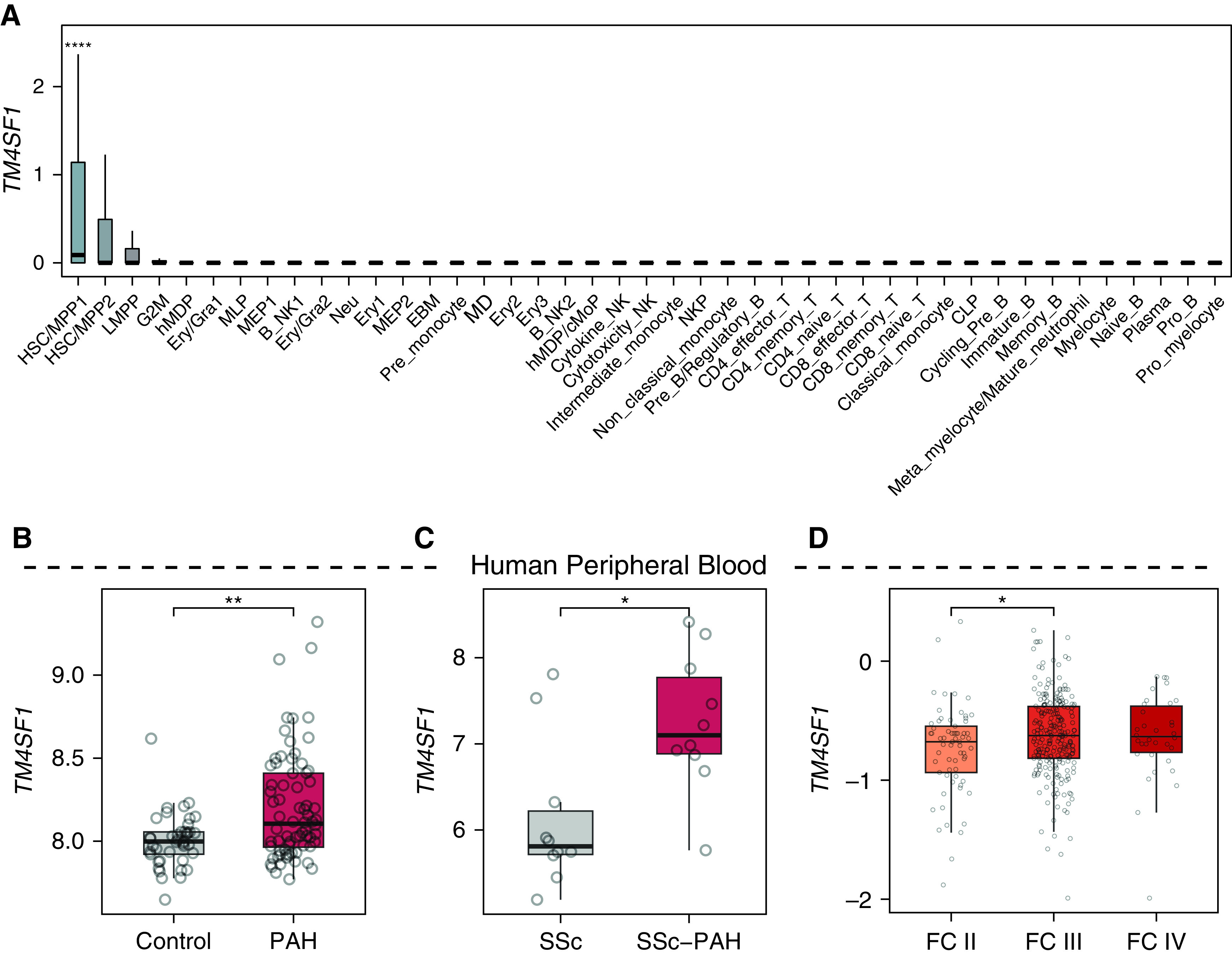
Tm4sf1 is a marker for hematopoietic stem cells and is upregulated in the peripheral blood of patients with PAH. (A) Box plots showing the expression of TM4SF1 in a data set of 7,551 human blood cell transcriptomes representing 43 cell type clusters derived from 21 healthy donors (29). (B–D) Box plots showing expression of TM4SF1 in peripheral blood mononuclear cells of (B) 72 patients with PAH compared with 41 healthy control subjects (30), (C) 10 patients with SSc-associated PAH compared with 10 patients with SSc without PAH (31), and (D) in whole blood of 65 patients with World Health Organization functional class (FC) II PAH, 239 with FC III PAH, and 34 with FC IV PAH. P values in B and C were obtained from Gene Expression Omnibus 2R and determined by Wilcoxon rank sum test in D: *P < 0.05 and **P < 0.01. CLP = common lymphoid progenitor; cMoP = common monocyte progenitor; EBM = erythroid-basophil-megakaryocytic; Ery = erythrocyte; Gra = granulocyte; hMDP = human monocyte–dendritic progenitor; HSC = hematopoietic stem cells; LMPP = lymphomyeloid-primed progenitor; MD = monocyte-dendritic; MEP = megakaryocytic–erythroid progenitor; MLP = multilymphoid progenitor; MPP = multipotent progenitor; NK = natural killer; NKP = natural killer lineage-restricted progenitor; SSc = systemic sclerosis.
Supporting the relevance of Tm4sf1 to PAH, we found in two independent microarray data sets that TM4SF1 was significantly upregulated in circulating peripheral blood mononuclear cells of patients with PAH compared with healthy control subjects (n = 72 and n = 41, respectively) (30) and in patients with systemic sclerosis (SSc)–associated PAH compared with those with SSc without PAH (n = 10 and n = 10, respectively) (31) (Figures 4B and 4C). Furthermore, in a third data set of whole-blood RNA-seq, patients with PAH with higher TM4SF1 expression were associated with worse World Health Organization functional class (n = 65, n = 239, and n = 34 for functional classes II, III, and IV, respectively) (Figure 4D) (32).
Tm4sf1 Is Downregulated in the Lungs of Patients with PAH and in Endothelial Cells from Patients with PAH and Models
Having established that the EA2 marker Tm4sf1 is upregulated in PAH peripheral blood and associated with disease severity, we next investigated whether Tm4sf1 is dysregulated in PAH lungs. Given that the power to detect PAH-specific differences in Tm4sf1 expression in our rat lung scRNA-seq data set may have been limited by relatively lower yield of endothelial cells from the tissue dissociation into single cells, we evaluated whether Tm4sf1 expression is dysregulated in other data sets. At the whole–lung tissue level, we found that Tm4sf1 was upregulated in both MCT and SuHx rats by bulk RNA-seq (Figure 5A). However, in human lung tissue, we found that TM4SF1 is downregulated in human PAH lungs (33–35) (Figures 5B–5E). At the cell-specific level, we found that contrary to the rat lung tissue level, Tm4sf1 was downregulated in a subpopulation of endothelial cells most similar to EA2 in an scRNA-seq data set we reanalyzed of lineage-traced endothelial cells sorted from SuHx mouse lungs (6) (Figures 5F and E6). In human endothelial cells, we found downregulation of TM4SF1 in patients with PAH and models from multiple studies (5, 36, 37), including cells derived from circulating EPCs from SSc-associated PAH as well as in cells isolated from explanted lungs of idiopathic PAH in an scRNA-seq study (Figures 5G–5J).
Figure 5.
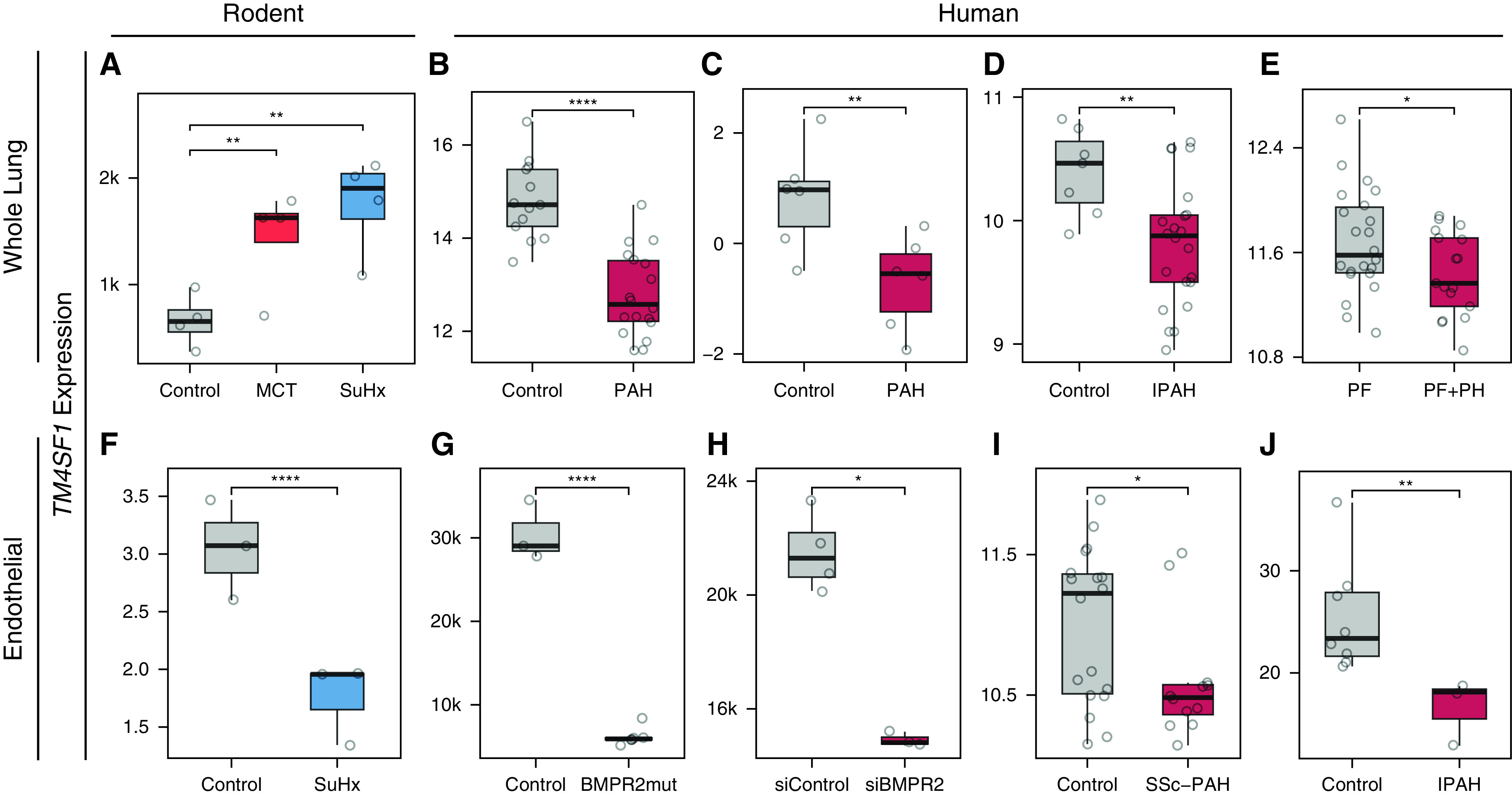
TM4SF1 is downregulated in the whole lung of patients with PAH and in endothelial cells from patients with PAH and models. (A) Box plot showing Tm4sf1 expression in control, MCT, and SuHx lungs by bulk RNA-seq (n = 4/group). (B–E) Box plots showing TM4SF1 expression in four independent human whole-lung microarray data sets: (B) 18 patients with PAH versus 13 control subjects (49), (C) 2 patients with PAH versus 2 control subjects profiled at three time points (0, 3, and 6 h) in an ex vivo lung perfusion study (Gene Expression Omnibus accession: GSE69416), (D) 22 female patients with idiopathic PAH versus 7 female control subjects (34), and (E) 17 patients with pulmonary fibrosis (PF) with severe pulmonary hypertension (PH) (mPAP > 40 mm Hg) versus 22 patients with PF without PH (mPAP < 20 mm Hg) (35). (F) Box plot showing expression of Tm4sf1 in a subcluster of 2,254 endothelial cells marked by Tm4sf1 from an scRNA-seq study of lung endothelial cells FACS purified from endothelial lineage-traced SuHx versus control mice (n = 3/group) (see Figure E6) (6). Expression is shown as an average across all cells within each animal. (G–J) Box plots showing TM4SF1 expression in four independent human endothelial data sets: (G) two CRISPR-generated BMPR2 mutants versus wild-type umbilical vein endothelial cells profiled by RNA-seq in three replicate in vitro experiments (36); (H) BMPR2- versus control siRNA–transfected pulmonary artery endothelial cells profiled by microarray in three and four replicate in vitro experiments, respectively (37); (I) endothelial progenitor cell–derived endothelial cells from peripheral blood of six patients with SSc-associated PAH and nine control subjects profiled by microarray at baseline and after hypoxic exposure in vitro (Gene Expression Omnibus accession: GSE73674); and (J) averaged expression of lung endothelial cells profiled by scRNA-seq from six PAH lungs versus three control subjects (5). P values were determined by DESeq2 for A and G; obtained from the National Center for Biotechnology Information’s Gene Expression Omnibus 2R for B–E, H, and I; and determined by Wilcoxon rank sum test for F and J: *P < 0.05 and **P < 0.01; ****FDR < 0.05. BMPR2mut = BMPR2 mutant; IPAH = idiopathic pulmonary arterial hypertension; mPAP = mean pulmonary arterial pressure; RNA-seq = RNA sequencing; siBMPR2 = BMPR2 siRNA; siControl = control siRNA.
EA2 Signature Genes Are Differentially Regulated in the Lungs of Patients with PAH and in PAH Models of Human Endothelial Cells
We next investigated whether other EA2 signature genes are also dysregulated in PAH lungs. We found that two other top upregulated genes in EA2, Procr (protein C receptor) and Sulf1 (sulfatase 1), were upregulated in the lungs of patients with PAH in two independent microarray data sets (Figures 6A–6D) (33, 34, 38).
Figure 6.
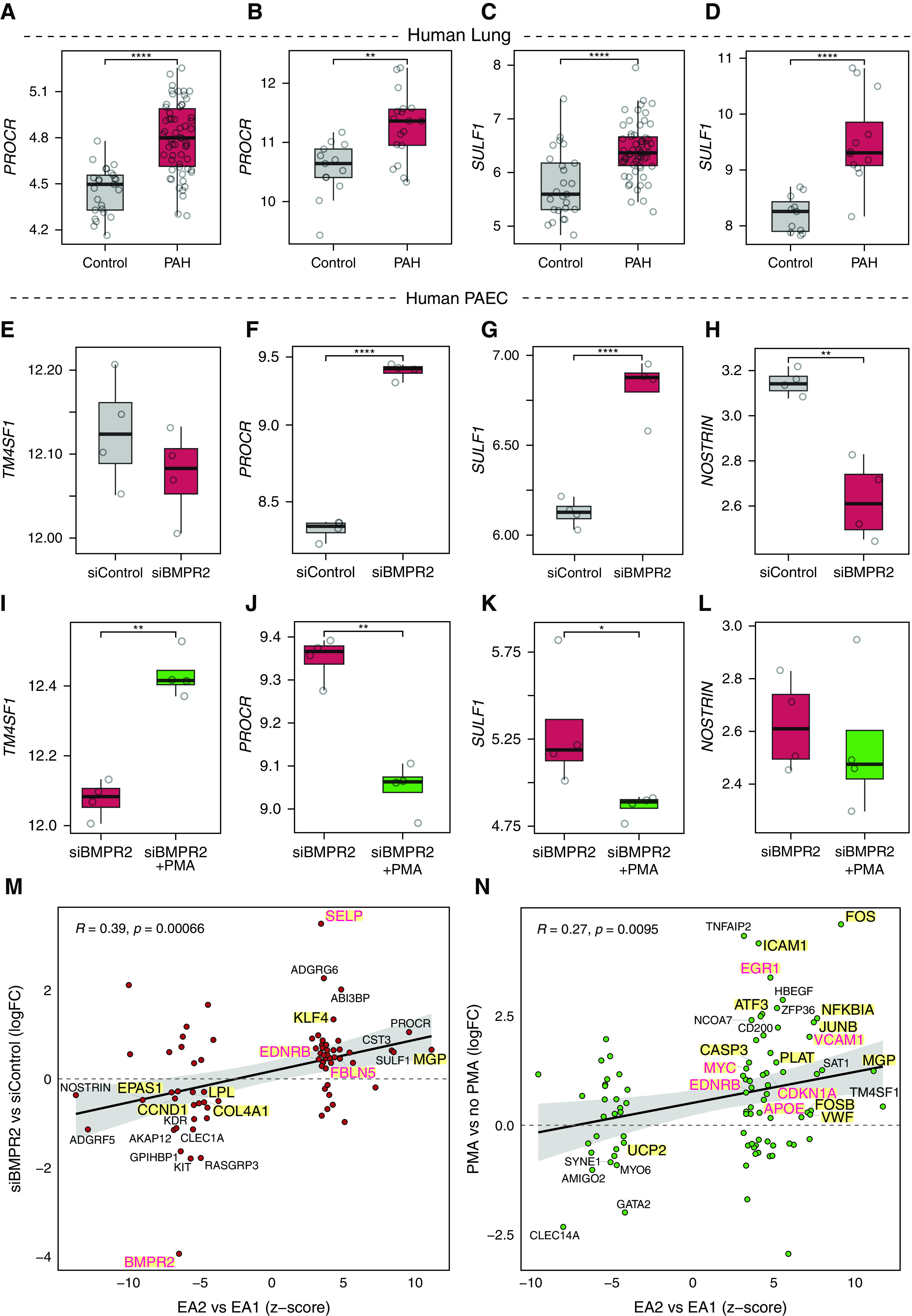
EA2 signature genes are differentially regulated in the lungs of patients with PAH and by PAH models in human endothelial cells. (A and C) Box plots showing expression of EA2 markers PROCR and SULF1 in 58 PAH lungs versus 25 control subjects (34). (B) Box plot showing expression of PROCR in 18 PAH lungs versus 13 control subjects (49). (D) Box plot showing expression of SULF1 in 12 PAH lungs versus 11 control subjects (38). (E–H) Box plots showing expression of (E) TM4SF1, (F) PROCR, (G) SULF1, and (H) NOSTRIN in four BMPR2- versus four control siRNA–transfected primary human PAECs from four donors (39). (I–L) Box plots showing expression of (I) TM4SF1, (J) PROCR, (K) SULF1, and (L) NOSTRIN in four PMA-exposed versus four nonexposed BMPR2-silenced PAECs from four donors (39). (M and N) Scatterplots showing correlation between DEGs in EA2 versus EA1 (x-axis) and DEGs from either (M) BMPR2-silenced PAECs (n = 8/group) or (N) PMA-exposed PAECs (n = 8/group) (39) with select top DEGs labeled by their gene names. DEGs highlighted in yellow represent human PAH-associated genes from either (black text) or both (red text) CTD and DisGeNET databases. P values were obtained from Gene Expression Omnibus 2R for B and D–N, determined by DESeq2 for A, and determined by Wilcoxon rank sum test for C: *P < 0.05 and **P < 0.01; ****FDR < 0.05. ABI3BP = ABI family member 3 binding protein; ADGRG6 = adhesion G protein-coupled receptor G6; CLEC1A = C-type lectin domain containing 1A; KIT = KIT proto-oncogene, receptor tyrosine kinase; logFC = log fold change; MYO6 = myosin VI; NCOA7 = nuclear receptor coactivator 7; PAEC = pulmonary artery endothelial cell; RASGRP3 = RAS guanyl releasing protein 3; SYNE1 = spectrin repeat containing nuclear envelope protein 1; TNFAIP2 = TNF alpha induced protein 2; ZFP36 = ZFP36 ring finger protein.
Having determined that top EA2 genes are dysregulated in the lungs of patients with PAH and given that Bmpr2, the most well-established causal gene in PAH, is downregulated in EA2 compared with EA1 (Figure 1G), we next asked whether EA2 genes are regulated by Bmpr2. We analyzed a microarray of siRNA knockdown of BMPR2 in primary human pulmonary artery endothelial cells (PAECs) in vitro (39) and found no change in TM4SF1, but PROCR and SULF1 were upregulated, consistent with upregulation in EA2 versus EA1 and PAH versus donor lungs (Figures 6E–6G). Furthermore, NOSTRIN, the top downregulated gene in EA2 versus EA1 (Figure 1G), was also downregulated in human PAECs upon BMPR2 knockdown (Figure 6H).
Given the strong connection between Tm4sf1 and various cancers (15), we then evaluated the response of Tm4sf1 and other signature genes of EA2 to PMA, a potent tumor promoter that activates PKC and downstream mitogen-activated protein kinase pathways. We found that TM4SF1 was upregulated in BMPR2-silenced human PAECs exposed to PMA, whereas PROCR, SULF1, and NOSTRIN were either downregulated or unchanged (Figures 6I–6L and E7). Overall, we found significant correlation between DEGs in EA2 (vs. EA1) and DEGs in human PAECs after either BMPR2 knockdown (Figure 6M) or PMA stimulation (Figure 6N) suggesting that portions of the EA2 signature can be explained by both downregulation of Bmpr2 and activation of PKC. Overlapping DEGs were enriched for known PAH genes (Figures 6M and 6N) and pathways (see Figure E8) in both conditions, such as EMT in BMPR2-silenced PAECs and TNF-α/NF-κB signaling in PMA-simulated PAECs.
TM4SF1+ Endothelial Cells Are Present in Distal Pulmonary Arteries and Form Tubules In Vitro
We next confirmed that TM4SF1 protein is expressed in situ in endothelial cells within both human and rat distal pulmonary arteries, the main site of pathology in PAH (Figures 7A and 7B). We then isolated TM4SF1+CD31+ endothelial cells from rat lungs by FACS while excluding EPCAM+ (epithelial cell adhesion molecule) cells given TM4SF1’s expression in lung epithelial cells (Figures 7C, 7D, and E11) (40). As TM4SF1 is also a marker of hematopoietic stem cells, we also assessed whether TM4SF1+CD31+ endothelial cells might also be positive for the hematopoietic marker CD45. We found that among CD31+ cells, 32% were positive for TM4SF1 and 9% were positive for CD45. Among CD45+CD31+ cells, 78% were also positive for TM4SF1, supporting a possible hemopoietic origin of EA2 cells. We then cocultured TM4SF1+CD31+ cells with human lung fibroblasts adapted from an assay developed to functionally test the angiogenic properties of EPCs (13). Using endothelial-specific markers Dil-acetylated low-density lipoprotein and isolectin GS-IB4 from Griffonia simplicifolia, we observed endothelial formation of tubules suggesting angiogenic capacity in EA2 (Figures 7E and E10).
Figure 7.
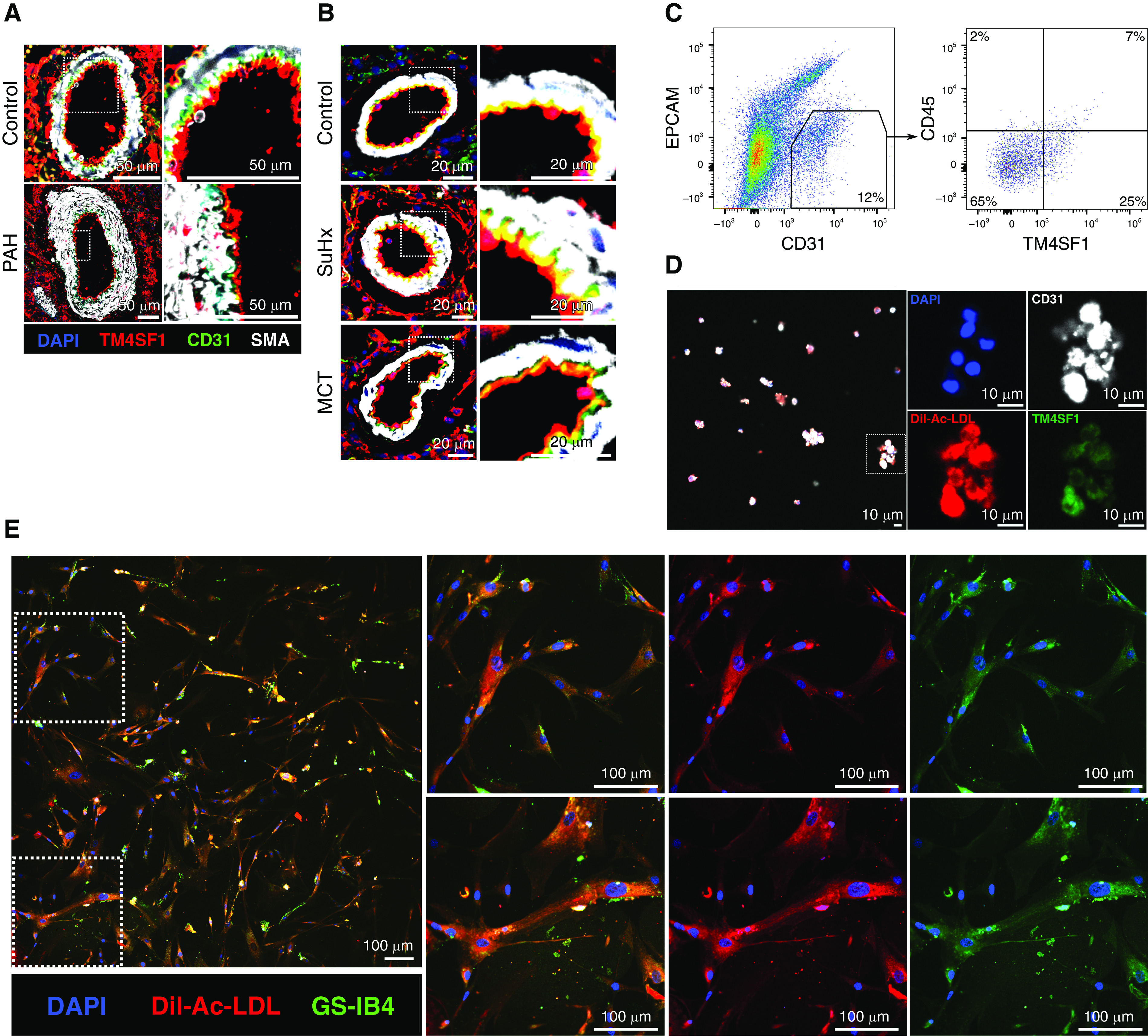
Visualization of TM4SF1+ endothelial cells in situ, after isolation by FACS, and after coculture with fibroblasts. (A and B) Representative immunofluorescence images of distal pulmonary arteries from lung sections of (A) a 31-year-old male donor control subject versus a 30-year-old man with idiopathic PAH and (B) control, SuHx, and MCT rats stained for TM4SF1 (red), CD31 (green), SMA (white), and DAPI (blue). Scale bars: A, 50 μm; B, 20 μm. (C) Gating strategy for the isolation of TM4SF1+CD31+EPCAM− (epithelial cell adhesion molecule) endothelial cells from rat lungs after gating for singlets and live cells. CD45 expression was also assessed. Percentages represent averages of three experiments. Full gating strategy is illustrated in Figure E9. (D) Representative confocal image of TM4SF1+CD31+ cells sorted from rat lungs and stained with antibodies to TM4SF1 (green) and CD31 (white) as well as Dil-acetylated low-density lipoprotein (Dil-Ac-LDL) (red) and DAPI (blue). Cells were pelleted after sorting, resuspended in a small volume of paraformaldehyde, and then left to dry on a coverslip before staining. Scale bars, 10 μm. (E) Representative confocal image of TM4SF1+CD31+ rat lung cells cocultured with human lung fibroblasts and stained after 6 hours with endothelial markers Dil-Ac-LDL (red), isolectin GS-IB4 (green), and DAPI (blue). Scale bars, 100 μm. SMA = smooth muscle actin.
Discussion
In this study, we uncover a Tm4sf1-marked subpopulation of rat lung endothelial cells and demonstrate its relevance to PAH through its distinct transcriptomic signature, cell signaling network, differential regulation of its marker genes in lung and blood, and angiogenic capacity in vitro.
Tm4sf1, the top marker of EA2, encodes a cell-surface protein and member of the transmembrane 4 superfamily that mediates signal transduction events that regulate cell development, activation, growth, and motility. TM4SF1 is highly expressed and associated with poor survival in a wide range of cancers (15). In cancer, TM4SF1 is known to promote PAH-relevant processes such as angiogenesis, but it has never previously been studied in PAH. We found that contrary to its high expression in cancer, TM4SF1 is downregulated in human lung tissue and endothelial cells from patients and PAH models, raising the question as to whether deficiency in TM4SF1 and/or EA2 cells contributes to the impairment of angiogenesis that plays a primary role in vascular remodeling and vessel rarefaction in PAH (41). Supporting this hypothesis, knockdown of TM4SF1 in PAECs leads to impaired angiogenesis and senescence (42), which has recently been shown to be causal in the transition from a reversible to the irreversible pulmonary vascular phenotype of end-stage PAH (43). Of note, transcriptional changes in Tm4sf1 in whole-lung tissue, including upregulation in PAH rat models, may be in part due to expression in other cell types, such as epithelial cells (see Figure E11), which has been previously reported (40).
Furthermore, we found that in contrast to PAH lung endothelial cells, TM4SF1 was upregulated in PAH circulation in multiple independent cohorts and associated with disease severity. We also found that TM4SF1 is a specific marker for HSCs, which is consistent with prior studies showing that circulating HSCs are increased (27, 44, 45) and correlate with disease severity in patients with PAH (27). HSCs reside in bone marrow, circulate in peripheral blood, and give rise to not only all blood cell lineages but also EPCs (46). Given that Tm4sf1 was also a marker for EA2 in our rat lung data set, this raises the possibility that EA2 might represent a stem/progenitor subpopulation of lung endothelial cells that derives from bone marrow via the circulating blood. Consistent with a potential stem/progenitor phenotype, our trajectory analysis predicted that EA2 cells are of an earlier differentiation state compared with other endothelial cells. Supporting this finding, recent studies have identified Tm4sf1 as a marker of multipotent endothelial precursor cells by scRNA-seq of lineage-traced mouse lungs (47) and a marker of alveolar epithelial progenitors in human and mouse lungs (40).
Although controversy and uncertainty persist in the definition and isolation of EPCs and their role in PAH (2, 3, 48), it is generally accepted there exist two distinct subsets: so-called late EPCs derived from endothelial lineage and early EPCs derived from hematopoietic lineage, with which EA2 shares features. Not only does EA2 share a novel marker with HSCs, suggesting the possibility of a hematopoietic origin, but we also found that a portion of TM4SF1+CD31+ cells isolated from rat lungs by FACS also expressed the hematopoietic marker CD45. Furthermore, the EA2 signature is highly enriched for myeloid signatures (Figure 1J), which is similarly noted in early EPCs. Renaming of early EPCs to “myeloid angiogenic cells” has been proposed to more precisely define their phenotype and function (3), such as secretion of key proangiogenic factors like CXCL12, a chemokine implicated in PAH (20, 21, 49), whose interactions with various target cells were unique to EA2 (Figure 2C). Consistent with our finding that EA2’s marker TM4SF1 is upregulated in PAH circulation, a previous study showed that early EPCs, but not late EPCs, are increased in PAH circulation as well (27). Among other studies of early EPCs, one revealed a trend toward an increase in PAH (26), and the other showed a decrease in PAH (28) but used an older isolation method now believed to be unreliable in EPC purification (48).
Further supporting EA2’s relevance in PAH, we observed a decrease in expression of Bmpr2 relative to EA1. We also found that a number of EA2 genes are differentially regulated after BMPR2 knockdown in human endothelial cells in the same direction as EA2’s signature, suggesting that these genes may be downstream of BMPR2 signaling. PAH-specific changes in BMPR2 and associated pathways within EA2 will require further investigation, such as with targeted transcriptional profiling of EA2 cells, to compare disease versus control lungs. For instance, although we observed the presence of an EA2-like subpopulation in a single-cell endothelial atlas spanning 73 human lungs, it was a relatively small subpopulation (see Figure E3).
Two of the genes that were upregulated in EA2 compared with EA1 and after BMPR2 knockdown were Sulf1 and Procr, which were also upregulated in explanted PAH lungs in multiple cohorts. In line with our findings, SULF1 was recently shown to be upregulated in remodeled pulmonary arterioles in patients with PAH and MCT and SuHx rats (50). Supporting the possibility of EA2’s stem/progenitor potential, Procr, encoding protein C receptor, was previously identified as a marker for murine HSCs (51) and blood vascular endothelial stem cells (52) which displayed EMT and angiogenic signatures. Whereas upregulated EA2 genes (relative to EA1) were enriched for EMT, genes downregulated in EA2 were enriched for angiogenesis (Figures 1H and 1I). For example, the top downregulated gene in EA2, NOSTRIN, is known to inhibit angiogenesis (53). Downregulation of NOSTRIN thus supports an angiogenic phenotype of EA2. In addition, TM4SF1 and many other EA2 signature genes were upregulated upon stimulation with PMA, which is an activator of PKC, an important promoter of angiogenesis (54). Moreover, when we evaluated TM4SF1+CD31+ rat lung cells using an assay developed to functionally characterize the angiogenic properties of EPCs (13), tubulogenesis was observed in coculture with human lung fibroblasts. The proangiogenic profile of EA2 and its top marker TM4SF1 suggests that they could be beneficial in PAH, but their therapeutic potential will require further investigation.
Given the challenges of enzymatically dissociating tissue into single cells that may be particularly fragile or tightly embedded, the lung dissociation for our original scRNA-seq data set yielded relatively few endothelial cells. Therefore, our statistical power to detect DEGs between PAH models versus control within subpopulations was limited, and we cannot exclude the possibility that other arterial endothelial subpopulations were not captured in our scRNA-seq data set. Alternative methods such as single-nucleus RNA-seq may provide a higher yield of difficult-to-dissociate cell types and a more precise cellular representation of tissue composition (55).
Conclusions
Our study provides an in-depth analysis of rat lung endothelial heterogeneity using unbiased single-cell computational methods to uncover a novel Tm4sf1-marked endothelial subpopulation with relevance to PAH supported by multiple independent human and animal data sets. Future experimental studies are warranted to further investigate the stem/progenitor potential and role of EA2 and Tm4sf1 in PAH.
Acknowledgments
Acknowledgment
The authors express gratitude to Dr. Douglas Arneson for his guidance with scRNA-seq analytical techniques, Drs. In Sook Ahn and Graciel Diamante for their help in performing droplet sequencing, and Drs. Christine Cunningham and May Bhetraratana for their contribution to lung tissue dissociation.
Footnotes
Supported by American Lung Association grant CA-675591 (J.H.), National Heart, Lung, and Blood Institute grant R01HL162124 (M.E.), and National Heart, Lung, and Blood Institute grant R01HL159865 (M.E.).
Author Contributions: J.H. contributed to the conception and design of the research and interpretation of the data. J.H. conducted or helped with all experiments, analyzed the data, made the figures, and wrote the manuscript. B.W. performed the immunofluorescence staining, flow cytometry and cell culture experiments, and helped with data interpretation, preparing figures, and writing the manuscript. S.U. generated the bulk RNA sequencing data. C.H. performed trajectory analysis. B.T. performed pathway enrichment analysis. G.R. and M.L. contributed to the cell culture experiments. G.R., X.Y., and M.E. provided intellectual input.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2022-0020OC on October 17, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Pu X, Du L, Hu Y, Fan Y, Xu Q. Stem/progenitor cells and pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol . 2021;41:167–178. doi: 10.1161/ATVBAHA.120.315052. [DOI] [PubMed] [Google Scholar]
- 2. Fadini GP, Avogaro A, Ferraccioli G, Agostini C. Endothelial progenitors in pulmonary hypertension: new pathophysiology and therapeutic implications. Eur Respir J . 2010;35:418–425. doi: 10.1183/09031936.00112809. [DOI] [PubMed] [Google Scholar]
- 3. Medina RJ, Barber CL, Sabatier F, Dignat-George F, Melero-Martin JM, Khosrotehrani K, et al. Endothelial progenitors: a consensus statement on nomenclature. Stem Cells Transl Med . 2017;6:1316–1320. doi: 10.1002/sctm.16-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Asosingh K, Comhair S, Mavrakis L, Xu W, Horton D, Taylor I, et al. Single-cell transcriptomic profile of human pulmonary artery endothelial cells in health and pulmonary arterial hypertension. Sci Rep . 2021;11:14714. doi: 10.1038/s41598-021-94163-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saygin D, Tabib T, Bittar HET, Valenzi E, Sembrat J, Chan SY, et al. Transcriptional profiling of lung cell populations in idiopathic pulmonary arterial hypertension. Pulm Circ . 2020;10:1–15. doi: 10.1177/2045894020908782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rodor J, Chen S-H, Scanlon JP, Monteiro JP, Caudrillier A, Sweta S, et al. Single-cell RNA sequencing profiling of mouse endothelial cells in response to pulmonary arterial hypertension. Cardiovasc Res . 2021;118:2519–2534. doi: 10.1093/cvr/cvab296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hong J, Arneson D, Umar S, Ruffenach G, Cunningham CM, Ahn IS, et al. Single-cell study of two rat models of pulmonary arterial hypertension reveals connections to human pathobiology and drug repositioning. Am J Respir Crit Care Med . 2021;203:1006–1022. doi: 10.1164/rccm.202006-2169OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hong J, Wong B, Huynh C, Tang B, Umar S, Yang X, et al. A Tm4sf1-marked subpopulation of endothelial stem/progenitor cells identified by lung single-cell omics of pulmonary arterial hypertension [abstract] Am J Respir Crit Care Med . 2022;203:A5803. [Google Scholar]
- 9. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol . 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol . 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat Protoc . 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 12. Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science . 2020;367:405–411. doi: 10.1126/science.aax0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sieveking DP, Buckle A, Celermajer DS, Ng MKC. Strikingly different angiogenic properties of endothelial progenitor cell subpopulations: insights from a novel human angiogenesis assay. J Am Coll Cardiol . 2008;51:660–668. doi: 10.1016/j.jacc.2007.09.059. [DOI] [PubMed] [Google Scholar]
- 14. Schupp JC, Adams TS, Cosme C, Jr, Raredon MSB, Yuan Y, Omote N, et al. Integrated single-cell atlas of endothelial cells of the human lung. Circulation . 2021;144:286–302. doi: 10.1161/CIRCULATIONAHA.120.052318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fu F, Yang X, Zheng M, Zhao Q, Zhang K, Li Z, et al. Role of transmembrane 4 L six family 1 in the development and progression of cancer. Front Mol Biosci . 2020;7:202. doi: 10.3389/fmolb.2020.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst . 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. HuBMAP Consortium. The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature . 2019;574:187–192. doi: 10.1038/s41586-019-1629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation . 2002;105:1672–1678. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- 19. Voelkel NF, Gomez-Arroyo J. The role of vascular endothelial growth factor in pulmonary arterial hypertension: the angiogenesis paradox. Am J Respir Cell Mol Biol . 2014;51:474–484. doi: 10.1165/rcmb.2014-0045TR. [DOI] [PubMed] [Google Scholar]
- 20. Bordenave J, Thuillet R, Tu L, Phan C, Cumont A, Marsol C, et al. Neutralization of CXCL12 attenuates established pulmonary hypertension in rats. Cardiovasc Res . 2020;116:686–697. doi: 10.1093/cvr/cvz153. [DOI] [PubMed] [Google Scholar]
- 21. McCullagh BN, Costello CM, Li L, O’Connell C, Codd M, Lawrie A, et al. Elevated plasma CXCL12α is associated with a poorer prognosis in pulmonary arterial hypertension. PLoS ONE . 2015;10:e0123709. doi: 10.1371/journal.pone.0123709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Le Hiress M, Tu L, Ricard N, Phan C, Thuillet R, Fadel E, et al. Proinflammatory signature of the dysfunctional endothelium in pulmonary hypertension. Role of the macrophage migration inhibitory factor/CD74 complex. Am J Respir Crit Care Med . 2015;192:983–997. doi: 10.1164/rccm.201402-0322OC. [DOI] [PubMed] [Google Scholar]
- 23. Toshner M, Voswinckel R, Southwood M, Al-Lamki R, Howard LSG, Marchesan D, et al. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med . 2009;180:780–787. doi: 10.1164/rccm.200810-1662OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Athanasiadis EI, Botthof JG, Andres H, Ferreira L, Lio P, Cvejic A. Single-cell RNA-sequencing uncovers transcriptional states and fate decisions in haematopoiesis. Nat Commun . 2017;8:2045. doi: 10.1038/s41467-017-02305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hayashi Y, Kuroda T, Kishimoto H, Wang C, Iwama A, Kimura K. Downregulation of rRNA transcription triggers cell differentiation. PLoS ONE . 2014;9:e98586. doi: 10.1371/journal.pone.0098586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Diller G-P, van Eijl S, Okonko DO, Howard LS, Ali O, Thum T, et al. Circulating endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic pulmonary arterial hypertension. Circulation . 2008;117:3020–3030. doi: 10.1161/CIRCULATIONAHA.108.769646. [DOI] [PubMed] [Google Scholar]
- 27. Asosingh K, Aldred MA, Vasanji A, Drazba J, Sharp J, Farver C, et al. Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am J Pathol . 2008;172:615–627. doi: 10.2353/ajpath.2008.070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Junhui Z, Xingxiang W, Guosheng F, Yunpeng S, Furong Z, Junzhu C. Reduced number and activity of circulating endothelial progenitor cells in patients with idiopathic pulmonary arterial hypertension. Respir Med . 2008;102:1073–1079. doi: 10.1016/j.rmed.2007.12.030. [DOI] [PubMed] [Google Scholar]
- 29. Xie X, Liu M, Zhang Y, Wang B, Zhu C, Wang C, et al. Single-cell transcriptomic landscape of human blood cells. Natl Sci Rev . 2021;8:nwaa180. doi: 10.1093/nsr/nwaa180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheadle C, Berger AE, Mathai SC, Grigoryev DN, Watkins TN, Sugawara Y, et al. Erythroid-specific transcriptional changes in PBMCs from pulmonary hypertension patients. PLoS ONE . 2012;7:e34951. doi: 10.1371/journal.pone.0034951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hsu E, Shi H, Jordan RM, Lyons-Weiler J, Pilewski JM, Feghali-Bostwick CA. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum . 2011;63:783–794. doi: 10.1002/art.30159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kariotis S, Jammeh E, Swietlik EM, Pickworth JA, Rhodes CJ, Otero P, et al. UK National PAH Cohort Study Consortium Biological heterogeneity in idiopathic pulmonary arterial hypertension identified through unsupervised transcriptomic profiling of whole blood. Nat Commun . 2021;12:7104. doi: 10.1038/s41467-021-27326-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rajkumar R, Konishi K, Richards TJ, Ishizawar DC, Wiechert AC, Kaminski N, et al. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am J Physiol Heart Circ Physiol . 2010;298:H1235–H1248. doi: 10.1152/ajpheart.00254.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stearman RS, Bui QM, Speyer G, Handen A, Cornelius AR, Graham BB, et al. Systems analysis of the human pulmonary arterial hypertension lung transcriptome. Am J Respir Cell Mol Biol . 2019;60:637–649. doi: 10.1165/rcmb.2018-0368OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mura M, Anraku M, Yun Z, McRae K, Liu M, Waddell TK, et al. Gene expression profiling in the lungs of patients with pulmonary hypertension associated with pulmonary fibrosis. Chest . 2012;141:661–673. doi: 10.1378/chest.11-0449. [DOI] [PubMed] [Google Scholar]
- 36. Hiepen C, Jatzlau J, Hildebrandt S, Kampfrath B, Goktas M, Murgai A, et al. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLoS Biol . 2019;17:e3000557. doi: 10.1371/journal.pbio.3000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alastalo T-P, Li M, Perez VdeJ, Pham D, Sawada H, Wang JK, et al. Disruption of PPARγ/β-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J Clin Invest . 2011;121:3735–3746. doi: 10.1172/JCI43382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao YD, Yun HZH, Peng J, Yin L, Chu L, Wu L, et al. De novo synthesize of bile acids in pulmonary arterial hypertension lung. Metabolomics . 2014;10:1169–1175. doi: 10.1007/s11306-014-0653-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Awad KS, Elinoff JM, Wang S, Gairhe S, Ferreyra GA, Cai R, et al. Raf/ERK drives the proliferative and invasive phenotype of BMPR2-silenced pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol . 2016;310:L187–L201. doi: 10.1152/ajplung.00303.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature . 2018;555:251–255. doi: 10.1038/nature25786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J . 2019;53:1801887. doi: 10.1183/13993003.01887-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shih S-C, Zukauskas A, Li D, Liu G, Ang L-H, Nagy JA, et al. The L6 protein TM4SF1 is critical for endothelial cell function and tumor angiogenesis. Cancer Res . 2009;69:3272–3277. doi: 10.1158/0008-5472.CAN-08-4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van der Feen DE, Bossers GPL, Hagdorn QAJ, Moonen J-R, Kurakula K, Szulcek R, et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci Transl Med . 2020;12:eaaw4974. doi: 10.1126/scitranslmed.aaw4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Montani D, Perros F, Gambaryan N, Girerd B, Dorfmuller P, Price LC, et al. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med . 2011;184:116–123. doi: 10.1164/rccm.201006-0905OC. [DOI] [PubMed] [Google Scholar]
- 45. Hashimoto R, Lanier GM, Dhagia V, Joshi SR, Jordan A, Waddell I, et al. Pluripotent hematopoietic stem cells augment α-adrenergic receptor-mediated contraction of pulmonary artery and contribute to the pathogenesis of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol . 2020;318:L386–L401. doi: 10.1152/ajplung.00327.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bailey AS, Jiang S, Afentoulis M, Baumann CI, Schroeder DA, Olson SB, et al. Transplanted adult hematopoietic stems cells differentiate into functional endothelial cells. Blood . 2004;103:13–19. doi: 10.1182/blood-2003-05-1684. [DOI] [PubMed] [Google Scholar]
- 47. 2020 Annual World Congress of the Pulmonary Vascular Research Institute. Pulm Circ . 2021;11:2045894020976949. doi: 10.1177/2045894020976949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yoder MC. Human endothelial progenitor cells. Cold Spring Harb Perspect Med . 2012;2:a006692. doi: 10.1101/cshperspect.a006692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yuan K, Liu Y, Zhang Y, Nathan A, Tian W, Yu J, et al. Mural cell SDF1 signaling is associated with the pathogenesis of pulmonary arterial hypertension. Am J Respir Cell Mol Biol . 2020;62:747–759. doi: 10.1165/rcmb.2019-0401OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Samokhin A, Yefang W, Wertheim BM, Alba GA, Arons E, Maron B. Abstract 12577: pulmonary endothelial sulfatase-1 is regulated by NEDD9 and increased in pulmonary arterial hypertension. Circulation . 2021;144:A12577. [Google Scholar]
- 51. Balazs AB, Fabian AJ, Esmon CT, Mulligan RC. Endothelial protein C receptor (CD201) explicitly identifies hematopoietic stem cells in murine bone marrow. Blood . 2006;107:2317–2321. doi: 10.1182/blood-2005-06-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yu QC, Song W, Wang D, Zeng YA. Identification of blood vascular endothelial stem cells by the expression of protein C receptor. Cell Res . 2016;26:1079–1098. doi: 10.1038/cr.2016.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chakraborty S, Ain R. Nitric-oxide synthase trafficking inducer is a pleiotropic regulator of endothelial cell function and signaling. J Biol Chem . 2017;292:6600–6620. doi: 10.1074/jbc.M116.742627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu H, Czerwinski P, Hortmann M, Sohn H-Y, Förstermann U, Li H. Protein kinase C α promotes angiogenic activity of human endothelial cells via induction of vascular endothelial growth factor. Cardiovasc Res . 2008;78:349–355. doi: 10.1093/cvr/cvm085. [DOI] [PubMed] [Google Scholar]
- 55. Wu H, Kirita Y, Donnelly EL, Humphreys BD. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol . 2019;30:23–32. doi: 10.1681/ASN.2018090912. [DOI] [PMC free article] [PubMed] [Google Scholar]