

Abstract
TAS2Rs (bitter taste receptors) are GPCRs (G protein–coupled receptors) expressed on human airway smooth muscle (HASM) cells; when activated by receptor agonists they evoke marked airway relaxation. In both taste and HASM cells, TAS2Rs activate a canonical Gβγ-mediated stimulation of Ca2+ release from intracellular stores by activation of PLCβ (phospholipase Cβ). Alone, this [Ca2+]i signaling does not readily account for relaxation, particularly since bronchoconstrictive agonists acting at Gq-coupled receptors also increase [Ca2+]i. We established that TAS2R14 activation in HASM promotes relaxation through F-actin (filamentous actin) severing. This destabilization of actin was from agonist-promoted activation (dephosphorylation) of cofilin, which was pertussis toxin sensitive. Cofilin dephosphorylation was due to TAS2R-mediated deactivation of LIM domain kinase. The link between early receptor action and the distal cofilin dephosphorylation was found to be the polarity protein partitioning defective 3 (Par3), a known binding partner with PLCβ that inhibits LIM kinase. The physiologic relevance of this pathway was assessed using knock-downs of cofilin and Par3 in HASM cells and in human precision-cut lung slices. Relaxation by TAS2R14 agonists was ablated with knock-down of either protein as assessed by magnetic twisting cytometry in isolated cells or intact airways in the slices. Blocking [Ca2+]i release by TAS2R14 inhibited agonist-promoted cofilin dephosphorylation, confirming a role for [Ca2+]i in actin-modifying pathways. These results further elucidate the mechanistic basis of TAS2R-mediated HASM relaxation and point toward nodal points that may act as asthma or chronic obstructive pulmonary disease response modifiers or additional targets for novel bronchodilators.
Keywords: bronchodilator, asthma, chronic obstructive pulmonary disease response modifiers, actin, calcium
Clinical Relevance
TAS2Rs (bitter taste receptors) are GPCRs (G protein–coupled receptors) expressed on airway smooth muscle cells, and when activated by agonists promote marked relaxation and bronchodilation. The mechanism of how these receptors signal to relaxation was explored, and was found to be due to its coupling through LIM kinase to activation (dephosphorylation) of cofilin, leading to destabilization and severing of F-actin (filamentous actin). These findings improve our understanding of how this potential new class of direct bronchodilators function within the context of human airway smooth muscle cells.
The family of GPCRs (G protein–coupled receptors) responsive to bitter-tasting compounds was identified and characterized 2 decades ago (1). These 24–25 TAS2Rs (bitter taste receptors) were believed to be exclusively expressed on taste cells in the tongue, acting as aversion receptors to protect against ingestion of toxins from plants. In 2010 we identified six TAS2Rs on human airway smooth muscle (HASM) cells (2). Several of these were expressed at a higher degree than β2AR (β2-adrenergic receptor), the current target for bronchodilator treatment in obstructive airway diseases. We found that TAS2R agonists caused marked cAMP-independent HASM relaxation and bronchodilation and showed that the signaling pathway diverged from what had been characterized in taste cells (2). Subsequently, TAS2Rs have been found to be expressed, and to have physiologic function, on many other tissues, including the pancreas, gastrointestinal tract, thyroid, and uterus (3, 4). The intracellular signaling pathways that lead to the cellular outcomes on these extraoral sites, however, have not been well defined. Indeed, because our initial characterization of the pathway differed from the accepted pathway in taste cells, the finding of functional TAS2Rs in cell types like HASM was met with some initial skepticism. For example, in the type II taste cells, TAS2Rs couple to the heterotrimeric G protein gustducin, with the released βγ interacting with PLCβ (phospholipase Cβ), resulting in the generation of IP3 (inositol 1,4,5-trisphosphate), which binds to the IP3 receptor on the endoplasmic reticulum (ER), releasing stored [Ca2+]i (see Figure E1 in the data supplement). This rise in Ca2+ activates a transient receptor potential channel, causing membrane depolarization, and the release of neurotransmitters from the cell, which activate receptors on the nearby type III taste cells that make up synaptic connections with nerves that signal to the brain (5). In contrast, ASM cells in humans do not appear to express gustducin (6), nor do these cells express the transient receptor potential channel. Furthermore, TAS2R agonists hyperpolarize the HASM membrane without a secondary release of neurotransmitters or communication to other cells or organs (Figure E1). Thus, the accepted pathway for TAS2Rs, established in type II taste cells, is not directly applicable to the physiologic response in HASM, and for that matter few of the other extraoral sites. On initial inspection, the divergence between pathways appears to be due to a compartmentalized [Ca2+]i pool that is used by TAS2Rs (2). However, this does not readily explain several aspects of how TAS2Rs signal to HASM relaxation and seems counterintuitive to the established paradigm that increased [Ca2+]i (at least by the Gq-coupled receptors in HASM) leads to contraction rather than relaxation. We have shown that TAS2Rs couple to Gi1,2,3 in HASM; these G proteins are highly expressed in HASM and are members of the pertussis toxin (PTX)–sensitive G protein family that includes gustducin. βγ antagonists, PLC inhibitors, and IP3 receptor antagonists inhibit [Ca2+]i signaling and attenuate relaxation by TAS2R agonists, so elements of the canonical pathway are involved, but the mechanism of the deep-pathway signaling of these receptors to physiologic relaxation remains unknown.
Smooth muscle cell contraction and relaxation are typically mediated by either MLC (myosin light chain) 20 phosphorylation/dephosphorylation or by F-actin (filamentous actin) stabilization/destabilization (7–9). MLC is phosphorylated by MLCK (myosin light chain kinase), with phosphorylated MLC subsequently interacting with actin to cause smooth muscle contraction (7). β-agonists act to relax HASM, in part, by inhibiting this mechanism or by activating MLCP (myosin light chain phosphatase), which dephosphorylates MLC (7). Another major actin-regulating protein is cofilin, which increases the off-rate of G-actin monomers from the pointed end of actin filaments and acts to sever F-actin (8, 9). This acts to rapidly and reversibly modulate actin dynamic equilibrium during contraction and relaxation of smooth muscle from various organs, although its activation/deactivation and mechanisms of modulation by GPCRs has not been explored in HASM cells. Dephosphorylated cofilin is the active form that interacts with F-actin, promoting its depolymerization and destabilization, leading to severing and relaxation. In contrast, inactive (phosphorylated) cofilin fails to bind to F-actin, inhibiting the relaxation imposed by F-actin severing, which leads to contraction. Cofilin deactivation is tightly regulated by LIMK (LIM domain kinase) (10), which is active in its phosphorylated state.
Here we have established an unexpected and highly efficient pathway that accounts for signaling from agonist activation of TAS2R14 (the most abundant TAS2R in HASM) through a G protein–mediated regulation of cofilin via a Par3/LIM kinase mechanism, leading to actin severing and HASM relaxation. The cascade incorporates the majority of the signals and cellular components identified to date into an integrated pathway, defining a mechanism by which these atypical HASM receptors bronchodilate and revealing nodal points for modulation of the pathway by disease or novel therapeutics.
Methods
Cell Culture, Transfections, Treatments
HASM cells were obtained from donor lungs from the National Disease Research Interchange or the International Institute for the Advancement of Medicine. The immortalized D9 HASM cell line (11) was also used, as indicated. Cells were grown and passaged as previously described (2, 6). siRNA transfections were performed on HASM cells in culture as previously described (6). The siRNAs for Par3, cofilin, β-arrestin1, and β-arrestin2 were from GE Dharmacon (Horizon Discovery). Transfections of siRNA (100 nM per 6-cm dish) for HASM cells were performed using Lipofectamine 2000 (Thermo Fisher Scientific) at 48 and again at 24 hours before experiments. For precision-cut human lung slices (PCLS), siRNA transfections were performed in the same manner, except in 24-well dishes (one slice per well), and the siRNA concentration was 400 nM. Confirmation of protein knock-down for HASM and PCLS was determined by western blots from total protein derived from the cells or from the pooled slices. Unless otherwise indicated, cells in culture were serum starved for 12 hours and treated with the TAS2R agonists diphenhydramine (DPD, 500 μM) or aristolochic acid (AA, 500 μM), without or with the brochoconstrictive agents endothelin-1 (ET-1, 100 nM) or histamine (1.0 μM), for 10 minutes. Because DPD is a pharmacologic antagonist at the histamine H1 receptor, the histamine + DPD combination was not tested. PTX (100 nM) pretreatment was for 12 hours, and BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid) (50 μM) pretreatment was for 30 minutes.
Western Blots and Immunocytochemistry
Cells or PCLS were washed and then solubilized by the addition of 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% NP-40, and 0.1% SDS (Radio-Immunoprecipitation Assay (RIPA) Buffer) with rotation at 4°C for 1 hour and clarified by centrifugation at 3,500 × g for 5 minutes. SDS-PAGE was performed using 10% SDS precast gels as previously described (12). The commercial sources of the antibodies were: p-cofilin (site 3, Cell Signaling Technology), cofilin (Santa Cruz Biotechnology), pMLC (sites 18 and 19) and MLC (Cell Signaling), Par3 (EMD Millipore), β-arrestin1 and β-arrestin2 (Cell Signaling Technology), pLIMK (site 508) and LIMK1/2 (Santa Cruz Biotechnology), p-SSH1 (site 978) and SSH1, Slingshot, from (ECM Bioscience), and actin (Cytoskeleton or Sigma). Proteins were transferred to nitrocellulose membranes. Primary antibody titers were 1:1,000 unless otherwise noted. The membranes were processed for enhanced chemiluminescence (Pierce–Thermo Fisher Scientific) according to the manufacturer’s protocol. Imaging was performed with a Fuji LAS-400 (GE Healthcare Life Sciences). Band density was determined with ImageJ software (National Institutes of Health) after subtraction of background. Reagents from Cytoskeleton were used to determine the F-actin/G-actin ratio according to the manufacturer’s protocol. In the figures, horizontal white lines demarcate images from membranes that were reprobed or were from separately run gels. Paraformaldehyde (4%) fixed cells were stained for immunocytochemistry as described (13), and the images were acquired using an Olympus FV10 or Zeiss LSM 880 confocal microscope at the indicated magnifications.
[Ca2+]i Measurements
Cells were loaded with FURA4-AM in 96-well plates and [Ca2+]i measured as described (14) at baseline and then every 2 seconds after addition of drug (or at other times as indicated) using a FlexStation 3 plate reader (Molecular Devices).
Magnetic Twisting Cytometry
Dynamic changes in cell stiffness of cultured HASM cells in response to agents added to the media were measured as an indicator of the single-cell contraction (increased stiffness) and relaxation (decreased stiffness) as previously described (2, 15, 16) (see also legend to Figure 4). In brief, RGD (Arg-Gly-Asp)-coated ferrimagnetic microbeads bound to integrin receptors are used as biosensors of the cytoskeleton. They were magnetized horizontally and then twisted in a vertically aligned magnetic field that varied sinusoidally in time. Lateral bead displacements in response to the resulting oscillatory torque from procontractile or prorelaxation agents were detected with a spatial resolution of ∼5 nm, and the ratio of specific torque to bead displacements (stiffness) was calculated.
Figure 4.
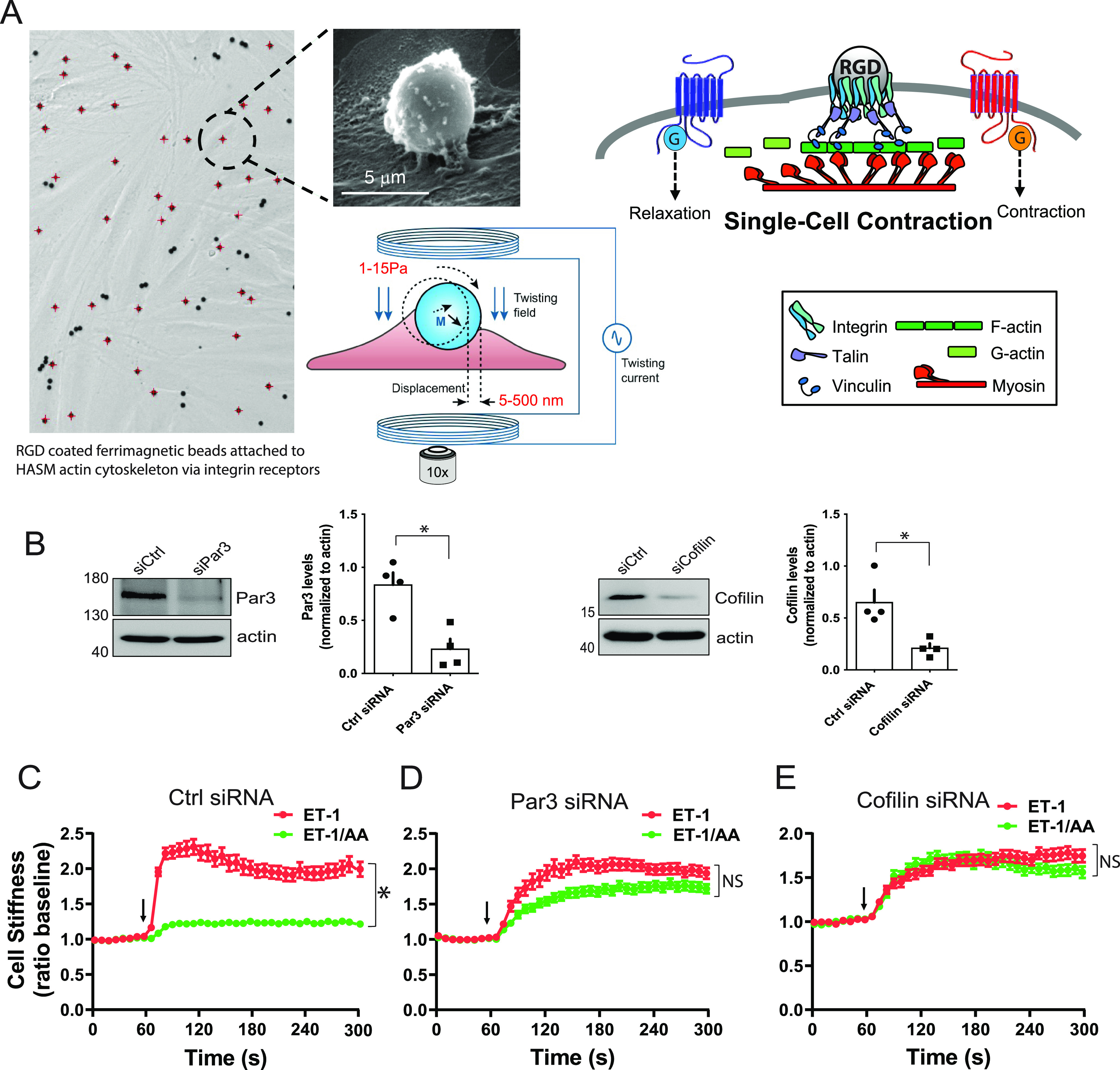
HASM cell mechanics with cofilin and Par3 knock-down identify their participation in a TAS2R14-mediated relaxation pathway. (A) MTC is performed by binding RGD (Arg-Gly-Asp)-coated ferrimagnetic beads (4.5 mm in diameter) to the actin cytoskeleton through the cell surface integrins on cultured HASM cells. Beads are magnetized horizontally (parallel to the plate surface) and then twisted in a vertically aligned homogenous magnetic field that is varying sinusoidally in time. This sinusoidal twisting field causes both a rotation and a pivoting displacement of the bead. As the bead moves, the cell develops internal traction forces that resist bead motions. The ratio of specific torque to lateral bead displacement is computed and is expressed as cell stiffness in Pascals (Pa)/nm. An increase or decrease in stiffness is the equivalent to cellular contraction or relaxation, respectively. (B) Representative western blots and results from four blots of HASM cell proteins using antibodies to Par3 and cofilin indicate >50% decrease in these two proteins from gene-specific siRNA transfections. *P < 0.01 versus control (scrambled) siRNA. (C) HASM cells contract to 100 mM ET-1 (red symbols, ET-1 alone), which is opposed by relaxation from the TAS2R14 agonist AA (green symbols, ET-1 + AA) in control siRNA-transfected cells. (D and E) Par3 or cofilin knock-down in HASM cells by gene-specific siRNAs ablate the TAS2R agonist effect (compare red and green symbols). ET-1 or ET-1 + AA were added at the 60-second time point (arrow), and the dynamic changes in HASM stiffness measured continuously over the next 240 seconds. *P < 0.001 versus ET-1 alone; n = 139–323 cells per condition. Scale bar, 5 μm.
PCLS
Human lung tissue samples were obtained from anonymous donors from National Disease Research Interchange and International Institute for the Advancement of Medicine. PCLS were prepared and studied as described (17). Briefly, lungs were inflated with low–melting temperature agarose, sectioned, and cored, and 350-μm slices made using an oscillating vibratome (see also legend to Figure 5). After siRNA transfection (see above for details), airways in the slices were constricted with 1.0 μM histamine, then relaxed with AA (1–100 μM), with multiple images taken after every treatment until the response stabilized. Luminal area of the airways was measured using ImageJ software and compared against baseline area to calculate percentage bronchodilation.
Figure 5.
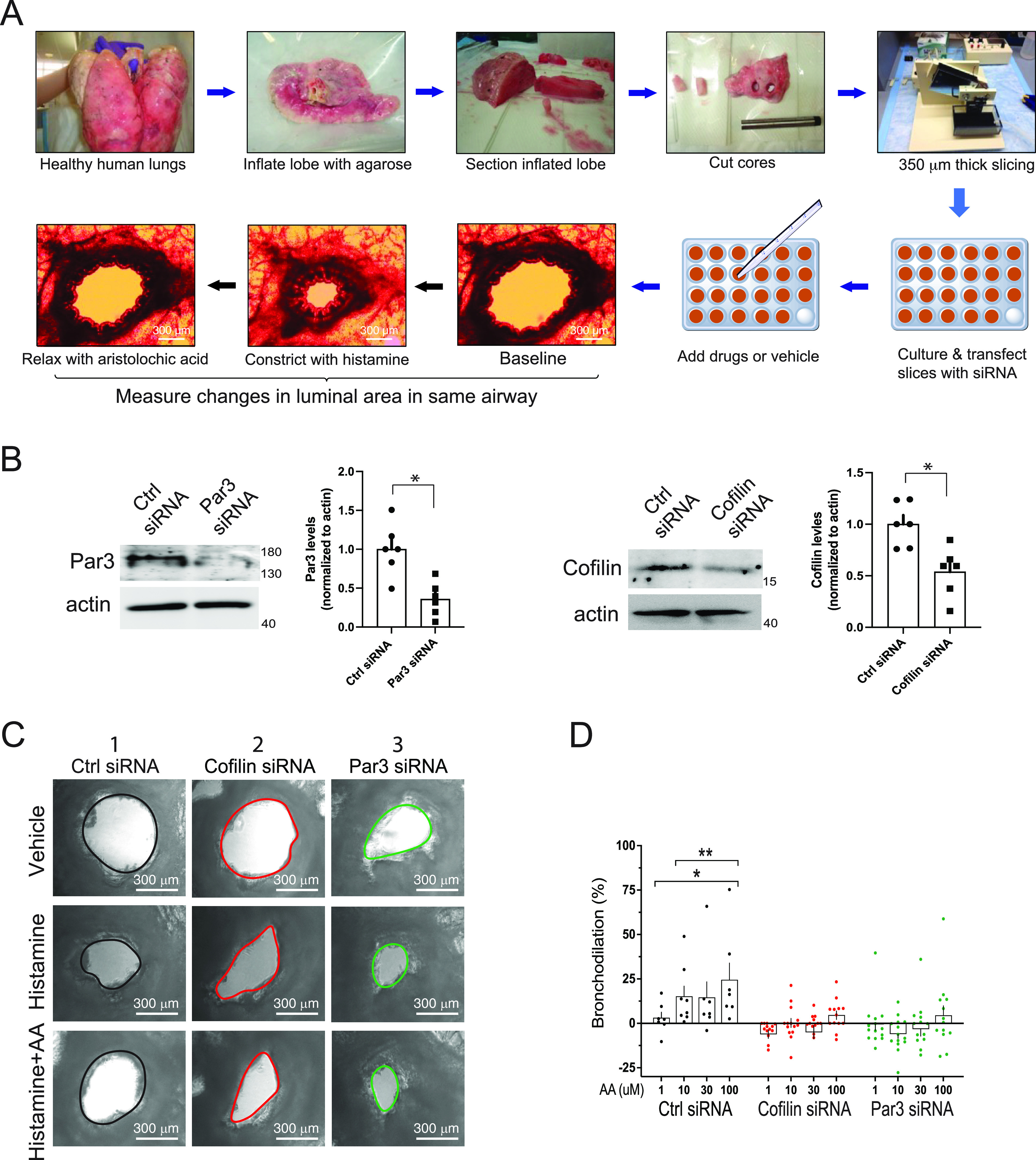
Human airway bronchodilation in precision-cut human lung slices (PCLS) by TAS2R agonist is ablated by knock-down of cofilin or Par3. (A) Human PCLS are studied by obtaining a core from a healthy donor lung that encompasses bronchi in parallel. Microtome cutting of the cores results in thin slices, which are placed in culture, with selected intact airways oriented parallel to the plating surface together with intact epithelium and interstitial tissue. Control (scrambled) or gene-specific siRNAs were transfected as described in Methods. Repeated images are acquired in response to potential bronchoactive agents and luminal area calculated using ImageJ software. (B) Gene-specific siRNA transfections of slices decreases cofilin or Par3 in protein derived from whole slices (representative western blots and results from multiple experiments are shown). *P < 0.01 versus control siRNA. (C) Representative images showing the same airway at baseline (vehicle) and in response to histamine or histamine with the TAS2R14 agonist AA. In the control siRNA condition (column 1), histamine constricts, while AA bronchodilates, as indicated by the increased airway area compared with the constriction evoked by histamine. In contrast, the cofilin (column 2) or Par3 (column 3) knock-downs show no effects of AA. (D) Results from measurements from four separate donor lungs, with a total of 12–15 slices per condition. *P < 0.05 versus a “0” baseline and **P < 0.01 versus responses from the respective doses under conditions of cofilin or Par3 knock-down. Scale bars, 300 μm; magnification, 40× .
Statistical and Other Calculations
The Tanimoto similarity between AA and DPD was calculated by the maximal common substructure method (https://chemminetools.ucr.edu, June 11, 2022). The ImageJ output from the PCLS studies were used to calculate percentage bronchodilation as described in detail (17). Statistical analysis was performed using Prism (GraphPad). Comparisons were performed using ANOVA followed by two-way post hoc t tests. In one aspect of the PCLS analysis, response data were compared with a baseline value using the two-way single-parameter t test. Significance was assigned when P < 0.05. Data in bar graphs show the individual data points from each experiment, the mean, and the SEM.
Results
TAS2R14 Agonists Activate the Actin-Severing Protein Cofilin in HASM Cells
We used two TAS2R14 agonists, DPD and AA, which have diverse structures (Tanimoto similarity score = 0.22) but have similar potencies (18), to study the relaxation pathway at both the molecular and physiologic levels. We first sought to address whether TAS2R14 agonism decreased MLC phosphorylation in cultured HASM cells. ET-1 couples to Gq, evokes HASM contraction and airway hyperreactivity, and is increased in asthmatic airways (19, 20). HASM exposure to ET-1 increased MLC phosphorylation as expected (Figures 1A and 1B). Neither the TAS2R agonist DPD or AA decreased ET-1–promoted MLC phosphorylation (Figures 1A and 1B).
Figure 1.
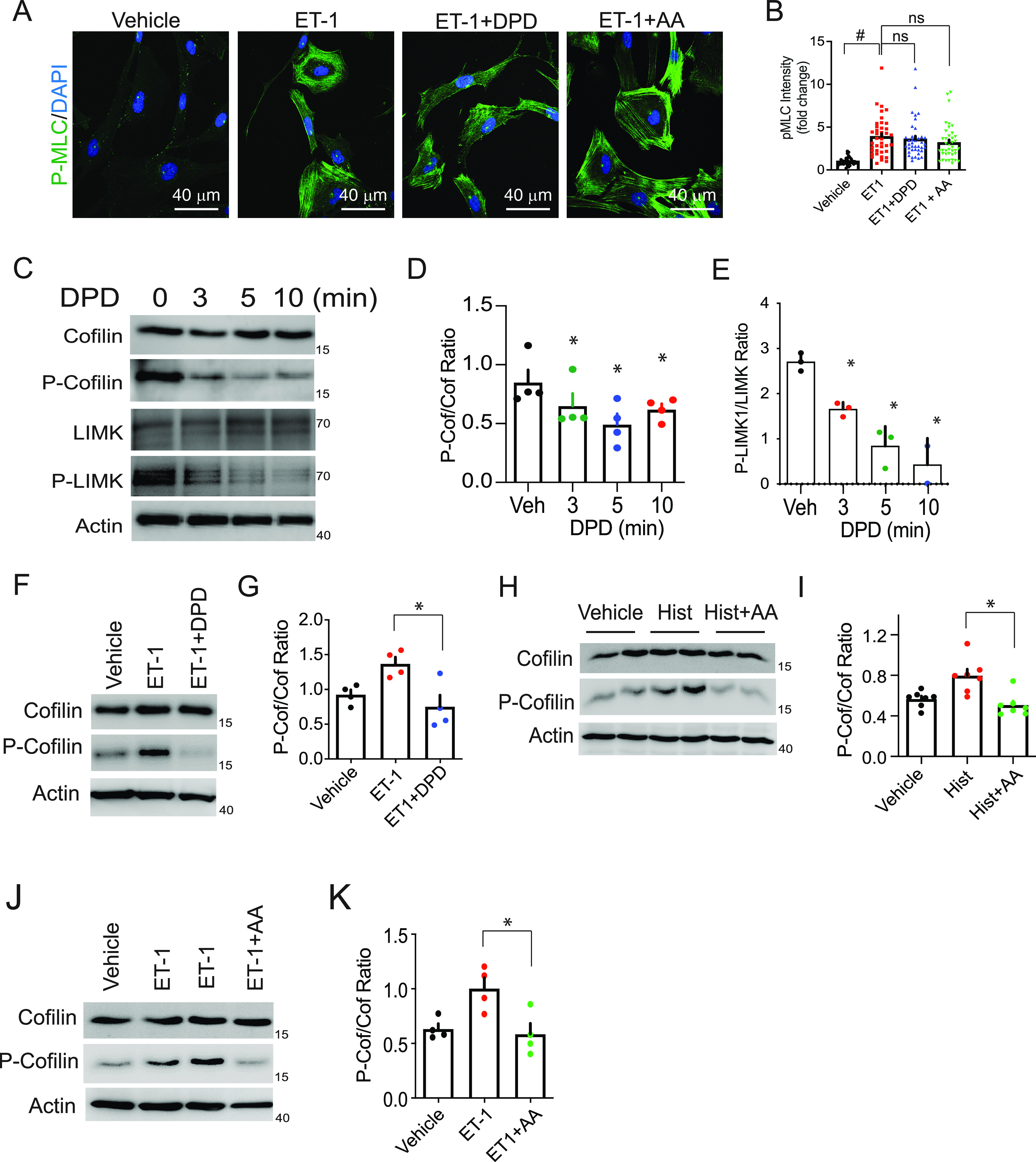
TAS2R14 (bitter taste receptor 14) signaling to elements of a proposed relaxation pathway in human airway smooth muscle. (A and B) The TAS2R14 agonists diphenhydramine (DPD) and aristolochic acid (AA) do not alter phosphorylation of MLC. Shown is a representative confocal image, and the graph shows quantitative results from four experiments (10 images/experiment). #P < 0.0001 versus vehicle. (C–E) Dephosphorylation of cofilin and LIMK by the agonist DPD. Shown is a representative western blot, and the bar graph shows results from three to four experiments. (F–K) Bronchoconstrictor (ET-1 [endothelin-1] and histamine)-mediated phosphorylation of cofilin is inhibited by AA and DPD. Because DPD is a pharmacologic antagonist at the histamine H1-receptor, the histamine + DPD combination was not tested. Representative western blot and a graph of four experiments are shown. *P < 0.01 versus ET-1 or histamine alone; white scale bar, 40 μm; magnification, 40×. Hist = histamine; MLC = myosin light chain; ns = not significant; Veh = vehicle.
Given this apparent lack of MLC dephosphorylation by these agonists, we investigated whether TAS2R14 agonists regulate the actin destabilization (depolymerization) pathway as introduced above, via modulation of cofilin activity to induce smooth muscle relaxation. We assessed whether the TAS2R14 agonist DPD regulates cofilin and/or LIMK in HASM cells by western blots using phospho-specific antibodies directed to these proteins. As introduced earlier, phosphorylated cofilin is its inactive form, and phosphorylated LIMK is its active form. A decrease in phosphorylation of both cofilin and LIMK was readily observed from baseline within 3 minutes of exposure of the cells to DPD (Figures 1C–1E) and was maximal by 5–10 minutes. For the remainder of the experiments, a 10-minute exposure to TAS2R agonist was used unless otherwise noted. The activated phosphatase SSH1 participates in dephosphorylation of cofilin by modulating LIMK phosphorylation. Dephosphorylated SSH1 is the active form. We thus assessed SSH1 phosphorylation by the TAS2R agonist AA and noted a small but significant decrease from basal levels (Figure E2). ET-1 did not increase SSH1 phosphorylation, and ET-1 + AA had no significant effect over ET-1 levels alone. Taken together, data from Figures 1A–1E indicate that TAS2R14 acts to relax HASM through activating cofilin by dephosphorylating LIMK within the actin network and that this receptor does not relax via modulating the phosphorylation of MLC.
TAS2R14 Agonists Inhibit ET-1– and Histamine-mediated Cofilin Inactivation in HASM Cells
Because TAS2R agonism promotes cofilin activation (dephosphorylation) and relaxation, we proceeded to ascertain the effects of DPD and AA on cofilin activation in the presence of two opposing constrictive agents, ET-1 and histamine, representing the status of HASM in vivo in asthma from the accumulation of these local endogenous procontractile agonists (21). ET-1 treatment significantly increased cofilin phosphorylation as shown (Figures 1F and 1G), shifting the equilibrium to the inactive state, which would promote contraction. Importantly, DPD fully blocked ET-1–mediated cofilin inactivation (Figures 1F and 1G). Cofilin phosphorylation levels under these conditions were typically even below basal (vehicle) levels, further indicating that TAS2R14 can evoke dephosphorylation of cofilin below baseline in the absence or presence of cotreatment by a contractile agent. These data are consistent with earlier observations that TAS2R agonists relax HASM from resting states when either intact bronchi or single cells were studied without preconstriction (2). We also found that the bronchoconstrictor histamine stimulates cofilin phosphorylation (i.e., inhibits activation) and that AA prevents histamine- and ET-1–mediated phosphorylation (Figures 1H–1K) in a pattern similar to what was observed with ET-1 + DPD. DPD is a pharmacologic antagonist at the histamine H1-receptor, so the histamine + DPD combination was not tested. Collectively, the data from Figures 1C–1K indicate that TAS2R14 agonism results in marked activation of cofilin (i.e., decreased cofilin phosphorylation), and this effect is observed regardless of whether the HASM cells are studied in the absence or presence of a procontractile agonist acting at Gq-coupled receptors.
TAS2R14 Inhibits ET-1–Mediated Cofilin:F-actin Colocalization and F-Actin Stabilization
Given that active cofilin potentially binds to F-actin in HASM, promoting severing and thus relaxation, we pursued the developing paradigm by testing whether TAS2R14 agonists promote the colocalization of cofilin and F-actin in these cells. Cells were treated with either ET-1 or histamine alone, ET-1 + DPD, ET-1 + AA, or histamine + AA and subsequently probed with cofilin antibody and phalloidin stain. We found TAS2R14 activation by both agonists markedly increased cofilin and F-actin colocalization in HASM cells (Figures 2A and 2B), in keeping with our findings that TAS2R14 agonists activate cofilin and promote cofilin interaction with F-actin. Because G-actin (globular actin) polymerizes to F-actin in smooth muscle, and activated cofilin depolymerizes F-actin to form G-actin, we performed F-actin/G-actin ratio assays in HASM cells to monitor the amount of F-actin content versus the free G-actin (Figures 2C and 2D). We first tested whether ET-1 promotes the expected increase in F-actin stability. Indeed, ET-1 markedly increased F-actin and decreased G-actin in HASM cells (Figure 2C). In contrast, cells treated with ET-1 plus the TAS2R14 agonist DPD revealed an F-actin/G-actin ratio equivalent to baseline (Figures 2C and 2D), compatible with a relaxation response, compared with ET-1 alone.
Figure 2.
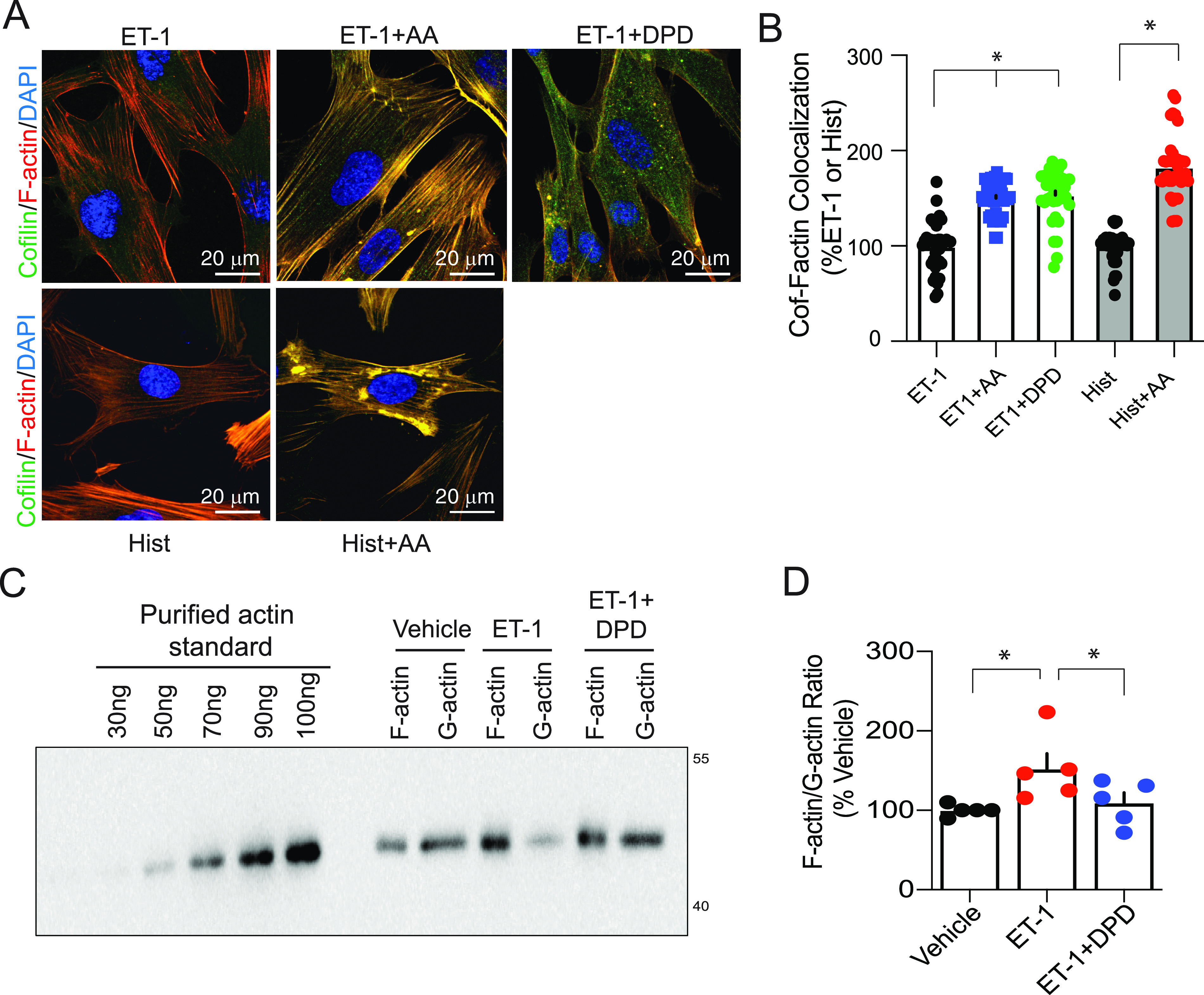
F-actin (filamentous actin) dynamics in human airway smooth muscle (HASM) are consistent with a TAS2R14-mediated relaxation effect. (A and B) F-actin colocalizes with cofilin in HASM exposed to the TAS2R14 agonists DPD and AA. Cells were treated with the procontractile agents ET-1 or histamine alone or with a procontractile agent and either AA or DPD. In the representative merged confocal image, the colors of the indicated proteins or DAPI staining are indicated, with yellow representing colocalization of F-actin and cofilin. The bar graph shows results from four experiments (multiple images/experiment). Because DPD is a pharmacologic antagonist at the histamine H1-receptor, the histamine + DPD combination was not tested. (C and D) The F-actin to G-actin ratio is altered by DPD. Representative western blot and graph of five experiments are shown. *P < 0.01 compared with vehicle; white scale bar, 20 μm; magnification, 40×.
TAS2R14 Signals by a Gi, [Ca2+]i, Par3, LIMK, Cofilin, F-Actin Pathway
To begin to unite the aforementioned changes in kinase activities, their substrates, and actin dynamics involved in TAS2R14-mediated relaxation, we probed the components of the early signal transduction complexes that might link TAS2R14 agonist binding to distal cofilin activation. PTX treatment of HASM cells ablated the TAS2R14 effect (dephosphorylation of cofilin; Figures 3A, 3B, and E3). These findings support our previous observations that TAS2Rs couple to the PTX-sensitive Gi1,2,3 proteins in HASM cells (6). Note that ET-1– and histamine-promoted phosphorylation of cofilin were not affected by the toxin, which is consistent with their respective receptors coupling to Gq. We have previously shown that TAS2R relaxation in HASM is dependent on the free βγ released from the Gi-heterodimer (2), which activates PLCβ. We recognize that one signaling partner within the activated PLCβ complex is the polarity protein Par3 (22). Par3 inhibits LIMK (23), which we show is regulated by TAS2R14 agonist (Figures 1C and 1D). To ascertain if the connection between TAS2R14 activation and cofilin activation was via Par3, we knocked down Par3 in HASM cells and determined the phosphorylation status of cofilin (Figures 3C and 3D). Under the control siRNA conditions, the DPD effect was observed as before. With Par3-specific siRNA transfection, Par3 protein levels were on average ∼80% reduced (Figure 3C). Under these knock-down conditions, the extent of ET-1–mediated phosphorylation of cofilin over baseline was decreased (Figure 3D). Thus, assessment of whether the phenotype observed with control siRNA on ET-1–stimulated phosphorylation of cofilin could not be assessed. However, because TAS2R agonists decrease levels of phosphorylation of even unstimulated cofilin, it is noteworthy that under Par3 knock-down the TAS2R agonist effect for this phenotype is indeed lost. Of note, this limitation was not observed in physiologic studies of HASM contraction/relaxation (see below), which clearly supported the concept of Par3 being the link between the TAS2R complex and LIMK/cofilin.
Figure 3.
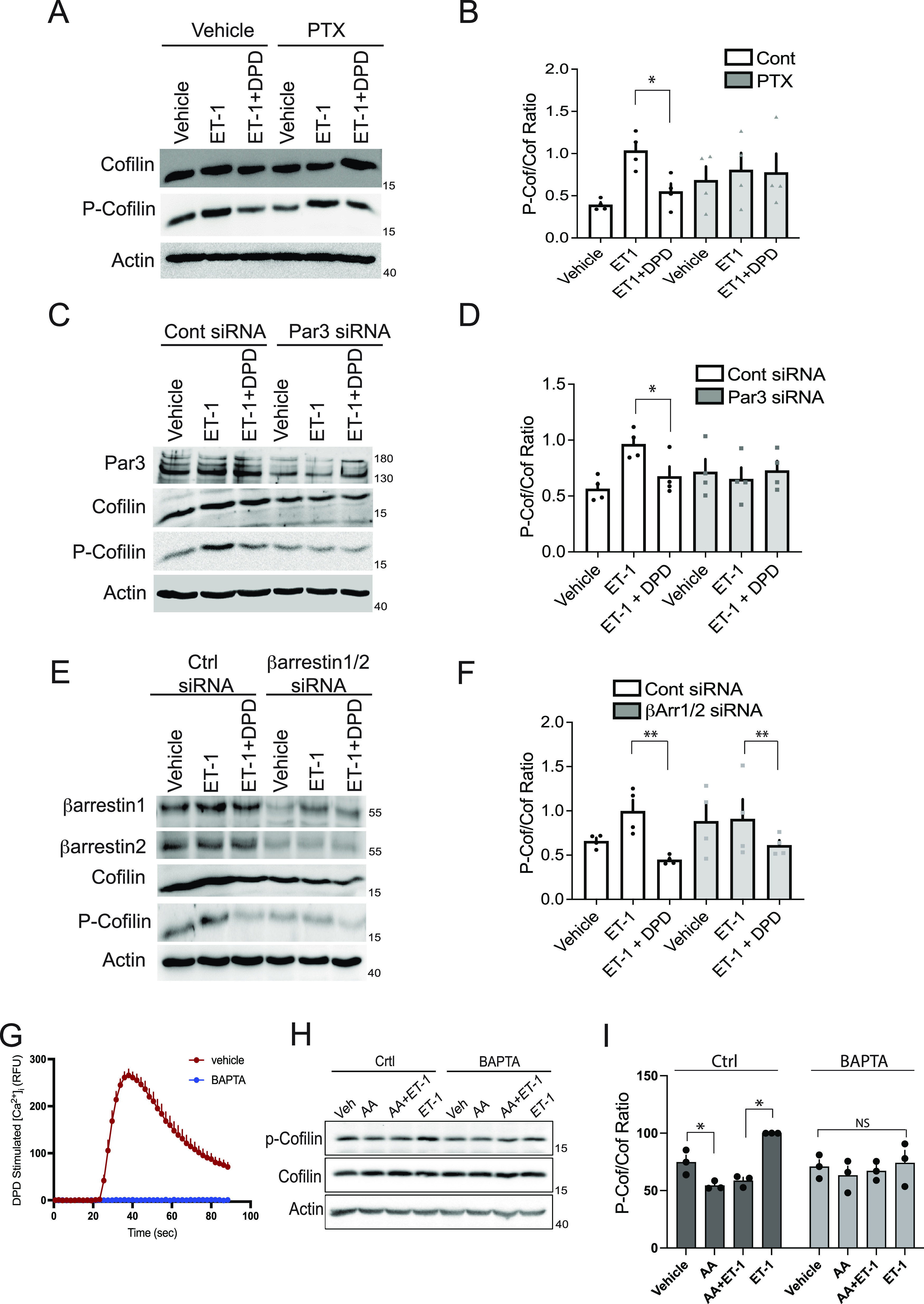
TAS2R14 signaling to cofilin in HASM cells is dependent on pertussis toxin (PTX)-sensitive G proteins, partitioning defective 3 (Par3) and [Ca2+]i, but not β-arrestin. (A and B) PTX (100 ng/ml) pretreatment of cells blocked DPD-mediated dephosphorylation of ET-1 phosphorylated cofilin. Representative western blot and graph of four experiments are shown. (C and D) Knock-down of Par3 eliminates the TAS2R agonist (DPD)-mediated dephosphorylation of cofilin. A representative western blot and graph of four experiments are shown. (E and F) Combined β-arrestin1/2 knock-down does not affect the TAS2R agonist (DPD)-mediated dephosphorylation of cofilin. Representative western blot and graph of four experiments are shown. (G–I) TAS2R14-mediated phosphorylation of cofilin is dependent on agonist-stimulated [Ca2+]i. DPD stimulates an increase in HASM cellular [Ca2+]i, which is blocked by BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid) (representative experiment of four performed). Under conditions of BAPTA preincubation, the effect of AA that decreases baseline (vehicle) or ET-1–stimulated p-cofilin is not observed. A representative western blot is shown, and a bar graph shows results from four experiments. *P < 0.01 and **P < 0.05, compared with ET-1–stimulated levels. Ctrl = control.
TAS2R14 is phosphorylated by G protein–coupled receptor kinases in an agonist-dependent manner (24). For many GPCRs, including TAS2Rs (24), this results in recruitment to the receptor (and nearby components) of β-arrestin1 and/or 2, which chaperone various proteins into signaling complexes (25). We considered that the β-arrestins might participate in the scaffolding of the components, which leads to receptor signaling to cofilin in an agonist-dependent manner. Knock-down of both β-arrestins revealed a preservation of the DPD-induced decrease in cofilin phosphorylation (Figures 3E and 3F), so this role for β-arrestins is not supported. TAS2R activation also increases [Ca2+]i (2), and, in fact, blocking TAS2R-mediated increases in [Ca2+]i ablates airway relaxation by agonists acting at these receptors (2). As indicated in Figure 3G, the agonist DPD causes a rapid rise in [Ca2+]i. Short-lived acute increases in [Ca2+]i are recognized as an initiator of downstream [Ca2+]i-mediated events in a number of signaling cascades (26). As indicated in Figure E4, HASM cell [Ca2+]i levels show a marked peak within seconds of TAS2R activation, and the levels continue to be elevated somewhat over baseline after the peak. Thus [Ca2+]i “pressure” remains after the peak to maintain relaxation (if necessary) during conditions of more prolonged agonist exposure. Preincubation with the [Ca2+]i chelator BAPTA eliminated the TAS2R14-evoked [Ca2+]i response in HASM cells (Figure 3G). With BAPTA, we found that the ET-1–dependent phosphorylation of cofilin over basal levels was no longer observed (Figures 3H and 3I). Regardless, we note that the decrease from basal (or ET-1) levels by TAS2R agonist was no longer evident under [Ca2+]i chelation.
Loss of TAS2R14-mediated HASM Relaxation with Par3 or Cofilin Knock-down Indicates Relevance in the Physiological End Response
We ascertained HASM function in two ways: single-cell mechanics using magnetic twisting cytometry (MTC) and PCLS with airway luminal diameter measurements. MTC measures the changes in cell stiffness from drug treatment of HASM cells tagged with ferrimagnetic beads grown on a matrix and placed within a magnetic field (see Figure 4A and legend). Thus, for MTC, effects of drugs or knock-downs are isolated to the cell type of interest with measurements that are physiologically relevant for that cell for treating obstructive airway disease. MTC was performed in early-passage HASM cells derived from a nonasthmatic donor lung, with siRNA knock-down of Par3 or cofilin exceeding 50% (Figure 4B). ET-1 evoked a ∼2.5-fold increase in stiffness, the equivalent of constriction in this assay, under the control siRNA conditions (Figure 4C, red symbols). The ET-1 + AA condition revealed marked attenuation of this response, consistent with the opposing relaxation from TAS2R14 activation (Figure 4C, green symbols). In marked contrast, Par3 knock-down virtually eliminated the pharmacologic effect of AA (Figure 4D). These results confirm that Par3 is at the interface between the activated receptor complex and cofilin. If so, we would also expect that cofilin knock-down would have a similar phenotype. And indeed, we found that cofilin knock-down HASM cells also failed to respond to the TAS2R14 agonist (Figure 4E). Interestingly, we note that the cofilin knock-down HASM had a smaller increase in stiffness from ET-1 over baseline (∼1.6-fold, red symbols of Figure 4E) compared with the control siRNA conditions, whereas Par3 knock-down had no statistically significant effect on contraction (∼2.2-fold vs. ∼2.4-fold). Because the Par3 component is localized to the TAS2R-promoted effector complex, its specificity for the relaxation component is congruent with the proposed pathway. On the other hand, cofilin is available for activation/inactivation by relaxation or constriction signaling, the latter represented by the ET-1 effect. Thus, under cofilin siRNA conditions, there is less available substrate for either response.
To confirm the findings with isolated HASM, we used PCLS, which has the advantage of measuring the coordinated action of airway smooth muscle (SM) cells in contraction and relaxation responses in slices of intact airways (see Figure 5A and legend). Studies of PCLS have the disadvantages of not specifically isolating drug actions to the SM alone, and the western blots showing the effects of the siRNAs are from protein extracted from all cell types within the slice. In the current study, siRNA knock-downs of Par3 or cofilin in the slices amounted to ∼50% (Figure 5B). A representative response to TAS2R agonist under control and targeted siRNA transfections is shown in Figure 5C. Control siRNA (column 1), shows bronchoconstriction (decrease in luminal area) by histamine, which is reversed by the relaxation effect of AA in the histamine + AA condition (see row labels). Cofilin knock-down (column 2) revealed constriction by histamine, but no counter-relaxation by AA on histamine-mediated constriction. Similarly, with Par3 knock-down (column 3), the TAS2R bronchodilation effect was not evident. In these experiments, doses of TAS2R agonist were studied across a 100-fold range, and although in the control siRNA condition dose-dependent bronchodilation was observed compared with baseline, the knock-downs showed no significant responses (Figure 5D). Taken together, these complimentary physiologic studies using two different techniques reveal that Par3, which is a proximal component to the receptor in the pathway, is necessary for HASM relaxation evoked by TAS2R14. In addition, the results corroborate that cofilin, which is distal to the initial receptor-effector complex, is a key downstream participant modulating F-actin in the TAS2R14 relaxation response.
Discussion
TAS2Rs are now recognized to be expressed on multiple cell types in the body. Although their “endogenous agonists,” if they exist, are not known, it is clear that their activation exerts physiologically important action (reviewed in References 3 and 4). In HASM, one of the first extraoral cell types that showed functional expression of these receptors, activation results in relaxation from the resting state and from intensely constricted states such as those evoked by spasmogens acting at Gq-coupled receptors. In many cases, the relaxation is greater in magnitude than that achieved by β-agonists, and unlike β-agonists the effect is not impaired in asthma (27). TAS2Rs couple to PTX-sensitive G proteins in the Gi-family, such as Ggust (gustducin) in taste cells and Gi1,2,3 in HASM cells. The released Gα subunit is known to activate phosphodiesterase, which would lower cAMP, and thus this pathway does not represent a mechanism for relaxation. The released Gβγ subunit has been shown to activate PLCβ, which results in production of IP3 and diacylglycerol. The latter has not been linked to the relaxation events. IP3 binding to the IP3 receptor on the sarcoplasmic reticulum (SR)/ER releases calcium from this intracellular depot. As indicated earlier, in taste cells this results in several steps leading to neurotransmitter release and communication with the central nervous system via afferent nerves (see also Figure E1). In contrast, HASM cells in isolation respond to TAS2R agonists, so there is no neuronal or paracrine action necessary for the physiologic response. Inhibition of Gi, βγ, PLCβ, and the IP3 receptor all block the [Ca2+]i response, as well as the relaxation effect. The unexpected effect of an increase in [Ca2+]i being associated with relaxation has remained a vexing issue, because such increases in HASM virtually always result in contraction. We found that TAS2R-stimulated [Ca2+]i occurred faster, was restricted to the slender tendrils of HASM, and was closely associated with the cell membrane, compared with a more global increase in [Ca2+]i found with histamine, indicative of a specialized [Ca2+]i pool, which was part of TAS2R-specific signaling that distinguishes it from Gq-receptor–based signaling (2). Indeed, it is recognized that many of the receptor-generated “second messengers,” such as [Ca2+]i (26) and cAMP (28), exist in distinct compartments, which appear to be colocalized with other components of a signaling complex. When SR/ER Ca2+ is depleted by agents such as thapsigargin, TAS2R-mediated relaxation is lost, further indicating a role for [Ca2+]i in the relaxation effect (2). However, the components of the signaling complex that leads to relaxation were not recognized before the current study; thus, there was an incomplete understanding of the mechanism of action of a target to which therapeutic agents are currently under investigation (29, 30).
Here we show that signaling from the G protein activates Par3 in HASM, which has previously been shown to be a binding partner to activated PLCβ (22). Par3 activation leads to inactivation of LIMK (23), which then favors dephosphorylation (activation) of cofilin, which destabilizes F-actin through severing, resulting in HASM relaxation (summarized in Figure 6). Importantly, we found that siRNA-mediated Par3 knock-down ablated the TAS2R agonist effect on cofilin. In addition, the critical findings were observed regardless of whether the HASM had been cotreated with Gq-receptor agonists, which constrict the airway. Thus, the idea that TAS2R agonists signal to relaxation strictly by “competitive” mechanisms with Gq-receptor actions (31) seems less likely, at least in human airway SM. Indeed, in prior work we showed that relaxation of human airway rings, and isolated HASM, was readily observed under resting (nonstimulated) conditions. Interestingly, in a recent report with mice, gustducin expression was detected in the mouse airway SM, and when deleted the TAS2R-evoked bronchodilation was lost (31). We find in HASM that this function is likely performed by Gi1,2,3 (6). We note that these G proteins and gustducin are closely related within the same family, are homologous in multiple structural regions, and are all PTX sensitive. Whether this difference between human and mouse TAS2R signaling to these G proteins constitutes a fundamental difference or represents minor species-specific variation with preservation of the main components leading to relaxation has yet to be addressed. The pathway we identified in the current work has been reported to be modulated by [Ca2+]i. This is apparent from the interplay between calcineurin and CaMK (calcium/calmodulin-dependent protein kinase) pathways. At low calcium concentrations, CaMKII phosphorylates SSH1 leading to inhibition, and also activates LIMK by phosphorylation, collectively driving cofilin phosphorylation and inactivation (32, 33). At higher intracellular [Ca2+]i, calcineurin-mediated dephosphorylation of SSH1 releases SSH1 from 14-3-3 inhibitory control, which activates SSH1 (34–36). Simultaneously, calcineurin deactivates CaMKII (32), thereby driving cofilin dephosphorylation. We found that the dephosphorylation of cofilin by TAS2R agonists is not apparent in the absence of TAS2R-stimulated Ca2+ release, nor does it occur when Par3 (or cofilin) expression is silenced. These findings, in conjunction with the known effects of [Ca2+]i on cofilin dynamics, suggest a convergence of the two signals (Figure 6). The collective data also indicate that both the stimulation of [Ca2+]i and the signaling through Par3/LIMK are necessary for TAS2R-mediated relaxation. The TAS2R-mediated actions of SSH1 on cofilin dephosphorylation are also expected to contribute to cofilin activation and to promote relaxation. To relate the molecular findings to airway physiology, we used MTC from isolated HASM and PCLS from human lungs to examine the relaxation effects of TAS2R agonists on HASM. These studies were performed under conditions of significant decreases in Par3 and cofilin expression from specific siRNA transfections of the HASM cells or the lung slices. The former protein represents an early (“proximal”) partner in the receptor–effector complex, whereas cofilin action is several steps removed and at the interface with actin. In studies using either MTC or PCLS, the knock-down of either of these two proteins had virtually the same effect: TAS2R-mediated relaxation was ablated.
Figure 6.

Summary of the mechanism of TAS2R-mediated HASM relaxation. Upon agonist occupancy, TAS2R14 couples to Gi, releasing the βγ subunit, which activates PLCβ (phospholipase Cβ). PLCβ interacts with Par3, which inhibits LIMK. The inhibition of LIMK (dephosphorylation; Figure 1) limits its kinase action on cofilin, leading to cofilin activation (dephosphorylation; Figures 1 and 3). Activated cofilin interacts with, and severs, F-actin (Figure 2), leading to HASM relaxation (Figures 4 and 5). PLCβ also increases IP3 (inositol 1,4,5-trisphosphate) levels, which bind IP3 receptors on the ER/sarcoplasmic reticulum (SR), releasing a specialized pool of [Ca2+]i, which promotes the net cofilin effect (Figure 3). Other mechanisms that may be involved are not shown; however, the loss of TAS2R-mediated relaxation with the knock-down of Par3 or cofilin (Figures 4 and 5) signifies that the depicted pathway is a major mechanism. ER = endoplasmic reticulum; PIP2 = phosphatidylinositol 4,5-bisphosphate; SSH = Slingshot. Created with BioRender.com.
As can be noted from our current and previous results (2, 37), and those of others (18), the effective concentrations of agonists that are necessary to fully activate TAS2Rs are relatively high (micromolar range). This is consistent with multiple studies using recombinant expression of TAS2Rs, which show low apparent affinities of bitter tastants for this receptor family (18). This may be due to evolutionary pressure to sense a broad range of exogenous bitter substances, which (in the case of the tongue) are in close contact with the receptor. We recognize that nonspecific effects are more likely when these higher concentrations are used in biochemical or physiologic studies. To minimize this potential, we studied two TAS2R14 agonists with diverse structures (low common substructure similarity score), yet with similar potencies. We obtained comparable results with either. The apparent lower affinity of many TAS2R agonists has not deterred preclinical drug discovery efforts, because receptor mutagenesis and structure-activity studies (29, 30, 38) have tentatively identified agonist binding sites and potentially higher-affinity agonists. These receptors appear to undergo less agonist-promoted desensitization (24) and may be more readily biased toward clinically favorable outcomes than β2AR (13, 16). Thus, inhaled TAS2R agonists may be effective therapy alone or in addition to β-agonists for treating or preventing reversible airflow obstruction in asthma or chronic obstructive pulmonary disease. In the current work we have revealed that these receptors lead to relaxation by signaling to cofilin and through modulation of the actin cytoskeleton. This provides additional clarification of the mechanism of action of TAS2Rs in the HASM cell and points to elements in the pathway between receptor activation and F-actin severing that may represent additional therapeutic targets and disease or response-modifying genes.
Acknowledgments
Acknowledgment
The authors thank Hannah R. Strzelinski and Siheun Lee for technical assistance and Himeshkumar N. Patel for assistance in manuscript preparation.
Footnotes
Supported by National Heart, Lung, and Blood Institute grants HL114471 and HL155532.
Author Contributions: J.A.W. and S.B.L.: wrote the paper, conception, design, and analysis. M.C., T.R.K., and J.L.: performed and analyzed experiments. C.J.K.-W., S.S.A., and D.K.: wrote the paper, conception, design, analysis, and performed and analyzed experiments. D.E.K.: conception, design, and analysis.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2022-0303OC on January 20, 2023
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Chandrashekar J, Mueller KL, Hoon MA, Adler E, Feng L, Guo W, et al. T2Rs function as bitter taste receptors. Cell . 2000;100:703–711. doi: 10.1016/s0092-8674(00)80706-0. [DOI] [PubMed] [Google Scholar]
- 2. Deshpande DA, Wang WC, McIlmoyle EL, Robinett KS, Schillinger RM, An SS, et al. Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nat Med . 2010;16:1299–1304. doi: 10.1038/nm.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lu P, Zhang CH, Lifshitz LM, ZhuGe R. Extraoral bitter taste receptors in health and disease. J Gen Physiol . 2017;149:181–197. doi: 10.1085/jgp.201611637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. D’Urso O, Drago F. Pharmacological significance of extra-oral taste receptors. Eur J Pharmacol . 2021;910:174480. doi: 10.1016/j.ejphar.2021.174480. [DOI] [PubMed] [Google Scholar]
- 5. Liggett SB. Bitter taste receptors in the wrong place: novel airway smooth muscle targets for treating asthma. Trans Am Clin Climatol Assoc . 2014;125:64–74. [PMC free article] [PubMed] [Google Scholar]
- 6. Kim D, Woo JA, Geffken E, An SS, Liggett SB. Coupling of airway smooth muscle bitter taste receptors to intracellular signaling and relaxation is via galphai1,2,3. Am J Respir Cell Mol Biol . 2017;56:762–771. doi: 10.1165/rcmb.2016-0373OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wirth A, Offermanns S. In: Muscle: fundamental biology and mechanisms of disease. Griendling KK, Kitsis RN, Stull JT, Sweeney HL, editors. Elsevier, Amsterdam, Netherlands; 2012. G-protein coupled receptors in smooth muscle; pp. 1145–1153. [Google Scholar]
- 8. Tang DD. Critical role of actin-associated proteins in smooth muscle contraction, cell proliferation, airway hyperresponsiveness and airway remodeling. Respir Res . 2015;16:134. doi: 10.1186/s12931-015-0296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gunst SJ, Zhang W. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol . 2008;295:C576–C587. doi: 10.1152/ajpcell.00253.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamill S, Lou HJ, Turk BE, Boggon TJ. Structural basis for noncanonical substrate recognition of cofilin/ADF proteins by LIM kinases. Mol Cell . 2016;62:397–408. doi: 10.1016/j.molcel.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gosens R, Stelmack GL, Dueck G, McNeill KD, Yamasaki A, Gerthoffer WT, et al. Role of caveolin-1 in p42/p44 MAP kinase activation and proliferation of human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol . 2006;291:L523–L534. doi: 10.1152/ajplung.00013.2006. [DOI] [PubMed] [Google Scholar]
- 12. Woo JA, Liu T, Fang CC, Castaño MA, Kee T, Yrigoin K, et al. β-Arrestin2 oligomers impair the clearance of pathological tau and increase tau aggregates. Proc Natl Acad Sci USA . 2020;117:5006–5015. doi: 10.1073/pnas.1917194117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Woo JA, Castaño M, Goss A, Kim D, Lewandowski EM, Chen Y, et al. Differential long-term regulation of TAS2R14 by structurally distinct agonists. FASEB J . 2019;33:12213–12225. doi: 10.1096/fj.201802627RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim D, Pauer SH, Yong HM, An SS, Liggett SB. Beta2-adrenergic receptors chaperone trapped bitter taste receptor 14 to the cell surface as a heterodimer and exert unidirectional desensitization of taste receptor function. J Biol Chem . 2016;291:17616–17628. doi: 10.1074/jbc.M116.722736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. An SS, Mitzner W, Tang WY, Ahn K, Yoon AR, Huang J, et al. An inflammation-independent contraction mechanophenotype of airway smooth muscle in asthma. J Allergy Clin Immunol . 2016;138:294–297.e4. doi: 10.1016/j.jaci.2015.12.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim D, Tokmakova A, Lujan LK, Strzelinski HR, Kim N, Najari Beidokhti M, et al. Identification and characterization of an atypical gαs-biased β(2)ar agonist that fails to evoke airway smooth muscle cell tachyphylaxis. Proc Natl Acad Sci USA . 2021;118:e2026668118. doi: 10.1073/pnas.2026668118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koziol-White C, Johnstone TB, Corpuz ML, Cao G, Orfanos S, Parikh V, et al. Budesonide enhances agonist-induced bronchodilation in human small airways by increasing cAMP production in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol . 2020;318:L345–L355. doi: 10.1152/ajplung.00393.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meyerhof W, Batram C, Kuhn C, Brockhoff A, Chudoba E, Bufe B, et al. The molecular receptive ranges of human TAS2R bitter taste receptors. Chem Senses . 2010;35:157–170. doi: 10.1093/chemse/bjp092. [DOI] [PubMed] [Google Scholar]
- 19. Hay DW. Putative mediator role of endothelin-1 in asthma and other lung diseases. Clin Exp Pharmacol Physiol . 1999;26:168–171. doi: 10.1046/j.1440-1681.1999.03009.x. [DOI] [PubMed] [Google Scholar]
- 20. Chalmers GW, Little SA, Patel KR, Thomson NC. Endothelin-1-induced bronchoconstriction in asthma. Am J Respir Crit Care Med . 1997;156:382–388. doi: 10.1164/ajrccm.156.2.9702066. [DOI] [PubMed] [Google Scholar]
- 21. Spina D. Asthma mediators: current views. J Pharm Pharmacol . 2000;52:125–145. doi: 10.1211/0022357001773779. [DOI] [PubMed] [Google Scholar]
- 22. Cai Y, Stafford LJ, Bryan BA, Mitchell D, Liu M. G-protein-activated phospholipase C-beta, new partners for cell polarity proteins Par3 and Par6. Oncogene . 2005;24:4293–4300. doi: 10.1038/sj.onc.1208593. [DOI] [PubMed] [Google Scholar]
- 23. Chen X, Macara IG. Par-3 mediates the inhibition of LIM kinase 2 to regulate cofilin phosphorylation and tight junction assembly. J Cell Biol . 2006;172:671–678. doi: 10.1083/jcb.200510061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim D, Castaño M, Lujan LK, Woo JA, Liggett SB. The short third intracellular loop and cytoplasmic tail of bitter taste receptors provide functionally relevant GRK phosphorylation sites in TAS2R14. J Biol Chem . 2021;296:100216. doi: 10.1074/jbc.RA120.016056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol . 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 26. Greotti E, De Stefani D. Biosensors for detection of calcium. Methods Cell Biol . 2020;155:337–368. doi: 10.1016/bs.mcb.2019.11.001. [DOI] [PubMed] [Google Scholar]
- 27. Robinett KS, Koziol-White CJ, Akoluk A, An SS, Panettieri RA, Jr, Liggett SB. Bitter taste receptor function in asthmatic and nonasthmatic human airway smooth muscle cells. Am J Respir Cell Mol Biol . 2014;50:678–683. doi: 10.1165/rcmb.2013-0439RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zaccolo M, Zerio A, Lobo MJ. Subcellular organization of the camp signaling pathway. Pharmacol Rev . 2021;73:278–309. doi: 10.1124/pharmrev.120.000086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim D, An SS, Lam H, Leahy JW, Liggett SB. Identification and characterization of novel bronchodilator agonists acting at human airway smooth muscle cell tas2r5. ACS Pharmacol Transl Sci . 2020;3:1069–1075. doi: 10.1021/acsptsci.0c00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang MY, Kim SK, Kim D, Liggett SB, Goddard WA., III Structures and agonist binding sites of bitter taste receptor tas2r5 complexed with Gi protein and validated against experiment. J Phys Chem Lett . 2021;12:9293–9300. doi: 10.1021/acs.jpclett.1c02162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou YW, Sun J, Wang Y, Chen CP, Tao T, Ma M, et al. Tas2R activation relaxes airway smooth muscle by release of Gαt targeting on AChR signaling. Proc Natl Acad Sci USA . 2022;119:e2121513119. doi: 10.1073/pnas.2121513119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao JW, Gao ZL, Ji QY, Wang H, Zhang HY, Yang YD, et al. Regulation of cofilin activity by CaMKII and calcineurin. Am J Med Sci . 2012;344:462–472. doi: 10.1097/MAJ.0b013e318244745b. [DOI] [PubMed] [Google Scholar]
- 33. Takemura M, Mishima T, Wang Y, Kasahara J, Fukunaga K, Ohashi K, et al. Ca2+/calmodulin-dependent protein kinase IV-mediated LIM kinase activation is critical for calcium signal-induced neurite outgrowth. J Biol Chem . 2009;284:28554–28562. doi: 10.1074/jbc.M109.006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davidson MM, Haslam RJ. Dephosphorylation of cofilin in stimulated platelets: roles for a GTP-binding protein and Ca2+ Biochem J . 1994;301:41–47. doi: 10.1042/bj3010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhan Q, Bamburg JR, Badwey JA. Products of phosphoinositide specific phospholipase C can trigger dephosphorylation of cofilin in chemoattractant stimulated neutrophils. Cell Motil Cytoskeleton . 2003;54:1–15. doi: 10.1002/cm.10079. [DOI] [PubMed] [Google Scholar]
- 36. Wang Y, Shibasaki F, Mizuno K. Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J Biol Chem . 2005;280:12683–12689. doi: 10.1074/jbc.M411494200. [DOI] [PubMed] [Google Scholar]
- 37. Deshpande DA, Robinett KS, Wang WC, Sham JS, An SS, Liggett SB. Bronchodilator activity of bitter tastants in human tissue. Nat Med . 2011;17:776–778. doi: 10.1038/nm0711-776b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nowak S, Di Pizio A, Levit A, Niv MY, Meyerhof W, Behrens M. Reengineering the ligand sensitivity of the broadly tuned human bitter taste receptor TAS2R14. Biochim Biophys Acta Gen Subj . 2018;1862:2162–2173. doi: 10.1016/j.bbagen.2018.07.009. [DOI] [PubMed] [Google Scholar]