

ABSTRACT
Monoclonal antibodies are powerful and versatile tools that enable the study of proteins in diverse contexts. They are often utilized to assist with identification of subcellular localization and characterization of the function of target proteins of interest. However, because there can be considerable sequence diversity between orthologous proteins in Xenopus and mammals, antibodies produced against mouse or human proteins often do not recognize Xenopus counterparts. To address this issue, we refined existing mouse monoclonal antibody production protocols to generate antibodies against Xenopus proteins of interest. Here, we describe several approaches for the generation of useful mouse anti-Xenopus antibodies to multiple Xenopus proteins and their validation in various experimental approaches. These novel antibodies are now available to the research community through the Developmental Study Hybridoma Bank (DSHB).
Keywords: Xenopus, Monoclonal Antibody, Hybridoma, Method
Summary: Novel monoclonal antibodies to Xenopus laevis proteins have been generated and characterized using refined hybridoma production methods suitable for basic science research laboratories.
INTRODUCTION
Monoclonal antibodies (mAbs) are produced by fusing B lymphocytes to immortal myeloma cells (Köhler and Milstein, 1975). Successful fusion creates a hybridoma cell line that produces a single heavy and light chain antibody specific to a particular antigenic determinant (epitope) of a given antigen. Like its myeloma parent, this cell line is immortal and can be propagated indefinitely. It can also be frozen, subsequently thawed, and expanded to produce large amounts of monospecific antibodies. Here, we make use of this powerful and versatile technology focused on monoclonal antibodies generated for the study of Xenopus laevis, a vertebrate model system widely used for cell and developmental biology research.
The Xenopus model system has been at the forefront of cell and developmental biology for decades and has led to the discovery of crucial developmental pathways that define the induction of embryonic tissues, the formation of primary axes, and the regulation of the cell cycle (Heasman, 2006). The large size of Xenopus embryos (∼1 mm) and the relative ease with which hundreds of synchronously developing embryos can be obtained make Xenopus ideally suited for studying the function and composition of protein complexes during early development (Exner and Willsey, 2021; Kostiuk and Khokha, 2021; Medina-Cuadra and Monsoro-Burq, 2021; Niehrs, 2022). Advances in genome annotation and genomic methods have allowed quantification of mRNA expression at most stages of embryo development and in most cell types (Gilchrist et al., 2020; Lindeboom et al., 2019). Advances in proteomics have also provided similar insights into protein expression and post-translational modifications at many developmental stages (Lindeboom et al., 2019; Lombard-Banek et al., 2019; Saha-Shah et al., 2019; Wasson et al., 2019). Although these discovery approaches are powerful, building upon them with mechanistic studies requires antibodies specific to individual proteins that can be used in studies to determine subcellular localization, confirm knockdown efficiency, and identify protein complexes formed during specific developmental stages.
Although some proteins, including histones and many kinases, are highly conserved between Xenopus and human, the average conservation of protein sequences between human and Xenopus is much lower (68%), which results in poor commercial monoclonal antibody cross-reactivity. Some approaches have taken advantage of the amenability of Xenopus embryos to large-scale protein purification to generate antibodies against proteins expressed in specific tissues or subcellular compartments (Nakazato and Ikenishi, 1989; Sakakibara et al., 2005). For example, hybridoma libraries have been produced against proteins isolated from both Xenopus and Pleurodeles oocyte nuclei and screened for their ability to recognize specific structures associated with lampbrush chromosomes (Lacroix et al., 1985; Roth and Gall, 1987).
Although polyclonal antibodies are relatively easy and inexpensive to make, they are a finite resource that should be carefully characterized because of variability in specificity between each batch. Examples abound in which a new lot of a commercial antibody no longer recognizes the original, specific target protein or begins to recognize additional, nonspecific targets. Consequently, recent efforts focus on clonal antibody production via either hybridoma fusion or using methods whereby immunoglobulin genes are cloned and expressed in cell lines (Ouisse et al., 2017).
Here, we describe a monoclonal antibody production pipeline utilizing ‘classical’ fusion methods paired with refinement strategies and recent advances in cell biology and cell culture. We outline both low-throughput techniques applicable to most laboratories as well as automated robotic improvements for medium-scale studies.
RESULTS AND DISCUSSION
Given the growing number of monoclonal antibodies and the convention of naming them according to the plate number and position (e.g. 9E10 for myc), we chose nomenclature that includes initials before the plate number, position and the target protein name (e.g. DA5H6sox3). All antibodies generated in this study are distributed via the Developmental Study Hybridoma Bank (DSHB; https://dshb.biology.uiowa.edu) at cost.
Antigen production
We have used three strategies to produce antigens: bacterial fusion proteins, proteins produced and purified from human Hek293T cells, and peptides. Although we were able to reliably produce mAbs with both bacterial fusion proteins and Hek293T proteins, we did not have any success with the peptides. For our initial antibody production, each protein antigen was a full-length protein tagged with FLAG. This was justified, as we could rapidly obtain these constructs from individual investigators in plasmids, such as pCS2, that allow both mRNA production via SP6 RNA polymerase and expression in cells by transfection via the CMV promoter. Following transfection in Hek293T cells, a typical construct would yield, on average, 10 μg of protein per μg of DNA. Thus, transfecting between five and ten 10-cm plates yielded sufficient protein for a full immunization schedule (200-1000 µg of protein). Xenopus XTC cells were also transfected with the same constructs, which enabled us to screen hybridomas by indirect immunofluorescence. This initial approach worked well for many proteins (Table 1), and, typically, four mice were injected with five proteins each, using either Freund or Adjuplex adjuvants. Although certain antigens were dominant in multiply immunized animals, reactivity against all five proteins was often found in the serum (Fig. 1). In general, the mice immunized with Freund's adjuvant gave a much stronger immune response than those immunized with Adjuplex, but both adjuvants generated useful hybridomas. To conserve limited resources, often only the ‘best mouse’ (defined by strong and broadly reactive serum titres to the target proteins) was selected for fusion rather than doing side-by-side comparisons. We observed that, in many cases, the variability of responses between mice was more significant than the variability between adjuvants, highlighting the difficulty of pinpointing ‘ideal’ protein/adjuvant/animal combinations. Nevertheless, each immunization produced hybridomas to nearly all targets that could recognize the protein by enzyme-linked immunosorbent assay (ELISA), western blot and immunofluorescence when tested on cells overexpressing the specific target. In addition, we also isolated two anti-FLAG hybridomas that will be useful to the community.
Table 1.
Antigens used to generate hybridomas

Fig. 1.

Immunofluorescence testing of serum. The serum from mouse M3 immunized with FoxD3-Flag, Twist-Flag, Six1-Flag, Slug-Flag and Sox3-Flag produced in Hek293T cells was tested by immunofluorescence on XTC cells transfected with each of the target or RFP-Flag as a negative control. A secondary Alexa 488-conjugated anti-mouse antibody was used to visualize the mouse primary antibodies (green). Notice the green nuclei in each of the transfections, with some non-transfected cells lacking signal.
Limitation of the Hek293T production approach
Although we were successful in generating many useful hybridomas using full-length proteins produced in Hek293T cells, there were cases where this proved problematic. First, we found that some proteins could not be produced or purified in sufficient quantity to be used for immunization (e.g. Adam11). Second, we found that for some proteins that are members of multiprotein families with significant sequence homology (e.g. FoxD3), all clones exhibited broad cross-reactivity with other members of those families. To circumvent these issues, histidine-tag fusion proteins were generated using less-conserved regions of these factors. These proteins were then expressed in and purified from bacteria. This approach was utilized for several transcription factors, including Slug, Twist, Six1, Prdm12, Eya1, Six3 and Mcrs1. This approach was also used to purify the cytoplasmic domains of transmembrane proteins (PCNS, PDGFRα, Adam13). For the most part, these proteins generated strong immune reactions and yielded many hybridomas. By contrast, bacterial fusion proteins often carried with them contaminants that were highly immunogenic and could hijack the reaction of the immune system. To avoid the detection of hybridomas that produce antibodies toward bacterial contaminants co-purified with the fusion proteins, we used extracts from Hek293T transfected with the full-length proteins for the ELISA assays (see ‘Primary screening’ section).
We found that, independently of the method of production and purification, we were able to produce hybridoma that specifically recognized the purified antigen. The complete list of antigens produced, the source of production and the identification of positive hybridomas is provided in Table 1.
Other antigens
We also tested three other types of antigens that will not be described in detail here. First, we used peptides corresponding to optimal sequences based on antigenicity and exposure on the folded protein coupled to keyhole limpet haemocyanin (KLH). Unfortunately, although we did get strong immune reactions from the injected mice, we did not identify useful hybridomas. We also produced mRNA and immunized mice using lipid nanoparticles, as previously described for the coronavirus (Gebre et al., 2022). In effect, in vitro transcribed mRNA is assembled into modified lipid vesicles that are used to deliver the mRNA intramuscular so that the foreign protein is produced in situ. As noted previously, most laboratories working with Xenopus embryos routinely inject in vitro-transcribed mRNA, making this approach extremely attractive. Unfortunately, the immunization of four mice with this strategy did not generate any immune response to the targets (Xenopus Adam9, Adam11, and Adam19). Finally, we also used Xenopus embryo extracts and Xenopus cell lines for immunizations. These gave solid immune responses and multiple hybridomas, but the targets were challenging to identify using mass spectrometry.
Hybridomas production
Although our initial fusions were performed using the classical polyethylene glycol (PEG) fusion with success, we moved to electrofusion, given its significantly higher efficiency (20-fold). In practice, this means that one mouse with an average spleen containing 100 million splenocytes that yielded an average of 1000 hybridomas with PEG instead yielded 20,000 hybridomas with electrofusion. Although screening 20,000 hybridomas for five targets is not routinely practical in a small laboratory, this allowed us to aliquot and freeze splenocytes for fusion at a later time should good hybridomas not be identified in the initial screening. It also meant that if one fusion was contaminated, we could easily repeat that fusion using frozen splenocytes and not lose the weeks of immunization. In general, two fusions were performed for each set of antigens using 10 million splenocytes and 10 million Sp2/0 per fusion that were then split into four 96-well plates. On average, each well contained one to three clones (400-1200 hybridomas/fusion).
Primary screening
To optimize the pipeline further, supernatants from four or eight plates (one or two fusions) were pooled for the primary screen to facilitate rapid identification of positive hybridomas with a minimum number of ELISA and immunofluorescence plates. In practice, at day ten post-fusion, 30 µl was collected from each well and pooled into a ‘master plate’, preserving the well identity (all A1 wells would be A1 in the master plate; see diagram in Fig. 2A). Although this can be done manually, we utilized an Opentrons robot (OT2; https://opentrons.com) to increase efficiency and reduce human error. Depending on the number of hybridomas per well, the supernatants were pooled from either four plates (if more than three clones per well) or eight plates (if fewer than three clones per well). The master plate was then used to apply the supernatant to ELISA plates coated with individual proteins as well as glass-bottom plates containing XTC cells transfected with plasmids coding each antigen (Fig. 2) paired with fluorescent markers. We assigned fluorescent markers as follows: our first construct was transfected with nuclear cherry, the second with membrane cherry, the third with nuclear sirius, the fourth with cytoplasmic sirius and the fifth with RFP. To facilitate screening, we typically paired nuclear fluorescent markers with transmembrane proteins and membrane fluorescent markers for nuclear proteins so that signals could readily be detected with a fluorescence microscope set on a triple filter channel (Zeiss Axiovert 200 M, red/blue/green). Once transfected individually, cells were pooled and plated in glass-bottom 96-well plates. Immunofluorescence was carried out using an Alexa 488-conjugated anti-mouse secondary antibody. Once positive wells were identified, for example, A6, that position was then tested on each fusion plate (1A6, 2A6, 3A6, 4A6) to identify the exact position of the positive colony (deconvolution, plate/line/column). In most cases, one well produced supernatant that recognized the same protein by both ELISA and immunofluorescence. In some cases, we identified wells that recognized all the proteins in both assays. These turned out to be directed against the FLAG-epitope tag used in the purification. We selected and characterized two of these anti-FLAG antibodies, which are now freely available from DSHB (https://dshb.biology.uiowa.edu/search?keywords=flag).
Fig. 2.
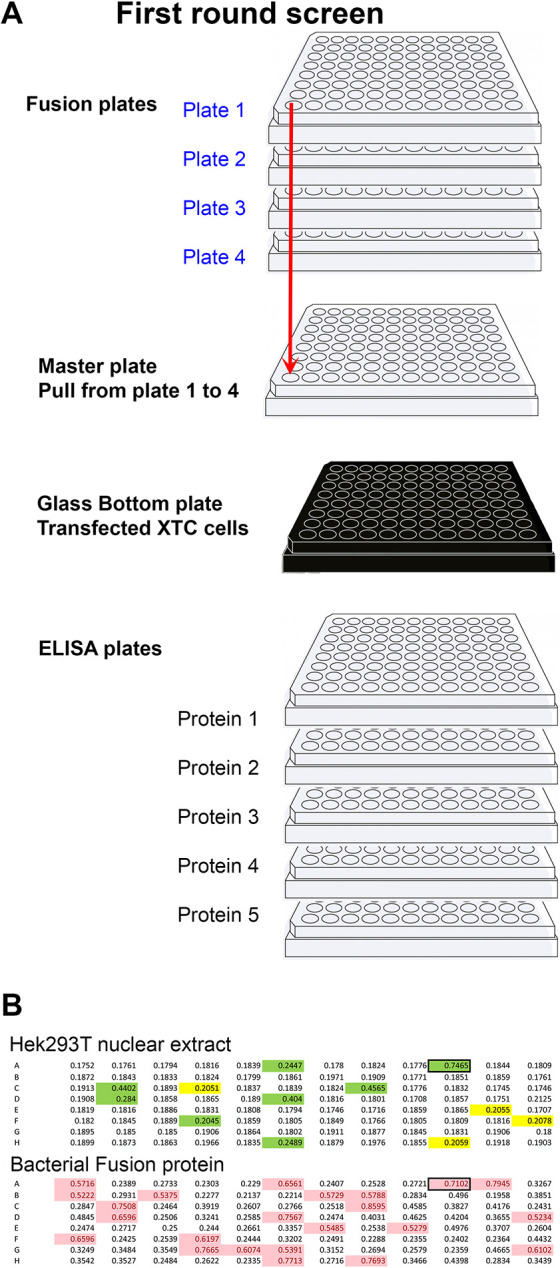
Primary screening method. (A) Diagram representing the strategy used to rapidly identify wells producing antibodies to multiple targets at once. Four plates per fusion were used. Thirty microlitres were taken from each well and placed in the master plate (total volume 120 µl). The master plate was diluted with 200 µl of TBST (TBS with 0.1% Tween 20). From the 320 µl of the master plate, 50 µl was added to each of the five ELISA plates plus the glass-bottom plate containing XTC cells transfected with each of the five targets. Simultaneously, 50 µl of supernatant was added to each well of five ELISA plates, each coated with individual proteins. Wells positive in either or both assays (A1 to H12) were then tested on each of the original plates (blue) to identify the plate number (deconvolution, 1A1 to 4H12). (B) Primary screen by ELISA using the cytoplasmic domain of Adam13/33 expressed in Hek293T cells (top) and his-tag bacterial fusion protein (bottom). The mice were immunized using the bacterial fusion protein. Highlighted wells are above the chosen background (0.2 for Hek293T and 0.5 for bacterial fusion protein). Wells highlighted in green are common to both ELISA and were selected for further screening.
When a western blotting antibody was preferred by an investigator, the primary screen was performed using ELISA, followed by a western blot on the protein expressed in Hek293T cells instead of immunofluorescence. This method prioritized the identification of antibodies to linear epitopes of denatured proteins rather than native conformations in fixed cells.
For the hybridomas that were produced against bacterial fusion proteins, we screened using the same proteins expressed in Hek293T cells. This was crucial when screening master plates, where every well corresponds to a pool of four wells, each with three hybridomas (12 potential colonies). In such cases, 75% of the colonies that appeared to detect the bacterial fusion protein did not detect the protein when expressed in Hek293T cells (Fig. 2B). This suggested that the hybridoma either recognized one of the plasmid expression tags (S-tag or his-tag in pet30a) or a co-purified bacterial protein contaminant. Being able to eliminate 75% of false-positive clones was essential for the successful identification of useful antibodies (in this example, eight hybridomas). For ELISAs, we found that protein purification from Hek293T cells was often not necessary. Instead, in this case, we either used raw nuclear extract (NE-per) or total protein extract (in PBS) to coat ELISA plates. This was especially convenient for proteins that were poorly expressed and/or purified. In the absence of detergent, the transfected proteins were sufficiently enriched to produce a detectable signal above the background.
Clone isolation
Once a positive well was identified, it was a race against time to identify the colony that produced a useful antibody. A typical well had three colonies of various sizes growing. In general, 50% of hybridomas do not produce immunoglobulin owing to the random distribution of chromosome sets during fusion. These tend to grow faster as they are not using their resources for antibody production. As cells multiply, the border between colonies becomes less obvious, and, if re-feeding is necessary, cell mixing becomes a real possibility. To maximize efficiency, we performed several rounds of screening from the original well to confirm the usefulness of the hybridoma before cloning (see ‘Secondary screening’ section below). We then hand-picked individual colonies from each positive well (Fig. 3). This step was critical, as it minimized the chances of losing the clone that was producing the antibodies of interest. In practice, a typical first-round screening identified 10-50 hybridomas per fusion. At three colonies per well, this led to 30-150 new wells after picking, a manageable number for the next series of screening. In the alternative scenario, if ultimate dilution was used directly, it would lead to 10-50 new 96-well plates, a number that far exceeds the capacity of most small laboratories. Another advantage of manually picking colonies is that the transferred colony will produce new supernatant ready to use within 3-5 days with minimum contamination from the original well (1/200 µl), which still contained the antibody produced during the first 10 days. Furthermore, by transferring the larger hybridomas to new wells, this technique provided a chance for smaller hybridoma colonies to grow to a size that allowed picking a few days later if none of the previously picked colonies produced the antibody of interest.
Fig. 3.

Manual clone picking. (A) Plates containing the hybridoma fusion were placed on a mirror to visualize large colonies. (B) Investigator pipetting a single colony guided by the mirror. (C) Photographs of the plate as seen in the mirror. (D) Higher magnification of four wells. Individual colonies are highlighted with red arrowheads.
Antibodies recognizing the full-length proteins expressed in cell lines by immunofluorescence and/or western blot were frozen and shared with individual investigators for further testing using the endogenous proteins and in the context of knockdown. In general, although we were able to produce monoclonal antibodies that recognized each of the targets that were used in our immunization, only a fraction (25/148; approximately 17%) were sufficiently selective or sensitive to detect the endogenous proteins. Antibodies that do not recognize the endogenous protein can still be used in transfection experiments or through overexpression to perform western blot, immunofluorescence and co-immunoprecipitation without the requirement for tags that may interfere with the function of the protein.
Secondary screening – confirmation of specificity in vivo
As our main goal was to produce antibodies that could recognize endogenous proteins expressed in Xenopus embryos, each positive hybridoma required further characterization. Promising antibodies were screened for their ability to detect endogenous protein by western blot, and, in some cases, they were further characterized by immunostaining or immunoprecipitation. Examples of each assay are given below.
Western blotting
We utilized a capillary western blotting system (WES ProteinSimpleTM) in which 24 individual hybridoma supernatants can be tested at once using only 10 µl of supernatant per sample. This meant that hybridomas could be tested before subcloning using the small amount of supernatant from the original well identified after deconvolution. Hybridomas were screened for those that could detect the overexpressed protein in Hek293T cells and that generated a similar size band in embryo lysates prepared from a stage at which the mRNA was expressed but detected no band at stages at which the mRNA was absent (https://www.xenbase.org/entry/). In cases in which a morpholino was available to deplete the protein of interest, we tested protein extracts from non-injected control embryos compared with embryos injected with 10 ng of the relevant morpholino. In our experience, this dose was sufficient to block translation effectively in all but the most abundant proteins (e.g. lbh) (Cousin et al., 2011; Weir et al., 2021). An example of this strategy is presented in Fig. 4A-C for the Xbra (Tbxt) monoclonal antibody.
Fig. 4.

Secondary screening. (A) RNA-sequencing (RNA-Seq) expression data for Xenopus brachyury (tbxt) from Xenbase. No mRNA is detected for this gene at stage 1 (fertilized egg), but it is strongly expressed during gastrulation (stage 12). (B) Capillary western blot (0.04 embryo equivalent) at stage 1 and stage 12 for three independent hybridomas (8F8, 12H2, 5D11). Note that bands are visible only at stage 12. (C) Signal intensity of the chemiluminescence for clone 12H2 at stages 1 and 12. Note that a single peak is present in stage 12 (green), but not stage 1 (blue), but the background signal is similar. (D) Whole-mount immunofluorescence of a neurula stage embryo. Sox3 is in magenta, and DAPI (to stain nuclei) is in blue. MAb DA5H6sox3 detects the nuclei of cells within the neural plate. (E,F) Chemiluminescence signal (E) from a capillary western blot (F) of embryo extracts (0.04 embryo equivalent per lane) incubated with antibodies to Sox3 (green), Ribophorin1 (Rpn1, red) and the muscle marker 12101 (blue). Antibodies were incubated either separately (lanes 1-3) or together (lane 4).
This step was critical to identify antibodies that recognized linear epitopes that are masked in the folded protein or in multiprotein complexes and might also be masked in immunoprecipitation and immunofluorescence assays.
Immunostaining
Our original intent was to test all positive clones by whole-mount immunostaining. However, we found that both alkaline phosphatase and peroxidase staining required significant optimization for each antibody, making it impractical to carry this out for each clone. Instead, the suitability of antibodies for use in immunostaining assays was tested on a case-by-case basis. Fig. 4 shows one such example for mAb DA5H6sox3 directed against the transcription factor Sox3 using immunofluorescence. This antibody readily detects nuclear Sox3 in the neural plate of Xenopus embryos (Fig. 4D). Importantly, it does not cross-react with the related SoxB1 factor Sox2 (Fig. S1), making this a valuable antibody for studies of neural development. MAb DA5H6sox3 also detects endogenous Sox3 in embryo extracts (Fig. 4E). Similarly, we were able to obtain whole-mount immunostaining (Fig. 5A) for brachyury (Xbra/Tbxt) and Adam13/33 (Fig. 5B). We also found that explants of cranial neural crest cells were an excellent tool to detect endogenous proteins present in these cells (Fig. 5C, DA1A8slug).
Fig. 5.

Immunostaining. (A) Whole-mount immunostaining using DA12H2xbra on bisected gastrula (Xbra/Tbxt). The dorsal side is to the left, and the animal pole is up. The antibody stains the nuclei in both the dorsal and ventral marginal zones (mesoderm). (B) Whole-mount immunostaining using a monoclonal antibody to Adam13/33 (DA8E6adam13) on embryos injected at the two-cell stage with a morpholino that blocks translation of Adam13. The cranial neural crest cells (black arrows) are visible on the non-injected (NI) side, but are absent on the injected side (MO13). (C) Immunofluorescence of a cranial neural crest explant using mAb DA1A8slug (green) and counterstained with DAPI (blue). Note that most of the nuclei are stained for Slug/Snai2 except for a small section on the left of the explant (dorsal) that is likely to be composed of neural plate cells.
Immunoprecipitation
In cases for which a commercial antibody (polyclonal) that recognized the Xenopus protein by western blot was available, we were able to test the hybridoma of interest for its utility in immunoprecipitation experiments. For these assays, the monoclonal antibody was used to pull down the target protein from an embryo extract, which was then detected by western blotting using the commercial antibody. Alternatively, immunoprecipitates were tested by tandem mass spectrometry to determine whether the expected protein target was pulled down (e.g. mAb Rpn1; Fig. S2). This step was critical to identify antibodies that may work in chromatin immunoprecipitation assays.
Conclusion and future directions
Animal model systems are critically important for the advancement of both basic science and biomedical research. The amphibian Xenopus laevis has been used for decades, first as a pregnancy test (human chorionic gonadotropin induces the laying of eggs) (Elkan, 1938) and then as a model to understand embryo development and evolution. Given the external development (embryos develop autonomously outside of the mother) that allows investigators to view and experiment on each stage, the ability to generate a large number of embryos, and advances in genomics, Xenopus is now a choice model system to test the effect of known human mutations causing diseases (Moody et al., 2015; Willsey et al., 2022). One of the resources that has lagged is the availability of high-quality monoclonal antibodies to study protein localization and function. Our laboratory has produced more than 100 monoclonal antibodies that recognize 25 endogenous protein targets in the embryo. These antibodies are available worldwide and are adding to the existing list of polyclonal (809) and monoclonal (767) antibodies that have been shown to function in Xenopus (https://www.xenbase.org/entry/reagents/antibody.do?resultsPerPage=50). When comparing the Xenopus tropicalis proteome (to avoid issues of duplicated genes) with that of humans, we found 36 identical proteins and 865 proteins with over 90% sequence identity (from 11,182 identified orthologues). We selected a cutoff of 90% as it is typically a good indication that epitopes might be sufficiently conserved to provide cross-reacting antibodies. This suggests that the number of cross-reacting commercial antibodies is unlikely to increase in the future unless companies design their epitopes to be conserved across multiple species.
Another approach that has been used extensively in the pharmaceutical industry is the use of recombinant antibodies (Maruthachalam et al., 2022; Panagides et al., 2022). Libraries of variable chain antibodies exist that can be used to screen and identify binders. Although this is an extremely efficient way of producing large amounts of a single antibody that can be sold and tailored to a specific application (for example, inserting the variable region into the human constant region), it is not a directly usable resource that can be provided to a typical size laboratory. It also requires the same amount of antigen to screen the libraries. We tested a phage display library to identify targets that would bind specifically to Xenopus proteins. Although we performed four rounds of enrichment, we did not find any individual clones that recognized our protein of interest. A similar approach using the single-chain camelid immunoglobulin to produce antibodies to Xenopus protein (Itoh et al., 2019) has only shown moderate success as well. As the technology improves, we expect that this will become a good solution for molecular biology laboratories that are used to screening phage libraries to produce their own recombinant antibodies.
MATERIALS AND METHODS
SP20 ATCC CRL-1581, SP2/0-Ag14
The SP2/0 myeloma cell line was grown from frozen stock in RPMI media supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 0.1 mg/ml streptomycin, 2 mM L-glutamine and 1 mM sodium pyruvate. Cells were grown for up to 1 week, maintaining a density of less than 1.106/ml. On the day before the fusion, cells were expanded into as many plates as needed with an expected density of 5.105-1.106/ml.
Hek293T ATCC CRL-3216
Hek293T cells were grown in RPMI media supplemented with 10% FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 2 mM L-glutamine and 1 mM sodium pyruvate. Cells were passaged the day before transfection to 30% confluence and transfected with polyethylenimine (PEI; 1 mg/ml in water) at a ratio of 2 million cells, 10 µl of PEI per μg of plasmid DNA. The DNA was mixed in pure RPMI (200 µl/µg of DNA) containing 10 µl of PEI (1 mg/ml). The solution was mixed using a vortex and incubated at room temperature (RT) for 15 min before being added dropwise to plated cells. Cells or supernatant were collected 36-48 h after transfection and extracted for either ELISA (see below) or protein purification. Most proteins utilized were FLAG-tagged and were purified on FLAG-affinity gel (Applied Biological Materials). Proteins were eluted using 100 mM glycine pH 2.8, and immediately neutralized using 1 M Tris pH 9.5 (50 mM final). Proteins were dialysed against PBS and quantified using a bicinchoninic acid (BCA) assay (Pierce). One aliquot was run on an SDS-PAGE gel, which was subsequently stained with Coomassie to evaluate protein purity.
Xenopus XTC cells
Xenopus XTC cells were grown in 67% L15 media supplemented with 10% FBS, 100 U/ml penicillin 0.1 mg/ml streptomycin, 2 mM L-glutamine, 50 µg/ml gentamycin and 1 mM sodium pyruvate at RT (20-25°C) without CO2. Transfection was performed using FuGENE HD (Sigma-Aldrich) according to the manufacturer's instructions. Typically, one well of a six-well plate (60-80% confluence) was transfected with 0.5-1 µg of DNA mixed with 1.5-3 µl of FuGENE, yielding 30-70% transfection efficiency.
Bacterial fusion protein Pet30a
Full cDNA or specific regions were selected for their comparative variability and cloned using Infusion (Takara Bio) into bacterial expression plasmid (Pet30a). The constructs were transformed into BL21 DE3 pLysS (Thermo Fisher Scientific) Escherichia coli, grown to 0.5 OD prior to stimulation with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and grown for an additional 3 h, after which the bacteria were pelleted. If the solubility of a fusion protein was unknown, native purification was performed first. The bacterial pellet was resuspended in sonication buffer containing 2 mM phenylmethylsulphonyl fluoride (PMSF) and sonicated three times for 60 s on ice. After the addition of Triton X-100 to 1% final concentration, the bacterial lysate was spun for 20 min at 10,000 g at 4°C to separate soluble and insoluble fractions. The insoluble pellet was then extracted in 8 M urea containing 100 mM NaH2PO4 and 10 mM Tris pH 8.0, mixed on a vortex, and rotated for 30 min at RT prior to spinning for 20 min at 10,000 g at RT. The supernatants were incubated with Ni-NTA agarose resin (HisPur, Sigma-Aldrich) overnight at 4°C (native) or 30 min at RT (denatured). Beads were washed in sonication buffer containing 20 mM imidazole (native) or 8 M urea pH 6.3 (denatured). Proteins were eluted with a step gradient of imidazole (100-400 mM) for the native purification or 8 M urea at pH 4.5 for the denatured proteins. All proteins were dialysed against PBS and quantified using a BCA assay (Pierce). The purity of fractions was visualized on SDS-PAGE using Coomassie Brilliant Blue (Thermo Fisher Scientific) before immunization.
Immunization
Eight-week-old female BalbC mice (The Jackson Laboratory) were immunized with either Freund's adjuvant (Sigma-Aldrich) or Adjuplex (Empirion LLC). The immunization schedule was the same independent of the adjuvant, with one primary immunization on day 1, followed by one boost on day 21. All immunizations were performed intraperitoneally with a maximum of 100 µg of purified protein representing either a single protein or 20 µg each of up to five different purified proteins. Serum was collected on day 30 and tested by western blotting against the protein expressed in Hek293T cells and by immunofluorescence against the protein expressed in Xenopus XTC cells. Mice whose serum displayed robust antigen recognition were selected for fusion. If antigen recognition was limited, another boost was performed 3 weeks after the previous boost. Prior to splenectomy, mice were rested for 1 month and immunized with the protein in PBS without adjuvant 3 days prior to sacrifice. Animal studies were approved by the University of Massachusetts, Amherst's Institutional Animal Care and Use Committee (IACUC protocol# 2800).
Fusion
Spleens were harvested aseptically in a biosafety cabinet. Splenocytes were isolated in RPMI media (Cytiva) and washed. Red blood cells were eliminated with RBC Lysis Buffer (Sigma-Aldrich) according to the manufacturer's instructions. Before fusion, SP20 cells were washed with RPMI and pelleted to remove any remaining serum. Initial fusions were performed using PEG (Roche) using up to 150 million splenocytes and 50 million SP20 using a standard protocol as described by Cousin et al. (2000). Most of our fusions were subsequently performed using electrofusion (BTX, Harvard Apparatus) using 10 million splenocytes and 10-30 million SP20 per fusion to obtain a similar number of hybridomas (∼1000). BTX settings were: 35 s AC alignment, 2 pulse at 10 µs at 1000 V, 9 s post fusion AC, using the Eppendorf iso-osmolar buffer. For each spleen, two-thirds of the splenocytes were aliquoted and frozen in 90% FBS and 10% DMSO for future fusions. Each fusion was resuspended in 25 ml of RPMI media containing 20% FBS, HAT (0.1 mM sodium hypoxanthine, 0.4 µM aminopterin, 16 µM thymidine), 100 U/ml penicillin, 0.1 mg/ml streptomycin, 2 mM L-glutamine, 1 mM sodium pyruvate and Hybridoma Fusion and Cloning Supplement (Roche) in a deep 10 cm plate (Fisher). One to three days later, the fusion was collected, spun down at 300 g for 5 min, and distributed into four 96-well plates with 200 µl of the same media per well.
Primary screening
At day ten post-fusion, hybridomas from 24 randomly selected wells were counted, and from there, the total number of colonies was extrapolated. The supernatant was then tested by ELISA on each antigen. For bacterial fusion protein immunogens, the primary screen was performed using the same protein produced in human Hek293T cells. In most cases, Hek293T cells were transfected with the target Xenopus protein (using PEI). The proteins were expressed for 48 h, and cells were washed with PBS and frozen. Protein extraction was performed using PBS with 5 mM EDTA and protease phosphatase inhibitor complex (Thermo Fisher Scientific) by passing cells through a 24 g needle ten times. Insoluble debris was pelleted for 10 min at 16,000 g at 4°C. The supernatant containing the transfected protein was directly coated onto ELISA plates without additional purification. For nuclear proteins, nuclear extractions were performed (Pierce NE-per) diluted and used directly in ELISA. Coating efficiency was tested using an anti-FLAG antibody (Sigma-Aldrich, M2).
Secondary screening
The optimal secondary screen was dependent on the desired endpoint application (e.g. western blot, immunofluorescence, immunoprecipitation). Here two main screens were utilized. The first screen used Xenopus XTC cells transfected with each target and a fluorescent marker (target 1 plus nuclear cherry, target 2 plus membrane cherry, etc.). After transfection, cells were collected, mixed and plated into 96-well glass-bottom plates (Cellvis) previously coated with bovine fibronectin (Sigma-Aldrich) at 5 µg/ml. Twenty-four hours after seeding, cells were fixed using 4% paraformaldehyde in 1× MBS (88.0 mM NaCl, 1.0 mM KCl, 2.4 mM NaHCO3, 15.0 mM HEPES pH 7.6, 0.3 mM CaNO3·4H2O, 0.41 mM CaCl2·6H2O, 0.82 mM MgSO4) for 30 min at RT. Cells were permeabilized in 1× MBS containing 0.5% Triton X-100 for 5 min at RT, washed with PBS, and blocked using PBS containing 1% bovine serum albumin (BSA) for at least 1 h at RT or overnight at 4°C. Plates were kept for up to 1 week at 4°C. The second screen was performed using an automatic capillary western blotting system (WES, ProteinSimpleTM) following the manufacturer's instructions.
Whole-mount immunostaining
Xenopus laevis embryos were obtained using in vitro fertilization according to a standard protocol (Sive and Harland, 2022) and raised to the appropriate stage in 0.1× MBS. For fluorescence, Xenopus laevis embryos were fixed in MEMFA (0.1 M MOPS pH 7.4, 2 mM EGTA pH 8, 1 mM MgSO4 and 4% paraformaldehyde), bleached, blocked in PBS with 1% BSA and 10% goat serum for 1 h at RT, and incubated overnight at 4°C with the primary antibody (1:100 mAb DA5H6sox3). Embryos were washed and incubated overnight at 4°C with an anti-mouse secondary antibody conjugated to an Alexa Fluor dye (1:500) and DAPI (1:5000). Embryos were imaged using a Nikon C2 upright confocal microscope. For peroxidase staining, Xenopus embryos were treated as above. The primary and secondary antibodies (goat anti-mouse HRP, 1/1000, Jackson ImmunoResearch) were incubated overnight at 4°C and washed six times for 10 min at RT with PBS 0.1% Tween 20 (PTW). KPL TrueBlue substrate was added to the embryos until the label was visible. Overstained embryos were washed in PTW and re-stained until the desired contrast was obtained.
Subcloning
Individual colonies from positive wells of the seeded 96-well plates were picked manually using a P2 PIPETMAN aspirating 1 µl and transferring into 200 µl of media in a new 96-well plate (Fig. 1). Briefly, 96-well plates were placed on a mirror in the biosafety cabinet (Fig. 1A), the positive well was identified, and the tip of a P2 PIPETMAN was slowly lowered to cover a single colony, which was aspirated (Fig. 1B). The procedure was repeated on each visible colony in each of the positive wells. Subclones were re-tested 3-5 days after transfer, expanded into six-well plates, and subsequently frozen. The best candidates were immediately sub-cloned by seeding one new 96-well plate at one cell per well (ultimate dilution) re-tested, expanded and frozen. Only hybridomas that went through the two rounds of sub-cloning were considered clonal.
Supplementary Material
Acknowledgements
We thank all Alfandari laboratory members for their discussion and suggestions; Dr Irini Topalidou and Dr Amy Burnside for proofreading the manuscript; Dr Inchul Yeo for his help in the characterization of the antibody to VegT; Dr Sergei Sokol, Takuya Nakayama and Helen Willsey for their help in the characterization of multiple monoclonal antibodies; all Xenopus contributors that provided plasmids to produce proteins; and the current and former staff at the Fred Hutch Antibody Technology Resource and the University of Massachusetts Amherst IALS microscopy and mass spectrometry facilities for their help. Mass spectral data were obtained at the University of Massachusetts Mass Spectrometry Core Facility (RRID:SCR_019063). Research reported in this publication was supported by the Office Of The Director, National Institutes Of Health of the National Institutes of Health under Award Number S10OD010645.
Footnotes
Author contributions
Conceptualization: D.A.; Methodology: B.H., D.A.; Validation: B.H., R.K., A.P., B.G.H., E.S., C.L., D.A.; Formal analysis: B.H., R.K., D.A.; Investigation: B.H., R.K., D.A.; Resources: D.A., B.G.H.; Data curation: D.A., B.H., R.K.; Writing - original draft: D.A.; Writing - review & editing: B.H., R.K., B.G.H., C.L., D.A.; Supervision: D.A.; Project administration: D.A.; Funding acquisition: D.A.
Funding
This work was supported by a grant from the National Institutes of Health (R24OD021485 from the office of the directorate to D.A.). D.A. is also supported by grants from the National Institute of Dental and Craniofacial Research (R01DE016289 to D.A. and R01DE026434 to S. Moody and D.A.). Deposited in PMC for release after 12 months.
Data availability
All relevant data can be found within the article and its supplementary information.
Peer review history
The peer review history is available online at https://journals.biologists.com/dev/lookup/doi/10.1242/dev.201309.reviewer-comments.pdf
References
- Cousin, H., Gaultier, A., Bleux, C., Darribere, T. and Alfandari, D. (2000). PACSIN2 is a regulator of the metalloprotease/disintegrin ADAM13. Dev. Biol. 227, 197-210. 10.1006/dbio.2000.9871 [DOI] [PubMed] [Google Scholar]
- Cousin, H., Abbruzzese, G., Kerdavid, E., Gaultier, A. and Alfandari, D. (2011). Translocation of the cytoplasmic domain of ADAM13 to the nucleus is essential for Calpain8-a expression and cranial neural crest cell migration. Dev. Cell 20, 256-263. 10.1016/j.devcel.2010.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkan, E. R. (1938). The Xenopus pregnancy test. Br. Med. J. 2, 1253-1274. 10.1136/bmj.2.4067.1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exner, C. R. T. and Willsey, H. R. (2021). Xenopus leads the way: frogs as a pioneering model to understand the human brain. Genesis 59, e23405. 10.1002/dvg.23405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebre, M. S., Rauch, S., Roth, N., Gergen, J., Yu, J., Liu, X., Cole, A. C., Mueller, S. O., Petsch, B. and Barouch, D. H. (2022). mRNA vaccines induce rapid antibody responses in mice. NPJ Vaccines 7, 88. 10.1038/s41541-022-00511-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist, M. J., Veenstra, G. J. C. and Cho, K. W. Y. (2020). Transcriptomics and proteomics methods for Xenopus embryos and tissues. Cold Spring Harb. Protoc. 2020, 098350. 10.1101/pdb.top098350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman, J. (2006). Patterning the early Xenopus embryo. Development 133, 1205-1217. 10.1242/dev.02304 [DOI] [PubMed] [Google Scholar]
- Itoh, K., Reis, A. H., Hayhurst, A. and Sokol, S. Y. (2019). Isolation of nanobodies against Xenopus embryonic antigens using immune and non-immune phage display libraries. PLoS One 14, e0216083. 10.1371/journal.pone.0216083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler, G. and Milstein, C. (1975). Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495-497. 10.1038/256495a0 [DOI] [PubMed] [Google Scholar]
- Kostiuk, V. and Khokha, M. K. (2021). Xenopus as a platform for discovery of genes relevant to human disease. Curr. Top. Dev. Biol. 145, 277-312. 10.1016/bs.ctdb.2021.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix, J. C., Azzouz, R., Boucher, D., Abbadie, C., Pyne, C. K. and Charlemagne, J. (1985). Monoclonal antibodies to lampbrush chromosome antigens of Pleurodeles waltlii. Chromosoma 92, 69-80. 10.1007/BF00327246 [DOI] [PubMed] [Google Scholar]
- Lindeboom, R. G. H., Smits, A. H., Perino, M., Veenstra, G. J. C. and Vermeulen, M. (2019). Mass spectrometry-based absolute quantification of single Xenopus embryo proteomes. Cold Spring Harb. Protoc. 2019, 479-485. 10.1101/pdb.prot098376 [DOI] [PubMed] [Google Scholar]
- Lombard-Banek, C., Moody, S. A., Manzini, M. C. and Nemes, P. (2019). Microsampling capillary electrophoresis mass spectrometry enables single-cell proteomics in complex tissues: developing cell clones in live Xenopus laevis and Zebrafish Embryos. Anal. Chem. 91, 4797-4805. 10.1021/acs.analchem.9b00345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruthachalam, B. V., Barreto, K., Hogan, D., Kusalik, A. and Geyer, C. R. (2022). Generation of synthetic antibody fragments with optimal complementarity determining region lengths for Notch-1 recognition. Front. Microbiol. 13, 931307. 10.3389/fmicb.2022.931307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Cuadra, L. and Monsoro-Burq, A. H. (2021). Xenopus, an emerging model for studying pathologies of the neural crest. Curr. Top. Dev. Biol. 145, 313-348. 10.1016/bs.ctdb.2021.03.002 [DOI] [PubMed] [Google Scholar]
- Moody, S. A., Neilson, K. M., Kenyon, K. L., Alfandari, D. and Pignoni, F. (2015). Using Xenopus to discover new genes involved in branchiootorenal spectrum disorders. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 178, 16-24. 10.1016/j.cbpc.2015.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazato, S. and Ikenishi, K. (1989). Monoclonal antibody production against a subcellular fraction of vegetal pole cytoplasm containing the germ plasm of Xenopus 2-cell eggs. Cell Differ. Dev. 27, 163-174. 10.1016/0922-3371(89)90697-7 [DOI] [PubMed] [Google Scholar]
- Niehrs, C. (2022). The role of Xenopus developmental biology in unraveling Wnt signalling and antero-posterior axis formation. Dev. Biol. 482, 1-6. 10.1016/j.ydbio.2021.11.006 [DOI] [PubMed] [Google Scholar]
- Ouisse, L. H., Gautreau-Rolland, L., Devilder, M. C., Osborn, M., Moyon, M., Visentin, J., Halary, F., Bruggemann, M., Buelow, R., Anegon, I.et al. (2017). Antigen-specific single B cell sorting and expression-cloning from immunoglobulin humanized rats: a rapid and versatile method for the generation of high affinity and discriminative human monoclonal antibodies. BMC Biotechnol. 17, 3. 10.1186/s12896-016-0322-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagides, N., Zacchi, L. F., De Souza, M. J., Morales, R. A. V., Karnowski, A., Liddament, M. T., Owczarek, C. M., Mahler, S. M., Panousis, C., Jones, M. L.et al. (2022). Evaluation of phage display biopanning strategies for the selection of anti-cell surface receptor antibodies. Int. J. Mol. Sci. 23, 8470. 10.3390/ijms23158470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth, M. B. and Gall, J. G. (1987). Monoclonal antibodies that recognize transcription unit proteins on newt lampbrush chromosomes. J. Cell Biol. 105, 1047-1054. 10.1083/jcb.105.3.1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha-Shah, A., Esmaeili, M., Sidoli, S., Hwang, H., Yang, J., Klein, P. S. and Garcia, B. A. (2019). Single cell proteomics by data-independent acquisition to study embryonic asymmetry in Xenopus laevis. Anal. Chem. 91, 8891-8899. 10.1021/acs.analchem.9b00327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara, K., Sato, K., Iwasaki, T., Kitamura, K. and Fukami, Y. (2005). Generation of an antibody specific to Xenopus fertilized eggs by subtractive immunization. Genes Cells 10, 345-356. 10.1111/j.1365-2443.2005.00838.x [DOI] [PubMed] [Google Scholar]
- Sive, H. L. and Harland, R. M. (2022). Obtaining Xenopus eggs and embryos. Cold Spring Harb. Protoc, 1-3. 10.1101/pdb.top106195 [DOI] [PubMed] [Google Scholar]
- Wasson, L., Amin, N. M. and Conlon, F. L. (2019). INTACT proteomics in Xenopus. Cold Spring Harb. Protoc. 2019, 473-478. 10.1101/pdb.prot098384 [DOI] [PubMed] [Google Scholar]
- Weir, E., Mclinden, G., Alfandari, D. and Cousin, H. (2021). Trim-Away mediated knock down uncovers a new function for Lbh during gastrulation of Xenopus laevis. Dev. Biol. 470, 74-83. 10.1016/j.ydbio.2020.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey, H. R., Guille, M. and Grainger, R. M. (2022). Modeling human genetic disorders with CRISPR technologies in Xenopus. Cold Spring Harb. Protoc. 2022, pdb.prot106997. 10.1101/pdb.prot106997 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.