

Abstract
Sickle cell disease (SCD) is due to a mutation in the β-globin gene causing production of the toxic sickle hemoglobin (HbS; α2βS2). Transplantation of autologous hematopoietic stem and progenitor cells (HSPCs) transduced with lentiviral vectors (LVs) expressing an anti-sickling β-globin (βAS) is a promising treatment; however, it is only partially effective, and patients still present elevated HbS levels. Here, we developed a bifunctional LV expressing βAS3-globin and an artificial microRNA (amiRNA) specifically downregulating βS-globin expression with the aim of reducing HbS levels and favoring βAS3 incorporation into Hb tetramers. Efficient transduction of SCD HSPCs by the bifunctional LV led to a substantial decrease of βS-globin transcripts in HSPC-derived erythroid cells, a significant reduction of HbS+ red cells, and effective correction of the sickling phenotype, outperforming βAS gene addition and BCL11A gene silencing strategies. The bifunctional LV showed a standard integration profile, and neither HSPC viability, engraftment, and multilineage differentiation nor the erythroid transcriptome and miRNAome were affected by the treatment, confirming the safety of this therapeutic strategy. In conclusion, the combination of gene addition and gene silencing strategies can improve the efficacy of current LV-based therapeutic approaches without increasing the mutagenic vector load, thus representing a novel treatment for SCD.
Keywords: MT: Non-coding RNAs, sickle cell disease, lentiviral vectors, gene silencing, gene therapy, microRNA, gene addition, βs silencing
Graphical abstract
Sickle cell disease is due to a mutation in the β-globin gene causing production of the toxic sickle hemoglobin. Brusson et al. developed a lentiviral vector expressing an anti-sickling β-globin and an artificial microRNA downregulating βS-globin expression; this vector reduces HbS levels and effectively corrects the sickling phenotype.
Introduction
Mutations in the adult β-globin (HBB) locus lead to abnormal production of the β-globin chain of the adult hemoglobin (HbA; α2β2) tetramers, causing β-hemoglobinopathies, the most common monogenic disorders worldwide. In β-thalassemia, reduced (β+) or absent (β0) β-globin chain production is responsible for precipitation of uncoupled α-globin chains, which, in turn, leads to apoptosis of erythroid precursors and impairment of erythroid differentiation (i.e., ineffective erythropoiesis) and hemolytic anemia.1 Sickle cell disease (SCD) is due to production of a mutant βS-globin forming sickle Hb (HbS; α2βS2), which has the propensity to polymerize under deoxygenated conditions, resulting in production of sickle-shaped red blood cells (RBCs). Sickle RBCs cause occlusion of small blood vessels, leading to impaired oxygen delivery to tissues, multiorgan damage, severe pain, and early mortality.2
Transplantation of autologous hematopoietic stem and progenitor cells (HSPCs) corrected by lentiviral vectors (LVs) expressing a transgene-encoded β-globin is a promising therapeutic option. However, gene addition strategies can be partially effective in correcting the clinical phenotype in patients with severe β-thalassemia (e.g., β0/β0 patients with no residual expression of the β-globin gene) and SCD (where high expression of βS-globin impairs incorporation of the therapeutic β-globin transgene into Hb).3,4,5,6,7,8 Total Hb levels are lower than 12 g/dL for the vast majority of patients, indicating death of uncorrected RBCs, and HbS levels remain high (representing >50% of total Hb).4,6 On the contrary, asymptomatic SCD carriers have only 30%–40% of HbS, and HbS levels are usually maintained below 30% (through RBC transfusion) in severe SCD patients to suppress SCD symptoms.9 However, the distribution of therapeutic Hb and HbS within RBCs is also an important determinant of the clinical outcome of SCD patients. By way of example, with HbF representing 20% of total Hb, the proportion of total RBCs that express sufficient therapeutic Hb levels (∼10 pg of HbF per RBC) to inhibit HbS polymerization varies between 1% and 24%.10 Almost 30% of HbF is required to have 70% of HbF+ RBCs protected from Hb polymerization.10
Moreover, in gene addition strategies, the high number of integration sites per cell required to express clinically relevant levels of the β-globin transgene is often limited by the poor LV transduction efficiency in HSPCs or can increase the potential genotoxic risks associated with integrating LVs.3,11,12
We developed a high-titer LV carrying a potent anti-sickling β-globin transgene (βAS3; anti-sickling amino acid substitutions: G16D for conferring to the transgene a competitive advantage over sickle β-globin in terms of interaction with the α-globin polypeptide, E22A for disrupting axial contacts in Hb polymers, and T87Q for blocking the lateral contact with valine 6 of the sickle β-globin chain) under control of the HBB promoter and a mini locus control region (LCR).13 However, despite the good gene transfer efficiency in SCD HSPCs, RBC sickling was only partially corrected, even at a high vector copy number per cell (VCN/cell), because of residual HbS expression.
The clinical severity of SCD and β-thalassemia is alleviated by co-inheritance of genetic mutations causing sustained fetal γ-globin (HBG1 and HBG2) chain production at adult age, a condition called hereditary persistence of fetal Hb (HPFH). SCD-HPFH patients with fetal Hb (HbF, α2γ2) representing more than 20% of total Hb show a less severe disease phenotype and improved survival.14 γ-Globin exerts an anti-sickling effect in SCD by displacing βS-globin from the Hb tetramer to form HbF or mixed Hb tetramers (α2βγ) that cannot polymerize.15 γ-Globin reactivation has been extensively explored as a therapeutic approach for β-hemoglobinopathies. A gene silencing strategy based on an LV expressing an artificial microRNA (amiRNA), consisting of a short hairpin RNA (shRNA) embedded in a miRNA backbone (shmiRNA), has been exploited to specifically downregulate the BCL11A extra-large (XL) transcript (BCL11A-XL) encoding the BCL11A isoform responsible for γ-globin repression in adulthood.16,17 The therapeutic benefit of HbF reactivation using LV strategies is currently under evaluation in clinical trials.18,19 Despite early promising clinical data, HbF levels were only ∼25%–∼40% reduced, HbS levels were modestly reduced, and Hb concentration was still less than 12 g/dL.19
Therefore, despite the undeniable progress in the field of gene therapy for β-hemoglobinopathies, additional improvements in LV design and LV-based gene therapy are required to obtain a clinical benefit in severe β-thalassemia and SCD without increasing the VCN.
In this study, we generated two novel LV vectors combining a βAS3 gene addition approach with an amiRNA-based gene silencing strategy aimed at down-regulating either BCL11A (βAS3/miRBCL11A) or βS-globin (βAS3/miRHBB) to boost therapeutic β-like globin levels without increasing the mutagenic vector load in HSPCs and correct the β-hemoglobinopathy phenotype. By downregulating BCL11A, amiRBCL11A re-activated expression of the endogenous anti-sickling fetal γ-globin in erythroid cells differentiated from β-thalassemia and SCD HSPCs. However, in SCD RBCs, HbF induction was not associated with a concomitant reduction of HbS expression levels required to correct the disease phenotype. On the contrary, the therapeutic strategy based on βS-globin downregulation combined with βAS3 expression led to a strong reduction of HbS levels and HbS+ RBCs derived from SCD HSPCs, which favored βAS3 incorporation in Hb tetramers, increased therapeutic Hb levels, and ameliorated the SCD phenotype.
Results
Development of LVs co-expressing the βAS3 transgene and miRBCL11A
To achieve therapeutic Hb levels for both β-thalassemia and SCD, we developed bifunctional LVs allowing HBG de-repression through an amiRNA targeting the HbF repressor BCL11A and concomitant expression of the βAS3 transgene. In particular, we used an amiRNA (miRBCL11A)16,17,18 targeting the XL BCL11A isoform (BCL11A-XL) responsible for HBG silencing,20,21,22 thus avoiding the potential side effects because of knockdown of other BCL11A isoforms. This amiRNA is composed of a shRNA embedded in the miR-223 backbone that has been extensively optimized to improve miRNA processing and reduce off-target binding by stringent strand selection16,17,23 (Figure 1A). More specifically, the miR-223 backbone favors selection of the guide strand (the strand that recognizes the target mRNA) over the passenger strand, which is instead degraded. In the case of the miRBCL11A, the guide strand of this amiRNA targets the 3′ end of the coding sequence of BCL11A-XL mRNA.
Figure 1.

Bifunctional LVs combining gene addition and gene silencing allow BCL11A-XL silencing and HbF reactivation
(A) The amiRNA (miRBCL11A) is composed of a shRNA embedded in the miR-223 backbone and targets BCL11A-XL mRNA after processing. The guide strand is responsible for recognition of the target mRNA when loaded on the RNA-induced silencing (RISC) complex. To create the novel bifunctional LVs, we inserted miRBCL11A into two distinct sites of intron 2 of the βAS3 transgene (in the βAS3 LV): Int2_del or Int2 (βAS3/miRBCL11A LVs). Δ, deleted HIV-1 U3 region; SD and SA, HIV splicing donor and acceptor sites, respectively; Ψ, HIV-1 packaging signal; RRE, HIV-1 Rev-responsive element; Ex, exons of the human HBB; β-p, promoter of HBB; HS2 and HS3, DNase I hypersensitive site 2 and 3 of human HBB LCR, respectively. Arrows indicate the mutations introduced in exon 1 (generating the G16D and E22A amino acid substitutions) and exon 2 (generating the T87Q amino acid substitution). (B) Infectivity (TU/ng p24) was calculated based on infectious (TUs per milliliter) and physical (p24 antigen ng/mL) titers (Figure S1A) (n = 1–3 independent LV productions for βAS3/miRCL11A_Int2_del and βAS3 and βAS3/miRBCL11A_Int2, respectively). (C) BCL11A-XL expression measured by qRT-PCR in mock- and LV-transduced HUDEP-2 cells during differentiation. mRNA levels were normalized to LMNB2 expression (n = 1 for mock transduced and n = 3 independent biological replicates for LV transduced). BCL11A-XL mRNA expression was not significantly different between cells transduced with the βAS3/miRBCL11A_Int2_del and Int2 LV (linear regression). (D) Globin expression determined by RP HPLC in mock-transduced (n = 1) and LV-transduced cells (n = 3 independent biological replicates per LV). The histogram shows β-like-/α-globin ratios. We observed increased expression of therapeutic γ-globin and similar expression of βAS3-globin in cells transduced with the bifunctional vectors compared with control cells (mock- or βAS3-transduced cells). (E–G) HSPCs from β-thalassemia (F) or SCD (G) donors were either mock transduced or transduced with control (βAS3 or βAS3/miRnt) or bifunctional (βAS3/miRBCL11A) vectors for 24 h. After transduction, cells were differentiated toward the erythroid lineage for 21 days. VCN/cell was measured 14 days after transduction by ddPCR in erythroblasts. Globin expression (HPLC) was evaluated in mature RBCs. βAS3- and βAS3/miRnt-transduced samples were pooled in all analyses (F and G). (F) Hb expression determined by CE HPLC in HSPC-derived RBCs from two β-thalassemia donors (β+/β+#1 and β0/β0#2) after 16 days of differentiation (n = 1–4 independent biological replicates per donor: n = 1 for mock per donor, n = 4 for βAS3 or βAS3/miRnt per donor, and n = 2 βAS3/miRBCL11A per donor). Linear regression of HbF expression vs. VCN/cell confirmed HbF reactivation in RBCs derived from βAS3/miRBCL11A-transduced HSPCs (linear regression, ∗p = 0.0396, n = 4–8 independent biological replicates). (G) HbAS3 (top left panel), HbF (top right panel), and HbS (bottom left panel) expression measured by CE HPLC in HSPC-derived RBCs after 20 days of differentiation (n = 3; two mobilized and one non-mobilized SCD donors). Linear regression for HbF expression, ∗∗∗∗p < 0.0001 (n = 3, 7, and 10 independent biological replicates for mock-, βAS3- or βAS3/miRnt-, and βAS3/miRBCL11A-transduced samples, respectively).
We have shown previously that amiRNAs can be expressed using RNA polymerase II (RNA Pol II) promoters.23 Therefore, we inserted miRBCL11A in the second 256-nt-long intron of the βAS3 transgene to express it under control of the HBB promoter and 2 potent enhancers derived from the HBB locus control region (βAS3 LV design13), thus reducing potential amiRNA toxicity by limiting its expression to the erythroid lineage (Figure 1A). To avoid negative effects on βAS3 RNA expression or processing (e.g., splicing and 3′ end formation), we tested only two different positions in the βAS3 intron 2 to insert the amiRNA (Int2_del and Int2). These regions are not known to be involved in β-globin RNA expression and splicing and far enough from the last 60 nt of intron 2 known to be required for efficient 3′ end mRNA formation24 (Figure 1A). Both physical and infectious titers of amiRNA-containing LVs (βAS3/miRBCL11A_Int2_del and _Int2) were similar to the original vector expressing only the βAS3 transgene (βAS3 LV) (Figure S1A). Therefore, insertion of the amiRNA did not impact viral particle production or infectivity (Figures S1A and 1B).
These LVs were then tested in a human erythroid progenitor cell line (HUDEP-2) (Figures 1C, 1D, and S1B–S1G). To assess the potential impact of the amiRNA on gene transfer efficiency, HUDEP-2 cells were transduced at increasing multiplicities of infection (MOI) with all LVs. VCN analysis by ddPCR revealed that neither insertion of the miRNA nor its position in intron 2 affects gene transfer efficiency (Figure S1C).
Mock- and LV-transduced HUDEP-2 cells were terminally differentiated into mature erythroblasts (Figure S1B). BCL11A-XL mRNA expression was decreased in HUDEP-2 cells transduced with LVs containing the amiRNA compared with control cells (mock transduced or transduced with βAS3 LV) (Figure 1C). BCL11A-XL mRNA downregulation led to a strong decrease of BCL11A-XL at the protein level in cells transduced at a high VCN/cell (Figure S1D). These results demonstrated that the amiRNA is expressed in the frame of the βAS3-expressing LVs and is able to reduce BCL11A-XL expression. Importantly, βAS3 mRNA was expressed at similar levels in samples transduced with amiRNA-containing LVs or with the control βAS3 LV, showing that neither insertion of the amiRNA nor its position in βAS3 intron 2 affects transgene expression (Figure S1E).
To evaluate whether BCL11A-XL silencing was associated with HBG re-activation, we measured HBG mRNA expression levels in terminally differentiated HUDEP-2 cells. HBG expression was substantially higher in mature erythroblasts transduced with amiRNA-expressing LVs than in cells transduced with the βAS3 LV (Figure S1E). Flow cytometry showed that the percentage of HbF+ populations and HbF content (measured as mean fluorescence intensity [MFI]) were increased in samples transduced with LVs expressing the miRNA targeting BCL11A compared with controls (Figure S1F). Reverse-phase (RP) high-performance liquid chromatography (HPLC) analysis of single globin chains showed increased γ-globin levels upon BCL11A-XL silencing and equivalent βAS3 expression in all transduced samples (Figures 1D and S1G). Importantly, we observed a decrease in the levels of endogenous adult β-globin (Figure 1D), which could be beneficial in the context of SCD to reduce expression of the mutant βS-globin.
Overall, these data showed that LVs expressing a βAS3 transgene and an amiRNA targeting BCL11A-XL could reactivate γ-globin expression without altering βAS3 production in an adult erythroid cell line model. These bifunctional LVs can be exploited to achieve therapeutic Hb levels in β-thalassemia and SCD. For further studies in primary patient cells and for development of bifunctional vectors expressing alternative amiRNAs (i.e., against HBB), we selected the Int2 insertion position. Although γ-globin expression was similar with the two vectors, βAS3/miRBCL11A_Int2 LV (hereafter called βAS3/miRBCL11A) tended to induce better BCL11A-XL downregulation even at low VCN/cell, suggesting better amiRNA production associated with the Int2 configuration (Figure 1C).
Downregulating BCL11A-XL modestly increases the therapeutic potential of βAS3-expressing LVs
First, we transduced adult granulocyte colony-stimulating factor (G-CSF)-mobilized HSPCs derived from two β-thalassemia patients (with a β+/β+ or a β0/β0 genotype) with the bifunctional LV harboring the amiRNA against BCL11A-XL (βAS3/miRBCL11A). As control vectors, we used βAS3 LV and an LV containing a non-targeting (nt) amiRNA (that does not recognize any human sequence) inserted in position Int2 of βAS3 intron 2 (βAS3/miRNAnt). Mock- and LV-transduced HSPCs were terminally differentiated into mature RBCs (Figures 1E and S2D). Transduction efficiency by βAS3/miRBCL11A and control vectors was similar in erythroid liquid cultures (VCN of ∼1 and ∼2 in the β+/β+ and the β0/β0 patient, respectively), indicating that infectivity was not affected by introduction of the amiRNA in primary HSPCs (Figure S2A). HSPCs were plated in clonogenic cultures (colony-forming cell [CFC] assay) allowing growth of erythroid (burst-forming unit erythroid [BFU-E]) and granulomonocytic (colony-forming unit for granulocytes and macrophages [CFU-GM]) progenitors. Importantly, LV transduction did not alter the growth and multilineage differentiation of hematopoietic progenitors (Figure S2B). VCNs in BFU-Es and CFU-GMs (∼1 and ∼2 in the β+/β+ and the β0/β0 patient, respectively) were similar to the values observed in the erythroid liquid culture, with ∼50% of transduced progenitors (Figures S2B and S2C).
To evaluate the therapeutic potential of this strategy, we measured globin expression in mock- and LV-transduced erythroid liquid cultures. Of note, the β0/β0 samples showed high basal HBG levels and a less severe in vitro phenotype compared with the β+/β+ samples (possibly because of in vitro culture conditions, stress erythropoiesis, or potential genetic modifiers of HbF expression present in this patient and a single 3.7-kb HBA gene deletion, which, taken together, possibly mitigate the β-thalassemic phenotype) (Figure S2E and S2F–S2K).25 However, γ-globin mRNA levels were higher in β+/β+ or a β0/β0 cells transduced with LVs containing the amiRNA against BCL11A-XL than in control cells transduced with control LVs (Figure S2E). Of note, βAS3 expression was not affected in cells expressing miRBCL11A, confirming no impact of amiRNA insertion on βAS3 mRNA production (Figure S2E). Moreover, HBG gene de-repression combined with βAS3 expression led to an improvement of the α/non-α globin mRNA ratio in cells transduced with the bifunctional LV compared with mock- or control LV-transduced cells (Figure S2E). The relative proportion of HbF, measured by cation exchange (CE) HPLC, was also increased as a consequence of HBG de-repression in cells expressing the amiRNA targeting BCL11A-XL (Figure 1F). Finally, flow cytometry revealed that the bifunctional LV increases the proportion of HbF+ RBCs (Figure S2F).
We then evaluated whether expression of Hb tetramers containing βAS3 (HbAS3) combined with HbF reactivation could improve β-thalassemia RBC clinical phenotypes. α-Globin precipitates, detected only in cells differentiated from the β+/β+ patient, were strongly decreased upon treatment with βAS3/miRBCL11A and control vectors (Figure S2G). Similarly, enucleation rate and RBC size, known to be reduced in β-thalassemia, were improved in samples derived from HSPCs transduced with either βAS3/miRBCL11A or the control LVs (Figures S2H and S2I). No changes in the expression of erythroid surface markers along differentiation were observed upon transduction with bifunctional LVs compared with control samples, as measured by flow cytometry (Figure S2J). Enucleated RBC parameters evaluated by phase microscopy were similar in samples derived from HSPCs transduced with either βAS3/miRBCL11A or the control LVs (Figure S2K). Of note, for the β0/β0 patient, RBC parameters were improved in LV-transduced samples compared with mock-transduced cells, while untreated β+/β+ cells could not be analyzed because of the low proportion of enucleated RBCs at the end of differentiation, which was rescued by LV transduction (Figures S2H, S2I, and S2K). In particular, for the β0/β0 patient, dry mass was increased in RBCs derived from transduced samples, reflecting amelioration of Hb content per cell, and RBC surface and perimeter (altered because of the anisocytosis and poikilocytosis typical of β-thalassemic cells) were less variable, revealing a more homogeneous cell population (Figure S2K).
Next, we applied this therapeutic strategy to SCD HSPCs. Expression of the anti-sickling βAS3- and γ-globins and the concomitant potentially decreased expression of adult βS-globin could be beneficial for SCD patients.26 SCD primary HSPCs were mock transduced or transduced with βAS3/miRBCL11A or control LVs (βAS3 or βAS3/miRNAnt) at increasing MOIs and differentiated into mature RBCs (Figures 1E and S3D). No impairment of erythroid differentiation or RBC enucleation was observed upon transduction with the bifunctional LV compared with control samples, as measured by flow cytometry (Figures S3E–S3G). In parallel, the CFC assay showed no impairment of HSPC viability or erythroid and granulo-monocytic differentiation by miRBCL11A (Figure S3A). VCN was similar for all vectors (i.e., ∼1 at an MOI of 1 and up to 2–4 at an MOI of 5 [up to ∼2 for βAS3/miRBCL11A LV and up to ∼4 for control vectors]), while an MOI of 25 was associated with a lower VCN (Figure S3B).
qRT-PCR revealed comparable βAS3 expression in all transduced samples and a VCN-dependent increase in γ-globin expression in cells transduced with the bifunctional LV compared with control samples (Figure S3H). However, no consequent βS-globin downregulation was induced upon HBG de-repression (Figure S3H). CE HPLC confirmed that HbAS3 was expressed at similar levels in all transduced samples harboring similar VCNs and that HbF was significantly increased in βAS3/miRBCL11A-transduced cells compared with control samples (Figure 1G). However, HbS expression was only modestly decreased in cells reactivating HbF more efficiently (VCN > 2; Figure 1G). Similar results were observed in BFU-E pools (Figure S3C). Similarly, we observed an increased proportion of HbF+ RBCs upon transduction with bifunctional LVs but no significant decrease in the proportion of HbS+ RBCs upon HbF reactivation (Figure S3I). Finally, we incubated mature RBCs at low oxygen concentration to induce RBC sickling. Bifunctional and control LVs led to a comparable decrease of the frequency of sickling cells, indicating that γ-globin reactivation associated with βAS3 expression was not sufficient to further ameliorate the SCD RBC phenotype compared with βAS3 expression alone (Figure S3J).
In summary, we created a bifunctional LV enabling simultaneous expression of a therapeutic transgene and an amiRNA down-regulating BCL11A-XL. The βAS3/miRBCL11A LV efficiently transduces HSPCs from β-thalassemia and SCD patients and leads to HbF reactivation without impairing βAS3 transgene production. HbF reactivation combined with βAS3 expression modestly improved the β-thalassemia phenotype in vitro, while in SCD cells the potential therapeutic benefit of this strategy was minimal.
miR7m efficiently downregulates β-globin expression in erythroid cell lines
The limited therapeutic benefit of the βAS3/miRBCL11A LV in SCD could be due to the persistence of high HbS levels preventing incorporation of therapeutic β-like globins in Hb tetramers and correction of the sickling RBC phenotype. To reduce HbS levels and correct the SCD cell phenotype, we used our bifunctional LV design to develop a strategy aimed at expressing βAS3 and concomitantly reducing βS-globin levels to favor incorporation of βAS3 into Hb tetramers.
To generate an effective miRNA targeting the β-globin gene, we adapted sequences from small interfering RNAs (siRNAs) and shRNAs targeting HBB or designed miRNAs using an online tool (https://felixfadams.shinyapps.io/miRN/27; Table 1; Figure S4A). These sequences were inserted into the miR-223 backbone, and the different amiRNAs targeting β-globin (miRHBB) were inserted within intron 2 of the βAS3 transgene (insertion site: Int2) as described previously for miRBCL11A (Figures 1A and S4B). The 6 miRHBBs generated from shRNA sequences (miRHBB: miR1, miR3, miR4, miR5, miR6, and miR7) were modified by removing 4 nt at the 5′ end of the guide strand and adding a GCGC motif at the 3′ end (miRHBBm: miR1m, miR3m, miR4m, miR5m, miR6m, and miR7m) (Table 1). These modifications were shown to further enhance selection of the guide strand over the passenger strand in the RISC complex because of improved 3′ end thermodynamic stability of the guide/passenger strand duplex and therefore could potentially increase βS-globin silencing.17 miR11 was adapted from a siRNA designed by Dykxhoorn et al.; this sequence contains a CC mismatched dinucleotide at the 5′ end of the guide strand and at the 3′ end of the passenger strand to increase silencing (Table 1).28
Table 1.
Guide strand sequences targeting the HBB mRNA
| ID | Origin | Guide strand sequence (5′-3′) | Type |
|---|---|---|---|
| miR1a | Samakoglu et al.33 | CTCCTCAGGAGTCAGATGC | shRNA |
| miR1ma | TCAGGAGTCAGGTGCGCGC | ||
| miR2a | Dykxhoorn et al.28 | AACTTCTCCTCAGGAGTCA | siRNA |
| miR3 | portals.broadinstitute.org | TCAGTGTGGCAAAGGTGCCCT | shRNA |
| miR3m | TGTGGCAAAGGTGCCCTGCGC | ||
| miR4 | portals.broadinstitute.org | ATAACAGCATCAGGAGTGGAC | shRNA |
| miR4m | CAGCATCAGGAGTGGACGCGC | ||
| miR5 | portals.broadinstitute.org | TTCATCCACGTTCACCTTGCC | shRNA |
| miR5m | TCCACGTTCACCTTGCCGCGC | ||
| miR6 | portals.broadinstitute.org | CAAAGAACCTCTGGGTCCAAG | shRNA |
| miR6m | GAACCTCTGGGTCCAAGGCGC | ||
| miR7 | portals.broadinstitute.org | CTTTCTTGCCATGAGCCTTCA | shRNA |
| miR7m | CTTGCCATGAGCCTTCAGCGC | ||
| miR8 | designed using the tool developed by Adams et al.27 | TGAAGTTCTCAGGATCCACGT | miR |
| miR9 | designed using the tool developed by Adams et al.27 | TTCTTTGCCAAAGTGATGGGC | miR |
| miR10 | Thermo Fisher Scientific | AAAGGCACCGAGCACTTTCTT | siRNA |
| miR11 | Dykxhoorn et al.28 | CCAGGGCCTCACCACCAAC | siRNA |
GCGC, motif added in miRm to improve the knockdown efficiency of the miRNA; CC, motif added to improve the knockdown efficiency of the miRNA; bold, matching sequence between the original miRNA (miRxx) and the modified version (miRxxm).
The miRs are designed to target only the WT β-globin mRNA. To target the βS-globin mRNA, the nucleotides indicated in italics need to be modified to match to the SCD mutation.
We generated a total of 17 LVs (βAS3/miRHBB) co-expressing the anti-sickling βAS3-globin and a different miRHBB. To select the most effective amiRNA, we transduced K562 erythroleukemic cells (which do not express endogenous β-globin) with the 17 LVs. For this amiRNA screening, we assessed the silencing effect of the amiRNA on the βAS3 transgene. The βAS3 transgene sequence was not yet modified to avoid targeting by the amiRNA and could be targeted by the 17 different amiRNAs because there are no mismatches between the miRHBB and their target sequences within the βAS3 transgene. Therefore, we compared βAS3 expression normalized per VCN in K562 cells transduced with either βAS3/miRHBB LVs or the control LVs (βAS3 or βAS3/miRnt). K562 cells transduced with βAS3/miR7, miR7m, miR9, and miR10 LVs showed a decrease of more than 50% in βAS3 mRNA expression compared with cells transduced with control LVs (Figure S4C). However, cells transduced with βAS3/miR9 LV have a very low VCN/cell (<0.5) in comparison with other LVs, which could reflect a low gene transfer efficiency that hampers its use as a therapeutic vector for SCD. In cells transduced with LV βAS3/miR1 and miR5m, we observed a reduction in βAS3 expression of ∼30% compared with control cells (Figure S4C). Overall, modification of the miRNA sequences17 did not increase gene silencing in K562 cells (Figure S4C). Based on these results we select LVs expressing miR1, miR5m, miR7, miR7m, or miR10 for further experiments.
To confirm that the selected amiRNAs downregulate the endogenous β-globin gene, we transduced HUDEP-2, an adult erythroid progenitor cell line. These cells can be differentiated into mature erythroid precursors expressing the endogenous α- and β-globin chains at the mRNA and protein levels. HUDEP-2 cells were transduced with control (βAS3 and βAS3/miRnt) and miRHBB LVs at a high (10) or low (2) MOI and differentiated to evaluate miRNA efficiency in down-regulating HBB and βAS3 expression (Figure S4D). We observed a strong reduction of the βAS3 transcripts per VCN (from ∼60% to 85%) in cells transduced with the LVs expressing miR7, miR7m, and miR10, confirming the results obtained in K562 cells (Figures S4C and S4E). The decrease in endogenous β-globin expression was correlated with VCN/cell. For miR7, miR7m, and miR10, we observed a reduction of β-globin expression ranging from 60% to 85% and from 30% to 70% at high and low VCN/cell, respectively (Figure S4E). On the contrary, miR1 and miR5m showed no or little activity on βAS3 and endogenous β-globin (Figure S4E). Of note, in HUDEP-2 cells, the modified version of miR7 (miR7m) outperformed miR7 in terms of β-like globin silencing.
The βAS3m/miR7m LV efficiently reduces HbS levels and corrects the sickling phenotype in RBCs differentiated from SCD primary HSPCs
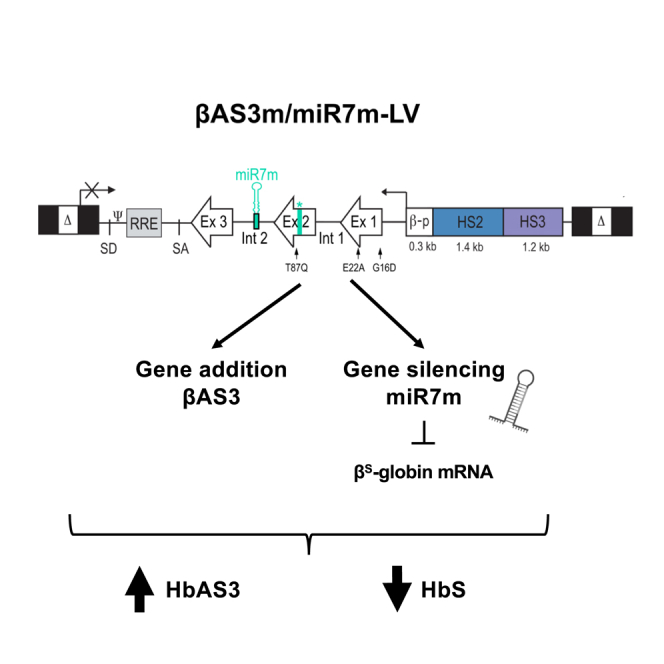
Based on our results, we selected miR7m as our best-performing miRNA in terms of β-globin silencing. Given the high sequence similarity between the βS-globin and the transgene, we modified the target sequence in βAS3 to avoid its silencing by the miRNA. To this aim, we introduced silent mutations in the βAS3 transgene sequence of βAS3/miR7m and βAS3/miRnt LV, choosing, when possible, codons commonly present in the HBB coding sequence (βAS3m, modified βAS3 transgene; Figure 2A). Overall, we introduced into the miR7m target region a number of mutations that reduce by 33% the complementarity between the miRNA and the βAS3 transcript (Figure 2A). Notably, vector titers and infectivity were comparable between βAS3m/miR7m and βAS3m/miRnt LVs and the original βAS3 LVs, demonstrating that transgene modification does not impact the viral titer (Figures 2B and S5A).
Figure 2.

The bifunctional βAS3m/miR7m vector efficiently and safely corrects the RBC sickling phenotype
(A) Structure of the βAS3m/miR7m LV. The sequence of miR7m aligned to the βAS3 transgene is shown in the top panel. The βAS3 transgene (βAS3 original) was modified to avoid its targeting by miR7m, thus generating the βAS3 modified transgene (βAS3m) harboring mismatches (green) with the βAS3 original transgene corresponding to silent mutations, as shown by the amino acid sequence (black boxes). (B) Infectivity (TU/ng p24) was calculated based on infectious (TU/mL) and physical (p24 antigen ng/mL) titers (Figure S5). Data are shown as mean ± SD. (n = 3 independent LV productions per LV). (C) Mobilized (2 donors) or non-mobilized (1 donor) peripheral blood HSPCs from SCD donors were either mock transduced (mock; n = 1 per donor) or transduced with the βAS3 or βAS3m/miRnt (n = 5–6 independent biological replicates per donor) or βAS3m/miR7m (n = 2–5 independent biological replicates per donor) LV at different MOIs for 24 h. After transduction, cells were differentiated in liquid culture toward the erythroid lineage. VCN/cell was measured 14 days after transduction by ddPCR in erythroblasts. Globin expression (qRT-PCR, HPLC, and flow cytometry) and RBC differentiation markers and properties (sickling assay) were evaluated in erythroblasts along differentiation or in mature RBCs (see also Figure S5). (D–G) βAS3- and βAS3m/miRnt-transduced samples were pooled in all analyses. VCN/cell is indicated below each graph. (F and G) 2 SCD donors. (D) HbAS3 and HbS expression in mature RBCs measured by CE HPLC. Linear regression, ∗∗∗p = 0.0005 (n = 3, 17, and 10 independent biological replicates for mock-, βAS3- or βAS3/miRnt-, and βAS3/miRBCL11A-transduced samples, respectively; 3 SCD donors). (E) Frequency of sickling RBCs after 1-h incubation at low oxygen tension (0% O2). Unpaired t test, ∗∗p < 0.01 (n = 2, 11, and 5 independent biological replicates for mock-, βAS3- or βAS3/miRnt-, and βAS3/miRBCL11A-transduced samples, respectively; 2 SCD donors). (F) Frequency of enucleated RBCs measured by flow cytometry on days 13 and 20 of erythroid differentiation. (G) Frequencies of (left) CD36+, (center) CD71+, and (right) CD235a+ erythroid cells measured by flow cytometry along differentiation. (H) Distribution of βAS3-LV or βAS3m/miR7m-LV integration sites in exonic, intronic, and intergenic genomic regions in HD HSPCs (n = 1). (I) mRNA-seq-based analysis comparing mRNA expression between two sample groups: βAS3 and mock (left), βAS3m/miRnt and βAS3 (center), and βAS3m/miR7m and βAS3m/miRnt (right) (2 SCD donors were used; n = 2 and 3 independent biological replicates for mock- and LV-transduced samples, respectively). Differentially expressed genes (DEGs) with an FDR of less than 0.05 and an absolute log2 fold change (logFC) greater than 1 are highlighted in red (upregulated [UP]) or blue (downregulated [DOWN]). Genes that are not differentially expressed are represented in gray (NotSig).
We transduced primary adult HSPCs derived from SCD donors with βAS3m/miR7m and control LVs (βAS3m/miRnt and βAS3) at increasing MOIs. Mock-transduced and transduced HSPCs were subjected to a CFC assay or differentiated in mature RBCs (Figure 2C). The number and proportion of BFU-Es and CFU-GMs were similar among the different samples (Figure S5B), indicating no impairment in erythroid and granulomonocytic cell growth and differentiation. VCN/cell in BFU-Es and erythroblasts was ∼2 in all samples even at a low MOI (1/2) (Figure S5C). Furthermore, we determined VCN/cell in single BFU-Es and showed a similar gene transfer efficiency in βAS3m/miR7m- and control LV-transduced samples (∼50% at an MOI of 1/2 and ∼65% at an MOI of 15; Figure S5D). Moreover, βAS3m/miR7m- and control LV-transduced samples displayed a similar VCN/cell distribution with the majority of BFU-Es harboring 1–2 VCN/cell (Figure S5D).
To evaluate the reversion of the SCD cell phenotype, SCD HSPCs were terminally differentiated into mature enucleated RBCs (Figure 2C). Efficient HSPC transduction by the βAS3m/miR7m LV led to a VCN-dependent decrease of βS-globin transcripts in HSPC-derived erythroid cells compared with cells transduced with control LVs (Figure S5G). Notably, the miRNA specifically down-regulated βS-globin without affecting βAS3 expression (Figure S5G). CE HPLC analysis showed that βS-globin gene downregulation led to a significant decrease of HbS that was associated with a substantial increase of the therapeutic Hb (Figure 2D). These results were confirmed in BFU-E pools (Figure S5E). We also measured β-like globin expression in individual BFU-Es derived from HD HSPCs transduced with the βAS3m/miR7m or control LVs. Importantly, the proportion of βAS3-globin transcripts was higher in BFU-Es transduced with the βAS3m/miR7m-LV than in BFU-Es transduced with the control vector at low or high VCN/cell (Figure S5F).
In liquid erythroid SCD cell cultures, we observed a significant reduction of the proportion of HbS+ cells in βAS3m/miR7m- compared with control LV-transduced samples as measured by flow cytometry (Figure S5H). The increased incorporation of βAS3 in Hb tetramers and the decrease in βS-globin led to better correction of the sickling phenotype in mature RBCs derived from HSPCs transduced with βAS3m/miR7m LV vs. control LVs (Figure 2E). Of note, the frequency of sickling cells obtained using our bifunctional LV approaches that observed in asymptomatic SCD carriers.8
Importantly, RBC enucleation and erythroid differentiation were not affected by βS-globin downregulation (Figures 2F, 2G, and S5I). Similarly, RBC parameters, such as dry mass, surface, and perimeter, evaluated by phase microscopy, showed no major change induced by βS downregulation (Figure S5J). We only observed a minor reduction in dry mass and surface in a small fraction of RBCs (13.7% ± 3.8% and 8.1% ± 6.2% for dry mass and surface, respectively) in samples obtained from HSPCs transduced with the bifunctional βAS3m/miR7m LV (Figure S5J). To evaluate whether the reduced dry mass could reflect a β-like-thalassemia phenotype, we measured, by RP HPLC, globin chain expression and evaluated the α/non-α globin ratio typically altered in β-thalassemia (HBB knockout [KO] sample; Figure S5K). Importantly, RBCs derived from HSPCs transduced with the βAS3m/miR7m or control LV showed a normal α/non-α globin ratio compared with β-thalassemic cells (Figure S5K).
The βAS3m/miR7m LV has safe integration and transcriptomics profiles in primary cells and does not affect engraftment and multilineage differentiation capability of HSPCs in immunodeficient mice
First, we mapped the integration sites of amiRNA-containing LVs by ligation-mediated (LM) PCR and next-generation sequencing (NGS) in healthy donor (HD) HSPCs. Our analyses revealed that the βAS3m/miR7m LV showed a standard, safe lentiviral integration profile essentially equivalent to that of the control βAS3 LV in terms of genome-wide distribution and top targeted genes (Figures 2H and S6A). Gene Ontology analysis confirmed that genes targeted by the bifunctional and control LVs have highly similar biological functions (Figure S6B). We also performed RNA sequencing (RNA-seq) and miRNA-seq on treated and non-treated erythroblasts differentiated from HSPCs derived from two SCD donors to confirm the safety of our approach and quantify expression of the transgene and miR7m (Figures 2I and S6D). Transduction with the βAS3-LV modestly affected the erythroid transcriptome (89 differentially expressed genes representing 0.6% of total genes compared with mock-transduced cells; Figure 2I). Addition of the miRnt did not further affect gene expression (no deregulated genes between βAS3- and βAS3m/miRnt-LV-transduced samples; Figure 2I). Only less than 0.1% of total genes were modestly deregulated upon transduction with the βAS3m/miRHBB LV compared with samples transduced with the βAS3m/miRnt LV (Figure 2I). As expected, endogenous HBB gene expression was significantly downregulated upon transduction with the βAS3m/miRHBB LV (2.2-fold decrease, false discovery rate [FDR] < 0.01; Figure S6C). Moreover, RNA-seq experiments confirmed that βAS3 expression in the transduced samples (βAS3 or βAS3m/miR7m) was not affected by miR7m (βAS3m/miR7m vs. βAS3) (Figure S6C). The pri-miR7m was detected only in βAS3m/miR7m-transduced samples (Figure S6C). Similarly, the miRNAome was not affected in samples transduced with the βAS3m/miRHBB LV compared with control samples transduced with the βAS3 and βAS3m/miRnt LV (Figure S6D). The mature miR7m was detected only in βAS3m/miR7m-transduced cells, and its expression was in the range of endogenous miRNAs involved in erythroid differentiation (e.g., miRNAs upregulated along erythropoiesis, such as miR15b-5p, miR-96-5p, and miR-22-3p, or miRNAs downregulated along erythropoiesis, such as miR-223-5p, miR-221-3p, miR-222-3p, and miR-181a-3p) (Figure S6E).29
Finally, we xenotransplanted human HSPCs from an SCD donor (either mock transduced or transduced with the βAS3- or the βAS3m/miR7m LV) in non-obese diabetic (NOD).Cg-KitW−41JTyr+PrkdcscidIl2rgtm1Wjl/ThomJ (NBSGW) mice (Figure 3A). The proportion of human CD45+ cells in the bone marrow, spleen, and thymus was similar between the three groups, confirming that the βAS3m/miR7m LV sustains the long-term engraftment capability of primary HSPCs (Figure 3B). Furthermore, engrafted βAS3m/miR7m-transduced HSPCs were able to generate all hematopoietic lineages (Figures 3C–3E). The VCN/cell in the bone marrow of mice receiving βAS3m/miR7m- or βAS3-transduced cells was not significantly different (1.5–1.7 for βAS3m/miR7m and 0.2–6.5 for βAS3; Figure 3F). Overall, these results validate the therapeutic potential and safety of this novel bifunctional LV.
Figure 3.

βAS3m/miR7m LV-transduced HSPCs engraft and differentiate in NBSGW mice
(A) Human SCD HSPCs were either mock-transduced (mock) or transduced with βAS3 or βAS3m/miR7m LVs at an MOI of 125 for 24 h. After transduction, cells were transplanted into NBSGW mice. After 18 weeks, chimerism, cell lineage reconstitution, and VCN/cell were evaluated. (B) Frequency (%) of human CD45+ (hCD45+) cells in the BM, spleen, and thymus of the transplant recipients (n = 4–6 mice per group). (C) Frequencies (%) of CD11b+, CD14+, and CD15+ cells in the hCD45+ population in the BM and in the spleen of the transplant recipients (n = 4–6 mice per group). (D) Frequencies (%) of CD3+ and CD19+ cells in the hCD45+ population in the BM and in spleen of the transplant recipients (n = 4–6). (E) Frequencies (%) of CD235a+ cells in the negative human (h) and mouse (m) CD45 population in the BM and spleen of the transplant recipients (n = 4–6 mice/group). (B–E) Kruskal-Wallis test, ns. (F) VCN/cell measured by ddPCR in the input cells (bulk of BFU-Es and CFU-GMs) and in the BM of transplant recipients (n = 3–6 mice/group). No significant statistical difference in VCN/cell was observed between the two LVs in BM cells (unpaired t test).
Discussion
Ongoing LV-based clinical trials for SCD reveal an amelioration of clinical phenotype associated with expression of therapeutic β-like globins via gene addition or gene silencing; however, Hb levels remain lower than the normal 12–18 g/dL range in the majority of SCD and β0/β0 thalassemic patients.4,5,6,19 In this study, we developed bifunctional LVs combining gene addition and gene silencing to improve gene therapy for β-thalassemia and SCD. Insertion of an amiRNA targeting BCL11A-XL or βS-globin in the second intron of the βAS3 transgene did not affect LV titer, gene transfer efficiency, or βAS3 expression in primary CD34+ HSPCs derived from β-hemoglobinopathy patients.
Combining βAS3 and BCL11A-XL down-regulation reactivated γ-globin but improved only the β-thalassemia phenotype compared with the mere gene addition strategy. At similar VCN, γ-globin levels were modest compared with those obtained by only expressing the shmiRNA targeting BCL11A-XL under control of similar regulatory elements,18,19 suggesting that processing of the intron-embedded amiRNA is less efficient or that βAS3 globin competes with γ-globin for incorporation in the Hb tetramers, at least in SCD cells. Interestingly, our results are in line with a recent study describing a similar LV simultaneously expressing the T87Q β-globin (carrying only one anti-sickling amino acid) and an amiRNA targeting BCL11A-XL.30 Of note, in the case of β-thalassemia, this strategy could be combined with introduction of a second amiRNA targeting α-globin, which has been shown recently to improve the α/β ratio in erythrocytes derived from β-thalassemia HSPCs.31 Another miRNA-based gene therapy strategy has been developed recently to boost HbF expression by expressing two amiRNAs targeting BCL11A or ZNF410 γ-globin repressors.32 However, the increase in HbF was limited, and this therapeutic strategy modestly improved the pathological phenotype in SCD or β-thalassemic erythroid cells compared with the individual approaches.
All of these therapeutic strategies are promising for β-hemoglobinopathies; however, their beneficial effect might be limited in SCD because of the competition between therapeutic globins and βS-globin for formation of Hb tetramers. Hence, to reduce HbS levels, still high after γ-globin reactivation and/or βAS3 gene addition, we replaced miRBCL11A with a miRNA targeting βS-globin. Among the different miRNAs, miR1/1m and miR2 were adapted from shRNA33 and siRNA,28 respectively, which efficiently silenced β-globin expression. The lower expression of the miRNA compared with high expression rates of shRNA/siRNA could in part explain the lower silencing efficacy of these miRNAs. Moreover, integration of shRNA or siRNA sequences in the miRNA backbone could affect miRNA processing and, therefore, silencing efficiency. Nevertheless, other miRNAs, such as miR7m and miR10, adapted from shRNAs, efficiently silenced βS expression. Overall, modification of the miRNA sequences described by Guda et al.17 did not increase βAS3 gene silencing in K562 cells, but in HUDEP-2 cells, miR7m outperformed miR7 in reducing βAS3 and wild-type (WT) β-globin expression, suggesting that this design can be explored to increase miRNA processing and ameliorate silencing of efficient amiRNA.
Next, we developed the βAS3m/miR7m LV containing the most efficient miRNA. βS-Globin downregulation induced by miR7m led to reduced expression of HbS and a significant decrease of HbS+ RBCs. Interestingly, despite modest BCL11A gene downregulation when using our vector design, β-globin gene silencing was highly efficient and finely tuned to avoid excessive β-globin downregulation. Importantly, neither miR7m nor introduction of silent mutations in exon 2 affected βAS3 expression. It has been reported in other systems that silent mutations could alter mRNA structure and half-life or impair protein production because of the low abundance of the cognate tRNA.34 On the contrary, elevated HbAS3 levels led to better correction of the sickling phenotype compared with control cells. Sickle β-globin downregulation was compensated by βAS3 expression, therefore avoiding generation of a β-thalassemic phenotype. A modest reduction in Hb content (i.e., dry mass) was observed in a small proportion of βAS3m/miR7m-treated RBCs, but the lower Hb concentration could further reduce its polymerization and improve the RBC phenotype.35,36,37 Moreover, beneficial effects of this strategy will be more pronounced in vivo because of positive selection of corrected RBCs, likely as a result of their prolonged lifespan compared with non-modified SCD RBCs.6
It is noteworthy that here we improved LVs to boost therapeutic β-like globin levels without increasing the mutagenic vector load in HSPCs. Increasing the vector load could favor therapeutic Hb formation and ameliorate the outcome of the current LV-based clinical trials; however, it potentially increases the genotoxicity risk associated with elevated LV integration sites per cell. This is particularly critical in SCD patients, who are characterized by a high mutational burden and inflammation that can be associated with clonal hematopoiesis. Recent studies reported that SCD patients have an increased risk of developing myeloid malignancies compared with the general population.38,39 Furthermore, three myelodysplasias associated with pro-oncogenic mutations and genomic rearrangements have been observed in an ongoing gene therapy trial for SCD.40,41 Even if these latter events were not associated with LV integration, it is desirable to minimize the VCN/cell. Hence, our strategy, based on two therapeutic solutions to boost therapeutic Hb, can reduce the VCN necessary to correct the SCD phenotype and, thus, LV-associated potential genotoxicity. Moreover, the lower VCN required to correct the SCD phenotype will also diminish the cost of LV production, making this strategy potentially widely available.42
Importantly, from a safety point of view, insertion of the amiRNA showed no impairment in HSPC viability and multilineage differentiation. The βAS3m/miR7m LV showed a standard lentiviral integration profile, did not alter the transcriptome or miRNAome of transduced erythroblasts, and did not impair HSPC long-term engraftment and hematopoietic lineage reconstitution (Figure 3). The VCN/cell in human bone marrow cells tended to be lower in cells transduced with the βAS3m/miR7m LV compared with control cells treated with the βAS3 vector but still remains therapeutically relevant (mean = 1.6; Figure 3F), considering the better performance of the bifunctional LV (Figure 2). The safety of our therapeutic strategy is also supported by an ongoing clinical trial using LV expressing a shmiRNA that showed stable gene marking over time without any adverse events associated with the drug product.19 Notably, a similar strategy was used by Samakoglu et al.33 by co-expressing γ-globin and an shRNA (inserted in the second intron of the γ-globin transgene) targeting βS-globin. However, γ-globin does not contain an aspartic acid in position 16 that increases affinity for the α-globin chain (and is indeed likely outcompeted by βAS3). Furthermore, the pri-miRNA needs to be processed to generate a mature, functional amiRNA, while the shRNA action is more immediate. In particular, the pri-miRNA does not saturate a specific step of the endogenous miRNA pathway.43,44 On the contrary, shRNA can cause toxicity associated with oversaturation of cellular small RNA pathways (namely, RISC incorporation), which could lead to alteration of the production of endogenous miRNAs. Importantly, in our study using an amiRNA expressed through an LV, we did not see relevant changes in the transcriptome or miRNAome. Finally, shRNA can trigger the innate immune response and cytotoxicity,45,46,47 while amiRNAs are known to be associated with lower toxicity.44,45,47,48,49 Of note, the interferon-responsive gene IRF/9ISGF3G was significantly upregulated in cells treated with the shRNA.33 On the contrary, we did not observe changes in genes involved in the innate immune response (Figure 2).
Genome editing strategies have also been developed by several groups, including ours,50,51,52 with early promising clinical results,53 but long-term efficacy and safety need to be demonstrated, particularly in light of the recent findings regarding CRISPR-Cas9-induced genotoxicity.54,55 On the contrary, our approach can be readily implemented in clinically accepted LV designs and eventually translated to the bedside after preclinical biosafety and biodistribution studies to change the goal of current gene therapy clinical trials from amelioration of clinical signs and reduction of blood transfusion to a complete cure of the disease.
Materials and methods
Generation of lentiviral constructs
The pCCL.b-AS3 plasmid (βAS3 LV) was used to generate all LV constructs used in this study.13 The HBB mini-gene (coding for the βAS3 transgene) contains three mutations determining three amino acid substitutions (G16D, E22A, and T87Q) and a 593-bp deletion in intron 2 removing a region located 85–679 bp downstream of HBB exon 2 (the total length of intron 2 is 257 bp). We inserted miRBCL11A between positions 85 and 86 (hereafter called Int2_del) or between 146 and 147 (hereafter called Int2) of βAS3 intron 2. We did not test additional positions in the short intron of the βAS3 transgene to avoid positions that are too close to the splicing junctions. We inserted the different shRNA-, siRNA-, or miRNA-derived sequences into the miR223 backbone23 and cloned the miRNA in βAS3 intron 2. The guide strand of miRBCL11A has been published previously,16 while those targeting HBB are displayed in Table 1. In particular, for miRNAs derived from shRNA, the sequence was either used as such or modified by adding a GCGC motif at the 3′ end of the guide strand and removing the 4 nt at the 5′ end (Table 1).17 We used miRnt as a control; it targets the LacZ mRNA (5′-AAATCGCTGATTTGTGTAGTC-3′).23 In the LV βAS3m/miR7m, containing the most efficient miRHBB, the βAS3 transgene was modified by inserting synonymous mutations as described in Figure 2.
LV production and titration
Third-generation LVs were produced by calcium phosphate-based transient transfection of HEK293T cells with the transfer vector, the packaging plasmid pHDMH gpm2 (encoding gag/pol), the Rev-encoding plasmid pBA Rev, and the vesicular stomatitis virus glycoprotein G (VSV-G) envelope-encoding plasmid pHDM-G. The physical titer of vector preparations was measured using the HIV-1 Gag p24 antigen immunocapture assay kit (PerkinElmer, Waltham, MA, USA) and expressed as p24 ng/mL. The viral infectious titer, expressed as transduction units per milliliter (TU/mL), was measured in HCT116 cells after transduction using serial vector dilutions. VCN/cell was measured by ddPCR. The LV titer was then calculated as follows: titer (TU/mL) = (number of transduced cells × VCN)/volume of vector used. Viral infectivity was calculated as the ratio between infectious and physical titers (TU/ng p24).
HUDEP-2 cell culture, differentiation, and transduction
HUDEP-2 cells were cultured and differentiated as described previously.50,56,57 HUDEP-2 cells were expanded in a basal medium composed of StemSpan SFEM (STEMCELL Technologies) supplemented with 10−6 M dexamethasone (Sigma), 100 ng/mL human stem cell factor (hSCF) (PeproTech), 3 IU/mL erythropoietin (EPO) Eprex (Janssen-Cilag, France), 100 U/ml L-glutamine (Life Technologies), 2 mM penicillin/streptomycin, and 1 μg/mL doxycycline (Sigma). HUDEP-2 cells were transduced at a cell concentration of 106 cells/mL in basal medium supplemented with 4 μg/mL protamine sulfate (Choay). After 24 h, cells were washed and cultured in fresh basal medium. Cells were differentiated for 9 days in Iscove’s modified Dulbecco’s medium (IMDM; Life Technologies) supplemented with 330 μg/mL holo-transferrin (Sigma), 10 μg/mL recombinant human insulin (Sigma), 2 IU/mL heparin (Sigma), 5% human AB serum (Eurobio AbCys), 3 IU/mL EPO, 100 ng/mL hSCF, 1 μg/mL doxycycline, 100 U/mL L-glutamine, and 2 mM penicillin/streptomycin.
K562 cell culture and transduction
K562 cells were maintained in RPMI 1640 medium (Lonza) containing glutamine and supplemented with 10% fetal bovine serum (Lonza), HEPES (Life Technologies), sodium pyruvate (LifeTechnologies), and penicillin/streptomycin (Life Technologies). K562 cells were transduced at a cell concentration of 5 × 105 cells/mL in the culture medium supplemented with 4 μg/mL Polybrene (Sigma). After 24 h, cells were washed and cultured in fresh medium.
HSPC purification, culture, and transduction
Peripheral blood plerixafor-mobilized or non-mobilized human adult HSPCs were obtained from SCD patients. Peripheral blood G-CSF-mobilized human adult HSPCs were obtained from HDs. Written informed consent was obtained from all subjects. All experiments were performed in accordance with the Declaration of Helsinki. The study was approved by the regional investigational review board (reference DC 2014-2272, CPP Île-de-France II “Hôpital Necker-Enfants malades,” Paris, France). SCD and HD HSPCs were purified by immunomagnetic selection (Miltenyi Biotec) after immunostaining using the CD34 MicroBead Kit (Miltenyi Biotec). Plerixafor/G-CSF-mobilized peripheral blood CD34+ cells were selected from patients affected by β-thalassemia upon signed informed consent approved by the ethics committee of the San Raffaele Hospital (Milan, Italy). Following mobilization and cell collection with the Spectra Cobe or Spectra Optia apheresis system (Terumo BCT), CD34+ cells were purified using immunomagnetic beads (CliniMACS, Miltenyi Biotec) by MolMed (Milan, Italy).58
SCD and HD CD34+ cells were thawed and cultured for 24 h at a concentration of 106 cells/mL in pre-activation medium (PAM) composed of X-VIVO 20 supplemented with penicillin/streptomycin (Gibco) and recombinant human cytokines: 300 ng/mL hSCF, 300 ng/mL Flt-3L, 100 ng/mL TPO, 20 ng/mL interleukin-3 (IL-3) (PeproTech), and 10 mM SR1 (STEMCELL Technologies). After pre-activation, cells (106 cells/mL) were cultured in PAM supplemented with 10 μM PGE2 (Cayman Chemical) on RetroNectin-coated plates (10 μg/cm2, Takara Bio) for at least 2 h. Cells (106 cells/mL) were then transduced for 24 h on RetroNectin-coated plates in PAM supplemented with 10 μM PGE2, protamine sulfate (4 μg/mL, Protamine Choay), and Lentiboost (1 mg/mL, Sirion Biotech). β-Thalassemia CD34+ cells were thawed and cultured for 24 h at a concentration of 106 cells/mL in PAM composed of CellGro (Corning) supplemented with 300 ng/mL hSCF, 300 ng/mL Flt-3L, 100 ng/mL TPO, and 20 ng/mL IL-3 (PeproTech) on RetroNectin-coated plates (10 μg/cm2, Takara Bio). Cells were then transduced in the same medium supplemented with 10 μM PGE2 (Cayman Chemical) and Lentiboost (1 mg/mL, Sirion Biotech) for 18 h in the presence of the LV at the indicated MOI.
VCN quantification by ddPCR
Genomic DNA was extracted from HCT116 cells 4 days after transduction, from K562 cells, HUDEP-2 cells, primary erythroblasts, BFU-E, CFU-GM colonies 14 days after transduction, and from human CD45+ (hCD45+) bone marrow (BM) cells from transplanted mice using the PureLink Genomic DNA Mini Kit (Invitrogen). DNA was digested using the DraI restriction enzyme (New England Biolabs) at 37°C for 30 min and then mixed with ddPCR reaction mix composed of 2× ddPCR SuperMix for probes (no dUTP) (Bio-Rad), forward (FOR) and reverse (REV) primers (at a final concentration of 900 nM) and probes (at a final concentration of 250 nM). We used probes and primers specific for (1) albumin (VIC-labeled ALB probe with a QSY quencher, 5′-CCTGTCATGCCCACACAAATCTCTCC-3′; FOR ALB primer, 5′-GCTGTCATCTCTTGTGGGCTGT-3′; REV ALB primer, 5′-ACTCATGGGAGCTGCTGGTTC-3′) and (2) the LV (FAM-labeled LV probe with an MGB quencher, 5′-CGCACGGCAAGAGGCGAGG-3′; FOR LV primer 5′-TCCCCCGCTTAATACTGACG-3′; REV LV primer 5′-CAGGACTCGGCTTGCTGAAG-3′ or FAM-labeled PRO-LV probe with an MGB quencher 5′-TCTCTAGCAGTGGCGCCCGAACAGG-3′; FOR PRO-LV primer: 5′-CACTCCCAACGAAGACAAGA-3′; REV PRO-LV primer: 5′-TCTGGTTTCCCTTTCGCTTT-3′ to measure VCN in BM cells59). The albumin gene was chosen as a reference locus to calculate the VCN per genome. Droplets were generated using a QX200 droplet generator (Bio-Rad) with droplet generation oil for probes (Bio-Rad) on a DG8 cartridge (Bio-Rad) and transferred to a semi-skirted 96-well plate (Eppendorf). After sealing with a pierceable foil heat seal using a PX1 PCR plate sealer (Bio-Rad), the plate was loaded on a SimpliAmp thermal cycler (Thermo Fisher Scientific) for PCR amplification using the following conditions: 95°C for 10 min, followed by 40 cycles at 94°C for 30 s and 60°C for 1 min and by a final step at 98°C for 10 min. The plate was analyzed using the QX200 droplet reader (Bio-Rad) (channel 1, FAM; channel 2, VIC) and analyzed using the QuantaSoft analysis software (Bio-Rad), which quantifies positive and negative droplets and calculates the starting DNA concentration using a Poisson algorithm. The average VCN/cell was calculated as (LV copies × x)/(albumin copies), where x = 2 for diploid cells and 4 for tetraploid cells.
To evaluate transduction efficiency in CFC-derived progenitors from β-thalassemia patients, single BFU-Es and CFU-GMs were lysed with QuickExtract lysis buffer (Epicentre). CFCs were incubated at 65°C for 20 min, followed by incubation at 98°C for 10 min and centrifugation at 13,000 rpm for 10 min. VCN was evaluated by ddPCR using the previously described HIV/Telo system.58
In vitro erythroid differentiation
Mature RBCs from mock- and LV-transduced CD34+ HSPCs were generated using a three-step protocol.13 Briefly, from day 0 to day 6, cells were grown in basal erythroid medium (BEM) supplemented with hSCF, IL-3, EPO (Eprex, Janssen-Cilag), and hydrocortisone (Sigma). From day 6 to day 20, cells were cultured on a layer of murine stromal MS-5 cells in BEM supplemented with EPO from day 6 to day 9 and without cytokines from day 9 to day 20. From day 13 to day 20, human AB serum was added to the BEM.
CFC assay
The number of hematopoietic progenitors was evaluated using a CFC assay. HSPCs were plated at a concentration of 5 × 102 cells/mL in a methylcellulose-containing medium (GFH4435, STEMCELL Technologies) under conditions supporting erythroid and granulo-monocytic differentiation. BFU-Es and CFU-GMs were scored after 14 days. BFU-Es and CFU-GMs were randomly picked and collected as bulk populations (containing at least 25 colonies) to evaluate transduction efficiency and globin expression.
qRT-PCR analysis
RNA was extracted from HUDEP-2 cells, K562 cells, primary erythroblasts, or BFU-Es using the RNeasy Micro Kit (QIAGEN). Reverse transcription of mRNA was performed using the SuperScriptIII First-Strand Synthesis System for RT-PCR (Invitrogen) with oligo(dT)20 primers. qPCR was performed using the SYBR Green detection system (Bio-Rad). We used the following primers: HBB FOR, 5′-AAGGGCACCTTTGCCACA-3′; HBB REV, 5′-GCCACCACTTTCTGATAGGCAG-3′; βAS3 FOR, 5′-GCCACCACTTTCTGATAGGCAG-3′; βAS3 REV, 5′-AAGGGCACCTTTGCCCAG-3′; BCL11A-XL FOR, 5′-ATGCGAGCTGTGCAACTATG-3′; BCL11A-XL REV, 5′-GTAAACGTCCTTCCCCACCT-3′; HBG1/2 FOR, 5′-CCTGTCCTCTGCCTCTGCC-3′; HBG1/2 REV, 5′-GGATTGCCAAAACGGTCAC-3′; LMNB2 FOR, 5′-AGTTCACGCCCAAGTACATC-3′; LMNB2 REV, 5′-CTTCACAGTCCTCATGGCC-3′; HBA FOR, 5′-CGGTCAACTTCAAGCTCCTAA-3′; HBA REV, 5′-ACAGAAGCCAGGAACTTGTC-3′; GAPDH FOR, 5′- GAAGGTGAAGGTCGGAGT-3′; GAPDH REV, 5′-GAAGATGGTGATGGGATTTC-3′. The samples were analyzed with the ViiA 7 real-time PCR system and software (Applied Biosystems).
Flow cytometry analysis
After 9 days of differentiation, HUDEP-2 cells were stained with a monoclonal mouse anti-human CD235a antibody (clone GA-R2, BD Biosciences), fixed and permeabilized with a fixation/permeabilization solution kit (BD Biosciences), and stained with a monoclonal mouse anti-human HbF antibody (clone HBF-1, Thermo Fisher Scientific). Cells were analyzed by flow cytometry using a BD LSRFortessa cell analyzer (BD Biosciences) and Diva (BD Biosciences) and FlowJo software.
In primary cell culture, expression of erythroid markers was monitored by flow cytometry using anti-CD36, anti-CD49d, anti-CD71, anti-CD235a (BD Biosciences), and anti-CD233 (band 3; IBGRL) antibodies and 7-AAD (BD Biosciences) for cell death assessment. The proportion of enucleated RBCs was measured using the nuclear dye DRAQ5 (eBioscience). The proportions of HbF- and HbS-positive RBCs were measured with an antibody recognizing HbF (Thermo Fisher Scientific for HUDEP-2 cells or BD Biosciences for primary cells) or HbS (BioMedomics), respectively. Briefly, RBCs were stained with a monoclonal mouse anti-human CD235a antibody (BD Biosciences), fixed with 0.05% glutaraldehyde for 10 min at room temperature (RT), permeabilized with 0.1% Triton X-100 for 10 min at RT, and stained with the HbF or the HbS antibody. Flow cytometry analyses were performed using a Gallios analyzer and Kaluza (Beckman-Coulter) and FlowJo software.
Western blot
HUDEP-2 cells, after 6 days of differentiation to detect BCL11A-XL, were lysed for 30 min at 4°C using a lysis buffer containing 10 mM Tris, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% SDS, 0.1% Na-deoxicholate, 140 mM NaCl (Sigma-Aldrich), and a protease inhibitor cocktail (Roche Diagnostics). Cell lysates were sonicated twice (50% amplitude, 10 s per cycle, pulse 9 s on/1 s off) and underwent 3 cycles of freezing/thawing (3 min at −80°C/3 min at 37°C). After centrifugation, the supernatant was collected, and protein concentration was measured using the Pierce BCA Protein Assay Kit (Thermo Scientific). After electrophoresis and protein transfer, BCL11A-XL, and GAPDH were detected using the antibodies ab19487 (Abcam) and sc-32233 (Santa Cruz Biotechnology), respectively. The bands corresponding to the different proteins were quantified using Chemidoc and Image Lab software (Bio-Rad).
HPLC
HPLC analysis was performed using a NexeraX2 SIL-30AC chromatograph (Shimadzu) and LC Solution software. Globin chains from differentiated HUDEP-2 cells, primary cells, or BFU-Es were separated by RP HPLC using a 250 × 4.6-mm, 3.6-μm Aeris Widepore column (Phenomenex). Samples were eluted with a gradient mixture of solution A (water:acetonitrile:trifluoroacetic acid, 95:5:0.1) and solution B (water:acetonitrile:trifluoroacetic acid, 5:95:0.1). The absorbance was measured at 220 nm. Hb tetramers from RBCs were separated by CE HPLC using a 2-cation exchange column (PolyCAT A, PolyLC, Columbia). Samples were eluted with a gradient mixture of solution A (20 mM Bis Tris, 2 mM KCN [pH 6.5]) and solution B (20 mM Bis Tris, 2 mM KCN, 250 mM NaCl [pH 6.8]). The absorbance was measured at 415 nm.
Sickling assay
On day 19 of terminal erythroid differentiation, RBCs were collected and incubated under hypoxic conditions to evaluate their sickling properties. Briefly, RBCs were resuspended in ID-CellStab stabilization solution for red cells (Bio-Rad) and exposed to an oxygen-deprived atmosphere for 60 min or longer. Images were captured using an AxioObserver microscope and Zen software (Carl Zeiss) at a magnification of 40×. Images were analyzed with ImageJ to determine the percentage of sickling RBCs per field of acquisition in the total RBC population.
Quantitative phase image microscopy of RBCs
At day 19–21 of terminal erythroid differentiation, RBCs were collected, resuspended in PBS, and placed in a 4 well μ-Slide (ibidi). Quantitative phase images of label-free RBCs were taken using a SID4 HR GE camera (Phasics, Saint-Aubin, France) with an inverted microscope (Eclipse Ti-E, Nikon) and a 40×/0.60 objective. For each image, a segmentation procedure was performed using BIO-Data R&D software (v.2.7.1.46) to isolate individual RBCs.51 For β-thalassemia samples, enucleated RBCs were selected based on their surface density (dry mass/surface), discarding nucleated cells with a surface density (pg/mm2) greater than 0.376 (βAS3) and greater than 0.4 (βAS3/miRBCL11A) for β+/β+#1 and greater than 0.358 (mock), greater than 0.368 (βAS3), and greater than 0.365 (βAS3/miRBCL11A) for β0/β0#2. Dry mass (pg), surface (mm2), and perimeter (mm) were measured for each enucleated RBC.
Vector integration site analysis
G-CSF-mobilized CD34+ cells from HD were thawed, cultured, and transduced as described above. After 6 days, DNA was extracted, and 3′ long terminal repeat (LTR) vector-genome junctions were amplified by LM PCR and sequenced as described in Poletti et al.60 Comparative Gene Ontology analysis (Gene Ontology [GO] biological processes BP5) was performed with the DAVID tool on target genes (defined by read count >95th percentile).
RNA-seq and miRNA-seq analysis
Total RNA was extracted from erythroblasts collected on day 13 of erythroid differentiation using the Quick-RNA MicroPrep Kit (Zymo Research). This protocol includes DNase treatment of the extracted samples. The concentration and purity of the total RNA were measured using an Xpose spectrophotometer (Trinean).
For the RNA-seq experiments, the integrity of the RNA was evaluated by capillary electrophoresis using a fragment analyzer (Agilent). Before preparing the RNA-seq libraries, total RNA was treated with heat-labile double-strand DNase (HL-dsDNase; ArcticZyme) to remove any potential residual genomic DNA contamination. Then, the Ovation Universal RNA-seq System (Tecan) was used to prepare the RNA-seq libraries from 35 ng of total RNA following the manufacturer’s protocol. Briefly, the reverse transcription and second-strand synthesis were followed by a fragmentation step and ligation of the Illumina-compatible indexed adaptor coupled to the strand selection enzymatic reaction to retain the information on the orientation of the transcripts. Next, insert-dependent adaptor cleavage (InDA-C)-specific primers were used to deplete human ribosomal RNA transcripts before PCR enrichment. Finally, an equimolar pool of the final indexed RNA-seq libraries was sequenced on an Illumina NovaSeq6000 (paired-end sequencing, 2× 100 bp) and ∼100 million paired-end reads per library were produced. Read quality was verified using FastQC (v.0.11.9; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Raw reads were trimmed for adapters and low-quality tails (quality < Q20) with BBDuk (v.38.92; sourceforge.net/projects/bbmap/); moreover, the first 10 nucleotides were force trimmed for low quality. Reads shorter than 35 bp after trimming were removed. Reads were subsequently aligned to the human reference genome (hg38) using STAR (v.2.7.9a61). Raw gene counts were obtained in R-4.1.1 using the featureCounts function of the Rsubread R package (v.2.6.462) and the GENCODE 38 basic gene annotation for the hg38 reference genome. Transgene sequences and annotations were added to the hg38 reference genome and annotation before performing read alignment and gene expression quantification. Gene counts were normalized to counts per million mapped reads (CPM) and to fragments per kilobase of exon per million mapped reads (FPKM) using the edgeR R package (v.3.34.163); only genes with a CPM greater than 1 in at least 2 samples were retained for differential analysis. Differential gene expression analysis was performed using the glmQLFTest function of the edgeR R package, using donor as a blocking variable. Genes with an FDR of less than 0.05 and absolute log2 fold change of 1 or greater were defined as differentially expressed.
The βAS3/βAS3m transgenes were distinguished from the endogenous gene thanks to the multiple mutations present in the transgenes compared with the endogenous gene. Of note, these mutations are distributed along the entire cDNA and are separated by only 100–150 nt; therefore, most of the 2×100-bp reads contain multiple mutations, enabling us to distinguish and correctly map reads coming from endogenous and transgene transcripts. Moreover, we used only uniquely mapped reads to quantify the expression of endogenous and transgene transcripts, discarding reads mapping on multiple loci (such as reads that map on unmodified sequences shared by endogenous and modified β-globin genes). This pipeline allowed us to have robust quantification and comparison of endogenous and transgene transcripts, even if it is possible that we are underestimating the absolute expression values.
For the miRNA-seq experiments, 50 ng of total RNA was used to prepare miRNA libraries using the QIAseq miRNA Library Kit (QIAGEN). The amount and size of the libraries were evaluated using the TapeStation 2100 (Agilent). Libraries were mixed in equimolar ratios and sequenced on a NextSeq500 sequencing instrument. Raw reads were demultiplexed, and FASTQ files were generated using bcl2fastq software (Illumina) Read quality was evaluated using FastQC (v.0.11.9; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). UMI extraction and adapter removal were performed using UMI-Tools (v.1.1.2); then, reads of less than 16 bp were removed using BBDuk (v.38.92; sourceforge.net/projects/bbmap/). Reads were subsequently aligned to the human reference genome (hg38) using STAR (v.2.7.9a;61). Alignments were then de-duplicated using UMI-Tools (v.1.1.2). UMI-based counts were obtained in R-4.1.1 using the featureCounts function of the Rsubread R package (v.2.6.462) and the miRBase annotation (v.2264) for the hg38 reference genome. The miR7m sequence and annotation were added to the hg38 reference genome and to the miRbase annotation before performing read alignment and gene expression quantification. UMI-based counts were normalized to CPM using the edgeR R package (v.3.34.1;63 only miRNAs with a CPM greater than 1 in at least 2 samples were retained for differential analysis. Differential miRNA expression analysis was performed using the glmQLFTest function of the edgeR R package, using donor as a blocking variable. miRNAs with an FDR of less than 0.05 and absolute log2 fold change of 1 or greater were defined as differentially expressed.
HSPC xenotransplantation in NBSGW mice
NBSGW mice were housed in a pathogen-free facility. Mock- or LV-transduced non-mobilized SCD CD34+ cells (5 × 105 cells/mouse) were transplanted into nonirradiated NBSGW male and female mice 5–7 weeks of age via retro-orbital sinus injection. NBSGW mice were conditioned with busulfan (Sigma, St. Louis, MO, USA) injected intraperitoneally (15 mg/kg body weight/day) 24 h before transplantation. 18 weeks after transplantation, NBSGW primary recipients were sacrificed. Cells were harvested from BM, thymus, and spleen and stained with antibodies against murine and human surface markers (murine CD45 [1/50 mCD45-VioBlue], Miltenyi Biotec; human CD45 [1/50 hCD45-APCvio770], Miltenyi Biotec; human CD3 [1/50 CD3-APC], Miltenyi Biotec; human CD14 [1/50 CD14-PE-Cy7], BD Biosciences; human CD15 [1/50 CD15-PE], Miltenyi Biotec; human CD19 [1/100 CD19-BV510]; human CD235a [1/50 CD235a-PE], BD Biosciences) and analyzed by flow cytometry using the MACSQuant analyzer (Miltenyi Biotec) and FlowJo software (BD Biosciences). Human BM CD45+ cells were sorted by immunomagnetic selection with AutoMACS (Miltenyi Biotec) after immunostaining with the CD45 MicroBead Kit (Miltenyi Biotec). All experiments and procedures were performed in compliance with the French Ministry of Agriculture’s regulations on animal experiments and approved by the regional Animal Care and Use Committee (APAFIS 2019061312202425_v4). Mice were housed in a temperature (20°C–22°C) and humidity (40%–50%)-controlled environment with a 12 h/12 h light/dark cycle and fed ad libitum with a standard diet.
Statistical analyses
Statistical analyses were performed when the total number of replicates for each group was 3 or more using GraphPad Prism v.9. We used a Shapiro-Wilk test to evaluate whether data follow a Gaussian distribution. For comparison between two groups or more following a Gaussian distribution, we used an unpaired t test to compare two averages or one- or two-way ANOVA followed by multiple comparisons to compare three or more averages. For non-Gaussian-distributed data, we used non-parametric tests; namely, Mann-Whitney test for comparing two groups and Kruskal-Wallis test for multiple comparison. p values are indicated with asterisks (∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001), and ns (not significant) reflects p > 0.05.
Acknowledgments
This work was supported by state funding from the Agence Nationale de la Recherche under the “Investissements d’avenir” program (ANR-10-IAHU-01 and ANR-20-CE17-0016), the Paris Île-de-France region under the “DIM Thérapie génique” initiative, and the Imagine Institute (Innogrant). We thank Christine Bole and the Imagine genomic facility for generation of the NGS data.
Author contributions
M.B. designed and conducted experiments, analyzed data, and wrote the paper. A.C., P.M., V.P., S.S., and S.R. conducted experiments and analyzed data. G.F., O.R., F.M., M.C., and M.A. contributed to the design of the experimental strategy. C.M. and O.R. analyzed NGS data. A.M. conceived the study, designed experiments. and wrote the paper.
Declaration of interests
M.B., F.M., M.C., M.A., and A.M. are the inventors of two patents describing bifunctional LVs for hemoglobinopathies.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.03.012.
Contributor Information
Mégane Brusson, Email: megane.brusson@institutimagine.org.
Annarita Miccio, Email: annarita.miccio@institutimagine.org.
Supplemental information
Data availability statement
For integration site analysis, data are available online in the NCBI Sequence Read Archive (SRA: PRJNA842958).
RNA-seq and miRNA-seq data are available in the Gene Expression Omnibus repository (GEO: GSE205074). The expression values of transgenes, vector-derived short RNAs, and the miR7m can be inspected in the uploaded gene expression matrices in the raws transgene_AS3, transgene_AS3m, immature_miR7m, and miR7m, respectively.
References
- 1.Thein S.L. Molecular basis of β thalassemia and potential therapeutic targets. Blood Cells Mol. Dis. 2018;70:54–65. doi: 10.1016/j.bcmd.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piel F.B., Steinberg M.H., Rees D.C. Sickle cell disease. N. Engl. J. Med. 2017;376:1561–1573. doi: 10.1056/NEJMra1510865. [DOI] [PubMed] [Google Scholar]
- 3.Cavazzana M., Bushman F.D., Miccio A., André-Schmutz I., Six E. Gene therapy targeting haematopoietic stem cells for inherited diseases: progress and challenges. Nat. Rev. Drug Discov. 2019;18:447–462. doi: 10.1038/s41573-019-0020-9. [DOI] [PubMed] [Google Scholar]
- 4.Kanter J., Walters M.C., Krishnamurti L., Mapara M.Y., Kwiatkowski J.L., Rifkin-Zenenberg S., Aygun B., Kasow K.A., Pierciey F.J., Bonner M., et al. Biologic and clinical efficacy of LentiGlobin for sickle cell disease. N. Engl. J. Med. 2021;386:617–628. doi: 10.1056/NEJMoa2117175. [DOI] [PubMed] [Google Scholar]
- 5.Locatelli F., Thompson A.A., Kwiatkowski J.L., Porter J.B., Thrasher A.J., Hongeng S., Sauer M.G., Thuret I., Lal A., Algeri M., et al. Betibeglogene autotemcel gene therapy for non-β0/β0 genotype β-thalassemia. N. Engl. J. Med. 2022;386:415–427. doi: 10.1056/NEJMoa2113206. [DOI] [PubMed] [Google Scholar]
- 6.Magrin E., Semeraro M., Hebert N., Joseph L., Magnani A., Chalumeau A., Gabrion A., Roudaut C., Marouene J., Lefrere F., et al. Long-term outcomes of lentiviral gene therapy for the β-hemoglobinopathies: the HGB-205 trial. Nat. Med. 2022;28:81–88. doi: 10.1038/s41591-021-01650-w. [DOI] [PubMed] [Google Scholar]
- 7.Magrin E., Miccio A., Cavazzana M. Lentiviral and genome-editing strategies for the treatment of β-hemoglobinopathies. Blood. 2019;134:1203–1213. doi: 10.1182/blood.2019000949. [DOI] [PubMed] [Google Scholar]
- 8.Ribeil J.-A., Hacein-Bey-Abina S., Payen E., Magnani A., Semeraro M., Magrin E., Caccavelli L., Neven B., Bourget P., El Nemer W., et al. Gene therapy in a patient with sickle cell disease. N. Engl. J. Med. 2017;376:848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- 9.Ware R.E., de Montalembert M., Tshilolo L., Abboud M.R. Sickle cell disease. Lancet. 2017;390:311–323. doi: 10.1016/S0140-6736(17)30193-9. [DOI] [PubMed] [Google Scholar]
- 10.Steinberg M.H., Chui D.H.K., Dover G.J., Sebastiani P., Alsultan A. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 2014;123:481–485. doi: 10.1182/blood-2013-09-528067. [DOI] [PubMed] [Google Scholar]
- 11.Cavazzana-Calvo M., Payen E., Negre O., Wang G., Hehir K., Fusil F., Down J., Denaro M., Brady T., Westerman K., et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goyal S., Tisdale J., Schmidt M., Kanter J., Jaroscak J., Whitney D., Bitter H., Gregory P.D., Parsons G., Foos M., et al. Acute myeloid leukemia case after gene therapy for sickle cell disease. N. Engl. J. Med. 2021 doi: 10.1056/NEJMoa2109167. [DOI] [PubMed] [Google Scholar]
- 13.Weber L., Poletti V., Magrin E., Antoniani C., Martin S., Bayard C., Sadek H., Felix T., Meneghini V., Antoniou M.N., et al. An optimized lentiviral vector efficiently corrects the human sickle cell disease phenotype. Mol. Ther. Methods Clin. Dev. 2018;10:268–280. doi: 10.1016/j.omtm.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Powars D.R., Weiss J.N., Chan L.S., Schroeder W.A. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood. 1984;63:921–926. [PubMed] [Google Scholar]
- 15.Akinsheye I., Alsultan A., Solovieff N., Ngo D., Baldwin C.T., Sebastiani P., Chui D.H.K., Steinberg M.H. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118:19–27. doi: 10.1182/blood-2011-03-325258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brendel C., Guda S., Renella R., Bauer D.E., Canver M.C., Kim Y.-J., Heeney M.M., Klatt D., Fogel J., Milsom M.D., et al. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J. Clin. Invest. 2016;126:3868–3878. doi: 10.1172/JCI87885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guda S., Brendel C., Renella R., Du P., Bauer D.E., Canver M.C., Grenier J.K., Grimson A.W., Kamran S.C., Thornton J., et al. miRNA-embedded shRNAs for lineage-specific BCL11A knockdown and hemoglobin F induction. Mol. Ther. 2015;23:1465–1474. doi: 10.1038/mt.2015.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brendel C., Negre O., Rothe M., Guda S., Parsons G., Harris C., McGuinness M., Abriss D., Tsytsykova A., Klatt D., et al. Preclinical evaluation of a novel lentiviral vector driving lineage-specific BCL11A knockdown for sickle cell gene therapy. Mol. Ther. Methods Clin. Dev. 2020;17:589–600. doi: 10.1016/j.omtm.2020.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esrick E.B., Lehmann L.E., Biffi A., Achebe M., Brendel C., Ciuculescu M.F., Daley H., MacKinnon B., Morris E., Federico A., et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N. Engl. J. Med. 2021;384:205–215. doi: 10.1056/NEJMoa2029392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu N., Hargreaves V.V., Zhu Q., Kurland J.V., Hong J., Kim W., Sher F., Macias-Trevino C., Rogers J.M., Kurita R., et al. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell. 2018;173:430–442.e17. doi: 10.1016/j.cell.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trakarnsanga K., Wilson M.C., Lau W., Singleton B.K., Parsons S.F., Sakuntanaga P., Kurita R., Nakamura Y., Anstee D.J., Frayne J. Induction of adult levels of β-globin in human erythroid cells that intrinsically express embryonic or fetal globin by transduction with KLF1 and BCL11A-XL. Haematologica. 2014;99:1677–1685. doi: 10.3324/haematol.2014.110155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu X., Wang Y., Pi W., Liu H., Wickrema A., Tuan D. NF-Y recruits both transcription activator and repressor to modulate tissue- and developmental stage-specific expression of human γ-globin gene. PLoS One. 2012;7:e47175. doi: 10.1371/journal.pone.0047175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amendola M., Passerini L., Pucci F., Gentner B., Bacchetta R., Naldini L. Regulated and multiple miRNA and siRNA delivery into primary cells by a lentiviral platform. Mol. Ther. 2009;17:1039–1052. doi: 10.1038/mt.2009.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antoniou M., Geraghty F., Hurst J., Grosveld F. Efficient 3’-end formation of human beta-globin mRNA in vivo requires sequences within the last intron but occurs independently of the splicing reaction. Nucleic Acids Res. 1998;26:721–729. doi: 10.1093/nar/26.3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sripichai O., Munkongdee T., Kumkhaek C., Svasti S., Winichagoon P., Fucharoen S. Coinheritance of the different copy numbers of alpha-globin gene modifies severity of beta-thalassemia/Hb E disease. Ann. Hematol. 2008;87:375–379. doi: 10.1007/s00277-007-0407-2. [DOI] [PubMed] [Google Scholar]
- 26.Breda L., Motta I., Lourenco S., Gemmo C., Deng W., Rupon J.W., Abdulmalik O.Y., Manwani D., Blobel G.A., Rivella S. Forced chromatin looping raises fetal hemoglobin in adult sickle cells to higher levels than pharmacologic inducers. Blood. 2016;128:1139–1143. doi: 10.1182/blood-2016-01-691089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams F.F., Heckl D., Hoffmann T., Talbot S.R., Kloos A., Thol F., Heuser M., Zuber J., Schambach A., Schwarzer A. An optimized lentiviral vector system for conditional RNAi and efficient cloning of microRNA embedded short hairpin RNA libraries. Biomaterials. 2017;139:102–115. doi: 10.1016/j.biomaterials.2017.05.032. [DOI] [PubMed] [Google Scholar]
- 28.Dykxhoorn D.M., Schlehuber L.D., London I.M., Lieberman J. Determinants of specific RNA interference-mediated silencing of human beta-globin alleles differing by a single nucleotide polymorphism. Proc. Natl. Acad. Sci. USA. 2006;103:5953–5958. doi: 10.1073/pnas.0601309103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Papasavva P.L., Papaioannou N.Y., Patsali P., Kurita R., Nakamura Y., Sitarou M., Christou S., Kleanthous M., Lederer C.W. Distinct miRNA signatures and networks discern fetal from adult erythroid differentiation and primary from immortalized erythroid cells. Int. J. Mol. Sci. 2021;22:3626. doi: 10.3390/ijms22073626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pires Lourenco S., Jarocha D., Ghiaccio V., Guerra A., Abdulmalik O., La P., Zezulin A., Smith-Whitley K., Kwiatkowski J.L., Guzikowski V., et al. Inclusion of a short hairpin RNA targeting BCL11A into a β-globin expressing vector allows concurrent synthesis of curative adult and fetal hemoglobin. Haematologica. 2021;106:2740–2745. doi: 10.3324/haematol.2020.276634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nualkaew T., Sii-Felice K., Giorgi M., McColl B., Gouzil J., Glaser A., Voon H.P.J., Tee H.Y., Grigoriadis G., Svasti S., et al. Coordinated β-globin expression and α2-globin reduction in a multiplex lentiviral gene therapy vector for β-thalassemia. Mol. Ther. 2021;29:2841–2853. doi: 10.1016/j.ymthe.2021.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu B., Brendel C., Vinjamur D.S., Zhou Y., Harris C., McGuinness M., Manis J.P., Bauer D.E., Xu H., Williams D.A. Development of a double shmiR lentivirus effectively targeting both BCL11A and ZNF410 for enhanced induction of fetal hemoglobin to treat β-hemoglobinopathies. Mol. Ther. 2022;30:2693–2708. doi: 10.1016/j.ymthe.2022.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samakoglu S., Lisowski L., Budak-Alpdogan T., Usachenko Y., Acuto S., Di Marzo R., Maggio A., Zhu P., Tisdale J.F., Rivière I., Sadelain M. A genetic strategy to treat sickle cell anemia by coregulating globin transgene expression and RNA interference. Nat. Biotechnol. 2006;24:89–94. doi: 10.1038/nbt1176. [DOI] [PubMed] [Google Scholar]
- 34.Kabra M., Moosajee M., Newby G.A., Molugu K., Saha K., Liu D.R., Pattnaik B.R. Comprehensive analysis of CRISPR base editing outcomes for multimeric protein. bioRxiv. 2022 doi: 10.1101/2022.06.20.496792. Preprint at. [DOI] [Google Scholar]
- 35.Das N., Xie L., Ramakrishnan S.K., Campbell A., Rivella S., Shah Y.M. Intestine-specific disruption of hypoxia-inducible factor (HIF)-2α improves anemia in sickle cell disease. J. Biol. Chem. 2015;290:23523–23527. doi: 10.1074/jbc.C115.681643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parrow N.L., Violet P.-C., George N.A., Ali F., Bhanvadia S., Wong R., Tisdale J.F., Fitzhugh C., Levine M., Thein S.L., Fleming R.E. Dietary iron restriction improves markers of disease severity in murine sickle cell anemia. Blood. 2021;137:1553–1555. doi: 10.1182/blood.2020006919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao K.R., Patel A.R., Honig G.R., Vida L.N., McGinnis P.R. Iron deficiency and sickle cell anemia. Arch. Intern. Med. 1983;143:1030–1032. [PubMed] [Google Scholar]
- 38.Brunson A., Keegan T.H.M., Bang H., Mahajan A., Paulukonis S., Wun T. Increased risk of leukemia among sickle cell disease patients in California. Blood. 2017;130:1597–1599. doi: 10.1182/blood-2017-05-783233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seminog O.O., Ogunlaja O.I., Yeates D., Goldacre M.J. Risk of individual malignant neoplasms in patients with sickle cell disease: English national record linkage study. J. R. Soc. Med. 2016;109:303–309. doi: 10.1177/0141076816651037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsieh M.M., Bonner M., Pierciey F.J., Uchida N., Rottman J., Demopoulos L., Schmidt M., Kanter J., Walters M.C., Thompson A.A., et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020;4:2058–2063. doi: 10.1182/bloodadvances.2019001330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaiser J. Gene therapy trials for sickle cell disease halted after two patients develop cancer. 2021. https://www.science.org/content/article/gene-therapy-trials-sickle-cell-disease-halted-after-two-patients-develop-cancer
- 42.Coquerelle S., Ghardallou M., Rais S., Taupin P., Touzot F., Boquet L., Blanche S., Benaouadi S., Brice T., Tuchmann-Durand C., et al. Innovative curative treatment of beta thalassemia: cost-efficacy analysis of gene therapy versus allogenic hematopoietic stem-cell transplantation. Hum. Gene Ther. 2019;30:753–761. doi: 10.1089/hum.2018.178. [DOI] [PubMed] [Google Scholar]
- 43.Castanotto D., Sakurai K., Lingeman R., Li H., Shively L., Aagaard L., Soifer H., Gatignol A., Riggs A., Rossi J.J. Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucleic Acids Res. 2007;35:5154–5164. doi: 10.1093/nar/gkm543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grimm D. The dose can make the poison: lessons learned from adverse in vivo toxicities caused by RNAi overexpression. Silence. 2011;2:8. doi: 10.1186/1758-907X-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boudreau R.L., Martins I., Davidson B.L. Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol. Ther. 2009;17:169–175. doi: 10.1038/mt.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Course M.M., Gudsnuk K., Desai N., Chamberlain J.R., Valdmanis P.N. Endogenous MicroRNA competition as a mechanism of shRNA-induced cardiotoxicity. Mol. Ther. Nucleic Acids. 2020;19:572–580. doi: 10.1016/j.omtn.2019.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McBride J.L., Boudreau R.L., Harper S.Q., Staber P.D., Monteys A.M., Martins I., Gilmore B.L., Burstein H., Peluso R.W., Polisky B., et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. USA. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borel F., van Logtenstein R., Koornneef A., Maczuga P., Ritsema T., Petry H., van Deventer S.J., Jansen P.L., Konstantinova P. In vivo knock-down of multidrug resistance transporters ABCC1 and ABCC2 by AAV-delivered shRNAs and by artificial miRNAs. J. RNAi Gene Silencing. 2011;7:434–442. [PMC free article] [PubMed] [Google Scholar]
- 49.Gröβl T., Hammer E., Bien-Möller S., Geisler A., Pinkert S., Röger C., Poller W., Kurreck J., Völker U., Vetter R., Fechner H. A novel artificial microRNA expressing AAV vector for phospholamban silencing in cardiomyocytes improves Ca2+ uptake into the sarcoplasmic reticulum. PLoS One. 2014;9:e92188. doi: 10.1371/journal.pone.0092188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Antoniani C., Meneghini V., Lattanzi A., Felix T., Romano O., Magrin E., Weber L., Pavani G., El Hoss S., Kurita R., et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood. 2018;131:1960–1973. doi: 10.1182/blood-2017-10-811505. [DOI] [PubMed] [Google Scholar]
- 51.Ramadier S., Chalumeau A., Felix T., Othman N., Aknoun S., Casini A., Maule G., Masson C., De Cian A., Frati G., et al. Combination of lentiviral and genome editing technologies for the treatment of sickle cell disease. Mol. Ther. 2022;30:145–163. doi: 10.1016/j.ymthe.2021.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weber L., Frati G., Felix T., Hardouin G., Casini A., Wollenschlaeger C., Meneghini V., Masson C., De Cian A., Chalumeau A., et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv. 2020;6:eaay9392. doi: 10.1126/sciadv.aay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.-S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 54.Boutin J., Cappellen D., Rosier J., Amintas S., Dabernat S., Bedel A., Moreau-Gaudry F. ON-target adverse events of CRISPR-cas9 nuclease: more chaotic than expected. CRISPR J. 2022;5:19–30. doi: 10.1089/crispr.2021.0120. [DOI] [PubMed] [Google Scholar]
- 55.Nahmad A.D., Reuveni E., Goldschmidt E., Tenne T., Liberman M., Horovitz-Fried M., Khosravi R., Kobo H., Reinstein E., Madi A., et al. Frequent aneuploidy in primary human T cells after CRISPR-Cas9 cleavage. Nat. Biotechnol. 2022;40:1807–1813. doi: 10.1038/s41587-022-01377-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Canver M.C., Smith E.C., Sher F., Pinello L., Sanjana N.E., Shalem O., Chen D.D., Schupp P.G., Vinjamur D.S., Garcia S.P., et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kurita R., Suda N., Sudo K., Miharada K., Hiroyama T., Miyoshi H., Tani K., Nakamura Y. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS One. 2013;8:e59890. doi: 10.1371/journal.pone.0059890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marktel S., Scaramuzza S., Cicalese M.P., Giglio F., Galimberti S., Lidonnici M.R., Calbi V., Assanelli A., Bernardo M.E., Rossi C., et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat. Med. 2019;25:234–241. doi: 10.1038/s41591-018-0301-6. [DOI] [PubMed] [Google Scholar]
- 59.Corre G., Seye A., Frin S., Ferrand M., Winkler K., Luc C., Dorange F., Rocca C.J., Galy A. Lentiviral standards to determine the sensitivity of assays that quantify lentiviral vector copy numbers and genomic insertion sites in cells. Gene Ther. 2022;29:536–543. doi: 10.1038/s41434-022-00315-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poletti V., Charrier S., Corre G., Gjata B., Vignaud A., Zhang F., Rothe M., Schambach A., Gaspar H.B., Thrasher A.J., Mavilio F. Preclinical development of a lentiviral vector for gene therapy of X-linked severe combined immunodeficiency. Mol. Ther. Methods Clin. Dev. 2018;9:257–269. doi: 10.1016/j.omtm.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 63.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kozomara A., Birgaoanu M., Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47:D155–D162. doi: 10.1093/nar/gky1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For integration site analysis, data are available online in the NCBI Sequence Read Archive (SRA: PRJNA842958).
RNA-seq and miRNA-seq data are available in the Gene Expression Omnibus repository (GEO: GSE205074). The expression values of transgenes, vector-derived short RNAs, and the miR7m can be inspected in the uploaded gene expression matrices in the raws transgene_AS3, transgene_AS3m, immature_miR7m, and miR7m, respectively.