

Abstract
Direct blockade of KRAS driver mutations in colorectal cancer (CRC) has been challenging. Targeting SOS1, a guanine nucleotide exchange factor, has arisen as an attractive approach for KRAS-mutant CRC. Here, we describe the development of novel SOS1 degraders and their activity in patient-derived CRC organoids (PDO). The design of these degraders as proteolysis-targeting chimera was based on the crystal structures of cereblon and SOS1. The synthesis used the 6- and 7-OH groups of a quinazoline core as anchor points to connect lenalidomide. Fifteen compounds were screened for SOS1 degradation. P7 was found to have up to 92% SOS1 degradation in both CRC cell lines and PDOs with excellent specificity. SOS1 degrader P7 demonstrated superior activity in inhibiting CRC PDO growth with IC50 5 times lower than that of SOS1 inhibitor BI3406. In summary, we developed new SOS1 degraders and demonstrated SOS1 degradation as a feasible therapeutic strategy for KRAS-mutant CRC.
INTRODUCTION
Colorectal cancer (CRC) remains the third most fatal cancer in the United States, despite extensive screening efforts and advances in therapies, especially for KRAS-mutant CRC.1 Approximately 41% of the patients with advanced CRC carry KRAS driver mutations, which have been associated with poor prognosis and serve as a negative predictive marker for epidermal growth factor receptor (EGFR) blockade.2 Intensive efforts to directly target KRAS have focused on developing allele-specific or pan-KRAS inhibitors.3 However, mutant KRAS inhibitor alone had only modest clinical activity in CRC.4 Approaches to indirectly inhibit KRAS by targeting downstream RAS effector pathways, namely MAPK and PI3K were largely unsuccessful due to feedback activation or incomplete pathway suppression.5 Thus, it is critical to develop novel strategies targeting KRAS-mutant CRC.
SOS1, as a guanine nucleotide exchange factor (GEF), not only promotes conversion of inactive GDP-bound KRAS to active GTP-bound KRAS at the catalytic site, but also enhances its own GEF function by binding GTP-bound KRAS at an allosteric site.6 Depletion of SOS1 decreases the survival of RAS-mutant CRC and other cancer cells.7–9 Given its direct interaction with mutant KRAS, targeting SOS1 also has advantages over approaches to indirectly inhibit KRAS by targeting downstream RAS effector pathways.5 Targeting SOS1 in KRAS-mutant cancers may be more effective as it is located upstream of mutant KRAS and less toxic due to its exclusive function as a GEF. Both small molecule inhibitors and agonists have been developed to modulate SOS1 function.10–17 Recently reported SOS1 inhibitors such as BAY293 and BI3406 were able to disrupt SOS1:KRAS interaction and demonstrated antiproliferative activity in various cancer lines.14,16 However, SOS1 inhibitors as a single agent were less effective in inhibiting RAS-driven CRC cell growth, which required combination with a MEK inhibitor for enhanced in vivo activity in CRC.16 SOS1 inhibitors in preclinical studies only showed an additive effect to enhance the activity of KRASG12C inhibitors in KRASG12C cancers.18 In addition, a series of SOS1 agonists targeting the same binding site with SOS1 inhibitors were designed and synthesized using a fragment-based drug discovery strategy.10–13,15,17 However, their binding affinity to SOS1 was much lower than existing SOS1 inhibitors. Some of these SOS1 agonists carried biphasic and concentration-dependent effects on KRAS signaling without further characterizations in disease-relevant cancer models.11–13
Hence, in the setting of insufficient single-agent activity of SOS1 inhibitor,14,16,18 the efforts to synthesize targeted SOS1 degraders and thorough evaluation of their cellular effects in relevant patient-derived models are crucial to significantly improve current therapeutic strategy in KRAS-mutant CRC. Targeted protein degradation of SOS1 as a therapeutic strategy offers several advantages over existing small molecule inhibitors of SOS1 in KRAS-mutant CRC. First, protein degradation with small molecules (i.e. degraders) adopts an event-driven pharmacology reducing oncoprotein function by dropping its cellular abundance.19 Second, the ability of degraders to rapidly and reversibly “knockdown” an oncoprotein of interest is complementary to genetic strategies. Compared to small molecule inhibitors, degraders often possess improved selectivity profile and efficacy.20 Preclinical work suggested that because degraders remove the entire targeted protein, their effects at lower concentrations are expected to be more profound and durable with less off-target toxicity. Finally, in addition to abrogating enzymatic activity of targeted protein, degraders may also remove its scaffolding function which contributes to the added efficacy.21 Here, we described the design and synthesis of SOS1 inhibitor-based degraders utilizing cereblon as the E3 ligase. Our SOS1 degrader achieved up to 92% SOS1 degradation in both CRC cell lines and PDOs with excellent specificity for SOS1. Our SOS1 degrader demonstrated superior activity to SOS1 inhibitor BI3406 in inhibiting growth of CRC PDOs. We demonstrated SOS1 degradation as a feasible and novel therapeutic strategy for targeting SOS1 in KRAS-mutant CRC.
RESULTS AND DISCUSSION
Design of SOS1 degrader
Recent advances in the discovery of small molecule SOS1 binders allowed us to design and synthesize heterobifunctional proteolysis targeting chimera (PROTAC) degraders with diverse molecular structures. Our design of SOS1 degrader was based on the structure-activity relationship of the KRASG12C–SOS1cat interaction inhibitor BAY29314 that has high binding affinity to SOS1 (Figure 1A). Further, our synthesis of SOS1 degraders was also informed by the crystal structure of SOS1 (PDB: 6SFR) in complex with SOS1 binder BI68BS (Figure 1B).16 This is an analog of SOS1 inhibitor BI3406 with 9.7 nM binding affinity to this GEF in preclinical studies16 and of SOS1 inhibitor BI1701963, currently in a phase I trial of patients with KRAS-mutant advanced solid tumors.22 Visual inspection and structural interaction fingerprint analysis of BI68BS with SOS123,24 demonstrated that in addition to the hydrophobic pocket and H-bond interaction with Asn879, 1) both the 6- and 7-methoxy groups are exposed to solvent; 2) π-π stacking between the quinazoline core of BI68BS and His905 is essential for its activity (Figure 1C). Also, it has been reported that this interaction can also be fulfilled by phthalazine and 2, 4, 6-triaza-2-indanyl groups in other reported SOS1 inhibitors.18,25
Figure 1.
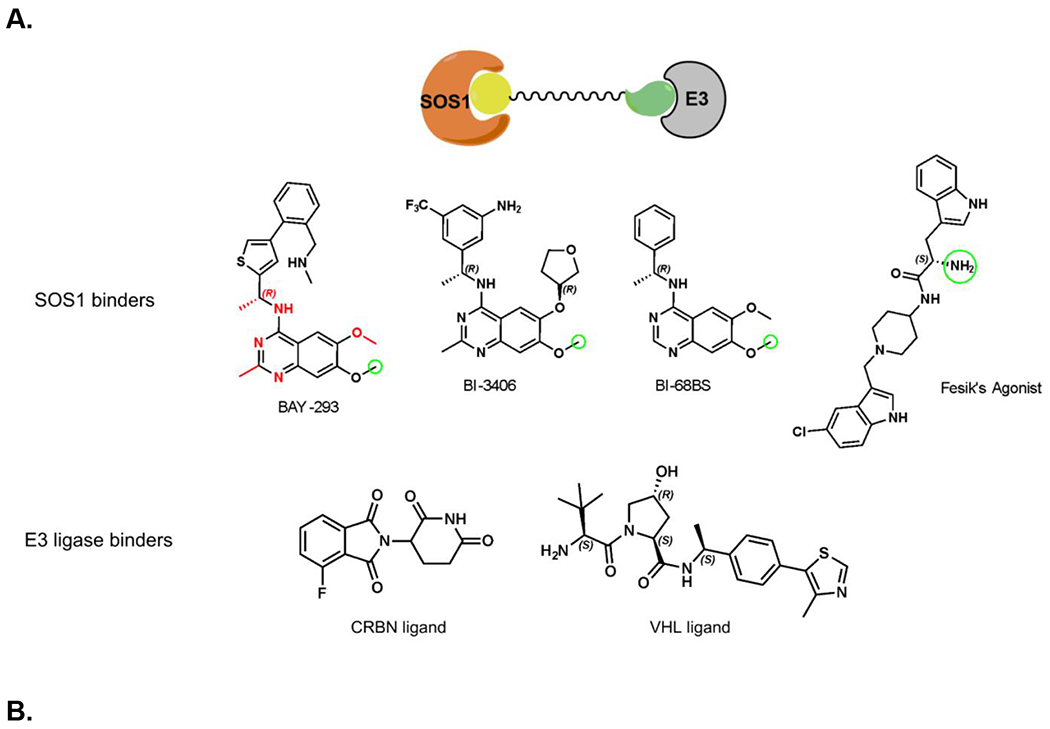
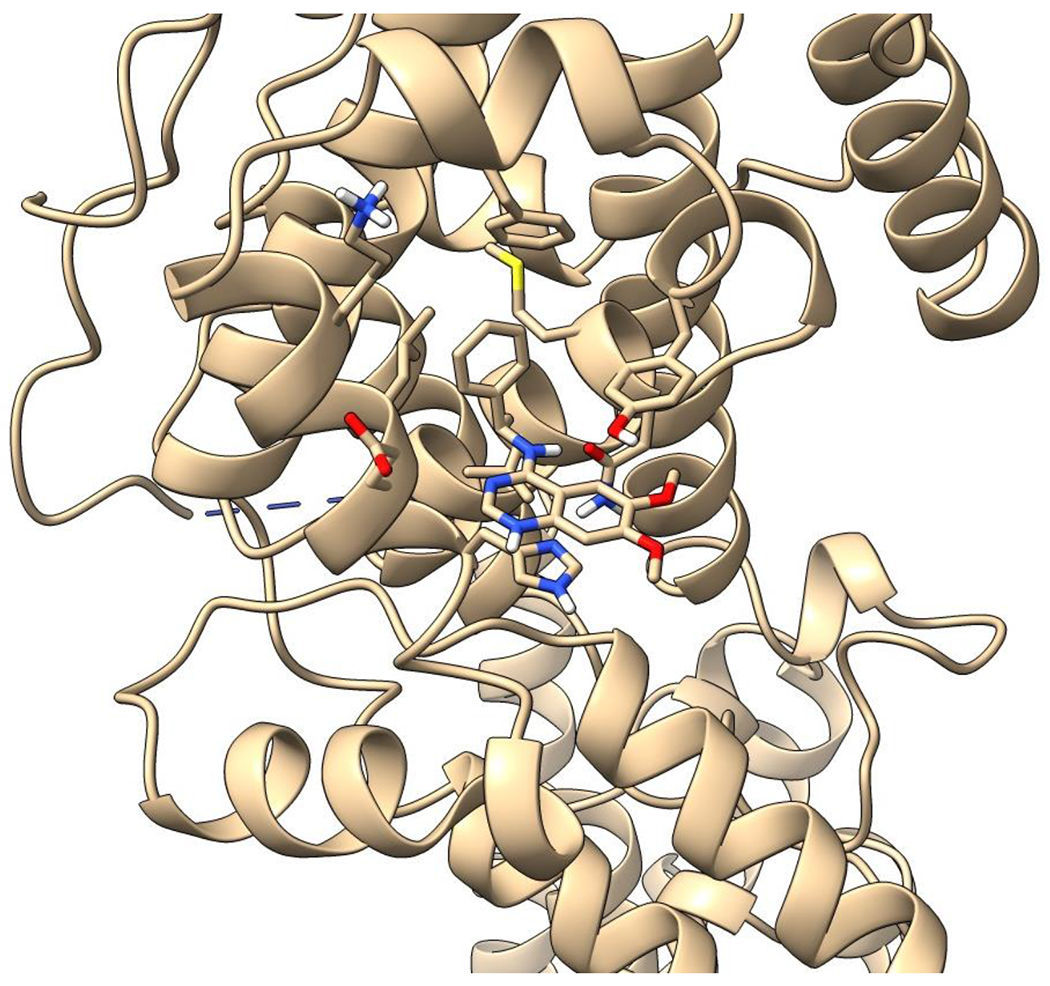
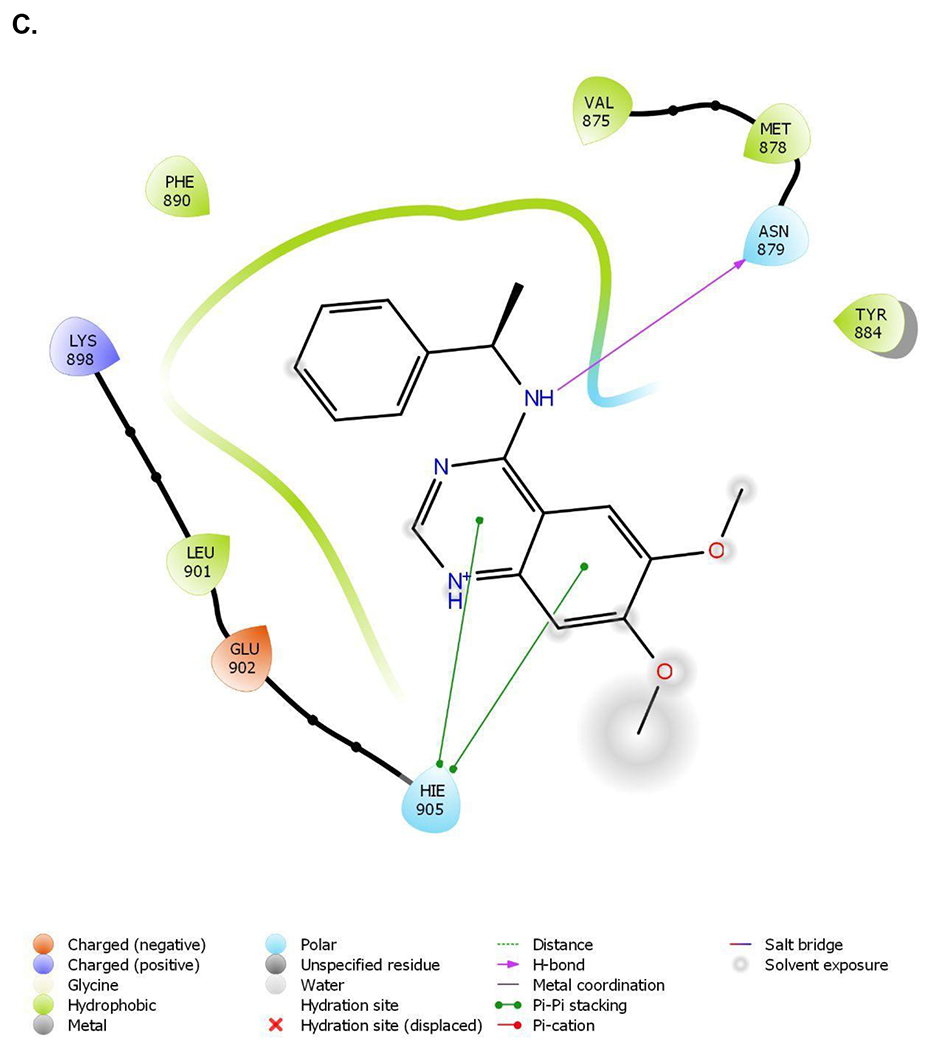
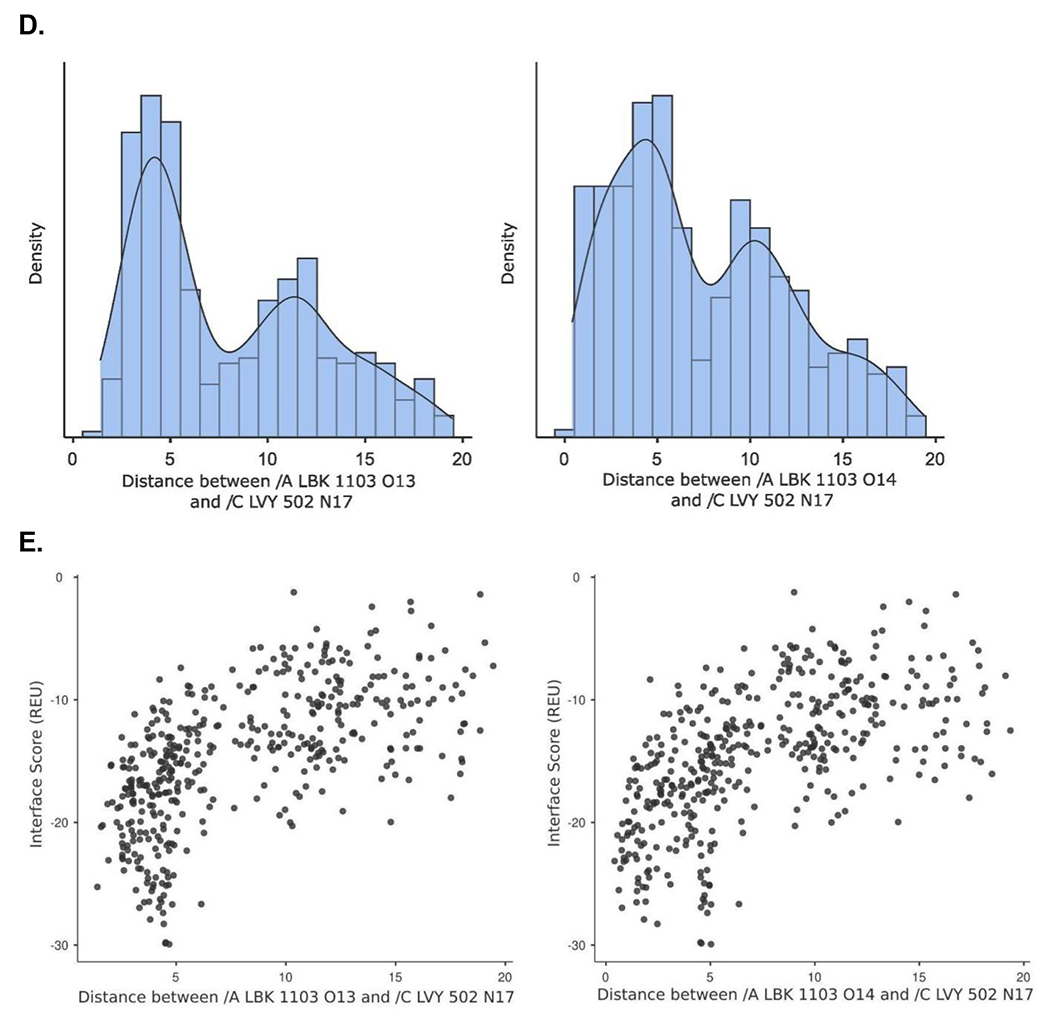
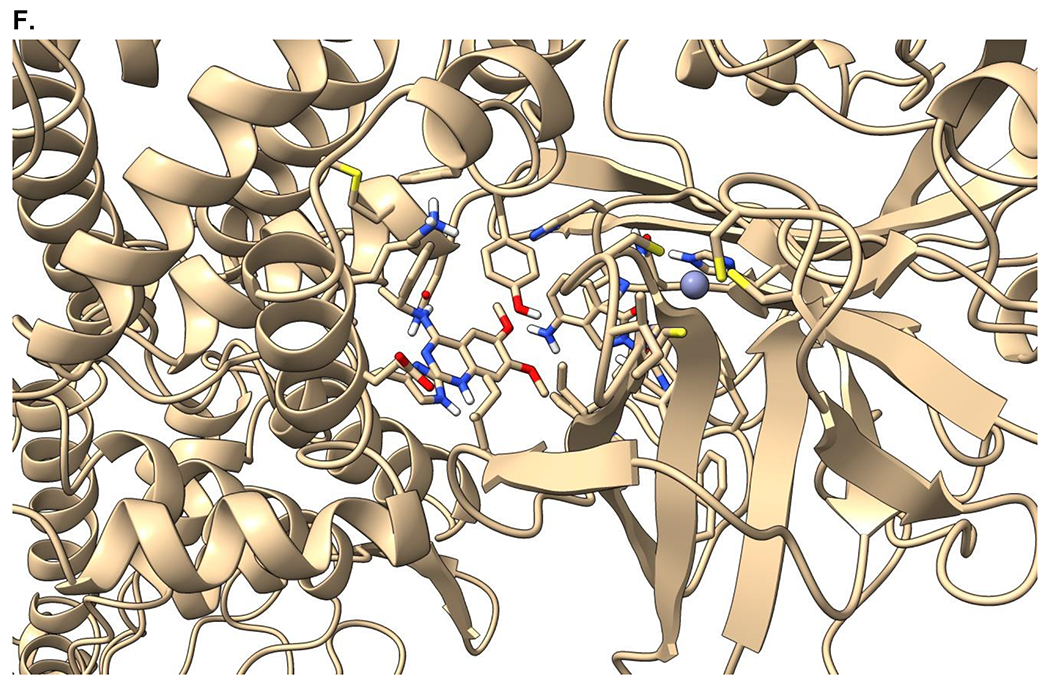
Crystal structure informed design of SOS1 PROTAC degraders. (A) A general scheme for the design of SOS1 degraders based on the PROTAC concept utilizing SOS1 binders and common E3 ligase binders. Atoms and groups in red are essential to retain the activity of SOS1 inhibition in BAY293. Groups circled in green are putative solvent exposure sites when the corresponding molecules are in complex with SOS1. (B) Crystal structure of SOS1 (PDB: 6SFR) in a complex with SOS1 binder BI68BS. (C) Structural interaction fingerprint analysis of BI68BS in a complex with SOS1 by Schrödinger-2020-3. (D) The distribution of the distance (Å) between /A LBK 1103 O13 (BI68BS 6-methoxy group) and /C LVY 502 N17 (lenalidomide amino group) and the distance (Å) between /A LBK 1103 O14 (BI68BS 7-methoxy group) and /C LVY 502 N17 in models from protein-protein docking of cereblon E3 ligase (PDB: 4TZ4) to SOS1 (PDB: 6SFR). (E) The relationship between interface score (Rosetta energy units) and the distance between /A LBK 1103 O13 and /C LVY 502 N17 and the distance between /A LBK 1103 O14 and /C LVY 502 N17 in models from the protein-protein docking. (F) An example of the most favorable conformations from the protein-protein docking. In this case, distance between /A LBK 1103 O13 and /C LVY 502 N17: 4.0 Å. Distance between /A LBK 1103 O14 and /C LVY 502 N17: 4.7 Å.
We then performed protein-protein docking of BI68BS-bound SOS1 with lenalidomide-bound cereblon to determine the optimal length and conformation of the linkers of SOS1 PROTAC degraders. In silico modeling of the interactions between proteins of interest and E3 ligases, especially cereblon favored conformations of ternary complexes, which are putative intermediates of targeted protein degradation induced by PROTAC.26 A protein-protein docking experiment27 provided 500 ranked conformation models of the BI68BS-bound SOS1 (PDB: 6SFR) and the lenalidomide-bound cereblon (PDB: 4TZ4). Among the models having the distance between BI68BS (6- or 7-methoxy group) and lenalidomide (amino group) within 20 Å and total energy less than −800 Rosetta energy units (REU), we found that the distances between lenalidomide amino group and BI68BS methoxy groups had bimodal distribution with a median of 6.2 Å for BI68BS 6-methoxy group and 6.1 Å for BI68BS 7-methoxy group (Figure 1D). Interface scores, which represent the energy of the interactions across the protein-protein interface, were also estimated from the docking experiments. The interface scores were lowest when plotted against the distances between BI68BS methoxy groups and the lenalidomide amino group, with favorable conformations with such distances at approximately 4-5 Å. (Figure 1E). In an example of the most favorable models (Figure 1F), BI68BS, as expected, can have the 6- and 7-methoxy groups serve as the sites for linker attachment. For lenalidomide, although the oxo-isoindolinyl sp2 carbons can serve as potential linker anchor points, the 4-amino group can certainly be utilized as a nucleophile for linker attachment.28 The distances between the potential linker attachment sites of BI68BS and lenalidomide could be as short as 4-5 Å, which prompted us to focus our initial syntheses of SOS1 degraders with relatively short linkers. In summary, our design of SOS1 degraders based on structural analysis and computational modeling has proven to be accurate in guiding the discovery of effective PROTAC degraders and identification of compounds with short linker for effective degradation activity and providing the potential advantage of rigid degrader and more defined ternary complex.
Chemical synthesis of SOS1 degrader
The design of our SOS1 PROTAC degrader was different from a recently reported agonist-based SOS1 degrader utilizing VHL E3 ligase.29 Our SOS1 degraders utilized cereblon as the E3 ligase binder and a SOS1 inhibitor as the SOS1 binder which could potentially have improved pharmacological properties and superior binding affinity to SOS1, respectively, compared to those used in the report by Zhu et al.29 Variations on the core structure in combination with different linkers and E3 ligase ligands led to 15 different SOS1 degraders (Figure 2). The synthesis of core structures is depicted in Scheme 1. Starting from commercially available methyl benzoate 2, iron mediated nitro group reduction afforded aniline 3 in 89% yield, which is subjected to acid catalyzed condensation with acetonitrile to create quinazolinone 4 in good yield. Tautomerization and sulfonate formation provided intermediate 5 and the subsequent SNAr reaction by addition of amines gave chiral 4-aminoquinazoline 8 and 9 in 86% and 76% yields, respectively. Hydrogenolysis released the free hydroxyl group, which was able to attach proper PROTAC linkers and E3 ligase ligands. Alternatively, quinazolinone 4 can be converted to chloride 13, which underwent SNAr to generate 15 for further modifications. To compare 2-methyl and 2-unsubstituted quinazolines on the degradation efficiency of SOS1 degraders, quinazolinone 10 was prepared by condensation of 3 with formamide in the presence of ammonium formate. One-pot SNAr was accomplished under the effect of phosphonitrilic chloride trimer. Core structure 11 and 12 were obtained in good to excellent yields. To evaluate the effect of linker at a different attachment point, compound 20 was prepared in analogous fashion. Commenced from 16 via sequential methylation, iodination, condensation, and chlorination, 18 was generated in overall good yield, which, upon SNAr substitution, proceeded to halogen-metal exchange and addition to carbonyl group to afford core structure 20.
Figure 2.
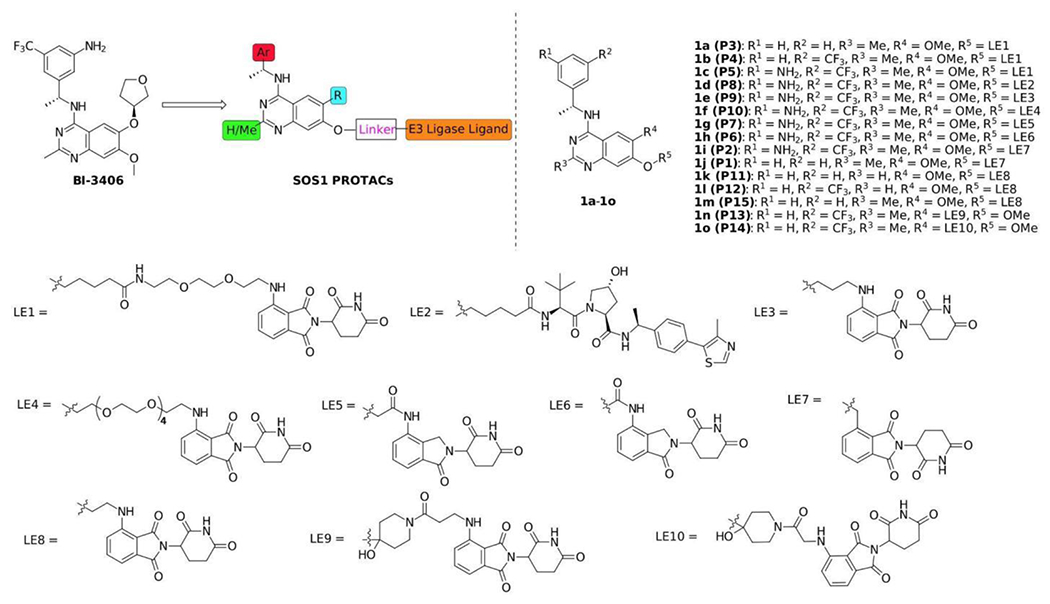
Chemical structures of synthesized SOS1 PROTAC degraders
Scheme 1.
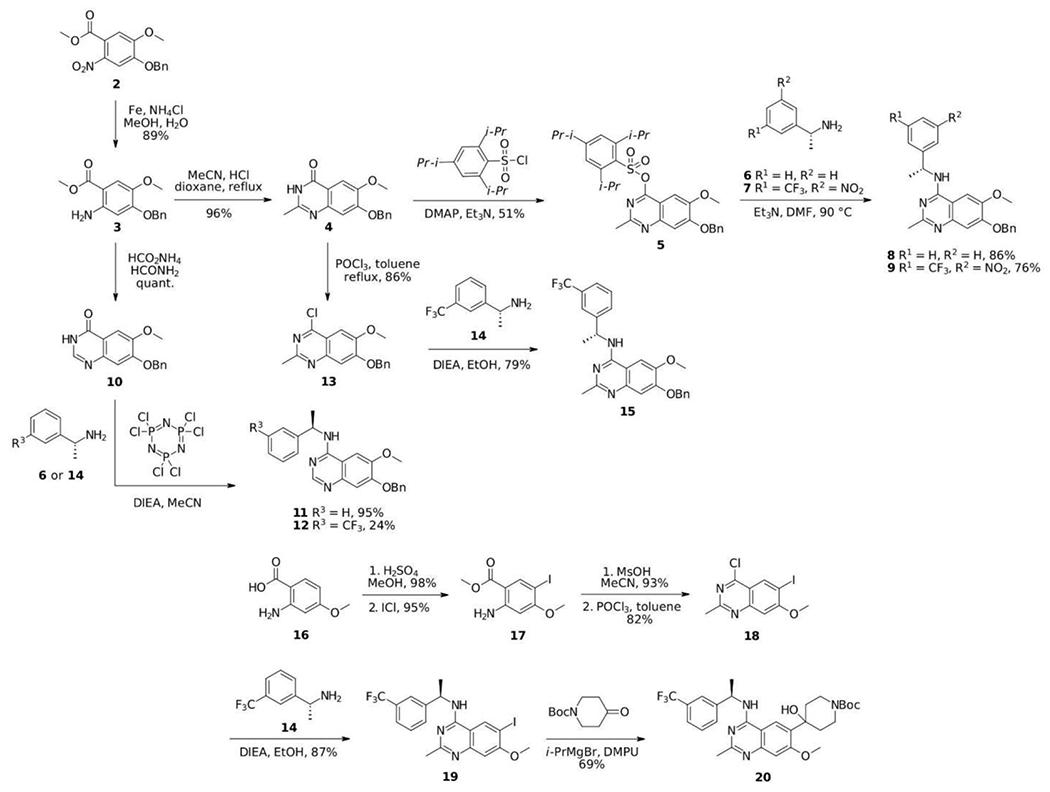
Synthesis of the core structures of SOS1 PROTAC degraders
With core compounds 8, 9, 11, 12, 15 and 20 in hands, the SOS1 degrader synthesis was performed according to Scheme 2 in a modular approach. Starting from compounds 8, 9 or 15, hydrogenation of the benzyl group followed by alkylation of tert-butyl bromocarboxylate furnished compounds 21-23, which were ready to couple E3 ligase ligands with an amino tether after acid treatment. Under the effects of carbodiimide and Oxymapure, SOS1 PROTACs 1a-1d were prepared in decent yields. Non-cleavable linkers such as 3-aminopropanol and 17-amino-3, 6, 9, 12, 15-pentaoxaheptadecan-1-ol were also used to create SOS1 PROTACs. As illustrated in Scheme 2, after hydrogenation of the benzyl group and alkylation of the corresponding linker tosylate with amino group protected with Boc, intermediates 24 and 25 were obtained in good yields, which were derivatized to 1e and 1f respectively upon successful SNAr reactions after acid treatment. SOS1 PROTACs 1g-1j and 1m bearing shorter linkers were synthesized by reacting compounds 9 or 28 with proper thalidomide or lenalidomide derivatives. Similarly, 1k and 1l were generated from the 2-unsubstituted 4-aminoquinazolines. The last two SOS1 PROTACs, 1n and 1o were prepared from 20 by reacting with thalidomide substituted carboxylic acids under standard amide coupling conditions. Thus, we adopted synthetic routes that can be easily adapted to the synthesis of multiple SOS1 PROTAC degrader candidates with full characterizations in Figure S1–S30. The flexibility in our synthetic approach allowed fast access to degraders with diverse linkerology enabling study of structural-activity relationships for SOS1 degradation.
Scheme 2.
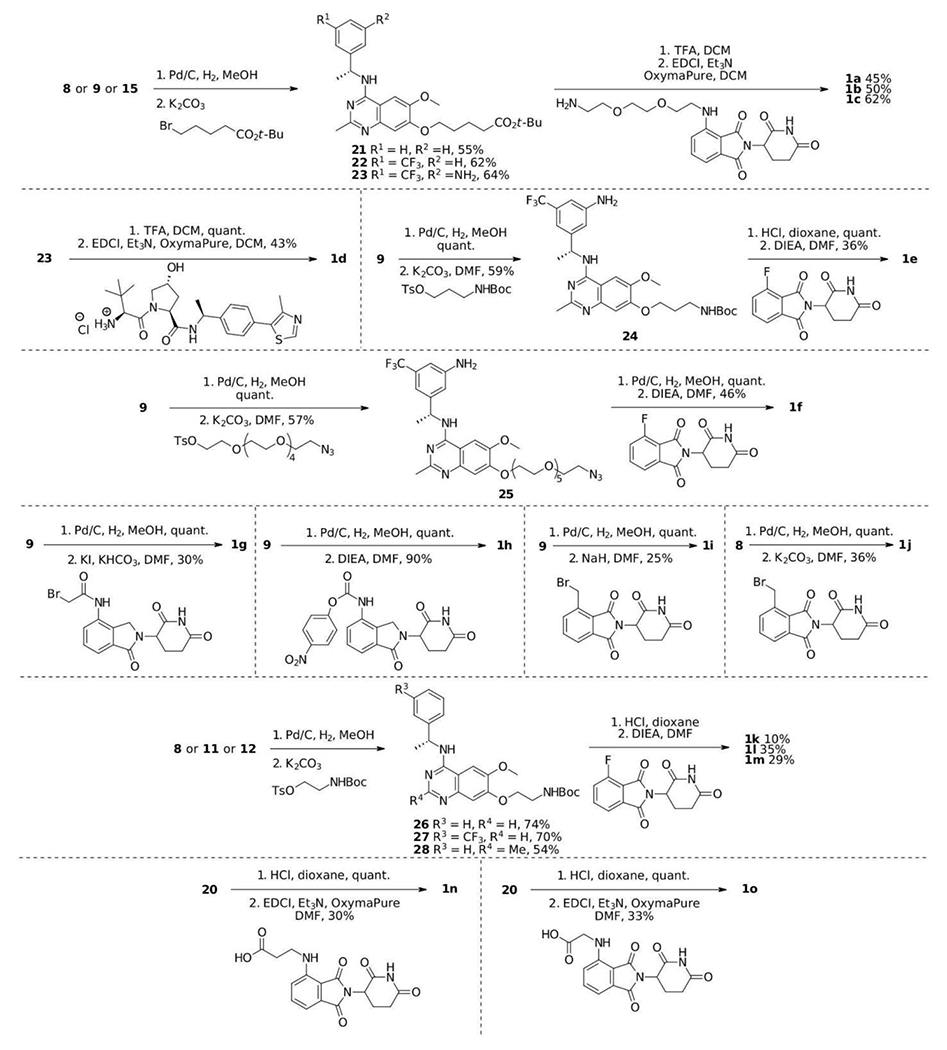
Synthesis of SOS1 PROTAC degraders.
Targeted SOS1 degradation in CRC
We next sought to screen the synthesized degrader candidates by immunoblotting. Treatment of CRC cell line SW620 for 6 hours with SOS1 degrader candidates showed that P7 among others at 1μM led to a 64% degradation of SOS1. The degree of SOS1 degradation by various SOS1 degrader candidates are summarized in Figure 3A and 3B. We observed that relatively short linker length, as suggested by our protein-protein docking analysis, provided high SOS1 degradation activity for compounds P7 and P11, and moderate activity for compounds P1 and P6. Thus, it suggested that structural variations of the SOS1 binder were important for SOS1 degradation, which warranted further structural-activity relationship studies. We then focused our subsequent evaluation on P7 given we did not synthesize P11 until much later in our study. As far as the difference in degradation activity between P7 and P11, we believed it could be due to their different degree of cellular uptake. We will likely directly compare P7 and P11 in further studies on structural-activity relationship and pharmacodynamics. P7 induced SOS1 degradation was compared to SOS1 inhibitor BI3406 and siRNA of SOS1, where BI3406 as expected did not induce SOS1 for degradation. The level of SOS1 degradation by P7 was comparable with that by genetic knockdown (Figure 4A). SOS1 degradation by P7 at 1μM was time-dependent. In contrast to some of the other protein PROTAC degraders,28 significant SOS1 degradation was observed at 24 hours or longer with 92% degradation at 48 hours. Such time-dependent SOS1 degradation was not observed in inactive compound P2 (Figure 4B). To investigate the effect of protein resynthesis on the relatively delayed SOS1 degradation by P7, we pretreated SW620 cells with protein synthesis inhibitor cycloheximide prior to the addition of P7, more than 75% SOS1 degradation was achieved starting at 3 hours and sustained for longer time period (Figure 4C). The delayed onset of SOS1 degradation by P7 could also be the result of compromised cellular uptake of P7 or increased drug efflux via p-glycoprotein. Some of the CRC cell lines are known to have high p-glycoprotein expression and P7 could be a p-glycoprotein substrate. This may be true as we did not see high remaining SOS1 protein levels upon treatment with P7. All these factors need further investigation in the future preclinical and clinical evaluations of SOS1 degraders. The mode of action of P7 for targeted SOS1 degradation was confirmed by the lack of significant SOS1 degradation by P7 after pretreatment with excessive amount of BI3406, lenalidomide, neddylation inhibitor MLN4924, and proteasome inhibitor MG132 (Figure 4D). Pre-treatment with high concentration of lenalidomide eliminated the ability of SOS1 degradation by P7 supported the need for cereblon binding for its degradation function. N-methylated lenalidomide would serve as an excellent control for P7 binding with cereblon in future studies. P7-induced SOS1 degradation was indeed mediated by the engagement of SOS1 and the ubiquitin-proteasome system. In addition to SW620, P7 also induced SOS1 degradation in other CRC cell lines HCT116, C2BB, and SW1417 in a concentration-dependent manner (Figure 4E). The half maximal degradation concentration (DC50) for SW620, HCT116, and SW1417 at 24 hours was 0.59μM (95%CI 0.23–1.42μM), 0.75μM (95%CI 0.27–1.98μM), and 0.19μM (95%CI 0.043–1.16μM), respectively. The maximal SOS1 degradation at 10μM for 24 hours was 87% (95%CI 71–100%), 76% (95%CI 61–94%), and 83% (95%CI 66–100%), respectively. These parameters for C2BB cells cannot be provided by nonlinear curve fitting but can be visualized in Figure 4E. We noticed differential degree of SOS1 degradation by P7 in different CRC cells, which could be attributed to many factors such as intrinsic resistance mechanism due to mutations in the ubiquitin-proteasome system or efficiency of cellular uptake among others. Further assessment on this phenomenon will likely inform discovery of predictive biomarkers to the sensitivity of SOS1 degrader in future preclinical and clinical studies.
Figure 3.
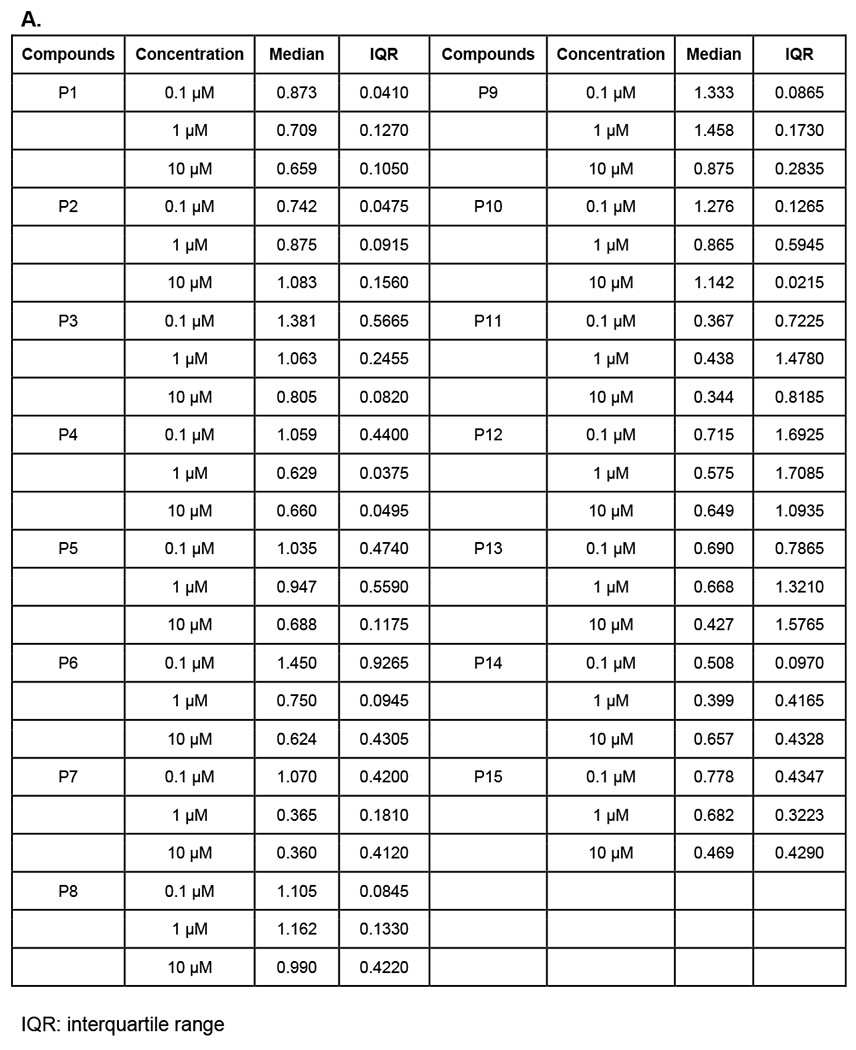
Screening of SOS1 PROTAC candidates for SOS1 degradation in SW620 after treatment for 6 hours. (A) SW620 cells were treated with the corresponding compounds for 6 hours followed by immunoblotting. SOS1 levels were normalized to that treated by DMSO as control with 1.0 as the absence of SOS1 degradation. (B) A summary of SOS1 degradation after treatment with the corresponding compounds for 6 hours in SW620 cells. The quantities of SOS1 were summarized as mean and standard deviation.
Figure 4.
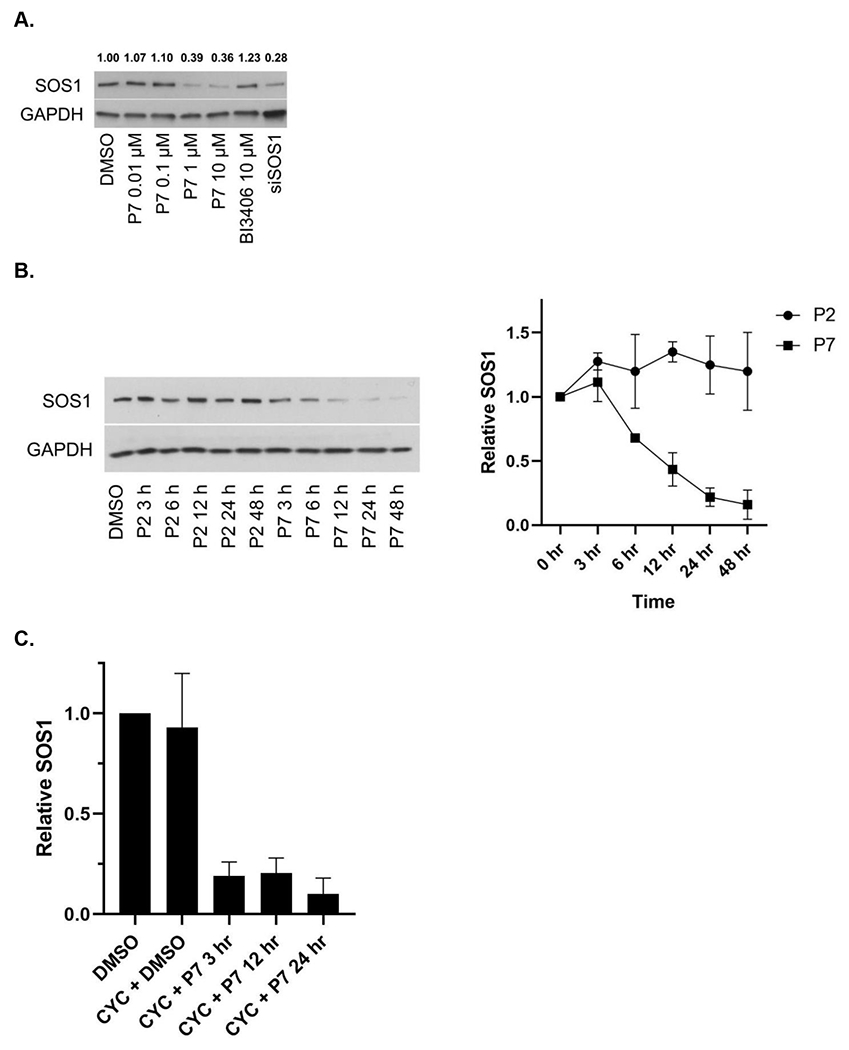
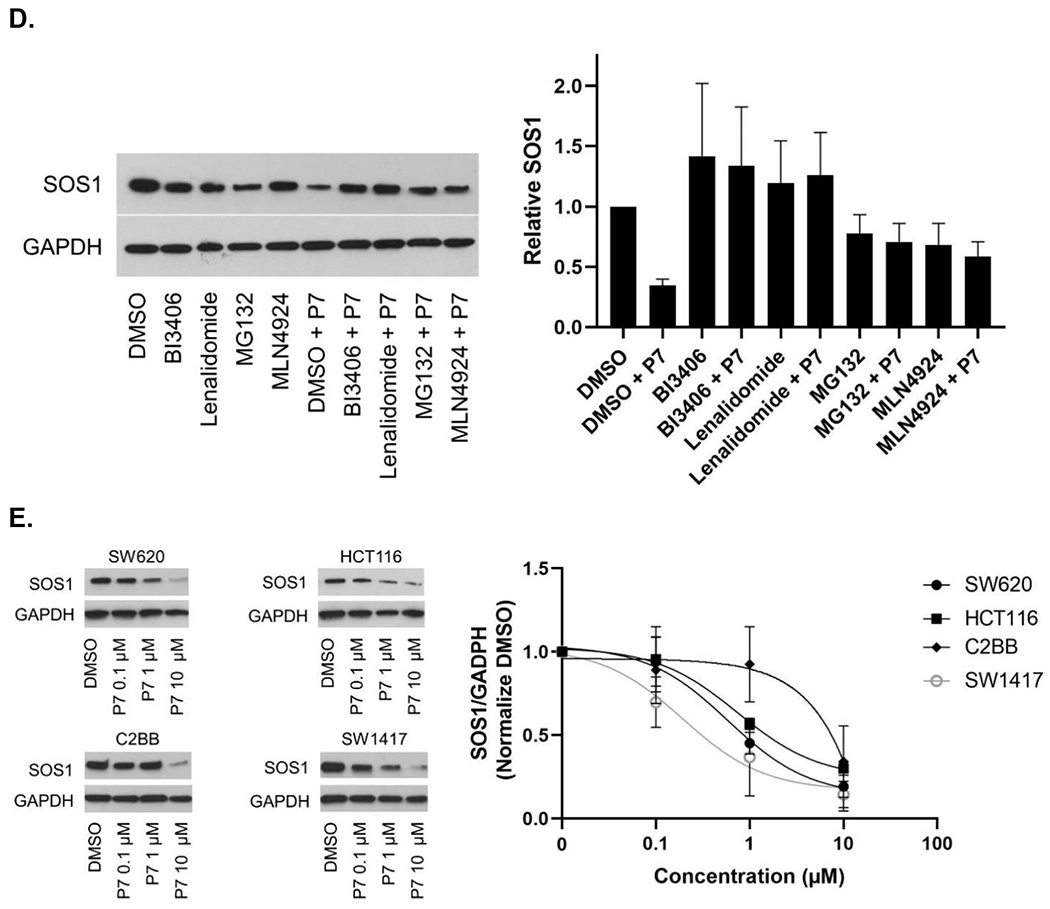
SOS1 degradation in colorectal cancer cell lines. (A) Immunoblots in SW620 cells treated with P7 for 6 hours in comparison to SOS1 inhibitor BI3406 and siSOS1. (B) Immunoblots in SW620 cells treated with SOS1 degrader P7 and inactive compound P2, with GAPDH used as the loading control at different time points; Cells were treated with 1 μM of P7 and P2 for indicated times, respectively. (C) SW620 cells were pre-treated with cycloheximide (CYC) for 1 hour at 100 μg/mL, followed by addition of DMSO or 1 μM P7. At the indicated times, cells were lysed and SOS1 levels were analyzed by western blot. (D) Mechanistic investigation of SOS1 degradation induced by P7 in SW620 cells. Cells were pretreated with BI3406 10 μM, lenalidomide 10 μM, MLN4924 0.5 μM, and MG132 3 μM for 1 hour, followed by a 24 h treatment with P7 at 1 μM. (E) SOS1 degradation in colorectal cancer cells treated with SOS1 degrader P7 at different concentrations for 24 hours.
We then assessed the specificity of SOS1 degradation by 1μM P7 at 24 hours by global proteomics analysis in SW620 cells. As shown in Figure 5, SOS1 was among the most degraded proteins without cereblon-binding lenalidomide induced degradation of GSPT130 and the zinc finger transcription factors such as IKZF1/3.31 We also found that several proteins are significantly upregulated after 24-hour treatment with P7. Both ABCG1 and SCAP were reported to be involved in cellular cholesterol homeostasis,32,33 which may be important in the survival of cancer cells with upregulated EGFR-RAS signaling.32 Although it has been reported that MEK1/2 inhibitors promoted protein degradation of ABCG1 in CHO and HuH7 cells,34 upregulation of ABCG1 upon P7-induced SOS1 degradation may serve as a potential resistance mechanism and target in CRC cells. Lack of known direct interaction between SOS1 and ABCG1 or SCAP suggested that their upregulation is a result of altered signaling changes and transcriptional effects due to targeted SOS1 degradation. Studies such as transcriptomic and phosphoproteomics studies could be proposed to evaluate these cellular adaptation mechanisms in the setting of acute SOS1 degradation. In summary, we identified P7 as the most effective compound for SOS1 degradation in multiple CRC cell lines. The effect of P7 was mediated by the ubiquitin-proteasome system. Our proteomics analysis not only showed that P7 is highly specific for SOS1 degradation, but also revealed upregulation of proteins that may contribute to cellular adaptation to SOS1 degradation.
Figure 5.
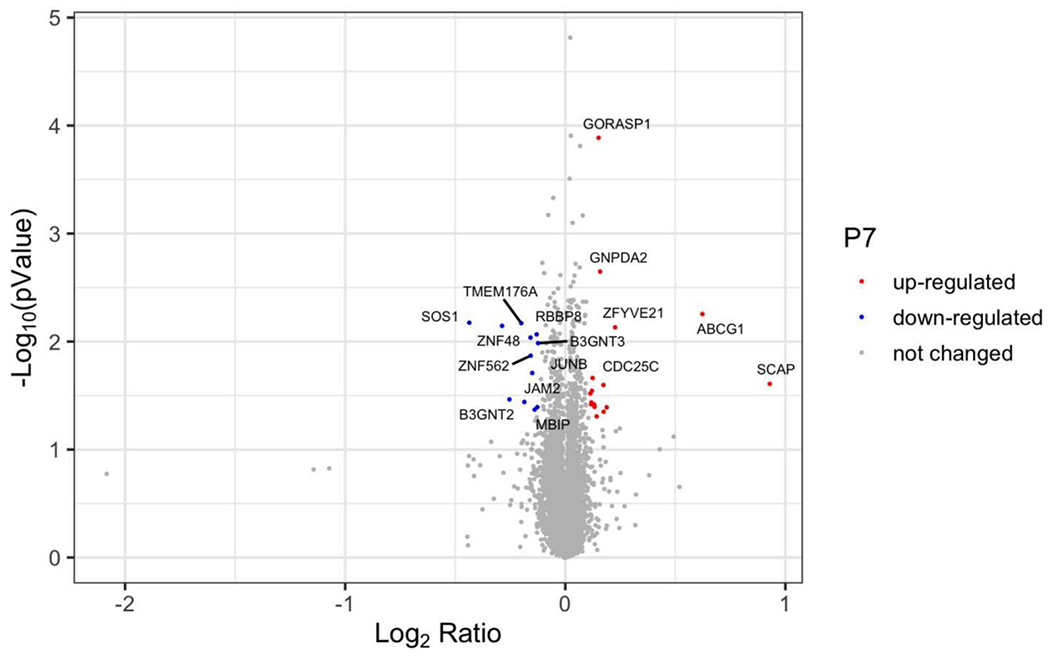
Specificity of P7 for SOS1 degradation. Volcano plot depicts the change in relative protein abundance in P7 (1 μM, 24 h)-treated SW620 cells compared with DMSO-treated cells. Protein abundance measurements were made using tandem mass spectrometry and the changes were assessed by Welch t test. The log2 (fold change) was represented on the x-axis. The negative log10 p value was represented on the y-axis. Three independent biological replicates were performed for each treatment.
Effect of SOS1 degradation in CRC biology
In our experiments, we have mainly used CRC cell lines to evaluate P7 degradation capacity (Figure 4E). To define the translational value of the SOS1 degrader P7, we evaluated its biological activity in CRC PDOs as these models closely represent the disease.35 PDOs are 3D tumor models resembling key molecular and biological features of the tumor origin and have better performance at predicting response to chemotherapy and radiation in CRC among many other cancer types compared to cell lines or xenograft models.36–38 For these reasons, we made an effort to adopt CRC PDOs early in the discovery process of our SOS1 degraders for the assessment of their activity instead of cell lines (Figure S31). To this end, CRC PDOs carrying KRAS G12A mutation (MCC19990-002, MCC19990-010, and MCC19990-013), and KRAS G12C mutation (MCC19990-006), were treated with 1μM P7 for 24 hours and immunohistochemically stained for SOS1 expression. As shown in Figure 6A, compared to the DMSO vehicle control, treatment with P7 led to significant SOS1 degradation at varying degrees in different cellular compartments of these CRC PDOs. In MCC19990-010, SOS1 degradation induced by 1μM P7 for 24 hours led to significant necrosis and loss of cellular structures compared to DMSO control (Figure 6B a, b) while in MCC19990-013, P7-induced a similar SOS1 degradation at 24 hours without sufficiently causing cell death but inducing structural alterations (Figure 6B c, d). Of note, we did not observe cytotoxicity of P7 in HPNE cells (normal human immortalized pancreas epithelial cells) as shown in Figure S32. P7-induced cellular effects were also supported by increased membranous expression of Annexin V in MCC19990-010 and MCC19990-013 after treatment with 1μM P7 for 24 hours as compared to DMSO control (Figure 6C). As shown in Figure 6D, MCC19990-002 was resistant to both P7 and BI3406. P7 had superior activity in MCC19990-006, MCC19990-010, and MCC19990-013 CRC PDOs. In MCC19990-006, P7 had an IC50 of 1.4 μM; BI3406 had an IC50 of 8.5 μM. In MCC19990-010, P7 had an IC50 of 0.48 μM; BI3406 had an IC50 of 1.9 μM. In MCC19990-013, P7 had an IC50 of 1.16μM; BI3406 had an IC50 of 6.7 μM. Finally, we measured phosphorylation of AKT (pAKT) and ERK (pERK) levels after treatment with P7 and BI3406 for 24 hours. We observed a more effective suppression of both pAKT and pERK by P7 compared to BI3406 (Figure S33). In addition to the evaluation of signaling activity, we also examined a potential cellular adaptation mechanism to SOS1 degradation by measuring SOS1 and SOS2 mRNA expression levels after treating SW620 with P7 at 0, 6, 24, 48 hours shown below in Figure S34. Both SOS1 and SOS2 mRNA expression increased upon treatment with P7. In summary, P7 demonstrated effective SOS1 degradation leading to morphologic alterations and apoptosis in ex vivo CRC PDOs. These cellular effects of P7-induced SOS1 degradation were different in CRC PDOs, which was largely due to tumor heterogeneity. Further, P7 had superior activity to SOS1 inhibitor BI3406 in SOS1 inhibition sensitive KRAS-mutant CRC PDOs.
Figure 6.
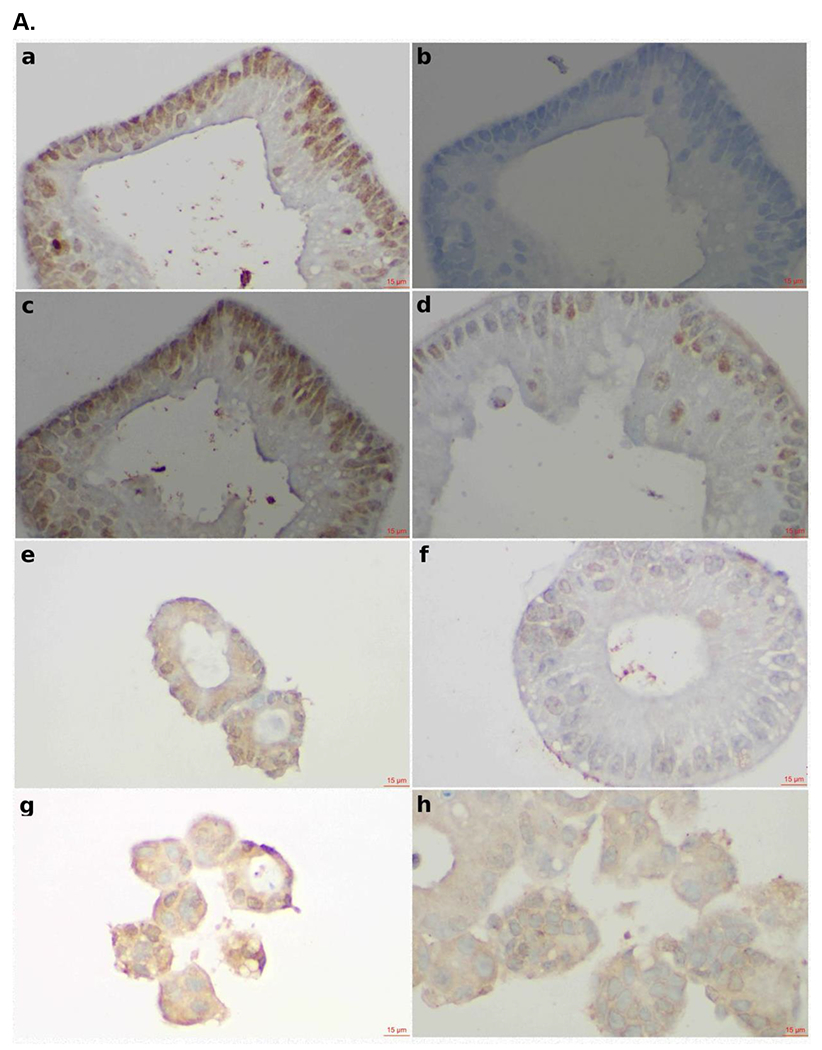
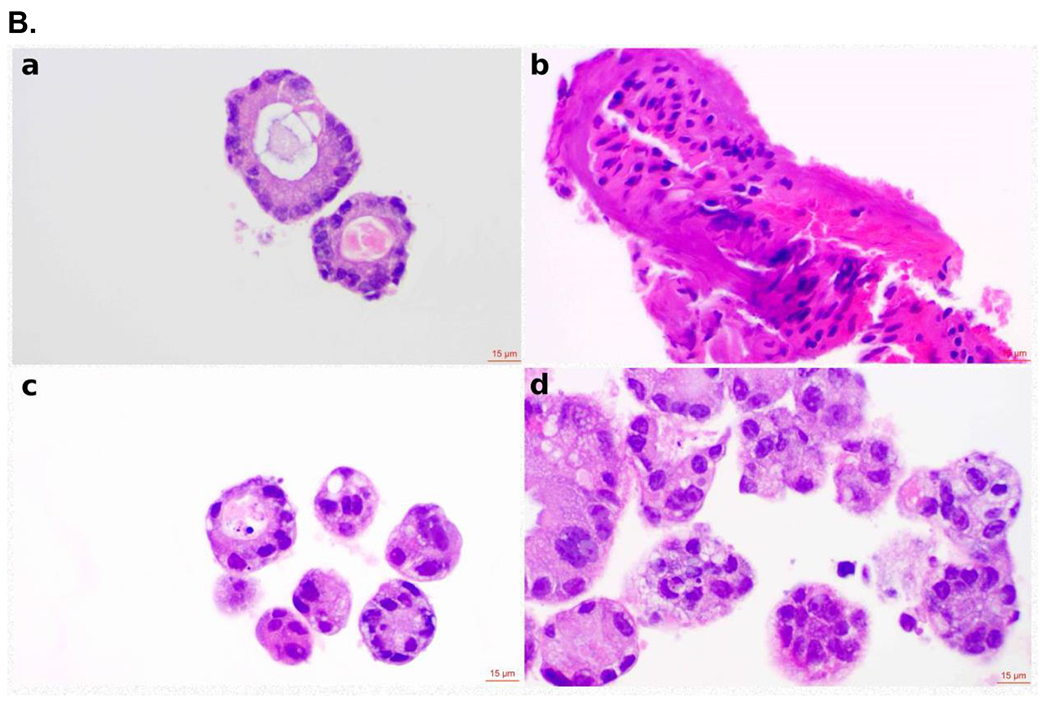
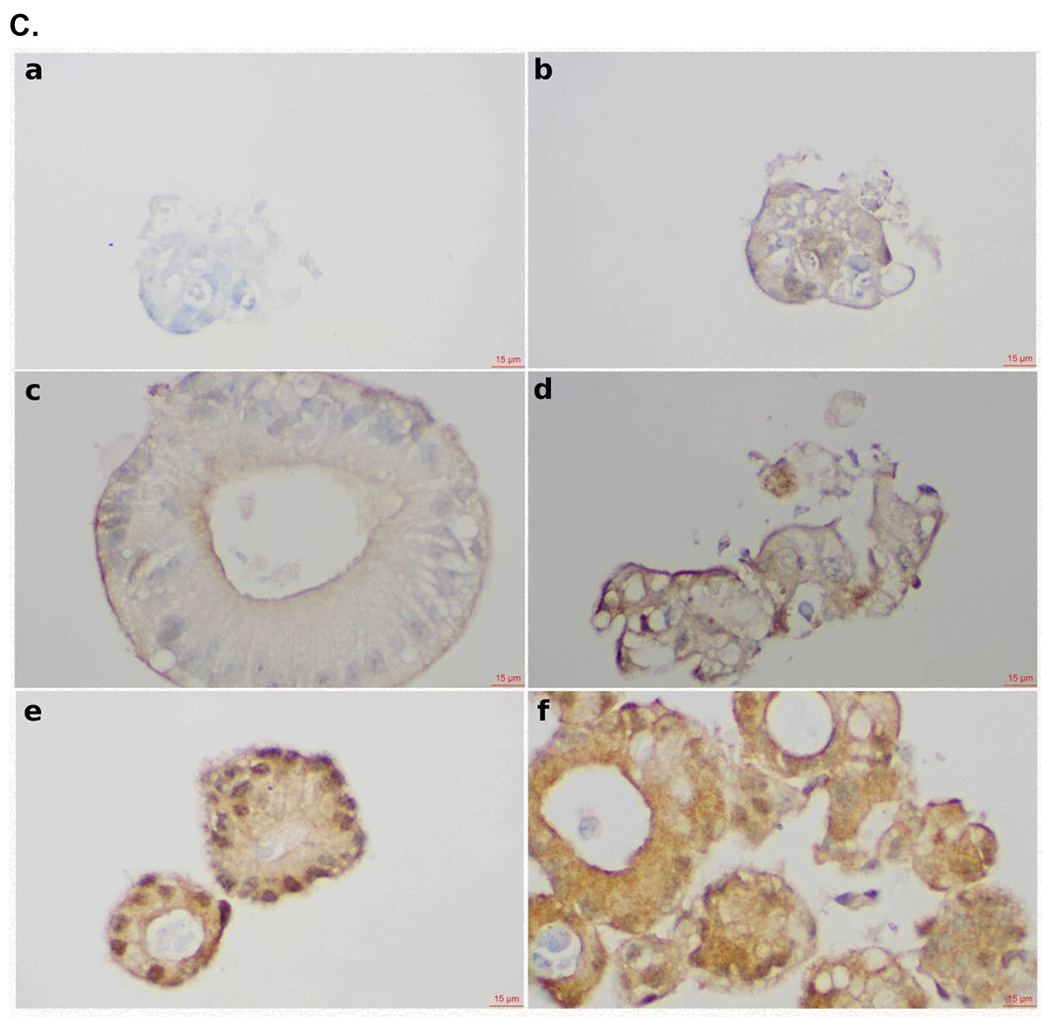
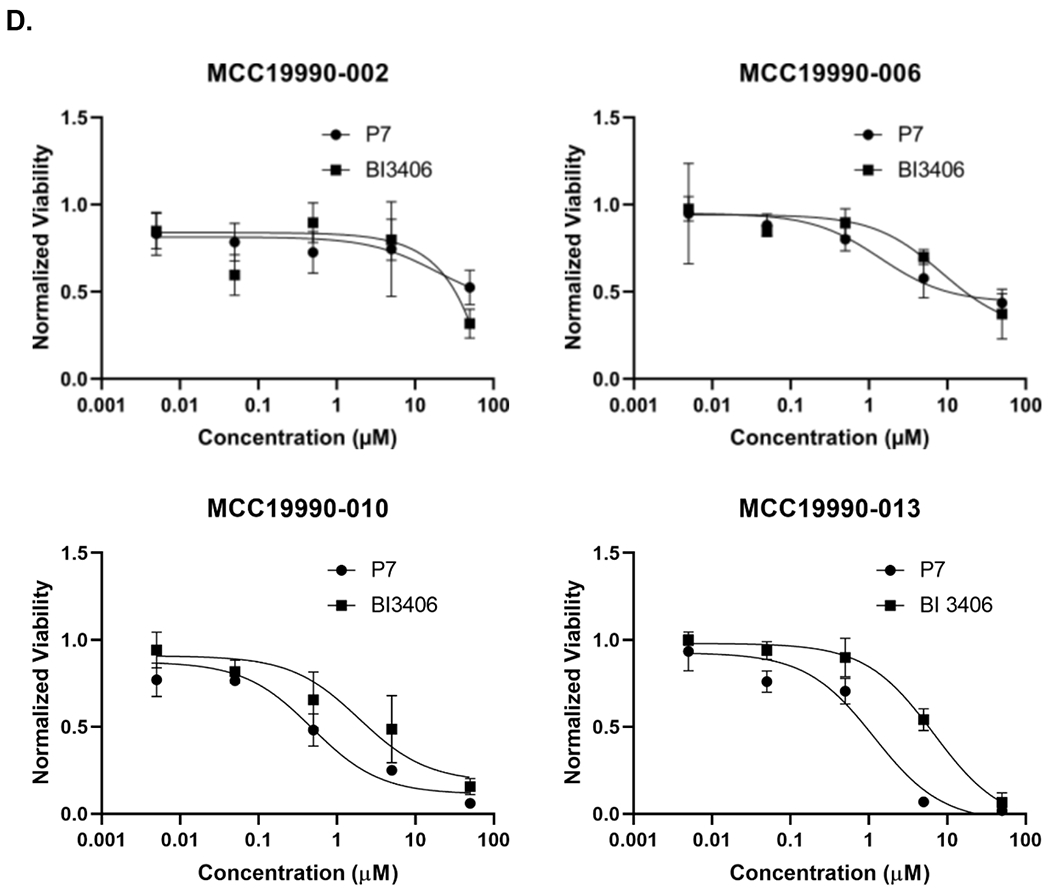
Effect of SOS1 degraders in colorectal cell lines and patient-derived organoid models. (A) SOS1 expression by IHC in colorectal cancer patient-derived organoid models. a. Positive control for SOS1 expression at 40x; b. Negative control for SOS1 expression at 40x. The negative controls used for the SOS1 IHC studies was the non-immune isotype-specific immunoglobulins; c. SOS1 expression of MCC19990-002 PDO after treatment with 0.2% DMSO for 24 hours at 40x; d. SOS1 expression of MCC19990-002 PDO after treatment with 1μM P7 for 24 hours at 40x; e. SOS1 expression of MCC19990-010 PDO after treatment with 0.2% DMSO for 24 hours at 40x; f. SOS1 expression of MCC19990-010 PDO after treatment with 1μM P7 for 24 hours at 40x; g. SOS1 expression of MCC19990-013 PDO after treatment with 0.2% DMSO for 24 hours at 40x; h. SOS1 expression of MCC19990-013 PDO after treatment with 1μM P7 for 24 hours at 40x. (B) H&E stain to assess morphology of colorectal cancer patient-derived organoid models. a. MCC19990-010 PDO after treatment with 0.2% DMSO for 24 hours at 40x; b. MCC19990-010 PDO after treatment with 1μM P7 for 24 hours at 40x; c. MCC19990-013 PDO after treatment with 0.2% DMSO for 24 hours at 40x; d. MCC19990-013 PDO after treatment with 1μM P7 for 24 hours at 40x. (C) Annexin V expression by IHC in colorectal cancer patient-derived organoid models. a. Negative control for Annexin V expression at 40x; b. Positive control (treated with FOLFIRI) for Annexin V expression at 40x; c. Annexin V expression of MCC19990-010 PDO after treatment with 0.2% DMSO for 24 hours at 40x; d. Annexin V expression of MCC19990-010 PDO after treatment with 1μM P7 for 24 hours at 40x; e. Annexin V expression of MCC19990-013 PDO after treatment with 0.2% DMSO for 24 hours at 40x; f. Annexin V expression of MCC19990-013 PDO after treatment with 1μM P7 for 24 hours at 40x. (D) Viability of patient-derived CRC organoid models after treatment with P7 compared to BI3406 at various concentrations for 72 hours. MCC19990-002 was resistant to both compounds. For MCC19990-006, P7 has IC50 1.4μM; BI3406 has IC50 8.5μM. For MCC19990-010, P7 has IC50 0.48μM; BI3406 has IC50 1.9μM. For MCC19990-013, P7 has IC50 1.16μM; BI3406 has IC50 6.7μM.
CONCLUSIONS
In this study, we reported the design and synthesis of a SOS1 degrader P7 which effectively targeted SOS1 for degradation through the ubiquitin-proteasome system. SOS1 degradation induced by P7 was time and concentration dependent with high specificity according to global proteomics analysis. P7 induced SOS1 degradation in multiple CRC cell lines and patient-derived PDO models. P7 also demonstrated superior cytotoxic activity compared to BI3406 in SOS1 inhibitor sensitive CRC PDO models. Compared to previously reported SOS1-agonist based degrader,29 although our degrader is slightly less potent and only underwent less rigorous in silico simulation of ternary complex formation, yet it has several advantages by using 1) a SOS1 inhibitor as warhead instead of SOS1 agonist. The former provides higher affinity and selectivity to SOS1 and avoids the possibility of SOS1 activation by an agonist; 2) a different E3 ligase cereblon instead of VHL. The former provides the advantage of possibly improved pharmacological properties such as oral availability; and 3) a different linkerology in terms of a very short linker that was designed by protein-protein docking, providing the advantage of rigid degrader and potentially more defined ternary complex. This has also helped in the synthesis of candidate degraders with short linkers. Together, our findings support further development of SOS1 degrader as a new class of agents targeting KRAS-driven CRC and as a tool to evaluate its activity either alone or in combination and to reveal cellular adaptation mechanisms to degraders in CRC.
EXPERIMENTAL SECTION
Chemistry
General Information. All chemicals were obtained from commercial suppliers and used as purchased without further purification. Reactions were monitored by LC/MS and thin layer chromatography (TLC). TLC was performed using SiliCycle Inc. silica plates, using short-wave UV light (254 nm, UVP, LLC) for visualization. Nuclear magnetic resonance (NMR) spectra were recorded on Bruker 400 and 500 MHz instruments and were calibrated using deuterated solvent (CDCl3: 1H NMR: 7.26 ppm, 13C NMR: 77.16 ppm, DMSO-d6: 1H NMR: 2.50 ppm, 13C NMR: 39.52 ppm, CD3OD: 1H NMR: 3.31 ppm, 13C NMR: 49.00 ppm). Data are reported as follows: chemical shift (δ), multiplicity, integrated intensity, and coupling constant (J) in hertz. Column chromatography was performed with silica gel (230–400 mesh) on the Yamazen AI580S EPCLC automated system. High performance liquid chromatography (HPLC) grade acetonitrile/water was obtained from Fisher Scientific International, Inc. High-resolution mass spectroscopy (HRMS) traces were obtained on an Agilent 6230 TOF/LC/MS. HPLC analysis was performed on an Agilent 1260 system using a ZORBAX C18 column (150 × 4.6 mm, 5 μm) at room temperature with a gradient elution using the mobile phase (A) nanopure water containing 0.1% formic acid and (B) acetonitrile containing 0.1% formic acid. All compounds used for biological evaluation have a purity of ≥ 95%.
Methyl 2-amino-4-(benzyloxy)-5-methoxybenzoate (3).
A mixture of 2 (3.0 g, 9.5 mmol), Fe (4.3 g, 76 mmol), NH4Cl (4.3 g, 80 mmol) in CH3OH/H2O (45.0 mL/22.5 mL) was stirred at 105 °C for 4 h. The mixture was filtered through celite and washed with EtOAc. The organic layer was washed with brine, dried over Na2SO4 and concentrated to afford the desired product 3 (2.4 g, 89% yield) as a brown solid. 1H NMR (400 MHz, CDCl3): δ 7.43 (dd, J = 8.1, 1.5 Hz, 2H), 7.40-7.36 (m, 2H), 7.36 (s, 1H), 7.34-7.30 (m, 1H), 6.40 (s, 1H), 5.15 (s, 2H), 3.86 (s, 3H), 3.84 (s, 3H).
6-Methoxy-2-methyl-7-(phenylmethoxy)-4(3H)-quinazolinone (4).
A mixture of 3 (2.3 g, 8.0 mmol), acetonitrile (21 mL), 4 M HCl in dioxane (42 mL) was refluxed for 15 h. The mixture was cooled and poured carefully into a cold NaHCO3(sat.) solution. The precipitate formed and was collected by filtration, washed extensively with water and air-dried to afford the desired product 4 (2.3 g, 96% yield) as a white solid. 1H NMR (400 MHz, CDCl3): δ 10.88 (s, 1H), 7.60 (s, 1H), 7.46 (d, J = 7.1 Hz, 2H), 7.39 (t, J = 7.3 Hz, 2H), 7.32 (t, J = 7.2 Hz, 1H), 7.12 (s, 1H), 5.27 (s, 2H), 4.01 (s, 3H), 2.52 (s, 3H).
7-(benzyloxy)-6-methoxy-2-methylquinazolin-4-yl 2,4,6-triisopropylbenzenesulfonate (5).
A mixture of 4 (748 mg, 2.53 mmol), 2,4,6-triisopropylbenzenesulfonyl chloride (918 mg, 3.03 mmol), DMAP (30.9 mg, 0.25 mmol) and Et3N (772 mg, 7.58 mmol) was suspended in DCM (35 mL). The reaction mixture was stirred at room temperature for 2 days. The reaction was diluted with DCM, extracted with NaHCO3 (sat.). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash chromatography using hexane/EtOAc to afford the desired product 5 (532 mg, 51% yield), recovered 4 200 mg, as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.46 (d, J = 7.0 Hz, 2H), 7.41-7.35 (m, 2H), 7.35-7.29 (m, 2H), 7.25 (s, 1H), 7.20 (s, 2H), 5.27 (s, 2H), 4.30-4.37 (m, 2H), 4.01 (s, 3H), 2.88-2.95 (m, 1H), 2.49 (s, 3H), 1.25 (d, J = 6.7 Hz, 18H).
General Procedure for Synthesis of Compounds 8 and 9. A mixture of 5 (1 equiv.), 6 or 716 (1.3 equiv.) and Et3N in DMSO was stirred at 90 °C for 12 h. The mixture was cooled to room temperature. The residue dissolved in DCM and extracted with water. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash chromatography to afford the desired product as a yellow solid.
(R)-7-(benzyloxy)-6-methoxy-2-methyl-N-(1-phenylethyl)quinazolin-4-amine (8).
1H NMR (400 MHz, CDCl3): δ 7.45 (d, J = 7.5 Hz, 2H), 7.41 (d, J = 7.8 Hz, 2H), 7.33 (dd, J = 5.7, 2.6 Hz, 4H), 7.28 (dd, J = 2.5, 1.4 Hz, 1H), 7.19 (s, 1H), 6.97 (s, 1H), 5.68-5.75 (m, 1H), 5.20 (s, 2H), 3.90 (s, 3H), 2.56 (s, 3H), 1.67 (d, J = 6.8 Hz, 3H).
(R)-7-(benzyloxy)-6-methoxy-2-methyl-N-(1-(3-nitro-5-(trifluoromethyl)phenyl)ethyl)q--uinazolin-4-amine (9).
1H NMR (400 MHz, CDCl3): δ 8.52 (s, 1H), 8.34 (s, 1H), 8.12 (s, 1H), 7.40 (d, J = 7.4 Hz, 2H), 7.28-7.35 (m, 3H), 7.20 (s, 1H), 7.09 (s, 1H), 5.61-5.68 (m, 1H), 5.20 (s, 2H), 4.00 (s, 3H), 2.48 (s, 3H), 1.76 (d, J = 6.5 Hz, 3H).
6-methoxy-7-benzyloxyquinazolin-4-ol (10).
A mixture of 3 (431 mg, 1.50 mmol), ammonium formate (85.1mg, 1.35 mmol) and formamide (3.6 mL) was stirred at 185 °C (oil bath temperature) for 1 h. The mixture was cooled to room temperature. The resulting precipitate was isolated, washed with water and dried to afford the desired product 10 (381 mg, quant.) as a brown solid. 1H NMR (400 MHz, CDCl3): δ 7.50-7.44 (m, 2H), 7.43-7.31 (m, 4H), 7.29 (s, 1H), 5.30 (s, 2H), 4.04 (s, 3H), 2.77 (s, 3H).
General Procedure for Synthesis of Compounds 11 and 12. A mixture of 10 (1 equiv.), phosphonitrilic chloride trimer (1 equiv.), DIPEA (5 equiv.) in MeCN were added to a nitrogen purged pressure tube. The reaction mixture was refluxed for 20 h as an activation time. The reaction was monitored by TLC. Then 6 or 1416 (2 equiv.) was added and the reaction mixture was refluxed for 16 h. After the mixture was concentrated under reduced pressure, the residue was purified by flash chromatography to afford the desired product as a yellow solid.
(R)-7-(benzyloxy)-6-methoxy-N-(1-phenylethyl)quinazolin-4-amine (11).
1H NMR (400 MHz, CDCl3): δ 7.55 (d, J = 7.6 Hz, 2H), 7.45 (d, J = 6.6 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.35-7.29 (m, 6H), 7.18 (s, 1H), 5.60-5,67 (m, 1H), 5.17 (s, 2H), 4.05 (s, 3H), 1.76 (d, J = 6.9 Hz, 3H).
(R)-7-(benzyloxy)-6-methoxy-N-(1-(3-(trifluoromethyl)phenyl)ethyl)quinazolin-4-amine (12).
1H NMR (400 MHz, CDCl3): δ 7.72 (s, 2H), 7.53 (d, J = 7.8 Hz, 1H), 7.47 (d, J = 8.2 Hz, 2H), 7.43 (d, J = 7.9 Hz, 2H), 7.38 (t, J = 7.5 Hz, 2H), 7.33-7.31 (m, 2H), 7.18 (s, 1H), 5.65-5.72 (m, 1H), 4.05 (s, 3H), 1.79 (d, J = 6.9 Hz, 3H).
7-(benzyloxy)-4-chloro-6-methoxy-2-methylquinazoline (13).
A mixture of 4 (200 mg, 0.67 mmol), POCl3 (1.03 g, 6.75 mmol) in toluene (1 mL) was refluxed for 16 h. The reaction mixture was cooled to room temperature and azeotroped with toluene. The mixture was dissolved in DCM and added with NaHCO3 (sat.) until a basic pH reached. The organic phase was collected by filtration, dried over Na2SO4 and concentrated to afford the desired product 13 (183 mg, 86% yield) as an orange solid. 1H NMR (400 MHz, CDCl3): δ 7.48 (d, J = 7.4 Hz, 2H), 7.37 (dd, J = 14.5, 8.6 Hz, 5H), 5.31 (s, 2H), 4.05 (s, 3H), 2.79 (s, 3H).
(R)-7-(benzyloxy)-6-methoxy-2-methyl-N-(1-(3-(trifluoromethyl)phenyl)ethyl)quinazolin-4-amine (15).
A mixture of 13 (183 mg, 0.58 mmol), DIPEA (150 mg, 1.16 mmol), 1416 (143 mg, 0.76 mmol) in EtOH (3 mL) was added to a nitrogen purged pressure tube and then was stirred at 100 °C for 16 h. The mixture was cooled to room temperature. After the mixture was concentrated under reduced pressure, the residue was purified by flash chromatography to afford the desired product 15 (214 mg, 79% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.72 (s, 1H), 7.66 (d, J = 7.7 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.44-7.37 (m, 3H), 7.35-7.27 (m, 3H), 7.24 (s, 1H), 7.17 (s, 1H), 5.66-5.73 (m, 1H), 5.18 (s, 2H), 3.95 (s, 3H), 2.53 (s, 3H), 1.71 (d, J = 6.9 Hz, 3H).
Methyl 2-amino-5-iodo-4-methoxybenzoate (17).
A mixture of 16 (10.0 g, 60 mmol), the concentrated sulfuric acid (18 mL) in methanol (100 mL) was refluxed for 22 h. The mixture was cooled to room temperature, quenched by NaHCO3 (sat.) to pH 6~7 and then extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, and concentrated to afford methyl 2-amino-4-methoxybenzoate (10.7 g, 98% yield) as a brown solid which was directly used for the next step without further purification. Methyl 2-amino-4-methoxybenzoate (725 mg, 4.00 mmol) was dissolved in EtOH (3.0 mL). Water (5.0 mL) and concentrated HCl (1.2 mL) was added and the mixture was cooled to 0 °C. A solution of iodine monochloride (714 mg, 4.40 mmol) in concentrated HCI (0.36 mL) was added dropwise and the reaction mixture was stirred at room temperature for 16 h. Then the mixture was quenched with water and the precipitate was filtered off. The crude product was washed with hexane to give the desired product 17 (1.14 g, 95% yield) as a brown solid. 1H NMR (400 MHz, CDCl3): δ 8.21 (s, 1H), 6.07 (s, 1H), 3.85 (s, 3H), 3.84 (s, 3H).
4-chloro-6-iodo-7-methoxy-2-methylquinazoline (18).
A mixture of 17 (307 mg, 1.00 mmol), methane sulfonic acid (0.52 mL) in CH3CN (2.6 mL) was added to a nitrogen purged pressure tube. The reaction mixture was refluxed for 18 h. After cooled to room temperature, the mixture was poured into NaHCO3 (sat.) solution and the precipitate filtered off to afford 6-iodo-7-methoxy-2- -methylquinazolin-4(3H)-one as a brown solid (295 mg, 93% yield) which was directly used for next step without further purification.
6-iodo-7-methoxy-2-methylquinazolin-4(3H)-one (722 mg, 2.28 mmol) in POCI3 (1.2 mL) and toluene (4.6 mL) was added Et3N (0.7 mL) dropwise. The mixture was stirred at 75 °C for 3 h. The reaction was quenched with ice water, washed with NaHCO3 (sat.) and extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography to afford the desired product 18 (627 mg, 82% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.66 (s, 1H), 7.21 (s, 1H), 4.05 (s, 3H), 2.80 (s, 3H).
(R)-6-iodo-7-methoxy-2-methyl-N-(1-(3-(trifluoromethyl)phenyl)ethyl)quinazolin-4-amine (19).
A mixture of 18 (335 mg, 1.00 mmol), 1439 (246 mg, 1.30 mmol), DIPEA (259 mg, 2.00 mmol) in EtOH (3 mL) was added to a nitrogen purged pressure tube. The mixture was heated to 100 °C for 16 h. After cooled to room temperature and concentrated under reduced pressure, the residue was dissolved in EtOAc and washed with NaHCO3 (sat.). The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography to afford the desired product 19 (426 mg, 87% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.15 (s, 1H), 7.72 (s, 1H), 7.63 (d, J = 7.6 Hz, 1H), 7.53 (d, J = 7.6 Hz, 1H), 7.46 (t, J = 7.7 Hz, 1H), 7.11 (s, 1H), 5.63-5.70 (m, 1H), 3.96 (s, 3H), 2.55 (s, 3H), 1.70 (d, J = 6.9 Hz, 3H).
(R)-tert-butyl 4-hydroxy-4-(7-methoxy-2-methyl-4-((1-(3-(trifluoromethyl)phenyl)ethyl)- -amino)quinazolin-6-yl)piperidine-1-carboxylate (20).
A mixture of 19 (24.5 mg, 0.05 mmol), DMPU (12.8 mg, 0.12 mmol) in dry THF was cooled to −78 °C. Then isopropylmagnesium bromide (15.4 mg, 0.15 mmol) was added dropwise and the reaction mixture was stirred at −78 °C for 1 h. Boc-4-piperidone (14.9 mg, 0.08 mmol) was added and the reaction mixture was stirred at room temperature for 12 h. The reaction was quenched with NH4CI (sat.) and extracted with DCM. The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography to afford the desired product 22 (19.5 mg, 69% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.78 (s, 1H), 7.74 (s, 1H), 7.67 (d, J = 7.7 Hz, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.44 (t, J = 7.7 Hz, 1H), 7.31 (d, J = 11.2 Hz, 1H), 5.70-5.78 (m, 1H), 4.01 (t, J = 11.2 Hz, 2H), 3.96 (s, 3H), 3.26 (t, J = 12.4 Hz, 2H), 2.59 (s, 3H), 2.13-2.04 (t, J = 11.4 Hz, 2H), 1.91 (t, J = 12.8 Hz, 2H), 1.74 (d, J = 7.0 Hz, 3H), 1.49 (s, 9H).
General Procedure for Synthesis of Compounds 1a, 1b, 1c, and 1d. A mixture of 8 or 9 or 15 (1 equiv.) and 10 wt% Pd/C (10 mol%) in methanol was stirred under H2 (1 atm) at room temperature for 3 h. After reaction completion detected by TLC, the mixture was filtered through celite and washed with EtOAc. Removal of organic solvent afforded the hydrogenation product as a yellow solid without further purification.
A mixture of hydrogenation product (1 equiv.), tert-butyl 5-bromopentanoate40 (2 equiv.) and K2CO3 (1.2 equiv.) in DMF was stirred at 70 °C for 3 h. The reaction was quenched with water and extracted with DCM. The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography to afford the desired product 21 or 22 or 23 as a yellow solid.
21 or 22 or 23 (1 equiv.) in DCM was treated with TFA (33 equiv.) and stirred at room temperature for 3 h. After reaction completion detected by TLC, the mixture was rinsed with DCM several times and concentrated under reduced pressure afforded the acid product as a yellow solid without further purification.
A mixture of acid (1 equiv.), EDCI (2 equiv.), OxymaPure (1.5 equiv.) and Et3N (5 equiv.) in DMF was stirred at room temperature for 30 min. Thalidomide-PEG2- -C2-NH2 hydrochloride41 (2 equiv.) or VHL ligand 2 hydrochloride (2 equiv.) was added and was stirred at room temperature overnight. The reaction was quenched with NH4Cl (sat.), and extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography to afford the desired product 1a-1d as a yellow solid.
N-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethyl)-5-((6-methoxy-2-methyl-4-(((R)-1-phenylethyl)amino)quinazolin-7-yl)oxy)pentanamide (1a).
1H NMR (400 MHz, CDCl3): δ 7.50-7.43 (m, 2H), 7.40-7.38 (m, 1H), 7.29 −7.27 (m, 1H), 7.22-7.19 (m, 1H), 7.18 (s, 1H), 7.00 (dd, J = 9.4, 7.1 Hz, 1H), 6.81 (dd, J = 8.6, 3.1 Hz, 1H), 6.68 (t, J = 5.6 Hz, 1H), 6.40-6.44 (m, 1H), 5.70 (q, J = 6.8 Hz, 1H), 4.80-4.89 (m, 1H), 3.97 (t, J = 6.0 Hz, 2H), 3.87 (s, 3H), 3.65 (t, J = 5.2 Hz, 2H), 3.59 (s, 4H), 3.55 (t, J = 5.3 Hz, 2H), 3.36-3.44 (m, 4H), 2.84-2.62 (m, 3H), 2.57 (s, 3H), 2.25-2.19 (m, 2H), 2.02-2.09 (m, 1H), 1.71-1.79 (m, 4H), 1.67 (d, J = 1.5 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 173.27, 171.67, 169.46, 169.20, 167.65, 161.24, 157.99, 154.33, 149.00, 146.75, 143.56, 136.14, 132.46, 128.57, 127.39, 126.85, 116.85, 111.72, 110.18, 105.96, 101.76, 70.63, 70.21, 69.94, 69.29, 69.15, 56.71, 50.55, 48.93, 45.05, 42.34, 39.41, 37.33, 36.03, 35.53, 32.89, 31.49, 29.78, 27.74, 24.66, 22.86, 21.49. HRMS (ESI): m/z [M + H]+ calcd for C42H49N7O9, 796.3592; found, 796.3594.
N-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethyl)-5-((6-methoxy-2-methyl-4-(((R)-1-(3-(trifluoromethyl)phenyl)ethyl)amino)quinazolin-7-yl)oxy)pentanamide (1b).
1H NMR (400 MHz, CDCl3): 7.77-7.73 (m, 1H), 7.71-7.66 (m, 1H), 7.46 (d, J = 7.9 Hz, 1H), 7.40-7.38 (m, 1H), 7.14 (s, 1H), 7.00-7.02 (m, 1H), 6.81 (d, J = 8.6 Hz, 1H), 6.65 (s, 1H), 6.43 (q, J = 5.2 Hz, 1H), 5.65-5.71 (m, 1H), 4.91-4.84 (m, 1H), 3.96 (d, J = 6.1 Hz, 2H), 3.88 (s, 3H), 3.65-3.68 (m, 2H), 3.61 (s, 4H), 3.56 (t, J = 5.2 Hz, 2H), 3.45-3.36 (m, 4H), 2.84-2.67 (m, 3H), 2.54 (d, J = 2.3 Hz, 3H), 2.22 (q, J = 6.3, 5.4 Hz, 2H), 2.11-2.04 (m, 1H), 1.81-1.72 (m, 4H), 1.71 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 173.30, 171.67, 169.51, 169.24, 167.70, 161.35, 157.90, 154.38, 149.05, 146.76, 144.90, 136.16, 132.47, 130.82, 130.49, 130.38, 129.05, 125.62, 124.12, 123.95, 122.92, 116.86, 111.75, 110.23, 106.03, 101.67, 70.68, 70.24, 69.95, 69.30, 69.16, 56.72, 50.42, 49.02, 42.36, 39.46, 36.06, 31.52, 29.81, 27.76, 24.79, 22.93, 21.65. HRMS (ESI): m/z [M + H]+ calcd for C43H48F3N7O9, 864.3466; found, 864.3468.
5-((4-(((R)-1-(3-amino-5-(trifluoromethyl)phenyl)ethyl)amino)-6-methoxy-2-methylquinazolin-7-yl)oxy)-N-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethyl)pentanamide (1c).
1H NMR (400 MHz, CDCl3): δ 8.01 (s, 1H), 7.84 (s, 1H), 7.40 (d, J = 9.2 Hz, 1H), 7.05-6.98 (m, 2H), 6.83 (d, J = 6.2 Hz, 1H), 6.73 (s, 1H), 6.43 (d, J = 7.5 Hz, 1H), 5.65 (s, 1H), 4.88 (s, 1H), 3.87-3.82 (m, 4H), 3.69 (t, J = 4.9 Hz, 3H), 3.63 (s, 3H), 3.60-3.55 (m, 3H), 3.39-3.44 (m, 4H), 2.70 (d, J = 11.4 Hz, 3H), 2.56 (d, J = 8.7 Hz, 3H), 2.17 (s, 2H), 2.09 (s, 1H), 1.74-1.68 (m, 4H), 1.63 (d, J = 5.1 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 173.56, 171.79, 171.75, 169.44, 167.69, 162.70, 158.97, 158.33, 155.42, 149.79, 146.78, 136.22, 132.45, 116.97, 111.68, 110.19, 105.19, 70.64, 70.29, 69.81, 69.30, 69.26, 58.59, 57.09, 51.40, 51.24, 49.00, 42.37, 39.53, 36.64, 35.87, 32.07, 31.59, 31.51, 29.85, 27.78, 22.97, 22.95, 22.84, 21.11, 18.58, 14.26, 1.16. HRMS (ESI): m/z [M + H]+ calcd for C43H49F3N8O9, 879.3575; found, 879.3582.
(2S,4R)-1-((S)-2-(5-((4-(((R)-1-(3-amino-5-(trifluoromethyl)phenyl)ethyl)amino)-6-methoxy-2-methylquinazolin-7-yl)oxy)pentanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (1d).
1H NMR (400 MHz, CDCl3): δ 8.65 (s, 1H), 7.54 (s, 2H), 7.39-7.30 (m, 4H), 7.08 (s, 1H), 7.02 (s, 1H), 6.71 (s, 1H), 5.63 (s, 1H), 5.10-5.02 (m, 1H), 4.71 (d, J = 9.3 Hz, 1H), 4.61 (d, J = 8.7 Hz, 1H), 4.47 (s, 1H), 4.06 (d, J = 11.2 Hz, 2H), 3.84 (s, 3H), 3.60 (t, J = 11.5 Hz, 2H), 2.50 (s, 2H), 2.48 (s, 3H), 2.35-2.26 (m, 2H), 2.24-2.12 (m, 2H), 1.74-1.64 (m, 5H), 1.45 (d, J = 6.9 Hz, 3H), 1.25 (s, 3H), 1.02 (s, 9H). 13C NMR (101 MHz, DMSO-d6): δ 171.87, 170.67, 169.60, 157.95, 154.71, 151.52, 149.55, 149.16, 147.77, 144.69, 131.15, 131.08, 129.71, 128.84, 126.41, 115.20, 109.66, 109.63, 108.39, 108.34, 105.12, 103.62, 103.59, 68.78, 68.55, 62.04, 58.58, 56.76, 56.46, 56.30, 49.84, 48.61, 47.73, 37.75, 35.21, 34.45, 29.05, 27.85, 26.47, 25.50, 24.84, 23.86, 22.45, 22.03, 21.23, 16.00, 13.97. HRMS (ESI): m/z [M + H]+ calcd for C47H57F3N8O6S, 919.4074; found, 919.4071.
General Procedure for Synthesis of Compounds 1e, 1f and 1k-1m. A mixture of 8 or 9 or 11 or 12 (1 equiv.) and 10 wt% Pd/C (10 mol%) in Methanol was stirred under H2 (1 atm) at room temperature for 3 h. After reaction completion detected by TLC, the mixture was filtered through celite and washed with EtOAc. Remove organic solvent to afford the hydrogenation product as a yellow solid without further purification.
A mixture of hydrogenation product (1 equiv.), 3-((tert-butoxycarbonyl)amino)propyl 4-methylbenzenesulfonate42 or 17-azido-3,6,9,12,15-pentaoxaheptadecyl 4-methylbenzenesulfonate40 or 2-((tert-butoxycarbonyl)amino)ethyl methanesulfonate (same way as 3-((tert-butoxycarbonyl)amino)propyl 4-methylbenzenesulfonate) (2 equiv.) and K2CO3 (1.2 equiv.) in DMF was stirred at 75 °C overnight. The reaction was quenched with water and extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography to afford the desired product 24-28 as a yellow solid.
24 or 26-28 (1 equiv.) in dioxane was treated with 4 M HCl in dioxane and stirred at room temperature for 1 h. After reaction completion detected by TLC, the mixture was rinsed with DCM several times and concentrated under reduced pressure afforded the amine product as a yellow solid without further purification.
A mixture of 25 (1 equiv.) and 10 wt% Pd/C (10 mol%) in Ethanol was stirred under H2 (1 atm) at room temperature for 6 h. After reaction completion detected by TLC, the mixture was filtered through celite and washed with EtOAc. Remove organic solvent to afford the amine product as a yellow solid without further purification.
A mixture of amine 24-28 (1 equiv.), DIEA, and thalidomide 4-fluoride43 in DMF was stirred at 75 °C overnight. The reaction was quenched with H2O, and extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography to afford the desired product 1e, 1f and 1k-1m as a yellow solid.
4-((3-((4-(((R)-1-(3-amino-5-(trifluoromethyl)phenyl)ethyl)amino)-6-methoxy-2-methylquinazolin-7-yl)oxy)propyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (1e).
1H NMR (400 MHz, CD3OD): δ 7.80 (s, 1H), 7.48-7.42 (m, 1H), 7.10 (d, J = 8.6 Hz, 1H), 6.95-6.99 (m, 4H), 6.83 (s, 1H), 5.74-5.68 (m, 1H), 5.05-5.00 (m, 1H), 4.27 (t, J = 5.6 Hz, 2H), 4.01 (s, 3H), 3.62-3.57 (m, 2H), 2.86-2.79 (m, 1H), 2.69-2.73 (m, 1H), 2.56 (s, 3H), 2.53-2.47 (m, 1H), 2.25-2.19 (m, 2H), 2.05-2.09 (m, 1H), 1.69 (d, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CD3OD): δ 174.61, 171.77, 170.62, 169.22, 161.21, 159.98, 156.92, 151.59, 150.42, 148.11, 146.61, 141.61, 139.07, 137.14, 133.86, 132.45, 126.93, 124.77, 117.94, 117.10, 112.55, 111.86, 111.18, 106.82, 103.72, 102.27, 68.01, 57.15, 52.02, 40.20, 32.17, 29.76, 23.77, 23.03, 21.38. HRMS (ESI): m/z [M + H]+ calcd for C35H34F3N7O6, 706.2523; found, 706.2523.
4-((17-((4-(((R)-1-(3-amino-5-(trifluoromethyl)phenyl)ethyl)amino)-6-methoxy-2-methylquinazolin-7-yl)oxy)-3,6,9,12,15-pentaoxaheptadecyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (1f).
1H NMR (400 MHz, CDCl3): δ 7.48-7.40 (m, 1H), 7.18 (s, 1H), 7.10-7.00 (m, 2H), 6.98 (s, 1H), 6.87 (dd, J = 8.5, 2.1 Hz, 1H), 6.76 (s, 1H), 6.41 (s, 1H), 4.90-4.84 (m, 1H), 3.95 (d, J = 3.1 Hz, 1H), 3.93 (s, 3H), 3.66 (dd, J = 10.5, 5.1 Hz, 9H), 3.59-3.47 (m, 10H), 3.43-3.37 (m, 4H), 3.35 (s, 1H), 2.89-2.78 (m, 4H), 2.76-2.67 (m, 2H), 2.10-2.06 (m, 1H), 1.70 (d, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 171.31, 171.29, 169.39, 169.37, 168.70, 168.65, 167.73, 157.94, 154.20, 149.00, 146.90, 136.18, 132.57, 132.54, 131.48, 131.21, 127.76, 125.54, 123.37, 116.91, 111.79, 110.55, 110.33, 70.99, 70.70, 70.58, 70.50, 70.38, 69.88, 69.58, 68.62, 68.30, 66.76, 56.53, 49.00, 42.47, 32.06, 31.54, 29.84, 29.50, 25.86, 22.88, 14.25, 1.15. HRMS (ESI): m/z [M + H]+ calcd for C44H52F3N7O11, 912.3677; found, 912.3667.
2-(2,6-dioxopiperidin-3-yl)-4-((2-((6-methoxy-4-(((R)-1-phenylethyl)amino)quinazolin-7-yl)oxy)ethyl)amino)isoindoline-1,3-dione (1k).
1H NMR (400 MHz, CD3OD): δ 8.57 (s, 1H), 7.89 (s, 1H), 7.55 (t, J = 7.8 Hz, 1H), 7.46 (d, J = 7.6 Hz, 2H), 7.35 (t, J = 6.6 Hz, 2H), 7.25 (d, J = 8.6 Hz, 2H), 7.15 (s, 1H), 7.04 (d, J = 6.9 Hz, 1H), 5.83 (d, J = 7.6 Hz, 1H), 4.97-5.04 (m, 1H), 4.41 (d, J = 4.8 Hz, 2H), 3.99 (s, 3H), 3.86 (d, J = 4.8 Hz, 2H), 2.85-2.76 (m, 1H), 2.73 (s, 1H), 2.66 (d, J = 18.5 Hz, 1H), 2.05 (s, 1H), 1.74 (d, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CD3OD): δ 174.57, 171.67, 170.60, 169.11, 160.31, 157.48, 152.61, 149.60, 147.97, 143.77, 137.08, 135.52, 133.74, 129.73, 128.64, 127.60, 118.68, 112.37, 111.56, 108.37, 104.27, 101.63, 70.25, 57.27, 52.89, 50.17, 42.64, 32.17, 30.74, 30.45, 23.77, 21.38. HRMS (ESI): m/z [M + H]+ calcd for C32H30N6O6, 595.2227; found, 595.2250.
2-(2,6-dioxopiperidin-3-yl)-4-((2-((6-methoxy-4-(((R)-1-(3-(trifluoromethyl)phenyl)ethyl)amino)quinazolin-7-yl)oxy)ethyl)amino)isoindoline-1,3-dione (1l).
1H NMR (400 MHz, CDCl3): δ 8.45 (s, 1H), 7.67 (s, 1H), 7.63 (d, J = 7.6 Hz, 1H), 7.48 (d, J = 7.9 Hz, 1H), 7.41 (s, 1H), 7.24 (s, 1H), 7.12 (s, 1H), 7.02 (t, J = 6.1 Hz, 1H), 6.93 (d, J = 8.6 Hz, 1H), 6.55 (d, J = 5.9 Hz, 1H), 5.67 (q, J = 6.9 Hz, 1H), 4.80-4.91 (m, 1H), 4.27 (d, J = 6.1 Hz, 2H), 3.87 (s, 3H), 3.70 (d, J = 5.9 Hz, 2H), 2.80 (s, 1H), 2.70 (dd, J = 11.8, 6.3 Hz, 2H), 2.09-2.02 (m, 1H), 1.68 (d, J = 4.2 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 171.59, 169.38, 169.07, 167.67, 162.70, 157.61, 153.57, 149.54, 146.63, 144.89, 136.07, 132.38, 131.09, 130.77, 130.25, 129.19, 125.59, 124.25, 122.88, 117.10, 112.08, 110.53, 108.91, 100.44, 67.82, 56.40, 50.02, 48.94, 41.79, 36.64, 31.50, 22.88, 22.05. HRMS (ESI): m/z [M + H]+ calcd for C33H29F3N6O6, 663.2101; found, 663.2100.
2-(2,6-dioxopiperidin-3-yl)-4-((2-((6-methoxy-2-methyl-4-(((R)-1-phenylethyl)amino)quinazolin-7-yl)oxy)ethyl)amino)isoindoline-1,3-dione (1m).
1H NMR (400 MHz, CDCl3): δ 7.46 (d, J = 7.7 Hz, 3H), 7.31 (t, J = 7.5 Hz, 2H), 7.22 (s, 1H), 7.12 (s, 1H), 7.04 (t, J = 8.0 Hz, 1H), 6.98 (dd, J = 8.6, 5.5 Hz, 1H), 6.57-6.48 (m, 1H), 5.74-5.64 (m, 1H), 4.93-4.77 (m, 1H), 4.26 (d, J = 5.1 Hz, 2H), 3.88 (s, 3H), 3.72 (s, 2H), 2.82 (dd, J = 13.0, 5.4 Hz, 1H), 2.76-2.64 (m, 2H), 2.57 (s, 3H), 2.11-2.00 (m, 1H), 1.69 (d, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 171.40, 169.31, 168.93, 167.57, 157.92, 154.00, 149.30, 146.63, 143.41, 136.14, 132.41, 128.71, 127.56, 126.86, 117.25, 112.07, 110.51, 106.51, 101.74, 67.99, 67.23, 56.80, 50.59, 48.98, 41.83, 31.48, 29.84, 24.83, 22.86, 21.58, 18.58, 14.26, 1.15. HRMS (ESI): m/z [M + H]+ calcd for C33H32N6O6, 609.2383; found, 609.2366.
2-((4-(((R)-1-(3-amino-5-(trifluoromethyl)phenyl)ethyl)amino)-6-methoxy-2-methylquinazolin-7-yl)oxy)-N-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)acetamide (1g).
A mixture of 9 (1 equiv.) and 10 wt% Pd/C (10 mol%) in methanol was stirred under H2 (1 atm) at room temperature for 3 h. After reaction completion detected by TLC, the mixture was filtered through celite and washed with EtOAc. Remove organic solvent to afford the hydrogenation product as a yellow solid without further purification.
A mixture of hydrogenation product (27.9 mg, 0.07 mmol), KI (1.18 mg, 0.01 mmol), KHCO3 (21.4 mg, 0.21 mmol) and 2-bromo-N-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)acetamide40 (32.5 mg, 0.09 mmol) in DMF (1 mL) was stirred at 60 °C for 12 h. The reaction was quenched with water and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography to afford the desired product 1g (15.1 mg, 30 %) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 11.03 (d, J = 2.9 Hz, 1H), 10.24 (s, 1H), 7.87-7.77 (m, 2H), 7.57-7.50 (m, 2H), 7.04 (s, 1H), 6.87 (d, J = 12.8 Hz, 2H), 6.71 (s, 1H), 5.63-5.57 (m, 1H), 5.56 (s, 1H), 5.14 (dd, J = 12.4, 4.8 Hz, 1H), 4.97 (s, 1H), 4.37 (d, J = 6.0 Hz, 1H), 4.32 (d, J = 3.9 Hz, 1H), 3.96 (s, 3H), 2.86-2.97 (m, 1H), 2.58 (t, J = 15.3 Hz, 1H), 2.39 (s, 3H), 2.27-2.16 (m, 1H), 2.04-1.97 (m, 1H), 1.58 (d, J = 7.1 Hz, 3H). 13C NMR (126 MHz, DMSO-d6): δ 172.85, 171.04, 167.75, 166.04, 157.71, 149.44, 134.28, 134.16, 132.90, 132.80, 129.82, 129.58, 128.89, 125.69, 125.54, 123.53, 119.77, 119.72, 115.10, 115.06, 109.62, 108.09, 106.51, 103.07, 69.80, 67.24, 56.42, 51.49, 46.30, 31.25, 30.72, 22.64, 22.62, 21.63. HRMS (ESI): m/z [M + H]+ calcd for C34H32F3N7O6, 692.2366; found, 692.2361.
4-(((R)-1-(3-amino-5-(trifluoromethyl)phenyl)ethyl)amino)-6-methoxy-2-methylquinazolin-7-yl (2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)carbamate (1h).
A mixture of 9 (1 equiv.) and 10 wt% Pd/C (10 mol%) in methanol was stirred under H2 (1 atm) at room temperature for 3 h. After reaction completion detected by TLC, the mixture was filtered through celite and washed with EtOAc. Remove organic solvent to afford the hydrogenation product as a yellow solid without further purification.
A mixture of hydrogenation product (14.3 mg, 0.04 mmol), DIEA (5.89 mg, 0.05 mmol) and 4-nitrophenyl (2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)carbamate45 (77.3 mg, 0.18 mmol) in DMF was stirred at room temperature for 3 days. The reaction was quenched with water and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography to afford the desired product 1h (30.5 mg, 90% yield) as a white solid. 1H NMR (400 MHz, CD3OD): δ 8.07-8.00 (m, 1H), 7.88 (d, J = 7.0 Hz, 1H), 7.80 (d, J = 8.6 Hz, 1H), 7.76 (d, J = 2.6 Hz, 1H), 7.51-7.48 (m, 1H), 7.46 (dd, J = 7.7, 2.2 Hz, 1H), 7.42 (s, 1H), 6.82 (s, 1H), 5.77 (q, J = 7.0 Hz, 1H), 5.16-5.12 (m, 1H), 4.57-4.47 (m, 2H), 3.98 (s, 3H), 3.66 (d, J = 3.3 Hz, 1H), 2.88 (dd, J = 11.7, 6.0 Hz, 1H), 2.79-2.72 (m, 1H), 2.54 (s, 3H), 2.22-2.13 (m, 1H), 1.74 (d, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 174.70, 172.17, 171.32, 160.14, 159.70, 154.66, 152.06, 147.20, 141.75, 139.23, 135.60, 133.54, 132.11, 130.15, 124.77, 124.21, 121.17, 119.26, 118.29, 114.99, 104.17, 103.24, 78.33, 56.98, 56.27, 53.63, 51.88, 32.34, 30.69, 24.00, 22.66, 21.54, 19.30. HRMS (ESI): m/z [M + H]+ calcd for C33H30F3N7O6, 678.2210; found, 678.2210.
4-(((4-(((R)-1-(3-amino-5-(trifluoromethyl)phenyl)ethyl)amino)-6-methoxy-2-methylquinazolin-7-yl)oxy)methyl)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (1i).
A mixture of 9 (1 equiv.) and 10 wt% Pd/C (10 mol%) in methanol was stirred under H2 (1 atm) at room temperature for 3 h. After reaction completion detected by TLC, the mixture was filtered through celite and washed with EtOAc. Remove organic solvent to afford the hydrogenation product as a yellow solid without further purification.
A mixture of hydrogenation product (20.0 mg, 0.05 mmol), 4-(bromomethyl)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione46 (35.8 mg, 0.10 mmol) and 60 wt% NaH (2.45 mg, 0.06 mmol) in DMF (1 mL) was stirred at 100 °C for 5 h. The reaction was quenched with water and extracted with DCM. The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography to afford the desired product 1i (8.40 mg, 25% yield) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.90 (d, J = 7.7 Hz, 1H), 7.71 (t, J = 6.6 Hz, 1H), 7.67-7.62 (m, 1H), 7.14 (s, 1H), 7.08 (d, J = 7.2 Hz, 1H), 7.01 (s, 1H), 6.97 (d, J = 5.7 Hz, 1H), 6.78 (s, 1H), 5.57 (s, 1H), 5.30 (s, 4H), 4.95-5.01 (m, 1H), 3.97 (s, 3H), 2.96-2.86 (m, 2H), 2.78 (d, J = 14.1 Hz, 1H), 2.54 (s, 3H), 2.22-2.15 (m, 1H), 1.66 (d, J = 6.8 Hz, 4H). 13C NMR (126 MHz, DMSO-d6): δ 172.80, 169.83, 167.17, 166.88, 157.82, 149.48, 135.15, 133.73, 131.78, 129.62, 128.41, 128.24, 127.79, 127.22, 125.67, 123.51, 123.17, 115.13, 109.62, 108.19, 103.38, 65.28, 56.61, 48.98, 33.57, 30.95, 29.02, 28.81, 24.75, 21.99, 21.47, 13.92, 2.00. HRMS (ESI): m/z [M + H]+ calcd for C33H29F3N6O6, 663.2101; found, 663.2100.
2-(2,6-dioxopiperidin-3-yl)-4-(((6-methoxy-2-methyl-4-(((R)-1-phenylethyl)amino)quinazolin-7-yl)oxy)methyl)isoindoline-1,3-dione (1j).
A mixture of 8 (1 equiv.) and 10 wt% Pd/C (10 mol%) in methanol was stirred under H2 (1 atm) at room temperature for 3 h. After reaction completion detected by TLC, the mixture was filtered through celite and washed with EtOAc. Remove organic solvent to afford the hydrogenation product as a yellow solid without further purification.
A mixture of hydrogenation product (31.8 mg, 0.11 mmol), 4-(bromomethyl)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (24.1 mg, 0.07 mmol) and K2CO3 (11.4 mg, 0.08 mmol) in DMF (1 mL) was stirred at 60 °C overnight. The reaction was quenched with water and extracted with DCM. The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography to afford the desired product 1j (14.3 mg, 36% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 11.14 (s, 1H), 8.25 (s, 1H), 7.93 (d, J = 4.1 Hz, 2H), 7.81 (s, 1H), 7.45 (d, J = 7.7 Hz, 2H), 7.33 (t, J = 7.5 Hz, 2H), 7.22 (t, J = 7.3 Hz, 1H), 7.09 (s, 1H), 5.66 (s, 2H), 5.17 (dd, J = 12.9, 5.4 Hz, 1H), 4.11 (d, J = 2.9 Hz, 1H), 3.94 (s, 3H), 2.88 (dd, J = 13.2, 5.0 Hz, 1H), 2.61 (d, J = 16.2 Hz, 1H), 2.36 (s, 3H), 2.12-2.05 (m, 1H), 2.03-1.96 (m, 1H), 1.60 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 172.77, 169.81, 167.16, 166.88, 161.13, 157.69, 152.58, 148.01, 144.66, 135.49, 135.06, 133.71, 131.75, 128.23, 127.73, 126.64, 126.28, 123.04, 106.61, 102.89, 73.26, 68.87, 65.06, 61.93, 56.40, 48.85, 33.56, 31.14, 28.86, 24.74, 21.98, 20.65. HRMS (ESI): m/z [M + H]+ calcd for C32H29N5O6, 580.2118; found, 580.2125.
General Procedure for Synthesis of Compounds 1n and 1o. Compound 20 in dioxane was treated with 4 M HCl in dioxane and stirred at room temperature for 3 h. After reaction completion detected by TLC, the mixture was rinsed with DCM several times and concentrated under reduced pressure afforded the amine product as a yellow solid without further purification.
A mixture of 3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)propanoic acid16 or 2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)acetic acid (2 equiv.), EDCI (2 equiv.), OxymaPure (1.5 equiv.) and Et3N (5 equiv.) in DMF was stirred at room temperature for 30 min. The amine product (1 equiv.) was added and was stirred at room temperature overnight. The reaction was quenched with NH4Cl (sat.), and extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography to afford the desired product 1n and 1o as a yellow solid.
2-(2,6-dioxopiperidin-3-yl)-4-((3-(4-hydroxy-4-(7-methoxy-2-methyl-4-(((R)-1-(3-(trifluoromethyl)phenyl)ethyl)amino)quinazolin-6-yl)piperidin-1-yl)-3-oxopropyl)amino)isoindoline-1,3-dione (1n).
1H NMR (400 MHz, CDCl3): δ 7.99 (s, 1H), 7.73 (s, 1H), 7.65 (s, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.41 (d, J = 7.4 Hz, 1H), 7.20 (s, 1H), 7.01 (d, J = 7.1 Hz, 1H), 6.89 (t, J = 7.3 Hz, 1H), 6.62-6.47 (m, 1H), 5.76-5.64 (m, 1H), 4.81 (d, J = 37.3 Hz, 1H), 4.45 (s, 1H), 3.87 (s, 3H), 3.54 (d, J = 32.0 Hz, 4H), 3.01 (s, 1H), 2.82-2.70 (m, 2H), 2.69-2.59 (m, 3H), 2.54 (s, 3H), 2.07 (d, J = 32.3 Hz, 3H), 1.80 (t, J = 17.0 Hz, 2H), 1.68 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 171.79, 169.51, 169.22, 167.75, 163.76, 161.45, 158.85, 146.65, 144.63, 136.34, 135.37, 132.62, 130.51, 130.35, 129.09, 125.61, 124.21, 123.90, 122.91, 120.02, 116.72, 111.69, 110.19, 106.29, 71.92, 56.17, 50.38, 49.00, 41.69, 38.66, 37.89, 36.04, 35.31, 32.62, 31.52, 29.83, 22.82, 21.51, 14.25, 1.15. HRMS (ESI): m/z [M + H]+ calcd for C40H40F3N7O7, 788.2941; found, 788.2957.
2-(2,6-dioxopiperidin-3-yl)-4-((2-(4-hydroxy-4-(7-methoxy-2-methyl-4-(((R)-1-(3-(trifluoromethyl)phenyl)ethyl)amino)quinazolin-6-yl)piperidin-1-yl)-2-oxoethyl)amino)isoindoline-1,3-dione (1o).
1H NMR (400 MHz, CD3OD): δ 8.46-8.42 (m, 1H), 7.78 (s, 1H), 7.72 (d, J = 7.3 Hz, 1H), 7.51 (q, J = 8.1, 6.6 Hz, 3H), 7.10-6.94 (m, 3H), 5.75-5.81 (m, 1H), 5.04 (dd, J = 12.5, 5.4 Hz, 1H), 4.48 (d, J = 12.9 Hz, 1H), 4.24-4.17 (m, 1H), 4.01 (s, 1H), 3.95 (s, 3H), 3.85 (s, 1H), 3.72-3.64 (m, 1H), 3.35 (s, 1H), 3.25 (d, J = 12.5 Hz, 1H), 2.89-2.80 (m, 1H), 2.76-2.65 (m, 2H), 2.65-2.54 (m, 1H), 2.52 (s, 3H), 2.13-2.05 (m, 1H), 1.89 (d, J = 14.0 Hz, 1H), 1.79 (d, J = 14.0 Hz, 1H), 1.70 (d, J = 2.1 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 174.65, 171.64, 170.47, 169.29, 168.48, 164.36, 163.40, 160.61, 147.02, 146.54, 138.06, 137.24, 133.80, 131.48, 130.32, 127.06, 124.93, 124.54, 122.28, 118.61, 112.22, 111.70, 107.18, 103.78, 72.71, 56.45, 53.53, 51.65, 44.95, 42.00, 39.73, 36.61, 35.94, 32.21, 30.70, 25.25, 24.37, 23.77, 21.56. HRMS (ESI): m/z [M + H]+ calcd for C39H38F3N7O7, 774.2785; found, 774.2782.
Crystal structure informed design of SOS1 PROTAC degraders
Coordinates for crystal structures of SOS1 in a complex with BI68BS (PDB: 6SFR) with a resolution of 1.92Å16 and cereblon in a complex with cereblon E3 ligase (PDB: 4TZ4) with a resolution of 3.01Å47 were retrieved from the RCSB Protein Data Bank. They were prepared using the protein preparation wizard in Maestro of Schrödinger Release 2020-3.48 In brief, hydrogens were added to all atoms; bond order and formal charges were added to heterogroups. Water molecules were not involved in protein-ligand interactions, thus were removed. To optimize the hydrogen bond interactions, hydroxyl and thiol hydrogens were sampled, 180° rotations of the terminal angle of Asn, Gln, and His residues were assigned, and His tautomers and ionization states were predicted. The conformation of missing side-chain atoms if present were predicted. A brief relaxation was performed using an all-atom constrained minimization to reduce steric clashes present in the original crystal structures.48 Structural interactions between SOS1 and BI68BS were outlined by the structural interaction fingerprint analysis function in Maestro.24
The prepared protein structures were used for protein-protein docking experiments in Rosetta 3.12.27 Protein-protein local docking protocol was applied to the structure coordinates in an input pdb file prepared for docking. The following scripts were used to generate 500 docking poses, from which total score and interface score were retrieved from the output file. The distance between /A LBK 1103 O13 (BI68BS 6-methoxy group) and /C LVY 502 N17 (lenalidomide amino group) and the distance between /A LBK 1103 O14 (BI68BS 7-methoxy group) and /C LVY 502 N17 were measured. Molecular graphics and analyses performed with UCSF ChimeraX49, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. Statistical analyses were performed using R packages50 and jamovi51.
$ROSETTA3/bin/docking_protocol.static.linuxgccrelease @flag_local_docking
flag_local_docking:
-in:file:s 6SFR_4TZ4_prepared.pdb
-unboundrot 6SFR_4TZ4_prepared.pdb
-nstruct 500
-partners A_C
-dock_pert 3 8
-ex1
-ex2aro
-out:path:all output_files
-out:suffix _local_dock
Cell line culture
SW620, HCT116, C2BB, and SW1417 were purchased from the American Type Culture Collection. The cells were cultured in modified McCoys’ 5A medium (Gibco, 16600082) supplemented with 10% FBS (ATCC, 30-2020) and 1% penicillin-streptomycin (ATCC, 30-2300). They were incubated under a 5% (v/v) CO2 atmosphere at 37 °C.
Patients
Patients were enrolled in one of several prospective studies approved for surgically resected tumor specimen collection at Moffitt Cancer Center (MCC). Tumor specimens used in this study were from either primary or metastatic CRC including but not limited to liver and peritoneal metastases as part of routine clinical care. These were approved under an umbrella protocol MCC20880 by the Institutional Review Board at MCC. Following collection of specimens, they were de-identified and assigned a lab number. Clinical information including, type and site of tumor specimen, patient’s treatment history, previous genetic information, and organoid initiation date were collected when available. Patient-derived xenografts (MCC IACUC approval: A4100-01) were used to generate and biobank tumor specimens. The tumor samples were subsequently used to generate CRC PDOs.
Patient-derived organoid culture
CRC organoids were generated and expanded using a protocol similar to previously published ones for CRC with modifications.52,38 In brief, the tumor specimen was placed in PBS and then fresh wash media, then minced into approximately 1 mm3 small fragments using sterile scalpels. Tissue fragments were placed in a Falcon tube with warmed digestion media and then incubated for 30-45 minutes in a 37°C shaker with agitation at 600 rpm to allow tissue to dissociate into single cells. Larger tissue fragments were allowed to settle under normal gravity for 2 minutes and the supernatant was transferred to a clean Falcon tube. An additional 3 mL of wash media with 10% FBS was added and then centrifuged. Cells were filtered through a 100μM mesh filter as well as additional 40μM filter to remove mouse fibroblast. Cell pellets suspended in 300 μL of ice-cold growth factor reduced Matrigel (Corning: 354230), then plated into 50µL domes in a 24-well pre-warmed culture plate, which was allowed to solidify for 15 min at 37 °C. When the Matrigel domes solidified, 500µL of pre-warmed complete growth media was added and incubated in 5% CO2 atmosphere at 37°C. The growth and quantity of the organoid cultures were monitored with fresh growth media added every 2 days. Organoid propagation started from adding digestion media with gentle mechanical digestion. The suspension was incubated in a Falcon tube for 30 minutes, inverted every 10 minutes, and then centrifuged at 1300 rpm for 6 minutes at 4°C. The cell pellet was resuspended in wash media with 0.1% BSA, followed by Matrigel, and then plated as domes. Wash media: advanced DMEM/F12 (Gibco: 12634010), 1M Hepes (Gibco: 15630080), 100X glutamine (Gibco: 25030149), primocin (Invivogen: ANTPM1). Digestion media: wash media/10% FBS, collagenase and dispase (Sigma: 10269638001). Growth media: wash media without FBS, 1X wnt3a/R-spondin/Noggin condition medium (L-WRN),53 1X B27 supplement (Gibco: 12587-010), 10 mM nicotinamide (Sigma Aldrich: N3376), 1.25 mM N-acetylcysteine (Sigma Aldrich: A9165), 100 μg/mL primocin (Invivogen: ANTPM1), 100ng/mL recombinant mouse Noggin (abcam: ab281818), 50ng/mL hEGF (R&D Systems: 236EG200), 100 ng/mL human FGF (R&D Systems: 233-FB), 10nM human gastrin I (R&D Systems: 3006/1), 500 nM A83-01 (Selleckchem: S7692), 10.5μM Y-267632 (Selleckchem: S1049), and 1μM PGE2 (R&D Systems: 2296/10).
Immunoblot
Cells were plated in a 100mm dish for incubation overnight before harvest for immunoblot. For drug treatment, cell confluency was checked prior to proceeding with the treatment. Cells were then harvested and lysed in a lysis buffer of 1x radioimmunoprecipitation assay (RIPA) lysis buffer (Thermo Scientific, 89900) and Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific, 78429) while maintained on ice. After centrifugation at 12,000 rpm for 15 minutes at 4°C, the protein concentration was determined using the BCA protein assay kit (Thermo Scientific, 23227). Approximately 40μg protein samples were loaded into each well and separated by SDS-PAGE gel. After protein transfer onto nitrocellulose membranes, 5% skim milk in TBST was used to block the membranes for 1 hour at room temperature prior to incubation with primary antibodies overnight at 4°C. At the completion, a horseradish peroxidase–conjugated secondary antibody was applied at room temperature for 1 hour. The bands were visualized using Western Lightning Plus-ECL (PerkinElmer). Primary antibodies used in this study included GAPDH (Cell Signaling, 2118), pERK (Cell Signaling, 4376), ERK (Cell Signaling, 9102), cleaved PARP (Cell Signaling, 5625), and SOS1 (Cell Signaling, 5890).
To prepare siRNA-treated samples for immunoblot, cells were plated with media for 24 hours, then treated with siRNA. 12μL lipofectamine RNAiMAX (Invitrogen: 13778150) was dilated in 200μL Opti-MEM (Gibco: 31985070); 4μL siRNA (10μM) was dilated in 200μL Opti-MEM. The mixture was incubated at room temperature for 5 minutes. After 48 hours, the cells were harvested and processed for immunoblot. siRNAs used: SOS1 (ambion: s13286, 4390824), GAPDH (ambion: s5572, 4390824), negative control (Invitrogen: AM4613).
Organoid drug sensitivity assay
Organoids were harvested with organoid cell recovery solution (Corning: 354253) and pipetted gently to dissolve Matrigel. AFter incubation for 15 minutes, the cells were collected and washed with 0.1%BSA/wash media. Cells were counted and resuspended in a mixture of 90% complete growth media and 10% Matrigel. 5000 cells in 30μL media were seeded into a 96-well plate in triplicates. Of note, the plate should be prepared with 30μL of 50% Matrigel and 50% complete growth media in each well and allowed for solidification at 37°C for 30 minutes. Once the organoids were visible (3-7 days), drugs were added and cultured for 3 days. 40μL of CellTiterGlo 3D (Promega: G9681) was added and chemiluminescence was read at 360/460nm on an Envision multi-well plate reader (PerkinElmer). After normalization of readings to DMSO-treated cells, dose-response curves were generated. IC50 values were calculated using GraphPad Prism 8.4.3.
Multiplexed quantitative global proteomics
Sample Preparation:
Approximately 10 millions of SW620 cells were treated with 1μM P7 or 0.2% DMSO for 24 hours, respectively. Triplicate samples for each condition were provided as cell pellets. Cells were lysed in denaturing lysis buffer containing 8M urea, 20 mM HEPES (pH 8), 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate and 1 mM β-glycerophosphate. A Bradford assay was carried out to determine the protein concentration. The proteins were reduced with 4.5 mM DTT and alkylated with 10 mM iodoacetamide. Trypsin digestion was carried out at room temperature overnight, and tryptic peptides were then acidified with 1% trifluoroacetic acid (TFA) and desalted with C18 Sep-Pak cartridges according to the manufacturer’s procedure. TMT Labeling: 100μg of peptide from each sample was labeled with TMTPro16plex reagent. The label incorporation was checked by LC-MS/MS and spectral counting. 98% or greater label incorporation was achieved for each channel. The samples were then pooled and lyophilized. High pH Reversed Phase Peptide Separation: After lyophilization, the peptides were re-dissolved in 250μL of 20mM ammonium formate (pH 10.0). The high pH reversed phase separation was performed on a Xbridge 4.6 mm x 100 mm column packed with BEH C18 resin, 3.5 µm, 130Å (Waters) The peptides were eluted as follows: 5% B (5 mM Ammonium Formate, 90% acetonitrile, pH 10.0) for 10 minutes, 5% - 15% B in 5 minutes, 15-40% B in 47 minutes, 40-100% B in 5 minutes and 100% B held for 10 minutes, followed by re-equilibration at 1% B. The flow rate was 0.6 ml/min, and 24 concatenated fractions were collected. Speedvac centrifuge was used to dry the peptides. LC-MS/MS: A nanoflow ultra high performance liquid chromatography (RSLC, Dionex, Sunnyvale, CA) coupled to an electrospray bench top orbitrap mass spectrometer (Orbitrap Exploris480, Thermo, San Jose, CA) was used for tandem mass spectrometry peptide sequencing experiments. The sample was first loaded onto a pre-column (2 cm x 100 µm ID packed with C18 reversed-phase resin, 5µm, 100Å) and washed for 8 minutes with aqueous 2% acetonitrile and 0.1% formic acid. The trapped peptides were eluted onto the analytical column, (C18, 75 µm ID x 25 cm, 2 µm, 100Å, Dionex, Sunnyvale, CA). The 120-minute gradient was programmed as: 95% solvent A (2% acetonitrile + 0.1% formic acid) for 8 minutes, solvent B (90% acetonitrile + 0.1% formic acid) from 5% to 38.5% in 90 minutes, then solvent B from 50% to 90% B in 7 minutes and held at 90% for 5 minutes, followed by solvent B from 90% to 5% in 1 minute and re-equilibrate for 10 minutes. The flow rate on the analytical column was 300 nl/min. nanoEasy source with FAIMS was used with spray voltage of 2100v. Data-dependent acquisition with two CV values (−45 and −65) were applied alternatively with 1.5 second cycle time each, with dynamic exclusion set at 45 seconds. The resolution settings were 120,000 and 45,000 for MS1 and MS/MS, respectively. The isolation window was 0.7 Th and HCD collision energy was set at 35%.
Data Analysis:
MaxQuant (version 1.6.14.0)54 was used to identify proteins and quantify the TMT reporter ion intensities with default settings. The protein database was downloaded in March 2021 from Uniprot.org. Samples were normalized within-plex using the Moffitt standard proteomics normalization pipeline iterative rank-order normalization (IRON).55 Median sample was determined to be Plex_1_TMT-131, DMSO_2. All normalized values and calculations are reported as Log2 abundances. Log2 ratios were calculated as the difference between average treatment – average DMSO. P-values were calculated using Welch’s 2-sided T-tests, assuming unequal variance. Differentially abundant proteins were determined by having a log2 ratio ≥ 2 standard deviations from the mean (+0.1089/−0.1131) and a P-value < 0.05. Pathway enrichment was conducted using the EnrichR R package.56
Immunohistochemistry of organoids
To one or two confluent wells of organoids was added 1:5 diluted 500μL of 5 units/mL dispase in HBSS (StemCell: 07913) and incubated at 37°C in 5% CO2 atmosphere. Organoids were harvested and transferred to a 1.5mL Eppendorf tube. The pellet was collected after centrifugation at 1500 rpm for 6 min, and washed with 1X PBS. The pellet was resuspended in 1mL of 4% PFA and incubated at 4°C overnight. The pellet was resuspended in 100μL of 37°C warmed histogel, centrifuged at 1000 rpm for 30 seconds to settle the organoids. The gel was allowed to polymerize at 4°C for 10 minutes and then the plug was transferred to a 5mL tube containing 2mL 70% ethanol, and sent to the histology lab. Organoids were paraffin-embedded and subjected to serial sections for hematoxylin and eosin staining and immunohistochemistry following a common procedure at the histology lab. The slides/sections were cut at 4 micron thickness using a Leica RM2245 microtome. Charged slides were used.
Immunohistochemical staining of SOS1 on organoids:
Slides were stained using a Ventana Discovery Ultra automated system (Ventana Medical Systems, Tucson, AZ) as per manufacturer’s protocol with proprietary reagents. Briefly, slides were deparaffinized on the automated system with Discovery Wash Solution (Ventana). Heat-induced antigen retrieval method was used in Cell Conditioning 1 Mild (Ventana). The rabbit primary antibody that reacts to SOS1 (Abcam ab140621, Cambridge, MA) was used at a 1:50 concentration in Dako antibody diluent (Carpinteria, CA) and incubated for 1.5 hours. The Ventana OmniMap anti-rabbit secondary antibody was applied for 16 minutes. The detection system used was the Ventana ChromoMap kit. Slides were counterstained with hematoxylin, then dehydrated and coverslipped as per normal laboratory protocol. Negative control was performed using substitution of serum or isotype-specific immunoglobulins at the same protein concentration as the primary antibody.
Immunohistochemical staining of Annexin V:
Slides were stained using a Ventana Discovery Ultra automated system (Ventana Medical Systems, Tucson, AZ) as per manufacturer’s protocol with proprietary reagents. Briefly, slides were deparaffinized on the automated system with Discovery Wash Solution (Ventana). Enzyme digestion method was used in Protease 1 for 4 minutes (Ventana). The rabbit primary antibody that reacts to Annexin V (Abcam ab108321, Cambridge, MA) was used at a 1:200 concentration in Dako antibody diluent (Carpinteria, CA) and incubated for 32 minutes. The Ventana OmniMap anti-rabbit secondary antibody was applied for 16 minutes. The detection system used was the Ventana ChromoMap kit and slides were then counterstained with hematoxylin. Slides were then dehydrated and coverslipped as per normal laboratory protocol. Positive controls were those organoids treated with 5-FU and SN38.
mRNA measurement and quantification
Cells were treated with drugs for indicated duration of time prior to harvest for RNA extraction and purification using Qiagen RNeasy mini kit (Qiagen: 74104). The RNA samples were quantified by optical density 260/280, 260/230 readings using a spectrophotometer (NanoDrop, Thermo Fisher Scientific, Waltham, MA, USA). We used the QuantiTect reverse transcription kit (Qiagen: 205311) for cDNA synthesis and gDNA removal in accordance with the manufacturer’s instructions. Quantitative RT-PCR was performed using the MX3000p Real-Time PCR System (Agilent Technologies, Santa Clara, CA, USA) to determine the mRNA expression levels of SOS1, SOS2, and GAPDH. Quantitative RT-PCR for each gene was performed using the TaqMan method with 5μL Taqman fast advanced master mix, 3.5μL nuclease free water, 1μL cDNA template with 0.5μL premade primer sets (SOS1: 4331182 Hs00893128_m1, SOS2: 4331182 Hs01127273_m1, GAPDH: 4331182 Hs03929097_g1; Thermo Scientific). This was followed by Amplitaq activation at 50°C for 2 minutes, 95°C for 2 minutes, and then 40 cycles at 95°C for 1 second for denaturing and at 60°C for 20 seconds for annealing and extension. We calculated ΔCt, defined as the difference between the crossover threshold (Ct) of the target gene and the Ct average of GAPDH for each sample.
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported by the Moffitt Cancer Center Support Grant to H. X. and the Tissue Core, Proteomics and Metabolomics Core, and the Biostatistics and Bioinformatics Shared Resource at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated Comprehensive Cancer Center (P30-CA076292). This study was also supported in part by University of Central Florida to Y. Y. CA265050 and Joan F. & Richard A. Abdoo Family Fund in Colorectal Cancer Research to M.E.F.-Z. The content is solely the responsibility of the authors and does not necessarily represent the official views of the sponsors or the H. Lee Moffitt Cancer Center & Research Institute or University of Central Florida, Mayo Clinic, and NCI.
ABBREVIATIONS USED
- ABCG1
ATP-binding cassette sub-family G member 1
- CHO
Chinese hamster ovary
- CI
confidence interval
- CRC
colorectal cancer
- DC50
half maximal degradation concentration
- DCM
dichloromethane
- DIEA
N,N-diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DMF
dimethylformamide
- DMPU
N,N′-dimethylpropyleneurea
- DMSO
dimethyl sulfoxide
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- EGFR
epidermal growth factor receptor
- GDP
guanosine diphosphate
- GEF
guanine nucleotide exchange factor
- GSPT1
eukaryotic peptide chain release factor GTP-binding subunit ERF3A
- GTP
guanosine triphosphate
- KRAS
Kirsten rat sarcoma virus
- IC50
half maximal inhibitory concentration
- ICI
iodine monochloride
- IKZF1/3
Ikaros family zinc finger protein 1/3
- MAPK
mitogen-activated protein kinase
- MCC
Moffitt Cancer Center
- MEK
mitogen-activated protein kinase kinase
- mRNA
messenger ribonucleic acid
- PDB
protein data bank
- PDO
patient-derived organoid
- pERK
phosphorylated extracellular signal-regulated kinase
- PI3K
phosphoinositide 3-kinase
- PROTAC
proteolysis-targeting chimera
- REU
Rosetta energy units
- SCAP
sterol regulatory element-binding protein cleavage-activating protein
- SHP2
Src homology-2 domain-containing protein tyrosine phosphatase-2
- siRNA
small interfering ribonucleic acid
- SOS1
son of sevenless homologue 1
- TFA
trifluoroacetic acid
- VHL
von Hippel-Lindau
Footnotes
Conflicts of Interest
None
Supporting information: Liquid chromatography traces, mass spectra, 1H, 19F NMR and 13C NMR spectra of compounds used for biological testing, additional figures for biological activity.
Supplemental Tables: Local protein-protein docking output file (Panel 1); Multiplexed quantitative global proteomics analyses (Panel 2–4); Molecular formula strings.
REFERENCES
- (1).Siegel RL; Miller KD; Jemal A Cancer Statistics, 2020. Ca Cancer J Clin 2020, 70 (1), 7–30. 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- (2).Lakatos G; Köhne C-H; Bodoky G Current Therapy of Advanced Colorectal Cancer According to RAS/RAF Mutational Status. Cancer Metast Rev 2020, 1–15. 10.1007/s10555-020-09913-7. [DOI] [PubMed] [Google Scholar]
- (3).Uprety D; Adjei AA KRAS: From Undruggable to a Druggable Cancer Target. Cancer Treat Rev 2020, 89, 102070. 10.1016/j.ctrv.2020.102070. [DOI] [PubMed] [Google Scholar]
- (4).Fakih MG; Kopetz S; Kuboki Y; Kim TW; Munster PN; Krauss JC; Falchook GS; Han S-W; Heinemann V; Muro K; Strickler JH; Hong DS; Denlinger CS; Girotto G; Lee M-A; Henary H; Tran Q; Park JK; Ngarmchamnanrith G; Prenen H; Price TJ Sotorasib for Previously Treated Colorectal Cancers with KRAS G12C Mutation (CodeBreaK100): A Prespecified Analysis of a Single-Arm, Phase 2 Trial. Lancet Oncol 2022, 23 (1), 115–124. 10.1016/s1470-2045(21)00605-7. [DOI] [PubMed] [Google Scholar]
- (5).Moore AR; Rosenberg SC; McCormick F; Malek S RAS-Targeted Therapies: Is the Undruggable Drugged? Nat Rev Drug Discov 2020, 19 (8), 533–552. 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Freedman TS; Sondermann H; Friedland GD; Kortemme T; Bar-Sagi D; Marqusee S; Kuriyan J A Ras-Induced Conformational Switch in the Ras Activator Son of Sevenless. Proc National Acad Sci 2006, 103 (45), 16692–16697. 10.1073/pnas.0608127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Depeille P; Henricks LM; Ven R. A. H. van de; Lemmens E; Wang C-Y; Matli M; Werb Z; Haigis KM; Donner D; Warren R; Roose JP RasGRP1 Opposes Proliferative EGFR–SOS1–Ras Signals and Restricts Intestinal Epithelial Cell Growth. Nat Cell Biol 2015, 17 (6), 804–815. 10.1038/ncb3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Jeng H-H; Taylor LJ; Bar-Sagi D Sos-Mediated Cross-Activation of Wild-Type Ras by Oncogenic Ras Is Essential for Tumorigenesis. Nat Commun 2012, 3 (1), 1168. 10.1038/ncomms2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).You X; Kong G; Ranheim EA; Yang D; Zhou Y; Zhang J Unique Dependence on Sos1 in KrasG12 D-Induced Leukemogenesis. Blood 2018, 132 (24), 2575–2579. 10.1182/blood-2018-09-874107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Abbott JR; Hodges TR; Daniels RN; Patel PA; Kennedy JP; Howes JE; Akan DT; Burns MC; Sai J; Sobolik T; Beesetty Y; Lee T; Rossanese OW; Phan J; Waterson AG; Fesik SW Discovery of Aminopiperidine Indoles That Activate the Guanine Nucleotide Exchange Factor SOS1 and Modulate RAS Signaling. J Med Chem 2018, 61 (14), 6002–6017. 10.1021/acs.jmedchem.8b00360. [DOI] [PubMed] [Google Scholar]
- (11).Abbott JR; Patel PA; Howes JE; Akan DT; Kennedy JP; Burns MC; Browning CF; Sun Q; Rossanese OW; Phan J; Waterson AG; Fesik SW Discovery of Quinazolines That Activate SOS1-Mediated Nucleotide Exchange on RAS. Acs Med Chem Lett 2018, 9 (9), 941–946. 10.1021/acsmedchemlett.8b00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Akan DT; Howes JE; Sai J; Arnold AL; Beesetty Y; Phan J; Olejniczak ET; Waterson AG; Fesik SW Small Molecule SOS1 Agonists Modulate MAPK and PI3K Signaling via Independent Cellular Responses. Acs Chem Biol 2019, 14 (3), 325–331. 10.1021/acschembio.8b00869. [DOI] [PubMed] [Google Scholar]
- (13).Burns MC; Sun Q; Daniels RN; Camper D; Kennedy JP; Phan J; Olejniczak ET; Lee T; Waterson AG; Rossanese OW; Fesik SW Approach for Targeting Ras with Small Molecules That Activate SOS-Mediated Nucleotide Exchange. Proc National Acad Sci 2014, 111 (9), 3401–3406. 10.1073/pnas.1315798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hillig RC; Sautier B; Schroeder J; Moosmayer D; Hilpmann A; Stegmann CM; Werbeck ND; Briem H; Boemer U; Weiske J; Badock V; Mastouri J; Petersen K; Siemeister G; Kahmann JD; Wegener D; Böhnke N; Eis K; Graham K; Wortmann L; Nussbaum F. von; Bader B Discovery of Potent SOS1 Inhibitors That Block RAS Activation via Disruption of the RAS–SOS1 Interaction. Proc National Acad Sci 2019, 116 (7), 2551–2560. 10.1073/pnas.1812963116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hodges TR; Abbott JR; Little AJ; Sarkar D; Salovich JM; Howes JE; Akan DT; Sai J; Arnold AL; Browning C; Burns MC; Sobolik T; Sun Q; Beesetty Y; Coker JA; Scharn D; Stadtmueller H; Rossanese OW; Phan J; Waterson AG; McConnell DB; Fesik SW Discovery and Structure-Based Optimization of Benzimidazole-Derived Activators of SOS1-Mediated Nucleotide Exchange on RAS. J Med Chem 2018, 61 (19), 8875–8894. 10.1021/acs.jmedchem.8b01108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hofmann MH; Gmachl M; Ramharter J; Savarese F; Gerlach D; Marszalek JR; Sanderson MP; Kessler D; Trapani F; Arnhof H; Rumpel K; Botesteanu D-A; Ettmayer P; Gerstberger T; Kofink C; Wunberg T; Zoephel A; Fu S-C; Teh JL; Böttcher J; Pototschnig N; Schachinger F; Schipany K; Lieb S; Vellano CP; O’Connell JC; Mendes RL; Moll J; Petronczki M; Heffernan TP; Pearson M; McConnell DB; Kraut N BI-3406, a Potent and Selective SOS1–KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov 2021, 11 (1), 142–157. 10.1158/2159-8290.cd-20-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sarkar D; Olejniczak ET; Phan J; Coker JA; Sai J; Arnold A; Beesetty Y; Waterson AG; Fesik SW Discovery of Sulfonamide-Derived Agonists of SOS1-Mediated Nucleotide Exchange on RAS Using Fragment-Based Methods. J Med Chem 2020, 63 (15), 8325–8337. 10.1021/acs.jmedchem.0c00511. [DOI] [PubMed] [Google Scholar]
- (18).Ketcham JM; Haling J; Khare S; Bowcut V; Briere DM; Burns AC; Gunn RJ; Ivetac A; Kuehler J; Kulyk S; Laguer J; Lawson JD; Moya K; Nguyen N; Rahbaek L; Saechao B; Smith CR; Sudhakar N; Thomas NC; Vegar L; Vanderpool D; Wang X; Yan L; Olson P; Christensen JG; Marx MA Design and Discovery of MRTX0902, a Potent, Selective, Brain-Penetrant, and Orally Bioavailable Inhibitor of the SOS1:KRAS Protein–Protein Interaction. J Med Chem 2022. 10.1021/acs.jmedchem.2c00741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kostic M; Jones LH Critical Assessment of Targeted Protein Degradation as a Research Tool and Pharmacological Modality. Trends Pharmacol Sci 2020, 41 (5), 305–317. 10.1016/j.tips.2020.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yang X; Yin H; Kim RD; Fleming JB; Xie H Preclinical and Clinical Advances of Targeted Protein Degradation as a Novel Cancer Therapeutic Strategy: An Oncologist Perspective. Target Oncol 2021, 16 (1), 1–12. 10.1007/s11523-020-00782-2. [DOI] [PubMed] [Google Scholar]
- (21).Wang M; Lu J; Wang M; Yang C-Y; Wang S Discovery of SHP2-D26 as a First, Potent, and Effective PROTAC Degrader of SHP2 Protein. J Med Chem 2020, 63 (14), 7510–7528. 10.1021/acs.jmedchem.0c00471. [DOI] [PubMed] [Google Scholar]
- (22).clinicaltrials.gov. A Study to Test Different Doses of BI 1701963 Alone and Combined With Trametinib in Patients With Different Types of Advanced Cancer (Solid Tumours With KRAS Mutation). https://clinicaltrials.gov/ct2/show/NCT04111458 (accessed 2022-07-06).
- (23).Singh J; Deng Z; Narale G; Chuaqui C Structural Interaction Fingerprints: A New Approach to Organizing, Mining, Analyzing, and Designing Protein–Small Molecule Complexes. Chem Biol Drug Des 2006, 67 (1), 5–12. 10.1111/j.1747-0285.2005.00323.x. [DOI] [PubMed] [Google Scholar]
- (24).Schrödinger Release 2020-3: Maestro, Schrödinger, LLC, New York, NY, 2020. [Google Scholar]
- (25).CREGG JJ; BUCKL A; Aay N; TAMBO-ONG AA; Koltun ES; Gill AL; THOMPSON S; GLIEDT MJ WO2020180770A1: Bicyclic heterocyclyl compounds and uses thereof. https://patents.google.com/patent/WO2020180770A1/en (accessed 2022-07-01).
- (26).Zaidman D; Prilusky J; London N PRosettaC: Rosetta Based Modeling of PROTAC Mediated Ternary Complexes. J Chem Inf Model 2020, 60 (10), 4894–4903. 10.1021/acs.jcim.0c00589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Gray JJ; Moughon S; Wang C; Schueler-Furman O; Kuhlman B; Rohl CA; Baker D Protein–Protein Docking with Simultaneous Optimization of Rigid-Body Displacement and Side-Chain Conformations. J Mol Biol 2003, 331 (1), 281–299. 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- (28).Donovan KA; Ferguson FM; Bushman JW; Eleuteri NA; Bhunia D; Ryu S; Tan L; Shi K; Yue H; Liu X; Dobrovolsky D; Jiang B; Wang J; Hao M; You I; Teng M; Liang Y; Hatcher J; Li Z; Manz TD; Groendyke B; Hu W; Nam Y; Sengupta S; Cho H; Shin I; Agius MP; Ghobrial IM; Ma MW; Che J; Buhrlage SJ; Sim T; Gray NS; Fischer ES Mapping the Degradable Kinome Provides a Resource for Expedited Degrader Development. Cell 2020. 10.1016/j.cell.2020.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhou C; Fan Z; Zhou Z; Li Y; Cui R; Liu C; Zhou G; Diao X; Jiang H; Zheng M; Zhang S; Xu T Discovery of the First-in-Class Agonist-Based SOS1 PROTACs Effective in Human Cancer Cells Harboring Various KRAS Mutations. J Med Chem 2021. 10.1021/acs.jmedchem.1c01774. [DOI] [PubMed] [Google Scholar]
- (30).Ishoey M; Chorn S; Singh N; Jaeger MG; Brand M; Paulk J; Bauer S; Erb MA; Parapatics K; Müller AC; Bennett KL; Ecker GF; Bradner JE; Winter GE Translation Termination Factor GSPT1 Is a Phenotypically Relevant Off-Target of Heterobifunctional Phthalimide Degraders. Acs Chem Biol 2018, 13 (3), 553–560. 10.1021/acschembio.7b00969. [DOI] [PubMed] [Google Scholar]
- (31).Matyskiela ME; Lu G; Ito T; Pagarigan B; Lu C-C; Miller K; Fang W; Wang N-Y; Nguyen D; Houston J; Carmel G; Tran T; Riley M; Nosaka L; Lander GC; Gaidarova S; Xu S; Ruchelman AL; Handa H; Carmichael J; Daniel TO; Cathers BE; Lopez-Girona A; Chamberlain PP A Novel Cereblon Modulator Recruits GSPT1 to the CRL4CRBN Ubiquitin Ligase. Nature 2016, 535 (7611), 252–257. 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- (32).Gabitova L; Restifo D; Gorin A; Manocha K; Handorf E; Yang D-H; Cai KQ; Klein-Szanto AJ; Cunningham D; Kratz LE; Herman GE; Golemis EA; Astsaturov I Endogenous Sterol Metabolites Regulate Growth of EGFR/KRAS-Dependent Tumors via LXR. Cell Reports 2015, 12 (11), 1927–1938. 10.1016/j.celrep.2015.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lee SH; Lee J-H; Im S-S The Cellular Function of SCAP in Metabolic Signaling. Exp Mol Medicine 2020, 52 (5), 724–729. 10.1038/s12276-020-0430-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mulay V; Wood P; Manetsch M; Darabi M; Cairns R; Hoque M; Chan KC; Reverter M; Álvarez-Guaita A; Rye K-A; Rentero C; Heeren J; Enrich C; Grewal T Inhibition of Mitogen-Activated Protein Kinase Erk1/2 Promotes Protein Degradation of ATP Binding Cassette Transporters A1 and G1 in CHO and HuH7 Cells. Plos One 2013, 8 (4), e62667. 10.1371/journal.pone.0062667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Drost J; Clevers H Organoids In Cancer Research. Nat Rev Cancer 2018,18, 407–418. 10.1038/s41568-018-0007-6. [DOI] [PubMed] [Google Scholar]
- (36).Pasch C; Favreau P; Yueh A; Babiarz C; Gillette A; Sharick J; Karim M; Nickel K; DeZeeuw A; Sprackling C; Emmerich P; DeStefanis R; Pitera R; Payne S; Korkos D; Clipson L; Walsh C; Miller D,; Carchman E; Burkard M; Lemmon K; Matkowskyj K; Newton M; Ong I; Bassetti M; Kimple R; Skala M; Deming D Patient-Derived Cancer Organoid Cultures to Predict Sensitivity to Chemotherapy and Radiation. Clin Cancer Res 2019, 25, 5376–5387. 10.1158/1078-0432.CCR-18-3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ooft S; Weeber F; Dijkstra K; McLean C; Kaing S; Werkhoven E; Schipper L; Hoes L; Vis D; Haar J; Prevoo W; Snaebjornsson P; Velden D; Klein M; Chalabi M; Boot H; Leerdam M; Bloemendal H; Beerepoot L; Wessels L; Cuppen E; Clevers H; Voest E Patient-derived Organoids Can Predict Response To Chemotherapy In Metastatic Colorectal Cancer Patients. Sci Transl Med 2019, 11, eaay2574. 10.1126/scitranslmed.aay2574. [DOI] [PubMed] [Google Scholar]
- (38).Vlachogiannis G; Hedayat S; Vatsiou A; Jamin Y; Fernández-Mateos J; Khan K; Lampis A; Eason K; Huntingford I; Burke R; Rata M; Koh D-M; Tunariu N; Collins D; Hulkki-Wilson S; Ragulan C; Spiteri I; Moorcraft SY; Chau I; Rao S; Watkins D; Fotiadis N; Bali M; Darvish-Damavandi M; Lote H; Eltahir Z; Smyth EC; Begum R; Clarke PA; Hahne JC; Dowsett M; Bono J. de; Workman P; Sadanandam A; Fassan M; Sansom OJ; Eccles S; Starling N; Braconi C; Sottoriva A; Robinson SP; Cunningham D;Valeri N Patient-Derived Organoids Model Treatment Response of Metastatic Gastrointestinal Cancers. Science 359, 920–926 (2018). 10.1126/science.aao2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kawaguchi M; Okabe T; Okudaira S; Nishimasu H; Ishitani R; Kojima H; Nureki O; Aoki J; Nagano T Screening and X-ray Crystal Structure-Based Optimization of Autotaxin (ENPP2) Inhibitors, Using a Newly Developed Fluorescence Probe. Acs Chem Biol 2013, 8 (8), 1713–1721. 10.1021/cb400150c. [DOI] [PubMed] [Google Scholar]
- (40).Liu J; Yuan L; Ruan Y; Deng B; Yang Z; Ren Y; Li L; Liu T; Zhao H; Mai R; Chen J Novel CRBN-Recruiting Proteolysis-Targeting Chimeras as Degraders of Stimulator of Interferon Genes with In Vivo Anti-Inflammatory Efficacy. J Med Chem 2022, 65 (9), 6593–6611. 10.1021/acs.jmedchem.1c01948. [DOI] [PubMed] [Google Scholar]
- (41).Devine WG; Diaz-Gonzalez R; Ceballos-Perez G; Rojas D; Satoh T; Tear W; Ranade RM; Barros-Álvarez X; Hol WGJ; Buckner FS; Navarro M; Pollastri MP From Cells to Mice to Target: Characterization of NEU-1053 (SB-443342) and Its Analogues for Treatment of Human African Trypanosomiasis. Acs Infect Dis 2017, 3 (3), 225–236. 10.1021/acsinfecdis.6b00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Clercq DJHD; Risseeuw MDP; Karalic I; Smet AD; Defever D; Tavernier J; Lievens S; Calenbergh SV Alternative Reagents for Methotrexate as Immobilizing Anchor Moieties in the Optimization of MASPIT: Synthesis and Biological Evaluation. Chembiochem 2015, 16 (5), 834–843. 10.1002/cbic.201402702. [DOI] [PubMed] [Google Scholar]
- (43).Chen H; Chen F; Pei S; Gou S Pomalidomide Hybrids Act as Proteolysis Targeting Chimeras: Synthesis, Anticancer Activity and B-Raf Degradation. Bioorg Chem 2019, 87, 191–199. 10.1016/j.bioorg.2019.03.035. [DOI] [PubMed] [Google Scholar]
- (44).Lu Y; Sun D; Xiao D; Shao Y; Su M; Zhou Y; Li J; Zhu S; Lu W Design, Synthesis, and Biological Evaluation of HDAC Degraders with CRBN E3 Ligase Ligands. Molecules 2021, 26 (23), 7241. 10.3390/molecules26237241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Liu J; Plewe M; Wang J; Han X; Chen L Cyclic-AMP Response Element Binding Protein (CBP) and/or Adenoviral E1A Binding Protein of 300 KDA (P300) Degradation Compounds and Methods of Use, March 9, 2020. [Google Scholar]
- (46).Robbins D; Peng G; Mihalic J; Sands A Bifunctional Compounds for Degrading BTK via Ubiquitin Proteasome Pathway, May 14, 2021. [Google Scholar]
- (47).Chamberlain PP; Lopez-Girona A; Miller K; Carmel G; Pagarigan B; Chie-Leon B; Rychak E; Corral LG; Ren YJ; Wang M; Riley M; Delker SL; Ito T; Ando H; Mori T; Hirano Y; Handa H; Hakoshima T; Daniel TO; Cathers BE Structure of the Human Cereblon–DDB1–Lenalidomide Complex Reveals Basis for Responsiveness to Thalidomide Analogs. Nat Struct Mol Biol 2014, 21 (9), 803–809. 10.1038/nsmb.2874. [DOI] [PubMed] [Google Scholar]
- (48).Schrödinger Release 2020-3: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2020; Impact, Schrödinger, LLC, New York, NY; Prime, Schrödinger, LLC, New York, NY, 2020. [Google Scholar]
- (49).Pettersen EF; Goddard TD; Huang CC; Meng EC; Couch GS; Croll TI; Morris JH; Ferrin TE UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci 2021, 30 (1), 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).R Core Team (2021). R: A Language and Environment for Statistical Computing. (Version 4.1) [Computer Software]. Retrieved from Https://Cran.r-Project.Org. (R Packages Retrieved from MRAN Snapshot 2022-01-01; ). [Google Scholar]
- (51).The Jamovi Project (2022). Jamovi. (Version 2.3) [Computer Software]. Retrieved from Https://Www.Jamovi.Org.
- (52).van de Wetering M; Francies HE; Francis JM; Bounova G; Iorio F; Pronk A; van Houdt W; van Gorp J; Taylor-Weiner A; Kester L; McLaren-Douglas A; Blokker J; Jaksani S; Bartfeld S; Volckman R; van Sluis P; Li VSW; Seepo S; Sekhar Pedamallu C; Cibulskis K; Carter SL; McKenna A; Lawrence MS; Lichtenstein L; Stewart C; Koster J; Versteeg R; van Oudenaarden A; Saez-Rodriguez J; Vries RGJ; Getz G; Wessels L; Stratton MR; McDermott U; Meyerson M; Garnett MJ; Clevers H Prospective Derivation of a Living Organoid Biobank of Colorectal Cancer Patients. Cell 2015, 161 (4), 933–945. 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Miyoshi H; Stappenbeck TS In Vitro Expansion and Genetic Modification of Gastrointestinal Stem Cells in Spheroid Culture. Nat Protoc 2013, 8 (12), 2471–2482. 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Cox J; Mann M MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat Biotechnol 2008, 26 (12), 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- (55).Welsh EA; Eschrich SA; Berglund AE; Fenstermacher DA Iterative Rank-Order Normalization of Gene Expression Microarray Data. Bmc Bioinformatics 2013, 14 (1), 153. 10.1186/1471-2105-14-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Xie Z; Bailey A; Kuleshov MV; Clarke DJB; Evangelista JE; Jenkins SL; Lachmann A; Wojciechowicz ML; Kropiwnicki E; Jagodnik KM; Jeon M; Ma’ayan A Gene Set Knowledge Discovery with Enrichr. Curr Protoc 2021, 1 (3), e90. 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.