

Abstract
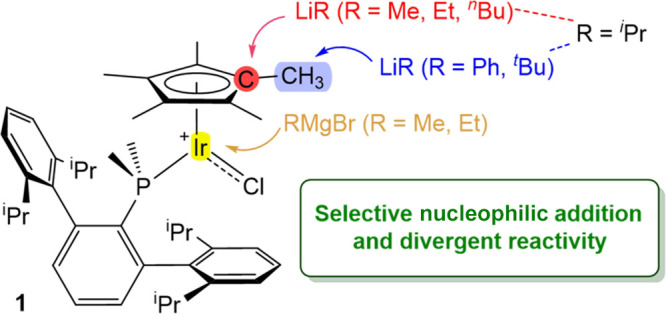
The divergent reactivity of the cationic iridium complex [(η5-C5Me5)IrCl(PMe2ArDipp2)]+ (ArDipp2 = C6H3–2,6-(C6H3–2,6-iPr2)2) toward organolithium and Grignard reagents is described. The noninnocent behavior of the Cp* ligand, a robust spectator in the majority of stoichiometric and catalytic reactions, was manifested by its unforeseen electrophilic character toward organolithium reagents LiMe, LiEt, and LinBu. In these unconventional transformations, the metal center is only indirectly involved by means of the Ir(III)/Ir(I) redox cycle. In the presence of less nucleophilic organolithium reagents, the Cp* ligand also exhibits noninnocent behavior undergoing facile deprotonation, which is also concomitant with the reduction of the metal center. In turn, the weaker alkylating agents EtMgBr and MeMgBr effectively achieve the alkylation of the metal center. These reactive iridium(III) alkyls partake in subsequent reactions: while the ethyl complex undergoes β-H elimination, the methyl derivative releases methane by a remote C–H bond activation. Computational studies, including the quantum theory of atoms in molecules (QTAIM), support that the preferential activation of the non-benzylic C–H bonds takes place via sigma-bond metathesis.
Short abstract
The divergent reactivity of the cationic iridium complex [(η5-C5Me5)IrCl(PMe2ArDipp2)]+ (ArDipp2 = C6H3−2,6-(C6H3−2,6-iPr2)2) toward organolithium and Grignard reagents is described.
Introduction
Since the serendipitous discovery of ferrocene in 1951,1,2 cyclopentadienyl ligands, [C5R5]−, have become indisputably one of the most important ligands in organometallic chemistry and homogeneous catalysis.3 In fact, their coordination complexes extend to virtually every metal in the periodic table.4−6 Their versatility is evidenced as well by their variable hapticity (from η1 to η5)7,8 and synthetic flexibility. Beyond the foremost and simplest [C5R5]− ligands, many versions have been developed, including mono and polyfunctionalized derivatives where R accounts for simple alkyl or aryl groups,9 or even bulky substituent to access extremely congested cyclopentadienyl ligands,10−16 heteroatom-containing fragments for cooperative reactivity with the metal,17,18 bridging anchors to access ansa-metallocenes,19−22 or chiral moieties to mediate asymmetric catalysis.23 However, the permethylated [C5Me5]− ligand (Cp*) is likely the one that has enjoyed the widest popularity.
A crucial driving force for the widespread use of cyclopentadienyl ligands is their robust spectator behavior, which is particularly strong in the case of Cp*. However, even for the later ligand, there are increasing examples of its noninnocent character (Figure 1). The methyl groups of Cp* can partake in several transformations, including, but not limited to, deprotonation by an external base or a bifunctional ligand,24−37 hydride abstraction which tends to proceed through single-electron processes,38−40 C–H oxidative addition to an adjacent transition metal in bimetallic structures,41−48 or direct and reversible methyl-to-metal hydride migration, which was soon identified in early transition metals49−52 and recently unlocked by our group as a viable process for late transition metals.53 In addition, the protonation of the internal ring has been exploited in proton-couple-electron-transfer (PCET) catalysis capitalizing on the reversible migration of the proton between the ring and the metal.54−60 Moreover, several radical routes have been identified for Cp*-containing species resulting as well in ligand functionalization.61
Figure 1.
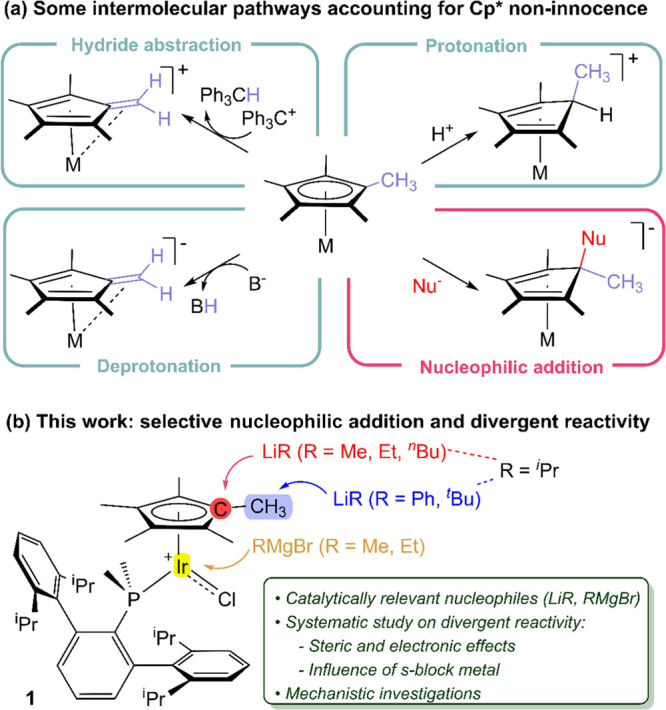
(a) Most common intermolecular pathways for the activation of the Cp* ligand in transition metal complexes; (b) Systematic study revealing divergent reactivity of complex 1 upon reaction with highly polarized carbon nucleophiles.
In contrast, the reactivity of the internal carbon centers of Cp* toward nucleophiles has only been observed in a limited number of cases,62 being more frequent on the less electron-rich and sterically hindered [C5H5]− upon addition of common highly polar reagents, typically organolithium and organomagnesium compounds.63−73 These transformations are of high relevance for a variety of catalytic processes involving cyclopentadienyl catalysts.74−82 For instance, cyclopentadienyl nickel and iron complexes are very active Kumada cross-coupling catalysts with organomagnesium reagents83−88 or for the polymerization of the latter.89 Organolithium and organomagnesium species are also used as initiators for olefin polymerization or diene isomerization with related catalysts.90,91 Besides, the use of organolithium and organomagnesium reagents in the presence of Cp*M complexes of both early92−100 and late-transition metals101−109 have been reported in many occasions, but the direct reactivity of the Cp* ligand has been overlooked in all cases. Cyclopentadienyl ligands, in particular Cp*, continue to be extensively employed in fundamental organometallic chemistry and homogeneous catalysis. On these bases, understanding these unforeseen reactions is crucial to avoid catalyst deactivation110 or undesired catalytic outcomes,111−113 and to further extend the utility of this platform beyond current capabilities, while gaining insight into the formation of active species from Cp*-bearing precatalysts in the presence of bases.114−116
With this goal, we have selected our recently published terphenyl phosphine iridium compound 1 [(η5-C5Me5)Ir(Cl)(PMe2ArDipp2)][BArF] (ArDipp2 = C6H3–2,6-(C6H3–2,6-iPr2)2)28,117 to carry out a systematic study of its reactivity toward highly polarized organolithium and organomagnesium reagents. This platform is particularly attractive for these endeavors because (i) it presents a vacant coordination site at the electrophilic Ir(III) center and a chloride ligand susceptible of participating in salt metathesis, yet both the ring and methyl groups of the Cp* can react preferentially toward nucleophiles and/or bases; (ii) the proven noninnocence of the Cp* ligand in this complex, encompassing deprotonation, reversible C–C bond formation and C–H bond breaking;28 (iii) its great stability toward cyclometallation;28 (iv) the possibility of accessing a bulkier analogue of Bergman’s complex [(η5-C5Me5)Ir(Me)(PMe3)(ClCH2Cl)]+;116,118 and (v) the in general prominent position of Cp*Ir complexes in the field of C–H bond activation.118−124
Results and Discussion
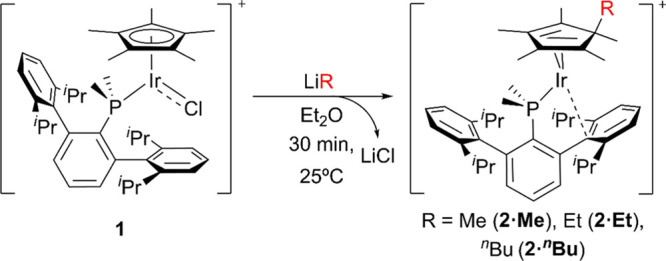
To start this systematic study, we first examined the equimolar reaction of complex 1 with the common nucleophile LiMe. As stated above, and considering the reduced size of the methyl anion, we anticipated the methyl group to either fill the vacancy of this unsaturated Ir(III) complex or replace the chloride to access a Bergman-type complex118 [(η5-C5Me5)Ir(Me)(PR3)(ClCH2Cl)]+. To our surprise, the only discernible product, which we fully characterized, is a cationic Ir(I) complex (2·Me) featuring a new methyl group bonded to one of the internal carbon atoms of the former Cp* ligand, as shown in Scheme 1. Analogous reactivity was found with lithium alkyls LiEt and LinBu (Scheme 1), whose equimolar addition to the iridium precursor 1 led, respectively, to compounds 2·Et and 2·nBu, in which a new hydrocarbyl fragment is installed in the exo-face of the parent Cp* ligand.
Scheme 1. Syntheses of Complexes 2·Me, 2·Et, and 2·nBu from 1 and LiMe, LiEt, and LinBu, Respectively.
The room temperature 1H NMR spectrum of complex 2·Me features broad resonances, suggestive of a dynamic solution process. This fluxional behavior arises from the rotation of the C5Me6 fragment, presumably through a tetrahedral coordination environment,124,125 and not from the exchange of the flanking Dipp rings of the phosphine ligand, according to exchange spectroscopy (EXSY) experiments (see Figure S4). However, at −20 °C, complex 2·Me exhibits a rigid solution structure providing sharp, well-resolved resonances. The absence of symmetry present in 2·Me results in a complex 1H NMR spectrum—six singlets, each with relative intensity corresponding to 3 H, are recorded in the 1.84–0.32 ppm range for the Me groups of the newly formed C5Me6 diene ligand. Likewise, the four Dipp iso-propyl substituents are inequivalent and originate corresponding multiplets centered at 2.64, 2.32, 2.15, and 2.00 ppm (see Section 2.1 of the SI and Figure S2) for the methine CHMe2 protons. As a means to compensate unsaturation, complex 2·Me features a secondary π-arene interaction125,126 with the metal center revealed by the low-frequency shift of one of the ipso carbon atoms of the flanking aryl rings (120.4 ppm, cf. the 135.7 ppm value for the corresponding carbon of the noncoordinated Dipp ring) and further supported by topological analysis and Energy Decomposition Analysis - Natural Orbital for Chemical Valence (EDA-NOCV)129 (see Sections 5.16.2 and 5.17.1 of the SI). One of the ortho carbon atoms of this ring seems to also participate in the bonding, resulting in η2-coordination of the arene, as its chemical shift (132.5 ppm) is significantly shifted to lower frequencies compared to its counterparts (141.5 ppm for the other ortho carbon within the same ring, and 146.5 and 146.9 ppm for the ones belonging to the nonbound Dipp). Interestingly, the 13C{1H} NMR spectrum of complex 2·Me also exhibits a clear difference in the chemical shift of the two pairs of carbons involved in the two formal double bonds of the C5Me6 unit (122.5 and 114.6 vs 78.3 and 61.9 ppm). This experimental evidence together with the longer C–C distance for the formal double bond trans to the phosphane (C34–C33: 1.431(6) vs C35–C36: 1.338(6) Å) and corresponding closer distance to the metal center (C34–Ir1: 2.118(5) and C33–Ir1: 2.163(4) vs C35–Ir1: 2.265(5) and C36–Ir1: 2.437(4) Å) support our hypothesis that the formal coordinated diene is closer in nature to a double bond and a metalacyclopropane. This can be explained by the stronger trans influence of the phosphane, also observed in a closely related system.126−128 EDA-NOCV studies further sustain this idea, revealing a major contribution of the carbon atoms with the longer C–C bond distance to the principal orbital interactions (see Section 5.17.2 of the SI). These spectroscopic features are similar to those found for compounds 2·Et and 2·nBu, whose full characterization is included in the Supporting Information.
The molecular formulation of the new compounds was corroborated by X-ray diffraction studies, confirming the exo attack on the Cp* and revealing a preferred η1-arene coordination in the solid state, rather than η2-binding as inferred from spectroscopic analysis. Thus, in complex 2·Me, the Ir–Carene bonding is characterized by an Ir–Cipso bond distance of 2.249(4) Å, and by significantly longer, and therefore weaker, Ir–Cortho interactions of length 2.544(5) and 2.686(4) Å (Figure 2). Similar geometric parameters are found in compounds 2·Et and 2·nBu, with notably shorter Ir–Cipso (2.231(5), 2·Et; 2.253(5) Å, 2·nBu) distances compared to Ir–Cortho (2.549(5), 2·Et; 2.593(6) Å, 2·nBu) interactions.
Figure 2.

ORTEP diagrams of the cation of complexes 2·Me, 2·Et, 2·iPr, and 2·nBu. Hydrogen atoms are excluded for clarity and thermal ellipsoids are set at 50% probability. Wireframe is used to represent the iso-propyl groups.
We carried out Density Functional Theory studies to gain insight into the mechanism of the reactions depicted in Scheme 1. For convenience, we focused on the relatively simpler LiMe. Our attempts to rationalize this reactivity through classical foregoing routes involving reductive coupling processes between the Cp* ligand and an Ir—Me functionality failed to provide energy barriers in agreement with experimental observations (see Figures S25 and S26). This led us to explore a more unconventional reaction pathway in which the metal center does not directly participate, and, instead, the direct attack of the LiMe molecule to the exo face of the Cp* moiety takes place. The transition state of the C–C bond formation step requires surmounting a barrier of only 8.0 kcal/mol and yields a neutral Ir(I) complex at −39.2 kcal/mol relative to the reactants. Subsequent chloride release assisted by the solvated lithium atom gave complex 2·Me through an accessible barrier (see Figures 3 and S24).
Figure 3.

Free energy profiles of LiMe attacking one of the internal carbon atoms of the Cp* moiety (left) and LiPh abstracting a proton from one of the methyl groups of the Cp* moiety (right). S = Me2O.
In contrast, the less nucleophilic lithium alkyls LiPh and LitBu acted instead as Brønsted-Lowry bases deprotonating one of the methyl groups of the Cp* moiety. As shown in Scheme 2, this event triggers a complex rearrangement involving reversible C–C bond formation that leads to a pseudoallylic structure, complex 3, previously reported by our group by the reaction with the much milder base NEt3.28 It is remarkable that the unexpected electrophilicity of the internal carbon atoms of the C5Me5 ring outcompetes the mild although well-known Brønsted-Lowry acidity of the C–H bonds, even with bases around 40 pKa units stronger than NEt3.
Scheme 2. Obtention of Complex 3, Final Product of the Reaction between Complex 1 and LitBu or LiPh.

Conditions: alkyl lithiums were added at −78 °C; solution was left to reach room temperature.
To rationalize such striking divergence in reactivity, DFT studies were performed to calculate the energy profiles for LiPh acting as a nucleophile or as a base. In agreement with the experimental observations, a lower energy barrier for the deprotonation step (6.3 kcal/mol, Figure 3) was obtained in comparison to the attack to an internal carbon atom of the Cp* (8.3 kcal/mol, see Figure S22). In both cases, square-planar Ir(I) species appear to be key intermediates. For completion, the energy barrier for the proton abstraction by the LiMe molecule was also calculated obtaining a TS at 11.5 kcal/mol, and thus, higher than the one belonging to the methylation pathway (see Figure S23).
Bearing in mind the contrasting reactivity of alkyl lithium reagents, in particular LinBu and LitBu, we wondered about the outcomes of an intermediate situation in terms of steric and electronic properties of the carbon nucleophile. Thus, we examined the reactivity of 1 with one equivalent of LiiPr. Not surprisingly, iso-propyl lithium finds its place between the two aforementioned cases, as the reaction between complex 1 and LiiPr yields a mixture of complex 2·iPr and complex 3 in a ca. 1:7 ratio, along with other minor unidentified species. Although complex 2·iPr could not be isolated in pure form, we could monitor its formation, along with that of 3, by 31P{1H} NMR spectroscopy (Figure S8) and characterize it crystallographically (Figure 2). The formation of a mixture is consistent with the close DFT-calculated barriers for the deprotonation and nucleophilic attack pathways (Figure S34).
As introduced earlier, organolithium reagents have been widely used in the chemistry of Cp*-containing complexes. Nonetheless, the weaker alkylating Grignard reagents have been even more commonly used and their implications in catalysis are broader.90,91 Therefore, we explored the reactivity of complex 1 toward less-polarized Grignard reagents. The addition of equimolar amounts of EtMgBr to diethyl ether solutions of the cationic chloride complex 1 resulted in an instantaneous color change from dark to orange due to the formation of a new species, complex 4 (Scheme 3). In stark contrast to the reactivity exhibited by organolithium reagents, the integrity of the Cp* ligand remains intact when milder Grignard reagents are used, which represents a remarkable divergent reactivity associated to common chemicals that are on many occasions used indistinctly. The coordinatively saturated complex 4 features a 31P{1H} NMR resonance at −27.0 ppm, therefore showing a large δ shift relative to that of complex 1 (6.6 ppm) and closer to free PMe2ArDipp2 (−41.3 ppm), supporting the absence of the aforementioned Ir–Carene π-interactions.126,129,130 A distinctive low-frequency doublet in the 1H NMR spectrum (δ −14.9 ppm, 2JHP = 30.2 Hz) indicates the presence of an iridium hydride, while a coordinated ethylene molecule gives rise to two resonances at 2.18 and 1.88 ppm. X-ray diffraction studies confirmed the proposed formulation and revealed a C–C bond length of 1.426(1) Å (Figure 4) for the ethylene ligand, as expected, longer than that of noncoordinated ethylene (1.3305 Å).129 A reasonable proposal for the mechanism of the reaction leading to complex 4 is the substitution of the chloride ligand by an ethyl group with concomitant precipitation of LiCl, followed by β-hydride elimination. Further insight into this proposed mechanism was obtained by DFT studies (see Figure S21). These revealed a low barrier (3.1 kcal/mol) for the formation of an agostic interaction131−133 between a C–H bond of the CH3 end of the ethyl group and the Ir atom, followed by almost barrierless β-hydride elimination (ΔG‡ = 0.1 kcal/mol, relative to the agostic complex). These low barriers are congruent with experimental observations, as attempts to spectroscopically detect the Ir–Et intermediate were unsuccessful even at low temperatures.
Scheme 3. Synthesis of Complex 4 from 1 and EtMgBr.
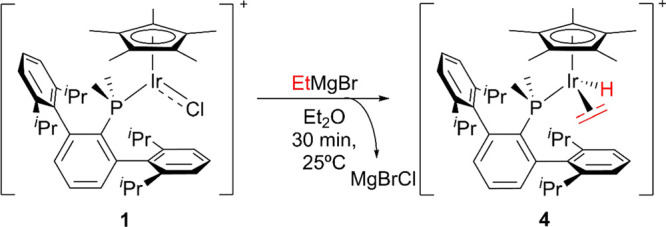
Figure 4.
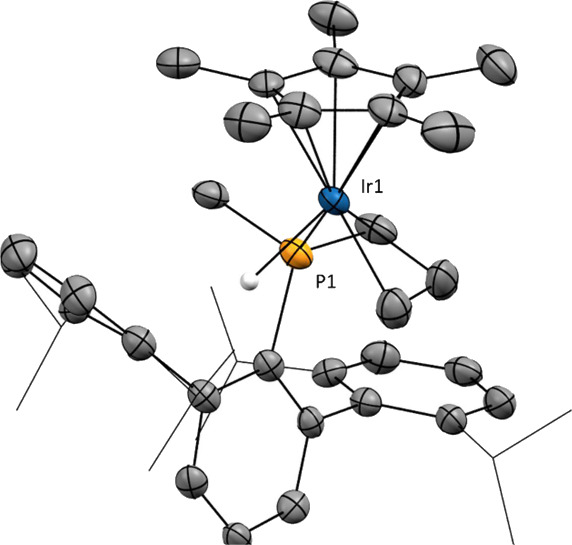
ORTEP diagram of the cation of complex 4. All hydrogen atoms but those of the hydride are excluded for clarity and thermal ellipsoids are set at 50% probability. Wireframe is used to represent the iso-propyl groups.
The results described above promised the obtention of the analogue of Bergman’s complex reacting MeMgBr with complex 1 due to the lack of hydrogen atoms in the β position in the expected Ir–Me complex. Once more, the use of a magnesium reagent circumvented the direct nucleophilic attack to the Cp* ring, which remained unaltered, yet the observed product of this reaction was complex 5, derived from the remote and selective activation of a non-benzylic C(sp3)–H bond of one iso-propyl group of a lateral terphenyl ring (Scheme 4). At variance with the analogous ethyl reagent, the methyl fragment does not remain at the structure of 5 and instead evolves as methane, which could be observed by careful NMR monitoring (1H NMR at 0.23 ppm). This reactivity also contrasts with the cyclometallation selectivity previously shown by this system, where the benzylic methine C–H bond is more amenable to activation.28 Complex 5 was fully characterized by multinuclear NMR spectroscopy. Three distinctive 1H multiplets, at 3.35, 0.73, and 0.22 ppm, each with relative intensity corresponding to 1 H, were assigned to the CH and the diastereotopic protons of the CH2 of the Ir–CH2CHCH3 moiety, respectively. The molecular structure was authenticated by X-ray diffraction studies, which also indicate that the metal center achieves coordinative saturation by means of an η2-arene interaction with the flanking arene (Figure 5). Other geometrical parameters are similar to previous complexes and do not require further discussion.
Scheme 4. Synthesis of Complex 5 from 1 and MeMgBr through Proposed Intermediate Complex A.
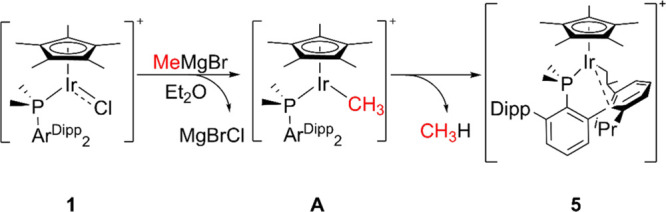
Figure 5.
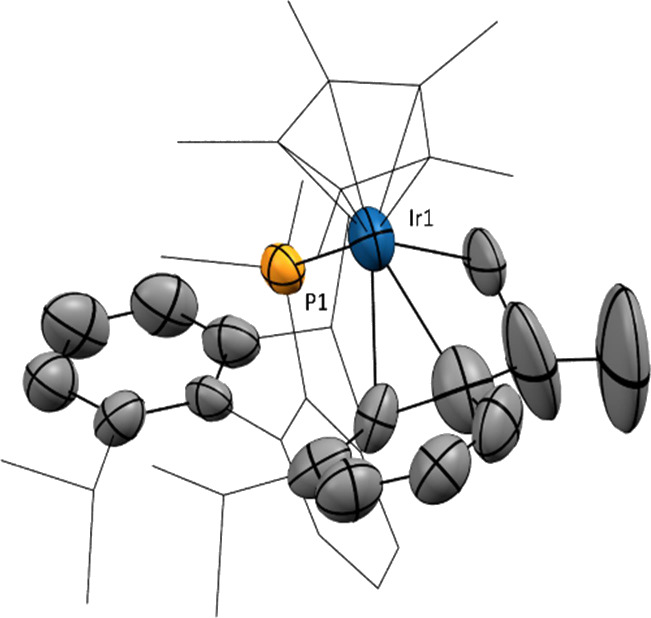
ORTEP diagram of the cation of complex 5. Hydrogen atoms are excluded for clarity and thermal ellipsoids are set at 50% probability. Wireframe is used to represent the Cp* ligand, the central aryl group of the phosphine, and the iso-propyl groups.
The mechanism of the reaction depicted in Scheme 4 was also studied through the DFT methodology. The direct attack of MeMgCl to the iridium center yielding a neutral Ir(I) complex was found to be inaccessible (Figure S28), which led us to explore an alternative mechanism. The formation of the Bergman’s type Ir–CH3 complex134 (A in Scheme 4) commences through the magnesium-assisted chloride release (ΔG‡ = 21.0 kcal/mol), yielding a dicationic Ir(III) complex at 11.4 kcal/mol. This readily reacts with the generated (MeMgCl2) moiety, alkylating the metal center with concomitant release of MgCl2 (ΔG‡ = 19.7 kcal/mol) and leading to intermediate A at −17.8 kcal/mol relative to the reactants (Figure S27). For comparison, the reaction pathway of MeMgCl attacking one of the internal carbon atoms of the Cp* was also calculated. This route involves a higher-in-energy TS (28.1 kcal/mol), which explains the selectivity of the reaction between 1 and MeMgCl (Figure S27).
We evaluated three different pathways for the release of methane from intermediate A, comprising the activation of either one of the two methyl termini of an iso-propyl group or the methine CH. Despite benzylic C–H bonds being usually more prone to metalate, the connectivity of 5 points in a different direction, as supported by our computational studies (Figure 6). As observed experimentally, the activation of the methyl groups is kinetically favored relative to the benzylic methine, despite the latter yielding the most stable product. Notably, the activation of the distinct methyl groups follows different mechanisms: in one case, formation of an agostic interaction133 leads to sigma bond metathesis (Figure S28 and Table S3). In contrast, the activation of the other methyl group, as well as for the methine CH, involved the formation of Ir(V) hydride complexes as intermediates.
Figure 6.

Three different reaction pathways for the elimination of methane from the proposed Ir(III) intermediate A. Zero energy corresponds to that of the optimized Ir(III) methylated complex. 5′ is a diastereoisomer of 5.
When a pure solution of complex 5 was able to evolve at room temperature for 5 days, its 31P{1H} NMR spectrum revealed the emergence of two new peaks at 9.4 and 8.2 ppm. After heating this solution at 80 °C for 5 h, conversion to the species resonating at 9.4 ppm, identified as the thermodynamically more stable complex 3, was complete. As the reaction between 1 and MeMgBr was originally carried out in a closed J. Young tube, one possible isomerization mechanism would be the reaction of 5 with CH4 leading to obtaining the most stable compound 3. However, DFT calculations showed that the energy barrier would be too high (ΔG‡ = 45.3 kcal/mol) (Figure 6), and experimentally, we found that isomerization also took place in the absence of CH4. Although the complex resonating at 8.2 ppm could not be isolated, its multinuclear NMR pattern perfectly fits with its assignment as a diastereoisomer of 5 (compound 5′ in Figure 6) resulting from the activation of the alternative methyl group (Figures S18–S20). DFT calculations for the isomerization process are currently ongoing.
Conclusions
In conclusion, we demonstrate that the noninnocent character of the widespread Cp* ligand in the presence of strongly polarized alkylating reagents is highly dependent on the nature of the carbon nucleophile. Thus, we identify up to three dissimilar reaction outcomes for the same Ir(III) precursor depending on the substrate employed. First, the Cp* displays an uncommon but clear electrophilic character toward unhindered lithium alkyls—LiMe, LiEt, LinBu—undergoing alkylation of one of the internal carbon atoms of the Cp* ring through a direct nucleophilic attack to its exo face, leading to the formal reduction of the metal toward Ir(I) complexes. In contrast, less nucleophilic lithium reagents such as LiPh and LitBu act as Brønsted bases, effecting the deprotonation of a methyl group of the Cp* ring. This event triggers a rearrangement that leads to the formation of a previously reported pseudoallylic structure. Interestingly, the use of LiiPr, with intermediate steric and electronic properties, leads to a mixture of the aforesaid structures. In stark contrast, the use of weaker alkylating magnesium agents, which tend to exhibit similar chemistry to organolithium compounds in the context of transition metal alkylations, enables the selective alkylation of the metal center, while the Cp* ligand remains intact. Moreover, a series of subsequent C–H bond activation events have been disclosed for the resulting iridium complexes.
Overall, the foregoing results represent a clear illustration of both the different reactivity of some of the most common reagents in organometallic chemistry, Grignard and organolithium reagents, in many cases exchangeable, and the noninnocent behavior of the Cp* ligand, which continues to be one of the most utilized ligands in organometallic chemistry. Gaining a deep understanding of reactions where Cp* ligand is not as a mere spectator, but an active contributor, is of crucial importance for the discovery of novel transformations and the development of future catalytic processes that rely on the use of this and related ligand frameworks. Indeed, the results of this study advise taking a fresh look at the comprehensive body of work on cyclopentadienyl-based transition metal catalysts that operate in the presence of strongly polarized organometallic reagents, nucleophiles, and bases.
Acknowledgments
This work has been supported by the European Research Council (ERC Starting Grant, CoopCat, 756575). We also thank Grant PID2019-110856GA-I00 funded by MCIN/AEI/ 10.13039/501100011033, Junta de Andalucía (P18-FR-4688) and US/JUNTA/FEDER, UE (US-1380849). J.J.M. thanks Junta de Andalucía for the postdoctoral program “Personal Investigador Doctor” (ref. DOC_00153). The authors gratefully acknowledge the financial support provided by the Consejo Superior de Investigaciones Científicas (studentship JAEINT_21_00695 granted to A.P.-M.) and the use of CESGA computational facilities.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.2c04381.
Author Contributions
A.P.-M., J.J.M., and M.G.A. synthesized and characterized all compounds. A.P. and J.J.M. carried out computational studies. M.G.A., C.M., and M.F.E. carried out XRD studies. A.P.-M. wrote the original draft. J.C. supervised the overall project. All authors contributed to review and editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Kealy T. J.; Pauson P. L. A New Type of Organo-Iron Compound. Nature 1951, 168, 1039–1040. 10.1038/1681039b0. [DOI] [Google Scholar]
- Miller A.; Tebboth J. A.; Tremaine F. Dicyclopentadienyliron. J. Chem. Soc. 1952, 632–635. 10.1039/JR9520000632. [DOI] [Google Scholar]
- Crabtree R. H. NHC Ligands versus Cyclopentadienyls and Phosphines as Spectator Ligands in Organometallic Catalysis. J. Organomet. Chem. 2005, 5451–5457. 10.1016/j.jorganchem.2005.07.099. [DOI] [Google Scholar]
- Poli R. Monocyclopentadienyl Halide Complexes of the d- and f-Block Elements. Chem. Rev. 1991, 91, 509–551. 10.1021/cr00004a004. [DOI] [Google Scholar]
- Budzelaar P. H. M.; Engelberts J. J.; van Lenthe J. H. Trends in Cyclopentadienyl–Main-Group-Metal Bonding. Organometallics 2003, 22, 1562–1576. 10.1021/om020928v. [DOI] [Google Scholar]
- Evans W. J. Tutorial on the Role of Cyclopentadienyl Ligands in the Discovery of Molecular Complexes of the Rare-Earth and Actinide Metals in New Oxidation States. Organometallics 2016, 35, 3088–3100. 10.1021/acs.organomet.6b00466. [DOI] [Google Scholar]
- O’Connor J. M.; Casey C. P. Ring-Slippage Chemistry of Transition Metal Cyclopentadienyl and Indenyl Complexes. Chem. Rev. 1987, 87, 307–318. 10.1021/cr00078a002. [DOI] [Google Scholar]
- Veiros L. F. The Role of Haptotropic Shifts in Phosphine Addition to Tricarbonylmanganese Organometallic Complexes: The Indenyl Effect Revisited. Organometallics 2000, 19, 3127–3136. 10.1021/om000195j. [DOI] [Google Scholar]
- Deck P. A. Perfluoroaryl-Substituted Cyclopentadienyl Complexes of Transition Metals. Coord. Chem. Rev. 2006, 250, 1032–1055. 10.1016/j.ccr.2005.11.001. [DOI] [Google Scholar]
- Ruspic C.; Moss J. R.; Schürmann M.; Harder S. Remarkable Stability of Metallocenes with Superbulky Ligands: Spontaneous Reduction of SmIII to SmII. Angew. Chem., Int. Ed. 2008, 47, 2121–2126. 10.1002/anie.200705001. [DOI] [PubMed] [Google Scholar]
- Meyer G. Superbulky Ligands and Trapped Electrons: New Perspectives in Divalent Lanthanide Chemistry. Angew. Chem., Int. Ed. 2008, 47, 4962–4964. 10.1002/anie.200801444. [DOI] [PubMed] [Google Scholar]
- Harder S.; Naglav D.; Schwerdtfeger P.; Nowik I.; Herber R. H. Metal Atom Dynamics in Superbulky Metallocenes: A Comparison of (CpBIG)2Sn and (CpBIG)2Eu. Inorg. Chem. 2014, 53, 2188–2194. 10.1021/ic4028546. [DOI] [PubMed] [Google Scholar]
- van Velzen N. J. C.; Harder S. Deca-Arylsamarocene: An Unusually Inert Sm(II) Sandwich Complex. Organometallics 2018, 37, 2263–2271. 10.1021/acs.organomet.8b00254. [DOI] [Google Scholar]
- Giesbrecht G. R.; Gordon J. C.; Clark D. L.; Scott B. L. Synthesis, Structure and Solution Dynamics of Lithium Salts of Superbulky Cyclopentadienyl Ligands. Dalton Trans. 2003, 3, 2658–2665. 10.1039/b302258g. [DOI] [Google Scholar]
- Orzechowski L.; Piesik D. F. J.; Ruspic C.; Harder S. Superbulky Metallocene Complexes of the Heavier Alkaline-Earth Metals Strontium and Barium. Dalton Trans. 2008, 35, 4742–4746. 10.1039/b809872g. [DOI] [PubMed] [Google Scholar]
- Harder S.; Ruspic C. Insight in Cyclopentadienyl Metal Complexes with Superbulky Ligands: The Crystal Structure of [CpBIGK]∞. J. Organomet. Chem. 2009, 694, 1180–1184. 10.1016/j.jorganchem.2008.09.073. [DOI] [Google Scholar]
- Blais M. S.; Rausch M. D. A new synthetic route to functionally substituted (η5-cyclopentadienyl) dicarbonyliridium compounds. J. Organomet. Chem. 1995, 502, 1. 10.1016/0022-328X(95)05627-2. [DOI] [Google Scholar]
- Conway B. G.; Rausch M. D. Formation and reactivity of halogen derivatives of (η5-cyclopentadienyl)thallium. Organometallics 1985, 4, 688. 10.1021/om00123a013. [DOI] [Google Scholar]
- Chen E. Y.-X.; Kruper W. J.; Roof G.; Wilson D. R. “Double Activation” of Constrained Geometry and Ansa-Metallocene Group 4 Metal Dialkyls: Synthesis, Structure, and Olefin Polymerization Study of Mono- and Dicationic Aluminate Complexes. J. Am. Chem. Soc. 2001, 123, 745–746. 10.1021/ja002654d. [DOI] [PubMed] [Google Scholar]
- Mitchell J. P.; Hajela S.; Brookhart S. K.; Hardcastle K. I.; Henling L. M.; Bercaw J. E. Preparation and Structural Characterization of an Enantiomerically Pure, C2-Symmetric, Single-Component Ziegler-Natta α-Olefin Polymerization Catalyst. J. Am. Chem. Soc. 1996, 118, 1045–1053. 10.1021/ja953419b. [DOI] [Google Scholar]
- Busico V.; Cipullo R. Influence of Monomer Concentration on the Stereospecificity of 1-Alkene Polymerization Promoted by C2-Symmetric Ansa-Metallocene Catalysts. J. Am. Chem. Soc. 1994, 116, 9329–9330. 10.1021/ja00099a060. [DOI] [Google Scholar]
- Wang B. Ansa-Metallocene Polymerization Catalysts: Effects of the Bridges on the Catalytic Activities. Coord. Chem. Rev. 2006, 250, 242–258. 10.1016/j.ccr.2005.05.012. [DOI] [Google Scholar]
- Mas-Roselló J.; Herraiz A. G.; Audic B.; Laverny A.; Cramer N. Chiral Cyclopentadienyl Ligands: Design, Syntheses, and Applications in Asymmetric Catalysis. Angew. Chem., Int. Ed. 2021, 60, 13198–13224. 10.1002/anie.202008166. [DOI] [PubMed] [Google Scholar]
- Bernechea M.; Berenguer J. R.; Lalinde E.; Torroba J. Facile Single or Double C-H Bond Activation on a Cp Ligand Promoted by the Presence of Alkynylphosphine Ligands. Organometallics 2009, 28, 312–320. 10.1021/om800867h. [DOI] [Google Scholar]
- Fan L.; Wei C.; Aigbirhio F. I.; Turner M. L.; Gusev O. V.; Morozova L. N.; Knowles D. R. T.; Maitlis P. M. Ring-Methyl Activation in Pentamethylcyclopentadienyl Complexes. 5. Syntheses and Structures of Tetramethylfulvene Complexes of Ruthenium(II). Organometallics 1996, 15, 98–104. 10.1021/om950462z. [DOI] [Google Scholar]
- Glueck D. S.; Bergman R. G. Deprotonation of a Cp* Methyl Group by an Iridium Anilide: Formation, Structure, and Solution Dynamics of an Η4-Tetramethylfulvene Complex. Organometallics 1990, 9, 2862–2863. 10.1021/om00161a006. [DOI] [Google Scholar]
- Caldwell H.; Pregosin P. S. Intramolecular Allylation of a Ru-Cp* Methyl Group. Organometallics 2008, 27, 1591–1595. 10.1021/om701248d. [DOI] [Google Scholar]
- Moreno J. J.; Espada M. F.; Campos J.; López-Serrano J.; Macgregor S. A.; Carmona E. Base-Promoted, Remote C-H Activation at a Cationic (η5-C5Me5)Ir(III) Center Involving Reversible C-C Bond Formation of Bound C5Me5. J. Am. Chem. Soc. 2019, 141, 2205–2210. 10.1021/jacs.8b11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto S.; Morita H.; Karitani K.; Fujiwara H.; Matsuzaka H. A Bimetallic Ru2Pt Complex Containing a Trigonal-Planar Μ3-Carbido Ligand: Formation, Structure, and Reactivity Relevant to the Fischer–Tropsch Process. J. Am. Chem. Soc. 2009, 131, 18026–18027. 10.1021/ja907387w. [DOI] [PubMed] [Google Scholar]
- Cloke F. G. N.; Green J. C.; Green M. L. H.; Morley C. P. Metal Atom Synthesis and Photochemistry of Bis(η-Pentamethylcyclopentadienyl)-Tungsten Dihydride. Chem. Commun. 1985, 14, 945–946. 10.1039/C39850000945. [DOI] [Google Scholar]
- Riley P. N.; Parker J. R.; Fanwick P. E.; Rothwell I. P. Formation of Tantalum “Tuck-in” Complexes by Activation of Methyl C–H Bonds in Pentamethylcyclopentadiene Groups by Carbazole Ligation. Organometallics 1999, 18, 3579–3583. 10.1021/om990388a. [DOI] [Google Scholar]
- Kupfer V.; Thewalt U.; Tišlerová I.; Štěpnička P.; Gyepes R.; Kubišta J.; Horáček M.; Mach K. Syntheses and Structures of Doubly Tucked-in Titanocene Complexes with Tetramethyl(Aryl)Cyclopentadienyl Ligands. J. Organomet. Chem. 2001, 620, 39–50. 10.1016/S0022-328X(00)00784-1. [DOI] [Google Scholar]
- Sun Y.; Spence R. E. V. H.; Piers W. E.; Parvez M.; Yap G. P. A. Intramolecular Ion–Ion Interactions in Zwitterionic Metallocene Olefin Polymerization Catalysts Derived from “Tucked-In” Catalyst Precursors and the Highly Electrophilic Boranes XB(C6F5)2 (X = H, C6F5). J. Am. Chem. Soc. 1997, 119, 5132–5143. 10.1021/ja970140h. [DOI] [Google Scholar]
- Mori Y.; Ando T.; Matsumoto T.; Yatabe T.; Kikkawa M.; Yoon K.-S.; Ogo S. Multifunctional Catalysts for H2O2-Resistant Hydrogen Fuel Cells. Angew. Chem., Int. Ed. 2018, 57, 15792–15796. 10.1002/anie.201810270. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Bárzano A.; Blacker A. J.; McGowan P. C. Synthesis and Characterisation of Tetramethylfulvene Complexes of Ruthenium. Dalton Trans. 2013, 42, 16669–16671. 10.1039/C3DT52747F. [DOI] [PubMed] [Google Scholar]
- Holland P. L.; Andersen R. A.; Bergman R. G.; Huang J.; Nolan S. P. Monomeric Cyclopentadienylnickel Methoxo and Amido Complexes: Synthesis, Characterization, Reactivity, and Use for Exploring the Relationship between H–X and M–X Bond Energies. J. Am. Chem. Soc. 1997, 119, 12800–12814. 10.1021/ja971829p. [DOI] [Google Scholar]
- Rüba E.; Mereiter K.; Schmid R.; Kirchner K.; Bustelo E.; Puerta M. C.; Valerga P. Reactions of RuCp and RuCp* Allyl Carbene Complexes: Products Derived from Activation of Phenyl, Cyclohexyl, and Methyl C–H Bonds in PPh3, PCy3, and Cp* Ligands. Organometallics 2002, 21, 2912–2920. 10.1021/om020094g. [DOI] [Google Scholar]
- Klet R. C.; Kaphan D. M.; Liu C.; Yang C.; Kropf A. J.; Perras F. A.; Pruski M.; Hock A. S.; Delferro M. Evidence for Redox Mechanisms in Organometallic Chemisorption and Reactivity on Sulfated Metal Oxides. J. Am. Chem. Soc. 2018, 140, 6308–6316. 10.1021/jacs.8b00995. [DOI] [PubMed] [Google Scholar]
- Li S.; Wang X.; Zhang Z.; Zhao Y.; Wang X. Isolation and Structural Characterization of a Mainly Ligand-Based Dimetallic Radical. Dalton Trans. 2015, 44, 19754–19757. 10.1039/C5DT03578C. [DOI] [PubMed] [Google Scholar]
- Meredith J. M.; Goldberg K. I.; Kaminsky W.; Heinekey D. M. η6 -Tetramethylfulvene and μ-Η3 : η3-Benzene Complexes of Iridium. Organometallics 2012, 31, 8459–8462. 10.1021/om3010492. [DOI] [Google Scholar]
- Einstein F. W. B.; Jones R. H.; Zhang X.; Yan X.; Nagelkerke R.; Sutton D. Structures of Cationic Di-Iridium Complexes Derived from (Η5-C5Me5)Ir(CO)2, Including the Dication [(η5-C5Me5)(CO)2Ir–Ir(CO)2(η5-C5Me5)]2+ and the Bridging Methylenetetramethylcyclopentadienyl (Tetramethylfulvene) Complex [(η5-C5Me5)(CO)Ir–Ir;(CO)2(η5-CH2C5Me4)]+. Chem. Commun. 1989, 19, 1424–1426. 10.1039/C39890001424. [DOI] [Google Scholar]
- Wang W.; Davis H. B.; Einstein F. W. B.; Pomeroy R. K. Stepwise C-H Cleavage of Two Methyl Groups of a Pentamethylcyclopentadienyl Ligand on a Tetraosmium Cluster. Organometallics 1994, 13, 5113–5121. 10.1021/om00024a062. [DOI] [Google Scholar]
- Evans W. J.; Perotti J. M.; Ziller J. W. Trialkylboron/Lanthanide Metallocene Hydride Chemistry: Polydentate Bridging of (HBEt3)- to Lanthanum. Inorg. Chem. 2005, 44, 5820–5825. 10.1021/ic0501061. [DOI] [PubMed] [Google Scholar]
- Takahashi Y.; Fujita K.; Yamaguchi R. Mild Oxidative Addition of C–H Bonds to a Hydrido-Bridged Dinuclear Complex of Iridium(II) Induced by the Coord.ination of Heteroatomic Ligands. Eur. J. Inorg. Chem. 2008, 28, 4360–4368. 10.1002/ejic.200800469. [DOI] [Google Scholar]
- Carbó J. J.; García-López D.; Gómez-Pantoja M.; González-Pérez J. I.; Martín A.; Mena M.; Santamaría C. Intermetallic Cooperation in C–H Activation Involving Transient Titanium-Alkylidene Species: A Synthetic and Mechanistic Study. Organometallics 2017, 36, 3076–3083. 10.1021/acs.organomet.7b00416. [DOI] [Google Scholar]
- Dyllick-Brenzinger R.; Olsen H. Conformational Mobility in 1,4-Bridged Cyclooctanes. Carbon-13 NMR Evidence for Facile Chirality Inversion. J. Am. Chem. Soc. 1981, 103, 704–706. 10.1021/ja00393a052. [DOI] [Google Scholar]
- Chung C.; Tseng W.-C.; Chi Y.; Peng S.-M.; Lee G.-H. Reactivity of the Tetrametallic Carbido Cluster (C5Me5)WOs3(Μ4-C)(μ-H)(CO)11 with Alkyne: Isomerization of an Allyl Fragment on a Tetrametallic Cluster Framework and Ring-Methyl Activation in the C5Me5 Ligand. Organometallics 1998, 17, 2207–2214. 10.1021/om9710677. [DOI] [Google Scholar]
- Shima T.; Hou Z. Rare Earth/d-Transition Metal Heteromultimetallic Polyhydride Complexes Based on Half-Sandwich Rare Earth Moieties. Organometallics 2009, 28, 2244–2252. 10.1021/om900024q. [DOI] [Google Scholar]
- McDade C.; Green J. C.; Bercaw J. E. A Kinetic and Mechanistic Study of the Thermolysis of Bis(Pentamethylcyclopentadienyl)Dimethyltitanium(IV). Organometallics 1982, 1, 1629–1634. 10.1021/om00072a015. [DOI] [Google Scholar]
- Bulls A. R.; Schaefer W. P.; Serfas M.; Bercaw J. E. Intramolecular Carbon-Hydrogen Bond Activation of Benzyl Ligands by Metalated Cyclopentadienyl Derivatives of Permethylhafnocene. Molecular Structure of (η5-C5Me5)(η5, η1-C5Me4CH2)HfCH2C6H5 and the Mechanism of Rearrangement to Its Hafnabenzocyclobutene Tautomer [Cyclic] (η5-C5Me5)2Hf(o-CH2C6H4). Organometallics 1987, 6, 1219–1226. 10.1021/om00149a016. [DOI] [Google Scholar]
- Bercaw J. E. Bis(Pentamethylcyclopentadienyl)Titanium(II) and Its Complexes with Molecular Nitrogen. J. Am. Chem. Soc. 1974, 96, 5087–5095. 10.1021/ja00823a012. [DOI] [Google Scholar]
- Cloke F. G. N.; Day J. P.; Green J. C.; Morley C. P.; Swain A. C. Bis(η-Pentamethylcyclopentadienyl) Complexes of Molybdenum, Tungsten and Rhenium via Metal Vapour Synthesis. Dalton Trans. 1991, 789–796. 10.1039/DT9910000789. [DOI] [Google Scholar]
- Alférez M. G.; Moreno J. J.; Hidalgo N.; Campos J. Reversible Hydride Migration from C5Me5 to RhI Revealed by a Cooperative Bimetallic Approach. Angew. Chem., Int. Ed. 2020, 59, 20863–20867. 10.1002/anie.202008442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley M. J.; del Castillo T. J.; Matson B. D.; Roddy J. P.; Peters J. C. Catalytic N2-to-NH3 Conversion by Fe at Lower Driving Force: A Proposed Role for Metallocene-Mediated PCET. ACS Cent. Sci. 2017, 3, 217–223. 10.1021/acscentsci.7b00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley M. J.; del Castillo T. J.; Matson B. D.; Peters J. C. Fe-Mediated Nitrogen Fixation with a Metallocene Mediator: Exploring PKa Effects and Demonstrating Electrocatalysis. J. Am. Chem. Soc. 2018, 140, 6122–6129. 10.1021/jacs.8b02335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley M. J.; Oyala P. H.; Peters J. C. Cp* Noninnocence Leads to a Remarkably Weak C–H Bond via Metallocene Protonation. J. Am. Chem. Soc. 2019, 141, 4721–4729. 10.1021/jacs.9b00193. [DOI] [PubMed] [Google Scholar]
- Quintana L. M. A.; Johnson S. I.; Corona S. L.; Villatoro W.; Goddard W. A.; Takase M. K.; VanderVelde D. G.; Winkler J. R.; Gray H. B.; Blakemore J. D. Proton–Hydride Tautomerism in Hydrogen Evolution Catalysis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 6409–6414. 10.1073/pnas.1606018113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman C. L.; Finster O. N. L.; Miller A. J. M. Cyclopentadiene-Mediated Hydride Transfer from Rhodium Complexes. Chem. Commun. 2016, 52, 9105–9108. 10.1039/C6CC00575F. [DOI] [PubMed] [Google Scholar]
- Pal S.; Kusumoto S.; Nozaki K. Dehydrogenation of Dimethylamine–Borane Catalyzed by Half-Sandwich Ir and Rh Complexes: Mechanism and the Role of Cp* Noninnocence. Organometallics 2018, 37, 906–914. 10.1021/acs.organomet.7b00889. [DOI] [Google Scholar]
- Kefalidis C. E.; Perrin L.; Burns C. J.; Berg D. J.; Maron L.; Andersen R. A. Can a Pentamethylcyclopentadienyl Ligand Act as a Proton-Relay in f-Element Chemistry? Insights from a Joint Experimental/Theoretical Study. Dalton Trans. 2015, 44, 2575–2587. 10.1039/C4DT02387K. [DOI] [PubMed] [Google Scholar]
- a Gusev O.; Denisovich L. I.; Peterleitner M. G.; Rubezhov A. Z.; Ustynyuk N. A.; Maitlis P. M. Electrochemical Generation of 19- and 20-Electron Rhodocenium Complexes and Their Properties. J. Organomet. Chem. 1993, 452, 219–222. 10.1016/0022-328X(93)83193-Y. [DOI] [Google Scholar]; b Gusev O. V.; Peterleitner M. G.; Ievlev M. A.; Kal’sin A. M.; Petrovskii P. V.; Denisovich L. I.; Ustynyuk N. A. Reduction of Iridocenium Salts [Ir(η5-C5Me5)(η5-L)]+ (L = C5H5, C5Me5, C9H7); Ligand-to-Ligand Dimerisation Induced by Electron Transfer. J. Organomet. Chem. 1997, 531, 95–100. 10.1016/S0022-328X(96)06675-2. [DOI] [Google Scholar]; c Blaha J. P.; Wrighton M. S. Relative Importance of Dissociative Loss of Carbon Monoxide and Formation of Benzyl Radicals from Photoexcitation of (η5-C5R5)Fe(CO)2(η1-CH2C6H5) and Evidence for Reaction of Carbon Monoxide with 17-Electron Radicals. J. Am. Chem. Soc. 1985, 107, 2694–2702. 10.1021/ja00295a023. [DOI] [Google Scholar]
- a Yao Z.-J.; Lin Y.-J.; Xu B.; Jin G.-X. Nucleophilic Addition of Carborane Anion to Ir, Rh-Coordinated Cp* Ring: C–C Bond Formation Accompanied by Reduction of Metal Center. Dalton Trans. 2014, 43, 4938–4940. 10.1039/C3DT51906F. [DOI] [PubMed] [Google Scholar]; b Moseley K.; Kang J. W.; Maitlis P. M. Pentamethylcyclopentadienyl-Rhodium and -Iridium Halides. Part II. Reactions with Mono-, Di-, and Tri-Olefins. J. Chem. Soc. A: Inorg., Phys., Theor. 1970, 2875–2883. 10.1039/J19700002875. [DOI] [Google Scholar]; c Razuvaev G. A.; Maryin V. P.; Vyshinskaya L. I.; Ya G.; Mal’kova Y.; Andrianov A.; Druzhkov O. N. Dokl. Akad. Nauk SSSR 1987, 294, 614. 10.1016/j.ejvs.2011.05.023. [DOI] [Google Scholar]; [Dokl. Chem., 1987,294 (Engl. Transl.)]; d Kochi T.; Nomura Y.; Tang Z.; Ishii Y.; Mizobe Y.; Hidai M. Synthesis and Reactivities of Ir2Ru Heterobimetallic Sulfido Clusters Derived from a Hydrogensulfido-Bridged Diiridium Complex. Dalton Trans. 1999, 15, 2575–2582. 10.1039/A902999K. [DOI] [Google Scholar]
- de Azevedo C. G.; Calhorda M. J.; de Carrondo M. A. A. F.; Dias A. R.; Duarte M. T.; Galvão A. M.; Gamelas C. A.; Gonçalves I. S.; da Piedade F. M.; Romão C. C. Nucleophilic and Electrophilic Reactions of C5 Cyclo-Polyenes Coordinated to the [CpMoL2]n+ Fragment (n = 1,2; L = 1/2dppe, PMe3, P(OMe)3, CO). J. Organomet. Chem. 1997, 544, 257–276. 10.1016/S0022-328X(97)00331-8. [DOI] [Google Scholar]
- Comte V.; Blacque O.; Kubicki M. M.; Moïse C. Reactivity of the Ansa-Bridged Metallocene Dichlorides [X(η5-C5H4)2]MCl2 (X = SiMe2, CMe2; M = Mo, W) toward Metallophosphide Anions [PPh2M’(CO)x]- (M’ = Cr, Mo, W, x = 5; M‘ = Fe, x = 4). Formation of Heterobimetallic Complexes by Nucleophilic Substitution on a Cyclopentadienyl Ligand or on the Metal M. Organometallics 1997, 16, 5763–5769. 10.1021/om970630i. [DOI] [Google Scholar]
- Hughes R. P.; Maddock S. M.; Guzei I. A.; Liable-Sands L. M.; Rheingold A. L. Reactions of Halofluorocarbons with Group 6 Complexes M(C5H5)2L (M = Mo, W; L = C2H4, CO). Fluoroalkylation at Molybdenum and Tungsten, and at Cyclopentadienyl or Ethylene Ligands. J. Am. Chem. Soc. 2001, 123, 3279–3288. 10.1021/ja003339u. [DOI] [PubMed] [Google Scholar]
- Hughes R. P.; Maddock S. M.; Rheingold A. L.; Liable-Sands L. M. Selective Fluoroalkylation of Cyclopentadienyl and Ethylene Ligands in Reactions of Perfluoroalkyl Iodides with Low-Valent Complexes of Molybdenum and Tungsten: Evidence for a Fluorocarbanion Mechanism. J. Am. Chem. Soc. 1997, 119, 5988–5989. 10.1021/ja970452k. [DOI] [Google Scholar]
- Liu R.; Zhou X. Selective Transformations of Cyclopentadienyl Ligands of Transition-Metal and Rare-Earth Metal Complexes. Chem. Commun. 2013, 49, 3171–3187. 10.1039/C2CC35637F. [DOI] [PubMed] [Google Scholar]
- Comte V.; Blacque O.; Kubicki M. M.; Moïse C. Regio- and Stereochemical Aspects of the Substitution Reaction between the Molybdenocene and Tungstenocene Dichlorides (Η5-C5H4-R)2MCl2 (R = CMe3, SiMe3; M = Mo, W) and Metallophosphide Anions [(CO)5M‘PPh2]Li (M‘ = Mo, W). Organometallics 2001, 20, 5432–5439. 10.1021/om010495g. [DOI] [Google Scholar]
- Forschner T. C.; Cooper N. J. Magnesium Dihalide Promoted Addition of Grignard Reagents to the Cyclopentadienyl Rings of Tungstenocene Dichloride. J. Am. Chem. Soc. 1989, 111, 7420–7424. 10.1021/ja00201a022. [DOI] [Google Scholar]
- Basato M.; Biffis A.; Buscemi G.; Callegaro E.; Polo M.; Tubaro C.; Venzo A.; Vianini C.; Graiff C.; Tiripicchio A.; Benetollo F. Reaction of Cyclopentadienyl Ruthenium Complexes with a Carborane Anion: Effect of the Spectator Ligands on the Substitution Site. Organometallics 2007, 26, 4265–4270. 10.1021/om700305e. [DOI] [Google Scholar]
- Basato M.; Biffis A.; Tubaro C.; Graiff C.; Tiripicchio A. Nucleophilic Substitution on a Ru-Coordinated Cp Ring by a Carborane Anion. Dalton Trans. 2004, 24, 4092–4093. 10.1039/B413803A. [DOI] [PubMed] [Google Scholar]
- Fischer E. O.; Herberich G. E. Über Die Reaktivitat Des Di-Cyclopentadienyl-Kobalt(III)-Kations. Chem. Ber. 1961, 94, 1517–1523. 10.1002/cber.19610940615. [DOI] [Google Scholar]
- Fischer E. O.; Angelici R. J. New Cyclopentadienyl Complexes of Rhodium. J. Am. Chem. Soc. 1963, 85, 3733. 10.1021/ja00906a004. [DOI] [Google Scholar]
- Millward D. B.; Waymouth R. M. Zirconocene-Mediated Cyclization of 2-Bromo α,ω-Dienes. Organometallics 1997, 16, 1153–1158. 10.1021/om960870i. [DOI] [Google Scholar]
- Suzuki N.; Kondakov D. Y.; Takahashi T. Zirconium-Catalyzed Highly Regioselective Carbon-Carbon Bond Formation Reactions. J. Am. Chem. Soc. 1993, 115, 8485–8486. 10.1021/ja00071a083. [DOI] [Google Scholar]
- Dzhemilev U. M.; D’yakonov V. A.; Khafizova L. O.; Ibragimov A. G. Cyclo- and Carbomagnesiation of 1,2-Dienes Catalyzed by Zr Complexes. Tetrahedron 2004, 60, 1287–1291. 10.1016/j.tet.2003.10.063. [DOI] [Google Scholar]
- Morken J. P.; Didiuk M. T.; Hoveyda A. H. Zirconium-Catalyzed Asymmetric Carbomagnesation. J. Am. Chem. Soc. 1993, 115, 6997–6998. 10.1021/ja00068a077. [DOI] [Google Scholar]
- Negishi E.-I.; Takahashi T. Patterns of Stoichiometric and Catalytic Reactions of Organozirconium and Related Complexes of Synthetic Interest. Acc. Chem. Res. 1994, 27, 124–130. 10.1021/ar00041a002. [DOI] [Google Scholar]
- Takahashi T.; Seki T.; Nitto Y.; Saburi M.; Rousset C. J.; Negishi E. Remarkably Pair- and Regioselective Carbon-Carbon Bond-Forming Reaction of Zirconacyclopentane Derivatives with Grignard Reagents. J. Am. Chem. Soc. 1991, 113, 6266–6268. 10.1021/ja00016a051. [DOI] [Google Scholar]
- Farády L.; Bencze L.; Markó L. Transition-Metal Alkyls and Hydrides: III. Alkyl-Olefin Exchange Reaction of Grignard Reagents Catalyzed by Nickel Chloride. J. Organomet. Chem. 1967, 10, 505–510. 10.1016/S0022-328X(00)83176-9. [DOI] [Google Scholar]
- Farády L.; Markó L. Transition Metal Alkyls and Hydrides X. Structure of Products Formed in the Reactions between Olefins and Grignard Reagents in the Presence of Nickel Chloride. J. Organomet. Chem. 1971, 28, 159–165. 10.1016/S0022-328X(00)84564-7. [DOI] [Google Scholar]
- Negishi E.; Rousset C. J.; Choueiry D.; Maye J. P.; Suzuki N.; Takahashi T. Zirconium-Catalyzed and Zirconium-Promoted Cyclization Reactions of Non-Conjugated Dienes with Alkylmagnesium Halides to Give Cycloalkylmethylmagnesium Derivatives. Inorg. Chim. Acta 1998, 280, 8–20. 10.1016/S0020-1693(98)00064-4. [DOI] [Google Scholar]
- Uemura M.; Yorimitsu H.; Oshima K. Synthesis of Cp*CH2PPh2 and Its Use as a Ligand for the Nickel-Catalysed Cross-Coupling Reaction of Alkyl Halides with Aryl Grignard Reagents. Chem. Commun. 2006, 45, 4726–4728. 10.1039/B612173J. [DOI] [PubMed] [Google Scholar]
- Fürstner A.; Martin R.; Krause H.; Seidel G.; Goddard R.; Lehmann C. W. Preparation, Structure, and Reactivity of Nonstabilized Organoiron Compounds. Implications for Iron-Catalyzed Cross Coupling Reactions. J. Am. Chem. Soc. 2008, 130, 8773–8787. 10.1021/ja801466t. [DOI] [PubMed] [Google Scholar]
- Monnereau L.; Sémeril D.; Matt D.; Toupet L.; Mota A. J. Efficient, Nickel-Catalysed Kumada–Tamao–Corriu Cross- Coupling with a Calix[4]Arene-Diphosphine Ligand. Adv. Synth. Catal. 2009, 351, 1383–1389. 10.1002/adsc.200800759. [DOI] [Google Scholar]
- Goetz A. E.; Garg N. K. Regioselective Reactions of 3,4-Pyridynes Enabled by the Aryne Distortion Model. Nat. Chem. 2013, 5, 54–60. 10.1038/nchem.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macklin T. K.; Snieckus V. Directed Ortho Metalation Methodology. The N,N-Dialkyl Aryl O-Sulfamate as a New Directed Metalation Group and Cross-Coupling Partner for Grignard Reagents. Org. Lett. 2005, 7, 2519–2522. 10.1021/ol050393c. [DOI] [PubMed] [Google Scholar]
- Teo W. J.; Wang Z.; Xue F.; Andy Hor T. S.; Zhao J. Cyclopentadienyl Nickel(Ii) N,C-Chelating Benzothiazolyl NHC Complexes: Synthesis, Characterization and Application in Catalytic C–C Bond Formation Reactions. Dalton Trans. 2016, 45, 7312–7319. 10.1039/C6DT00252H. [DOI] [PubMed] [Google Scholar]
- Tamba S.; Fuji K.; Meguro H.; Okamoto S.; Tendo T.; Komobuchi R.; Sugie A.; Nishino T.; Mori A. Synthesis of High-Molecular-Weight Head-to-Tail-Type Poly(3-Substituted-Thiophene)s by Cross-Coupling Polycondensation with [CpNiCl(NHC)] as a Catalyst. Chem. Lett. 2013, 42, 281–283. 10.1246/cl.2013.281. [DOI] [Google Scholar]
- Yanlong Q.; Lu; Jiaqui; Weihua X. Studies on Olefin Isomerization Catalyzed by Transition Metals: Part IV. Isomerization of 1,5-Cyclooctadiene Catalyzed by (R-Cp)2TiCl2/R’MgX Systems. J. Mol. Catal. 1986, 34, 31–38. 10.1016/0304-5102(86)87035-3. [DOI] [Google Scholar]
- Tao X.; Qian F.; Yong L.; Qian Y. Substituent Effect on Oligomerization of Isoprene Catalyzed by Ring-Substituted (RCp)2TiCl2/i-C3H7MgCl System. J. Mol. Catal. A: Chem. 2000, 156, 121–126. 10.1016/S1381-1169(99)00418-5. [DOI] [Google Scholar]
- Dysard J. M.; Tilley T. D. Synthesis and Reactivity of Η5-Silolyl, Η5-Germolyl, and Η5-Germole Dianion Complexes of Zirconium and Hafnium. J. Am. Chem. Soc. 2000, 122, 3097–3105. 10.1021/ja993563n. [DOI] [Google Scholar]
- Mohapatra S. K.; Büschel S.; Daniliuc C.; Jones P. G.; Tamm M. Selective Lithiation and Phosphane-Functionalization of [(Η7-C7H7)Ti(Η5-C5H5)] (Troticene) and Its Use for the Preparation of Early-Late Heterobimetallic Complexes. J. Am. Chem. Soc. 2009, 131, 17014–17023. 10.1021/ja9080056. [DOI] [PubMed] [Google Scholar]
- Chiu M.; Hoyt H. M.; Michael F. E.; Bergman R. G.; van Halbeek H. Synthesis, Structural Characterization, and Quantitative Basicity Studies of Lithium Zirconimidate Complexes. Angew. Chem., Int. Ed. 2008, 47, 6073–6076. 10.1002/anie.200801463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín A.; Mena M.; Morales-Varela M. D. C.; Santamaría C. Deprotonation of Μ3-Methylidyne Groups on a Ti3O3 Support: A Way to Build Oxotitanocubanes Containing Alkali and Alkaline-Earth Metals. Eur. J. Inorg. Chem. 2004, 9, 1914–1921. 10.1002/ejic.200300840. [DOI] [Google Scholar]
- Hernán-Gómez A.; Martín A.; Mena M.; Santamaría C. Contact and Solvent-Separated Ion Pair Aluminium “Ate” Complexes on a Titanium Oxide Molecular Model. Dalton Trans. 2013, 42, 5076–5084. 10.1039/C3DT32725F. [DOI] [PubMed] [Google Scholar]
- Visser C.; Meetsma A.; Hessen B. Synthesis and Trimethylsilyl C–H Activation Processes of 14-Electron Cp*Hf(2,3-Dimethyl-1,3-Butadiene)Trimethylsilylmethyl Complexes. Organometallics 2002, 21, 1912–1918. 10.1021/om011062g. [DOI] [Google Scholar]
- Kirchbauer F. G.; Pellny P.-M.; Sun H.; Burlakov V. V.; Arndt P.; Baumann W.; Spannenberg A.; Rosenthal U. Synthesis and Reactions with Carbon Dioxide of Mono(σ-Alkynyl) Titanocene(III) Complexes Cp*2Ti(C⋮CR) (R = Me, t-Bu) and the Corresponding “Ate” Complexes [Cp*2Ti(C⋮CR)2Li(THF)n] (R = SiMe3, t-Bu, Ph). Organometallics 2001, 20, 5289–5296. 10.1021/om010404f. [DOI] [Google Scholar]
- García-Castro M.; Martín A.; Mena M.; Yélamos C. Coord.ination of [{Ti(η5-C5Me5)(μ-NH)}3(Μ3-N)] to Metal Cyclopentadienides: Cyclopentadienyl Azaheterometallocubanes. Organometallics 2004, 23, 1496–1500. 10.1021/om030554l. [DOI] [Google Scholar]
- Schock L. E.; Marks T. J. Organometallic Thermochemistry. Metal Hydrocarbyl, Hydride, Halide, Carbonyl, Amide, and Alkoxide Bond Enthalpy Relationships and Their Implications in Pentamethylcyclopentadienyl and Cyclopentadienyl Complexes of Zirconium and Hafnium. J. Am. Chem. Soc. 1988, 110, 7701–7715. 10.1021/ja00231a020. [DOI] [Google Scholar]
- Golden J. T.; Peterson T. H.; Holland P. L.; Bergman R. G.; Andersen R. A. Adduct Formation and Single and Double Deprotonation of Cp*(PMe3)Ir(H)2 with Main Group Metal Alkyls and Aryls: Synthesis and Structure of Three Novel Ir–Al and Ir–Mg Heterobimetallics. J. Am. Chem. Soc. 1998, 120, 223–224. 10.1021/ja973230v. [DOI] [Google Scholar]
- Bretschneider-Hurley A.; Winter C. H. Pentamethylpentalithioruthenocene and Decalithioruthenocene. J. Am. Chem. Soc. 1994, 116, 6468–6469. 10.1021/ja00093a071. [DOI] [Google Scholar]
- Ohashi M.; Matsubara K.; Iizuka T.; Suzuki H. Trinuclear Ruthenium Polyhydride Complexes with a Triply Bridging Ligand: [{(η5-C5Me5)Ru}3(Μ3-M)(μ-H)3(Μ3-H)] (M=Li, MgiPr, and ZnEt) and [{(η5-C5Me5)Ru}3(Μ3-M)(μ-H)3] (M=AlEt and GaMe). Angew. Chem., Int. Ed. 2003, 42, 937–940. 10.1002/anie.200390249. [DOI] [PubMed] [Google Scholar]
- Seneviratne K. N.; Bretschneider-Hurley A.; Winter C. H. Synthesis, Spectroscopic Characterization, and Reactivity of Ruthenocenes Bearing Pentamagnesiated Cyclopentadienyl Ligands. J. Am. Chem. Soc. 1996, 118, 5506–5507. 10.1021/ja960444o. [DOI] [Google Scholar]
- Chao S.; Robbins J. L.; Wrighton M. S. A New Ferrocenophane Surface Derivatizing Reagent for the Preparation of Nearly Reversible Electrodes for Horse Heart Ferri-/Ferrocytochrome c: 2,3,4,5-Tetramethyl-1-[(Dichlorosilyl)Methyl][2]Ferrocenophane. J. Am. Chem. Soc. 1983, 105, 181–188. 10.1021/ja00340a006. [DOI] [Google Scholar]
- Sun X.; Singh A. K.; Yadav R.; Jin D.; Haimerl M.; Scheer M.; Roesky P. W. Triple-Decker Complexes Incorporating Three Distinct Deck Architectures. Chem. Commun. 2022, 58, 673–676. 10.1039/D1CC06182H. [DOI] [PubMed] [Google Scholar]
- Weng W.; Bartik T.; Brady M.; Bartik B.; Ramsden J. A.; Arif A. M.; Gladysz J. A. Synthesis, Structure, and Redox Chemistry of Heteropolymetallic Carbon Complexes with MC2M’, MC4M’, and MC4M’C4M Linkages. Transmetalations of Lithiocarbon Complexes (η5-C5Me5)Re(NO)(PPh3)(C≡CLi) and (η5-C5Me5)Re(NO)(PPh3)(C≡CC≡CLi). J. Am. Chem. Soc. 1995, 117, 11922–11931. 10.1021/ja00153a014. [DOI] [Google Scholar]
- Herring F. G.; Legzdins P.; Richter-Addo G. B. Organometallic Nitrosyl Chemistry. 40. Reduction Behavior of the Complexes Cp’M(NO)X2 (Cp’ = Cp (η5-C5Me5) or Cp*(η5-C5Me5); M = Mo or W; X = Cl, Br, or I): Synthesis and Characterization of the [Cp’Mo(NO)X2]· Radical Anions. Organometallics 1989, 8, 1485–1493. 10.1021/om00108a019. [DOI] [Google Scholar]
- Glassman T. E.; Liu A. H.; Schrock R. R. Bimetallic Hydrazido(3- and 4-) and Nitrido Complexes of Tungsten Containing the W(η5-C5Me5)Me3 Core. Inorg. Chem. 1991, 30, 4723–4732. 10.1021/ic00025a010. [DOI] [Google Scholar]
- Crabtree R. H. Deactivation in Homogeneous Transition Metal Catalysis: Causes, Avoidance, and Cure. Chem. Rev. 2015, 115, 127–150. 10.1021/cr5004375. [DOI] [PubMed] [Google Scholar]
- Maimone T. J.; Milner P. J.; Kinzel T.; Zhang Y.; Takase M. K.; Buchwald S. L. Evidence for in Situ Catalyst Modification during the Pd-Catalyzed Conversion of Aryl Triflates to Aryl Fluorides. J. Am. Chem. Soc. 2011, 133, 18106–18109. 10.1021/ja208461k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner P. J.; Maimone T. J.; Su M.; Chen J.; Müller P.; Buchwald S. L. Investigating the Dearomative Rearrangement of Biaryl Phosphine-Ligated Pd(II) Complexes. J. Am. Chem. Soc. 2012, 134, 19922–19934. 10.1021/ja310351e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather A. C.; Lee H. G.; de La Rosa V. Y.; Yang Y.; Müller P.; Buchwald S. L. A Fluorinated Ligand Enables Room-Temperature and Regioselective Pd-Catalyzed Fluorination of Aryl Triflates and Bromides. J. Am. Chem. Soc. 2015, 137, 13433–13438. 10.1021/jacs.5b09308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen J. M.; Sheehan S. W.; Hashmi S. M.; Campos J.; Hintermair U.; Crabtree R. H.; Brudvig G. W. Electrochemical Activation of Cp* Iridium Complexes for Electrode-Driven Water-Oxidation Catalysis. J. Am. Chem. Soc. 2014, 136, 13826–13834. 10.1021/ja5068299. [DOI] [PubMed] [Google Scholar]
- Sharninghausen L. S.; Campos J.; Manas M. G.; Crabtree R. H. Efficient Selective and Atom Economic Catalytic Conversion of Glycerol to Lactic Acid. Nat. Commun. 2014, 5, 5084. 10.1038/ncomms6084. [DOI] [PubMed] [Google Scholar]
- Campos J.; Hintermair U.; Brewster T. P.; Takase M. K.; Crabtree R. H. Catalyst Activation by Loss of Cyclopentadienyl Ligands in Hydrogen Transfer Catalysis with Cp*IrIII Complexes. ACS Catal. 2014, 4, 973–985. 10.1021/cs401138f. [DOI] [Google Scholar]
- Moreno J. J.; Espada M. F.; Maya C.; Campos J.; López-Serrano J.; Macgregor S. A.; Carmona E. Isomerization of a Cationic (η5-C5Me5)Ir(III) Complex Involving Remote C–C and C–H Bond Formation. Polyhedron 2021, 207, 115363 10.1016/j.poly.2021.115363. [DOI] [Google Scholar]
- Arndtsen B. A.; Bergman R. G. Unusually Mild and Selective Hydrocarbon C-H Bond Activation with Positively Charged Iridium (III) Complexes. Science 1995, 270, 1970–1973. 10.1126/science.270.5244.1970. [DOI] [Google Scholar]
- Labinger J. A.; Bercaw J. E. Understanding and Exploiting C–H Bond Activation. Nature 2002, 417, 507–514. 10.1038/417507a. [DOI] [PubMed] [Google Scholar]
- Balcells D.; Clot E.; Eisenstein O. C—H Bond Activation in Transition Metal Species from a Computational Perspective. Chem. Rev. 2010, 110, 749–823. 10.1021/cr900315k. [DOI] [PubMed] [Google Scholar]
- Klei S. R.; Tilley T. D.; Bergman R. G. The Mechanism of Silicon–Hydrogen and Carbon–Hydrogen Bond Activation by Iridium(III): Production of a Silylene Complex and the First Direct Observation of Ir(III)/Ir(V) C–H Bond Oxidative Addition and Reductive Elimination. J. Am. Chem. Soc. 2000, 122, 1816–1817. 10.1021/ja992954z. [DOI] [Google Scholar]
- Carlsen R.; Wohlgemuth N.; Carlson L.; Ess D. H. Dynamical Mechanism May Avoid High-Oxidation State Ir(V)–H Intermediate and Coord.ination Complex in Alkane and Arene C–H Activation by Cationic Ir(III) Phosphine. J. Am. Chem. Soc. 2018, 140, 11039–11045. 10.1021/jacs.8b05238. [DOI] [PubMed] [Google Scholar]
- Hoyano J. K.; Graham W. A. G. Oxidative Addition of the Carbon-Hydrogen Bonds of Neopentane and Cyclohexane to a Photochemically Generated Iridium(I) Complex. J. Am. Chem. Soc. 1982, 104, 3723–3725. 10.1021/ja00377a032. [DOI] [Google Scholar]
- Janowicz A. H.; Bergman R. G. C-H Activation in Completely Saturated Hydrocarbons: Direct Observation of M + R-H → M(R)(H). J. Am. Chem. Soc. 1982, 104, 352–354. 10.1021/ja00365a091. [DOI] [Google Scholar]
- Marinelli G.; Rachidi I. E. I.; Streib W. E.; Eisenstein O.; Caulton K. G. Alkyne Hydrogenation by a Dihydrogen Complex: Synthesis and Structure of an Unusual Iridium-Butyne Complex. J. Am. Chem. Soc. 1989, 111, 2346–2347. 10.1021/ja00188a083. [DOI] [Google Scholar]
- Knies M.; Kaiser M.; Isaeva A.; Müller U.; Doert T.; Ruck M. Front Cover: The Intermetalloid Cluster Cation (CuBi8)3+. Chem. Eur. J. 2018, 24, 1. 10.1002/chem.201705257. [DOI] [PubMed] [Google Scholar]
- Moreno J. J.; Espada M. F.; Krüger E.; López-Serrano J.; Campos J.; Carmona E. Ligand Rearrangement and Hemilability in Rhodium(I) and Iridium(I) Complexes Bearing Terphenyl Phosphanes. Eur. J. Inorg. Chem. 2018, 2018, 2309–2321. 10.1002/ejic.201800169. [DOI] [Google Scholar]
- Martin R.; Buchwald S. L. Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling Reactions Employing Dialkylbiaryl Phosphine Ligands. Acc. Chem. Res. 2008, 41, 1461–1473. 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- Marín M.; Moreno J. J.; Navarro-Gilabert C.; Álvarez E.; Maya C.; Peloso R.; Nicasio M. C.; Carmona E. Synthesis, Structure and Nickel Carbonyl Complexes of Dialkylterphenyl Phosphines. Chem. – Eur. J. 2019, 25, 260–272. 10.1002/chem.201803598. [DOI] [PubMed] [Google Scholar]
- Ortega-Moreno L.; Fernández-Espada M.; Moreno J. J.; Navarro-Gilabert C.; Campos J.; Conejero S.; López-Serrano J.; Maya C.; Peloso R.; Carmona E. Synthesis, Properties, and Some Rhodium, Iridium, and Platinum Complexes of a Series of Bulky m-Terphenylphosphine Ligands. Polyhedron 2016, 116, 170–181. 10.1016/j.poly.2016.04.023. [DOI] [Google Scholar]
- Craig N. C.; Groner P.; McKean D. C. Equilibrium Structures for Butadiene and Ethylene: Compelling Evidence for Π-Electron Delocalization in Butadiene. J. Phys. Chem. A 2006, 110, 7461–7469. 10.1021/jp060695b. [DOI] [PubMed] [Google Scholar]
- Brookhart M.; Green M. L. H.; Parkin G. Agostic Interactions in Transition Metal Compounds. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6908–6914. 10.1073/pnas.0610747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Moreno L.; Fernández-Espada M.; Moreno J. J.; Navarro-Gilabert C.; Campos J.; Conejero S.; López-Serrano J.; Maya C.; Peloso R.; Carmona E. Synthesis, Properties, and Some Rhodium, Iridium, and Platinum Complexes of a Series of Bulky m-Terphenylphosphine Ligands. Polyhedron 2016, 116, 170–181. 10.1016/j.poly.2016.04.023. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.