

Abstract
Three central cell populations play roles in morphogen action, the cells that produce, the cells that distribute, and the cells that respond to the morphogen. Taking advantage of the properties of embryonic stem cell to aggregate and readily differentiate into neural progenitor tissue, we describe an approach using genetically modified murine stem cell lines to individually address the contribution of these cells in the establishment and response to a morphogenetic gradient in mosaic spinal cord organoids.
Keywords: Non-cell autonomous Sonic Hedgehog signaling, Genetic mosaicism, Spinal cord organoids, Embryonic stem cells, Neural development
1. Introduction
Sonic Hedgehog (Shh) is a morphogen. Morphogens are critical signaling molecules that induce the formation of a pattern in a homogeneous cell population in a concentration dependent manner (Andrews et al. 2019). The morphogen concentration is proposed to be established by the distribution away from the sites of synthesis, resulting in high morphogen concentrations close to the source, and ever lower concentrations away from the source. Combining this graded distribution of the morphogen with certain threshold levels for induction of specific cell types in the responding tissue would result in a stereotypic pattern (Kerszberg and Wolpert 1998). The mechanism by which morphogens are distributed remains contentious, as this might not be achieved solely by diffusion. For example, many aspects of morphogenetic signaling in Drosophila can be explained by the detection of morphogen on the source cells via cellular processes (cytonemes) sent out by the responding cells (Kornberg 2014). As more distal cells are expected to have fewer processes that reach the source than proximal cells, a pattern can be generated by a stereotypic response in each cell possibly determined by the number of processes that contact the source.
In the developing vertebrate embryo Shh is thought to move away from the sites of synthesis, thus establishing a gradient away from its site of synthesis. This Shh gradient is interpreted by local cells that differentiate into a stereotypic pattern. The mechanism by which Shh is transported along the morphogenetic gradient to induce a measured response at a distance away from the source field remains poorly understood. Perhaps not surprisingly, several molecular interactions have been described that affect Shh release from the source, as well as subsequent distribution and response. Dedicated molecules are required for the correct release of Shh from the source cells and several components of the extracellular matrix interact with Shh to facilitate or inhibit its distribution as well as affect Shh binding to its cognate receptors.
The establishment of a morphogenetic gradient involves three central events. 1) the production of the signaling molecule, 2) the distribution of the morphogen in a gradient away from the source, and 3) the graded response that results in patterning. In a morphogenetic field cells might be involved in more than one of these events simultaneously. The independent interrogation of these populations of cells that mediate production and transport of, and the response to Shh is critical in the mechanistic study of Shh-mediated morphogenesis. In vitro approaches can address the mechanism how cells respond to different concentrations of added morphogen, while in vivo approached are useful in addressing the sufficiency and requirements of signaling molecules in morphogenetic signaling. However, both approaches are limited in addressing all function of the three central functions in parallel. Here we describe an experimental approach that allows the interrogation of cell populations for their role in either production or transport of, or the response to Shh.
Using mosaic spinal cord organoids to assess Shh morphogenesis
We use an approach that employs mosaic spinal cord organoids (SCOs) made by aggregating cells with distinct genotypes. Formation of SCOs starts with the generation of embryoid bodies (EBs), each an aggregate of around 104 embryonic stem cells. In a completely defined and serum free medium these aggregates are differentiated into tissues resembling the developing spinal cord, a classic model for Shh morphogenesis (Wichterle et al. 2002). A wealth of markers is available that define many cells types that appear at stereotypical positions in relation to the Shh source in response to distinct Shh concentrations.
The aggregation of around 104 cells into an EB allows us to mix ESCs with distinct genotypes, and even cells with a specific genotype present at only a few percent are reliably incorporated into the EBs. In general, this approach allows us to assess the patterning consequences of changing the properties of the Shh source cells, the Shh transporting cells, and the cells that respond to Shh.
1. Materials
All tissue culture media should be prepared under sterile conditions in a biosafety cabinet.
1.1. Adherent Embryonic stem cell culture
Culture of ES cells is performed under standard conditions. We have used several lines of ES cells without noticeable difference in these aggregation and differentiation procedures.
-
1.1.1
Embryonic stem cell medium: mouse embryonic stem cells are cultured in DMEM supplemented with 20% FBS, 2 mM L-Glutamine (Gibco), 1X MEM non-essential amino acids (Gibco), 1X nucleosides for ES cells (EMD Millipore), 1nM Leukemia Inhibitory Factor (LIF), and .55 μM 2-Mercaptoethanol (Gibco) in a standard humidified incubator at 37° C and 5% CO2. Culture conditions can be adjusted for specific cell lines as needed (see Notes 4.9).
-
1.1.2.
Before use, Tissue Culture dishes are treated for at least 10 min with 0.1% w/v Gelatin in PBS. This solution is aspirated right before adding cells to the wet dish.
-
1.1.3
0.25% trypsin with EDTA
1.2. Generation of spinal cord organoids
-
1.2.1
Sterile PBS
-
1.2.2
Cell counter or hemocytometer
-
1.2.3
Sterile bacterial grade Petri dishes, or any other “low adherence/hydrophobic” type of dish.
-
1.2.4
DFNB medium: 25% DMEM, 25% HAMF12 and 50% Neurobasal medium, supplemented with 1.5 mM L-Glutamine (Gibco) and 2% (volume) B27 supplement (Gibco).
-
1.2.5
Retinoic Acid in DMSO (10 mM). Stock is stable at −20°C.
-
1.2.6
Rotating platform in incubator, rotation speed of about 60 rpm (1Hz)
-
1.2.7
Optional: Lineage tracer like DiI/DiO or CMFDA
1.3. Immunostaining for neural markers
-
1.3.1
PBS with 0.1% Triton X-100 (PBS-T)
-
1.3.2
Fixation solution: 4% Paraformaldehyde in PBS
-
1.3.3
Blocking and antibody dilution solution: 10% Heat-inactivated goat or donkey serum in PBS-T
-
1.3.4
Primary antibody of choice diluted at the recommended concentration in antibody dilution solution
-
1.3.5
Secondary antibody directed against the host species of the primary antibody and coupled to a fluorophore, diluted at the recommended concentration in antibody dilution solution
-
1.3.6
Mounting medium of choice (see Notes 4.11).
-
1.3.7
Microscope slides and cover slips
1.4. LacZ reporter assay for transcriptional response
-
1.4.1
Galacton™ Light Detection System or other system to measure Ptch1:LacZ (Goodrich et al. 1997) expression
-
1.4.2
PBS
-
1.4.3
Microcentrifuge tubes and centrifuge
-
1.4.4
Bradford reagent diluted 1:5 with ultrapure water
-
1.4.5
Protein standards (e.g. 0.1 – 0.5 mg/ml BSA)
-
1.4.6
96 well assay plates
-
1.4.7
Luminometer and spectrophotometer
2. Methods
2.1. Embryonic stem cell culture
-
2.1.1
Plate approximately 2×104 – 3×104 mES cells/cm2 in tissue culture plates coated with gelatin.
-
2.1.2
Exchange medium every 1–2 days.
-
2.1.3
Passage cells every other day into a new tissue culture plate (see Notes 4.1)
2.2. Generation of spinal cord organoids
-
2.2.1
Remove cells with the desired genotypes from the tissue culture plates by incubating with trypsin for 5–10 min, followed by trituration until a single cell suspension (assessed by microscopy) is obtained.
-
2.2.2
Collect the cells in separate conical tubes and wash at least twice with sterile PBS to remove any residual medium. Do so by centrifuging (<1000rpm), aspirating the supernatant, resuspending in 4–5 ml PBS, and centrifuging again. After washing resuspend the cells in DNFB.
-
2.2.3
Determine the cell number with a hemocytometer or cell counter.
-
2.2.4
Optional: Lineage tracing (see Notes 4.10)
-
2.2.5
Pipet the desired cell number the desired genotypes in the desired ratio into a sterile Petri dish (see Table 1). We recommend a total cell number of 500,000 for a 60 mm dish and 200,000 per well of an uncoated (suspension culture) 6 well plate (see Notes 4.3). This will yield around 100 EBs.
-
2.2.6
Add DFNB to a total volume of 5 ml for 60 mm dishes and 2 ml per well of a 6 well plate.
-
2.2.7
Place the dish on a rotating platform inside an incubator and rotate at 0.8 to 1 Hz (see Notes 4.4).
-
2.2.8
Incubate on the rotating platform for 48h. Small aggregates should be visible under the microscope after one night of incubation (see Notes 4.2).
-
2.2.9
Optional: If the medium turns yellow, carefully remove about half of the medium and replenish with fresh medium. In order to prevent removal of embryoid bodies when removing medium, the embryoid bodies can be collected in the middle of the plate by gentle swirling. This requires some practice, but collection of the EBs in the center of the plate can be easily observed against a black background.
-
2.2.10
Add 2 μM of Retinoic Acid (5000x dilution of 10mM stack) 48 h after aggregation.
-
2.2.11
Returns plates back on the rotating platform (see Notes 4.5).
-
2.2.12
Incubate until the mosaic SCOs are collected for analysis. Exchange the medium (including RA) whenever it turns peach/yellow. Genetically encoded transcriptional reporter systems like Ptch1:LacZ expression can be measured 24h-48h after the addition of RA and immunostaining for neural progenitors 48h-72h after the addition of RA (see Table 2).
Table 1: Composition of mosaic SCOs.
Cells in mosaic SCOs can exert desired functions (left column) according to their genotype (middle column). The percentages of cells with a specific genotype or function in the mosaic SCO is shown in the right column.
| type | genotype | percentage of cells in mosaic SCO |
|---|---|---|
| source | Shh transgene | 3–5 % |
| reporter | HB9::GFP | 5–10 % |
| Sim1::mCherry | 10 % | |
| Ptchl LacZ/LacZ | 20–50 % | |
| intermediate | any genotype of interest | 50–90 % |
Table 2: Examples of neural markers for immunostaining of SCOs.
SCOs start expressing either ventral or dorsal markers of neural progenitor cells depending on their genotype and environment. These markers can be stained with fluorescently labeled antibodies (or fluorescent reporters) at the indicated recommended time points of in vitro culture.
| identity | neural marker | recommended SCO incubation time |
|---|---|---|
| ventral | Nkx2.2 | 4 days |
| Olig2 | 4 days | |
| Isl1/2 | 4–5 days | |
| Hb9 | 5 days | |
| Sox2 | 3 days | |
| dorsal | Pax7 | 2 days |
2.3. Immunostaining for neural progenitor markers
-
2.3.1
Incubate the mosaic SCO according to expression time points specified in Table 2.
-
2.3.2
Collect the mosaic SCO in the middle of the plate by gentle swirling and transfer them to a 1.5 ml centrifuge tube with a p1000 pipet.
-
2.3.3
Let the SCOs settle at the bottom of the tube by gravity for 1–2 min and carefully aspirate the supernatant (see Notes 4.6).
-
2.3.4
Add 1 ml of PBS, let SCOs settle, and aspirate supernatant.
-
2.3.5
Fix SCOs with 4% PFA in PBS in a fume hood for 10 min on ice. The volume of fixation solution depends on the number of SCOs but 100 μl should suffice to cover the SCOs. Gently flick the tube after addition of the fixation solution and after 5 min of incubation. Do not invert the tube to avoid SCOs being stuck at the wall of the tube!
-
2.3.6
Remove fixation solution and wash twice with PBS-T. At this point 0.1% goat serum can be added to prevent sticking and clumping of the SCOs
-
2.3.7
Add 100 μl blocking solution (2% goat serum works generally well) and incubate at room temperature for 30 min.
-
2.3.8
Remove blocking solution and add 100 μl of primary antibody solution. Incubate for at least 1 h at room temperature; an incubation overnight at 4 degrees Celsius is recommended.
-
2.3.9
Remove primary antibody solution and wash 3 times with PBS-T. Each wash step should be 5–10 min.
-
2.3.10
Add secondary antibody solution and incubate for at least 1 h at room temperature in the dark; an incubation overnight at 4° C is recommended.
-
2.3.11
Remove secondary antibody solution and wash 3 times with PBS-T. Each wash step should be 5–10 min, and one long wash (> 6h) is recommended.
-
2.3.12
Wash one last time with PBS, ideally for 30 min to 1h (see Notes 4.7).
-
2.3.13
Before mounting, make sure microscope slides are clean and free of dust. Pipet 12 μl of mounting medium in a circle on the microscope slide to form a donut shape.
-
2.3.14
“Wash” a pipet tip in heat-inactivated goat/donkey serum to prevent the organoids from sticking to the tip. This reduces the number of SCOs getting stuck inside the pipet tip while pipetting.
-
2.3.15
Transfer 30–50 SCOs into the center of the prepared donut shape of mounting medium using the “washed” tip. The volume of transferred PBS should not exceed 15 μl.
-
2.3.16
Gently place a cover slip on top (see Notes 4.8).
-
2.3.17
Remove excessive liquid from the sides of the cover slip.
-
2.3.18
Optional: Seal the cover slips onto the microscope slide with nail polish. This reduces evaporation and enables long-term storage of the slides.
-
2.3.19
Let the prepared slides dry overnight in a slide folder at room temperature prior to microscopy.
2.4. LacZ reporter assay for transcriptional response
-
2.4.1
Incubate the mosaic SCOs for 3–4 days.
-
2.4.2
Collect the SCOs in the middle of the plate by gentle swirling and transfer them to a 1.5 ml centrifuge tube with a p1000 pipet.
-
2.4.3
Let SCOs settle by gravity, aspirate supernatant, and add 1 ml PBS. Let SCOs settle again and aspirate supernatant (see Notes 4.6).
-
2.4.4
Add 100 μl lysis buffer and break up cells by vigorously pipetting up and down.
-
2.4.5
Clear lysate of cell debris by centrifugation at maximum speed for 3 min in a microcentrifuge. Transfer supernatant to a new tube (see Notes).
-
2.4.6
Total protein measurement: Pipet 1–3 μl of lysate or protein standard into a well of a 96 well plate and add 100 μl of diluted Bradford Reagent. Read the absorbance of each sample and protein standard at 595 nm. The amount of total protein per sample can be calculated with the help of the protein standard curve.
-
2.4.7
Quantification of Ptch1:LacZ expression: Follow instructions of the Galacton Light Detection Kit for suspension cells and measure Ptch1:LacZ expression levels in a luminometer. The obtained values should be divided by total protein measurement of the same sample for normalization.
3. Examples of mosaic SCOs
We have successfully used mosaic SCOs to investigate aspects of Shh signaling. We showed that cells lacking Ptch1 and Ptch2 facilitate signaling between small fractions of Shh producing cells and Shh responding cells (Roberts et al. 2016). Similarly, we showed that the loss of glypican5 in the Shh-transporting cells facilitates signaling between fractions of Shh producing cells and Shh responding cells (Guo and Roelink 2019). When using lineage tracing to assess the extent of mosaicism and found that mosaic aggregates had a typical “salt and pepper” distribution of cells. In most instances this patter was maintained throughout the experiments, but in some cases cells with the same genotype clustered over time (Figure 2B, 24h). in this example Shh−/− cells that also lacked the gene coding for an enzyme involved in cholesterol synthesis (Sc5d−/−) clustered in a background of Ptch1LacZ/LacZ;Ptch2−/−;Shh−/− cells over a 24h period.
Figure 2: Examples of mosaic SCOs.
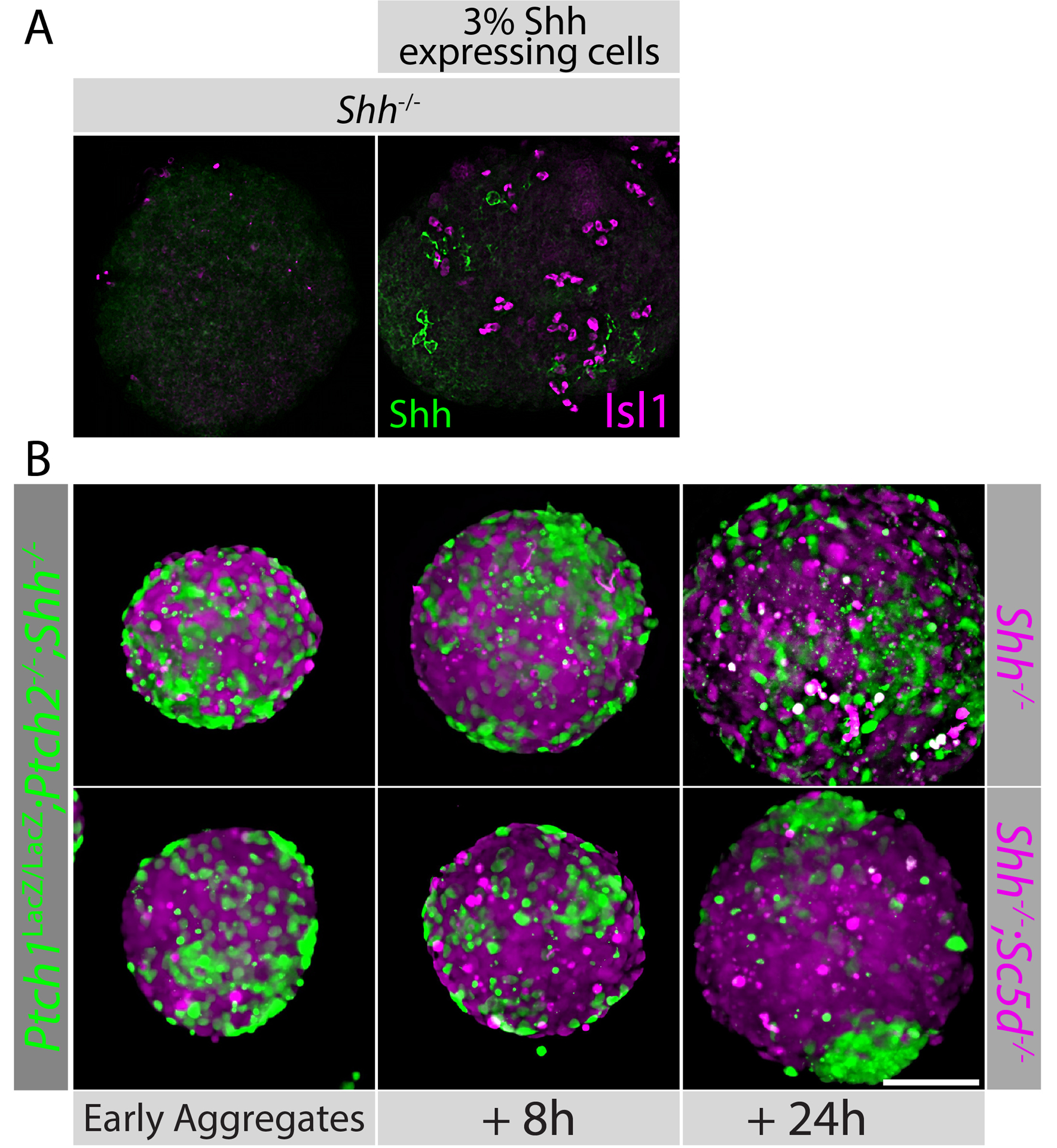
A: Shh−/− SCOs were generated with or without 3% of Shh-expressing cells (indicated). The presence of the Shh expressing cells (labeled for Shh in green) induces the localized expression of the motor neuron marker Isl1 (magenta). B: Ptch1LacZ/LacZ;Ptch2−/−;Shh−/− mES cells were loaded with CellTracker™ CMFDA (green) dye and Shh−/− (left panel) or Shh−/−;Sc5d−/− (bottom row) mES cells with CellTracker™ CMAC (magenta) prior to aggregation. Images were taken at the indicated time points of aggregation. Earliest aggregates can be observed approximately 15 h after cells were mixed and placed on a rotating platform.
4. Notes
-
4.1
In general, mES cells deposit low amounts of extracellular matrix. We find that re-plating the cells into the same tissue culture dish for up to 5 passages can help in adhering the cells.
-
4.2
It frequently happens that embryoid bodies clump together or form networks after one or two days of incubation. These bigger aggregates can be broken up by pipetting up and down or using a razor blade without negatively impacting downstream analysis. For prevention of clumps/networks forming see Notes 4.3 and 4.4.
-
4.3
The total number of cells can be reduced if embryoid bodies clump together or form aggregates that are too big.
-
4.4
The frequency of rotation affects clumping and the size of aggregates. Optimization is empirical. Larger culture dishes should generally be rotated at a lower frequency than smaller ones.
-
4.5
If needed, embryoid bodies can be incubated without rotation after one night. EBs should, however, be dispersed throughout the dish and agitated regularly to prevent adhesion to the culture dish and outgrowth of cells.
-
4.6
The “pellet” in the tube is fragile and easily aspirated. Careful removal of medium using a disposable transfer pipette is recommended. Centrifugation to collect SCOs in a more stable “pellet” is not recommended as this promotes clumping.
-
4.7
As SCOs are three-dimensional objects, antibody diffusivity through the tissue is decreased. Longer wash times are therefore recommended to reduce background staining. In general, the bigger the SCOs, the longer the washing step should be. It is suggested to do the final wash overnight.
-
4.8
Mounted SCOs are easily flattened. For normal analysis this is desired as it significantly improves microscopic analysis. However, if analysis of spherical SCOs is necessary, use coverslip standoffs, or analyze without coverslips.
-
4.9
If possible EB culture is performed without antibiotics.
-
4.10
Lineages should be traced to ensure that cells with different genotypes do not grow disproportionally in the SCOs. Ideal are genetically encoded (fluorescent) markers but we have achieved satisfactory results with DiI/DiO or short-term CellTracker™ (Thermo Fisher) dyes. Cells can be loaded separately with the dye of choice prior to aggregation (between steps 3.2.3 and 3.2.5). Cells should be incubated at 37°C with DiI/DiO for 5 min and with CellTracker™ dyes for 30 min and washed at least twice with PBS. Re-count cells if necessary.
-
4.11
Mounting medium containing DAPI is not recommended. ESCs have a high nucleus:cytoplasm ratio, so staining DNA usually provides little information. Furthermore, exposure of DAPI to UV light causes it to fluoresce at longer wavelengths, often interfering with green fluorescent dyes.
Figure 1: Diagram of experimental approach.
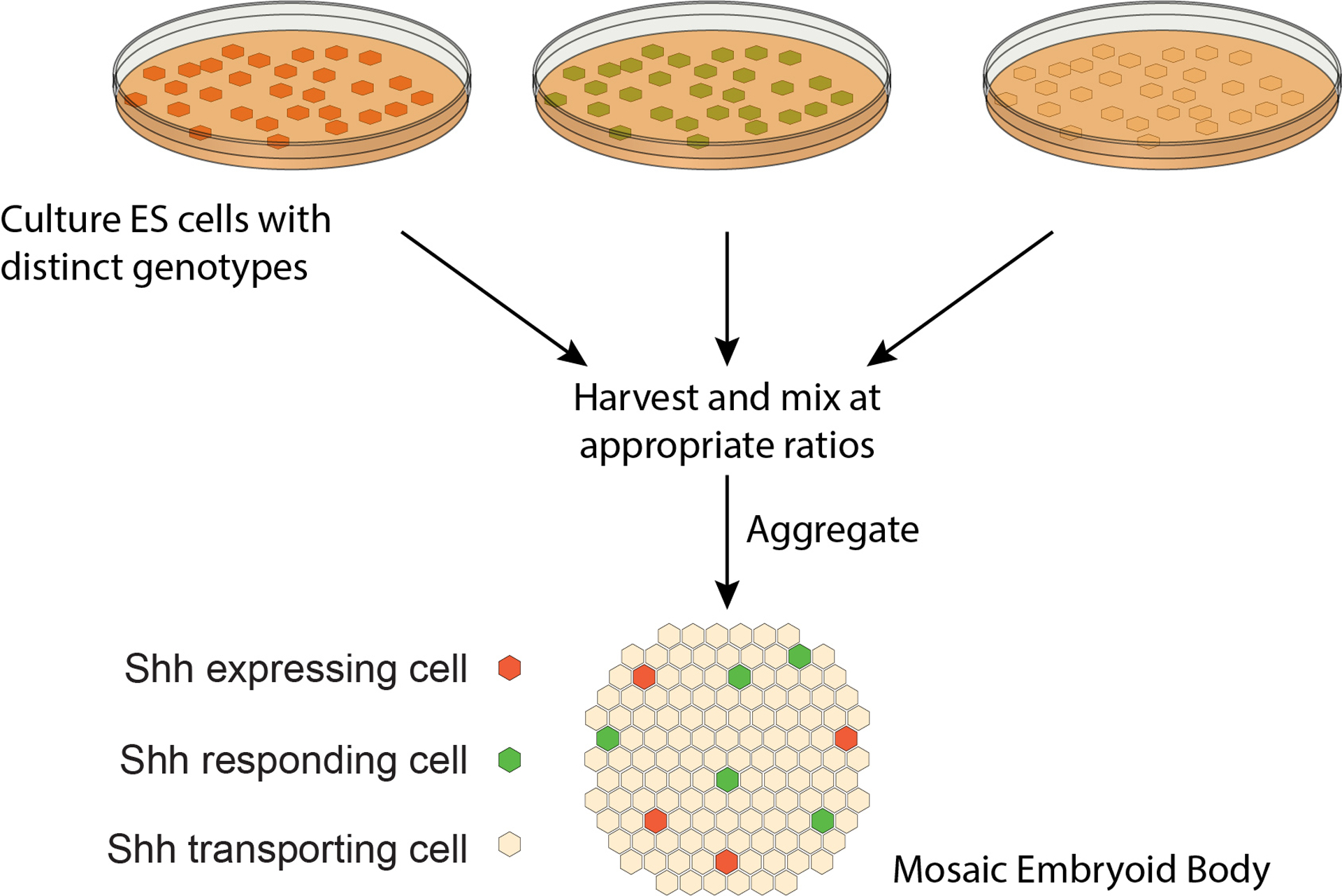
Embryonic stem (ES) cells with distinct genotypes are harvested and mixed at appropriate ratios. The resulting aggregates, called mosaic embryoid bodies, contain Shh expressing, responding, and transporting cells, and can be differentiated into tissue resembling that of the developing spinal cord.
References
- Andrews MG, Kong J, Novitch BG, Butler SJ (2019) New perspectives on the mechanisms establishing the dorsal-ventral axis of the spinal cord. Current topics in developmental biology 132:417–450. doi: 10.1016/bs.ctdb.2018.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP (1997) Altered neural cell fates and medulloblastoma in mouse patched mutants. Science (New York, NY 277:1109–1113. [DOI] [PubMed] [Google Scholar]
- Guo W, Roelink H (2019) Loss of the Heparan Sulfate Proteoglycan Glypican5 facilitates long-range Shh signaling. Stem cells (Dayton, Ohio). doi: 10.1002/stem.3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerszberg M, Wolpert L (1998) Mechanisms for positional signalling by morphogen transport: a theoretical study. Journal of theoretical biology 191:103–114. [DOI] [PubMed] [Google Scholar]
- Kornberg TB (2014) Cytonemes and the dispersion of morphogens. Wiley Interdiscip Rev Dev Biol 3:445–463. doi: 10.1002/wdev.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts B, Casillas C, Alfaro AC, et al. (2016) Patched1 and Patched2 inhibit Smoothened non-cell autonomously. Elife 5:e17634. doi: 10.7554/eLife.17634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM (2002) Directed differentiation of embryonic stem cells into motor neurons. Cell 110:385–397. [DOI] [PubMed] [Google Scholar]