

Abstract
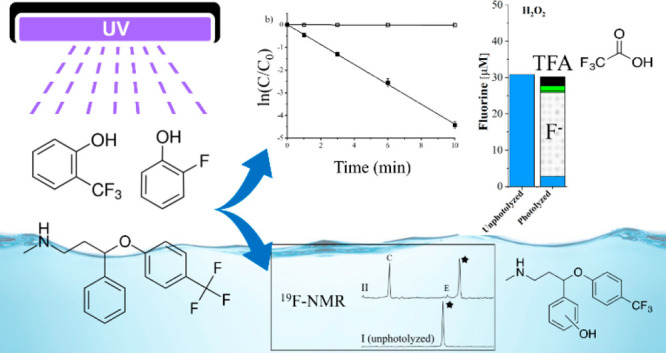
Fluorine incorporation into organic molecules has increased due to desirable changes in the molecular physiochemical properties. Common fluorine motifs include: aliphatic fluorines and −CF3, or −F containing groups bonded directly onto an aromatic (Ar–CF3 and Ar–F) or heteroaromatic ring. Photolysis of these compounds, either in natural or engineered systems, is a potential source of new fluorinated byproducts. Given the potential persistence and toxicity of fluorinated byproducts, monitoring of product formation during photolysis of various fluorinated motifs is needed. 19F-NMR is a means to detect and quantify these species. Ar–CF3 and Ar–F model compounds (2-, 3-, and 4-(trifluoromethyl)phenol, 2-, 3-, 4-fluorophenol, and 2,6-, 3,5-difluorophenol) were photolyzed under a variety of aqueous conditions: pH 5, pH 7, pH 10, 1 mM H2O2 at pH 7 to form •OH, and 0.5 mM SO32– at pH 10 to form eaq–. Pharmaceuticals with the Ar–CF3 (fluoxetine) and Ar–F plus pyrazole-CF3 (sitagliptin) motifs were treated similarly. Parent molecule concentrations were monitored with high pressure liquid chromatography with a UV detector. Fluorine in the parent and product molecules was quantified with 19F-NMR and complete fluorine mass balances were obtained. High resolution mass spectrometry was used to further explore product identities. The major product for Ar–F compounds was fluoride. The Ar–CF3 model compounds led to fluoride and organofluorine products dependent on motif placement and reaction conditions. Trifluoroacetic acid was a product of 4-(trifluoromethyl)phenol and fluoxetine. Additional detected fluoxetine products identified using mass spectrometry resulted from addition of −OH to the aromatic ring, but a dealkylation product could not be distinguished from fluoxetine by 19F-NMR. Sitagliptin formed multiple products that all retained the pyrazole-CF3 motif while the Ar–F motif produced fluoride. 19F-NMR, mass spectrometry, and chromatography methods provide complementary information on the formation of fluorinated molecules by modification or fragmentation of the parent structure during photolysis, allowing screening for fluorinated photoproducts and development of fluorine mass balances.
Keywords: 19F-NMR, photolysis, pharmaceuticals, mass balance, mass spectrometry, oxidation, reduction
Introduction
Fluorinated compounds derived from natural processes are rare in the environment, despite fluorine being the 13th most abundant element in the Earth’s crust.1,2 The majority of fluorine is found in its mineral form of fluorite which is not bioavailable.2,3 While roughly 3700 organohalides naturally occur in the environment, only 30 contain fluorine.1,4 Contrary to naturally derived molecules, the presence of fluorine is widespread in anthropogenic compounds, and its use is becoming more common. In 2010, 20% of administered drugs contained fluorine, with an increasing trend.5−7 In 2018, 17 new pharmaceuticals that contained fluorine were approved by the U.S. Food and Drug Administration, which was the most ever to date.7,8 Since the first fluorinated drug, fludrocortisone, came to market in 1954, over 150 other fluorinated drugs have followed. Some of the most common drugs, fluoxetine (Prozac), atorvastatin (Lipitor), and ciprofloxacin (Ciprobay), contain fluorine and generate billions of dollars in revenue indicating widespread use.9,10
The incorporation of fluorine in pharmaceuticals and agrochemicals affects the physiochemical properties.8,11 The addition of fluorine to a molecule yields desirable effects including metabolic stability, thermal stability, change in pKa values, increased lipophilicity and polarity, and increased membrane permeability.12 Substituting a hydrogen for a fluorine atom in a drug can alter the biological function of the molecule8,13 and minimally changes the steric and geometric structure relative to the entire molecule.14 Fluorine, being a highly electronegative atom, forms a strong bond with carbon.15 This strong bond dissociation energy leads to favorable properties such as high resistance to oxidative and thermal stresses16 and a longer half-life within the body due to increased metabolic stability and thus an increased time over which the drug is effective.17 Fluorine can be added to a variety of functional groups within a molecule. These motifs include aryl-F, benzylic-CF3, heteroaromatic fluorine (e.g., fluorinated pyrazoles), −O–CF3, −S–CF3, and aliphatic-fluorine (Figure 1).5,18 Ar–CF3, Ar–F, and alkyl-F contribute to about 45.3, 14.6, and 14.9% of total organofluorine present in pharmaceuticals.6
Figure 1.
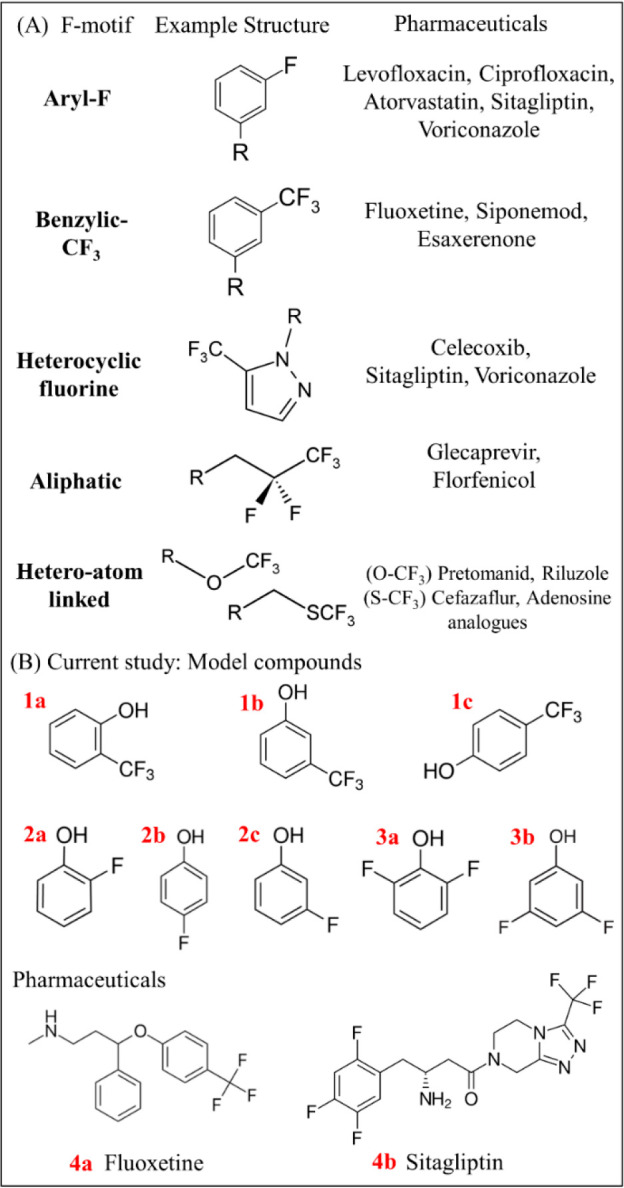
(A) Examples of fluorinated motif structures commonly found in pharmaceuticals and (B) model fluorinated compounds and pharmaceuticals chosen for the current study. The model compounds are 2-(trifluoromethyl)phenol (1a), 3-(trifluoromethyl)phenol (1b), 4-(trifluoromethyl)phenol (1c), 2-fluorophenol (2a), 4-fluorophenol (2b), 3-fluorophenol (2c), 2,6-difluorophenol (3a), 3,5-difluorophenol (3b), and the pharmaceuticals fluoxetine (4a) and sitagliptin (4b).
Organofluorine compounds found in surface water, sediment, and groundwater can be attributed to human activity because few fluorine-containing molecules naturally occur.3 Poly- and perfluoroalkyl substances (PFAS) are highly fluorinated chemicals used throughout industry with a wide variety of applications. PFAS are largely persistent and are transported globally,19 with few known biotic or abiotic transformations. While PFAS have received considerable attention within environmental research, many other compounds are of interest such as fluorinated agrochemicals and pharmaceuticals. The persistence and environmental degradation products of these fluorinated molecules remains less well-studied. Furthermore, pharmaceutical residues have been detected in almost all environmental matrices such as groundwater and surface water, including the scarcely populated polar regions.20 Wastewater treatment plants (WWTP) are a point source of pharmaceuticals, including antidepressants (e.g., fluoxetine) and antidiabetic medications (e.g., sitagliptin), in the environment.20 Hospital effluent is another source that is rich in pharmaceuticals that may be discharged to surface waters. A study in Japan showed detection of 41 pharmaceuticals, antiseptics, and other healthcare drugs in a hospital effluent.21 The study showed eight compounds in a downstream WWTP linked to the hospital waste. Commonly used fluorinated pharmaceuticals such as fluoxetine and sitagliptin have been found to be present and persist in surface waters (e.g., levels of 1–100 ng g–1 on suspended sediments in rivers).22 In rural areas, agriculture, animal husbandry, and aquaculture are the likely the main sources of (fluorinated) anthropogenic compounds in surface water.23
While biotic degradation of pharmaceuticals can occur, abiotic transformation by photolysis is an important degradation pathway that leads to the formation of largely unknown byproducts.24 Because pharmaceuticals are designed to be stable in aqueous solutions and biological systems, photolysis is an important degradation pathway to consider in the environment and in water treatment systems. In natural waters, direct photolysis occurs when the compound of interest is capable of absorbing solar photons, leading to chemical transformations. Indirect photolysis may occur when other reactive species, such as hydroxyl radical (•OH), are generated and react with the compound of interest, leading to transformation rate constants typically near diffusion-controlled limits (109–1010 M–1 s–1).25 Additionally, •OH is relevant in advanced oxidation processes (AOPs) used in water treatment plants to remove and transform organic micropollutants.26,27 The hydrated electron (eaq–), generated when sulfite ions are activated by UV, is a highly reactive and short-lived species, but it is of importance in reductive degradation of highly oxidized species. Recent work has shown that eaq– generated in advance reductive processes (ARPs) are able to defluorinate highly stable compounds, such as PFAS.28,29
The identity and toxicity of the reaction products produced from fluorinated compounds also need to be studied. For example, trifluoroacetic acid (TFA) is formed from certain fluorinated pharmaceuticals during the UV/ozonation process at WWTPs.30 While TFA was previously believed to be naturally occurring, recent studies show that this may be due to inadequate analytical techniques.31 In Germany it was noted that the concentration of TFA in rivers was higher than expected (>100 μg/L). It was found that in WWTPs, the highest percentage of TFA formation on a molar basis were from pharmaceuticals with a trifluoromethyl group attached to a benzene ring (Ar–CF3).30 This was thought to be the reason for the high concentration of TFA in German rivers. TFA was not found to be a risk to human health at the concentrations being detected in surface waters.32 It should be noted, however, that TFA has little to no degradation pathways in the environment and can accumulate.33 It is of concern whether photolysis reactions lead to minor structural modification with retention of the fluorinated motif, new persistent fluorinated compounds, or fluoride.
Identification of fluorinated reaction products from photolysis processes has only received limited attention.34−37 Thus, fluorinated products formed via degradation of these compounds may pose unforeseen consequences such as production of benzoic acids via the photolysis of trifluoromethylphenyl dervatives.38 To better understand the degradation pathways of these fluorinated compounds in the environment, reliable detection methods must first be established. Nuclear magnetic resonance (NMR) is a means to identify chemical structures of compounds, and using NMR to detect 19F, which is 100% abundant, gives 19F-NMR an advantage over NMR analyses of other nuclei in terms of both sensitivity and lack of background signals from solvents and buffers. Using 19F-NMR allows rapid identification of the chemical environment of the fluorine, and thus whether degradation occurs via structural modification of the molecule (and retention of the fluorine functional group), production of new fluorine-containing functional groups, or defluorination to produce fluoride based on the chemical shifts observed in the NMR spectrum over time. Quantification of organofluorine and fluoride concentrations using NMR is possible using standards with known concentrations of fluorine. The use of a capillary tube for an internal standard has been adopted and is a viable way to obtain quantitative information from NMR spectra.39,40
19F-NMR has been previously used to track the herbicide trifluralin in a matrix containing organic matter.41 It was determined that some soils did not degrade the parent compound while other soil types completely transformed the parent into various metabolites. In the study, the 19F peak broadened after being exposed to the soil organic matter, suggesting that covalent bonding was the mechanism of sorption to the tested soils.41,42 The use of 19F-NMR was also used for the detection and quantification of fluorinated acids in rainwater, including trifluoroacetic acid.43 The 19F-NMR analysis of the environmental samples matched the concentrations found using gas chromatography–mass spectrometry, further proving that 19F-NMR can be used to both rapidly identify and quantify fluorinated compounds in environmental samples.43
Here we focus our study on several fluorinated phenol model compounds (Figure 1) covering key motifs (Ar–F and Ar–CF3) and two pharmaceutical compounds (fluoxetine and sitagliptin) under direct and indirect photolysis/advanced treatment conditions that produce •OH or eaq–. The fluorinated phenol model compounds were chosen because they have sufficient solubility for study, have slow hydrolysis reactions, and represent substructures present in pharmaceutical compounds. We evaluated and quantified all fluorine photoproducts at the end of photolysis for each compound in each aqueous matrix. Kinetics of degradation were studied by monitoring parent compound concentrations. The varied effects of each photolysis condition on each studied fluorine motif were observed with respect to byproducts formed. Finally, liquid chromatography high resolution mass spectrometry was used to compare detected byproducts to 19F-NMR results.
Experimental Section
Chemicals
Chemical sources and purities are given in the Supporting Information (SI; Section 1).
Photolysis Experiments
Acetate (pH 5), phosphate (pH 7), and borate (pH 10) buffers, each at 10 mM, were prepared and used in the photolysis experiments. Stock solutions of compounds of interest at 1–2 mM were prepared in Milli-Q water and stored in the dark at 4 °C. The stock solutions were diluted into the buffer matrix to a concentration of 10 μM. Hydrogen peroxide was added to achieve a concentration of 1 mM into the pH 7 buffer matrix to study the reaction with •OH. Sodium sulfite was added at a concentration of 0.5 mM into the pH 10 buffer matrix to study the reaction with eaq–.44−48
Irradiation was performed using a medium pressure polychromatic-source mercury vapor lamp apparatus consisting of a 450-W UV immersion lamp, a quartz immersion well with cooling water circulation, and a Pyrex 7740 absorption sleeve with a cutoff value of 280 nm (Ace Glass) such that the wavelengths are environmentally relevant and sufficient to generate the desired reactive species. A carousel was placed around the lamp such that the test tubes were rotated around the center of the lamp to ensure uniform light exposure on each test tube.
The compounds of interest at a concentration of 10 μM in a pH 5 buffer, pH 7 buffer, pH 10 buffer, pH 7 buffer with an excess of 1 mM hydrogen peroxide, or pH 10 buffer with an excess of 0.5 mM sodium sulfite were poured into quartz test tubes, one of which was wrapped in aluminum foil and used as a dark control. Samples were capped with cork stoppers, which did not touch the aqueous solution, and placed in the carousel. Each experiment was performed in triplicate.
Photolysis Kinetics
A minimum of 5 time points were taken to monitor parent compound concentrations using high pressure liquid chromatography (HPLC). An Agilent 1100 series HPLC with a variable wavelength UV detector was used for analysis. Sampling procedures and HPLC conditions for each compound of interest are provided in the SI (Section 2, Table S1). First-order rate constants were determined by plotting ln(C/C0) with respect to time. Note that measurements of direct photolysis quantum yields and bimolecular reaction rate constants with reactive radicals were not a goal of this study.
Fluorine Quantification using 19F- NMR
Wilmad 7-in. length class A glass 500 MHz NMR tubes with 5 mm outer diameter were used for analysis. All aqueous NMR samples contained 10% D2O (v/v). In the case for the phenolic model compounds in the pH 10 matrix, an additional 1.2% (v/v) of 1 M HCl was added to the NMR tube to prevent hydrolysis of any residual parent or photoproducts. Samples analyzed were an unphotolyzed sample and a sample at the completion of photolysis, where at least 1 to 2 log reduction in concentration was observed with HPLC. Hexafluorobenzene (HFB) was enclosed in a sealed melting-point capillary tube, placed in the NMR tube, and used as a chemical shift reference (−164.9 ppm) and an internal standard to quantify the fluorine in each sample. A 600 MHz Avance Neo NMR equipped with a 5 mm three channel TCI inverse cryoprobe for 19F NMR spectral acquisition without 1H decoupling was used to determine and quantify the fluorinated parent compound and any fluorinated products from the photolysis experiments. A delay time of 10 s, acquisition time of 0.9 s, and number of scans of 1024 were experimentally determined to give a sufficient signal-to-noise ratio and complete longitudinal relaxation for accurate and reproducible quantitative measurements. For a more detailed explanation of NMR conditions and parameters, see Section 3 of the SI.
Liquid Chromatography-High Resolution Mass Spectrometry (LC-HRMS)
A Velos HRAM LC MS Orbitrap system equipped with a Luna C18 nanocolumn was used for mass spectrometry analysis. The mobile phase was a mixture of HPLC grade water and acetonitrile, both with 0.1% formic acid. The starting ratio was 98:2 water to acetonitrile by volume, which switched to 2:98 water to acetonitrile over the course of 33 min. The flow rate was constant at 0.1 mL/min. The injection volume was set to 4.0 μL, and the typical detection rage was from a molecular weight of 100 to 800 m/z. A smaller range (65–300 m/z) was used to detect smaller molecules (e.g., phenols, TFA). The blank sample containing only the unspiked photolysis aqueous matrix was injected to obtain a baseline. Then the unphotolyzed and photolyzed samples were run on the instrument. Additional details and the product identification process, which was largely restricted to molecular formulas containing fluorine for the pharmaceuticals, are described in the SI (Section 4). Retention times, areas, and the most abundant ion masses of peaks were recorded.
Results and Discussion
Photolysis of (Trifluoromethyl)phenol Model Compounds (1a–c)
Three (trifluoromethyl)phenols (1a–c) were first selected for an initial evaluation of fluorinated functional group effects on compound stability during photolysis. Photolysis rates of all (trifluoromethyl)phenol model compounds were pH dependent, with the rate constants for direct photolysis at pH 10 > pH 7 > pH 5 (Table 1, representative kinetic plot for model 1a in Figures 2; S4 and S6 for 1b and 1c). For example, the direct photolysis rate constant for the representative model compound 1a at pH 10 was 2 orders of magnitude larger than that at pH 7 (Table 1; Figure 2). The shift of equilibrium toward the deprotonated form of compounds 1a–c in the pH 10 buffer (pKa values 7.74, 8.08, and 8.27 for 1a, 1b, and 1c, respectively49) increases the rate of photolysis of (trifluoromethyl)phenols due to changes in the absorption spectra leading to ready excitation of the deprotonated anion (see UV–visible spectra in Figures S1–S3).37 Given that the pKa of 1a is 7.74, the presence of some deprotonated 1a at pH 7 explains the faster rate constant compared to 1b and 1c.
Table 1. First-Order Photolysis Rate Constants for 2-, 3-, and 4-(Trifluoromethyl)phenols, 2-, 3-, 4-Fluorophenols, 2,6-, 3,5-Difluorophenols, Fluoxetine, and Sitagliptin Using a 450-W UV Immersion Lamp with a 280 nm Cutoff Value in Various Aqueous Photolysis Matricesa.
| compound | photolysis conditions | rate constant (h–1) |
|---|---|---|
| (Trifluoromethyl)phenols | ||
| 1a | pH 5 | 3.52 ± 0.07 |
| pH 7 | 26.4 ± 0.64 | |
| pH 10 | 3.34 × 102 ± 9.34 × 101 | |
| 1 mM H2O2 | 30.0 ± 1.47 | |
| 0.5 mM SO32– | 4.22 × 102 ± 9.38 | |
| 1b | pH 5 | 2.72 ± 0.06 |
| pH 7 | 3.27 ± 0.63 | |
| pH 10 | 2.08 × 102 ± 7.51 | |
| 1 mM H2O2 | 7.80 ± 0.20 | |
| 0.5 mM SO32– | 2.26 × 102 ± 1.36 × 101 | |
| 1c | pH 5 | 2 × 10–2 ± 5.5 × 10–4 |
| pH 7 | 0.10 ± 0.02 | |
| pH 10 | 1.58 ± 0.71 | |
| 1 mM H2O2 | 6.85 ± 0.93 | |
| 0.5 mM SO32– | 2.38 ± 0.45 | |
| Fluorophenols | ||
| 2a | pH 5 | 0.21 ± 0.01 |
| pH 7 | 0.85 ± 0.03 | |
| pH 10 | 16.0 ± 0.37 | |
| 2b | pH 5 | 0.17 ± 2.5 × 10–3 |
| pH 7 | 0.36 ± 0.01 | |
| pH 10 | 10.2 ± 0.29 | |
| 2c | pH 5 | 4.28 ± 0.10 |
| pH 7 | 4.17 ± 0.11 | |
| pH 10 | 25.8 ± 1.66 | |
| Difluorophenols | ||
| 3a | pH 5 | 2.4 × 10–2 ± 8 × 10–4 |
| pH 7 | 0.32 ± 6.2 × 10–3 | |
| pH 10 | 1.08 ± 0.02 | |
| 1 mM H2O2 | 6.56 ± 0.51 | |
| 0.5 mM SO32– | 1.0 ± 0.02 | |
| 3b | pH 5 | 2.1 × 10–2 ± 1 × 10–3 |
| pH 7 | 9.8 × 10–2 ± 1 × 10–3 | |
| pH 10 | 1.03 ± 0.04 | |
| 1 mM H2O2 | 2.68 ± 0.25 | |
| 0.5 mM SO32– | 1.07 ± 0.19 | |
| Pharmaceuticals | ||
| 4a | pH 7 | 0.27 ± 0.01 |
| pH 10 | 0.56 ± 0.15 | |
| 1 mM H2O2 | 12.1 ± 1.54 | |
| 0.5 mM SO32– | 5.79 ± 0.95 | |
| 4b | pH 7 | 9.6 × 10–3 ± 3.8 × 10–4 |
| pH 10 | 3 × 10–2 ± 1.4 × 10–3 | |
| 1 mM H2O2 | 1.21 ± 0.04 | |
| 0.5 mM SO32– | 0.43 ± 0.06 | |
The structures and nomenclature are provided in Figure 1. The pH values were set by acetate, phosphate, and borate for pH 5, 7, and 10, respectively, and the buffered pH for H2O2 and SO32– were 7 and 10, respectively. Reported errors are 95% confidence intervals determined by a weighted average of triplicate samples. Photolysis rate constants are corrected for losses in dark controls.
Figure 2.

Photochemical degradation kinetic plots for 2-(trifluoromethyl)phenol (model compound 1a) with photolysis (filled shapes) and dark controls (equivalent hollow shapes) in (a) pH 5 buffer (black squares), (b) pH 7 buffer (black circles) and pH 7 buffer with 1 mM H2O2 (red triangles), and (c) pH 10 buffer (black inverted triangles) and pH 10 buffer with 0.5 mM sulfite (blue diamonds). The photolysis rate constants calculated from the slopes are 3.52 ± 0.07 h–1 for pH 5, 26.4 ± 0.64 h–1 for pH 7, 29.99 ± 1.47 h–1 for H2O2, 334.1 ± 93.45 h–1 for pH 10, and 422.4 ± 9.38 h–1 for sulfite. Error bars represent the standard deviation of triplicate samples. Reported rate constant errors represent the average 95% confidence interval determined by regression statistics. Note the change in scales/units along both the x- and y-axes.
With the addition of H2O2 to the pH 7 solution, an increase in the rate constant was observed for compounds 1a–c. The addition of SO32– had minimal effect in a pH 10 solution for compounds 1b and 1c but aided in the photolysis of compound 1a. These results indicate that the oxidizing conditions increased the rate of degradation of Ar–CF3 motifs, while the reducing conditions are effective to a lesser extent. The increase in the rate constant with addition of H2O2 is most pronounced for 1c (68-fold increase) as compared to 1b (2.3-fold) and least for 1a (1.1-fold). All three compounds likely react with •OH at close to the diffusion controlled limit, and the increment in the rate constant is 4 to 5 h–1 for each. The much faster direct photolysis of 1a and 1b leads to a lesser overall impact of H2O2 addition. One limitation of the experimental conditions is that experiments with SO32– are usually performed with 254 nm excitation wavelengths. Thus, in these experiments, we are relying on the small amount of light <280 nm that the cutoff filter does not completely eliminate to generate eeq–. This choice was made because irradiating with 254 nm light was likely to lead to rapid direct photolysis. Because there is acceleration of the reaction with the addition of SO32–, this is strong evidence that some eeq– was generated, but further studies with more efficiently generated eeq– will be needed to fully assess the utility of ARPs for the compounds studied herein.
The NMR spectra in Figure 3 show three panels with different chemical shift ranges at which products are observed for the initial and photolyzed samples in each matrix (compare Figure 3 for 1a to Figures S5 and S7 for 1b and 1c). Fluorinated photoproduct formation varied with both the position of −CF3 and pH. Using the 19F-NMR spectra and a hexafluorobenzene internal standard, mass balances were made for a visual representation of quantification of photolysis products of 1a–1c (panel d in Figure 3). At pH 5, compound 1a produced two reaction products: one that is an unidentified singlet but with chemical shift consistent with an aryl-F50 (product A at −157.8 ppm, broad singlet) and the other product was fluoride (−121.5 ppm). The variation in the chemical shifts of fluoride are attributed to interactions with F– caused by pH changes or other conditions. Prior observations have shown that such variation can be >1 ppm.51
Figure 3.

Representative 19F-NMR spectra (a–c) and fluorine mass balance (d) plots for initial (unphotolyzed) and final (photolyzed) samples. Panels a–c are NMR spectra for 2-(trifluoromethyl)phenol (compound 1a) before photolysis (I, unphotolyzed) and after photolysis in pH 5 acetate buffer (II), pH 7 phosphate buffer (III), pH 10 borate buffer (IV), pH 7 buffer with 1 mM H2O2 (V), and pH 10 with 0.5 mM SO32– (VI). The parent compound 1a (black star) and product A (NMR shift −82.0 ppm) are shown in panel a, fluoride (F–) production is shown in panel b, and fluorinated photoproduct singlet peaks with NMR shifts of −157.8 ppm (product B) are shown in panel c. Similar 19F-NMR spectral plots for 1b and 1c are in the SI. The y-axes scales have been adjusted in panels a–c to display full peaks. The internal standard peak at −164.9 ppm for HFB is not shown. Panel d displays fluorine quantification for compounds 1a–1c for each matrix (pH 5, pH 7, pH 10, pH 7 with 1 mM H2O2, and pH 10 with 0.5 mM SO32–) for unphotolyzed (initial) and photolyzed (final) samples. Fluoride is the major photolysis product in all cases. Peaks for products A and B from 1a and products X, Y, and Z from 1c are fluorinated organic products. TFA is only formed from 1c at pH 5 and pH 7 conditions. Errors associated with the mass balances are shown in Tables S4–S6.
At pH 7 with and without H2O2 for compound 1a, fluoride was the only end-product. At pH 10 and with SO32–, fluoride was the major photoproduct, but another unidentified product peak that was a broad singlet was found at a shift of −82 ppm. Different photoproducts for pH 5 and 10 indicate different degradation mechanisms for the protonated and deprotonated phenol forms, and previous studies have shown that mechanism and product formation is influenced by the formation of quinone intermediates and their protonation state, with protonated forms (lower pH) leading to TFA formation.37 Fluoride was the only photoproduct for compound 1b under all reaction conditions (Figure 3d). TFA was only observed when −CF3 was in para-position (1c) for pH 5, 7, and H2O2 experiments but not for pH 10 and sulfite conditions (Figure 3d). The percentage of total initial fluorine in 1c that was converted to TFA was 5%, 1.5%, and 3% for pH 5, 7, and H2O2 matrices, respectively. Results also indicate that TFA may not be formed from the deprotonated phenolic species, which is consistent with past results.37 Further work is needed to assess how functional group position influences TFA formation and yield. As with 1a, additional photoproducts (X, Y, and Z, Figure S7) were observed at pH 5 and 10 for 1c, but the products were near the parent compound’s chemical shift, i.e. they retained the −CF3 motif. We conclude from these studies and previous work on (trifluoromethyl)phenols that fluoride is the primary photoproduct for all compounds, with TFA only being produced when the −CF3 motif is in the para position.38
Photolysis of Fluorophenol Model Compounds
Rate constants for the three fluorophenols (2a–c) are given in Table 1, with 4-fluorophenol reacting most rapidly, and pH 10 giving the fastest rate constant for all compounds. By 19F-NMR, the only photoproduct observed for all fluorophenol compounds under the conditions studied was fluoride (−121.5 ppm). It must be noted that the fluoride peak may appear broad in some buffered matrices due to buffer interactions, inhomogeneities in magnetic field, or shimming of the instrument, but these issues did not affect the calculated mass balances.42 It is known that the phenolate can undergo ring condensation as a photodegradation mechanism. Under basic conditions, the Ar–F motif can remain on the ring structure.52 Two degradation mechanisms involving ring-condensation form either fluoride or an organofluorine compound, both requiring the formation of phenolate, either by the fluorine removing the hydroxyl hydrogen, or deprotonation above the pKa value respectively (see Schemes S1 and S2).52,53 The 19F-NMR spectra of the fluorophenol model compounds only show fluoride formation at pH values even below the pKa, suggesting that, if ring-condensation occurs, it is through the mechanism of hydroxyl hydrogen abstraction by fluoride thereby forming hydrofluoric acid.52
The difluorophenols (3a, 3b) were photolyzed to examine the effects of more diversely substituted fluorophenols. Photolysis rate constants were again highly pH dependent, increasing ∼10-fold from pH 5 to 7 and from pH 7 to 10. The addition of •OH increased the rate constant for both compounds by an order of magnitude compared to the direct photolysis at pH 7, while generation of eaq– had a minimal effect. These results indicate that the presence of hydroxyl radicals increase the rate of loss of aromatic difluorophenols, while eaq– have little to no effect. The 19F-NMR spectra showed fluoride production under all conditions, except at pH 5 where the extent of transformation was limited. For 3a (pH 7 conditions) and 3b (pH 7 and sulfite conditions), minor products (Product K for 3a and L and M for 3b, singlet peaks, Figures S18–S22) close to the parent compound shift were present indicating a modification of the ring structure that retained an Ar–F motif. No other products or modified structures were observed for the fluorophenols. Kinetic plots, NMR spectra, and fluorine mass balances for the five fluorophenols are compiled in the SI (Figures S8–S22; Tables S7–S11).
Photolysis of Fluoxetine
Based on our model system studies, photolysis of the pharmaceutical fluoxetine (4a) was studied to be compared to the three Ar–CF3 compounds (1a–c). The photolysis rate was pH dependent (the fluoxetine pKa is 9.8). At pH 10, the rate was nearly twice that at pH 7. The addition of H2O2 and SO32– increased the rate roughly 45- and 10-fold, respectively (Table 1), suggesting that both the oxidizing and reducing conditions can increase the photolysis rate of fluoxetine. Fluoride was the most abundant product formed under all conditions, but four fluorinated compounds with chemical shifts distinct from fluoxetine were observed, one being TFA (Figure 4). The use of Ar–CF3 model compounds to better understand TFA formation allowed insight into the TFA formation from fluoxetine. With 4-(trifluoromethyl)phenol (1c) being a proposed intermediate photoproduct for TFA formation, TFA was formed from fluoxetine at pH 7 (with and without H2O2), similar to the findings for 1c. The NMR shift of Product C is also consistent with that of 1c. No TFA was formed under basic conditions, with and without the addition of SO32–, suggesting that the position of the Ar–CF3 motif and protonation state are important factors in TFA formation. The model compounds, however, may not be a perfect representation of fluoxetine. At pH 10, small amounts of organofluorine products were observed for 1c, whereas only fluoride is formed from fluoxetine.
Figure 4.

(a–c) 19F-NMR spectra and (d) fluorine mass balances for fluoxetine (compound 4a) for all aqueous matrices. Panels a–c show spectra before photolysis (I, unphotoyzed) and after photolysis in pH 7 phosphate buffer (II), pH 10 borate buffer (III), pH 7 buffer with 1 mM H2O2 (IV), and pH 10 with 0.5 mM SO32– (V). The parent compound 1a (black star), fluoride (F–), and fluorinated photoproduct singlet peaks with NMR shifts −62.8, −63.0, −63.1, and −77.3 ppm (C, D, E and TFA respectively) are divided into panels a–c based on their respective shifts. (d) Fluorine mass balance for the same matrices. An additional product (modified fluoxetine) was identified with the same shift as the parent compound fluoxetine. The modified fluoxetine product has the same shift as that of the parent compound fluoxetine because the modification is too distant from the −CF3 to affect the NMR shift. The modified fluoxetine compound has been identified by LC-MS/MS as norfluoxetine. The y-axes scales for panels a–c are different and have been adjusted to show full peaks. The internal standard peak for HFB is not shown in this figure. Error associated with the mass balances are shown in Table S12.
The three other products observed via NMR had similar shifts to the parent fluoxetine compound, suggesting that the products retained the Ar–CF3 motif. Product C was formed at pH 7, both with and without the addition of H2O2, Product D was only formed with H2O2, and Product E was formed at pH 7. No organic fluorine photoproduct was formed via direct photolysis at pH 10; however, Product E was also detected with the addition of SO32– at pH 10 (Figure 4). Kinetic plots and further NMR details are in the SI (Figure S23; Table S12).
While quantification using NMR showed parent compound peaks to be present in the solution at specific concentrations, product tracing via HPLC gave different concentrations, and a greater extent of transformation of fluoxetine after photolysis. The difference in concentrations indicated a product of photolysis with the same NMR shift as that of fluoxetine. In this case, we surmise that the product was a modified fluoxetine product (Figure 4d) with a similar structure to that of fluoxetine, but the modification was too far from the fluorine motif in the structure to change its shift in the NMR spectrum. LC-MS/MS analysis revealed a demethylation reaction during photolysis in all aqueous matrices forming norfluoxetine (Table S14), where the removal of a methyl group occurred at the other end of the molecule from the fluorine motif. The presence of norfluoxetine has also been confirmed in previous photolysis studies.36 This result demonstrates that the NMR data must be complemented with HPLC analysis to fully assess removal of the parent compounds because, in some cases, structural modifications may not lead to a change in shifts in the NMR spectra.
Because of the presence of several products, including at least on with an identical NMR shift to the parent, LC-HRMS and MS/MS fragmentation data were also used to identify products and attempt to link them to the NMR results (Table S14). The major product fluoride is formed through defluorination and formation of nonfluorinated carboxylic acid Product 3, which was identified (Table S14). Cleavage of the C–O bond in fluoxetine also primarily yields 4-trifluoromethylphenol (1c; Product C) as a minor product for pH 7 and H2O2 conditions. The presence of TFA only at pH 7 conditions indicates that TFA formation occurs only through 1c. Products detected that arise from the addition of the OH group to the benzene ring (Table S14; m/z 326.1362) are consistent with the two fluorinated products D and E where the NMR shift has changed slightly due to the addition of the OH group. The byproducts formed by N-dealkylation (norfluoxetine) and hydroxylation mechanisms (Products D and E in Table S14) match with direct photolysis transformation products observed in the works of Tisler et al. and Lam et al.34,36 These results confirm that product screening by 19F-NMR provides useful information regarding the formation of fluorinated organic products and the fluorinated motifs they retain.
Photolysis of Sitagliptin
Photolysis of sitagliptin (4b), with three Ar–F and one Py-CF3 motif, expanded the range of studied motifs. At pH 7, the addition of H2O2 increased the photolysis rate nearly 145-fold, suggesting that the presence of •OH effectively increases photolysis rates of sitagliptin (Table 1, Figure S24). Photolysis in the presence of SO32– was about 15-fold faster than the direct photolysis rate at pH 10 (Table 1, Figure S24), suggesting that sitagliptin is susceptible to reaction with eaq– as well.
Multiple organofluorine peaks were observed in every photolyzed matrix. All the fluorinated products were singlets and had shifts close to the parent Py–CF3 shift (Figure 5a). The change in concentrations in Py–CF3 and Ar–F motifs while fluoride was forming relative to the parent concentrations was determined to observe which motif was producing fluoride (Figure 5). The same pattern was observed for each reaction condition except for direct photolysis at pH 10. Unlike fluoxetine, the concentrations of sitagliptin found with NMR and HPLC matched, indicating no structures that might have the same shift as the parent. The Ar–F motifs decreased as the fluoride appeared, and the fluorine from the Py-CF3 motif stayed relatively constant throughout photolysis. This suggests that for pH 7/H2O2/SO32– conditions, fluoride is being produced from the Ar–F motifs and for direct photolysis at pH 10, fluoride is also being produced from the Py–CF3 motif. The overall faster reaction with SO32– likely shifts the balance between the two pathways, and any product of Py–CF3 photolysis is at too low of a concentration to be observed when SO32– is present given the different time scales of the reactions with and without SO32– (Figure S23).
Figure 5.

(a–c) 19F-NMR spectra and (d) fluorine mass balance plots for sitagliptin (compound 4b) before photolysis (I) and after photolysis in pH 7 phosphate buffer (II), pH 10 borate buffer (III), pH 7 buffer with 1 mM H2O2 (IV), and pH 10 with 0.5 mM SO32– (V). Panels a–c show that the parent compound had four fluorine motifs i.e. one Ar–CF3 (blue star, F1 doublet) and three Ar–F (brown star F2a and red star F2b from a single fluorine, green star F3 singlet, and an orange star F4 doublet). Product fluorine peaks include F, G, H, I, and J all in 1a. The gray circles indicate other smaller product fluorine peaks and details can be found in the SI. (d) Fluorine mass balance plots for initial (unphotolyzed) and final (photolyzed) samples of sitagliptin for each photolysis matrix. Products F–J and other products shown in the figure are all near the F1 peak (Py-CF3 peak). The y-axes scales for panels a–c are different and have been adjusted to show full peaks. The internal standard peak for HFB is not shown in this figure. Peak 2b and F– occur together in panel b for V conditions but could be separated for mass balances because F2a and F2b had the same concentrations in all samples. Errors associated with the mass balances are shown in Table S13.
Because there is no hydroxyl group on the aromatic ring on which the Ar–F motifs reside, mineralization mechanisms may differ from the fluorophenol model compounds. All reaction conditions had multiple common products. Two unique products were identified, Product O (−64 ppm) at pH 10 and Product Q (−64.8 ppm) with the addition of H2O2, both retaining the Py–CF3 motif like all other products observed. All products were within 2 ppm of each other in the NMR spectrum, suggesting that the unique products formed were similar in structure to the other photoproducts.
Possible fluorinated photoproducts formed from sitagliptin at pH 7 were identified with LC-MS/MS (Table S15). The addition of oxygen occurred in each photoproduct and the Py-CF3 motif was retained. One photoproduct replaced an Ar–F with a hydroxyl group, providing a route to fluoride mineralization. Others added a hydroxyl group to the ring, replacing a single hydrogen. One product replaced the −NH2 group with a ketone. Oxidation continued with this product as two more oxygen atoms were added to the aromatic ring as hydroxyl groups (Figure S24). All products observed with LC-MS/MS were consistent with the chemical shifts observed in the 19F-NMR spectra.
Implications
Compared to PFAS, the fluorinated molecules studied herein, and presumably other fluorinated pharmaceuticals and pesticides, are susceptible to direct photolysis and/or accelerated reactions with photochemically produced reactive intermediates such as •OH and eaq–. Detailed studies of reaction kinetics and product formation are needed, because this reactivity will lead to a variety of byproducts and fluoride. 19F-NMR allows for rapid screening of fluorinated byproducts and gives information on the type of fluorine moieties on the transformation products. The large spectral range and 100% abundance of the 19F isotope made it possible to detect all fluorinated photoproducts and parent compounds with good peak separation and obtain a fluorine mass balance. Using 19F-NMR can give information on whether a natural or engineered reaction pathway would lead to desirable formation of fluoride or produce other organic fluorinated compounds. Because the retention of the fluorine moiety likely leads to greater overall persistence and potential uptake by biota, these reaction products are potential targets for study regarding environmental or toxicological impacts. Additionally, understanding which specific fluorinated motifs are most amenable to mineralization via photolysis, and treatment via AOP and/or ARP conditions will assist in future development of fluorinated compounds that mineralize to fluoride and have reduced potential for environmental persistence. Overall, the use of integrated analysis using HPLC, 19F-NMR, and untargeted, high resolution liquid chromatography–mass spectrometry methods allows for identification of all fluorinated reaction products including small modifications of the parent compound, quantification of these products, and assessment of the possible reaction pathways that lead to these byproducts.
Acknowledgments
Funding for this project was provided by the Minnesota Environment and Natural Resources Trust Fund as recommended by the Legislative-Citizen Commission on Minnesota Resources (LCCMR) and by a graduate fellowship to A.P.B. from the University of Minnesota College of Science and Engineering. Thanks to Jiaqian Li for help finalizing the NMR conditions and Samuel Lombardo for running preliminary experiments. We thank the Minnesota NMR Center for access to instrumentation. Funding for NMR instrumentation was provided by the Office of the Vice President for Research, the Medical School, the College of Biological Science, NIH, NSF, and the Minnesota Medical Foundation. Thanks to Peter Villalta, Yingchun Zhao, and Jingfang Huang at the analytical biochemistry mass spectrometry services shared resource at the Masonic Cancer Center, University of Minnesota for the help with LC-MS/MS instrumentation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsenvironau.1c00057.
List of chemicals, analytical procedures, detailed kinetics and NMR spectra, and mass balance data for all model compounds and pharmaceuticals tested and additional mass spectrometry results (PDF)
Author Contributions
○ A.P.B., T.F.M., and Q.T.W. contributed equally to the work and may each list themselves as first author.
The authors declare no competing financial interest.
Supplementary Material
References
- Ghosh A.; Mukherjee K.; Ghosh S. K.; Saha B. Sources and Toxicity of Fluoride in the Environment. Res. Chem. Intermed. 2013, 39, 2881–2915. 10.1007/s11164-012-0841-1. [DOI] [Google Scholar]
- Harnisch J.; Eisenhauer A. Natural CF4 and SF6 on Earth. Geophys. Res. Lett. 1998, 25 (13), 2401–2404. 10.1029/98GL01779. [DOI] [Google Scholar]
- Harper D. B.; O’Hagan O. The Fluorinated Natural Products. Nat. Prod. Rep. 1994, 11, 123–133. 10.1039/np9941100123. [DOI] [PubMed] [Google Scholar]
- Gribble G. W.Naturally Occurring Organofluorines. In Organofluorines. The Handbook of Environmental Chemistry (Vol. 3 Series: Anthropogenic Compounds); Neilson A. H., Eds.; Springer: Berlin, Heidelberg, 2002; Vol 3N. [Google Scholar]
- Zhou Y.; Wang J.; Gu Z.; Wang S.; Zhu W.; Acenã J. L.; Soloshonok V. A.; Izawa K.; Liu H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II-III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116 (2), 422–518. 10.1021/acs.chemrev.5b00392. [DOI] [PubMed] [Google Scholar]
- Inoue M.; Sumii Y.; Shibata N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5 (19), 10633–10640. 10.1021/acsomega.0c00830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Liu A.; Dhawan G.; Mei H.; Zhang W.; Izawa K.; Soloshonok V. A.; Han J. Fluorine-Containing Pharmaceuticals Approved by the FDA in 2020: Synthesis and Biological Activity. Chin. Chem. Lett. 2021, 32 (11), 3342–3354. 10.1016/j.cclet.2021.05.042. [DOI] [Google Scholar]
- Mei H.; Han J.; Fustero S.; Medio-Simon M.; Sedgwich D. M.; Santi C.; Ruzzicni R.; Soloshonok V. A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem.—Eur. J. 2019, 25, 11797–11819. 10.1002/chem.201901840. [DOI] [PubMed] [Google Scholar]
- Müller K.; Faeh C.; Diederich F. Fluorine in Pharmaceuticals: Looking beyond Intuition. Science 2007, 317 (5846), 1881–1886. 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- Wenthur C. J.; Bennett M. R.; Lindsley C. W. Classics in Chemical Neuroscience: Fluoxetine (Prozac). ACS Chem. Neurosci. 2014, 5, 14–23. 10.1021/cn400186j. [DOI] [Google Scholar]
- Maienfisch P.; Hall R. G. The Importance of Fluorine in the Life Science Industry. Fluor. Life Sci. Ind. 2004, 58 (3), 93–99. 10.2533/000942904777678091. [DOI] [Google Scholar]
- Braun M.; Eicher J. The Fluorine Atom in Health Care and Agrochemical Applications: A Contribution to Life Science. Modern Synthesis Processes and Reactivity of Fluorinated Compounds: Progress in Fluorine Science 2017, 7–25. 10.1016/B978-0-12-803740-9.00002-0. [DOI] [Google Scholar]
- Park B K.; Kitteringham N. R; O'Neill P. M Metabolism of Fluorine -Containing Drugs. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–470. 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
- Stabel A.; Dasaradhi L.; O'Hagan D.; Rabe J. P. Scanning Tunneling Microscopy Imaging of Single Fluorine Atom Substitution in Stearic Acid. Langmuir 1995, 11, 1427–1430. 10.1021/la00005a003. [DOI] [Google Scholar]
- Saunders B. C.Some Aspects of the Chemistry and Toxic Action of Organic Compounds Containing Phosphorus and Fluorine; Cambridge University Press: Cambridge, 1957. [Google Scholar]
- Shaughnessy M. J.; Harsanyi A.; Li J.; Bright T.; Murphy C. D.; Sandford G. Targeted Fluorination of a Nonsteroidal Anti-Inflammatory Drug to Prolong Metabolic Half-Life. ChemMedChem. 2014, 9, 733–736. 10.1002/cmdc.201300490. [DOI] [PubMed] [Google Scholar]
- Swallow S. Fluorine in Medicinal Chemistry. Progress in Medicinal Chemistry 2015, 54, 65–133. 10.1016/bs.pmch.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- Martin J.; Smithwick M.; Braune B.; Hoekstra P.; Muir D.; Mabury S. Identification of Long-Chain Perfluorinated Acids in Biota from the Canadian Arctic. Environ. Sci. Technol. 2004, 38, 373–380. 10.1021/es034727+. [DOI] [PubMed] [Google Scholar]
- Patel M.; Kumar R.; Kishor K.; Mlsna T.; Pittman C. U.; Mohan D. Pharmaceuticals of Emerging Concern in Aquatic Systems: Chemistry, Occurrence, Effects, and Removal Methods. Chem. Rev. 2019, 119, 3510–3673. 10.1021/acs.chemrev.8b00299. [DOI] [PubMed] [Google Scholar]
- Azuma T.; Arima N.; Tsukada A.; Hirami S.; Matsuoka R.; Moriwake R.; Ishiuchi H.; Inoyama T.; Teranishi Y.; Yamaoka M.; Mino Y.; Hayashi T.; Fujita Y.; Masada M. Detection of Pharmaceuticals and Phytochemicals Together with Their Metabolites in Hospital Effluents in Japan, and Their Contribution to Sewage Treatment Plant Influents. Sci. Total Environ. 2016, 548–549, 189–197. 10.1016/j.scitotenv.2015.12.157. [DOI] [PubMed] [Google Scholar]
- Boulard L.; Dierkes G.; Schlüsener M. P.; Wick A.; Koschorreck J.; Ternes T. A. Spatial Distribution and Temporal Trends of Pharmaceuticals Sorbed to Suspended Particulate Matter of German Rivers. Water Res. 2020, 171, 115366. 10.1016/j.watres.2019.115366. [DOI] [PubMed] [Google Scholar]
- Lin A. Y.; Yu T.; Lin C. Pharmaceutical Contamination in Residential, Industrial, and Agricultural Waste Streams: Risk to Aqueous Environments in Taiwan. Chemosphere 2008, 74, 131–141. 10.1016/j.chemosphere.2008.08.027. [DOI] [PubMed] [Google Scholar]
- Schwarzenbach R. P.; Gschwend P. M.; Imboden D. M.. Environmental Organic Chemistry, 3rd Ed.; Wiley: Hoboken, NJ, 2017. [Google Scholar]
- Buxton G. V.; Greenstock C. L.; Helman W. P.; Ross A. B. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (·OH /·O–) in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17 (2), 513–885. 10.1063/1.555805. [DOI] [Google Scholar]
- Litter M. I. Introduction to Photochemical Advanced Oxidation Processes for Water Treatment. Environmental Photochemistry Part II 2005, 2, 325–366. 10.1007/b138188. [DOI] [Google Scholar]
- Barbusi K.; Filipek K. Use of Fenton’s Reagent for Removal of Pesticides from Industrial Wastewater. Polish J. Environ. Stud. 2001, 10 (4), 207–212. [Google Scholar]
- Cui J.; Gao P.; Deng Y. Destruction of Per- and Polyfluoroalkyl Substances (PFAS) with Advanced Reduction Processes (ARPs): A Critical Review. Environ. Sci. Technol. 2020, 54 (7), 3752–3766. 10.1021/acs.est.9b05565. [DOI] [PubMed] [Google Scholar]
- Vellanki B. P.; Abdel-wahab A.; Batchelor B. Advanced Reduction Processes: A New Class of Treatment Processes. Environ. Eng. Sci. 2013, 30 (5), 264–271. 10.1089/ees.2012.0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheurer M.; Nodler K.; Freeling F.; Janda J.; Happel O.; Riegel M.; Muller U.; Storck F. R.; Fleig M.; Lange F. T.; Brunsch A.; Brauch H.-J. Small, Mobile, Persistent: Trifluoroacetate in the Water Cycle - Overlooked Sources, Pathways, and Consequences for Drinking Water Supply. Water Res. 2017, 126, 460–471. 10.1016/j.watres.2017.09.045. [DOI] [PubMed] [Google Scholar]
- Joudan S.; De Silva A. O.; Young C. J. Insufficient Evidence for the Existence of Natural Trifluoroacetic Acid. Environ. Sci. Process. Impacts 2021, 23 (11), 1641–1649. 10.1039/D1EM00306B. [DOI] [PubMed] [Google Scholar]
- Boutonnet J. C.; Bingham P.; Calamari D.; Rooij C. de; Franklin J.; Kawano T.; Libre J.-M.; McCul-loch A.; Malinverno G.; Odom J. M.; Rusch G. M.; Smythe K.; Sobolev I.; Thompson R.; Tiedje J. M. Environmental Risk Assessment of Trifluoroacetic Acid. Hum. Ecol. Risk Assess. An Int. J. 1999, 5 (1), 59–124. 10.1080/10807039991289644. [DOI] [Google Scholar]
- Kim B. R.; Suidan M. T.; Wallington T. J.; Du X. Biodegradability of Trifluoroacetic Acid. Environ. Eng. Sci. 2000, 17 (6), 337–342. 10.1089/ees.2000.17.337. [DOI] [Google Scholar]
- Petrovic M.; Petrovic M.; Barcelo D. LC-MS for Identifying Photodegradation Products of Pharmaceuticals in the Environment. Trends Anal. Chem. 2007, 26 (6), 486–493. 10.1016/j.trac.2007.02.010. [DOI] [Google Scholar]
- Lam M. W.; Young C. J.; Mabury S. A. Aqueous Photochemical Reaction Kinetics and Transformations of Fluoxetine. Environ. Sci. Technol. 2005, 39 (2), 513–522. 10.1021/es0494757. [DOI] [PubMed] [Google Scholar]
- Tisler S.; Zindler F.; Freeling F.; Nödler K.; Toelgyesi L.; Braunbeck T.; Zwiener C. Transformation Products of Fluoxetine Formed by Photodegradation in Water and Biodegradation in Zebrafish Embryos (Danio Rerio). Environ. Sci. Technol. 2019, 53 (13), 7400–7409. 10.1021/acs.est.9b00789. [DOI] [PubMed] [Google Scholar]
- Ellis D. A.; Mabury S. A. The Aqueous Photolysis of TFM and Related Trifluoromethylphenols. An Alternate Source of Trifluoroacetic Acid in the Environment. Environ. Sci. Technol. 2000, 34 (4), 632–637. 10.1021/es990422c. [DOI] [Google Scholar]
- Manfrin A.; Hänggli A.; van den Wildenberg J.; McNeill K. Substituent Effects on the Direct Photolysis of Benzotrifluoride Derivatives. Environ. Sci. Technol. 2020, 54 (18), 11109–11117. 10.1021/acs.est.9b07429. [DOI] [PubMed] [Google Scholar]
- Koch K. R. Quantitative Determination of Aluminium in Tea by Means of Aluminium-27 Nuclear Magnetic Resonance Spectroscopy. Analyst 1990, 115 (6), 823–825. 10.1039/an9901500823. [DOI] [PubMed] [Google Scholar]
- Ycas P. D.; Wagner N.; Olsen N. M.; Fu R.; Pomerantz W. C. K. 2 - Fluorotyrosine Is a Valuable but Understudied Amino Acid for Protein - Observed 19F NMR. J. Biomol. NMR 2020, 74 (1), 61–69. 10.1007/s10858-019-00290-0. [DOI] [PubMed] [Google Scholar]
- Strynar M.; Dec J.; Benesi A.; Jones A. D.; Fry R. A.; Bollag J.-M. Using 19F NMR Spectroscopy to Determine Trifluralin Binding to Soil. Environ. Sci. Technol. 2004, 38 (24), 6645–6655. 10.1021/es0403110. [DOI] [PubMed] [Google Scholar]
- Achtnich C.; Fernandes E.; Bollag J.-M.; Knackmuss H.-J.; Lenke H. Covalent Binding of Reduced Metabolites of [15N3] TNT to Soil Organic Matter during a Bioremediation Process Analyzed by N NMR Spectroscopy. Environ. Sci. Technol. 1999, 33 (24), 4448–4456. 10.1021/es990427+. [DOI] [Google Scholar]
- Ellis D. A.; Martin J. W.; Muir D. C. G.; Mabury S. A. Development of an 19F NMR Method for the Analysis of Fluorinated Acids in Environmental Water Samples. Anal. Chem. 2000, 72 (4), 726–731. 10.1021/ac9910280. [DOI] [PubMed] [Google Scholar]
- Chawla O. P.; Arthur N. L.; Fessenden R. W. An Electron Spin Resonance Study of the Photolysis of Aqueous Sulfite Solutions. J. Phys. Chem. 1973, 77 (6), 772–776. 10.1021/j100625a008. [DOI] [PubMed] [Google Scholar]
- Deister U.; Warneck P. Photooxidation of SO32- in Aqueous Solution. J. Phys. Chem. 1990, 94 (17), 2191–2198. 10.1021/j100368a084. [DOI] [Google Scholar]
- Dogliotti L.; Hayon E. Flash Photolysis Study of Sulfite, Thiocyanate, and Thiosulfate Ions in Solution. J. Phys. Chem. 1968, 72 (5), 1800–1807. 10.1021/j100851a073. [DOI] [Google Scholar]
- Fischer M.; Warneck P. Photodecomposition and Photooxidation of Hydrogen Sulfite in Aqueous Solution. J. Phys. Chem. 1996, 100 (37), 15111–15117. 10.1021/jp953236b. [DOI] [Google Scholar]
- Hayon E.; Treinin A.; Wilf J. Electronic Spectra, Photochemistry, and Autoxidation Mechanism of the Sulfite-Bisulfite-Pyrosulfite Systems. SO2–, SO3–, SO4–, and SO5– Radicals. J. Am. Chem. Soc. 1972, 94 (1), 47–57. 10.1021/ja00756a009. [DOI] [Google Scholar]
- Dearden J. C.; Weeks G. R. Relationship of Physicochemical Properties of Isomeric Trifluoromethylphenols with Their Bactericidal Effect on Escherichia Coli. J. Pharm. Sci. 1973, 62 (5), 843–844. 10.1002/jps.2600620540. [DOI] [PubMed] [Google Scholar]
- Dolbier W. R., Jr.Guide to Fluorine NMR for Organic Chemists; John Wiley & Sons, 2016. [Google Scholar]
- Hudlicky M. Chemical shifts of fluorine in hydrogen fluoride and fluoride ion. J. Fluor. Chem. 1985, 28 (4), 461–472. 10.1016/S0022-1139(00)81136-7. [DOI] [Google Scholar]
- Chatterjee P.; Ghosh A. K.; Chakraborty T. Hydrogen Bond Induced HF Elimination from Photoionized Fluorophenol Dimers in the Gas Phase. J. Chem. Phys. 2017, 146, 084310. 10.1063/1.4976988. [DOI] [PubMed] [Google Scholar]
- Boule P.; Guyon C.; LeMaire J. Photochemistry and the Environment. VIII. Photochemical Behavior Of Dichlorophenols in a Dilute Aqueous Solution. Chemosphere 1984, 13 (5), 603–612. 10.1016/0045-6535(84)90197-8. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.