

Abstract
This Review focuses on the structure–function relationship of radical S-adenosyl-l-methionine (SAM) enzymes involved in the assembly of metallocofactors corresponding to the active sites of [FeFe]-hydrogenase and nitrogenase [MoFe]-protein. It does not claim to correspond to an extensive review on the assembly machineries of these enzyme active sites, for which many good reviews are already available, but instead deals with the contribution of structural data to the understanding of their chemical mechanism (Buren et al. Chem. Rev. 2020, 142 ( (25), ) 11006−11012; Britt et al. Chem. Sci. 2020, 11 ( (38), ), 10313–10323). Hence, we will present the history and current knowledge about the radical SAM maturases HydE, HydG, and NifB as well as what, in our opinion, should be done in the near future to overcome the existing barriers in our understanding of this fascinating chemistry that intertwine organic radicals and organometallic complexes.
Keywords: Nitrogenase, [FeFe]-hydrogenase, X-ray crystallography, Metalloproteins, Iron−sulfur clusters, Radicals, Active site assembly
Introduction
Trace elements and more specifically transition metals are essential to life, notably because they confer unique properties to proteins and expand the repertoire of chemical reactions afforded by the use of amino acids alone. Among these metals, V, Mn, Fe, Co, Ni, Cu, Zn, Mo, or W, are key elements of many bioinorganic cofactors, often constituting unique and complex active sites capable of catalyzing difficult reactions such as hydrogen oxidation and nitrogen or carbon dioxide reduction. Consequently, the resulting metalloenzymes are found at key steps in many fundamental biological processes including respiration or photosynthesis. Iron–sulfur ([FeS]) clusters are probably the most common inorganic cofactors existing either as [Fe2S2] rhombs or [Fe4S4] cubes (Figure 1a and b, respectively).3,4 When bound to proteins, they can act as redox centers to wire electrons and/or perform redox catalysis. They can also be involved in nonredox catalysis, hence acting as Lewis acids,5 play a role in structural function, or be used as receptors to sense signals from the environment.6 Yet, metallocofactors can be even more sophisticated as illustrated by the active sites of the carbon monoxide dehydrogenase/acetyl coenzyme-A synthase (CODH/ACS),7 the [FeFe]-hydrogenase,8 or the nitrogenase [MoFe]-protein1 (Figures 1c–f, respectively). Their structural resemblance to some minerals led to the hypothesis for a mineral surface origin of life.9 These metallocofactors would therefore correspond to a vestige or legacy from the past.
Figure 1.

Representation of (a) [Fe2S2] cluster (PDB 1QT9(10)), (b) [Fe4S4] cluster (PDB 1HFE(11)), (c,d) active sites of CODH/ACS, respectively (PDB 1OAO(12)), (e) [FeFe]-hydrogenase (PDB 6N59(13)), and (f) [MoFe]-nitrogenase (PDB 1M1N(14)). Transition metals and sulfide ions are depicted as balls and sticks, while the metal ligands are shown as sticks. Fe, Mo, Ni, N, C, O, and S are colored in dark red, cyan, green, blue, gray, red, and yellow, respectively.
Despite being constituted of simple pieces such as sulfide ions and transition metals, it appeared very early that these active sites are not spontaneously assembled and rather require the well-orchestrated action of dedicated multiprotein machineries.4 For instance, the pioneer work of Dean et al. on the cysteine desulfurase NifS15 led to the characterization of the NIF (nitrogen fixation) regulon, responsible for the assembly of the nitrogenase FeMo-cofactor in addition to the ISC (iron–sulfur cluster) and SUF (sulfur assimilation) [FeS] cluster assembly machineries and paved the way to a highly active new field of research dedicated to [FeS] cluster biogenesis.16
In 2001, Sofia and co-workers identified a set of [Fe4S4] cluster containing proteins as member of a superfamily sharing specific motifs and suggesting they would also share functional features, despite a very low primary structure conservation, usually below 10% identity.17 Time confirmed their assumption, and in a 20 year period this family turned out to correspond to one of the most crowded superfamilies of enzymes using a conserved structural core domain.18,19 With only very few exceptions, they indeed share functional properties while covering a broad range of chemical reactions catalyzed on various substrates ranging from small organic molecules to large macromolecules including proteins, RNA, and DNA. The first available crystal structures highlighted that the conserved motifs within the family were either responsible for [Fe4S4] cluster and S-adenosyl-l-methionine (SAM) binding or would correspond to key structural elements defining the radical SAM core domain fold.20−22 Conversely, motifs specific to a given function diverge within the family. They are located down the SAM binding site and are likely responsible for substrate recognition. Radical SAM enzymes all use a reduced [Fe4S4]+ cluster to perform the reductive cleavage of one of the SAM carbon-sulfonium bonds, usually the C5′-S bond, leading to the production of a highly reactive 5′-deoxyadenosyl radical species, which, in turn, will, in most of the cases, abstract a hydrogen atom of the substrate and trigger the radical-based reaction (Figure 2).
Figure 2.

Depiction of the reductive S-adenosyl-l-methionine (SAM) cleavage into 5′-deoxyadenosyl radical (5′dA·) and l-methionine (l-Met) and the substrate hydrogen abstraction catalyzed by radical SAM enzymes.
Soon after, in 2004, Posewitz, King, and co-workers identified two new radical SAM enzymes termed HydE and HydG as responsible for the assembly of the [FeFe]-hydrogenase active site.23 In 2006, Curatti, Rubio, and co-workers succeeded in purifying the radical SAM enzyme NifB,24 allowing for the first time to in vitro activate the nitrogenase [MoFe]-protein starting only from simple elements and purified proteins.25 The conjunction of these three events afforded many breakthroughs in our understanding of the function of these metallocofactor assembly machineries. Today, crystal structures of the three radical SAM proteins HydE, HydG, and NifB are available (Figure 3).26−30 In this Review, we will discuss how structural data have contributed to the understanding of their respective mechanisms. We will also briefly discuss how structural biology will continue to contribute to this field.
Figure 3.

Crystal structures of the radical SAM proteins. (a) HydE from Thermotoga maritima (PDB 3CIW(28)), (b) HydG from Thermoanaerobacter italicus (Ti) (PDB 4WCX(27)), and (c) NifB from Methanothermobacter thermautotrophicus (PDB 7BI7(30)). The β-strands and the α-helices of the β-barrel, common to all radical SAM proteins, are depicted in light yellow and blue, respectively; differences between the three proteins are shown in transparent gray. The radical SAM (RS) [Fe4S4] cluster of all three proteins, the auxiliary (Aux) [Fe4S4] cluster of HydG, and the K-cluster of NifB are represented as balls and sticks with Fe and S atoms colored in dark red and yellow, respectively.
[FeFe]-Hydrogenase
[FeFe]-Hydrogenase catalyzes the reversible reaction of hydrogen oxidation.8 The crystal structures solved at the end of the 1990s11,31 were decisive to our current understanding of the structure and function of the active site and have inspired many chemists in the development of bioinspired catalysts useful for hydrogen oxidation. This active site, also termed H-cluster, is deeply buried in the protein matrix. It corresponds to a unique two-iron subcluster ([2Fe]H) bridged to a regular [Fe4S4] cluster by a conserved cysteine residue (Figure 4). The [2Fe]H subcluster is constituted of two iron atoms, each bound to a carbon monoxide and a cyanide ion. In addition, both iron atoms are bridged by an azadithiolate molecule and an additional carbon monoxide. The distal iron has an open site corresponding to where the heterolytic hydrogen splitting reaction occurs. The azadithiolate bridgehead nitrogen atom serves as a base32 to polarize the H–H bond and affords efficient proton transfer through a relay that connects the active site to the surface of the protein. Therefore, the presence of this [2Fe]H subcluster is necessary to produce an active enzyme. Early work on the heterologous expression of the [FeFe]-hydrogenase from Desulfovibrio vulgaris Hildenborough in Escherichia coli showed that only the [Fe4S4] clusters are inserted into the protein, leading to an inactive enzyme.33
Figure 4.
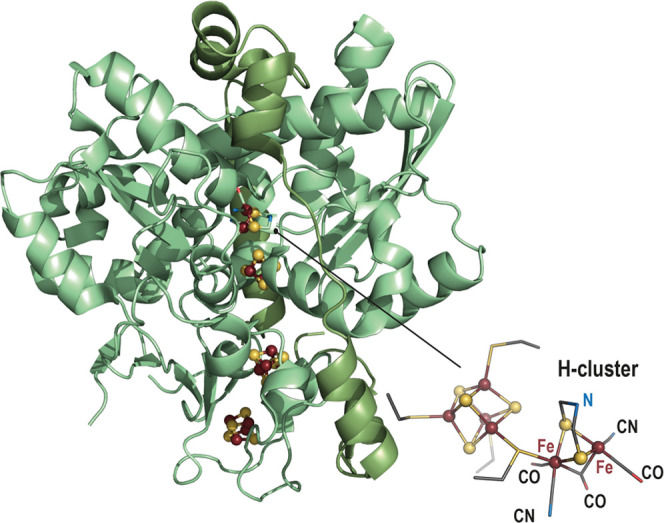
Ribbon representation (light green) of the [FeFe]-hydrogenase crystal structure (PDB 1HFE(11)). [Fe4S4] clusters are represented as balls and sticks with the Fe and the S atoms colored in dark red and yellow, respectively. A zoom-in of the H-cluster is represented as balls and sticks for the iron and sulfide ions; the ligands are represented as sticks with N, C, O and S atoms colored in blue, gray, red and yellow, respectively.
King, Posewitz, and co-workers showed that, upon addition of three genes coding for the metalloproteins HydF, HydE, and HydG, E. coli produces an active hydrogenase.23 Mulder, Peters, and co-workers demonstrated that these Hyd proteins are responsible for the assembly of the [2Fe]H subcluster (Figure 5).34 The H-cluster [Fe4S4] cluster is produced by the housekeeping iron sulfur cluster assembly machinery instead and must be loaded first to afford the [2Fe]H subcluster insertion and hydrogenase activation.35 HydF corresponds to a metalloprotein containing one [Fe4S4] cluster and a GTPase domain.36 It was identified earlier as a scaffold/insertase, onto which the [2Fe]H subcluster would be built prior to its insertion into the [Fe4S4] cluster preloaded hydrogenase. Using a [2Fe]H synthetic precursor Fe2(SCH2NHCH2S)(CO)4(CN)2, Fontecave and co-workers confirmed that HydF is indeed an insertase capable of transferring a [2Fe]H precursor into the hydrogenase.37 Crystal structures of apo and [Fe4S4] cluster-bound HydF were solved, but no further structural information is available to date regarding both the [2Fe]H precursor binding site and the mechanism for its insertion into the hydrogenase.38−41 Furthermore, whether the [2Fe]H subcluster is built stepwise onto HydF remains an open question to address and the role for GTP is unknown. Yet, because HydF is not a radical SAM enzyme, it is beyond the scope of this Review and the authors suggest recent reviews on this topic for interested readers.2 Conversely, HydG and HydE belong to the radical SAM superfamily.42 They are also thoroughly discussed in another review in the current volume. Early amino acid sequence comparisons suggested that HydG would be related to the 2-iminoacetate synthase ThiH43 whereas HydE would be related to biotin synthase BioB,21 two pioneer members of the radical SAM superfamily. The former uses l-tyrosine to produce 2-iminoacetate and p-cresol.44 The latter inserts a sulfur atom between two unreactive sp3 carbon atoms in desthiobiotin.45 In the latter, the sulfur atom is provided by a [Fe2S2] cluster, which is destroyed upon reaction and needs to be rebuilt to afford multiple turnovers. It was therefore proposed that, starting from substrates yet to be identified, HydG and HydE would be responsible for CO/CN– and azadithiolate biosynthesis, respectively.
Figure 5.

Scheme of the [2Fe]H-subcluster biosynthesis highlighting the role of maturase HydG, HydE, and HydF. Steps before the [2Fe]H insertion into apo-hydrogenase (HydA) are discussed in the text. Recently, Rao et al. showed that the carbon and nitrogen atoms of the azadithiolate ligand both originate from l-serine through a pathway yet to be determined.46 However, this does not seem to involve either HydE or HydG and therefore is not further discussed in the present review.
HydG: A 2-Iminoacetate Synthase and More..
HydG shares about 25% amino acid sequence identity with ThiH (Figure 6a).43 Multiple sequence alignments forecast these conservations as radical SAM protein general features and motifs specific to substrate recognition, suggesting HydG would also use l-tyrosine as substrate. We confirmed that HydG can process l-tyrosine in vitro to release p-cresol.43 In the meantime, Swartz et al. developed the first in vitro [FeFe]-hydrogenase reconstitution assay using clarified lysates and showed that l-tyrosine stimulates hydrogenase maturation.47 It was subsequently shown by Driesener, Roach, and co-workers that, starting from l-tyrosine, HydG produces p-cresol and cyanide in a 1:1 ratio.48 Soon after, Shepard, Broderick, and co-workers demonstrated that HydG also produces carbon monoxide.49 Using stable isotope labeled l-tyrosine, it was shown that cyanide and carbon monoxide are derived from its Cα-N and carboxyl moieties, respectively, and that all five carbon monoxide and cyanide ligands of the [2Fe]H subcluster originate from l-tyrosine, further confirming HydG is a masterpiece in the hydrogenase assembly machinery.50
Figure 6.

(a) Sequence alignment of TiHydG and EcThiH primary structures. Red and white boxes indicate conserved and similar amino acids in the two proteins. TiHydG secondary structure elements are depicted on top. (b) SAXS-generated envelope for (left) a HydG mutant lacking the C-terminal domain, HydGΔ91 from Thermoanaerobacter tengcongensis (Tte), and (right) wild-type TteHydG.51 The TiHydG crystal structure27 without (left, blue ribbon representation) and with (right, blue and green ribbon representation) the C-terminal domain and auxiliary cluster (see main text) were placed in the envelope. [FeS] clusters are represented as van der Waals spheres.
In addition to the radical SAM domain reminiscent of ThiH, HydG exhibits a long C-terminal stretch that contains three conserved cysteine residues and is able to accommodate an additional auxiliary [Fe4S4] cluster.42,51 Removing this C-terminal domain leads to a ThiH-like protein producing p-cresol and mainly 2-iminoacetate.51,52 Yet, a minute amount of cyanide but no carbon monoxide is detected, supporting that 2-iminoacetate fragmentation would not require the C-terminal domain. Small angle X-ray scattering (SAXS) experiments using either wild-type HydG or a C-terminal domain depleted variant indicated this domain is located at the bottom of the radical SAM domain β-barrel (Figure 6b).51 Homology modeling suggested superposition of layers along the barrel: residues dedicated to [Fe4S4] cluster binding and SAM cleavage on top; l-tyrosine binding site and place of radical-based chemistry in the middle; and channel specific to HydG where cyanide is produced at the bottom. l-Tyrosine activation by the 5′-deoxyadenosyl radical species was also a topic for discussion.
Early on, because phenoxyl oxygen-centered radicals were prevalent in biology, it was postulated that initial hydrogen atom abstraction would take place at that position. Therefore, the subsequent detection and characterization of an oxidobenzyl radical in HydG using rapid freeze-quench electron paramagnetic (EPR) spectroscopy led to the proposal for an heterolytic cleavage of the l-tyrosine Cα-Cβ bond upon activation by the primary 5′-deoxyadenosyl radical.53 Yet, despite a lack of structural data for l-tyrosine-bound HydG, the crystal structure of the L-tryptophan-bound radical SAM tryptophan lyase NosL,54 which also shares about 25% amino acid sequence identity with HydG, highlighted that, in this protein, the atom closest to the 5′-deoxyadenosine C5′ atom is actually the amino-nitrogen atom (Figure 7), supporting a similar binding mode for l-tyrosine in HydG. This assumption was further supported by the subsequent characterization of a C2-centered radical by EPR when using 4-hydroxyphenyl propanoic acid as alternative substrate.55
Figure 7.
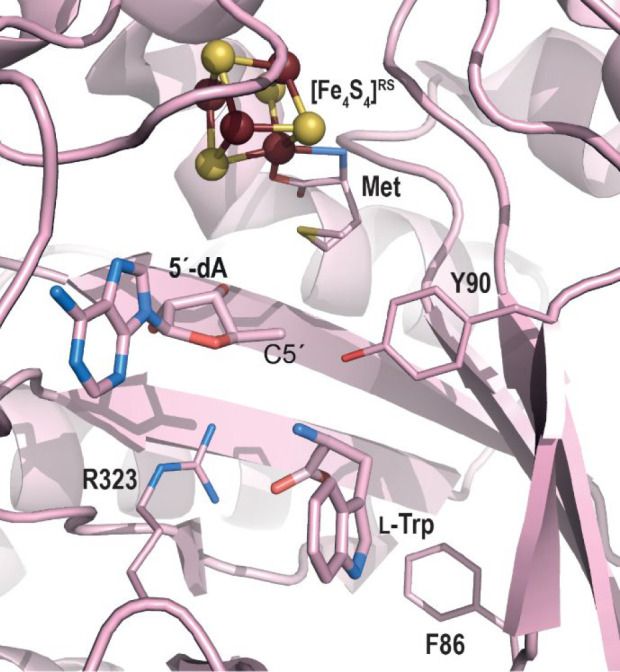
Zoom in of the active site of l-tryptophan-bound NosL from Streptomyces actuosus.54 The overall protein crystal structure is represented as pink ribbons, while key residue side chains are shown with thin sticks. 5′-dA and the l-tryptophan substrate are represented with thicker sticks. C, O, and N atoms are colored in pink, blue, and red, respectively. The radical SAM [Fe4S4] cluster is depicted as balls and sticks with iron and sulfur atoms colored in dark red and yellow, respectively.
A major contribution in our understanding of the HydG mechanism came from the crystal structure of HydG from Thermoanaerobacter italicus (TiHydG) by Dinis and Roach in 2015.27 This structure and that from Carboxydothermus hydrogenoformans solved by us26 confirmed the location of the C-terminal domain at the bottom of the barrel and the existence of a narrow channel that connects the l-tyrosine radical fragmentation site and the [Fe4S4]Aux cluster. More importantly, it revealed the unexpected presence of an additional iron atom, termed “dangler iron”, bridged to the [Fe4S4]Aux cluster by a sulfur atom.27 After reinvestigation of the crystallographic data, it appeared that this dangler iron displays an octahedral six-coordination geometry with a conserved histidine residue, two water molecules, and a κ3-l-homocysteine as ligands (Figure 8).56 Advanced EPR spectroscopy analyses identified this iron as a ferrous ion.27 Inspired by this crystal structure, Suess, Britt, and co-workers identified that l-cysteine and not homocysteine is the best dangler iron ligand, leading to higher HydG activity, establishing that the structure of the HydG auxiliary cluster is in fact an [Fe4S4] bound to a [κ3-l-cysteine-FeII(His)(OHx)2] species.57,58 The two water molecules/hydroxyl ions point toward the 2-iminoacetate delivery channel and were proposed to be the site for carbon monoxide/cyanide binding and/or synthesis.56 This delivery channel is filled with two nonconnected water molecules networks separated by a crown of hydrophobic residues proposed to act as a backflow valve supporting HydG as a nano factory assembly line.56 Identification of a conserved anion binding site in the bottom cavity suggested this site would correspond to transient cyanide binding site during turnover.
Figure 8.
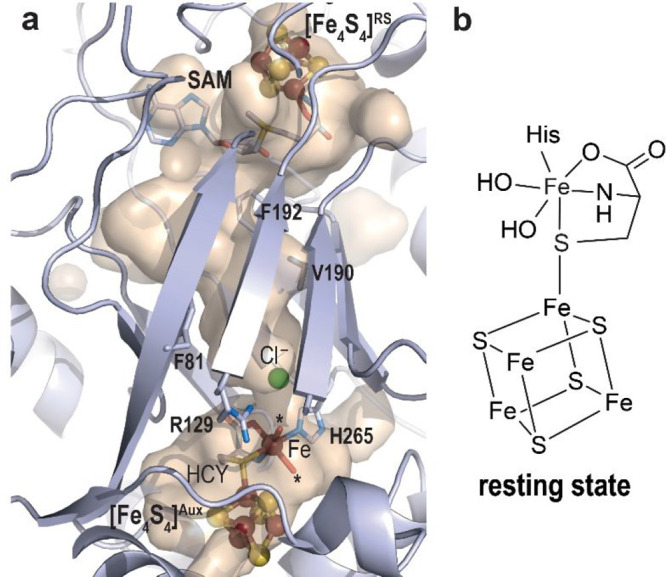
(a) Revisited model of TiHydG56 (see main text). The overall protein is represented as light blue ribbons while residues delineating the channel connecting both active sites are depicted as sticks. The active site where 2-iminoacetate is produced is indicated by the [Fe4S4]RS cluster and SAM shown as balls and sticks and sticks, respectively. The auxiliary cluster is shown as balls and sticks for the [Fe4S4] moiety and the dangler iron and sticks for the ligands. Fe, N, C, O, and S are colored in dark red, blue, light blue, red and yellow, respectively. The stars indicate the two water molecules discussed in the main text and the chloride ion site is depicted as a green ball. (b) Scheme of the [Fe4S4]Aux cluster and [κ3-l-cysteine-FeII(His)(OHx)2] species.
In a very elegant experiment, using stable isotope labeled l-tyrosine, Rao, Britt, and co-workers demonstrated that upon a single turnover, the dangler iron binds one carbon monoxide and one cyanide ligand leading to a [κ3-l-cysteine-FeII(His)(CN)(CO)] species also termed complex-A.59 A second turnover leads to the formation of [l-cysteine-FeII(CN)(CO)2] named complex-B. The second cyanide ion would induce complex-B release upon binding to the unique iron of the auxiliary [Fe4S4] cluster.57 The HydG reaction mechanism, based on all available experimental data, is presented in Figure 9. Using syn-B, a synthetic complex-B, it was then possible to produce an active hydrogenase without the need for HydG, confirming complex-B is the final HydG product.60 With the use of stable isotope labeled syn-B, Rao, Rauchfuss, and co-workers showed that, in addition to carbon monoxide and cyanide, the dangler iron and the l-cysteine sulfur atom also end in the [2Fe]H hydrogenase subcluster.60 Besides confirming the importance of complex-B as a structured block, its existence raises questions about its subsequent transfer and transformation. Indeed, syn-B is not stable in water and decays rapidly, supporting specific contacts between HydG and its partner would occur to prevent unnecessary degradation. In a very recent study, Shepard, Broderick, and co-workers reported the intriguing observation that HydG would release free carbon monoxide and cyanide and that the dangler iron would not be essential for activity.61 Yet, as will be discussed in the next section, HydE turned out to take complex-B as the substrate to perform radical-based chemistry. How 2-iminoacetate is converted into cyanide and carbon monoxide is probably the last important gray area in our understanding of the reaction. Indeed, this part has not undergone experimental investigations so far. Removing the C-terminal domain or the dangler iron completely abolishes carbon monoxide production but not that of cyanide. Meanwhile, in these conditions, formate was detected instead, suggesting that 2-iminoacetate fragmentation would take place in the delivery channel and carbon monoxide would take place at the dangler iron site.56 Because the latter and the 4-iron containing auxiliary cluster resemble the active site of carbon monoxide dehydrogenase, a similar mechanism has been proposed to split a –COOH intermediate species into CO and a water molecule bound to the dangler iron. However, how 2-iminoacetate is fragmented, how complex-A and complex-B are produced, and how the latter is transferred to HydE are the remaining questions to address. While writing this Review, Chen, Britt, and co-workers reported an investigation of the different steps of HydG reaction using density functional theory calculation.62 In this work, they suggest that p-cresyl· would abstract a hydrogen atom from 2-iminoacetate that would be key to the fragmentation into cyanide and a ·COO– radical. Yet, such a proposed 2-iminoacetyl radical has not been detected while the p-cresyl· EPR signal decayed over time.53 Hence, further experimental evidence is needed to validate the proposal.
Figure 9.

Scheme of the reactions catalyzed by HydG in both active sites (see also Figure 8).
HydE: A Radical SAM Cysteine Desulfurase?
Despite a less than 20% amino acid sequence identity with biotin synthase, in 2001, HydE primary structures were misidentified as BioB proteins in most of the databases. Meanwhile, HydE remained without a known substrate or function until 2020, fueling speculations about its role in [FeFe]-hydrogenase maturation, even suggesting at some point that it was not essential to the assembly of the [2Fe]H subcluster.63 During all this time, HydE was always associated somehow with sulfur chemistry. Multiple sequence alignments pointed out the existence of two subsets of HydE, those with and those without an auxiliary cluster in addition to the radical SAM [Fe4S4] cluster.28 Based on these early observations and because HydG turned out to be responsible for the carbon monoxide and cyanide ligand synthesis, HydE was first proposed to be involved in the production of the azadithiolate ligand, notably in radical-based sulfur addition to sp3 carbon atoms.42
The first crystal structure of HydE from Thermotoga maritima (TmHydE) was determined by us in 2008, and it corresponds to a (βα)8 β-barrel fold very similar to biotin synthase.28 The active site cavity defined by the eight β-strands is rather broad, suggesting either a large or multiple substrates. Initial cavity mapping using different ions highlighted three anion binding sites termed S1–3 and spread along the cavity (Figure 10). A thiocyanate molecule was observed in the S3 site, further supporting the hypothesis that HydE would synthesize the S–C–N segments of the [2Fe]H-subcluster azadithiolate ligand. Site-directed mutagenesis experiments further supported that the S3 site is important for HydE function. Yet, only a few residues surrounding the cavity would be directly involved in the chemistry and would rather play a role in substrate recognition or intermediate stability. In addition, a crown of hydrophobic residues divides the cavity into two pockets and is proposed to correspond to a backflow valve to direct the reaction from top to bottom, as a nanofactory assembly line alike HydG.
Figure 10.
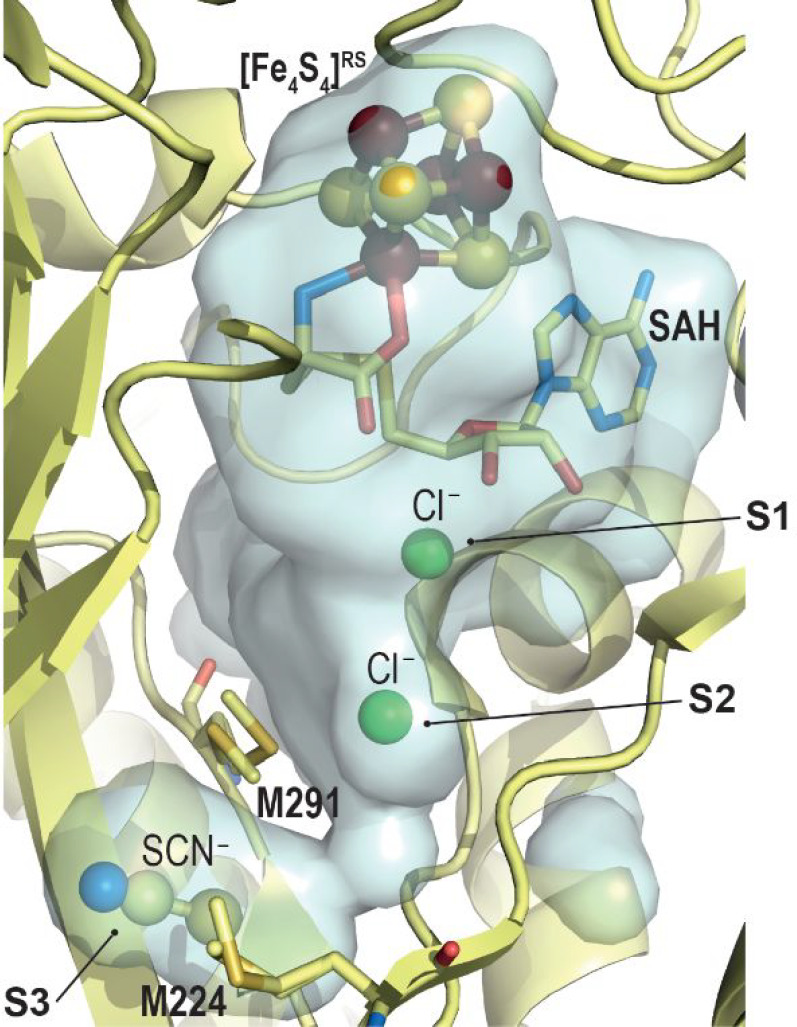
Zoom in of the TmHydE active site cavity.28 The overall protein is represented as light yellow ribbons. In this crystal structure, Sites S1 and S2 are occupied by a chloride ion (green ball, S1 and S2) while a thiocyanate molecule (ball-and-stick) is located at the S3 site. Methionine residues discussed in the text are depicted as sticks. The [Fe4S4]RS-cluster and SAM are shown as balls and sticks and sticks, respectively. Fe, N, C, O, and S are colored in dark red, blue, light yellow, red, and yellow, respectively.
TmHydE belongs to the auxiliary [Fe4S4] cluster-containing subset.42 The crystal structure highlighted this cluster is solvent-exposed at the surface of the protein and is very sensitive to degradation in vitro.28 Sequential reconstitution, oxidation/reduction in HydE crystals unveiled an unexpected plasticity for this cluster with associated structural rearrangements limited to a specific loop that covers the active site cavity as a lid.64 However, early [FeFe]-hydrogenase maturation assays indicated that it was nonessential for activity.28 Hence, its presence in a large subset of HydE remains unexplained.
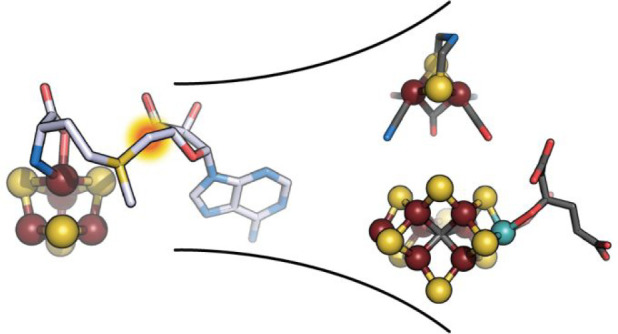
Betz, Broderick, and co-workers reported that natural and non-natural thiol-containing molecules can stimulate SAM cleavage, supporting at least transient binding in the HydE cavity.65 Yet, despite our many attempts, no structure of complexes between these molecules and the protein was obtained. Meanwhile, we performed an extensive structure-based molecular docking screening with various databases, including the KEGG E. coli metabolite database, to obtain leads for HydE substrate identification. The main challenge came from the fact that molecular docking programs are designed to maximize scores for high affinity ligands, which is usually not the case for substrates. Nevertheless, among the putative ligands, several molecules led to HydE-compound structures allowing the identification of thiazolidine derivatives as non-natural substrates.66 Cocrystallizing HydE with various thiazolidine, thiazinane, and pyrrolidine derivatives unveiled a site distinct from sites S1–3 and that seems to strongly favor sulfur atoms. Hence, using thiazolidine ligands as the substrate, it was possible to trigger SAM cleavage in crystals and track the ensuing radical-based reaction, which corresponds to the attack of the thiazolidine sulfur atom by the 5′-deoxyadenosyl radical producing S-adenosyl-l-cysteine and pyruvate (Figure 11).66 In spite of not being HydE’s natural substrate, (2R,4R)-2-methyl-1,3-thiazolidine-2,4-dicarboxylic acid (Figure 12a) structurally resembles complex-B (Figure 12b). Tao, Britt, and co-workers showed that HydE can indeed react with syn-B and produces two successive EPR-active intermediate species.67 The former is a S-adenosyl-l-cysteine–FeI(CO)2CN compound resulting from the addition of the 5′-deoxyadenosyl radical to the l-cysteine S atom of complex-B. This intermediate is most probably produced via a mechanism reminiscent of that previously reported when using thiazolidine as substrate. The second intermediate results from the release of the l-cysteine −CH2–CHNH2COO– fragment and is proposed be a 5′-thioadenosyl–FeI(CO)2CN species. Because previous isotope labeling experiments showed that S, Fe, CO, and CN end in the [2Fe]H hydrogenase subcluster, how the C5′-S bond is broken remains a hot question.
Figure 11.

(a) X-ray model of (top) SAM-bound and (bottom) [5′-dA+Met]-bound TmHydE with the corresponding 2Fo-Fc electron density maps. Map contours were drawn at the 1σ level.68 The [Fe4S4]RS cluster is shown as balls and sticks; SAM or [5′-dA+Met] and cysteine ligands are depicted as sticks. Fe, N, C, O, and S are colored in dark red, blue, light yellow, red, and yellow, respectively. (b) Scheme of the proposed S-adenosyl-l-cysteine (SAC) formation mechanism from (2R,4R)-2-methyl-1,3-thiazolidine-2,4-dicarboxylic acid ((2R,4R)-MeTDA) catalyzed by HydE.66 The SAM C5′–Sγ bond is reductively cleaved with concomitant formation of the C5′–S1 bond and disruption of the (2R,4R)-MeTDA S1–C2 bond. A subsequent one-electron oxidation of the C2-centered radical species into an imine, followed by hydrolysis of the latter would lead to SAC formation. The high-resolution structures and the minimal changes observed during the reaction in crystals allowed us to follow the electronic structure from SAM to the formation of SAC using quantum mechanical/molecular mechanical (QM/MM) calculations.66
Figure 12.

Crystal structures of (a) (2R,4R)-2-methyl-1,3-thiazolidine-2,4-dicarboxylic acid bound, (b) complex-B bound, and (c) R2SM224-Fe(CO)2CNCl bound TmHydE. The overall protein is represented as light yellow ribbons. Bound ligands are represented with sticks with the exception of the Fe atom depicted as a dark red sphere; SAM, [5′-dA+Met] or SAH and M224 and M291 residues, discussed in the main text, are shown with thinner sticks. The [Fe4S4]RS cluster is shown as balls and sticks. Fe, N, C, O, and S are colored in dark red, blue, light yellow, red, and yellow, respectively.
Stimulated by this important breakthrough, we determined the crystal structure of complex-B-bound TmHydE.69 This high-resolution structure establishes the identity of complex-B as a (κ3-cysteinate)FeII(CN)(CO)2 center, whose stereochemistry corresponds to a facial l-cysteinate tridentate, a cyanide trans to the sulfur atom, and two carbon monoxides trans to the amine and carboxyl groups (Figure 12b). Complex-B binds with an induced-fit of the active site cavity surrounding residues, leading to a close contact with the substrate, when compared to the resting state structure. In this orientation, the complex-B sulfur atom is facing the C5′-atom of 5′-deoxyadenosine at the same position as that of the sulfur atom of thiazolidine ligands, further highlighting a site with a high affinity for sulfur in front of SAM. Tracking the radical-based reaction directly in crystals showed formation of S-adenosyl-l-cysteine, confirming the direct attack of the complex-B sulfur atom by the 5′-deoxyadenosyl radical. Yet, no species corresponding to any of the intermediates previously identified by EPR was detected. This absence was interpreted as a combination of their fleeting character and probable low occurrence that impaired detection in the corresponding electron density map. Conversely, triggering the reaction in solution right before crystallization led to a new crystal structure that contains a new iron-containing species at the S3 site. This species was best-modeled as a trigonal bipyramid iron center bound to two carbon monoxide and one cyanide ligands in addition to a thioether from the conserved M224 residue and a chloride ion from solvent (R2SM224-Fe(CO)2CNCl) (Figure 12c).69
Despite resulting from the decay of the t = 10 s EPR active intermediate, this unique species likely mimics a HS-Fe(CO)2CN species corresponding to either HydE end-product or an intermediate awaiting a second turnover to dimerize into a putative Fe2S2(CO)4(CN)2 species, before being transferred to HydF (Figure 13). Preliminary structural analyses indicated that the HydE active site cavity is broad enough to house such di-iron species in a region between M224 residue and another conserved methionine residue, M291, which is part of the backflow valve (Figure 12b and c). M291 displays alternative conformations and was proposed to ease the HS-Fe(CO)2CN species transfer from the upper pocket to the S3 site thanks to thioether-Fe bond exchanges. A careful analysis around the S3 site highlighted specific conserved residues, including a threonine and two arginine residues that may define another chalcogen binding site. Mutating one of these residues significantly impairs HydE ability to maturate hydrogenase, confirming their importance to HydE activity, despite being far from complex-B binding site.
Figure 13.

Scheme of the reaction catalyzed by HydE based on recent results (see main text). It is still unknown whether the fusion of the HS-Fe(CO)2CN species occurs within HydE or, after being transferred, in HydF.
The S3 site is located near the surface of the protein, and in that region the cavity is covered by a short C-terminal stretch that follows the (βα)8-barrel domain. Among the many crystal structures of TmHydE available in the Protein Data Bank and corresponding to different space groups and different crystal packing, this stretch was observed in all but one structure, where it is disordered, leaving the cavity wide open, thus suggesting a leaving path for the HydE end product to be delivered to HydF. Likewise, a putative channel that may involve the auxiliary cluster, when present, was proposed to afford complex-B delivery from HydG to HydE (Figure 14). Therefore, the overall HydE architecture supports the enzyme functions as a nanofactory assembly line, similar to the case of HydG. The questions regarding the true HydE product, its production mechanism involving C5′-S bond breaking, and substrate/product delivery from/to partners remain to be addressed.
Figure 14.
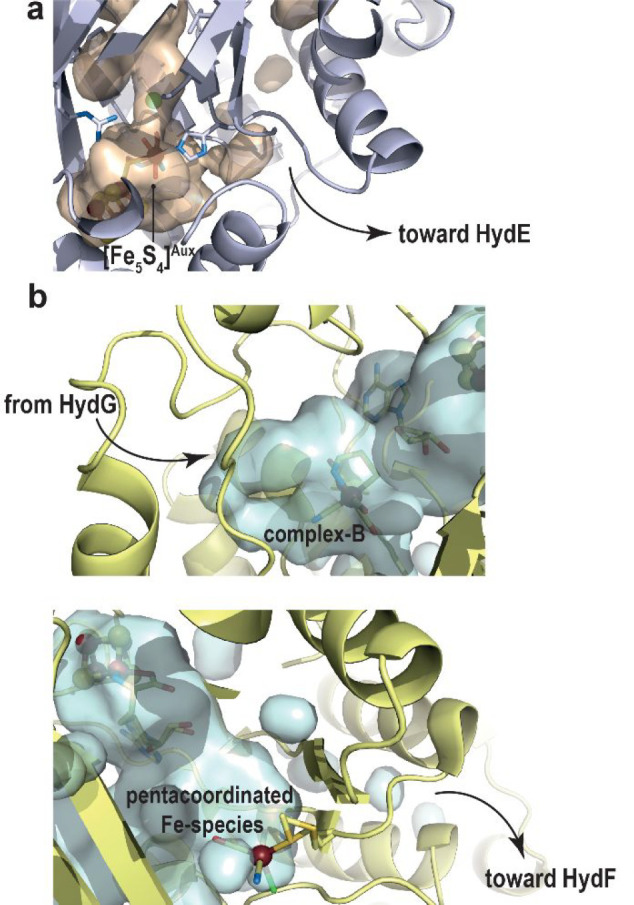
(a) HydG auxiliary-cluster site close to the surface showing a putative product exit pathway. The overall HydG protein is represented as light blue ribbons and the cavity shown in light orange was calculated retaining only the protein residues. Ligands of the auxiliary cluster are represented as sticks. (b) (top) HydE complex-B site showing its putative entrance, coming from HydG; [5′-dA+Met] and complex-B are depicted as sticks; (bottom) new iron-containing species found at the S3 site of HydE displaying an escape route of HydE product toward HydF; SAH and the ligands of the new species are shown as sticks. The overall HydE protein (top and bottom) is represented as light yellow ribbons, and the cavity represented in light blue was calculated retaining only the protein residues. In all figures, the iron atoms and sulfide ions from the [FeS] clusters are shown as dark red and yellow balls, respectively.
Nitrogenase
Nitrogenase catalyzes the reduction of dinitrogen into ammonia affording nitrogen assimilation by living organisms, hence acting as a key player in the global nitrogen cycle.1,70 Breaking the N2 triple bond, one of the strongest bonds in nature, is highly energy-demanding. Yet, nitrogenase performs the following reaction: N2 + 8 e– + 8 H+ + 16 MgATP → 2 NH3 + H2 + 16 MgADP + 16 Pi at ambient temperature and normal pressure due to the use of highly sophisticated organometallic cofactors.71 The molybdenum nitrogenase is the most prominent one and consists of two components: an electron donor (NifH) and a catalytic moiety (NifDK). The former couples ATP hydrolysis and electron transfer at the right redox potential to effectively reduce N2 into NH3. The latter corresponds to an α2β2-tetramer, which contains two unique metalloclusters per αβ-dimer. The [Fe8S7] P-cluster is responsible for the successive electron transfers from NifH to the FeMo-co, the actual catalytic site cofactor, which is constituted of a unique [MoFe7S9C-R-homocitrate] cluster (Figure 1f).72 The FeMo-co can be best described as the combination of [MoFe3S3] and [Fe4S3] modules, connected via three μ2-sulfide and one μ6-carbide ions. However, its exact structure was gradually unveiled. Indeed, the first crystal structures of the nitrogenase [MoFe]-protein afforded the description of the active site as a [MoFe7S9-R-homocitrate] center, missing the presence of an interstitial lighter atom.14,72,73 Its presence and nature were first suspected and proposed to be either a nitride N3–, an oxide O2–, or a carbide C4–, initially favoring a nitride for mechanistic reasons. Yet, many different spectroscopic studies early on excluded this atom as a nitrogen species.74−80 With the atomic resolution structure of [MoFe]-protein combined to electron spin echo envelope modulation spectroscopy, in a very elegant analysis, Spatzal, Einsle, and co-workers succeeded in characterizing this interstitial atom as a carbide ion, hence establishing the exact structure of FeMo-co as a [MoFe7S9C-R-homocitrate] organometallic center.72 Soon after Wiig, Ribbe, and co-workers demonstrated that this carbide ion originates from a SAM molecule methyl group provided by the radical SAM enzyme NifB.81
Synthesis and insertion of FeMo-co in nitrogenase requires a set of accessory proteins usually gathered in what is known as the Nif (Nitrogen-Fixing) regulon. An excellent review extensively describes both nitrogenase mechanism and FeMo-co biosynthesis,1 and in this Review we will mainly focus on the key radical SAM protein NifB. However, we will briefly describe the overall function of the Nif assembly machinery.
First, a cysteine desulfurase NifS abstracts a sulfur atom from l-cysteine and transfers it to a scaffold protein NifU, where an [Fe2S2] cluster is built stepwise using ferrous ions from a still unknown source and electrons provided by a ferredoxin. [Fe2S2] cluster are subsequently combined to form [Fe4S4] centers prior to be transferred to the radical SAM enzyme NifB. The latter then catalyzes the unique fusion of two [Fe4S4] clusters combined to the production and insertion of a carbide ion C4–. The subsequent insertion of a ninth sulfide ion leads to an [Fe8S9C] center also termed NifB-co that constitutes the core of the FeMo-co. NifB-co is then transferred to the scaffold protein NifEN, where molybdenum and R-homocitrate are added to produce the final FeMo-co to be integrated into the apo-nitrogenase [MoFe]-protein (Figure 15). Therefore, NifB appears as a key component of the machinery as it catalyzes a unique and highly complex radical-based reaction to convert regular [Fe4S4] clusters into the NifB-co [Fe8S9C] center. Yet, the protein is rather small, with less than 300 amino acids, raising substantial mechanistic questions.
Figure 15.

Nif assembly machinery with corresponding crystal structures that are shown with β-strands and α-helices depicted in light yellow and blue, respectively; [FeS] clusters are represented as balls and sticks with Fe and S atoms colored in dark red and yellow, respectively. The crystal structures of NifS, NifB, NifEN ,and NifDK correspond to PDB 5WT2,827BI7,303PDI,83 and 1M1N,14 respectively.
The Radical SAM Carbide Synthase NifB
Five years after the identification of the radical SAM protein superfamily by Sofia, Curatti, Rubio, and co-workers succeeded in purifying NifB from Azotobacter vinelandii, paving the way to subsequent biochemical characterizations and to the development of nitrogenase in vitro reconstitution assays.24,25 In addition to the radical SAM [Fe4S4] cluster ([Fe4S4]RS) bound to the conserved CX3CX2C motif, NifB can accommodate two additional [Fe4S4] clusters, termed K1 and K2, that were initially proposed to be modules of a larger K-cluster.84,85 However, these clusters cannot be considered as cofactors as it is usually the case because they correspond to substrates, explaining their high instability. As mentioned above, radioactive isotope labeling experiments performed by Wiig, Ribbe, and co-workers demonstrated NifB provides the FeMo-co interstitial carbide ion.81 The protein uses at least two SAM molecules per turnover. The first one provides its methyl group. Site-directed mutagenesis experiments to sequentially cancel either K1 or K2 combined to EPR experiments supported a methyl transfer to one of the sulfide ions of the K2-cluster via an SN2 nucleophilic substitution mechanism, releasing S-adenosyl-l-homocysteine.85,86 The second SAM molecule is reductively cleaved into a 5′-deoxyadenosyl radical species to abstract one hydrogen-atom from the above-mentioned methyl group and trigger carbide synthesis. The two remaining hydrogen atoms were proposed to be subsequently abstracted through deprotonation, but no experimental evidence has been provided to support this mechanism to date.81 Site-directed mutagenesis experiments combined with advanced EPR spectroscopy analyses afforded identification of three conserved cysteine residues and one histidine as ligands of the K1-cluster.84,85,87 Two conserved residues and a semiconserved cysteine residue, in addition to a putative fourth ligand, yet to be identified, were also proposed to bind to the K2-cluster. Strikingly, in vitro assays showed that NifB must be fully reduced by low-potential electrons before SAM binding and methyl transfer occur.85
Multiple sequence alignments highlighted that most of the NifB sequences contain an additional NifX-like domain at the N-terminus. This extra domain is presumably responsible for NifB-co storage, before being transferred to NifEN via the facultative carrier protein NifX. Yet, shorter NifB sequences lacking this NifX-like domain were also identified in some methanogens.88 These simpler NifB proteins also display significant activity to activate nitrogenase. This discovery turned out to be crucial to obtain pure protein suitable for X-ray crystallography analyses. Inspired by contributions of the different structural genomic consortiums, we screened for numbers of shorter NifB from different thermophilic organisms and determined the crystal structure of NifB from the archaeon Methanotrix thermoacetophila PT (MtNifB) loaded with the [Fe4S4]RS and K1-clusters (Figure 16a).29 The absence of the K2-cluster, the most labile one, induced significant flexibility at the C-terminal stretch, which is invisible in the corresponding electron density map. Yet, this first crystal structure provided significant insights into the mechanism of the enzyme. At odds with what was initially proposed from site-directed mutagenesis, K1 is bound to only two cysteine, one histidine and one glutamate residues. The H42 and C128 residues (MtNifB notation) belong to β-strands that constitute the radical SAM structural core domain and thus strongly anchor K1 into the protein. Conversely, cysteine C29 belongs to a short N-terminal stretch that caps the radical SAM partial β-barrel groove at its N-terminal tip. The E65 residue belongs to a long loop inserted into the radical SAM domain and specific to NifB that goes in the active site cavity (in red in Figure 16a). Unexpectedly, another conserved cysteine residue, C62, that belongs to the same loop binds the [Fe4S4]RS cluster where SAM should bind, hence impairing any binding.
Figure 16.

Crystal structures of the available NifB structures containing (a) the RS- and K1-clusters (PDB 6Y1X(29)) and (b) the RS- and proposed K-clusters (PDB 7BI7(30)). In (a) and (b), the overall protein is shown as peach ribbons, except for the plug loop (see main text), and N-terminal and C-terminal domains are colored in red, blue, and green, respectively. A zoom in of the K1- and K-clusters is shown with the corresponding residues that are ligands. The [FeS] clusters are shown as balls and sticks, and their ligands as sticks. Fe, N, C, O, and S are colored in dark red, blue, peach, red, and yellow, respectively.
Site-directed mutagenesis experiments showed that these two residues actually prevent futile SAM cleavage cycles.29 The corresponding loop was hence proposed to act as a plug to avoid unwanted SAM binding and cleavage before K1 and K2 were correctly loaded. We therefore proposed that this crystal structure corresponds to an early stage of the reaction awaiting K2-cluster loading to proceed further. Soon after, Kang, Hu, and co-workers reported two crystal structures of NifB from Methanobacterium thermoautotrophicum (MthNifB) either depleted (apo-) or presumably with the full complement of [FeS] clusters (holo-).89 In the apo-MthNifB crystal structure, all the regions expected to interact with [Fe4S4]RS, K1, or K2, including the CX3CX2C motif and the N- and C-terminal stretches, were disordered and therefore absent in the corresponding electron density. Such disorder may reflect the structural dynamics necessary to assist [Fe4S4]-cluster fusion and the associated ligand coordination exchanges upon NifB-co synthesis. Unfortunately, the original holo-MthNifB structure turned out to be poorly refined and exhibited highly improbable cluster geometry and coordination. After a thorough reinvestigation, our newly refined MthNifB crystal structure unveiled the presence of a unique [Fe8S8] cluster presumably corresponding to the true K-cluster (Figure 16b).30 Its geometry and coordination sphere are reminiscent to that of the P-cluster of the nitrogenase [MoFe]-protein in its reduced PN state.90 This K-cluster can be best described as the combination of two [Fe4S3] modules, connected via one μ2-sulfide, one μ6-sulfide, and one bridging cysteine residue. The latter corresponds to C18 (MthNifB numbering) from the flexible N-terminal stretch. In addition, each [Fe4S3] module is connected to two side chain residues: C115 and H31 (K1-cluster) or C260 and C263 (K2-cluster) (MthNifB notation). In this structure, the plug loop is pulled out, opening the cavity for SAM access (Figure 16b). The C-terminal stretch, which contains residues C260 and C263, is now visible and covers the K-cluster from the solvent. The μ2-sulfide ion is pointing toward the SAM binding site at a distance suitable for methyl transfer. Hence, the K-cluster structure agrees with the previous observation that NifB must be fully reduced beforehand to afford methyl transfer and places cluster fusion before methyl transfer.85,86 In addition, this new crystal structure provided key insights into the NifB mechanism, notably about the role of the protein matrix in the tight control of the reaction, leading to the following sequence of events (Figure 17). As mentioned above, the K1-cluster is bound to two residues (H31 and C115) that belong to β-strands of the radical SAM core domain, tightly anchoring K1 into the protein matrix and supporting it to be loaded first.29,30 Upon binding, both the N-terminal stretch that contains residue C18 and the plug loop would fold around K1, strengthening its interaction with the protein. This resting state would correspond to the MtNifB crystal structure. At this stage, SAM binding and cleavage would be put on hold due to the presence of the plug loop inside the active site cavity. The K2-cluster would be subsequently loaded with concomitant folding of the C-terminal stretch that contains residues C260 and C263, acting like a strap and closing the active site cavity. Upon reduction, cluster fusion would then occur, pushing away the plug loop. Such release would allow SAM binding and methyl transfer to the K-cluster μ2-sulfide ion. Ligand exchange at the [Fe4S4]RS cluster would lead to SAH release and binding of a second SAM molecule. Subsequent SAM cleavage would produce the 5′-deoxyadenosyl radical leading to abstraction of one methyl hydrogen atom, facilitating subsequent deprotonations. The carbide ions would subsequently exchange with the μ6-sulfide ion, that would become μ2-bound, leading to the previously characterized [Fe8S8C] L*-cluster. Upon such conversion, additional ligand exchanges are necessary. The K-cluster structure observed in MthNifB supports that, upon K- to L*-cluster conversion, residues H31 and C263 would be driven out by the new μ2-sulfide ion. In the L*-cluster, the third μ2-belt position would be filled with residue C18 from the N-terminal stretch, located at the tip of the active site groove. The apo-MthNifB structure demonstrated that this domain is indeed flexible and may therefore be displaced by a protein partner to afford ligand exchange and provide the missing ninth sulfide ion.89 Whereas sulfite has been proposed to provide such a sulfide in vitro,91 a more typical sulfur transferase protein would play this role in vivo. Indeed, in Azotobacter vinelandii, a rhodanese-like gene possibly encoding a sulfur transferase is found in the same transcriptional unit as nifB.92 Such an enzyme would therefore provide a sulfide ion as a persulfide in a mechanism reminiscent to that of the [Fe2S2]-cluster assembly in NifU or IscU and achieve NifB-co synthesis.93,94
Figure 17.

Proposed mechanism for the reaction catalyzed by NifB based on experimental data available to date (see main text). Note that this mechanism is not charge balanced, pending further spectroscopic evidence to identify the oxidation states of the clusters.
This whole mechanism remains highly speculative and needs further experimental data. Notably, a crystal structure of the K-cluster at a higher resolution combined with a spectroscopic characterization is urgently needed to confirm its unique structure and binding mode. Indeed, previous EPR analyses of dithionite-reduced NifB reported a three-[Fe4S4]+ state of the enzyme.84,85 The time-scale difference between these EPR and X-ray crystallography experiments (seconds versus days) might allow for complete reduction and cluster fusion to occur before crystallization. In light of this K-cluster structure, new advanced spectroscopic NifB characterizations are therefore required. However, the available structural data already provide key insights into the succession of events and better explain how the protein matrix would handle tremendous ligand exchanges triggered by this complex cluster fusion while minimizing conformational changes.
Perspective and Future Challenges
Despite being structurally and functionally different, [FeFe]-hydrogenase and nitrogenase share several features: (i) they both use sophisticated organometallic species as active sites (H-cluster and FeMo-co, respectively); (ii) these organometallic complexes are built stepwise by multiprotein machineries that act as highly elaborate assembly lines; (iii) the final product synthesized in a scaffold protein is subsequently transferred as a single piece to the apoprotein; and (iv) these assembly machineries require the specific action of dedicated radical SAM enzymes to catalyze essential steps in the process. In this Review, we presented the current knowledge about the role of these enzymes and highlighted the contribution of structural biology to this understanding. Yet, much remains to be done to fully address their mechanism.
For instance, regarding HydG, a crystal structure of a l-tyrosine-bound protein would confirm the postulated orientation of the substrate when inside the active site cavity. Likewise, crystal structures of the protein with the correct [Fe4S4] cluster, including either the [κ3-l-cysteine-FeII(His)(OHx)2] species or complex-A, are important milestones in our quest to decipher its mechanism. However, 2-iminoacetate transfer and fragmentation, in addition to carbon monoxide synthesis, remain the darkest points and are likely the most difficult steps to address experimentally. Indeed, they correspond neither to initial nor to final steps in the time course of the reaction. Furthermore, they involve fleeting intermediates and most probably require dynamics of the protein matrix to lead the reaction to completion. Therefore, we envision that addressing such mechanistic questions would require a combination of advanced spectroscopic techniques, advances in crystallographic approaches, and thorough theoretical calculations. Indeed, considering the recent development of time-resolved serial crystallography, implementing such a technique to anaerobic systems will certainly allow us to go one step further in the structural investigation of radical SAM enzymes. For instance, in HydE, one key question that remains to be understood is how the C5′–S bond is broken to release the “SFe(CO)2CN” piece that would constitute the [2Fe]H subcluster. Initial in-crystal kinetic experiments using syn-B highlighted that the reaction is faster than that in solution and the intermediate quickly decays. Therefore, serial crystallography may be more suitable to follow what happens in the active site cavity. Another important question regarding HydE would be to establish the exact nature of its final product, corresponding to either a monoiron or a di-iron species.
Regarding NifB, in addition to a high-resolution crystal structure of the K-cluster bound protein, a [K1 + K2]-bound structure would also provide important insights into the cluster fusion reaction. It also seems important to determine a SAM-bound structure to address methyl transfer and hydrogen-atom abstraction reactions. L*-Cluster and NifB-co bound NifB structures are also important milestones. Again, serial crystallography would significantly contribute to understanding this fascinating reaction, notably regarding the complex carbide ion insertion. Yet, because significant structural changes are envisioned, several steps of the reaction may not be directly experimentally accessible. Therefore, quantum mechanical (QM)95 and hybrid quantum mechanical/molecular mechanical method (QM/MM) approaches96,97 may help in filling the gap between the different structures. Yet, considering NifB, but this is also true for HydE and HydG, using computational methods is challenging because the substrate, intermediates, and product involve modeling complex transition metal-containing systems and radical chemistry.98 In addition, modeling the dynamics of the protein matrix in the process, including larger motions such as intermediate transfer between the different cavities observed in both HydE and HydG, remains an important challenge in the field.99 Including such dynamics can be, in part, addressed by experimental approaches like time-resolved serial crystallography. Likewise, substrate/product transfers between partners, for instance, between HydG and HydE, HydE and HydF, NifU and NifB, or NifB and NifEN, are also an important challenge where structural biology can significantly contribute, using both standard X-ray crystallography and cryo-electron microscopy, provided suitable setups are available to prepare samples under anaerobic conditions.
We therefore consider that, as structural biology has significantly contributed to our current knowledge of radical SAM enzyme function, it will continue to provide key information to decipher their mechanisms. The rise of advanced techniques such as cryo-electron microscopy or serial crystallography and their implementation to proteins sensitive to oxygen will certainly lead to new breakthroughs serving to elucidate the mechanism of these fascinating enzymes and push the boundaries of our knowledge about how radical SAM enzymes tightly control the radical-based reaction. Ongoing development of theoretical tools100−102 including machine learning103,104 will also significantly contribute to the field. Yet, these approaches will always need top-quality biochemistry to prepare pure and homogeneous protein complexes and expert chemists to synthesize organometallic synthons. The latter have proven to be essential in unscrambling the role of each protein, notably in tracking specific atoms that constitute the [FeFe]-hydrogenase [2Fe]H subcluster.
Acknowledgments
The work from our group, described here, was supported by the Radis-Bio contract from the CEA/DRF-Impulsion program, the ADOMET, SELAROM and CARBONARA contracts from the French National Research Agency (ANR-08-BLAN-0224-01, ANR-12-BSV8-0013, ANR-16-CE29-0019, respectively). Our recent studies on HydE and NifB were sustained by the French National Research Agency in the framework of the Investissement d’Avenir program (ANR-15-IDEX-02), through the funding of the “Origin of Life” project of the Univ. Grenoble-Alpes. P.A. appreciates the help from the staff of the computing facility provided by the Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA/DRF/GIPSI), Saclay and CCRT, Bruyères-le-Châtel.
The authors declare no competing financial interest.
References
- Buren S.; Jimenez-Vicente E.; Echavarri-Erasun C.; Rubio L. M. Biosynthesis of Nitrogenase Cofactors. Chem. Rev. 2020, 120, 4921–4968. 10.1021/acs.chemrev.9b00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt R. D.; Rao G.; Tao L. Biosynthesis of the Catalytic H-Cluster of [FeFe] Hydrogenase: The Roles of the Fe-S Maturase Proteins HydE, HydF, and HydG. Chem. Sci. 2020, 11 (38), 10313–10323. 10.1039/D0SC04216A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinert H.; Holm R. H.; Münck E. Iron-Sulfur Clusters: Nature’s Modular, Multipurpose Structures. Science 1997, 277 (5326), 653–659. 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- Johnson D. C.; Dean D. R.; Smith A. D.; Johnson M. K. Structure, Function, and Formation of Biological Iron-Sulfur Clusters. Annu. Rev. Biochem. 2005, 74, 247–281. 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- Castro L.; Tórtora V.; Mansilla S.; Radi R. Aconitases: Non-Redox Iron–Sulfur Proteins Sensitive to Reactive Species. Acc. Chem. Res. 2019, 52 (9), 2609–2619. 10.1021/acs.accounts.9b00150. [DOI] [PubMed] [Google Scholar]
- Crack J. C.; Gray E.; Le Brun N. E. Sensing Mechanisms of Iron–Sulfur Cluster Regulatory Proteins Elucidated Using Native Mass Spectrometry. Dalton Trans. 2021, 50 (23), 7887–7897. 10.1039/D1DT00993A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfano M.; Cavazza C. Structure, Function, and Biosynthesis of Nickel-Dependent Enzymes. Protein Sci. 2020, 29 (5), 1071–1089. 10.1002/pro.3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontecilla-Camps J. C.; Volbeda A.; Cavazza C.; Nicolet Y. Structure/Function Relationships of [NiFe]- and [FeFe]-Hydrogenases. Chem. Rev. 2007, 107 (10), 4273–4303. 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- Wachtershauser G. Before Enzymes and Templates: Theory of Surface Metabolism. Microbiol. Rev. 1988, 52 (4), 452–484. 10.1128/mr.52.4.452-484.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R.; Charon M.-H.; Hudry-Clergeon G.; Pétillot Y.; Norager S.; Medina M.; Frey M. Refined X-Ray Structures of the Oxidized, at 1.3 Å, and Reduced, at 1.17 Å, [2Fe-2S] Ferredoxin from the Cyanobacterium Anabaena PCC7119 Show Redox-Linked Conformational Changes. Biochemistry 1999, 38 (48), 15764–15773. 10.1021/bi991578s. [DOI] [PubMed] [Google Scholar]
- Nicolet Y.; Piras C.; Legrand P.; Hatchikian C. E.; Fontecilla-Camps J. C. Desulfovibrio desulfuricans Iron Hydrogenase: The Structure Shows Unusual Coordination to an Active Site Fe Binuclear Center. Structure 1999, 7 (1), 13–23. 10.1016/S0969-2126(99)80005-7. [DOI] [PubMed] [Google Scholar]
- Darnault C.; Volbeda A.; Kim E. J.; Legrand P.; Vernède X.; Lindahl P. A.; Fontecilla-Camps J. C. Ni-Zn-[Fe4-S4] and Ni-Ni-[Fe4-S4] Clusters in Closed and Open α Subunits of Acetyl-CoA Synthase/Carbon Monoxide Dehydrogenase. Nat. Struct. Mol. Biol. 2003, 10 (4), 271–279. 10.1038/nsb912. [DOI] [PubMed] [Google Scholar]
- Artz J. H.; Zadvornyy O. A.; Mulder D. W.; Keable S. M.; Cohen A. E.; Ratzloff M. W.; Williams S. G.; Ginovska B.; Kumar N.; Song J.; McPhillips S. E.; Davidson C. M.; Lyubimov A. Y.; Pence N.; Schut G. J.; Jones A. K.; Soltis S. M.; Adams M. W. W.; Raugei S.; King P. W.; Peters J. W. Tuning Catalytic Bias of Hydrogen Gas Producing Hydrogenases. J. Am. Chem. Soc. 2020, 142 (3), 1227–1235. 10.1021/jacs.9b08756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einsle O.; Tezcan F. A.; Andrade S. L. A.; Schmid B.; Yoshida M.; Howard J. B.; Rees D. C. Nitrogenase MoFe-Protein at 1.16 A Resolution: A Central Ligand in the FeMo-Cofactor. Science 2002, 297 (5587), 1696–1700. 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- Zheng L.; White R. H.; Cash V. L.; Jack R. F.; Dean D. R. Cysteine Desulfurase Activity Indicates a Role for NIFS in Metallocluster Biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 1993, 90 (7), 2754–2758. 10.1073/pnas.90.7.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc B.; Gerez C.; Ollagnier de Choudens S. Assembly of Fe/S Proteins in Bacterial Systems: Biochemistry of the Bacterial ISC System. Biochim. Biophys. Acta, Mol. Cell Res. 2015, 1853 (6), 1436–1447. 10.1016/j.bbamcr.2014.12.009. [DOI] [PubMed] [Google Scholar]
- Sofia H. J.; Chen G.; Hetzler B. G.; Reyes-Spindola J. F.; Miller N. E. Radical SAM, a Novel Protein Superfamily Linking Unresolved Steps in Familiar Biosynthetic Pathways with Radical Mechanisms: Functional Characterization Using New Analysis and Information Visualization Methods. Nucleic Acids Res. 2001, 29 (5), 1097–1106. 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick J. B.; Duffus B. R.; Duschene K. S.; Shepard E. M. Radical S-Adenosylmethionine Enzymes. Chem. Rev. 2014, 114 (8), 4229–4317. 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolet Y. Structure-Function Relationships of Radical SAM Enzymes. Nat. Catal. 2020, 3 (4), 337–350. 10.1038/s41929-020-0448-7. [DOI] [Google Scholar]
- Layer G.; Moser J.; Heinz D. W.; Jahn D.; Schubert W.-D. Crystal Structure of Coproporphyrinogen III Oxidase Reveals Cofactor Geometry of Radical SAM Enzymes. EMBO J. 2003, 22 (23), 6214–6224. 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovitch F.; Nicolet Y.; Wan J. T.; Jarrett J. T.; Drennan C. L. Crystal Structure of Biotin Synthase, an S-Adenosylmethionine-Dependent Radical Enzyme. Science 2004, 303 (5654), 76–79. 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolet Y.; Drennan C. L. AdoMet Radical Proteins--from Structure to Evolution--Alignment of Divergent Protein Sequences Reveals Strong Secondary Structure Element Conservation. Nucleic Acids Res. 2004, 32 (13), 4015–4025. 10.1093/nar/gkh728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posewitz M. C.; King P. W.; Smolinski S. L.; Zhang L.; Seibert M.; Ghirardi M. L. Discovery of Two Novel Radical S-Adenosylmethionine Proteins Required for the Assembly of an Active [Fe] Hydrogenase. J. Biol. Chem. 2004, 279 (24), 25711–25720. 10.1074/jbc.M403206200. [DOI] [PubMed] [Google Scholar]
- Curatti L.; Ludden P. W.; Rubio L. M. NifB-Dependent in Vitro Synthesis of the Iron-Molybdenum Cofactor of Nitrogenase. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (14), 5297–5301. 10.1073/pnas.0601115103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curatti L.; Hernandez J. A.; Igarashi R. Y.; Soboh B.; Zhao D.; Rubio L. M. In Vitro Synthesis of the Iron-Molybdenum Cofactor of Nitrogenase from Iron, Sulfur, Molybdenum, and Homocitrate Using Purified Proteins. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (45), 17626–17631. 10.1073/pnas.0703050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolet Y.; Pagnier A.; Zeppieri L.; Martin L.; Amara P.; Fontecilla-Camps J. C. Crystal Structure of HydG from Carboxydothermus hydrogenoformans: A Trifunctional [FeFe]-Hydrogenase Maturase. ChemBioChem 2015, 16 (3), 397–402. 10.1002/cbic.201402661. [DOI] [PubMed] [Google Scholar]
- Dinis P.; Suess D. L. M.; Fox S. J.; Harmer J. E.; Driesener R. C.; De La Paz L.; Swartz J. R.; Essex J. W.; Britt R. D.; Roach P. L. X-Ray Crystallographic and EPR Spectroscopic Analysis of HydG, a Maturase in [FeFe]-Hydrogenase H-Cluster Assembly. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (5), 1362–1367. 10.1073/pnas.1417252112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolet Y.; Rubach J. K.; Posewitz M. C.; Amara P.; Mathevon C.; Atta M.; Fontecave M.; Fontecilla-Camps J. C. X-Ray Structure of the [FeFe]-Hydrogenase Maturase HydE from Thermotoga Maritima. J. Biol. Chem. 2008, 283 (27), 18861–18872. 10.1074/jbc.M801161200. [DOI] [PubMed] [Google Scholar]
- Fajardo A. S.; Legrand P.; Payá-Tormo L. A.; Martin L.; Pellicer Martínez M. T.; Echavarri-Erasun C.; Vernède X.; Rubio L. M.; Nicolet Y. Structural Insights into the Mechanism of the Radical SAM Carbide Synthase NifB, a Key Nitrogenase Cofactor Maturating Enzyme. J. Am. Chem. Soc. 2020, 142 (25), 11006–11012. 10.1021/jacs.0c02243. [DOI] [PubMed] [Google Scholar]
- Jenner L. P.; Cherrier M. V.; Amara P.; Rubio L. M.; Nicolet Y. An Unexpected P-Cluster like Intermediate En route to the Nitrogenase FeMo-Co. Chem. Sci. 2021, 12 (14), 5269–5274. 10.1039/D1SC00289A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J. W.; Lanzilotta W. N.; Lemon B. J.; Seefeldt L. C. X-Ray Crystal Structure of the Fe-Only Hydrogenase (CpI) from Clostridium pasteurianum to 1.8 Angstrom Resolution. Science 1998, 282 (5395), 1853–1858. 10.1126/science.282.5395.1853. [DOI] [PubMed] [Google Scholar]
- Nicolet Y.; de Lacey A. L.; Vernède X.; Fernandez V. M.; Hatchikian E. C.; Fontecilla-Camps J. C. Crystallographic and FTIR Spectroscopic Evidence of Changes in Fe Coordination Upon Reduction of the Active Site of the Fe-Only Hydrogenase from Desulfovibrio desulfuricans. J. Am. Chem. Soc. 2001, 123 (8), 1596–1601. 10.1021/ja0020963. [DOI] [PubMed] [Google Scholar]
- Voordouw G.; Hagen W. R.; Krüse-Wolters K. M.; van Berkel-Arts A.; Veeger C. Purification and Characterization of Desulfovibrio vulgaris (Hildenborough) Hydrogenase Expressed in Escherichia Coli. Eur. J. Biochem. 1987, 162 (1), 31–36. 10.1111/j.1432-1033.1987.tb10537.x. [DOI] [PubMed] [Google Scholar]
- Mulder D. W.; Ortillo D. O.; Gardenghi D. J.; Naumov A. V.; Ruebush S. S.; Szilagyi R. K.; Huynh B.; Broderick J. B.; Peters J. W. Activation of HydA(DeltaEFG) Requires a Preformed [4Fe-4S] Cluster. Biochemistry 2009, 48 (26), 6240–6248. 10.1021/bi9000563. [DOI] [PubMed] [Google Scholar]
- Mulder D. W.; Boyd E. S.; Sarma R.; Lange R. K.; Endrizzi J. A.; Broderick J. B.; Peters J. W. Stepwise [FeFe]-Hydrogenase H-Cluster Assembly Revealed in the Structure of HydA(DeltaEFG). Nature 2010, 465 (7295), 248–251. 10.1038/nature08993. [DOI] [PubMed] [Google Scholar]
- Brazzolotto X.; Rubach J. K.; Gaillard J.; Gambarelli S.; Atta M.; Fontecave M. The [Fe-Fe]-Hydrogenase Maturation Protein HydF from Thermotoga maritima Is a GTPase with an Iron-Sulfur Cluster. J. Biol. Chem. 2006, 281 (2), 769–774. 10.1074/jbc.M510310200. [DOI] [PubMed] [Google Scholar]
- Berggren G.; Adamska A.; Lambertz C.; Simmons T. R.; Esselborn J.; Atta M.; Gambarelli S.; Mouesca J. M.; Reijerse E.; Lubitz W.; Happe T.; Artero V.; Fontecave M. Biomimetic Assembly and Activation of [FeFe]-Hydrogenases. Nature 2013, 499 (7456), 66–69. 10.1038/nature12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech I.; Silakov A.; Lubitz W.; Happe T. The [FeFe]-Hydrogenase Maturase HydF from Clostridium acetobutylicum Contains a CO and CN- Ligated Iron Cofactor. FEBS Lett. 2010, 584 (3), 638–642. 10.1016/j.febslet.2009.12.016. [DOI] [PubMed] [Google Scholar]
- Czech I.; Stripp S.; Sanganas O.; Leidel N.; Happe T.; Haumann M. The [FeFe]-Hydrogenase Maturation Protein HydF Contains a H-Cluster like [4Fe4S]-2Fe Site. FEBS Lett. 2011, 585 (1), 225–230. 10.1016/j.febslet.2010.11.052. [DOI] [PubMed] [Google Scholar]
- Cendron L.; Berto P.; D’Adamo S.; Vallese F.; Govoni C.; Posewitz M. C.; Giacometti G. M.; Costantini P.; Zanotti G. Crystal Structure of HydF Scaffold Protein Provides Insights into [FeFe]-Hydrogenase Maturation. J. Biol. Chem. 2011, 286 (51), 43944–43950. 10.1074/jbc.M111.281956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caserta G.; Pecqueur L.; Adamska-Venkatesh A.; Papini C.; Roy S.; Artero V.; Atta M.; Reijerse E.; Lubitz W.; Fontecave M. Structural and Functional Characterization of the Hydrogenase-Maturation HydF Protein. Nat. Chem. Biol. 2017, 13 (7), 779–784. 10.1038/nchembio.2385. [DOI] [PubMed] [Google Scholar]
- Rubach J. K.; Brazzolotto X.; Gaillard J.; Fontecave M. Biochemical Characterization of the HydE and HydG Iron-Only Hydrogenase Maturation Enzymes from Thermotoga Maritima. FEBS Lett. 2005, 579 (22), 5055–5060. 10.1016/j.febslet.2005.07.092. [DOI] [PubMed] [Google Scholar]
- Pilet E.; Nicolet Y.; Mathevon C.; Douki T.; Fontecilla-Camps J. C.; Fontecave M. The Role of the Maturase HydG in [FeFe]-Hydrogenase Active Site Synthesis and Assembly. FEBS Lett. 2009, 583 (3), 506–511. 10.1016/j.febslet.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Kriek M.; Martins F.; Challand M. R.; Croft A.; Roach P. L. Thiamine Biosynthesis in Escherichia coli: Identification of the Intermediate and by-Product Derived from Tyrosine. Angew. Chem., Int. Ed. 2007, 46 (48), 9223–9226. 10.1002/anie.200702554. [DOI] [PubMed] [Google Scholar]
- Fugate C. J.; Stich T. A.; Kim E. G.; Myers W. K.; Britt R. D.; Jarrett J. T. 9-Mercaptodethiobiotin Is Generated as a Ligand to the [2Fe-2S]+ Cluster during the Reaction Catalyzed by Biotin Synthase from Escherichia coli. J. Am. Chem. Soc. 2012, 134 (22), 9042–9045. 10.1021/ja3012963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao G.; Tao L.; Britt R. D. Serine Is the Molecular Source of the NH(CH2)2 Bridgehead Moiety of the in Vitro Assembled [FeFe] Hydrogenase H-Cluster. Chem. Sci. 2020, 11 (5), 1241–1247. 10.1039/C9SC05900H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchenreuther J. M.; Stapleton J. A.; Swartz J. R. Tyrosine, Cysteine, and S-Adenosyl Methionine Stimulate in Vitro [FeFe] Hydrogenase Activation. PLoS One 2009, 4 (10), e7565 10.1371/journal.pone.0007565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driesener R. C.; Challand M. R.; McGlynn S. E.; Shepard E. M.; Boyd E. S.; Broderick J. B.; Peters J. W.; Roach P. L. [FeFe]-Hydrogenase Cyanide Ligands Derived from S-Adenosylmethionine-Dependent Cleavage of Tyrosine. Angew. Chem., Int. Ed. 2010, 49 (9), 1687–1690. 10.1002/anie.200907047. [DOI] [PubMed] [Google Scholar]
- Shepard E. M.; Duffus B. R.; George S. J.; McGlynn S. E.; Challand M. R.; Swanson K. D.; Roach P. L.; Cramer S. P.; Peters J. W.; Broderick J. B. [FeFe]-Hydrogenase Maturation: HydG-Catalyzed Synthesis of Carbon Monoxide. J. Am. Chem. Soc. 2010, 132 (27), 9247–9249. 10.1021/ja1012273. [DOI] [PubMed] [Google Scholar]
- Kuchenreuther J. M.; George S. J.; Grady-Smith C. S.; Cramer S. P.; Swartz J. R. Cell-Free H-Cluster Synthesis and [FeFe] Hydrogenase Activation: All Five CO and CN– Ligands Derive from Tyrosine. PLoS One 2011, 6 (5), e20346 10.1371/journal.pone.0020346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tron C.; Cherrier M. V.; Amara P.; Martin L.; Fauth F.; Fraga E.; Correard M.; Fontecave M.; Nicolet Y.; Fontecilla-Camps J. C. Further Characterization of the [FeFe]-Hydrogenase Maturase HydG. Eur. J. Inorg. Chem. 2011, 2011 (7), 1121–1127. 10.1002/ejic.201001101. [DOI] [Google Scholar]
- Driesener R. C.; Duffus B. R.; Shepard E. M.; Bruzas I. R.; Duschene K. S.; Coleman N. J.-R.; Marrison A. P. G.; Salvadori E.; Kay C. W. M.; Peters J. W.; Broderick J. B.; Roach P. L. Biochemical and Kinetic Characterization of Radical S-Adenosyl-L-Methionine Enzyme HydG. Biochemistry 2013, 52 (48), 8696–8707. 10.1021/bi401143s. [DOI] [PubMed] [Google Scholar]
- Kuchenreuther J. M.; Myers W. K.; Stich T. A.; George S. J.; Nejatyjahromy Y.; Swartz J. R.; Britt R. D. A Radical Intermediate in Tyrosine Scission to the CO and CN- Ligands of FeFe Hydrogenase. Science 2013, 342 (6157), 472–475. 10.1126/science.1241859. [DOI] [PubMed] [Google Scholar]
- Nicolet Y.; Zeppieri L.; Amara P.; Fontecilla-Camps J. C. Crystal Structure of Tryptophan Lyase (NosL): Evidence for Radical Formation at the Amino Group of Tryptophan. Angew. Chem., Int. Ed. 2014, 53 (44), 11840–11844. 10.1002/anie.201407320. [DOI] [PubMed] [Google Scholar]
- Sayler R. I.; Stich T. A.; Joshi S.; Cooper N.; Shaw J. T.; Begley T. P.; Tantillo D. J.; Britt R. D. Trapping and Electron Paramagnetic Resonance Characterization of the 5′dAdo• Radical in a Radical S-Adenosyl Methionine Enzyme Reaction with a Non-Native Substrate. ACS Cent. Sci. 2019, 5 (11), 1777–1785. 10.1021/acscentsci.9b00706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnier A.; Martin L.; Zeppieri L.; Nicolet Y.; Fontecilla-Camps J. C. CO and CN- Syntheses by [FeFe]-Hydrogenase Maturase HydG Are Catalytically Differentiated Events. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (1), 104–109. 10.1073/pnas.1515842113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suess D. L. M.; Bürstel I.; De La Paz L.; Kuchenreuther J. M.; Pham C. C.; Cramer S. P.; Swartz J. R.; Britt R. D. Cysteine as a Ligand Platform in the Biosynthesis of the FeFe Hydrogenase H Cluster. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (37), 11455–11460. 10.1073/pnas.1508440112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suess D. L. M.; Pham C. C.; Bürstel I.; Swartz J. R.; Cramer S. P.; Britt R. D. The Radical SAM Enzyme HydG Requires Cysteine and a Dangler Iron for Generating an Organometallic Precursor to the [FeFe]-Hydrogenase H-Cluster. J. Am. Chem. Soc. 2016, 138 (4), 1146–1149. 10.1021/jacs.5b12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao G.; Tao L.; Suess D. L. M.; Britt R. D. A [4Fe-4S]-Fe(CO)(CN)-L-Cysteine Intermediate Is the First Organometallic Precursor in [FeFe] Hydrogenase H-Cluster Bioassembly. Nat. Chem. 2018, 10 (5), 555–560. 10.1038/s41557-018-0026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao G.; Pattenaude S. A.; Alwan K.; Blackburn N. J.; Britt R. D.; Rauchfuss T. B. The Binuclear Cluster of [FeFe] Hydrogenase Is Formed with Sulfur Donated by Cysteine of an [Fe(Cys)(CO)(2)(CN)] Organometallic Precursor. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (42), 20850–20855. 10.1073/pnas.1913324116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard E. M.; Impano S.; Duffus B. R.; Pagnier A.; Duschene K. S.; Betz J. N.; Byer A. S.; Galambas A.; McDaniel E. C.; Watts H.; McGlynn S. E.; Peters J. W.; Broderick W. E.; Broderick J. B. HydG, the “Dangler” Iron, and Catalytic Production of Free CO and CN(−): Implications for [FeFe]-Hydrogenase Maturation. Dalton Trans. Camb. Engl. 2003 2021, 50 (30), 10405–10422. 10.1039/D1DT01359A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N.; Rao G.; Britt R. D.; Wang L.-P. Quantum Chemical Study of a Radical Relay Mechanism for the HydG-Catalyzed Synthesis of a Fe(II)(CO)(2)(CN)Cysteine Precursor to the H-Cluster of [FeFe] Hydrogenase. Biochemistry 2021, 60 (40), 3016–3026. 10.1021/acs.biochem.1c00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchenreuther J. M.; Britt R. D.; Swartz J. R. New Insights into [FeFe] Hydrogenase Activation and Maturase Function. PLoS One 2012, 7 (9), e45850 10.1371/journal.pone.0045850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolet Y.; Rohac R.; Martin L.; Fontecilla-Camps J. C. X-Ray Snapshots of Possible Intermediates in the Time Course of Synthesis and Degradation of Protein-Bound Fe4S4 Clusters. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (18), 7188–7192. 10.1073/pnas.1302388110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz J. N.; Boswell N. W.; Fugate C. J.; Holliday G. L.; Akiva E.; Scott A. G.; Babbitt P. C.; Peters J. W.; Shepard E. M.; Broderick J. B. [FeFe]-Hydrogenase Maturation: Insights into the Role HydE Plays in Dithiomethylamine Biosynthesis. Biochemistry 2015, 54 (9), 1807–1818. 10.1021/bi501205e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohac R.; Amara P.; Benjdia A.; Martin L.; Ruffie P.; Favier A.; Berteau O.; Mouesca J.-M.; Fontecilla-Camps J. C.; Nicolet Y. Carbon-Sulfur Bond-Forming Reaction Catalysed by the Radical SAM Enzyme HydE. Nat. Chem. 2016, 8 (5), 491–500. 10.1038/nchem.2490. [DOI] [PubMed] [Google Scholar]
- Tao L.; Pattenaude S. A.; Joshi S.; Begley T. P.; Rauchfuss T. B.; Britt R. D. Radical SAM Enzyme HydE Generates Adenosylated Fe(I) Intermediates En route to the [FeFe]-Hydrogenase Catalytic H-Cluster. J. Am. Chem. Soc. 2020, 142 (24), 10841–10848. 10.1021/jacs.0c03802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolet Y.; Amara P.; Mouesca J.-M.; Fontecilla-Camps J. C. Unexpected Electron Transfer Mechanism upon AdoMet Cleavage in Radical SAM Proteins. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (35), 14867–14871. 10.1073/pnas.0904385106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohac R.; Martin L.; Liu L.; Basu D.; Tao L.; Britt R. D.; Rauchfuss T. B.; Nicolet Y. Crystal Structure of the [FeFe]-Hydrogenase Maturase HydE Bound to Complex-B. J. Am. Chem. Soc. 2021, 143 (22), 8499–8508. 10.1021/jacs.1c03367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B. M.; Lukoyanov D.; Yang Z.-Y.; Dean D. R.; Seefeldt L. C. Mechanism of Nitrogen Fixation by Nitrogenase: The next Stage. Chem. Rev. 2014, 114 (8), 4041–4062. 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einsle O.; Rees D. C. Structural Enzymology of Nitrogenase Enzymes. Chem. Rev. 2020, 120 (12), 4969–5004. 10.1021/acs.chemrev.0c00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatzal T.; Aksoyoglu M.; Zhang L.; Andrade S. L. A.; Schleicher E.; Weber S.; Rees D. C.; Einsle O. Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334 (6058), 940. 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J. W.; Stowell M. H.; Soltis S. M.; Finnegan M. G.; Johnson M. K.; Rees D. C. Redox-Dependent Structural Changes in the Nitrogenase P-Cluster. Biochemistry 1997, 36 (6), 1181–1187. 10.1021/bi9626665. [DOI] [PubMed] [Google Scholar]
- Xie H.; Wu R.; Zhou Z.; Cao Z. Exploring the Interstitial Atom in the FeMo Cofactor of Nitrogenase: Insights from QM and QM/MM Calculations. J. Phys. Chem. B 2008, 112 (36), 11435–11439. 10.1021/jp803616z. [DOI] [PubMed] [Google Scholar]
- George S. J.; Igarashi R. Y.; Xiao Y.; Hernandez J. A.; Demuez M.; Zhao D.; Yoda Y.; Ludden P. W.; Rubio L. M.; Cramer S. P. Extended X-Ray Absorption Fine Structure and Nuclear Resonance Vibrational Spectroscopy Reveal That NifB-Co, a FeMo-Co Precursor, Comprises a 6Fe Core with an Interstitial Light Atom. J. Am. Chem. Soc. 2008, 130 (17), 5673–5680. 10.1021/ja0755358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelmenschikov V.; Case D. A.; Noodleman L. Ligand-Bound S = 1/2 FeMo-Cofactor of Nitrogenase: Hyperfine Interaction Analysis and Implication for the Central Ligand X Identity. Inorg. Chem. 2008, 47 (14), 6162–6172. 10.1021/ic7022743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukoyanov D.; Pelmenschikov V.; Maeser N.; Laryukhin M.; Yang T. C.; Noodleman L.; Dean D. R.; Case D. A.; Seefeldt L. C.; Hoffman B. M. Testing If the Interstitial Atom, X, of the Nitrogenase Molybdenum-Iron Cofactor Is N or C: ENDOR, ESEEM, and DFT Studies of the S = 3/2 Resting State in Multiple Environments. Inorg. Chem. 2007, 46 (26), 11437–11449. 10.1021/ic7018814. [DOI] [PubMed] [Google Scholar]
- Yang T.-C.; Maeser N. K.; Laryukhin M.; Lee H.-I.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. The Interstitial Atom of the Nitrogenase FeMo-Cofactor: ENDOR and ESEEM Evidence That It Is Not a Nitrogen. J. Am. Chem. Soc. 2005, 127 (37), 12804–12805. 10.1021/ja0552489. [DOI] [PubMed] [Google Scholar]
- Lee H.-I.; Benton P. M. C.; Laryukhin M.; Igarashi R. Y.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. The Interstitial Atom of the Nitrogenase FeMo-Cofactor: ENDOR and ESEEM Show It Is Not an Exchangeable Nitrogen. J. Am. Chem. Soc. 2003, 125 (19), 5604–5605. 10.1021/ja034383n. [DOI] [PubMed] [Google Scholar]
- Dance I. The Consequences of an Interstitial N Atom in the FeMo Cofactor of Nitrogenase. Chem. Commun. 2003, 3, 324–325. 10.1039/b211036a. [DOI] [PubMed] [Google Scholar]
- Wiig J. A.; Hu Y.; Chung Lee C.; Ribbe M. W. Radical SAM-Dependent Carbon Insertion into the Nitrogenase M-Cluster. Science 2012, 337 (6102), 1672–1675. 10.1126/science.1224603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura R.; Hikita M.; Ogawa S.; Takahashi Y.; Fujishiro T. Snapshots of PLP-Substrate and PLP-Product External Aldimines as Intermediates in Two Types of Cysteine Desulfurase Enzymes. FEBS J. 2020, 287 (6), 1138–1154. 10.1111/febs.15081. [DOI] [PubMed] [Google Scholar]
- Kaiser J. T.; Hu Y.; Wiig J. A.; Rees D. C.; Ribbe M. W. Structure of Precursor-Bound NifEN: A Nitrogenase FeMo Cofactor Maturase/Insertase. Science 2011, 331 (6013), 91–94. 10.1126/science.1196954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcoxen J.; Arragain S.; Scandurra A. A.; Jimenez-Vicente E.; Echavarri-Erasun C.; Pollmann S.; Britt R. D.; Rubio L. M. Electron Paramagnetic Resonance Characterization of Three Iron-Sulfur Clusters Present in the Nitrogenase Cofactor Maturase NifB from Methanocaldococcus infernus. J. Am. Chem. Soc. 2016, 138 (24), 7468–7471. 10.1021/jacs.6b03329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettberg L. A.; Wilcoxen J.; Lee C. C.; Stiebritz M. T.; Tanifuji K.; Britt R. D.; Hu Y. Probing the Coordination and Function of Fe4S4Modules in Nitrogenase Assembly Protein NifB. Nat. Commun. 2018, 9 (1), 2824. 10.1038/s41467-018-05272-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiig J. A.; Hu Y.; Ribbe M. W. Refining the Pathway of Carbide Insertion into the Nitrogenase M-Cluster. Nat. Commun. 2015, 6, 8034. 10.1038/ncomms9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettberg L. A.; Wilcoxen J.; Jasniewski A. J.; Lee C. C.; Tanifuji K.; Hu Y.; Britt R. D.; Ribbe M. W. Identity and Function of an Essential Nitrogen Ligand of the Nitrogenase Cofactor Biosynthesis Protein NifB. Nat. Commun. 2020, 11 (1), 1757. 10.1038/s41467-020-15627-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay A. W.; Wiig J. A.; Lee C. C.; Hu Y. Identification and Characterization of Functional Homologs of Nitrogenase Cofactor Biosynthesis Protein NifB from Methanogens. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (48), 14829–14833. 10.1073/pnas.1510409112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W.; Rettberg L.; Stiebritz M.; Jasniewski A.; Tanifuji K.; Lee C. C.; Ribbe M. W.; Hu Y. Crystallographic Analysis of NifB with a Full Complement of Clusters: Structural Insights into the Radical SAM-Dependent Carbide Insertion during Nitrogenase Cofactor Assembly. Angew. Chem., Int. Ed. 2021, 60, 2364. 10.1002/anie.202011367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keable S. M.; Zadvornyy O. A.; Johnson L. E.; Ginovska B.; Rasmussen A. J.; Danyal K.; Eilers B. J.; Prussia G. A.; LeVan A. X.; Raugei S.; Seefeldt L. C.; Peters J. W. Structural Characterization of the P1+ Intermediate State of the P-Cluster of Nitrogenase. J. Biol. Chem. 2018, 293 (25), 9629–9635. 10.1074/jbc.RA118.002435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanifuji K.; Lee C. C.; Sickerman N. S.; Tatsumi K.; Ohki Y.; Hu Y.; Ribbe M. W. Tracing the “ninth Sulfur” of the Nitrogenase Cofactor via a Semi-Synthetic Approach. Nat. Chem. 2018, 10 (5), 568–572. 10.1038/s41557-018-0029-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez J. A.; George S. J.; Rubio L. M. Molybdenum Trafficking for Nitrogen Fixation. Biochemistry 2009, 48 (41), 9711–9721. 10.1021/bi901217p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuvaniyama P.; Agar J. N.; Cash V. L.; Johnson M. K.; Dean D. R. NifS-Directed Assembly of a Transient [2Fe-2S] Cluster within the NifU Protein. Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (2), 599. 10.1073/pnas.97.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinoni E. N.; de Oliveira J. S.; Nicolet Y.; Raulfs E. C.; Amara P.; Dean D. R.; Fontecilla-Camps J. C. (IscS-IscU)2 Complex Structures Provide Insights into Fe2S2 Biogenesis and Transfer. Angew. Chem., Int. Ed. 2012, 51 (22), 5439–5442. 10.1002/anie.201201708. [DOI] [PubMed] [Google Scholar]
- Himo F. Recent Trends in Quantum Chemical Modeling of Enzymatic Reactions. J. Am. Chem. Soc. 2017, 139 (20), 6780–6786. 10.1021/jacs.7b02671. [DOI] [PubMed] [Google Scholar]
- Senn H. M.; Thiel W. QM/MM Methods for Biomolecular Systems. Angew. Chem., Int. Ed. 2009, 48 (7), 1198–1229. 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- Liu M.; Wang Y.; Chen Y.; Field M. J.; Gao J. QM/MM through the 1990s: The First Twenty Years of Method Development and Applications. Isr. J. Chem. 2014, 54 (8–9), 1250–1263. 10.1002/ijch.201400036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himo F.; Siegbahn P. E. M. Quantum Chemical Studies of Radical-Containing Enzymes. Chem. Rev. 2003, 103 (6), 2421–2456. 10.1021/cr020436s. [DOI] [PubMed] [Google Scholar]
- Blue T. C.; Davis K. M. Computational Approaches: An Underutilized Tool in the Quest to Elucidate Radical SAM Dynamics. Molecules 2021, 26 (9), 2590. 10.3390/molecules26092590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W.-J.; Qian H.-X.; Wang W.-J.; Liao R.-Z. Computational Understanding of the Selectivities in Metalloenzymes. Front. Chem. 2018, 6, 638. 10.3389/fchem.2018.00638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechay M. R.; Valdez C. E.; Alexandrova A. N. Computational Treatment of Metalloproteins. J. Phys. Chem. B 2015, 119 (19), 5945–5956. 10.1021/acs.jpcb.5b00028. [DOI] [PubMed] [Google Scholar]
- Jing Z.; Liu C.; Cheng S. Y.; Qi R.; Walker B. D.; Piquemal J.-P.; Ren P. Polarizable Force Fields for Biomolecular Simulations: Recent Advances and Applications. Annu. Rev. Biophys. 2019, 48 (1), 371–394. 10.1146/annurev-biophys-070317-033349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-J.; Khorshidi A.; Kastlunger G.; Peterson A. A. The Potential for Machine Learning in Hybrid QM/MM Calculations. J. Chem. Phys. 2018, 148 (24), 241740. 10.1063/1.5029879. [DOI] [PubMed] [Google Scholar]
- Böselt L.; Thürlemann M.; Riniker S. Machine Learning in QM/MM Molecular Dynamics Simulations of Condensed-Phase Systems. J. Chem. Theory Comput. 2021, 17 (5), 2641–2658. 10.1021/acs.jctc.0c01112. [DOI] [PubMed] [Google Scholar]