

A General Overview of the Radical S-Adenosylmethionine Superfamily
In 2001, Heidi Sofia and colleagues published a groundbreaking bioinformatics study of a superfamily of enzymes that use S-adenosylmethionine (SAM or AdoMet) to carry out a wide variety of reactions that proceed through mechanisms of catalysis involving organic radicals.1 This superfamily of enzymes is denoted by their ability to catalyze a reductive cleavage of SAM to methionine and a 5′-deoxyadenosyl 5′-radical (5′-dA·) (Figure 1).1−4 These “Radical SAM” (RS) enzymes number over 700,000 unique sequences and catalyze over 100 distinct reactions, including the formation of stable protein radicals, complex rearrangements, methylation and thiolation of unactivated carbon centers, methylation of phosphinate phosphorus atoms, epimerization, carbon–carbon bond formation between sp2- and sp3-hybridized carbon centers or two sp3-hybridized carbon centers, steps in the biosynthesis of complex metallocofactors, oxidative decarboxylation, hydroxylation, cyclopropanation, and dehydrogenation, among other reaction types (Figure 2).5−9 However, a large portion of the radical SAMeome is currently unannotated.6 Given the potential for a remaining reservoir of novel transformations, a major challenge is to develop strategies to annotate enzymes within the radical SAMeome. Moreover, the finding that many of these enzymes catalyze key reactions in bacteria that constitute the human microbiome, suggests the importance of the radical SAMeome in human health and disease.
Figure 1.

Formation of the 5′-deoxyadenosyl 5′-radical via intermediate omega.
Figure 2.

Representative transformations catalyzed by RS enzymes.
RS superfamily members all contain at least one [Fe4S4]2+ cluster that is ligated by three cysteine residues (one for each of three Fe ions) that are most often found in a CxxxCxxC motif.9 This spacing of cysteines is conserved in at least 90% of all RS proteins and is one of the major determinants used to identify RS proteins bioinformatically.1,9 SAM associates to the fourth (unique) Fe ion in a bidentate fashion through its amino and carboxylate groups.10−12 When the cluster is reduced to the [Fe4S4]+ state, it induces the fragmentation of SAM to yield the 5′-dA·. In almost all RS reactions—except for the reaction catalyzed by TsrM and most likely similar reactions on analogous substrates13−15—the role of the 5′-dA· is to abstract hydrogen atoms (H·) from a substrate, which typically initiates turnover. Studies from the Broderick and Hoffman laboratories have provided evidence for an intermediate that precedes 5′-dA· formation (Figure 1). This intermediate, termed omega, contains methionine bound to the unique iron ion of the [Fe4S4] cluster and a bond between the unique iron and the 5′-carbon of 5′-deoxyadenosine.16,17 This discovery highlights a similarity between this radical generating system and 5′-deoxyadenosyl 5′-cobalamin (AdoCbl), the other biological cofactor that is used to generate the 5′-dA·.18,19 The 5′-dA· had never been observed for many decades despite myriad attempts to do so by various investigators. In 1999, Magnusson, Reed, and Frey reported the use of S-3′,4′-anhydroadenosylmethionine, an allylic analogue of SAM, which led to the formation of a stable 5′-deoxy-3′,4′-anhydroadenosyl radical in the lysine 2,3-aminomutase (KAM) reaction.20 This radical was then characterized extensively by electron paramagnetic resonance (EPR) spectroscopy.20 More recently, the groups of Broderick and Hoffman and that of Britt developed clever strategies to observe the 5′-dA· directly from SAM in the RS enzymes pyruvate formate-lyase activase (PFL-AE) and HydG.21,22
The Genesis of the Radical SAM Superfamily
The genesis of RS enzymology has been described in a 2001 review by Frey and Booker, although the RS superfamily had not been established in name at that time.2 Pioneering studies on KAM and pyruvate formate-lyase (PFL) indicated that their reactions had an unusual requirement for SAM. KAM catalyzes a 1,2-cross migration between the 2-amino group and the 3-proR hydrogen of lysine to afford β-lysine, a reaction that is typical of enzymes that use AdoCbl in catalysis.2,18,23 In fact, a similar enzyme, d-lysine 5,6-aminomutase, which generates 2,5-diaminohexanoate, uses AdoCbl in catalysis.24 However, isolated KAM does not contain AdoCbl and its activity is not stimulated by treatment with exogenous AdoCbl. Instead, activity is stimulated by the addition of pyridoxal 5′-phosphate (PLP), a thiol-reducing agent, iron, and SAM to the reaction.25 This requirement for SAM was puzzling because the reaction does not involve the transfer of a methyl group or any of the other groups bonded to SAM’s sulfur center. Around the same time, Knappe’s laboratory reported that PFL exists in active and inactive states and that the inactive state of the enzyme could be activated by treatment with PFL-AE, SAM, and a reducing system. They also noted that SAM is reductively cleaved to 5′-deoxyadenosine (5′-dAH) and methionine in the process and that this reaction is accompanied by the production of a free radical on the protein.26−28 We now know that the reaction catalyzed by PFL-AE results in the abstraction of the 2-proS H· from Gly-734 of PFL to yield a stable glycyl radical cofactor.28−30 Similar studies were carried out on the anaerobic ribonucleotide reductase (ARR) and its corresponding activase (ARR-AE). In this instance, the radical is located on Gly-681 of the ARR.31,32
In 1987, Moss and Frey published a key study that defined the role of SAM in RS reactions, establishing its similarity to AdoCbl in function. In these experiments, SAM labeled with 3H either in its adenine ring, methyl moiety, or at its C-5′ position was incubated with KAM. Upon the reaction reaching equilibrium and reisolating the substrate and product, 3H was found associated both with the lysine substrate and with the β-lysine product but only when SAM labeled at C-5′ was used. Given the large amounts of tritium transferred and the extent of transfer depending on the concentration of enzyme in the reaction, it was concluded that (1) SAM does not readily exchange on and off of KAM, and (2) the C-5′–sulfur bond must be broken during catalysis to allow the C-5′carbon to rotate freely.33 These findings suggested that a 5′-dA· is formed from SAM, which can then function like the 5′-dA· from AdoCbl-dependent reactions.
As described in the Perspective by Vahe Bandarian, I too had a front-row seat in observing the evolution of the RS superfamily.24 Both he and I were at the Institute for Enzyme Research at the University of Wisconsin, Madison in the mid-1990s; he was a graduate student with George Reed, while I was a postdoctoral fellow with Perry Frey, arguably the father of RS enzymology. Heidi Sofia and Perry knew each other well. In fact, she had been a graduate student at Wisconsin. She contacted Perry before submitting her work for publication and asked him what he thought about the name radical SAM for this emerging superfamily, a conversation that he relayed to me. At the time, we had just completed an X-ray absorption spectroscopy (XAS) study in collaboration with Bob Scott’s laboratory at the University of Georgia and found that, upon the reductive cleavage of SAM in the KAM reaction, the sulfur of methionine (in this instance selenomethionine) coordinates to the cluster, moving from ∼3.2 Å to the unique iron in the X-ray crystal structure with SAM to ∼2.7 Å with selenomethionine as determined by XAS.34,35 We decided to submit both manuscripts to Science simultaneously, but each was returned without review. The manuscript by Heidi Sofia has garnered over 1065 citations at this writing, according to Google Scholar.
In addition to KAM and the activating enzymes of PFL and ARR, the founding members of the RS superfamily, established in the 2001 paper by Sofia et al., included biotin synthase (BioB) and lipoyl synthase (LipA), two RS enzymes that catalyze the attachment of sulfur atoms to aliphatic carbons.1 At this time, these enzymes were five of over 600 other proteins that were believed to share the ability to use an [Fe4S4] cluster and SAM to catalyze the formation of a 5′-dA· intermediate. A select number of these proteins were tentatively assigned a function based on available biochemical data. Many were involved in the biosynthesis of natural products, such as spectinomycin, subtilosin, nikkomycin, mitomycin C, oxetanocin, fortimicin, fosfomycin, bialaphos, the desosamine moiety of erythromycin, oleandomycin, methymycin, neomethymycin, narbomycin, and pikromycin. Some, like spore-photoproduct lyase, were involved in DNA repair, while others were involved in generating important cellular cofactors or coenzymes such as biotin, lipoic acid, heme, molybdopterin, thiamin, bacteriochlorophyll, pyrroloquinoline quinone, and the complex nitrogenase metallocofactor. Moreover, some were involved in the modification of nucleic acids (e.g., MiaB family), while others, like AtsB, were involved in protein modification.1 One RS protein, which has gained tremendous attention, was reported to be interferon-inducible and to display antiviral properties. This protein, first designated as cig5 but now better known as viperin, was ultimately discovered to catalyze a radical-dependent dehydration of cytidine triphosphate to give 3′-deoxy-3′,4′-didehydro-CTP (ddhCTP), which is an inhibitor of many viral RNA-dependent RNA polymerases (Figure 3).36
Figure 3.

Formation of 3′-deoxy-3′,4′-didehydro-CTP (ddhCTP) by viperin.
From 2001 to Radical SAM.Org
By 2008, the RS superfamily had grown to over 2800 unique sequences of proteins.8 This growth, undoubtedly spawned by advances in technology that enabled organismal genomes to be sequenced rapidly and cost-effectively, created a problem. The RS superfamily was evolving at a rate that vastly outpaced the ability to annotate the reactions that they catalyzed. In September 2012, John Gerlt, the director of the Enzyme Function Initiative (EFI), organized a workshop on RS enzymes at the University of California at San Francisco (UCSF) that involved many of the key stakeholders in the field. The EFI was a large-scale collaborative project funded by the National Institutes of General Medical Sciences (NIGMS) to develop strategies to annotate the large number of proteins discovered through genomic sequencing projects. This pivotal meeting led to the incorporation of the RS superfamily into the Structure–Function Linkage Database (SFLD), which was curated by Patricia Babbitt and colleagues at UCSF.37 The EFI was interested in the RS superfamily because of the superfamily’s size and the large potential to aid the scientific community in the discovery of novel and mechanistically rich transformations and biochemical pathways. Moreover, given that RS enzymes are abundant in the bacteria that constitute the human gut microbiome, the potential for impacting human health and disease was substantial. In fact, Emily Balkus’ laboratory had shown just prior to the meeting that a glycyl radical enzyme (GRE) is responsible for the transformation of choline to trimethylamine. The further oxidation of trimethylamine produces trimethylamine N-oxide, which is implicated in several diseases, such as nonalcoholic fatty liver disease, atherosclerosis, and the metabolic disorder trimethylaminuria (fish malodor syndrome).38
One of the key tools at the EFI’s disposal was protein sequence similarity networks (SSNs), developed in part by Babbitt, which provide a visual method to observe the interrelatedness of proteins in large data sets through all-by-all BLAST pairwise alignments.39,40 At the San Francisco meeting, the EFI and SFLD provided the first initial glimpse of an SSN of the RS superfamily, which was generated from 113,776 full-length superfamily sequences in the SFLD database. Twenty subgroups (http://sfld.rbvi.ucsf.edu/archive/django/superfamily/29/index.html), identified by clustering, were annotated based on some level of biochemical characterization of representative proteins in each cluster. A detailed analysis of the RS SSN has been published, which describes all the annotated RS reactions at that time.9 A more current SSN is shown in Figure 4. Extensive reviews of RS reactions have also been published.5,7 One caveat, however, is that the superfamily continues to grow at a rapid pace, necessitating frequent literature updates that describe new and novel reactions.
Figure 4.

A full SSN of the RS Superfamily highlighting five megagroups (1–5) composed of multiple subgroups and five single subgroups. See radicalSAM.org and ref (6) for details on generating the SSN.
A major advance in the study of RS enzymes that grew out of the SFLD and the EFI is the recent creation of RadicalSAM.org, which is described in the Perspective by Oberg, Precord, Mitchell, and Gerlt in this virtual issue of ACS Bio & Med Chem Au.6 The RS superfamily had grown so large (∼700,000 unique protein sequences) by this time that it would be challenging to construct and display SSNs for the entire superfamily and even for some of the subgroups on typical desktop computers. RadicalSAM.org rectifies this problem by providing predetermined SSNs for the entire superfamily and the various subgroups. In addition, users can pivot directly from a given protein sequence in RadicalSAM.org to its genome neighborhood, which can provide additional clues in determining a protein’s function and even an entire pathway in which it participates. Other features include the ability to quickly identify what RS proteins exist in any given organism (I asked for that personally), to quickly identify conserved cysteines in a multiple sequence alignment of any of the subclasses of RS proteins, and to download a multiple sequence alignment and/or WebLogo of any of the subclasses of RS proteins. RadicalSAM.org is consistently updated, and users are encouraged to reach out to the administrative team with questions or requests for additional capabilities that they would like to see incorporated into the resource.6
Contributions to the Radical SAM Virtual Issue in ACS Bio & Med Chem Au
In addition to the genesis of RadicalSAM.org, this virtual issue of ACS Bio & Med Chem Au highlights some of the major discoveries and advances in RS enzymology since the establishment of the superfamily in 2001. Tetrapyrrole (e.g., heme, heme d1, and bacteriochlorophyll) biosynthesis and metabolism is a recurring theme in RS enzymology, and several RS enzymes in these pathways were highlighted by Sofia et al. One of the key steps in the biosynthesis of heme is the oxidative decarboxylation of two propionate side chains of coproporphyrinogen III to give protoporphyrinogen IX, which is also an early precursor of chlorophylls and bacteriochlorophylls. This reaction is catalyzed in some bacteria by HemN, which is now known as CgdH.41 HemN was the first RS enzyme to have its three-dimensional structure determined,42 which was followed soon after by the structure of BioB.43 Additional RS enzymes involved in tetrapyrrole biosynthesis include NirJ, AhbC, and AhbD. AhbD catalyzes a reaction that is like that of CgdH, while AhbC catalyzes the removal of acetate moieties from a 12,18-didecarboxysiroheme intermediate. The precise reaction catalyzed by NirJ is currently unknown. The Perspective by Gunhild Layer and colleagues beautifully compares and contrasts these reactions and highlights the structure of CdgH from E. coli, which she, as first author of the work, published back in 2003.42 Also discussed is the RS enzyme BchE, which catalyzes an amazing six-electron oxidation of Mg-protoporphyrin IX monomethylester (Mg-PME) to protochlorophyllide, a key step in the anaerobic pathways for the biosynthesis of bacteriochlorophylls (Figure 5). Formally, this reaction entails the oxygenation of the C-131 methylene carbon to an oxo group and a C–C linkage between C-132 and C-15 to form a ring. Importantly, the added oxygen atom is derived from water rather than molecular oxygen, which distinguishes BchE from AcsF, its aerobic counterpart. Indeed, this enzyme has sometimes been referred to as a “Magic Enzyme.”
Figure 5.
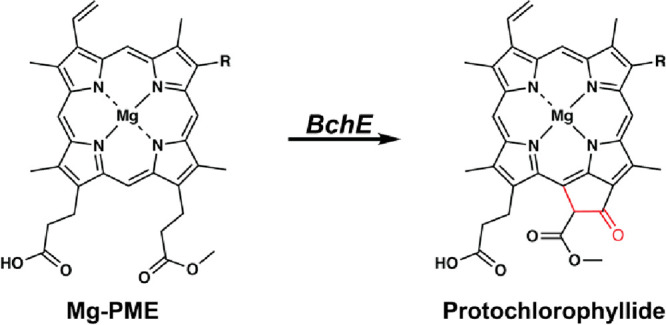
Reaction catalyzed by Mg-protoporphyrin IX monomethylester cyclase (BchE). Only the Mg-PME substrate and protochlorophyllide products are shown.
HemN-like enzymes constitute one of the largest subfamilies of RS enzymes.1,9 Structurally, these enzymes contain a C-terminal HemN domain, believed to be involved in substrate recognition, in addition to a partial triose phosphate isomerase (TIM) barrel that is characteristic of most RS enzyme structures.44 HemN-like enzymes are functionally diverse, as detailed in the Perspective from Qi Zhang’s laboratory.45 The structure of HemN (now CdgH), unlike structures of any other RS enzyme, shows the presence of two bound SAM molecules. One (SAMI) binds to the [Fe4S4]RS cluster in the fashion determined in the classic experiments by Broderick and colleagues, while the second SAM (SAMII) binds a few angströms away from SAMI.42 The initial steps in the mechanisms of HemN-like enzymes are believed to involve an H· abstraction from the methyl moiety of SAMII by a 5′-dA· derived from SAMI. In CdgH, the resulting radical then abstracts an H· from the substrate, which is followed by loss of an electron to an external acceptor and decarboxylation.46 CdgH is one of many enzymes in the HemN subfamily. Others can catalyze the methylation or cyclopropanation of sp2-hybridized carbon centers and additional reactions, as described in the Review from Bill Lanzilotta’s lab.47 In these alternative reactions that have been characterized biochemically, the resulting SAMII methylene radical then adds to an sp2-hybridized carbon atom of the target substrate, which is followed by loss of S-adenosylhomocysteine (SAH) to yield an intermediate that has several difficult outcomes. In one reaction, catalyzed by NosN, this intermediate is formed after a C1 transfer to an aromatic carbon of a 3-methylindolic acid moiety. Loss of an electron leads to a highly electrophilic species, which is then trapped by a glutamyl residue of the peptide substrate to form the side ring of the antibiotic nosiheptide.45 In some instances, a similar intermediate is reduced by an electron, which results in the methylation of the sp2-hybridized carbon center. In ChuW and HutW, reactions studied by the Lanzilotta laboratory, this intermediate is formed during the anaerobic degradation of heme by certain pathogenic bacteria for iron acquisition.47
Several key bioinformatics studies were published by Daniel Haft and colleagues and reported that RS proteins are often found closely associated with gene clusters that encode a ribosomally synthesized structural peptide that is modified by tailoring enzymes.48−50 These RS enzymes typically contain a cysteine-rich C-terminal motif—named a SPASM domain—that ligates up to two additional iron–sulfur (FeS) clusters. An X-ray crystallographic structural analysis of an anaerobic sulfatase maturating enzyme, which catalyzes a dehydrogenation reaction, defined the SPASM domain.51 A subsequent structural analysis of BtrN, an RS dehydrogenase that acts on a small molecule during the biosynthesis of the antibiotic butirosin, showed that it contains a truncated SPASM domain, which was named “twitch”.52 According to RadicalSAM.org, RS enzymes containing SPASM or twitch domains represent the largest subclass of the superfamily (Figure 4).6,9 Many of these enzymes are involved in the maturation of ribosomally produced and post-translationally modified peptides, or RiPPs, which often exhibit antibiotic or antitumor activity. The Review from the Latham laboratory offers a nice overview of the rise of this RS subfamily, highlighting how members are involved in C–C, C–S, and C–O bond formation during the maturation of RiPPs.53 C–C bonds are difficult to make, especially when the bond is formed from precursors that contain one or more unactivated sp3-hybridized carbon centers.
The Perspective from the Seyedsayamdost laboratory provides a nice introduction to some of the early studies that helped to define the RS superfamily and highlights RS enzymes involved in the maturation of RiPPs in bacteria found in the human microbiome.54 RS enzymes involved in RiPP maturation have historically been identified after the discovery of the RiPP natural product and then the identification of the biosynthetic gene cluster (BGC) for the RiPP. One goal of the Seyedsayamdost laboratory is to reverse that process, by developing strategies to identify natural products from the BGCs that encode them. One advantage of annotating proteins involved in RiPP maturation is that their substrates are genetically encoded in BGCs and are readily identifiable. In this Perspective, the Syedsayamdost laboratory details how the natural product streptide was identified upon characterizing a BGC in Streptococci.54
The Perspective by Yokoyama, Li, and Pang describes heroic studies by the Yokoyama lab, and others, to dissect a complex transformation by MoaA, an RS enzyme, and a partner protein, MoaC, during the biosynthesis of molybdopterin.55 Molybdopterin is an essential redox cofactor used in molybdenum-containing enzymes. The inability to synthesize the cofactor in humans results in several potentially fatal diseases. Moreover, the cofactor is essential in a number of pathogenic bacteria, suggesting that its biosynthesis may be a good target for the development of antibacterial agents. MoaA, which also has a twitch domain, was the third RS enzyme to have its structure determined; however, the reaction mechanism and even the actual reaction that the enzyme catalyzes were not readily discernible from the structure.56 Work from the Yokoyama lab showed that the reaction catalyzed by MoaA is not as complex as originally thought. It takes GTP and forms 3′,8-cyclco-7,8-dihydroguanosine 5′-triphosphate. Then, MoaC catalyzes the complex rearrangement of the product of the MoaA to cyclic pyranopterin monophosphate, a precursor to molybdopterin.
The versatility of RS enzymes is extended by their ability to bind and use additional cofactors to expand the repertoire of reactions they catalyze. Indeed, in addition to PLP, RS enzymes have been shown to bind auxiliary Fe/S clusters, flavins, and cobalamin.57 The cobalamin-dependent RS enzymes most often catalyze the methylation of unactivated carbon centers and in some cases phosphinate phosphorus centers.58 However, there are several known cobalamin-dependent RS enzymes that do not catalyze methylation reactions, such as BchE, mentioned above, and OxsB, which is involved in the biosynthesis of oxetanocin-A.59 The study of cobalamin-dependent RS enzymes, especially the methylases, has been the subject of deep interest because they are found in myriad pathways for the biosynthesis of clinically important antibiotics and herbicides. However, their characterization was initially hampered largely by extreme insolubility upon overproduction.60 Early challenges associated with the purification and study of cobalamin-dependent enzymes are outlined in the Perspective from the Townsend laboratory, which also highlights their outstanding work on RS enzymes involved in the biosynthesis of the clinically important carbapenem antibiotics.58 Other systems discussed include polytheonamides, which are heavily modified RiPP natural products (Figure 6). Their maturation involves up to 18 C-methylations and 21 epimerizations that are catalyzed by just three RS enzymes! The Perspective by Bridwell-Rabb, Li, and Drennan is a beautiful overview of cobalamin-dependent RS enzymes and the extensive scope of reactions that they catalyze. Some of the enzymes catalyze C-methylations of both sp2- and sp3-hybridized carbon centers, P-methylation, and sequential C-methylations to generate ethyl, isopropyl, or t-butyl groups, while others, like OxsB and BchE, catalyze ring formations. In particular, this Perspective focuses on three enzymes that had been structurally characterized at the time: OxsB, TsrM, and TokK.59
Figure 6.

Structure of polytheonamide A, a RiPP natural product consisting of up to 18 methylations and 21 epimerizations catalyzed by just three RS enzymes (PoyB, PoyC, and PoyD). RS-dependent methylations are highlighted in orange (both bonds and one-letter codes), and epimerizations, to generate the D-configured residue, are shown in red (both bonds and one-letter codes).
Auxiliary Fe/S clusters also serve as additional cofactors in myriad RS enzymes, as described above for RS enzymes containing SPASM or twitch domains. However, Fe/S clusters in RS enzymes can also be used as substrates. Two examples include the RS enzymes BioB and LipA. BioB contains an [Fe2S2] cluster in addition to the radical SAM ([Fe4S4]RS) cluster, while LipA contains an additional [Fe4S4] cluster. In each of these enzymes, the auxiliary cluster is sacrificed (i.e., degraded) during the reaction to supply the appended sulfur atom, limiting the enzyme to only one turnover. Recent studies have shown that the auxiliary cluster in LipA can be regenerated after each catalytic event by an Fe/S cluster carrier protein, NfuA.61 An Article in this virtual issue from the Booker laboratory describes the characterization of the human form of LipA, called LIAS, as well as the human NfuA analogue, NFU1. This Article reports that NFU1 binds tightly to LIAS and, like NfuA’s effect on LipA, can regenerate the auxiliary cluster of LIAS, allowing the protein to conduct multiple turnovers.100 A second Article contributed by the Booker laboratory describes studies on a completely new system for generating the lipoyl cofactor that is found predominantly in archaea. Unlike the canonical system (i.e., LipA and LIAS), which requires only a protein composed of a single polypeptide, this new system requires two proteins to insert both sulfurs. These two proteins, called LipS1 and LipS2, had been annotated as biotin synthases. LipS2 inserts a sulfur atom at C8 of an octanoyl chain, while LipS1 inserts a sulfur atom at C6 of an octanoyl chain. The work by Booker and colleagues showed that like the reaction catalyzed by Escherichia coli BioB, sulfur insertion takes place first at the terminal carbon of the substrate, while in canonical lipoyl synthases sulfur insertion takes place first at the internal (C6) carbon (Figure 7). Unlike E. coli BioB, however, both LipS1 and LipS2 contain auxiliary [Fe4S4] clusters.62
Figure 7.

Two pathways for the biosynthesis of the lipoyl cofactor. The classical pathway involves a protein composed of a single polypeptide (LIAS or LipA), which attaches sulfur at C6 first and then C8. The alternative pathway involves two proteins. LipS2 installs sulfur first at C8, while LipS1 subsequently installs sulfur at C6. In both pathways, octanoyllysyl-H-protein serves as the starting substrate.
One of the areas of RS enzymology that highlights the extreme power and versatility of these enzymes is in the generation of complex metallocofactors, such as the H-cluster of the [FeFe]-hydrogenase and the molybdenum–iron (MoFe) cofactor of nitrogenase (Figure 8). The [FeFe] hydrogenase is one of three subclasses of hydrogenases, which catalyze the reversible reduction of protons to H2. The H-cluster is composed of a two-iron subcluster, [2Fe]H, bridged to an [Fe4S4] cluster by a conserved cysteinyl residue. The [2Fe]H subcluster is composed of two iron ions, each of which is ligated by one carbon monoxide (CO) and one cyanide ion (CN).63−65 In addition, both iron ions are bridged by an azadithiolate ligand and an additional molecule of CO (Figure 8B). Two RS enzymes are required for the assembly of the [2Fe]H subcluster. The first is HydG, which, in addition to the [Fe4S4]RS cluster, contains an auxiliary [Fe4S4] cluster connected to a free cysteine-coordinated mononuclear (dangler) iron through the sulfur atom of the cysteine amino acid. HydG catalyzes an amazing dissection of l-tyrosine to yield p-cresol and the CO and CN ligands for the [2Fe]H subcluster as well as the modification of its auxiliary cluster to an [Fe(CN)(CO)2(cysteinate)]− organometallic complex called complex B. HydE, the second RS enzyme involved in the synthesis of the H-cluster, binds complex B and catalyzes its transformation into an adenosylated intermediate through the attack of the 5′-dA· on the sulfur atom of the cysteinate ligand. HydE further transforms this intermediate into a five-coordinate [Fe(I)S(CO)2CN] complex that is proposed to be dimerized before binding to HydF, which is not an RS protein. Thus, the [Fe2S2] core of the [2Fe]H comes from complex B through two rounds of catalysis by HydG.
Figure 8.

Structures of the [MoFe]-nitrogenase (PDB: 6N59) (A) and the [FeFe]-hydrogenase (PDB: 1M1N) (B). Insets depict the metallocofactor that requires the participation of one or more RS enzymes (NifB for the MoFe cofactor and HydG/HydE for the H-cluster) for its assembly.
In contrast to the [FeFe]-hydrogenase, the biosynthesis of the MoFe cofactor of nitrogenase requires only one RS enzyme. However, like HydG and HydE, this enzyme, NifB, catalyzes an amazing but complex sequence of events. Nitrogenase catalyzes the reduction of N2 to ammonia, allowing nitrogen to be taken up by living systems. Nitrogen reduction takes place on the MoFe cofactor, which has the chemical formula [MoFe7S9C-R-homocitrate]. It is typically viewed as a combination of [MoFe3S3] and [Fe4S3] modules that are connected through three μ2-sulfides and one μ6-carbide. The identity of the carbide and the mechanism by which it is installed into the cofactor have been the subject of intense investigation by numerous laboratories. NifB contains two auxiliary clusters (called K1 and K2) in addition to the [Fe4S4]RS cluster. It catalyzes the initial transfer of a methyl group from SAM to one of the sulfide ions of the K2 cluster via a traditional SN2 mechanism. A second SAM molecule is used to abstract an H· from the resulting methyl moiety, which triggers the synthesis of the carbide ion via a mechanism that is not well established along with the formation of the [Fe8S8C] L*-cluster. The incorporation of the last “belt” sulfide results in the [Fe8S9C] L-cluster, a precursor to the MoFe cofactor.
The Perspective by Britt et al. discusses in detail how the HydG reaction mechanism was established. In particular, it focuses on how advanced electron paramagnetic resonance (EPR), Mössbauer, and Fourier transform infrared (FTIR) spectroscopic techniques were used to elucidate many of the key details of the assembly process. The Review by Nicolet, Cherrier, and Amara discusses both HydG and HydE mechanisms from a structural point of view as well as the role of NifB in generating the MoFe cofactor of nitrogenase. Both Reviews are exciting reads!
Summary and Outlook
The discovery of the RS superfamily has contributed enormously to the basic understanding of how radicals can be used in a controlled fashion to catalyze kinetically challenging reactions that can generate enormously complex molecules. Indeed, RS enzymology has been at the leading edge of bioinorganic chemistry as well as bioorganic chemistry, especially as it pertains to the biosynthesis of complex natural products, and has spawned new research areas for beginning investigators as well as those who are more seasoned. The reactions that RS enzymes catalyze continue to amaze and even stun. Recently, studies by Zeng et al. and Lloyd et al. showed that RS enzymes can even be used to join two sp3-hybridized carbon centers in completely unactivated aliphatic carbon chains, reactions that were theretofore unprecedented in nature (Figure 9).66,67 There are many unanswered questions associated with the myriad RS enzymes that have been already identified and studied, and many of these questions will require collaborative efforts to address. However, one of the burning questions that relates to the entire superfamily is how can we develop strategies to go from gene sequence to protein function?
Figure 9.

Reactions catalyzed by GDGT-MAS, GrsA, and GrsB, three RS enzymes that form bonds between two aliphatic sp3-hybridized carbon centers on hydrocarbons.
Acknowledgments
Our work on Radical SAM enzymes is supported by the National Institutes of Health (GM-122595 and AI-160172), the National Science Foundation (MCB- 1716686), and the Eberly Family Distinguished Chair in Science. S.J.B. is an investigator of the Howard Hughes Medical Institute.
Views expressed in this editorial are those of the authors and not necessarily the views of the ACS.
Notes
This article is subject to HHMI’s Open Access to Publications policy. HHMI lab heads have previously granted a nonexclusive CC BY 4.0 license to the public and a sublicensable license to HHMI in their research articles. Pursuant to those licenses, the author-accepted manuscript of this article can be made freely available under a CC BY 4.0 license immediately upon publication.
References
- Sofia H. J.; et al. Radical, SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001, 29, 1097–1106. 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey P. A.; Booker S. J. Radical mechanisms of S-adenosylmethionine-dependent enzymes. Adv. Protein Chem. 2001, 58, 1–45. 10.1016/S0065-3233(01)58001-8. [DOI] [PubMed] [Google Scholar]
- Wang S. C.; Frey P. A. S-adenosylmethionine as an oxidant: the radical SAM superfamily. Trends Biochem. Sci. 2007, 32, 101–110. 10.1016/j.tibs.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Fontecave M.; Mulliez E.; Ollagnier-de Choudens S. Adenosylmethionine as a source of 5′-deoxyadenosyl radicals. Curr. Opin. Chem. Biol. 2001, 5, 506–511. 10.1016/S1367-5931(00)00237-4. [DOI] [PubMed] [Google Scholar]
- Broderick J. B.; et al. Radical S-adenosylmethionine enzymes. Chem. Rev. 2014, 114, 4229–4317. 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberg N.; et al. RadicalSAM.org: A Resource to Interpret Sequence-Function Space and Discover New Radical SAM Enzyme Chemistry. ACS Bio Med. Chem. Au 2022, 2 (1), 22–35. 10.1021/acsbiomedchemau.1c00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challand M. R.; Driesener R. C.; Roach P. L. Radical S-adenosylmethionine enzymes: mechanism, control and function. Nat. Prod. Rep. 2011, 28, 1696–1721. 10.1039/c1np00036e. [DOI] [PubMed] [Google Scholar]
- Frey P. A.; Hegeman A. D.; Ruzicka F. J. The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 63–88. 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- Holliday G. L.; et al. Atlas of the radical SAM superfamily: divergent evolution of function using a “plug and play″ domain. Methods Enzymol 2018, 606, 1–71. 10.1016/bs.mie.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs C.; et al. Coordination of adenosylmethionine to a unique iron site of the [4Fe-4S] of pyruvate formate-lyase activating enzyme: a Mössbauer spectroscopic study. J. Am. Chem. Soc. 2002, 124, 912–913. 10.1021/ja017562i. [DOI] [PubMed] [Google Scholar]
- Walsby C. J.; et al. An anchoring role for FeS clusters: chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe-4S] cluster of pyruvate formate-lyase activating enzyme. J. Am. Chem. Soc. 2002, 124, 11270–11271. 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- Walsby C. J.; et al. Spectroscopic approaches to elucidating novel iron-sulfur chemistry in the ″radical-SAM″ protein superfamily. Inorg. Chem. 2005, 44, 727–741. 10.1021/ic0484811. [DOI] [PubMed] [Google Scholar]
- Pierre S.; et al. Thiostrepton tryptophan methyltransferase expands the chemistry of radical SAM enzymes. Nat. Chem. Biol. 2012, 8 (12), 957–959. 10.1038/nchembio.1091. [DOI] [PubMed] [Google Scholar]
- Blaszczyk A. J.; et al. Spectroscopic and Electrochemical Characterization of the Iron-Sulfur and Cobalamin Cofactors of TsrM, an Unusual Radical S-Adenosylmethionine Methylase. J. Am. Chem. Soc. 2016, 138 (10), 3416–3426. 10.1021/jacs.5b12592. [DOI] [PubMed] [Google Scholar]
- Knox H. L.; et al. Structural basis for non-radical catalysis by TsrM, a radical SAM methylase. Nat. Chem. Biol. 2021, 17, 485–491. 10.1038/s41589-020-00717-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byer A. S.; et al. Paradigm Shift for Radical S-Adenosyl-l-methionine Reactions: The Organometallic Intermediate Omega Is Central to Catalysis. J. Am. Chem. Soc. 2018, 140 (28), 8634–8638. 10.1021/jacs.8b04061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horitani M.; et al. Radical SAM catalysis via an organometallic intermediate with an Fe-[5′-C]-deoxyadenosyl bond. Science 2016, 352 (6287), 822–5. 10.1126/science.aaf5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey P. A.; Ballinger M. D.; Reed G. H. S-adenosylmethionine: a ’poor man’s coenzyme B12’ in the reaction of lysine 2,3-aminomutase. Biochem. Soc. Trans. 1998, 26 (3), 304–10. 10.1042/bst0260304. [DOI] [PubMed] [Google Scholar]
- Broderick W. E.; Hoffman B. M.; Broderick J. B. Mechanism of Radical Initiation in the Radical S-Adenosyl-l-methionine Superfamily. Acc. Chem. Res. 2018, 51 (11), 2611–2619. 10.1021/acs.accounts.8b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson O. T.; Reed G. H.; Frey P. A. Spectroscopic Evidence for the Participation of an Allylic Analogue of the 5′-Deoxyadenosyl Radical in the Reaction of Lysine 2,3-Aminomutase. J. Am. Chem. Soc. 1999, 121, 9764–9765. 10.1021/ja9925507. [DOI] [Google Scholar]
- Yang H.; et al. The Elusive 5′-Deoxyadenosyl Radical: Captured and Characterized by Electron Paramagnetic Resonance and Electron Nuclear Double Resonance Spectroscopies. J. Am. Chem. Soc. 2019, 141 (30), 12139–12146. 10.1021/jacs.9b05926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayler R. I.; et al. Trapping and Electron Paramagnetic Resonance Characterization of the 5′dAdo (*) Radical in a Radical S-Adenosyl Methionine Enzyme Reaction with a Non-Native Substrate. ACS Cent Sci. 2019, 5 (11), 1777–1785. 10.1021/acscentsci.9b00706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey P. A.; Magnusson O. T. S-Adenosylmethionine: a wolf in sheep’s clothing, or a rich man’s adenosylcobalamin?. Chem. Rev. 2003, 103, 2129–2148. 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- Bandarian V. Journey on the Radical SAM road as an accidental pilgram. ACS Bio Med. Chem. Au 2022, 2, 187–195. 10.1021/acsbiomedchemau.1c00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirpich T. P.; et al. Lysine 2,3-aminomutase. Purification and properties of a pyridoxal phosphate and S-adenosylmethionine-activated enzyme. J. Biol. Chem. 1970, 245 (7), 1778–89. 10.1016/S0021-9258(19)77160-9. [DOI] [PubMed] [Google Scholar]
- Knappe J.; et al. Pyruvate Formate-Lyase Reaction in Escherichia coli: The Enzymatic System Converting an Inactive Form of the Lyase into the Catalytically Active Enzyme. Eur. J. Biochem. 1969, 11, 316–327. 10.1111/j.1432-1033.1969.tb00775.x. [DOI] [PubMed] [Google Scholar]
- Conradt H.; et al. Pyruvate formate-lyase (inactive form) and pyruvate formate-lyase activating enzyme of Escherichia coli: isolation and structural properties. Arch. Biochem. Biophys. 1984, 228 (1), 133–42. 10.1016/0003-9861(84)90054-7. [DOI] [PubMed] [Google Scholar]
- Knappe J.; et al. Post-translational activation introduces a free radical into pyruvate formate-lyase. Proc. Natl. Acad. Sci. U. S. A. 1984, 81 (5), 1332–5. 10.1073/pnas.81.5.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. F.; Frey M.; Neugebauer F. A.; Schafer W.; Knappe J.; et al. The free radical in pyruvate formate-lyase is located on glycine-734. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 996–1000. 10.1073/pnas.89.3.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey M.; et al. Adenosylmethionine-dependent synthesis of the glycyl radical in pyruvate formate-lyase by abstraction of the glycine C-2 pro-S hydrogen atom. J. Biol. Chem. 1994, 269 (17), 12432–12437. 10.1016/S0021-9258(18)99892-3. [DOI] [PubMed] [Google Scholar]
- Logan D. T.; et al. A glycyl radical site in the crystal structure of a class III ribonucleotide reductase. Science 1999, 283 (5407), 1499–504. 10.1126/science.283.5407.1499. [DOI] [PubMed] [Google Scholar]
- Sun X.; et al. The free radical of the anaerobic ribonucleotide reductase from Escherichia coli is at glycine 681. J. Biol. Chem. 1996, 271, 6827–6831. 10.1074/jbc.271.12.6827. [DOI] [PubMed] [Google Scholar]
- Moss M.; Frey P. A. The role of S-adenosylmethionine in the lysine 2,3-aminomutase reaction. J. Biol. Chem. 1987, 262 (31), 14859–62. 10.1016/S0021-9258(18)48103-3. [DOI] [PubMed] [Google Scholar]
- Lepore B. W.; et al. The x-ray crystal structure of lysine-2,3-aminomutase from Clostridium subterminale. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 13819–13824. 10.1073/pnas.0505726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosper N. J.; et al. Direct FeS cluster involvement in generation of a radical in lysine 2,3-aminomutase. Biochemistry 2000, 39, 15668–15673. 10.1021/bi0022184. [DOI] [PubMed] [Google Scholar]
- Gizzi A. S.; et al. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 2018, 558 (7711), 610–614. 10.1038/s41586-018-0238-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiva E.; et al. The Structure-Function Linkage Database. Nucleic Acids Res. 2014, 42 (Database issue), D521–D530. 10.1093/nar/gkt1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craciun S.; Balskus E. P. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. U S A 2012, 109, 21307–21312. 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot R.; Oberg N.; Gerlt J. A. The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 2019, 58 (41), 4169–4182. 10.1021/acs.biochem.9b00735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson H. J.; et al. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS One 2009, 4 (2), e4345 10.1371/journal.pone.0004345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer G.; et al. Radical SAM enzymes involved in tetrapyrrole biosynthesis and insertion. ACS Bio Med. Chem. Au 2022, 2, 196–204. 10.1021/acsbiomedchemau.1c00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer G.; et al. Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of Radical SAM enzymes. EMBO J. 2003, 22, 6214–6224. 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovitch F.; et al. Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science 2004, 303, 76–79. 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vey J. L.; Drennan C. L. Structural insights into radical generation by the radical SAM superfamily. Chem. Rev. 2011, 111, 2487–2506. 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J.; et al. Functional diversity of HemN-like proteins. ACS Bio Med. Chem. Au 2022, 2, 109–119. 10.1021/acsbiomedchemau.1c00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X.; et al. Revisiting the Mechanism of the Anaerobic Coproporphyrinogen III Oxidase HemN. Angew. Chem., Int. Ed. Engl. 2019, 58 (19), 6235–6238. 10.1002/anie.201814708. [DOI] [PubMed] [Google Scholar]
- Mathew L. G.; Brimberry M.; Lanzilotta W. N. Class C Radical SAM methyltransferases involved in anaerobic heme degradation. ACS Bio Med. Chem. Au 2022, 2, 120–124. 10.1021/acsbiomedchemau.1c00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft D. H.; Basu M. K. Biological systems discovery in silico: radical S-adenosylmethionine protein families and their target peptides for posttranslational modification. J. Bacteriol. 2011, 193 (11), 2745–2755. 10.1128/JB.00040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft D. H.; et al. TIGRFAMS and genome properties in 2013. Nucleic Acids Res. 2012, 41, D387–D395. 10.1093/nar/gks1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft D. H. Bioinformatic evidence for a widely distributed, ribosomally produced electron carrier precursor, its maturation proteins, and its nicotinoprotein redox partners. BMC Genomics 2011, 12, 21. 10.1186/1471-2164-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman P. J.; et al. X-ray structure of an AdoMet radical activase reveals an anaerobic solution for formylglycine posttranslational modification. Proc. Natl. Acad. Sci. U S A 2013, 110, 8519–8524. 10.1073/pnas.1302417110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman P. J.; et al. X-ray analysis of butirosin biosynthetitic enzyme BtrN redefines structural motifs for AdoMet radical chemistry. Proc. Natl. Acad. Sci. U S A 2013, 110, 15949–15954. 10.1073/pnas.1312228110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendauletova A.; et al. How a subfamily of radical S-adenosylmethionine enzymes became a mainstay of ribosomally synthesized and post-translationally modified peptide discovery. ACS Bio Med. Chem. Au 2022, 2, 53–59. 10.1021/acsbiomedchemau.1c00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K. A.; Bushin L. B.; Seyedsayamdost M. R. RaS-RiPPs in Streptococci and the Human Microbiome. ACS Bio Med. Chem. Au 2022, 2 (4), 328–339. 10.1021/acsbiomedchemau.2c00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama K.; Li D.; Pang H. Resolving the Multidecade-Long Mystery in MoaA Radical SAM Enzyme Reveals New Opportunities to Tackle Human Health Problems. ACS Bio Med. Chem. Au 2022, 2 (2), 94–108. 10.1021/acsbiomedchemau.1c00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann P.; Schindelin H. Crystal structure of the S-adenosylmethionine-dependent enzyme MoaA and its implications for molybdenum cofactor deficiency in humans. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 12870–12875. 10.1073/pnas.0404624101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A. P.; Bandarian V. Eukaryotic TYW1 Is a Radical SAM Flavoenzyme. Biochemistry 2021, 60 (27), 2179–2185. 10.1021/acs.biochem.1c00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinner E. K.; Marous D. R.; Townsend C. A. Evolution of Methods for the Study of Cobalamin-Dependent Radical SAM Enzymes. ACS Bio Med. Chem. Au 2022, 2 (1), 4–10. 10.1021/acsbiomedchemau.1c00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridwell-Rabb J.; Li B.; Drennan C. L. Cobalamin-Dependent Radical S-Adenosylmethionine Enzymes: Capitalizing on Old Motifs for New Functions. ACS Bio Med. Chem. Au 2022, 2 (3), 173–186. 10.1021/acsbiomedchemau.1c00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz N. D.; et al. Enhanced Solubilization of Class B Radical S-Adenosylmethionine Methylases by Improved Cobalamin Uptake in Escherichia coli. Biochemistry 2018, 57 (9), 1475–1490. 10.1021/acs.biochem.7b01205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy E. L.; Booker S. J. Destruction and reformation of an iron-sulfur cluster during catalysis by lipoyl synthase. Science 2017, 358 (6361), 373–377. 10.1126/science.aan4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warui D. M. In vitro demonstration of human lipoyl synthase (LIAS) catalytic activity in the presence of NFU1. ACS Bio & Med Chem Au 2022, 2, 456–468. 10.1021/acsbiomedchemau.2c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neti S. S.; et al. Characterization of LipS1 and LipS2 from Thermococcus kodakarensis: Proteins Annotated as Biotin Synthases, which Together Catalyze Formation of the Lipoyl Cofactor. ACS Bio Med. Chem. Au 2022, 2 (5), 509–520. 10.1021/acsbiomedchemau.2c00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnier A.; et al. [FeFe]-Hydrogenase In Vitro Maturation. Angew. Chem., Int. Ed. Engl. 2022, 61, e202212074. 10.1002/anie.202212074. [DOI] [PubMed] [Google Scholar]
- Britt R. D.; et al. Proposed Mechanism for the Biosynthesis of the [FeFe] Hydrogenase H-Cluster: Central Roles for the Radical SAM Enzymes HydG and HydE. ACS Bio Med. Chem. Au 2022, 2 (1), 11–21. 10.1021/acsbiomedchemau.1c00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolet Y.; Cherrier M. V.; Amara P. Radical SAM enzymes and metallocofactor assembly: A structural point of view. ACS Bio Med. Chem. Au 2022, 2, 36–52. 10.1021/acsbiomedchemau.1c00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd C. T.; et al. Discovery, structure and mechanism of a tetraether lipid synthase. Nature 2022, 609 (7925), 197–203. 10.1038/s41586-022-05120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z.; et al. GDGT cyclization proteins identify the dominant archaeal sources of tetraether lipids in the ocean. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (45), 22505–22511. 10.1073/pnas.1909306116. [DOI] [PMC free article] [PubMed] [Google Scholar]