

Abstract

UV-advanced reduction processes (UV-ARP) are an advanced water treatment technology characterized by the reductive transformation of chemical contaminants. Contaminant abatement in UV-ARP is most often accomplished through reaction with hydrated electrons (eaq–) produced from UV photolysis of chemical sensitizers (e.g., sulfite). In this Review, we evaluate the photochemical kinetics, substrate scope, and optimization of UV-ARP. We find that quantities typically reported in photochemical studies of natural and engineered systems are under-reported in the UV-ARP literature, especially the formation rates, scavenging capacities, and concentrations of key reactive species like eaq. The absence of these quantities has made it difficult to fully evaluate the impact of operating conditions and the role of water matrix components on the efficiencies of UV-ARP. The UV-ARP substrate scope is weighted heavily toward contaminant classes that are resistant to degradation by advanced oxidation processes, like oxyanions and per- and polyfluoroalkyl substances. Some studies have sought to optimize the UV-ARP treatment of these contaminants; however, a thorough evaluation of the impact of water matrix components like dissolved organic matter on these optimization strategies is needed. Overall, the data compilation, analysis, and research recommendations provided in this Review will assist the UV-ARP research community in future efforts toward optimizing UV-ARP systems, modeling the eaq–-based chemical transformation kinetics, and developing new UV-ARP systems.
Keywords: photochemistry, UV-ARP, water treatment, hydrated electron, PFOA, PFOS, PFAS, reduction, water quality, contaminants
1. Introduction
Ultraviolet-based advanced reduction processes (UV-ARP) have received significant attention in recent years for the treatment of several classes of recalcitrant chemical contaminants in water.1−7 Advanced reduction processes are based on production of highly reducing hydrated electrons (eaq–, Figure 1), which exhibit fast bimolecular reaction rate constants with inorganic and organic compounds.8 In UV-ARP, eaq are produced by the illumination of eaq– sensitizers with UV lamps. Photoproduced eaq can react with target contaminants (TC), leading to TC transformation. Given its high reactivity, eaq– is also scavenged by various chemical species naturally present in water.
Figure 1.

Summary of UV-advanced reduction processes (UV-ARP) generation of hydrated electrons (eaq–).
The need for UV-ARP is motivated by the fact that many pollutants commonly found in contaminated waters are not readily treated by UV-advanced oxidation processes (UV-AOP), one of the most widely used degradative technologies in advanced water treatment. The need for an alternative to in situ oxidation processes has been recognized for some time. A patent by Bolton and Cater in 1993 detailing the treatment of wastewater or groundwater containing chloroform as a model halogenated organic with UV/iodide, UV/thiosulfate, UV/sulfite, and UV/iodide/thiosulfate as the eaq– source was one of the first reports of this technology.9 Subsequent UV-ARP studies have focused on substrates that can undergo reduction by eaq reaction such as oxyanions,6,10−19 highly oxidized elements (e.g., Cr(VI)),20,21 halogenated disinfection byproducts,5,6,16 and per- and polyfluoroalkyl substances (PFAS).22−34 Several bench-scale studies22−31 and a recent pilot-scale32 demonstration suggest that UV-ARP is one of the more promising technologies for degrading PFAS, which are resistant to direct photolysis and UV-AOP.22,23,35
Despite the promise of UV-ARP, implementation of these technologies at the large scale has not been realized. Given the high level of research activity of UV-ARP for the eaq–-based transformation of contaminants over the past decade, there is a critical need to understand the limitation of these technologies from both chemical and engineering perspectives. While recent publications have reviewed various aspects of UV-ARP, like specific eaq sensitizer systems (e.g., UV-sulfite3,36,37) or applicability toward certain contaminant classes (e.g., PFAS2,38), a comprehensive review of UV-ARP technologies is lacking. The objective of this Review is therefore to critically evaluate the factors influencing the effectiveness of UV-ARP for the degradation of contaminants in water. We summarize and review the UV-ARP literature for treatment of some important inorganic and organic contaminants, identify the knowledge gaps and potential limitations for these technologies, and suggest paths forward for research in this field. This is accomplished by presenting the photochemical kinetics of eaq– production and scavenging in UV-ARP, reviewing the sensitizers that have been employed to generate eaq in UV-ARP, examining the classes of contaminants for which UV-ARP treatment has been studied, and highlighting novel UV-ARP technologies that seek to increase the rate of target contaminant degradation. The data compilation and perspectives provided in this review will assist the UV-ARP research community in future efforts to optimize UV-ARP systems, model the eaq–-based transformation kinetics, and develop novel UV-ARP systems.
2. Production and Scavenging of Hydrated Electrons in UV-ARP
In UV-ARP, target contaminants are degraded through direct photolysis or through reaction with photochemically generated eaq–9,39 (Figure 2). Considering the latter, indirect, photolysis pathway, the rate of target contaminant transformation in UV-ARP systems is a function of both the intrinsic reactivity of the contaminant with eaq (its bimolecular reaction rate constant) and the concentration of eaq–. The bimolecular rate constant for a target contaminant with eaq is a constant value under specific solution conditions (e.g., temperature, ionic strength). Thus, strategies to optimize UV-ARP must aim to increase the exposure of the contaminant to eaq–, by increasing either the irradiation time or the hydrated electron concentration, [eaq]. Increasing [eaq–] can be accomplished through multiple means, such as using higher wattage lamps, increasing sensitizer concentrations, and minimizing the rate of interfering eaq scavenging reactions. Understanding the factors that influence [eaq–] is therefore an important consideration in UV-ARP studies. The sections below address these factors by using established photochemical kinetic models to describe the rate of eaq-based contaminant transformation, the chemistry of known eaq– sensitizers, and the scavenging kinetics of eaq in water. We end this section by focusing on knowledge gaps in this area, primarily the lack of a standardized measurement protocol for quantifying [eaq–] in UV-ARP.
Figure 2.

Direct and indirect (i.e., sensitized) photochemical transformation of a target contaminant in UV-ARP. The sensitizer charge (n) increases by +1 upon photoionization.
2.1. Transformation of a Target Contaminant by Hydrated Electrons
The rate law for the loss of a target contaminant (TC) in UV-ARP, which can be due both to direct photolysis and eaq–- sensitized reactions, is given by
| 2.1 |
where kd′ is the pseudo-first-order rate constant of TC by direct photolysis (s–1), kTC,eaq– is the TC-eaq bimolecular reaction rate constant (M–1 s–1), [TC] is the concentration (M) of TC remaining, and [eaq–] is the concentration (M) of hydrated electron.40Equation 2.1 can be simplified by assuming that [eaq] remains at a steady-state throughout the treatment process, resulting in the expression
| 2.2 |
where kTC′ is a first-order rate constant (kTC = kTC,eaq–[eaq–]ss). Equations 2.1 and 2.2 forms the basis of all photochemical kinetic models for describing the time dependence of [TC]. Equation 2.1 can be further specified by including equations that describe the formation and scavenging of eaq. For example, if [eaq–] cannot be approximated as steady-state, then the time-dependent [eaq] can be described by
| 2.3 |
where Rf,teaq– represents the formation rate of eaq (M s–1) and kS,t′ represents the total eaq scavenging capacity, all as a function of time t.
In UV-ARP, eaq– is typically formed by illumination of a chemical sensitizer by UV lamps. Under monochromatic conditions (e.g., a low-pressure Hg lamp), eq 2.3 can be expanded to eq 2.4
| 2.4 |
where Φeaq– is the eaq– quantum yield of the sensitizer (sens), I0 is the photon irradiance (mmol photons cm–2 s–1), εsens is the molar absorption coefficient (M–1 cm–1) of the sensitizer, [sens]t is the sensitizer’s concentration at time t (M), αt is the absorption of the background water matrix (cm–1) at time t, kSi,eaq– is the bimolecular reaction rate constant for transformation of scavenger Si (M–1 s–1), [Si]t is the scavenger concentration (M) at time t, and l is the path length (cm).41 The units in eq 2.4 are consistent because the conversion from mmol to mol and cm3 to L cancel each other.41
Integrating eq 2.1 results in a complete expression (eq 2.5), which can be employed to model the change in [TC] as a function of time.
| 2.5 |
In eq 2.5, ∫0t[eaq]t dt is called the eaq– exposure and incorporates both Rf,t and kS,t′ for any given background water matrix conditions. While the above expressions are time-based, it is also possible to present target contaminant transformation kinetics using fluence-based rate constants, which has been discussed extensively by Bolton et al.42
Direct photolysis plays a minor but non-negligible role in the transformation of some target contaminants. The contribution of direct photolysis to contaminant transformation in UV-ARP can be calculated in terms of the direct photolysis rate constant, kd′
| 2.6 |
where ΦTC is the reaction quantum yield for direct photolysis of the TC, εTC is the molar absorption coefficient of the TC (M–1 cm–1), and other terms ([TC], I0, l, and αt) are as defined previously.41 A key point is that there must be overlap between the spectra of the light source and TC for direct photolysis to occur (Grotthus–Draper law43). Contaminants containing chromophores that absorb in the UVC (e.g., nitrate, nitrite, and halogenated aromatic compounds) undergo direct photolysis.44−46 Given the typical minor role of direct photolysis in UV-ARP, the factors influencing this pathway are not discussed extensively in this Review. Section 3 will specify contaminant classes in which direct photolysis plays a role.
2.2. Kinetics of Hydrated Electron Scavenging Reactions
Background water matrix components that react with eaq– decrease its availability for reaction with the target contaminant (TC) according to the following competition
| 2.7 |
| 2.8 |
where S represents the sum of eaq– scavengers other than the TC. The expected impact of eaq scavengers on target contaminant transformation can be estimated by determining the first-order scavenging capacity (kS,t′, s–1), which is defined as the sum product of bimolecular eaq scavenging rate constants (kSi,eaq–) and [Si]t (eq 2.9)
| 2.9 |
Bimolecular rate constants for eaq– reactions depend on temperature and aqueous ionic strength according to the Arrhenius (eq 2.10) and Brønsted–Bjerrum equations (eq 2.11), respectively.8
| 2.10 |
| 2.11 |
In these equations, k2 represents the bimolecular rate constant at two different temperatures (T1 or T2, units of K) or ionic strengths (I = 0 or I, units of M), Ea is the activation energy (kJ/mol), R is the gas constant, and ZAZB is the product of the charges of the reactants. Because eaq– bears a negative charge, its reactivity with anionic species will be enhanced at higher ionic strengths because the increased shielding of like-charged reactants decreases the distance over which Coulombic forces repel the ions. Many of the contaminant classes highlighted in this Review are anionic at pH ranges used for UV-ARP, so increasing ionic strength will increase the eaq bimolecular rate constant.
Figure 3 shows the relative change in the eaq– bimolecular rate constant as a function of temperature (10–40 °C) and ionic strength (0.001–1 M), which covers the range of values that could be anticipated in waters treated by UV-ARP. An activation energy of 15 kJ/mol and ZAZB of 1 were assumed for these calculations. The bimolecular rate constant is more sensitive to temperature than ionic strength, with much higher ionic strengths (hundreds of mol/L) needed to achieve the same impact on the rate constant as a 10–40 °C temperature increase. Treatment processes that concentrate contaminants (e.g., by filtration or ion exchange) prior to UV-ARP readily achieve high ionic strengths. Thus, bimolecular rate constants will need to be modified accordingly (eq 2.11) when applying quantitative kinetic models (e.g., eq 2.5) in these high ionic strength systems.
Figure 3.
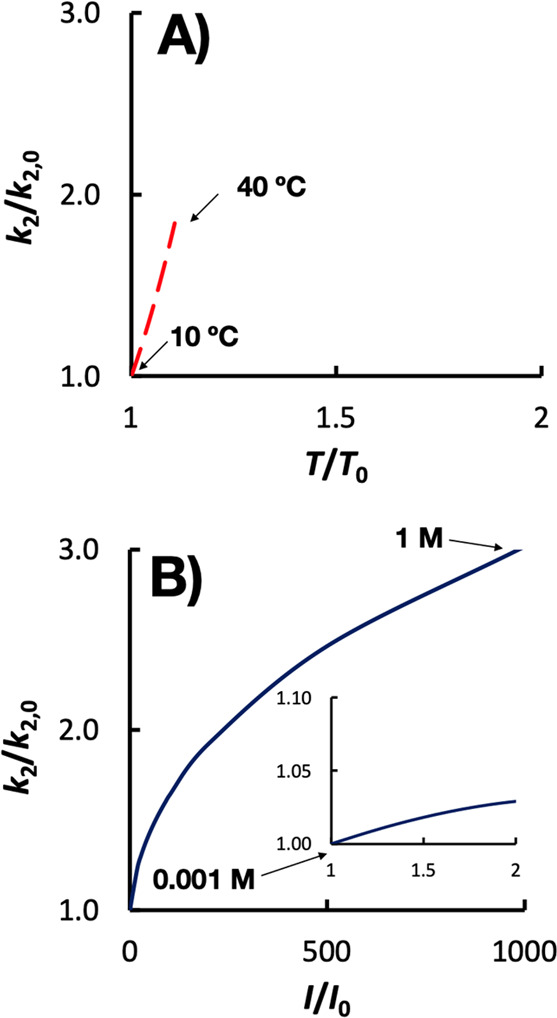
Relative change in bimolecular rate constant (k2) as a function of the relative change in A) temperature (T) or B) ionic strength (I). Note the difference in scale of the x axis. The temperature dependence calculation assumes an activation energy of 15 kJ/mol, which is typical for radical reactions. The ionic strength dependence calculation assumes that ZAZB = 1 as this would cause an increase in k2 with increasing ionic strength. The inset in (B) has the same axes labels as the outer figure.
2.2.1. Hydrated Electron Scavenging Reactions in Real-World Waters
In UV-ARP systems, there are many eaq– scavengers, all with varying concentrations and reactivities toward eaq. Scavengers can arise from background water matrix components (H+, O2, NO3–, NO2–, and DOM) and even the eaq– sensitizer itself (see section 2.3). A list of eaq bimolecular rate constants used extensively in this Review is provided in Table 1. This list does not describe all eaq– reactions or those initiated by eaq that could be occurring in UV-ARP. We refer readers to the Notre Dame Radiation Laboratory/National Institute of Standards and Technology solution kinetics database (https://kinetics.nist.gov/solution/) as a source of bimolecular rate constants for eaq– and other radical species.
Table 1. Bimolecular Rate Constants for Reaction of eaq– with Species Discussed in This Reviewa.

Values obtained from the NDRL/NIST solution kinetics database (https://kinetics.nist.gov/solution/) unless otherwise noted. The reaction products are shown only if present in the NDRL/NIST database. See Table 4 for tabulated eaq– bimolecular rate constants for PFAS. The Supporting Information contains a larger list of eaq bimolecular rate constants for aliphatic and aromatic compounds.
From, eq 2.4, we can see that [eaq–] is inversely proportional to kS,t. In principle, it is possible to calculate kS,t′ a priori provided the concentrations and rate constants for all important eaq scavengers are known. Figure 4 illustrates this fact by showing the scavenging capacity for selected water matrix components as a function of their concentration (eq 2.9) calculated using the values in Table 1. While bimolecular eaq– rate constants are widely available for important hydrated electron scavengers,8 conducting a full water chemistry analysis to determine scavengers' concentrations is not always practical. In addition, Rosenfeldt and Linden demonstrated that calculated •OH scavenging capacities in UV-AOP differed substantially from values measured with p-chlorobenzoic acid,40 suggesting that the same may be true for UV-ARP.
Figure 4.

Scavenging capacities of (kS,t=0′) as a function of species concentration for some eaq scavengers in UV-ARP. Bimolecular rate constants used for these calculations are shown in Table 1. A value of 1.2 × 108 MC–1 s–1 (∼104 L mgC–1 s–1) was used for the DOM-eaq– reaction based on the DOM-•OH rate constant for SRFA.50 Filled circles represent the scavenging capacity of each species under the following conditions: [H+] = 10–7 M, [O2] = 250 × 10–6 M (8 mg/L), [NO3–] = 1.61 × 10–5 M (1 mg/L), [NO2–] = 2.17 × 10–5 M (1 mg/L), [DOM] = 1.66 × 10–4 molC L–1 (2 mgC/L), and [HCO3–] = 10–3 M (61 mg/L).
These scavenging capacities allow for comparison of the impact of different water matrix components on [eaq–]. For example, while the fastest bimolecular rate constant is for eaq reaction with H+, the typical [H+] (pH > 9) is optimized to minimize this pathway, making the eaq– scavenging capacity of H+ a small contribution to kS,t in most UV-ARP.48 Dissolved oxygen (∼250 μM in aerated water) is a significant scavenger of eaq–. In UV-ARP treatment, removal of dissolved oxygen is accomplished through mechanical deaeration (sparging with N2 or Ar) or reactive quenching by sulfite,32,51 which also functions as an eaq sensitizer. Figure 4 demonstrates that dissolved oxygen will exert a significant scavenging capacity (kS,t=0′ = 4.75 × 106 s–1 for [O2] = 250 μM) if not removed prior to treatment, which is an important limitation of UV-ARP (see section 2.2.2). Nitrate is a potent eaq scavenger, and the concentration of this species in natural waters (∼1 mg/L for 50% of natural waters in the United States52) will exert a significant scavenging capacity (kS,t=0′ = 2.02 × 105 s–1), thus limiting the effectiveness of UV-ARP. This idea is illustrated by Figure 5 which shows that UV/sulfite treatment of perfluorooctanoic acid (PFOA) in a contaminated groundwater is limited until nearly all nitrate and nitrite (formed during nitrate photoreduction) have been degraded.
Figure 5.

Time courses of nitrate, nitrite, and perfluorooctanoic acid (PFOA) during UV/sulfite treatment of a contaminated groundwater. Line at ∼200 min illustrates the point at which eaq– scavenging by the water matrix becomes low enough for PFOA-eaq reactions to occur. Experimental conditions: 18 W low-pressure Hg lamp, pH0 = 9.5, 10 mM sulfite, 30 min of sparging with industrial grade N2, ACE glass water-jacketed immersion well reactors. PFOA concentration was measured according to previously described literature methods.32
Another probable eaq– scavenger is dissolved organic matter (DOM). Despite the ubiquity of DOM in waste streams for UV-ARP treatment, the reactivity between eaq and DOM remains unexplored. DOM contains several oxygenated functional groups,53 which, as individual chemical moieties, are known to have high reactivity with eaq–. These moieties include carbonyl-containing compounds (e.g., aldehydes, ketones, and quinones), alkoxyphenols, and amino acids.8,54 The estimated value of 104 L mgC–1 s–1 (∼108 L molC–1 s–1) shown in Table 1 is based on the bimolecular rate constant for the reaction between DOM isolates and the hydroxyl radical reported by Westerhoff et al.50
2.2.2. Hydrated Electron Scavenging by Dissolved Oxygen in Laboratory Studies
UV-ARP studies are often performed in buffered, laboratory grade water that has been sparged with an inert gas like nitrogen to remove dissolved O2. However, prior research shows that a residual [O2] of approximately 0.2 mg/L (∼6 μM) remains even after sparging with nitrogen (25 mL/s) for 1 h.55 An [O2] of 6 μM in deaerated solutions will still exhibit a high eaq– scavenging capacity (kS,t=0 = 1.2 × 105 s–1), which will impact the initial [eaq–] and thereby the rate of target contaminant transformation. Therefore, variations in [O2] in laboratory studies with different sparging methods may substantially impact the observed kinetics for target contaminant transformation, making it difficult to compare UV-ARP data for different sensitizer systems and experimental setups. In addition, the use of different eaq sensitizers may impact the residual [O2] since reducing species like sulfite can remove O2.32,51
The impact of residual [O2] will also depend on the solution conditions and target contaminant. Residual [O2] will have the largest impact on the degradation of contaminants that have an eaq– bimolecular rate constant comparable to O2 and thus react on similar time scales. Examples include nitrate, nitrite, and most halogenated organic compounds. On the other hand, target contaminants with slower eaq bimolecular rate constants will be less impacted because the residual [O2] will be reduced by eaq– at early reaction times. For example, Bentel et al. observed similar degradation kinetics for PFOA in the UV/sulfite system regardless of whether the solution was initially sparged with nitrogen.30 This concept is further illustrated in Figure 6, which shows the calculated concentration profiles for the transformation of several compounds in a typical UV-ARP. Fast-reacting compounds like oxygen, nitrate, and nitrite are completely removed within a few minutes, while perfluorooctanoic acid (PFOA) and perfluoroctanesulfonate (PFOS), two representative PFAS, are degraded at much longer time scales.
Figure 6.
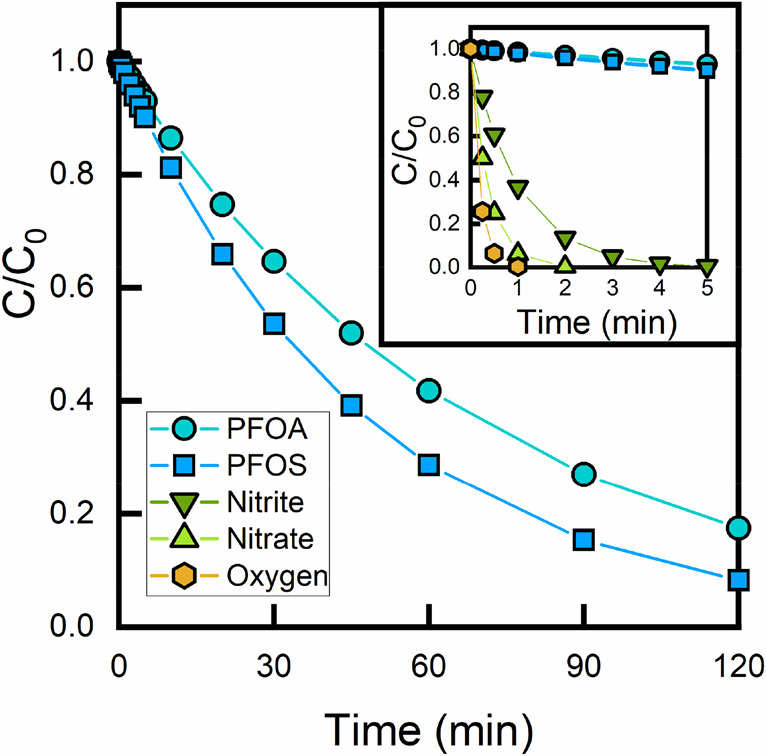
Calculated concentration profiles of typical UV-ARP target contaminants based on eq 2.5, assuming [eaq–] = 4.76 × 10–12 M, no direct photolysis for nitrate and nitrite, and bimolecular rate constants (M–1 s–1) of kPFOA,eaq– = 5.1 × 107, kPFOS,eaq– = 7.3 × 107, kNO3–,eaq– = 9.7 × 109 M–1 s–1, kNO2–,eaq– = 3.5 × 109 M–1 s–1, kO2,eaq– = 1.9 × 1010 M–1 s–1.
2.3. Chemical Sensitizers for Hydrated Electron Production
It is possible to generate eaq– from the photolysis of many organic and inorganic compounds if the appropriate illumination wavelengths are used.56 However, only a subset of the chemicals capable of producing eaq under UV irradiation have been explored in UV-ARP. Absorption of photons by sensitizer (sens) molecules will result in a portion of them ejecting an electron (eq 2.12).56
| 2.12 |
The ejected e– initially forms an ion contact pair with sensn+1, and only a fraction of e– eventually escapes the contact pair to form hydrated electrons, eaq–. The overall efficiency of this process can be described by the quantum yield of eaq formation (Φeaq–), which represents the rate of eaq– formation (Rf, M s–1) divided by the rate of light absorption by the sensitizer (photons L–1 s–1). Employing sensitizers with higher Φeaq– therefore represents one possible approach to enhancing [eaq–] in UV-ARP. The UV photochemistry of many of these eaq sensitizers was recently reviewed by Cui et al.2
Table 2 provides a summary of eaq– sensitizers that have been examined in the literature, their molar absorption coefficient (ϵ) at selected wavelengths, Φeaq– values, and the method of Φeaq– determination. There is some variation in ϵ and Φeaq– and some sensitizers for which no data are available. Reported Φeaq– values for sulfite vary by less than a factor of 2. The Φeaq– value of 0.116 at 254 nm determined by steady-state photolysis57 is in good agreement with the recent value of 0.108 at 248 nm measured by transient absorption spectroscopy.48 Given the agreement between these two values, we expect that a Φeaq– of ≈0.1 for sulfite is reliable. Uncertainty in Φeaq– values impact the ability of kinetic models to accurately predict the change in [TC] with time according to eq 2.5.
Table 2. Summary of Hydrated Electron (eaq–) Sensitizers Used in UV-ARP, Their Quantum Yields (Φeaq–), and the Method of Φeaq– Determination if Available62,63,65−67.

Referenced study refers to application of the sensitizer in UV-ARP systems.
ϵ254 estimated using Web Plot Digitizer (https://apps.automeris.io/wpd/).
Measured for this Review.
Value is for indole (not indoleacetic acid) in ethanol.
Value is for indole (not indoleacetic acid) at 30 °C.
Measurements are conducted in deaerated solutions.
To help address the uncertainty in ϵ254 values, we have measured molar absorption coefficients for 3-indoleacetic acid (IAA), nitrilotriacetic acid (NTA), iodide, and sulfite (Figure 7), four of the eaq– sensitizers employed most frequently in UV-ARP studies. Our results agree well with most with literature data, where available. For sulfite, ϵ254 values agree within ∼15%, and we suggest that the value measured by Tenorio et al. (17.8 M–1cm–1) is reliable.34 Our measured ϵ254 for iodide (172 M–1cm–1) agrees well with one literature report (162 M–1cm–1)61 but is substantially different from another (220 M–1cm–1).60
Figure 7.
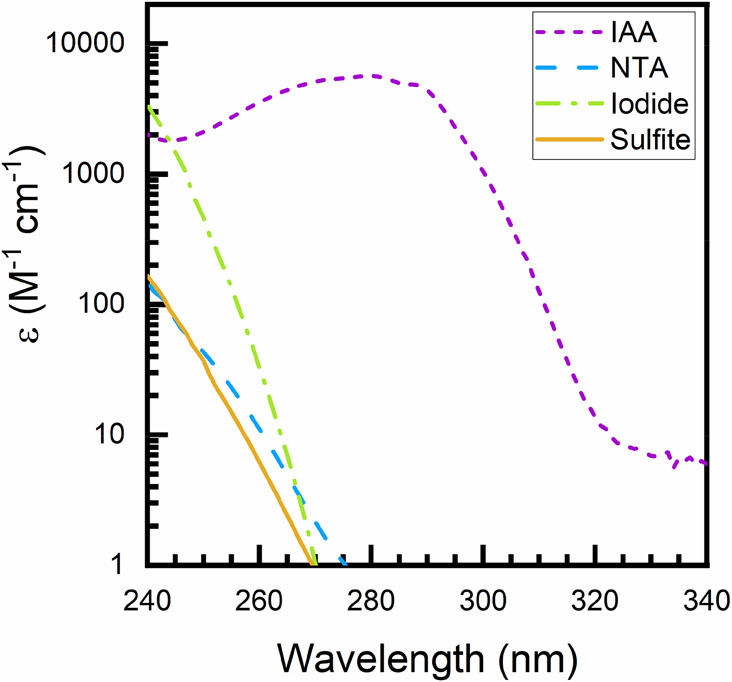
Absorption spectra for four commonly used hydrated electron sensitizers in UV-ARP studies, including 3-indoleacetic acid (IAA), nitrilotriacetic acid (NTA), iodide, and sulfite. Molar absorption coefficients at 254 nm (ϵ254) are 17.6 M–1cm–1 (sulfite), 26.3 M–1cm–1 (NTA), 172 M–1cm–1 (iodide), and 2576 M–1cm–1 (IAA) and are listed in Table 2. Experimental conditions: All solutions besides iodide (potassium iodide) were prepared in pH 10 borate buffer (10 mM), whereas potassium iodide was prepared in unbuffered laboratory grade water (18.2 MΩ·cm). Sulfite solutions were prepared in N2 sparged (30 min) water. Molar absorption coefficients are derived from a linear fit of absorbance to molar concentration with an absorbance (optical density) less than 2.0 measured with a Cary-100 Bio dual beam spectrophotometer in a 1 cm path length quartz cuvette.
2.3.1. Sulfite
Sulfite is one of the most studied eaq– sensitizers in UV-ARP systems. Sulfite absorbs in the UV region of the spectrum (ε254 ≈ 18 M–1cm–1)57 and its Φeaq– has been measured in several previous studies48,57,68
| 2.13 |
In the absence of oxygen, dithionate (S2O62–) and sulfate are the major end products of this reaction, while in oxygenated solutions only sulfate is observed.68
eaq– generation in the UV/sulfite ARP is much less efficient at pH values near and below the pKa of HSO3– (7.19) because photolysis of HSO3– has a low Φeaq–.69 In addition, HSO3– is a scavenger of eaq.48
| 2.14 |
Moreover, sulfite itself reacts with eaq–. Although the SO32–-eaq reaction is slow (the upper limit for the rate constant is 1.6 × 106 M–1 s–1),8 the high sulfite concentrations typically employed in UV-ARP (ca. 10 mM) could mean that this reaction pathway is significant. For example, Figure 8 shows the calculated eaq– formation rate (Rf), scavenging capacity (kS,t=0′), and [eaq] expected in an aqueous solution containing monochloroacetate (MCAA at a fixed concentration of 20 μM) and sulfite at various concentrations (between 0 and 20 mM). The eaq– scavenging capacity exerted by sulfite increases linearly with increasing sulfite concentration (eq 2.9). However, Rf increases asymptotically due to the (1–10–(εsens[sens]t + αt)l) term in eq 2.4. Overall, this results in an asymptotic increase in [eaq–] with increasing sulfite concentration. Although there is not a consensus on an optimal sulfite concentration in UV-ARP, previous literature demonstrates diminishing increases in target contaminant transformation kinetics above ∼10 mM sulfite.13,23,34 This is consistent with the calculations presented in Figure 8.
Figure 8.

Plot of calculated eaq– formation rate (Rf,t=0), scavenging capacity (kS,t=0′), and [eaq]t=0 (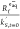 ) as a function of initial sulfite concentration.
It is assumed that sulfite functions as the sole light absorber while
both sulfite and another target compound (MCAA) act as eaq– scavengers.
Conditions: I0 = 1.13 × 10–8 Es cm–2 s–1, Φeaq– =
0.116 mol Es–1, εsens = 18.14 M–1 cm–1, l = 2.85
cm, ksulfite,eaq– = 1.3 × 106 M–1 s–1, kMCAA,eaq– =
1.0 × 109 M–1 s–1, [MCAA]0 = 20 μM, pH0 = 9.5.
) as a function of initial sulfite concentration.
It is assumed that sulfite functions as the sole light absorber while
both sulfite and another target compound (MCAA) act as eaq– scavengers.
Conditions: I0 = 1.13 × 10–8 Es cm–2 s–1, Φeaq– =
0.116 mol Es–1, εsens = 18.14 M–1 cm–1, l = 2.85
cm, ksulfite,eaq– = 1.3 × 106 M–1 s–1, kMCAA,eaq– =
1.0 × 109 M–1 s–1, [MCAA]0 = 20 μM, pH0 = 9.5.
2.3.2. Iodide
Iodide photolysis produces eaq– with a Φeaq– of ≈0.2 at 248 nm.59 This is approximately a factor of 2 higher than the Φeaq– for sulfite. Iodide also has a higher molar absorption coefficient at 254 nm than sulfite (Table 2). However, as will be discussed further below, several UV-ARP studies have demonstrated that target contaminant transformation rates are faster in the UV/sulfite system compared to the UV/iodide system. This indicates that the [eaq] is higher in the UV/sulfite system, despite iodide’s higher Φeaq– and ϵ254 values. The lower [eaq–] in the UV/iodide system is likely due to eaq scavenging reactions70,71 of I-containing species, like iodine and triiodide, which are formed during photooxidation of iodide.
| 2.15 |
| 2.16 |
| 2.17 |
2.3.3. Nitrilotriacetic Acid
Nitrilotriacetic acid (NTA) produces eaq– at UV irradiation wavelengths, but with an unknown quantum yield. It has been proposed that the mechanism of eaq formation from nitrilotriacetic acid photolysis does not involve photoionization of nitrilotriacetic acid (eq 2.1).56 Instead, electronically excited nitrilotriacetic acid is thought to sensitize the photooxidation of water, concomitantly producing eaq–, •OH, and •H.56,64
| 2.18 |
| 2.19 |
Photoproduced •OH can react with the C–H bonds in nitrilotriacetic acid through H atom abstraction (eq 2.20),64
| 2.20 |
which reduces the •OH-eaq– recombination rate (k = 2.8 × 1010 M–1 s–1)72 thereby allowing more eaq to react with target contaminants.
Historically, the mechanism proposed for nitrilotriacetic acid is an alternative to the photooxidation mechanism previously described (eq 2.12) and may in fact be operative for other eaq– sensitizers. The main point is that both mechanisms (photoionization and NTA-sensitized H2O ionization) yield eaq.
2.3.4. Indole derivatives
It has been suggested that indoles produce eaq– through the photoionization mechanism described in eq 2.12 with Φeaq– of ≈0.1 (Table 2). Given the high molar absorption coefficient of 3-indoleacetic acid at 254 nm (≈ 2600 M–1 cm–1) and Φeaq– value comparable to that of sulfite, indoles should have high rates of eaq formation in UV-ARP systems. However, previous studies of UV/indoleacetic acid treatment of PFOA show no difference in degradation rates between direct photolysis controls and in the presence of 1 mM 3-indoleacetic acid.25 This observation could indicate a high rate of scavenging between eaq– and indole itself (k = 2.6 × 108 M–1 s–1)49 or indole photoproducts that limits the [eaq] available for reaction with PFOA. As discussed later (see Section 3.2.3), indoleacetic acid does effectively defluorinate PFOA in a heterogeneous UV-ARP involving an organomontmorillonite clay surface.25,26
2.3.5. Vacuum UV
An alternative to the use of eaq– sensitizers is vacuum UV photons (VUV, 100 nm < λ < 200 nm), which are capable of directly ionizing water molecules to form eaq, H•, and •OH.73 For example, studies have shown that photolysis of aqueous PFAS with Hg lamps not manufactured to block VUV degrades these contaminants in the absence of any added eaq– sensitizer,24,74 implying that photoionization of water by the small amount of VUV emitted from these lamps is occurring. Practically, VUV is challenging to implement, as VUV photons are absorbed within the first ∼5 mm of water.73
2.3.6. Factors Influencing the Choice of Chemical Sensitizer
Several studies have attempted to optimize experimental conditions to maximize the rate of eaq– generation from these sensitizers. For example, Yu et al. demonstrated that reductive dechlorination of monochloroacetate in the UV/iodide system is enhanced by adding sulfite.60 In this case, it is believed that sulfite reduces reactive iodine species (e.g., I2, I3–) back to iodide, whereas in the absence of sulfite, these reactive iodine species significantly scavenge eaq (eqs 2.15–2.17). In the Yu et al. study, monochloroacetate dechlorination was fastest in the UV/sulfite/iodide system, when compared to UV/iodide and UV/sulfite.60 Similarly, Sun et al. observed faster degradation of perfluorooctanesulfonate by UV/nitrilotriacetic acid when compared to UV/sulfite.64 In this case, nitrilotriacetic acid was hypothesized to concomitantly sensitize the formation of eaq– and scavenge photoproduced •OH (eq 2.18–2.20).
Another important aspect of selecting an eaq– sensitizer for UV-ARP is the sensitizer’s expected degradation products. For example, iodide photolysis generates iodine and triiodide,75 which absorb light in the visible region of the spectrum. These iodine species would need to be removed (e.g., through reduction to iodide) after the UV-ARP. Photolysis of nitrilotriacetic acid produces a foul odor characteristic of amine compounds.32 Conversely, sulfite photooxidation produces inert sulfate. Although high concentrations of sulfate in drinking water should be avoided,76 this anion is relatively benign compared to iodide and nitrilotriacetic acid photolysis products. Another benefit to using sulfite is that it can be regenerated following its oxidation to SO3•-. This is inferred from the fact that sulfite persists in UV-ARP systems remain high even after many hours of light exposure.34 Conversely, when used alone, sensitizers like iodide and nitrilotriacetic acid are degraded quickly.64
Collectively, sensitizer concentration, eaq– quantum yield, and molar absorption coefficient at the actinic wavelength employed all impact the rate of eaq formation and are important for sensitizer selection in UV-ARP. Byproducts of eaq– sensitizer photolysis are also an important consideration. Taken as a whole, using sulfite as a sensitizer in UV-ARP appears to have the optimal benefits for treatment applications.
2.4. Knowledge Gaps
Recent UV-ARP studies have employed an array of eaq– sensitizers, varying intensity UV light sources,77,78 and optimized solution conditions79 in an attempt to enhance degradation rates of target contaminants. However, progress in UV-ARP research has been limited by the lack of a systematic framework for evaluating treatment performance between these individual studies and the effect of process variables such as the UV-ARP irradiation source, sensitizer identity and concentration, and water quality. Overall, there is a need for the development of quantitative tools to allow comparison between UV-ARP studies, such as methods for quantifying [eaq] and modeling target contaminant degradation kinetics.
2.4.1. Probe Compounds for Studying UV-ARP
There is a need to apply eaq– probe compounds to quantify photochemical parameters related to UV-ARP systems. Although several eaq probes have been used in the broader chemical literature (vide infra), and some have been applied in UV-ARP studies, they have not been used extensively to quantify eaq– formation rates (Reaq–), eaq– concentrations ([eaq]), or eaq– scavenging capacities (kS,t) in UV-ARP. This contrasts with research on UV-AOP, which has made extensive use of p-chlorobenzoic acid as an •OH probe,40,80,81 allowing for a comparisons of treatment performance.82−85 When measured using a validated probe compound, Rfeaq–, [eaq], and kS,t′ provide metrics for assessing the effects of different water matrices, solution conditions, and process variables on UV-ARP treatment performance.
eaq– probe compounds used in the literature include chloroacetate (monitoring the [parent] by ion chromatography or gas chromatography),57 chloroethanol (monitoring the product [Cl–] by ion chromatography or an ion selective electrode),86 nitrous oxide (monitoring the [N2] by gas chromatography), and 3-amino-2,2,5,5-tetramethyl-1-pyrrolidinyloxy (monitoring the derivatized reaction product).87
Chloroethanol86 and the nitroxide probe87 were used for characterizing eaq– formation from chromophoric dissolved organic matter at UV–B and UV-A wavelengths. However, to be a successful eaq probe in UV-ARP, the nitroxide would need to be stable under UV–C irradiation (e.g., at 254 nm, where the nitroxide is highly absorbing). Another issue with the nitroxide probe is that it requires millimolar concentrations of dimethyl sulfoxide, which absorbs at 254 nm. For these reasons it is unlikely that the nitroxide probe is suitable for quantifying [eaq–] in UV-ARP. Because chloroethanol relies on measurement of [Cl–], which has a high background concentration in real-world waters, it is also unsuitable for quantifying [eaq] in UV-ARP. However, chloroethanol could be employed to measure fundamental photochemical parameters like Φeaq– in laboratory grade water containing low background [Cl–].
A few studies have demonstrated success using chloroacetate as an eaq– probe.57,60,88,89 Chloroacetate can be quantified via ion chromatography in aqueous aliquots withdrawn from UV-ARP experiments down to ∼2 μM.57 Lower concentrations can be quantified via derivatization and gas chromatography.90 The availability of these analytical methods has important implications for the chosen probe compound concentration. If ion chromatography is the only method available, this limits the initial probe concentration to the micromolar range, where it can still exert a significant scavenging capacity in the system.
When employing chloroacetate as an eaq– probe, it is also possible to quantify chloride as an eaq reduction product, but this would again be impractical in natural waters with high background [Cl–]. Alternatively, the use of fluoroacetate as an eaq– probe would enable detection of fluoride since the fluoride concentration in natural waters is typically orders of magnitude lower than that of chloride.52 Fluoroacetate is much less reactive with eaq (k ≤ 1.2 × 106 M–1 s–191) than chloroacetate (k = 1.0 × 109 M–1 s–1). Thus, higher concentrations of fluoroacetate could be employed (aiding with detection limit issues) with minimal impacts to the natural eaq– scavenging capacity of the background water matrix. Use of fluoroacetate as an eaq would require accurate measurement of its eaq– bimolecular rate constant (the current value is an upper limit) and the fluoride reaction yield (see Section 3.2.2).
While development of eaq– probe compounds is needed, several challenges unique to UV-ARP have made this difficult. First, many UV-ARP studies are conducted in buffered solutions of laboratory-grade water where there is minimal background scavenging of eaq. This makes the system sensitive to potential eaq–-reactive impurities like trace levels of O2 not removed during prior deaeration.92 Another potential challenge for a standard eaq probe compound is the possibility that reactive species besides eaq– generated in UV-ARP systems could cause probe transformation. For example, SO3•– formed upon sulfite photooxidation has been reported to act as both an oxidant and reductant (see also section 3.3.2).13 If SO3•– were to reduce the employed probe compound like eaq, then the probe would not be selective. One way to overcome this challenge is to verify the selectivity of the probe using an eaq– quencher. Compounds like nitrous oxide, nitrate, and nitrite have been widely employed for this purpose in prior studies.8,57,60 For example, N2O reacts quickly with eaq (k = 9.1 × 109 M–1 s–1) but not with SO3•– (k < 1 × 106 M–1 s–1) (Table 1 and https://kinetics.nist.gov/solution/).
2.4.2. Computer Kinetic Models
An alternative approach for gaining a quantitative description of UV-ARP chemistry is by modeling the kinetics of the system through computer numerical integration, which has received considerable attention in UV-AOP systems.93,94 The process involves collecting all the known reactions occurring in a system and their rate constants and inputting this information into a computer program. Free (e.g., Kinetcus95) and commercial programs (e.g., FACSIMILE) are available for building and running these kinetic models. If all parameters are known, running such programs to produce time profiles of target contaminants, intermediates, and reactive radicals is straightforward. Alternatively, if there are some unknown parameters (e.g., bimolecular rate constants, quantum yields) the kinetic model can be fit to a given data set to estimate the unknown parameters.10,96 Despite the abundance of bimolecular eaq– rate constants available,8 examples of such numerical computer models being applied in UV-ARP are limited.60
One potential limitation of computer kinetics models is that they may not be readily transferable across different UV-ARP. They may also fail to adequately describe experimental data in complex reaction systems where it is simply not possible to know all the reactions.97 For these reasons, computer kinetics models may be most beneficial for estimating unknown parameters for a specific UV-ARP and understanding the impact of design and process variables on treatment performance.
2.4.3. The Role of Dissolved Organic Matter
While some prior research has evaluated the impact of dissolved organic matter (DOM) on target contaminant transformation in UV-ARP, more work is needed to fully understand the role of DOM in eaq–-based treatment systems. While it is expected that DOM is an important eaq scavenger, specific bimolecular rate constants are not yet established. Dissolved oxygen, nitrate, and nitrite, which all exhibit high reactivity with eaq–, will likely dominate the eaq scavenging at low eaq– exposures (Figures 5 and 6). However, given the variety of structural moieties present, DOM may exert a significant eaq scavenging capacity long into the UV-ARP process. In addition, the impact of eaq– exposure on DOM light absorption in UV-ARP systems needs to be further evaluated, although prior studies indicate that various reduction methods (borohydride,98 electrochemical,99 and UV/sulfite12) decreases DOM absorbance. Changes in DOM absorbance at the actininic wavelength(s) will impact the rate of light absorption by the sensitizer and therefore the rate of eaq formation in the system.
3. UV-ARP Substrate Scope
Previous research has evaluated the suitability of UV-ARP for degradation of both inorganic and organic contaminants. Target contaminants that have been considered for UV-ARP treatment typically have a high oxidation state central atom (e.g., N in NO3–) or one or more carbon–halogen bonds as part of their structure. Indeed, previous computational studies showed that bimolecular eaq– reaction rate constants were inversely proportional to the energy of the lowest unoccupied molecular orbital (ELUMO) for a large set (>100) of substituted aliphatic compounds.100,101
As part of this Review, we have compiled literature data from UV-ARP treatment studies of important inorganic and organic contaminants. Table 3 presents a summary of these UV-ARP studies separated by compound class, which includes details like lamp power and type (low-, medium-, or high-pressure Hg), eaq– sensitizer, solution conditions, and degradation rates (final % degraded and reaction time). A searchable copy of Table 3 including additional details (e.g., average UV fluence rate and sparging conditions) is included in the Supporting Information. The sections below highlight key results from this analysis for oxyanions, halogenated organics, and PFAS.
Table 3. UV-ARP Organic and Inorganice Treatment Studies Summarya.
| ref | compound class | [compound]0 (μM) | heterogeneous or homogeneous system | UV lamp | UV sensitizer | water characteristics | parent compound degradation % (time) |
|---|---|---|---|---|---|---|---|
| (102) | halogenated organic | 10 μM trichloroacetic acid | heterogeneous (TiO2) | LP-Hg 10 W | 4.0 mM formate | ultrapure water (pH 6.5; 23 °C) | 100% (150 min) |
| (45) | halogenated organic | 10 μM 4-bromophenol | homogeneous | LP-Hg 10 W | 1.0 mM sulfite | ultrapure water (pH 9.2; 20 °C) | 100% (10 min) |
| (103) | halogenated organic | 20 μM 1,2-dichlororoethane | homogeneous | LP-Hg 36 W | 1.0 mM dithionite; 1.2 mM sulfite; 0.4 mM sulfide; 4.0 mM ferrous iron | ultrapure water (pH 11, 11, 9, 9; 25 °C) | 100% (varies) |
| (104) | halogenated organic | 10 μM decabromodiphenyl ethers | homogeneous | LP-Xe 50 W and LED | none | reaction performed in methanol | 90% (60 min) |
| (105) | halogenated organic | 10 μM MCAA | homogeneous | n.r. | 1.0 mM sulfite; 1.0 g L–1 bismuth oxyiodide | ultrapure water (pH 7; 25 °C) | 100% (20 min) |
| (57) | halogenated organic | 50 μM MCAA | homogeneous | LP-Hg 10 W | 1.0 mM sulfite | ultrapure water (pH 9.2; 25 °C) | 100% (15 min) |
| (88) | halogenated organic | 50 μM MCAA | homogeneous | LP-Hg 15 W | 0.2 mM phenol | ultrapure water (pH 12; 25 °C) | 100% (6 min) |
| (60) | halogenated organic | 50 μM MCAA | homogeneous | LP-Hg 10 W | 1.0 mM sulfite; 0.1 mM iodide | ultrapure water (pH 9.2; 25 °C) | 100% (5 min) |
| (89) | halogenated organic | 50 μM MCAA | homogeneous | LP-Hg 10 W | 1.0 mM sulfite (UPW); 2.0 mM sulfite (river) | ultrapure water; Yangtze River, China (pH 9.2; 25 °C) | 100% (15 min in ultrapure water; 75 min in river water) |
| (106) | halogenated organic | 300 μM Diclofenac | homogeneous | LP-Hg (n.r. W) | 8.0 mM sulfite | ultrapure water (pH 9.2; room temp) | 98% (30 min) |
| (107) | halogenated organic | 600 μM 2,4-dichlorophenol | homogeneous | LP-Hg (n.r. W) | 8.0 mM sulfite | ultrapure water (pH 10; n.r °C) | 99.45% (40 min) |
| (39) | halogenated organic | 0.6 mg L–1 vinyl chloride | homogeneous | LP-Hg 36 W | 20 mg L–1 sodium dithionite; 12.5 mg L–1 sodium sulfite; 8.4 mg L–1 sodium hydrosulfide; 49 mg L–1 ferrous sulfate | ultrapure water (pH 3,7,10; n.r. °C) | varies |
| (108) | halogenated organic | 1 mg L–1 vinyl chloride | homogeneous | LP-Hg 36 W | 0.16 mM sulfite | ultrapure water (pH 9; 34 °C) | 100% (90 min) |
| (109) | halogenated organic | 10 mg L–1 difluoroacetic acid | homogeneous | LP-Hg 8 W | 1.0 mg L–1 iodide | ultrapure water (pH 9; 20 °C) | n.r. |
| (110) | halogenated organic | 10 mg L–1 trichloroethylene | homogeneous | LP-Hg 36 W | 10.0 mM sulfite | ultrapure water (pH 10; 20 °C) | 100% (60 min) |
| (46) | halogenated organic | 250 mg L–1 2, 4, 6-trichlorophenol | homogeneous | LP-Hg (n.r. W) | 6.2 mM sulfite | ultrapure water (pH 7; 20 °C) | 100% (80 min) |
| (111) | halogenated organic | 30 mg L–1 Acid Yellow 17 | homogeneous | LP-Hg 6 W | 1.5 mM dithionite | ultrapure water (pH 9; n.r. °C) | 90% (5 min) |
| (112) | halogenated organic | 5 mg L–1 tetrachloromethane | homogeneous | LP-Hg 14 W | 10.0 mM sulfite | ultrapure water (pH 9; n.r. °C) | 100% (5 min) |
| (113) | halogenated organic | 50 mg L–1 pentachlorophenol | homogeneous | LP-Hg 16 W | 100 mg L–1 dithionite; 100 mg L–1 phenol | ultrapure water (pH 7; n.r. °C) | 96% (180 min) |
| (114) | halogenated organic | 2 μM iopamidol | homogeneous | LP-Hg 10 W | 0.5 mM sulfite | ultrapure water (pH 7; 20 °C) | 80% (30 min) |
| (115) | organic | 21.16 μM carbamazepine | homogeneous | LP-Hg (n.r. W) | 1.6 mM sulfite | ultrapure water (pH 8.4; room temp) | 100% (10 min) |
| (116) | organic | 30 μM dinitrotoluene | homogeneous | LP-Hg 10 W | 0.05 mM toluene | ultrapure water (pH 7.5; 25 °C) | 100% (20 min) |
| (20) | oxidized transition metal | 5 mg L–1 Cr(VI) | homogeneous | MP-Hg 300 W | 5.0 mM sulfite | ultrapure water (pH 5–10.5; 20 °C) | 100% (20 min with pH 5) |
| (21) | oxidized nonmetal | 10 mg L–1 Se(VI) | heterogeneous (Fe(III) coagulation) | MP-Hg 300 W | 5.0 mM sulfite | ultrapure water (pH 9.2; 20 °C) (HA DOM spike) | 99% (20 min without HA); 72% (20 min with HA) |
| (19) | oxyanion | 100 μM bromate | heterogeneous (Fe(II)–Ti(IV) LDH Nanoparticles) | LP-Hg 15 W | 0.5 g L–1 Fe(II)–Ti(IV) | tap water (pH 7; 25 °C) | 90% (1 h) |
| (16) | oxyanion | 4 μM bromate | homogeneous | LP-Hg 8 W; MP-Hg 150 W | varying sulfite | ultrapure water (pH varies; 26 °C) | 100% (varies) |
| (14) | oxyanion | 6.6 μM bromate | heterogeneous (superparamagnetic BiOCl) | LP-Hg 10 W | 4.0 mM formate | ultrapure water (pH 7; 25 °C) | 79% (15 min) |
| (117) | oxyanion | 10 μM bromate | homogeneous | LP-Hg 10 W | 1.0 mM sulfite/0.1 mM iodide | ultrapure water (pH 9.2; 20 °C) | 100% (5 min) |
| (15) | oxyanion | 39 μM bromate | homogeneous | LP-Hg 15 W | 0.78 mM sulfite | ultrapure water (pH 9.8; n.r. °C) | 86% (4 h) |
| (18) | oxyanion | 100 μM bromate | homogeneous | LP-Hg 15 W; MP-Hg 100 W | 2.0 mM sulfur(IV) | ultrapure water; tap water (pH 9; 25 °C) | 82% (20 min in tap water); 99% (20 in ultrapure water) |
| (6) | oxyanion | 2.75 μM bromate | homogeneous | LP-Hg 36 W | 25.6 mg L–1 sulfite | ultrapure water (pH 7, n.r. °C) | 100% (125 min in LP) |
| (118) | oxyanion | 100 μM bromate | homogeneous | LP-Hg 10 W | 1.0 mM sulfite | ultrapure water (pH 7; 23 °C) (HA DOM spike) | 20% (40 min) |
| (119) | oxyanion | 160 μM nitrate | homogeneous | LP-Hg 36 W | 8.4 mM sulfite | ultrapure water (pH 9; 27 °C) | 100% (20 min) |
| (113) | oxyanion | 50 mg L–1 nitrate | homogeneous | LP-Hg 16 W | 100 mg L–1 dithionite; 100 mg L–1 phenol | ultrapure water (pH 7; n.r. °C) | 94% (180 min) |
| (11) | oxyanion | 50 mg L–1 nitrate | homogeneous | LP-Hg (n.r. W) (254 and 185 nm) | none | ultrapure water (pH 6; 25 °C) | 99% (80 min) |
| (120) | oxyanion | 2 μM nitrate | homogeneous | LP-Hg 9 W | 60 mM sulfite | ultrapure water (pH 9.2; 25 °C) | 100% (90 min) |
| (12) | oxyanion | 100 μM perchlorate | homogeneous | LP-Hg 36 W | 11.0 mM sulfite | ultrapure water (pH 11; n.r. °C) (SRHA DOM spike) | 10% (25 h, 50 mg/L SRHA) 80% (25 h, 0 mg/L SRHA) |
| (13) | oxyanion | 100 μM perchlorate | homogeneous | LP-Hg 36 W | 3.76 mM sulfite | ultrapure water (pH 11; 30 °C) | 20% (10 h) |
| (64) | PFAS | 10 μM PFOS | homogeneous | LP-Hg 14 W | 2.0 mM nitrilotriacetic acid | ultrapure water (pH 10; 30 °C) | 85.4% (10 h) |
| (121) | PFAS | 20 μM PFOA | homogeneous | LP-Hg 15 W | 0.6 mM iodide | ultrapure water (pH 9.2; 20, 25, 40 °C) | 98% (6 h), 99% (6 h), 100% (6 h) |
| (28) | PFAS | 24 μM PFOA | heterogeneous (self-assembled micelle with cationic surfactant) | HP-Hg 500 W | 1.0 mM indoleacetic acid | ultrapure water (pH 4–8; 25 °C) | 90% (2.5 h) |
| (22) | PFAS | 25 μM PFOA | homogeneous | LP-Hg 15 W | 0.3 mM iodide | ultrapure water (pH 9; room temp) | 95% (6 h) |
| (122) | PFAS | 25 μM PFOA | homogeneous | LP-Hg 15 W | 0.25 mM iodide | ultrapure water (pH 5, 7, 10; n.r. °C) | 9.1% (2 h), 74.6% (2 h), 97.9% (2 h) |
| (123) | PFAS | 25 μM PFCA | homogeneous | LP-Hg 18 W | 10.0 mM sulfite | ultrapure water (pH 12; 20 °C) | varies |
| (33) | PFAS | 25 μM PFCAs, PFSAs, fluorotelomers | homogeneous | LP- Hg 18 W | 10.0 mM sulfite | ultrapure water (pH 12; 20 °C) | varies (4 h) |
| (78) | PFAS | 32 μM PFOS | homogeneous | HP-Hg 250 W | 10.0 mM sulfite | ultrapure water (pH 9.2; 25 °C) | 98% (0.5 h) |
| (124) | PFAS | 37.2 μM PFOS | homogeneous | MP- Hg 500 W | none | ultrapure water (pH 7.4; 90 °C) | 90% (11 days) |
| (77) | PFAS | 38.7 μM PFOA | homogeneous | HP-Hg 250 W | 10.0 mM sulfite | ultrapure water (pH 9.2; 25 °C) | 100% (10 min) |
| (25) | PFAS | 10 mg L–1 PFOA; 10 mg L–1 PFOS | heterogeneous (organo-montmorillonite) | LP-Hg 36 W | 1.0 mM 3-indoleacetic acid | ultrapure water (pH 3–11; 25 °C) | 100% (5 h) |
| (1) | PFAS | 10 mg L–1 PFOS, PFOA, PFHS, PFBS, PFHA, PFBA | homogeneous | LP-Hg 8 W | 10.0 mM iodide | ultrapure water (pH 6–8; n.r. °C) | 5–30% (150 min) |
| (34) | PFAS | Varying AFFF (15 PFAS) | homogeneous | LP-Hg 18 W | 10.0 mM sulfite | ultrapure water (pH 9.5; 20 °C) | varies |
| (23) | PFAS | PFOA | homogeneous | LP-Hg 10 W | 10.0 mM sulfite | ultrapure water (pH 10.3; 25 °C) | 100% (1 h) |
| (30) | PFAS | 25 μM PFCAs | homogeneous | LP-Hg 18 W | 10.0 mM sulfite | ultrapure water (pH 9.5; 20 °C) | 100% (8–12 h) |
| (31) | PFAS | 25 μM PFECAs | homogeneous | LP-Hg 18 W | 10.0 mM sulfite | ultrapure water (pH 9.5; 20 °C) | varies |
| (125) | PFAS | 30 μM PFOA | homogeneous | LP-Hg 14 W | 0.3 mM iodide | ultrapure water (pH 10; 25 °C) (HA DOM spike) | 67.5% (1.5 h) |
| (58) | PFAS | 30 μM PFOS | homogeneous | LP-Hg 14 W | 0.3 mM iodide | ultrapure water (pH 10; 25 °C) (HA DOM spike) | 86% (1.5 h) |
| (24) | PFAS | 37.2 μM PFOS | homogeneous | LP-Hg 10 W (254 and 185 nm) | 20 mM sulfite | ultrapure water (pH 10; 25 °C) (HA DOM spike) | 97.3% (4 h without HA); 85% (4 h with HA) |
| (27) | PFAS | 24 μM PFOA | heterogeneous (self-assembled micelle with cationic surfactant) | LP-Hg 36 W | 1.0 mM indole-acetic acid | ultrapure water (pH 6; 25 °C) (SRHA and SRFA DOM spike) | 100% (6 h) |
| (29) | PFAS | 24 μM PFOA | homogeneous | LP-Hg 36 W | 1.0 mM indole-3-acetic acid; 1.0 mM indole-1-acetic acid; 1.0 mM indole-3-carboxylic acid; 1.0 mM indole; 1.0 mM skatole; 1.0 mM donaxine | ultrapure water (pH 6; 25 °C) | 35.7% (8 h); 31.4% (8 h); 24.4% (8 h); 100% (8 h); 100% (8 h); 100% (8 h) |
| (26) | PFAS | 24.1 μM PFOA | heterogeneous (organo-montmorillonite) | LP-Hg 36 W | 1.0 mM 3-indole-acetic acid; 1.0 mM indole; 1.0 mM 3-indole-carboxylic acid; 1.0 mM 3-methylindole | ultrapure water (pH 6; 25 °C) (SRHA DOM spike) | 100% (5 or 8 h with DOM spike; 3-indole-acetic acid) |
n.r. is used to indicate if information was not reported by authors. UV sensitizer specified as “none” refers to vacuum UV studies.
3.1. Oxyanions
UV-ARP technologies have been studied for the degradation of oxyanions such as NO3–, NO2–, ClO4–, and BrO3–,6,10−19 which contain a highly oxidized central atom. The reaction mechanism involves eaq– addition to the central atom and loss of O•- (the conjugate base of •OH). The conversion of bromate to bromite is shown in Figure 9 as an example.
Figure 9.

Reaction of eaq– with bromate (BrO3–) to form bromite (BrO2–) and O•-. Although shown as a concerted process, a one-electron adduct (BrO32–) may be formed prior to Br–O bond homolysis.
Many oxyanions have been shown to be degraded quickly by eaq– generated in UV-ARP (e.g., NO3–, NO2–, BrO3–),16,18,120 while ClO4– reacts more slowly.12,13,126 Conversion of oxyanions to benign end products requires further reduction of intermediate products.119,127 For example, NO3– reacts with eaq to produce NO2– which is reduced further through additional reactions with eaq– to eventually produce N2.
| 3.1 |
Given sufficient eaq– exposure, it may be possible to quantitatively produce the free halides of halogen-containing oxyanions.13,15,18 For example, bromite is completely degraded during UV/sulfite treatment and yields bromide as the only product. Conversely, even at higher [SO32–], higher light intensity, and longer reaction times, ClO4– treatment by UV/sulfite treatment has not achieved 100% abatement. However, Vellanki et al. showed that perchlorate degradation during UV/sulfite treatment was significantly enhanced at pH 12 (∼75% abatement) relative to pH 9 (∼25% abatement7). In the case of ClO4–, Cl– and ClO3– were the only transformation products observed. This suggests that the rate limiting step is either eaq reduction of ClO4– or ClO3–, which is consistent with very slow eaq– bimolecular rate constants for these species (k < 106 M–1 s–1) and the much faster values for ClO2– and ClO– (>109 M–1 s–1). Consequently, entries in Table 3 reveal huge differences in UV-ARP treatment effectiveness for BrO3– and ClO4–, consistent with the fact that ClO4– is frequently employed as an “inert” electrolyte.
The reactivity of oxyanions with eaq– depends on several key factors. The affinity for oxyanion reduction by eaq is based primarily on the availability of an electron orbital offered by the central atom, not the compound’s one-electron redox potential.127 For example, NO2– reacts with eaq– more slowly (k = 3.5 × 109 M–1 s–1)128 than NO3– (k = 9.7 × 109 M–1 s–1),8 which is opposite of what one would expect from the NO2–/NO redox couple129 (E° = 0.46 V) vs (NO3–/NO2– of 0.10 V130). The oxyanion’s molecular geometry and charge of the central atom can further impact their reduction by eaq. For example, NO3– is more reactive with eaq– than ClO4– (k < 1.6 × 106 M–1 s–1).129 While the Cl atom in ClO4– is at a higher oxidation state (+7) than N in NO3– (+5), the slower reactivity of eaq may be attributed to the planar structure of NO3–, facilitating eaq– interaction versus the inhibiting tetrahedral structure of ClO4–.127
Additional radical species can also contribute to oxyanion degradation occurring in UV-ARP. For example, Vellanki and Batchelor proposed that SO3•- is the main reactive species responsible for the reduction of ClO4– in the UV/sulfite system.13 In this case, it was proposed that SO3•- abstracts an oxygen atom from ClO4– to form the sulfate radical and chlorate.13 Similarly, the carbon dioxide radical anion (CO2•-) was proposed to be the main reducing agent responsible for BrO3– and NO3– reduction in the UV/formate system.10,14 Moussavi and Shekoohiyan discovered that hydrogen atoms (H•) are responsible for a small percentage of NO3– reduction under VUV irradiation (185 nm radiation wavelength).11 In addition, H• are the main reactive species responsible for BrO3– degradation under acidic conditions6,18 due to increased formation of H• from the reaction of H+ and eaq– (Table 1).
3.1.1. Other Inorganic Species
UV-ARP have also been studied for the removal of oxidized metal ions such as chromium(VI) and nonmetals like selenium(IV).20,21 The eaq– reduction lowers the oxidation state of these contaminants, which decreases their solubility and often induces precipitation. For example, UV/dithionite treatment completely removed selenite through this combined reduction/precipitation process after ∼120 min.21 In the case of chromium, UV/sulfite treatment between pH 5 and 10 effectively reduced Cr(VI) to Cr(III), followed by precipitation of Cr(III) salts.20 While other approaches can also be used for reductive precipitation of Cr(VI) and Se(IV), reduction by eaq occurs readily at neutral and basic pH, while other reductants are limited by the decreased reduction potential of Cr(VI) and Se(IV) with increasing pH.
3.2. Organic Compounds
In addition to inorganic contaminants, a number of studies have evaluated UV-ARP for the degradation of halogenated contaminants. Below, we discuss UV-ARP based treatment for halogenated organic compounds, including chlorinated and brominated contaminants. A separate section is devoted to PFAS given the currently high level of interest in eaq–-based treatment for these compounds. For readers interested in further understanding the reactivity of eaq with organic compounds beyond halogenated contaminants, the book chapter by Anbar entitled “The Reactions of Hydrated Electrons with Organic Compounds” provides a thorough discussion of eaq– reactivity with various organic functional groups.131
3.2.1. Halogenated Organic Compounds
The reactivity of organic compounds with eaq– in terms of bimolecular rate constants has been studied for many decades.8,131,132 Bimolecular rate constants for eaq with organic compounds vary widely depending on compound class and the presence of halogen atoms (Figure 10). For example, aliphatic compounds bearing only N and O atoms, regardless of the functional group, have substantially lower reactivity than aliphatic compounds also bearing halogen atoms. Halogen atom substitution on carbonylic and aromatic compounds also generally increases their reactivity with eaq–. An exception is that, for nonaromatic compounds, substituting −H with −F tends to decrease the reactivity with eaq (e.g., acetone vs fluoroacetone, Figure 10A). For aromatic compounds, substituting −H with −F slightly increases its reactivity with eaq– and this is further enhanced when multiple −F are substituted (e.g., benzene, fluorobenzene, 1,4-difluorobenzene, Figure 10B).
Figure 10.

Bimolecular rate constants for reaction of hydrated electron with nonaromatic (A) and aromatic (B) compounds. Rate constants are from Li et al.100 and are tabulated by compound class in the Supporting Information. Compound classes are differentiated by the presence of O, N, and X = F, Cl, Br, I atoms. The number of compounds in each class is represented by n. Selected compounds are shown within each class to compare trends in reactivity.
3.2.1.1. Haloaliphatic Compounds
In a homologous series of halogenated aliphatic compounds, the reactivity with eaq– typically follows the trend F ≪ Cl < Br < I.131 For example, the relative reactivity of fluoroacetate, chloroacetate, bromoacetate, and iodoacetate is 0.002 < 1.0:6.2:12.0 (Figure 11).8 This trend demonstrates that the C–X bond is more reactive than the carboxyl group.91 Indeed, acetate’s reactivity with eaq (k = 1.1 × 106 M–1 s–1) is comparable to that of fluoroacetate.8
Figure 11.

Relative reactivity of monosubstituted haloacetate ions with hydrated electron in terms of their bimolecular reaction rate constant.
Reductive dehalogenation of haloaliphatic compounds is thought to occur via a two-step reaction, in which a reduced intermediate, RX•-, is first formed which subsequently undergoes unimolecular dissociation:131
| 3.2 |
| 3.3 |
with the products of the second step being a carbon centered radical (R•) and halide (X–). The fate of the carbon centered radical depends on the water matrix composition. Since UV-ARP seek to maintain low [O2], the formation of peroxyl radicals is not expected to be a significant sink of R•. As an example, the following reaction pathway for UV/sulfite transformation of chloroacetate in laboratory grade water has been established (eqs 3.4–3.757)
| 3.4 |
| 3.5 |
| 3.6 |
| 3.7 |
where the percent yields in parentheses are based on the observed rates of product formation relative to the rate of chloroacetate degradation. Chloride is formed at near 100% yield. Several additional carbon-containing products were also observed with varying yields, including succinic acid (formed from coupling of two •CH2COO– radicals), sulfoacetic acid (radical coupling of SO3•- and •CH2COO–), and acetate (possibly due to reduction of •CH2COO– by SO3•-).
For haloaliphatic compounds, UV-ARP studies have largely focused on haloacetic acids and vinyl chloride.39,57,60,89,102,108 Direct photolysis is typically an unimportant pathway for haloaliphatic compounds. Liu et al. studied the dechlorination of trichloroacetic acid using UV/titanium dioxide with formate and suggested that both CO2•– and eaq– reactions were occurring.102 Other dechlorinated products such as di- and monochloroacetic acid are formed during the reaction between trichloroacetic acid and eaq. Vinyl chloride can be degraded by UV-ARP through both direct photolysis (due to its unsaturated carbon–carbon bond) and reaction with radical species.39,108 In the UV/sulfite ARP, Liu et al. found that the rate of vinyl chloride dechlorination was inversely proportional to solution pH.108 At pH 9, UV/sulfite treatment resulted in ∼88% dechlorination. Since eaq– is the main product of sulfite photolysis at this pH (see Section 2.3), this result suggests that at pH 9 the major mechanism of vinyl chloride degradation is reductive dechlorination induced by eaq. Conversely, at pH 7, only ∼18% of the total [Cl–] possible was produced. Liu et al. proposed that the low dechlorination of vinyl chloride at pH 7 was due to the reaction of SO3•- or H• (the products HSO3– photolysis) with the C=C bond to produce chloroethane (identified by GC-MS).108
3.2.1.2. Haloaromatic Compounds
Haloaromatic compounds exhibit a similar trend in their reactivity with eaq– as haloaliphatic compounds, but the fluorinated analogues tend to be more reactive than their aliphatic counterparts. This is demonstrated in Figure 12, which shows the bimolecular reaction rate constants for the aryl halide series. Iodobenzene is approximately 3 orders of magnitude more reactive than the fluorobenzene (like haloacetic acids in Figure 11). However, the difference in reactivity between fluorobenzene and chlorobenzene (factor of ∼30) is less than for the haloacetate ions (factor of ∼1000).
Figure 12.

Relative reactivity of aryl halides with hydrated electron in terms of their bimolecular reaction rate constant.
Halogenated phenols such as 4-bromophenol, 2,4-dichlorophenol, 2,4,6-trichlorophenol, and pentafluorophenol can also be degraded by UV-ARP.45,46,104,107,133,134 Direct photolysis contributes to the transformation of these compounds in UV-ARP systems.45,46 The eaq–-induced reaction pathways for halophenols have been studied in detail,135,136 with halides being produced quantitatively upon reductive electron capture by the aromatic ring.136 Phenoxyl radicals are believed to be the initial nonhalogenated products following halide loss.135
From a treatment perspective, Xie et al. found that the UV/sulfite ARP completely debrominated 4-bromophenol within 10 min,45 and quenching experiments with nitrate and nitrite suggested that this degradation was induced by the reaction with eaq–. Similarly, Yu et al. observed that 2,4-dichlorophenol could be degraded with the UV/sulfite ARP, demonstrating that the reaction with eaq is the main reactive species responsible for its degradation.107 For fluorinated phenols, Shoute et al. reported that the rate constant between eaq– and pentafluorophenol was fast (2.4 × 1010 M–1 s–1) and that a fluoride ion was eliminated to yield a tetrafluorophenoxyl radical.134 This study only evaluated the bimolecular rate constant for eaq reaction with pentafluorophenol and not its degradation or defluorination percentage under steady-state UV-ARP treatment.
3.2.2. Halide Yields
While bimolecular rate constants provide some important information regarding the eaq–-based transformation of contaminants in water, the halide ion yield (i.e., mol of halide formed per mol of eaq-substrate reaction) is also an important factor, as this parameter defines the overall reaction efficiency. The rate of formation of the reduction product (e.g., halide) is given by137
| 3.8 |
where RfX– is the rate of halide ion formation (M s–1), Rf is the rate of eaq– formation (M s–1), γeaq–,X– is the halide ion yield, and kTC,eaq– and kTC,eaq–q are the bimolecular rate constants (M–1 s–1) for transformation of eaq by the target contaminant and quenching of the target contaminant by eaq–, respectively. eaq bimolecular rate constants are typically measured by directly observing the decay kinetics of the eaq– transient in the near-infrared, which yields the quenching rate constant kTC,eaq–. For radical reactions, it is typically assumed that kTC,eaq–t is equivalent to kTC,eaq–.137
γeaq–,X– values are not readily available for most of the substrates (organic or inorganic) for which UV-ARP are considered a possible treatment technology. However, some halide ion yields are available from the radiation chemistry literature (Figure 13).138 For benzyl halides, γeaq–,X– depends strongly on the halogen substituent and solution pH. At acidic pH (4.2), there is a clear trend of decreasing halide ion yield with increasing electronegativity of the halogen substituent. This trend largely vanishes at basic pH, with fluoro-, chloro-, and bromobenzene having near 100% halide ion yields.
Figure 13.

Halide ion yields for aryl halide reactions with hydrated electron.138 Yields are calculated by GX–/Geaq– where GX– is μmol X–/Gy radiation and Geaq– is the eaq– yield from water radiolysis (0.27 μmol/Gy8).
Halide ion yields may also depend on the location of the halogen with respect to other substituents on an aromatic ring (Figure 13). For example, the chloride yield from the reaction of eaq– with chloroaniline isomers decreases with an increasing number of sigma bonds between the amino and chloro substituents at circumneutral pH. However, at basic pH, γeaq–,X– are ∼100% for all isomers.
It is also possible to evaluate γeaq–,X– in steady-state photochemical experiments by measuring RfX– and Rf. For example, the yield of chloride from reaction with eaq– is ∼0.94 based on the rates of chloroacetate degradation (3.76 ± 0.07 μM/min) and chloride formation (3.52 ± 0.01 μM/min) in the UV/sulfite process (see eq 3.4).57 The experimental conditions (110 μM chloroacetate, 1 mM sulfite, lab grade water) at which these rates were measured are such that most eaq should be scavenged by chloroacetate (kS,t=0′ = 110 000 s–1). Thus, for γeaq–,Cl– ≈ 1, the initial rate of chloroacetate degradation should be equal to Rf, as observed.
3.2.3. Per- and Polyfluorinated Alkyl Substances
Perfluoroalkyl substances are “aliphatic substances for which all of the H atoms attached to C atoms in the nonfluorinated substance from which they are notionally derived have been replaced by F atoms, except those H atoms whose substitution would modify the nature of any functional groups present”.139 Polyfluoroalkyl substances are “aliphatic substances for which all H atoms attached to at least one (but not all) C atoms have been replaced by F atoms, in such a manner that they contain the perfluoroalkyl moiety CnF2n+1”.139 These per- and polyfluoroalkyl substances (PFAS) are characterized by extremely strong C–F bonds, which, combined with their properties as surfactant molecules, makes them extremely difficult to remove from, or degrade in, water.140
UV-ARP have increasingly been proposed as a promising technology for eaq–-based transformation of PFAS, although it is interesting to note that Anbar originally described fluorinated aliphatics as “unreactive” toward eaq due to fluorine’s high electronegativity.131 Previous reports have demonstrated that it is possible to achieve near 100% reductive defluorination (production of nontoxic fluoride) of some PFAS through UV-ARP.22,27,30,33 Although studies of PFOA and PFOS are more common, UV-ARP technology has now been evaluated for many PFAS subclasses, including short- and long-chain perfluoroalkanesulfonates (PFSA),1,33 perfluoroalkylcarboxylic acids (PFCA),30,31 perfluoroethercarboxylic acids (PFECA),31 and fluorotelomer sulfonates and carboxylic acids (Figure 14).33 Additionally, Tenorio et al. investigated UV/sulfite treatment of aqueous film-foaming foam (AFFF), which contains thousands of PFAS, diluted in sodium carbonate buffer at a ratio of 1:60 000.34 They observed that pseudo-first-order rate constants for degradation of specific PFAS in diluted AFFF were nearly identical to single-compound mixtures in the same sodium bicarbonate buffer system, suggesting that scavenging of eaq– by AFFF components did not significantly impact [eaq].34
Figure 14.

PFAS subclasses referred to frequently in this review, including their acronym and a representative structure.
In one of the earliest UV-ARP PFAS studies, Park et al. investigated the reductive defluorination of perfluorocarboxylic acids and perfluorosulfonates in the UV/iodide system.1 They found that the headgroup (e.g., −SO3– vs COO–) and −(CF2)n– chain length influenced the reductive defluorination kinetics. Specifically, the degradation rate of perfluorosulfonates increased with increasing chain length whereas perfluorocarboxylic acid chain length had little impact on the observed kinetics. Interestingly, Park et al. observed that UV/iodide degradation of PFOS occurred faster than that of PFOA, but several later studies employing UV/sulfite demonstrate an opposite trend in reactivity.30,34 This result from Park et al. is probably the result their PFAS/KI solutions being at different initial pHs, where the initial pH of PFOS solution was ∼7 while for PFOA it was ∼4.5. Therefore, quenching of eaq– by H+ in PFOA solution (pH 4.5, see Table 1) is a likely explanation of its slower transformation than PFOS. Conversely, UV/sulfite studies buffer solution at pH > 9 to maintain SO32– as the major S(IV) species,23,34 which also minimizes eaq quenching by H+.
3.2.3.1. PFAS Transformation Mechanisms
The mechanistic understanding of PFAS reductive defluorination has improved in the past few years. In their investigations of UV/sulfite treatment of PFAS sub classes,30,31,33 Bentel et al. proposed that reductive defluorination can proceed by one of two main mechanisms, as summarized in Figure 15.
Figure 15.

Proposed reaction pathways for the transformation of perfluorocarboxylic acids (PFCA), perfluorosulfonates (PFSA), and fluorotelomercarboxylic acids or fluorotelomer sulfonates (FTCA + FTSA) by hydrated electron (solid, black lines) and hydroxyl radical reactions (dashed, gray lines). Based on work of Bentel et al.2,30,79 and Liu, Bentel et al.33
The first pathway is H/F atom exchange, in which a −CF2– group is transformed to a −CH2– group. This reaction takes two eaq– and two H+ per H/F exchange. For perfluorocarboxylic acids (like PFOA), H/F exchange is thought to occur preferentially at the carbon atom adjacent to the carboxylate headgroup. For perfluorosulfonates (like PFOS), the H/F exchange is thought to occur toward the middle of the chain. This H/F exchange mechanism is supported by the formation of polyfluorinated intermediates identified through nontarget analysis during UV/sulfite treatment of PFAS.30 Computational chemistry results are also supportive of this mechanism. For example, based on spin density isosurfaces, Van Hoomissen and Vyas showed that an excess electron added to PFOA is localized around the carboxylate headgroup whereas for PFOS•2– the extra electron is distributed throughout the fluoroalkyl chain in a fashion similar to perfluorooctane.141 The location of eaq reaction during H/F exchange also relates to the positive correlation between chain length and perfluorosulfonate reactivity with eaq– reported in several studies.1,30,34 For example, aqueous vertical attachment energies correlated positively to the chain length of perfluorosulfonates but not perfluorocarboxylic acids.141 Additionally, Bentel et al. demonstrated that perfluorosulfonate C–F bonds with the lowest bond dissociation energy tended to be toward the middle of the perfluoro chain whereas the lowest energy C–F bonds in perfluorocarboxylic acids tended to be adjacent to the carboxylate group.30
The product of H/F exchange is a polyfluorinated substance and UV/sulfite studies show that polyfluorinated substances like fluorotelomercarbxylic acids (FTCA) are less reactive toward eaq–.30,34 Thus, reaction via H/F exchange limits the complete defluorination of PFAS. To address this problem, Liu, Bentel et al. recently demonstrated that greater reductive defluorination of PFAS could be achieved by performing both reductive and oxidative treatments in sequence.33 These reaction pathways are highlighted in the context of UV-ARP treatment in Figure 15 with additional details relevant to the study of Liu, Bentel et al.33 shown in Figure 16. In brief, polyfluorinated compounds formed during pH 12 UV/sulfite treatment of perfluorocarboxylic acids or perfluoroalkanesulfonates (H/F exchange) could be further defluorinated by subsequent •OH oxidation via high temperature NaOH + K2S2O8. When employing fluorotelomer sulfonates and carboxylates as the starting contaminant, a sequence of oxidation–reduction–oxidation was able to achieve near 100% defluorination for most chain lengths (Figure 16).
Figure 16.

Reaction scheme for the oxidation–reduction–oxidation treatment scheme developed by Liu, Bentel et al. for the near complete defluorination of fluorotelomer carboxylates and sulfonates.33
The second reaction pathway is decarboxylation-hydroxylation-elimination-hydrolysis (DHEH) and is thought to occur only for perfluorocarboxylic acids. The DHEH mechanism is proposed to shorten the perfluoroalkyl chain by one −CF2– group in each step. The decarboxylation leads to a perfluorinated alcohol, which is susceptible to HF elimination. The DHEH mechanism results in chain shortening and so-called “deeper” levels of defluorination than H/F exchange.30 Note that perfluorosulfonates do not undergo DHEH directly but could do so after desulfonation, which has been proposed to explain the formation of PFOA and other perfluorocarboxylic acids during UV/sulfite treatment of PFOS.30 The mechanism is supported in part by the significantly faster kinetics and greater defluorination percentage of perfluorocarboxylic acid abatement during UV/sulfite treatment at pH 12 relative to pH 9.5.123 Perfluoroether carboxylic acids (e.g., GenX) were proposed to undergo a similar reduction pathway, with cleavage of the C–O ether bond helping to enhance formation of the unstable perfluoroalcohol.31
Collectively, the reductive defluorination mechanisms discussed above suggest that the abatement of parent PFAS and ultimate fluoride production is determined by more than just the availability of eaq–. The reactivity of PFAS transformation products with eaq appears to be an important controlling factor. Thus, biasing solution conditions to favor the DHEH reaction pathway (e.g., high pH) could enhance reductive defluorination.
3.2.3.2. Optimizing PFAS Treatment by UV-ARP
Multiple studies have investigated the conditions needed to optimize PFAS treatment by increasing its eaq– exposure.1,2,22−25,58,64,121,122,124,125 For example, UV irradiation of sulfite, iodide, nitrilotriacetic acid, and several indoleacetic acid derivatives have all been shown to degrade PFOA or PFOS through reductive defluorination by eaq, all with varying kinetic rates.22,23,25,64 However, no single study has compared all these eaq– sensitizers under equivalent photochemical conditions. Sun et al. demonstrated that the UV/nitrilotriacetic ARP led to faster defluorination of PFOS than UV/sulfite at an equivalent concentration (2 mM), but it is important to note that nitrilotriacetic acid has a higher molar absorption coefficient (ε254 = 26.3 M–1 cm–1 at pH 1064) at the actinic wavelength (254 nm) than sulfite (ε254 ≈ 18 M–1 cm–157) (see Table 2). In addition, Yu et al. showed that combining sulfite and iodide into one system yielded faster rates of reductive dechlorination of a monochloroacetate system60 (this UV/sulfite/iodide system has not yet been applied to PFAS degradation). The UV/indoleacetic acid system is ineffective at degrading PFOA in bulk, aqueous solution (see further below).25
Additional treatment conditions studied for PFOA and PFOS degradation includes the initial solution pH, temperature, and the presence of eaq– scavenging species like O2, NO3–, NO2–, and DOM.1,2,22−24,58,121,122,124,125 Higher temperatures resulted in faster degradation kinetics for PFOA and PFOS, with apparent activation energies of ∼50 and 25 kJ/mol, respectively, in the UV/iodide system.121,124 Qu et al. demonstrated the major scavenging impact of O2 on PFOA degradation; when continuously sparging solutions with O2, the percent defluorination decreased from ∼98% (under N2 saturation) to less than 5% (O2-saturated solution).22 However, Bentel et al. showed that initial sparging of reaction solutions with N2 made no difference in the removal kinetics of PFOA,30 which could be explained by O2 removal through reaction with sulfite, either through a thermal51 or photochemical process68 at early UV-ARP treatment times (see also Figure 6).
Gu et al. found that PFOS degradation in the UV/sulfite ARP was significantly inhibited by humic acid (≥2.64 mgC/L) and NO3– (≥0.5 mM NO3–), whereas HCO3– had minimal impact at a concentration of 1.0 mM.24 The decreased reactivity of PFOS in the UV/sulfite system in the presence of humic acid (a surrogate for DOM) could be due to the following three mechanisms: (1) shielding of the eaq– sensitizer from UV light by DOM chromophores, (2) DOM molecules acting as a scavenger of eaq, or (3) through the production of eaq–-quenching reactive oxygen species (e.g., •OH, 1O2, or H2O2) by DOM photolysis.142 Interestingly, in the UV/iodide system,58 Aldrich Humic Acid enhanced the degradation and defluorination rate of PFOS when present at ≤10 mg/L, but it had an inhibitory effect at 30 mg/L. Sun et al. provided several hypotheses for this enhancement,58 including the possibility that phenolic electron donors reduce photochemically formed reactive iodine species (I2, I3–) back to I–, thereby decreasing the eaq scavenging by these species (eq 2.15–2.17). Although DOM also generates eaq–, this pathway is likely an insignificant source of eaq in UV-ARP due to its low quantum yield (Φeaq– ∼ 10–5).143 Further research is needed to identify the role of DOM in UV-ARP treatment of PFAS, especially in terms of eaq– scavenging and potential DOM-PFAS interactions.58,144
One
of the more significant factors influencing PFAS degradation
efficiencies is solution pH. Multiple studies have reported a clear
trend of increasing degradation rates and improved defluorination
percentages with increasing pH for perfluorocarboxylic acids and perfluorosulfonates,2,122 with parent contaminant abatement defluorination percentages being
especially rapid at pH 12.33,79 The [H+]
influences multiple aspects of UV-ARP systems, including eaq– production
from the sensitizer (see section 2.3), eaq scavenging (see Table 1 and eq 2.14), and secondary chemistry following the initial PFAS-eaq– reduction
event. Scavenging of eaq by H+ does not contribute significantly
to 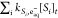 at
pH 7 or higher (Figure 4). At circumneutral pH, HSO3– quenching
reaction may be the main eaq– scavenger in UV/sulfite
ARP,48 where scavenging by H+ should be minimal. In addition, HSO3– has a low quantum yield of eaq generation, so eaq– formation rates in the
UV-sulfite system will be reduced with decreasing pH. Finally, it
is interesting to observe the pH dependence of γeaq–,X– values for halobenzenes and chloro-substituted
anilines (Figure 12), in light of the discussion above on the effect of pH on the DHEH
mechanism for reductive PFAS defluorination.
at
pH 7 or higher (Figure 4). At circumneutral pH, HSO3– quenching
reaction may be the main eaq– scavenger in UV/sulfite
ARP,48 where scavenging by H+ should be minimal. In addition, HSO3– has a low quantum yield of eaq generation, so eaq– formation rates in the
UV-sulfite system will be reduced with decreasing pH. Finally, it
is interesting to observe the pH dependence of γeaq–,X– values for halobenzenes and chloro-substituted
anilines (Figure 12), in light of the discussion above on the effect of pH on the DHEH
mechanism for reductive PFAS defluorination.
3.2.3.3. Nonhomogeneous UV-ARP
So far, we have discussed UV-ARP studies in which eaq– and PFAS are homogeneously distributed throughout aqueous solution. An alternative approach that has received attention recently is to enhance PFAS transformation by creating a microheterogeneous system.25−28 In principle, these microheterogeneous reactors enhance the rate of reductive defluorination relative to the homogeneous system by bringing the eaq and PFAS in closer proximity, thereby increasing the frequency of eaq–-PFAS reactions (Figure 17).
Figure 17.

Illustration of the microheterogeneous reductive defluorination of perfluorooctanoic acid (PFOA) via a cationic micelle with a negatively charged hydrated electron sensitizer.
Reported UV-ARP microheterogeneous technologies include UV/organomontmorillonite clay and UV/micelle systems, both of which use indoleacetic acid as the eaq– sensitizer.25,26,28,29 The UV/organomontmorillonite clay system works by first adsorbing indoleacetic acid and PFAS on the cationic clay surface.25,26 Tian et al. demonstrated that the UV/organomontmorillonite clay system allows indoleacetic acid to generate eaq and radical cations within the clay layer.25 This system increased the reductive defluorination of PFOA from approximately 20% (PFOA and indoleacetic acid only, no different from direct photolysis control) to 90% (PFOA, indoleacetic acid, and organomontmorillonite clay) in 10 h. The authors suggested that the clay system further stabilized the radical cations produced from 3-indoleacetic acid and thus minimized radical cations recombination with eaq–.25 In the UV/micelle system, a self-assembled micelle was created by a cationic surfactant (e.g., cetyltrimethylammonium bromide), indoleacetic acid (the eaq sensitizer), and PFOA.28 The self-assembled micelle increased the reductive defluorination of PFOA from approximately 10% (PFOA and indoleacetic acid only) to 90% (PFOA, indoleacetic acid, and cationic surfactant) in 2.5 h. Both microheterogeneous technologies demonstrate enhanced reductive defluorination kinetics when compared to the homogeneous UV/sensitizer system. Both technologies also reduce the eaq– scavenging impacts of H+, allowing treatment over a wider range of pH conditions (pH 4 to 8). Chen et al. found that the UV/micelle system could still be effective for PFOA transformation in the presence of high ionic strength and low concentrations of fulvic acid (1 mg/L).27
A related microheterogeneous approach is the combination of electrochemical and UV methods for ARP degradation of PFAS. Recent work by Rao et al. demonstrated that PFAS defluorination by eaq– is enhanced by PFAS sorption to carbon nanotube electrodes at low applied potentials (−0.58 V vs Ag/AgCl).74 It was hypothesized that this reaction occurs via a two-electron process in which the first electron (derived from the cathode) reduces the C–F bond strength, thereby allowing the second electron (from VUV photolysis of water) to more effectively defluorinate the PFAS than UV treatment alone. In this combined electrochemical/photochemical ARP, PFAS defluorination exhibited a chain-length dependence similar to UV-ARP, with degradation rates of perfluorocarboxylic acids being relatively independent of chain length compared to perfluorosulfonates.1,30,34
While these microheterogeneous reactors appear promising for enhanced PFAS treatment, additional research is needed to investigate the application of these systems in actual surface-and groundwater matrices, especially given the impact of water matrix components (e.g., nitrate, DOM) that could also interact with micelles, minerals, and carbon nanotube electrode materials. It is challenging to compare the treatment effectiveness between microheterogeneous systems as experimental conditions vary significantly. Future research should explore utilizing a probe compound to measure key parameters such as Rfeaq–, [eaq], and kS,t′ to compare between systems.
3.3. Knowledge Gaps
Although UV-ARP have been studied for the eaq–-based transformation of organic and inorganic compounds has been studied for over a decade, several knowledge gaps are still apparent.
3.3.1. Degradation Byproducts
A question that all degradative advanced water treatment technologies need to address is “are the target contaminants completely mineralized and, if not, are the end products more toxic, less toxic, or just as toxic as initial substrate?” UV-ARP processes also need to address this question. Given the focus on halogen-containing contaminants, the goal of UV-ARP is not complete mineralization but to reduce all halogens to a −1 oxidation state. For inorganic oxyanions, the reaction pathways are well established, with each eaq– reaction leading to successive reduction of the central atom; for example, bromate can be reduced all the way to bromide given sufficient eaq exposure.117 More work is needed to establish the operating conditions required to achieve the eaq– exposure necessary for complete transformation of the less-reactive ClO4– to Cl– ion.
Haloaliphatic compounds react by reductive electron capture according to eqs 3.2 and 3.3, the products of which are X– and a carbon-centered radical (R•). Halogen atoms will be the main site of eaq– reactivity until all C–X bonds are reduced, with the fate of the carbon centered radical being highly dependent on the water matrix. The reaction pathways for haloaromatic compounds are also established, with halides being produced upon reductive electron capture by the aromatic ring.136 The secondary chemistry of the aromatic is dependent on additional (nonhalogen substituents). For example, eaq reduction of chlorophenol is believed to produce phenoxy radicals.135 Nonhalogenated organic products (e.g., phenol) tend to be less reactive with eaq– and thus more persistent following dehalogenation of the initial contaminant. For halogenated aromatic and aliphatic compounds, a remaining knowledge gap is the ultimate fate of nonhalogenated end products.
As highlighted herein, recent reports have demonstrated that a variety of PFAS structures (see Figure 14) can be degraded with near 100% defluorination by sequential UV-ARP and oxidation reactions. Continued research efforts are needed to evaluate how these proposed treatment schemes are impacted by water matrix components such as dissolved organic matter.
3.3.2. Role of Additional Reactive Species in UV-ARP Systems
Prior literature indicates that reactive species other than eaq– may play an important role in contaminant degradation in UV-ARP.22,108,145−149 For example, SO3•- was hypothesized to be responsible for the transformation of vinyl chloride at acidic pH, resulting in a lower percentage dechlorination than obtained using eaq.108 This observation is consistent with the view of Neta and Huie150 who suggested that SO3•- may act as a mild oxidant (eq 3.9)
| 3.9 |
| 3.10 |
where E is the reduction potential vs NHE. Equation 3.10 demonstrates that SO3•- has a potential comparable to that of eaq– (−2.9 V). Based on computational chemistry estimates of one-electron reduction potentials of perfluoropentanoic acid and perfluorobutanesulfonate made by Van Hoomissen and Vyas (E < −2.0 V),141 reductive defluorination of PFAS by SO3•- should be thermodynamically feasible. Future research should evaluate this possibility.
The superoxide radical anion (O2•-) is another reactive species of interest in eaq–-based UV-ARP145 since this species is formed by reaction of O2 with eaq. There is disagreement in the literature about the role of O2•- in the degradation of perfluorocarboxylic acids (like PFOA146−148), with some reports suggesting that O2•- can displace fluorine in C–F bonds yielding F–. Yet other studies demonstrate that O2•- is completely unreactive with PFOA.149 The latter conclusion is consistent with the low oxidation potential of O2•- (E = 0.16 V) and low nucleophilicity of O2•- in water.151 The UV-ARP studies employing eaq– surveyed in this Review indicate that O2•- is not involved in the reductive defluorination of PFAS. If it were, then significant PFAS abatement should be expected in UV-ARP systems containing dissolved oxygen. However, this is not the case; for example, Qu et al. observed minimal changes in PFOA concentration when continuously sparging O2 (which should favor O2•- formation) in the UV/iodide ARP over a 10 h irradiation period.22
3.3.3. Availability of Hydrated Electron Bimolecular Rate Constants
Bimolecular rate constants for eaq– and many of the organic and inorganic contaminants listed in Table 3 are widely available. There are at least two circumstances in which this is not the case. First, some eaq reactions with contaminants (e.g., perchlorate), background scavengers (e.g., bicarbonate), or eaq– sensitizers (e.g., sulfite) have reported rate constant values that can be considered only as upper limits. This situation arises when the species of interest is “unreactive” toward eaq, which makes the transient absorption monitoring of eaq– more subject to errors from eaq reactive impurities present in either the solvent used (i.e., pure water) or the added chemical solutes.8 The importance of these slow-reacting species in the total scavenging capacity will increase with increasing eaq– exposure as more reactive eaq scavengers are abated. Thus, the lack of solid eaq– bimolecular rate constants for these species limits estimation of the scavenging capacity under these conditions.
Second, although many studies have demonstrated reductive defluorination of diverse PFAS structures by UV-ARP, absolute bimolecular rate constants for eaq–-PFAS reactions are limited. For the few values that have been reported, there is substantial variability between different studies, even when using the same experimental methods (Table 4). For example, Huang et al. using laser flash photolysis and potassium ferricyanide (K4Fe(CN)6) as an eaq sensitizer reported bimolecular rate constants for eaq– and PFCA of ∼106–7 M–1 s–1 (1.7 × 107 M–1 s–1 for PFOA).152 A recent report by Maza et al. employed the same eaq sensitizer and detection method but measured much faster rate constants, not only for perfluorocarboxylic acids (6.4 × 108 M–1 s–1 for PFOA) but also for perfluorosulfonates (2.4 × 109 M–1 s–1 for PFOS).132 A pulse-radiolysis study measured PFOA-eaq– and PFOS-eaq bimolecular rate constants of 5.1 × 107 and 7.3 × 107 M–1 s–1,153 respectively. The pulse radiolysis values are faster than the value measured by Huang et al.152 but still an order of magnitude slower than measured by Maza et al.132
Table 4. Summary of Literature Hydrated Electron (eaq–) Bimolecular Rate Constants for Per- And Polyfluoroalkyl Substances (PFAS).
| compd | acronym (chain length) | biomolecular rate constant, k (M-1s-1) | method | ref |
|---|---|---|---|---|
| trifluoroacetic acid | TFA (2C) | 1.9 × 106 | LFPa,b | (152) |
| 5.0 × 108 | LFPa | (132) | ||
| perfluoropropanoic acid | PFPrA (3C) | 5.8 × 108 | LFPa | (132) |
| perfluorobutanoic acid | PFBA (4C) | 7.1 × 106 | LFPa,b | (152) |
| 5.4 × 108 | LFPa | (132) | ||
| perfluoropentanoic acid | PFPA (5C) | 5.2 × 108 | LFPa | (132) |
| perfluorohexanoic acid | PFHxA (6C) | 5.4 × 108 | LFPa | (132) |
| perfluoroheptanoic acid | PFHpA (7C) | 7.5 × 108 | LFPa | (132) |
| perfluoroocatanoic acid | PFOA (8C) | 1.7 × 107 | LFPa | (152) |
| 5.1 × 107 | PRc | (153) | ||
| 7.1 × 108 | LFPa | (132) | ||
| perfluorononanoic acid | PFNA (9C) | 6.4 × 108 | LFPa | (132) |
| perfluorobutanesulfonate | PFBS (4C) | 1.8 × 109 | LFPa | (132) |
| perfluorohexanesulfonate | PFHxS (6C) | 1.7 × 109 | LFPa | (132) |
| perfluorooctanesulfonate | PFOS (8C) | 2.4 × 109 | LFPa | (132) |
| 7.3 × 107 | PRc,d | (153) |
Laser flash photolysis, K4Fe(CN)6 as eaq– sensitizer, transient absorption monitoring of eaq.
Corrected to zero ionic strength.
Pulse-radiolysis generation of eaq–, transient absorption monitoring of eaq.
Measured at concentration greater than critical micellular concentration.
It is difficult to understand the wide variability in these reported values between different studies using similar experimental approaches. This was noted by Maza et al. in their recent study,132 who commented that the reason for the discrepancy was not immediately apparent and probably not due to differences in ionic strength, which tend to produce only small differences in eaq– bimolecular rate constants within the ranges studied (see also Figure 3). Maza et al. did point out that Huang et al. did not report their range of [PFOA] used in their study, which could have facilitated a closer comparison. It is interesting to note, however, that the eaq bimolecular rate constant measured for trifluoroacetic acid by Huang et al. (1.9 × 106 M–1 s–1) agrees with the upper limit reported previously by Anbar and Hart (<1.4 × 106 M–1 s–191), whereas Maza et al.’s value is 2 orders of magnitude faster (5.0 × 108 M–1 s–1). One potential reason for this scatter could be the level of residual [O2] in these solutions; as noted previously, some dissolved oxygen remains no matter what deaeration techniques are used. This [O2] might not have been consistent between studies. Based on the fast rate constant of O2 reduction, even micromolar levels will dominate the PFAS-eaq– chemistry. Alternatively, the PFAS purity might be different, with even trace impurities impacting the PFAS reduction chemistry. However, the disparate values for trifluoroacetic acid suggest that the variability in eaq bimolecular rate constants for longer chain PFAS are not caused by the presence of branched isomers, which tend to be more reactive with eaq–.34
Practically, if values from the recent report by Maza et al.132 are correct and eaq– rate constants are on the order of 108–109 M–1 s–1, then it is difficult to understand the slow degradation kinetics of PFAS in UV-ARP systems. These data suggest that there is a need to resolve the uncertainty in absolute eaq bimolecular rate constants for PFAS.
For PFAS, recommended approaches to measuring eaq– bimolecular rate constants, such as laser flash photolysis or pulse radiolysis, are only possible for those compounds that have high-purity sources at high concentrations. This represents a small number of the thousands of relevant PFAS structures that may be present in contaminated waters. Another consideration is the isomeric form of the compound of interest. Branched PFAS isomers are known to be much more reactive with eaq,34 and varying percentages of branched isomers have been quantified in commercially available PFAS reagents.154 If branched and linear isomers are present, then measured eaq– bimolecular rate constants will represent an average of individual values weighted by the fractional composition of each isomer.
One path forward apart from experimental methods is the use of computational chemistry tools for the prediction of eaq– bimolecular rate constants. This could include the use of quantitative structure–activity relationships (QSARs) to relate known eaq rate constants to molecular descriptors calculated by quantum chemical techniques. Li et al. recently employed this approach and found that the energy of the lowest unoccupied molecular orbital (ELUMO) was a good predictor of kTC,eaq–.100,101 Similarly, for hydroxyl radical reactions, density functional theory (DFT)-based methods have been used by Minakata et al. to calculate the aqueous-phase free energies of activation,155 which in turn were correlated to measured bimolecular reaction rate constants through linear free energy relationships. This latter approach has been utilized extensively for UV-AOP but has not yet been applied to UV-ARP, highlighting this possible area for future research.
3.3.4. Fluoride Yields during Reductive Defluorination of PFAS
The efficiency of defluorination of PFAS by eaq– is limited by three main factors: (1) the amount of eaq generated for the reaction, (2) the fraction of eaq– reacting with PFAS relative to water matrix species, and (3) the yield of fluoride per eaq-PFAS reaction event. This latter probability can be described as a fluoride ion yield. Despite the importance of these yields, no data have been reported for eaq–-PFAS reactions. Acquiring these data are important, because the fluoride ion yield describes the intrinsic limits of eaq-based PFAS transformation efficiency. It is important to note that the commonly reported percent defluorination (number of F– produced per total F in parent PFAS) is different from the fluoride yield as it does not account for the reaction efficiency. This is illustrated by the equations below for the two terms. For % defluorination (deF%)
| 3.11 |
where [F–] is the molar fluoride concentration at the end of the UV-ARP, [PFAS]0 is the initial molar concentration of the parent PFAS, and NC–F is the number of C–F bonds in the parent compound. When using steady-state gamma radiolysis, the fluoride yield is calculated as
| 3.12 |
where GF– and Geaq– are the radiolysis yields of fluoride (unknown) and eaq– (0.27 μmol Gy–18), respectively. The assumption in eq 3.12 is that all eaq formed in the system are scavenged by the PFAS. This typically requires high concentrations of reagents (millimolar) so that all radiolytically formed eaq– are scavenged by the added substrate, in which case it is important that the reagents are free as possible from eaq-reactive impurities. To isolate eaq– reactions, solutions can be spiked with an alcohol such as tert-butanol (to scavenge •OH), sparged with N2 (to limit the eaq-O2 reaction), and aliquots withdrawn at varying time intervals (irradiation doses) to measure the concentration of target contaminant and/or transformation product(s).
Apart from radiolysis methods, it may be possible to estimate γeaq–,F– from UV/sulfite PFAS transformation data. Using this approach, γeaq–,F– can be calculated from the ratio of the eaq– formation rate (Rf,t=0) and rate of fluoride ion formation (Rf,t=0F–), the latter of which can be obtained from the commonly reported deF%). We used the PFOA deF% data from Bentel et al.30 to calculate Rf,t=0 and then calculated Rf,t=0eaq– from Figure 8 (similar reactors, sulfite concentrations, and lamp wattages were used) to estimate γeaq–,F– for PFOA (eqs 3.13–3.15):
| 3.13 |
| 3.14 |
| 3.15 |
The low fluoride ion yield calculated here for PFOA is based on several assumptions (e.g., all eaq– are scavenged by 25 μM PFOA) and future research measuring fluoride yields from PFAS of diverse structure is needed. Further, given the pH-dependence of halide ion yields for halobenzenes and haloanilines (Figure 13), it is possible that a similar trend of increasing yield with increasing pH could be occurring for PFAS defluorination. Acquiring these yields for different PFAS would enhance our understanding of the reductive defluorination chemistry.
4. Synthesis of Existing Information to Optimize UV-ARP Treatment
Development of an optimized UV-ARP system for eaq–-based contaminant transformation is needed to advance these technologies. The above sections have reviewed literature on eaq generation (in terms of the specific sensitizer) and eaq– scavenging in UV-ARP systems. The following optimized experimental conditions were common to all substrates reviewed.
Sensitizer selection: Sulfite is the most used sensitizer and appears to have the optimal benefits for UV-ARP treatment. A concentration of ∼10–15 mM likely provides the maximum level of eaq– generation
pH conditions: Maintaining a pH above 7 is highly desired to mitigate the impact of scavenging by H+ while maintaining at least 2 pH units above 7 when using sulfite as a sensitizer is essential to eliminate the scavenging impact of HSO3–.48 UV/sulfite treatment at pH 11–12 seems especially rapid for PFAS and perchlorate, and it would expedite treatment if pH adjustment is feasible.123
Minimizing scavengers: For substrates with a fast-biomolecular rate constant (≥109 M–1 s–1), it is important to remove dissolved oxygen and minimize other scavengers with fast biomolecular rate constants (e.g., NO3–, NO2–). Additional research is necessitated to optimize UV-ARP treatment for real-world waters with DOM and to evaluate the impacts of changing water background matrix conditions on substrate degradation.
Recent PFAS treatment literature has focused on the electrical energy per order (EEO) as a metric for comparing degradative technologies.32,156EEO is defined as the energy needed to reduce the concentration of contaminant by 1 order of magnitude in 1 m3 of water.157 Comparing EEO values between different UV-ARP systems in this way reveals the differences in efficiency of these processes. For example, although the microheterogeneous UV/indoleacetic acid/micelle system reported recently was described as “efficient,” a very high-powered lamp (500 W) was used to treat only 10 mL of solution,28 resulting in a high EEO (Table 5). A recent study by Liu et al. combined nanofiltration (NF) with UV-sulfite treatment at the pilot scale and found an EEO of 3.7 kWh m–3 for PFOA,32 which is approximately 4-fold lower than the EEO for PFOA in a bench-scale photochemical system (15.8 kWh m–3) reported by Bentel et al.30,31 The much lower value reported by Liu et al. results from the fact that they considered the total volume of water treated in the NF-UV/sulfite process, which was approximately an order of magnitude greater than the volume treated by UV-sulfite. EEO values for PFOA treatment via UV-ARP in Table 5 tend to be lower than most other degradative technologies.156 UV-ARP at pH 12.0 is comparable in efficiency to electrochemical oxidation; for example, electrochemical oxidation with a Ti4O7 electrochemical membrane yielded EEO values of 5.1 and 6.7 kWh m–3 for PFOA and PFOS, respectively.156
Table 5. Electrical Energy per Order (EEO) for the Degradation of Perfluorooctanoic Acid (PFOA) for Different UV-ARP.
| technology | EEO (kWh/m3)a | ref |
|---|---|---|
| UV/sulfite, pH 12.0, 18 W, 600 mL, kobs = 4.4 h–1 (bench) | 15.8 | (79) |
| UV/sulfite, pH 11.2, 1320 W, 568 L, kobs = 1.5 h–1 (pilot) | 3.7 | (32) |
| UV/indoleacetic acid/micelle, 500 W lamp, 10 mL treated, kobs = 1.7 h–1 (bench) | 67 600 | (28) |
EEO calculated as EEO = 2.303/kobs × power/volume, where kobs (h–1) is the first-order rate constant, power is the lamp power in kW, and volume is the treated volume in m3.
Minimizing the background scavenging of eaq– by water matrix components will enhance [eaq] and thereby increase the rate of target contaminant degradation. Such minimization may only be important when the treatment time scale is short (less than a few hours). For example, eaq–-based transformation of PFAS occurs over a period of >12 h during which fast-reacting eaq scavengers (e.g., O2, NO3–) will be abated quickly through their reaction with eaq–. Additional background scavengers with slow bimolecular rate constants, such as HCO3– and CO32–, may become more impactful for PFAS treatment in groundwater systems with high alkalinity, as these compounds become the main scavengers once the fast-reacting eaq scavengers are removed. Liu et al. recently employed nanofiltration (NF) to remove PFAS from contaminated groundwater while concomitantly producing a concentrated waste stream that was treated by UV-sulfite.32 The advantage of NF compared to other membranes like reverse osmosis is that NF does not substantially concentrate ions like nitrate and nitrite.158 Thus, concentrating PFAS in a small-volume waste stream while at the same time not concentrating eaq– scavengers would effectively treat more mass per unit time than if the UV-ARP were applied to the contaminated groundwater directly.
Another way to optimize the rate of target contaminant degradation in UV-ARP is to increase the rate of eaq– formation. This can be done by selecting sensitizers with a higher eaq quantum yield or by increasing the fraction of incoming photons absorbed by the sensitizer. Most eaq– sensitizers absorb in the UV region of the spectrum, where background absorbance by DOM is significant. However, an alternative approach developed by Goez and co-workers is to utilize visible-absorbing eaq sensitizers.159 These visible-absorbing eaq– generation systems utilize low-cost green light emitting diodes, a ruthenium catalyst ([Ru(bpy)32+]), and ascorbate (Asc2–) as the electron donor. Following absorption, [Ru(bpy)32+] rapidly forms a triplet metal-to-ligand charge transfer state that oxidizes Asc2–. The one-electron reduction product of [Ru(bpy)32+] is encapsulated in a cationic micelle, which facilitates lifetimes long enough to undergo another green photon absorption, thereby producing eaq and reforming ground state [Ru(bpy)32+]. This photocatalyst system demonstrates one possible route toward a more energy-efficient generation of eaq– by using a green light emitting diode; however, it is not practical for water treatment at this stage given the need for a transition-metal-containing catalyst and self-assembled cationic micelle. Future research efforts to generate visible-absorbing eaq sensitizers could improve the energy efficiency of UV-ARP.
5. Outlook and Recommendations for Future Research
UV-ARP technologies offer a unique advantage to treating recalcitrant chemical contaminants in water that are resistant to UV-AOP or other oxidative processes. Numerous UV-ARP studies have demonstrated effective degradation of oxyanions, high valence transition metals, disinfection byproducts, and PFAS. Yet, the utilization of these technologies in real-world waters is limited. Based on this review of the UV-ARP research, we suggest the following research needs and opportunities to advance UV-ARP research and clarify the role of this technology in water treatment. Note that these research needs are discussed in depth in sections 2.4 and 3.3.
Create standard methods for measuring eaq-in UV-ARP. Future research should focus on developing a probe compound capable of quantifying photochemical parameters important in UV-ARP, such as [eaq–], background eaq scavenging capacity, and rates of eaq– formation. The deployment of a probe compound will provide researchers a way to compare treatment effectiveness between studies and optimize treatment in real-world waters. Measuring the [eaq] will also facilitate modeling target contaminant transformation rates in UV-ARP.
Understand the role of DOM. The impact of DOM on the eaq– scavenging capacity and light absorbing capacity in UV-ARP systems needs further investigation. Future research should aim to determine eaq biomolecular rate constants for DOM and the impact of eaq– exposure on DOM’s absorbance. Addressing these research gaps will help to evaluate the impact of DOM on UV-ARP treatment of real-world waters where DOM is an important eaq scavenger and light absorber.
Clarify the role of additional reactive species in UV-ARP. Studies have suggested that SO3•- contributes to target contaminant transformation in the UV/sulfite system, for example, perchlorate and nitrate. Future research should explore whether reactions with SO3•- contribute to the reductive defluorination of PFAS. This reaction is thermodynamically favorable based on the reduction potentials of SO3•- (experimental)150 and perfluorobutanesulfonate (computational).141 Direct, time-resolved kinetic measurements of bimolecular rate constants between PFAS and SO3•- could help answer this question. A computational assessment of the reactivity between PFAS and SO3•- may also provide insight.
Resolve the uncertainty in eaq-bimolecular rate constants for PFAS. There is an abundance of bimolecular rate constants for eaq– and various organic and inorganic contaminants. PFAS are an exception to this, however, due the large number of PFAS and the difficulty of obtaining high quantities of pure compounds needed for eaq kinetic experiments. In addition, available PFAS-eaq– rate constants in the literature are highly variable (k = 107–109 M–1 s–1 for PFOA; see Table 4), and thus, the intrinsic reactivity of PFAS with eaq is still a major knowledge gap. Future work should investigate this discrepancy, potentially by incorporating the reaction yield (eq 3.12), as has been done previously for halogenated aromatic compounds (e.g., Figures 11 and 12). We suggest that direct, time-resolved methods such as laser flash photolysis and pulse radiolysis be employed for measuring eaq– bimolecular rate constants given the complex background photochemistry occurring with eaq sensitizers. In addition, computational chemistry approaches should be explored to discern structural trends in biomolecular rate constants with the more than thousands of relevant PFAS structures that may be present in contaminated waters.
Determine PFAS fluoride yields. Quantifying the fluoride ion yield for eaq–-PFAS reactions is important, as it describes the intrinsic limits of eaq-PFAS transformation efficiency. Equation 3.12 demonstrates how these values can be obtained from steady-state gamma radiolysis experiments. While steady-state gamma radiolysis is our recommended method for measuring these values, other experimental approaches may be possible. For example, the preliminary calculations presented above using UV/sulfite data suggest that the fluoride ion yield for PFOA is ≈5%. Future work to verify this value and expand fluoride yield measurements to additional PFAS structures is needed.
We would like to conclude by giving some final thoughts on the question of whether there is there any practical role for UV-ARP in water treatment. This question is important considering the attention given to UV-ARP for treatment of PFAS. While eaq–-based processes appear to be one of the more promising methods for PFAS transformation, it is difficult to see how UV-ARP could be implemented at the same scale as UV-AOP. The main barriers include the large UV doses required, the undesirable byproducts from eaq sensitizers in the treated water, and the rapid scavenging of eaq– by O2. The use of UV-ARP for contaminant treatment appears to be most promising for groundwater applications with low background eaq scavengers (e.g., little to no dissolved oxygen or nitrate present). Alternatively, the treatment of small volumes of concentrated contaminant waste streams, like from NF, RO filtration or ion exchange, could justify the large energy requirements of UV-ARP. Additional applications could include modular pump and treat systems for contaminated sites or temporary cleanup from chemical spills. Exploring these UV-ARP applications requires continued bench level experiments and more emphasis on pilot studies. Future research should seek to determine where UV-ARP and other eaq–-based treatment schemes fit into the management of chemical contaminants in water, most notably PFAS.
Acknowledgments
G.M. and S.P.M. gratefully acknowledge support from the U.S. National Science Foundation (CBET #2050934 and #2050882). G.M. also acknowledge the Texas Hazardous Waste Research Center for financial support. B.D.F. gratefully acknowledges financial support from the Texas A&M University Graduate Merit Fellowship, Stantec & AWWA Water Equation, and the Texas Engineering Foundation.
Glossary
Abbreviations and Symbols Used
- α
(absorption coefficient of background water matrix, cm–1)
- CO2•–
(carbon dioxide radical anion)
- eaq–
(hydrated electron)
- εsens
(molar absorption coefficient, M–1cm–1)
- EEO
(electrical energy per order, kWh m–3)
- kd′
(first-order direct photolysis rate constant, s–1)
- kS,t′
(eaq– scavenging capacity, s–1)
- kTC,eaq–q
(bimolecular rate constant for eaq– quenching by the target contaminant, M–1 s–1)
- kTC,eaq–t
(bimolecular rate constant for target contaminant transformation, M–1 s–1)
- kTC,eaq–
(bimolecular target contaminant-hydrated electron rate constant, M–1 s–1)
- I0
(photon irradiance, mmol photons cm–2s–1)
- IAA
(3-indoleacetic acid)
- •H
(hydrogen atom)
- l
(path length, cm)
- LED
(light emitting diode)
- LP-M
(low-pressure mercury lamp)
- MP-M
(medium-pressure mercury lamp)
- MCAA
(monochloroacetic acid)
- NTA
(nitrilotriacetic acid)
- •OH
(hydroxyl radical)
- Φeaq–
(quantum yield of eaq– formation)
- PFAS
(per- and polyfluoroalkyl substances)
- PFCA
(perfluorocarboxylic acid)
- PFECA
(perfluoroethercarboxylic acid)
- PFSA
(perfluorosulfonate)
- Rfeaq–
(hydrated electron formation rate, M s–1)
- RfX–
(halide formation rate, M s–1)
- sens
(sensitizer)
- SO3•–
(sulfite radical anion)
- S
(scavenger)
- TC
(target contaminant)
- UV
(ultraviolet)
- UV-ARP
(UV-Advanced Reduction Process)
- VUV
(vacuum ultraviolet)
- W
(watt)
- γeaq–,X–
(halide yield)
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsenvironau.1c00042.
Excel sheets containing bimolecular rate constants for hydrated electron reaction with organic compounds and a summary of UV-ARP treatment literature (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- Park H.; Vecitis C. D.; Cheng J.; Choi W.; Mader B. T.; Hoffmann M. R. Reductive Defluorination Of Aqueous Perfluorinated Alkyl Surfactants: Effect of Ionic Headgroup and Chain Length. J. Phys. Chem. A. 2009, 113, 690–696. 10.1021/jp807116q. [DOI] [PubMed] [Google Scholar]
- Cui J.; Gao P.; Deng Y. Destruction of Per- and Polyfluoroalkyl Substances (PFAS) with Advanced Reduction Processes (ARPs): A Critical Review. Environ. Sci. Technol. 2020, 54 (7), 3752–3766. 10.1021/acs.est.9b05565. [DOI] [PubMed] [Google Scholar]
- Yang L.; He L.; Xue J.; Ma Y.; Shi Y.; Wu L.; Zhang Z. UV/SO32– Based Advanced Reduction Processes of Aqueous Contaminants: Current Status and Prospects. Chemical Engineering Journal 2020, 397, 125412. 10.1016/j.cej.2020.125412. [DOI] [Google Scholar]
- Kaushik V.; Duan Y.; Jung B.; Batchelor B.; Abdel-Wahab A. Arsenic Removal Using Advanced Rreduction Process with Dithionite/UV—A Kinetic Study. Journal of Water Process Engineering 2018, 23, 314–319. 10.1016/j.jwpe.2018.04.012. [DOI] [Google Scholar]
- Jung B.; Sivasubramanian R.; Batchelor B.; Abdel-Wahab A. Chlorate Reduction by Dithionite/UV Advanced Reduction Process. International Journal of Environmental Science and Technology 2017, 14, 123–134. 10.1007/s13762-016-1132-y. [DOI] [Google Scholar]
- Botlaguduru V. S. V.; Batchelor B.; Abdel-Wahab A. Application of UV–Sulfite Advanced Reduction Process to Bromate Removal. Journal of Water Process Engineering 2015, 5, 76–82. 10.1016/j.jwpe.2015.01.001. [DOI] [Google Scholar]
- Vellanki B. P.; Batchelor B.; Abdel-Wahab A. Advanced Reduction Processes: A New Class of Treatment Processes. Environ. Eng. Science 2013, 30 (5), 264–271. 10.1089/ees.2012.0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxton G. V.; Greenstock C. L.; Helman W. P.; Ross A. B. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (·OH/·O– in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17 (2), 513–886. 10.1063/1.555805. [DOI] [Google Scholar]
- Bolton J. R.; Cater S. R.. Treatment of Contaminated Waste Waters and Groundwaters With Photolytically Generated Hydrated Electrons. US 5,258,124 C1, 1991.
- Chen G.; Hanukovich S.; Chebeir M.; Christopher P.; Liu H. Nitrate Removal via a Formate Radical-Induced Photochemical Process. Environ. Sci. Technol. 2019, 53 (1), 316–324. 10.1021/acs.est.8b04683. [DOI] [PubMed] [Google Scholar]
- Moussavi G.; Shekoohiyan S. Simultaneous Nitrate Reduction and Acetaminophen Oxidation Using the Continuous-flow Chemical-less VUV Process as an Integrated Advanced Oxidation and Reduction Process. J. Hazard Mater. 2016, 318, 329–338. 10.1016/j.jhazmat.2016.06.062. [DOI] [PubMed] [Google Scholar]
- Duan Y.; Batchelor B. Impacts of Natural Organic Matter on Perchlorate Removal by an Advanced Reduction Process. J. Environ. Sci. Health A Tox Hazard Subst Environ. Eng. 2014, 49 (6), 731–40. 10.1080/10934529.2014.865462. [DOI] [PubMed] [Google Scholar]
- Vellanki B. P.; Batchelor B. Perchlorate Reduction by the Sulfite/Ultraviolet Light Advanced Reduction Process. Journal Hazardous Materials 2013, 262, 348–56. 10.1016/j.jhazmat.2013.08.061. [DOI] [PubMed] [Google Scholar]
- Liu X.; Wang L.; Sun Z.; Shao Y.; Yu T. Treatment of Aqueous Bromate by Superparamagnetic BiOCl-Mediated Advanced Reduction Process. Catalysts 2017, 7 (12), 131. 10.3390/catal7050131. [DOI] [Google Scholar]
- Nawaz S.; Shah N. S.; Khan J. A.; Sayed M.; Al-Muhtaseb A. a. H.; Andersen H. R.; Muhammad N.; Murtaza B.; Khan H. M. Removal Efficiency and Economic Cost Comparison of Hydrated Electron-mediated Reductive Pathways for Treatment of Bromate. Chemical Engineering Journal 2017, 320, 523–531. 10.1016/j.cej.2017.03.011. [DOI] [Google Scholar]
- Jung B.; Nicola R.; Batchelor B.; Abdel-Wahab A. Effect of Low- and Medium-pressure Hg UV Irradiation on Bromate Removal in Advanced Reduction Process. Chemosphere 2014, 117, 663–72. 10.1016/j.chemosphere.2014.09.086. [DOI] [PubMed] [Google Scholar]
- Xiao Q.; Yu S.; Li L.; Wang T.; Liao X.; Ye Y. An Overview of Advanced Reduction Processes for Bromate Removal from Drinking Water: Reducing Agents, Activation Methods, Applications and Mechanisms. J. Hazard Mater. 2017, 324 (Pt B), 230–240. 10.1016/j.jhazmat.2016.10.053. [DOI] [PubMed] [Google Scholar]
- Xiao Q.; Wang T.; Yu S.; Yi P.; Li L. Influence of UV Lamp, Sulfur(IV) Concentration, and pH on Bromate Degradation in UV/Sulfite Systems: Mechanisms and Applications. Water Res. 2017, 111, 288–296. 10.1016/j.watres.2017.01.018. [DOI] [PubMed] [Google Scholar]
- Xiao Q.; Yu S.; Li L.; Zhang Y.; Yi P. Degradation of Bromate by Fe(II)Ti(IV) Layered Double Hydroxides Nanoparticles Under Ultraviolet Light. Water Res. 2019, 150, 310–320. 10.1016/j.watres.2018.11.067. [DOI] [PubMed] [Google Scholar]
- Xie B.; Shan C.; Xu Z.; Li X.; Zhang X.; Chen J.; Pan B. One-step Removal of Cr(VI) at Akaline pH by UV/Sulfite Process: Reduction to Cr(III) and in situ Cr(III) Precipitation. Chemical Engineering Journal 2017, 308, 791–797. 10.1016/j.cej.2016.09.123. [DOI] [Google Scholar]
- Wang X.; Liu H.; Shan C.; Zhang W.; Pan B. A Novel Combined Process for Efficient Removal of Se(VI) from Sulfate-rich Water: Sulfite/UV/Fe(III) Coagulation. Chemosphere 2018, 211, 867–874. 10.1016/j.chemosphere.2018.07.159. [DOI] [PubMed] [Google Scholar]
- Qu Y.; Zhang C.; Li F.; Chen J.; Zhou Q. Photo-reductive Defluorination of Perfluorooctanoic Acid in Water. Water Res. 2010, 44 (9), 2939–47. 10.1016/j.watres.2010.02.019. [DOI] [PubMed] [Google Scholar]
- Song Z.; Tang H.; Wang N.; Zhu L. Reductive Defluorination of Perfluorooctanoic Acid by Hydrated Electrons in a Sulfite-mediated UV Photochemical System. J. Hazard Mater. 2013, 262, 332–8. 10.1016/j.jhazmat.2013.08.059. [DOI] [PubMed] [Google Scholar]
- Gu Y.; Liu T.; Wang H.; Han H.; Dong W. Hydrated Electron Based Decomposition of Perfluorooctane Sulfonate (PFOS) in the VUV/Sulfite System. Sci. Total Environ. 2017, 607–608, 541–548. 10.1016/j.scitotenv.2017.06.197. [DOI] [PubMed] [Google Scholar]
- Tian H.; Gao J.; Li H.; Boyd S. A.; Gu C. Complete Defluorination of Perfluorinated Compounds by Hydrated Electrons Generated from 3-Indole-Acetic-Acid in Organomodified Montmorillonite. Sci. Rep. 2016, 6, 32949. 10.1038/srep32949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H.; Gu C. Effects of Different factors on Photodefluorination of Perfluorinated Compounds by Hydrated Electrons in Organo-montmorillonite System. Chemosphere 2018, 191, 280–287. 10.1016/j.chemosphere.2017.10.074. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Mi N.; Li C.; Teng Y.; Chen Y.; Gu C. Effects of Different Variables on Photodestruction of Perfluorooctanoic Acid in Self-assembled Micelle System. Sci. Total Environ. 2020, 742, 140438. 10.1016/j.scitotenv.2020.140438. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Li C.; Gao J.; Dong H.; Chen Y.; Wu B.; Gu C. Efficient Reductive Destruction of Perfluoroalkyl Substances under Self-Assembled Micelle Confinement. Environ. Sci. Technol. 2020, 54 (8), 5178–5185. 10.1021/acs.est.9b06599. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Teng Y.; Mi N.; Jin X.; Yang D.; Wang C.; Wu B.; Ren H.; Zeng G.; Gu C. Highly Efficient Hydrated Electron Utilization and Reductive Destruction of Perfluoroalkyl Substances Induced by Intermolecular Interaction. Environ. Sci. Technol. 2021, 55 (6), 3996–4006. 10.1021/acs.est.0c07927. [DOI] [PubMed] [Google Scholar]
- Bentel M. J.; Yu Y.; Xu L.; Li Z.; Wong B. M.; Men Y.; Liu J. Defluorination of Per- and Polyfluoroalkyl Substances (PFASs) with Hydrated Electrons: Structural Dependence and Implications to PFAS Remediation and Management. Environ. Sci. Technol. 2019, 53 (7), 3718–3728. 10.1021/acs.est.8b06648. [DOI] [PubMed] [Google Scholar]
- Bentel M. J.; Yu Y.; Xu L.; Kwon H.; Li Z.; Wong B. M.; Men Y.; Liu J. Degradation of Perfluoroalkyl Ether Carboxylic Acids with Hydrated Electrons: Structure-Reactivity Relationships and Environmental Implications. Environ. Sci. Technol. 2020, 54 (4), 2489–2499. 10.1021/acs.est.9b05869. [DOI] [PubMed] [Google Scholar]
- Liu C. J.; McKay G.; Jiang D.; Tenorio R.; Cath J. T.; Amador C.; Murray C. C.; Brown J. B.; Wright H. B.; Schaefer C.; Higgins C. P.; Bellona C.; Strathmann T. J. Pilot-Scale Field Demonstration of a Hybrid Nanofiltration and UV-Sulfite Treatment Train for Groundwater Contaminated by Per- and Polyfluoroalkyl Substances (PFASs). Water Res. 2021, 205, 117677. 10.1016/j.watres.2021.117677. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Bentel M. J.; Yu Y.; Ren C.; Gao J.; Pulikkal V. F.; Sun M.; Men Y.; Liu J. Near-Quantitative Defluorination of Perfluorinated and Fluorotelomer Carboxylates and Sulfonates with Integrated Oxidation and Reduction. Environ. Sci. Technol. 2021, 55 (10), 7052–7062. 10.1021/acs.est.1c00353. [DOI] [PubMed] [Google Scholar]
- Tenorio R.; Liu J.; Xiao X.; Maizel A.; Higgins C. P.; Schaefer C. E.; Strathmann T. J. Destruction of Per- and Polyfluoroalkyl Substances (PFASs) in Aqueous Film-Forming Foam (AFFF) with UV-Sulfite Photoreductive Treatment. Environ. Sci. Technol. 2020, 54 (11), 6957–6967. 10.1021/acs.est.0c00961. [DOI] [PubMed] [Google Scholar]
- Javed H.; Lyu C.; Sun R.; Zhang D.; Alvarez P. J. J. Discerning the Inefficacy of Hydroxyl Radicals During Perfluorooctanoic Acid Degradation. Chemosphere 2020, 247, 125883. 10.1016/j.chemosphere.2020.125883. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Qiu W.; Li J.; Jiang J.; Pang S. Review on UV/Sulfite Process for Water and Wastewater Treatments in the Presence or Absence of O2. Sci. Total Environ. 2021, 765, 142762. 10.1016/j.scitotenv.2020.142762. [DOI] [PubMed] [Google Scholar]
- Wu S.; Shen L.; Lin Y.; Yin K.; Yang C. Sulfite-based Advanced Oxidation and Reduction Processes for Water Treatment. Chemical Engineering Journal 2021, 414, 128872. 10.1016/j.cej.2021.128872. [DOI] [Google Scholar]
- Trojanowicz M.; Bojanowska-Czajka A.; Bartosiewicz I.; Kulisa K. Advanced Oxidation/Reduction Processes Treatment for Aaqueous Perfluorooctanoate (PFOA) and Perfluorooctanesulfonate (PFOS) – A Review of Recent Advances. Chemical Engineering Journal 2018, 336, 170–199. 10.1016/j.cej.2017.10.153. [DOI] [Google Scholar]
- Liu X.; Yoon S.; Batchelor B.; Abdel-Wahab A. Photochemical Degradation of Vinyl Chloride with an Advanced Reduction Process (ARP) – Effects of Reagents and pH. Chemical Engineering Journal 2013, 215–216, 868–875. 10.1016/j.cej.2012.11.086. [DOI] [Google Scholar]
- Rosenfeldt E. J.; Linden K. G. The ROH, UV Concept to Characterize and the Model UV/H2O2 Process in Natural Waters. Environ. Sci. Technol. 2007, 41 (7), 2548–2553. 10.1021/es062353p. [DOI] [PubMed] [Google Scholar]
- Apell J. N.; McNeill K. Updated and Validated Solar Irradiance Reference Spectra for Estimating Environmental Photodegradation Rates. Environ. Sci. Process Impacts 2019, 21 (3), 427–437. 10.1039/C8EM00478A. [DOI] [PubMed] [Google Scholar]
- Bolton J. R.; Mayor-Smith I.; Linden K. G. Rethinking the Concepts of Fluence (UV Dose) and Fluence Rate: The Importance of Photon-based Units - A Systemic Review. Photochem. Photobiol. 2015, 91 (6), 1252–62. 10.1111/php.12512. [DOI] [PubMed] [Google Scholar]
- Leifer A.The Kinetics of Environmental Aquatic Photochemistry Theory and Practice; American Chemical Society: 1988. [Google Scholar]
- Mack J.; Bolton J. R. Photochemistry of Nitrite and Nitrate in Aqueous Solution: A Review. J. Photochem. Photobiol., A 1999, 128 (1–3), 1–13. 10.1016/S1010-6030(99)00155-0. [DOI] [Google Scholar]
- Xie B.; Li X.; Huang X.; Xu Z.; Zhang W.; Pan B. Enhanced Debromination of 4-Bromophenol by the UV/Sulfite Process: Efficiency and Mechanism. J. Environ. Sci. (China) 2017, 54, 231–238. 10.1016/j.jes.2016.02.001. [DOI] [PubMed] [Google Scholar]
- Yazdanbakhsh A.; Eslami A.; Moussavi G.; Rafiee M.; Sheikhmohammadi A. Photo-assisted Degradation of 2, 4, 6-Trichlorophenol by an Advanced Reduction Process Based on Sulfite Anion Radical: Degradation, Dechlorination and Mineralization. Chemosphere 2018, 191, 156–165. 10.1016/j.chemosphere.2017.10.023. [DOI] [PubMed] [Google Scholar]
- Nash K.; Mulac W.; Noon M.; Fried S.; Sullivan J. C. Pulse Radiolysis Studies of U(VI) Complexes in Aqueous Media. Journal of Inorganic and Nuclear Chemistry 1981, 43 (5), 897–899. 10.1016/0022-1902(81)80146-7. [DOI] [Google Scholar]
- Maza W. A.; Breslin V. M.; Plymale N. T.; DeSario P. A.; Epshteyn A.; Owrutsky J. C.; Pate B. B. Nanosecond Transient Absorption Studies of the pH-dependent Hydrated Electron Quenching by HSO3–. Photochem. Photobiol. Sci. 2019, 18 (6), 1526–1532. 10.1039/C9PP00063A. [DOI] [PubMed] [Google Scholar]
- Shetiya R. S.; Rao K. N.; Shankar J. Determination of rate constants for the reactions of H, OH and e aq with indole-3-acetic acid and other plant hormones. Radiation Effects 1972, 14 (3–4), 185–189. 10.1080/00337577208231199. [DOI] [Google Scholar]
- Westerhoff P.; Mezyk S.; Cooper W. J.; Minakata D. Electron Pulse Radiolysis Determination of Hydroxyl Radical Rate Constants with Suwannee River Fulvic Acid and Other Dissolved Organic Matter Isolates. Environ. Sci. Technol. 2007, 41 (13), 4640–4646. 10.1021/es062529n. [DOI] [PubMed] [Google Scholar]
- Wong G. T. F.; Zhang L. Chemical Removal of Oxygen with Sulfite for the Polarographic or Voltammetric Determination of Iodate or Iodide in Seawater. Marine Chemistry 1992, 38 (1–2), 109–116. 10.1016/0304-4203(92)90070-Q. [DOI] [Google Scholar]
- Brezonik P. L.; Arnold W. A.. Water Chemistry: An Introduction to the Chemistry of Natural and Engineered Aquatic Systems; Oxford University Press, Inc.: 2011. [Google Scholar]
- What are Humic Substances? https://humic-substances.org/ (accessed October 5, 2021).
- NDRL/NIST Solution Kinetics Database on the Web, 2002 ed.; National Institute of Standards and Technology: Notre Dame Radiation Laboratory, 2021. https://kinetics.nist.gov/solution/ (accessed October 5, 2021).
- Trojanowicz M.; Bobrowski K.; Szostek B.; Bojanowska-Czajka A.; Szreder T.; Bartoszewicz I.; Kulisa K. A Survey of Analytical Methods Employed for Monitoring of Advanced Oxidation/Reduction Processes for Decomposition of Selected Perfluorinated Environmental Pollutants. Talanta 2018, 177, 122–141. 10.1016/j.talanta.2017.09.002. [DOI] [PubMed] [Google Scholar]
- Grossweiner L. I.; Swenson G. W.; Zwicker E. F. Photochemical Generation of the Hydrated Electron. Science 1963, 141 (3583), 805–806. 10.1126/science.141.3583.805. [DOI] [PubMed] [Google Scholar]
- Li X.; Ma J.; Liu G.; Fang J.; Yue S.; Guan Y.; Chen L.; Liu X. Efficient Reductive Dechlorination of Monochloroacetic Acid by Sulfite/UV Process. Environ. Sci. Technol. 2012, 46 (13), 7342–9. 10.1021/es3008535. [DOI] [PubMed] [Google Scholar]
- Sun Z.; Zhang C.; Chen P.; Zhou Q.; Hoffmann M. R. Impact of Humic Acid on the Photoreductive Degradation of Perfluorooctane Sulfonate (PFOS) by UV/Iodide Process. Water Res. 2017, 127, 50–58. 10.1016/j.watres.2017.10.010. [DOI] [PubMed] [Google Scholar]
- Sauer M. C.; Crowell R. A.; Shkrob I. A. Electron Photodetachment from Aqueous Anions. 1. Quantum Yields for Generation of Hydrated Electron by 193 and 248 nm Laser Photoexcitation of Miscellaneous Iorganic nions. Journal Physical Chemistry A 2004, 108 (25), 5490–5502. 10.1021/jp049722t. [DOI] [Google Scholar]
- Yu K.; Li X.; Chen L.; Fang J.; Chen H.; Li Q.; Chi N.; Ma J. Mechanism and Efficiency of Contaminant Reduction by Hydrated Electron in the Sulfite/Iodide/UV Process. Water Res. 2018, 129, 357–364. 10.1016/j.watres.2017.11.030. [DOI] [PubMed] [Google Scholar]
- Awtrey A. D.; Connick R. E. The Absorption Spectra of I2, I3–, I–, IO3–, S4O6= and S2O3=. Heat of the Reaction I3– = I2 + I–. J. Am. Chem. Soc. 1951, 73 (4), 1842–1843. 10.1021/ja01148a504. [DOI] [Google Scholar]
- Iwata A.; Nakashima N.; Kusaba M.; Izawa Y.; Yamanaka C. Quantum Yields of Hydrated Electrons by UV Laser Irradiation. Chem. Phys. Lett. 1993, 207 (2–3), 137–142. 10.1016/0009-2614(93)87004-M. [DOI] [Google Scholar]
- Deister U.; Neeb R.; Helas G.; Warneck P. Temperature Dependence of the Equilibrium CH2(OH)2 + HSO3– = CH2(OH)SO3– + H2O in Aqueous Solution. J. Phys. Chem. 1986, 90 (14), 3213–3217. 10.1021/j100405a033. [DOI] [Google Scholar]
- Sun Z.; Zhang C.; Xing L.; Zhou Q.; Dong W.; Hoffmann M. R. UV/Nitrilotriacetic Acid Process as a Novel Strategy for Efficient Photoreductive Degradation of Perfluorooctanesulfonate. Environ. Sci. Technol. 2018, 52 (5), 2953–2962. 10.1021/acs.est.7b05912. [DOI] [PubMed] [Google Scholar]
- Tian H.; Guo Y.; Pan B.; Gu C.; Li H.; Boyd S. A. Enhanced Photoreduction of Nitro-aromatic Compounds by Hydrated Electrons Derived from Indole on Natural Montmorillonite. Environ. Sci. Technol. 2015, 49 (13), 7784–92. 10.1021/acs.est.5b01026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper J. M.; Gebhard O.; Van Den Berg C. J.; Veldstra H. Studies on Indolepyruvic Acid. Arch. Biochem. Biophys. 1963, 103 (3), 475–487. 10.1016/0003-9861(63)90441-7. [DOI] [PubMed] [Google Scholar]
- Zechner J.; Kohler G.; Getoff N.; Tatischeff I.; Klein R. Wavelength Effects on the Photoprocesses of Indole and Derivatives in Solution. Photochem. Photobiol. 1981, 34 (2), 163–168. 10.1111/j.1751-1097.1981.tb08980.x. [DOI] [Google Scholar]
- Deister U.; Warneck P. Photooxidation of SO32- in Aqueous Solution. J. Phys. Chem. 1990, 94, 2191–2198. 10.1021/j100368a084. [DOI] [Google Scholar]
- Fischer M.; Warneck P. Photodecomposition and Photooxidation of Hydrogen Sulfite in Aqueous Solution. J. Phys. Chem. 1996, 100, 15111–15117. 10.1021/jp953236b. [DOI] [Google Scholar]
- Lee J. Y.; Baugher J. F.; Grossweiner L. I. Pairwise Recombination in the Decay of Photochemical Hydrated Electrons. Photochem. Photobiol. 1979, 29 (5), 867–874. 10.1111/j.1751-1097.1979.tb07784.x. [DOI] [Google Scholar]
- Schwarz H. A.; Gill P. S. Diffusion-Limited Solvated Electron Reactions in Ethanol and Water. J. Phys. Chem. 1977, 81 (1), 22–25. 10.1021/j100516a006. [DOI] [Google Scholar]
- Elliot A. J.; Ouellette D. C. Temperature Dependence of the Rate Constant for the Reaction eaq– + OH in Water up to 150 C. J. Chem. Soc., Faraday Trans. 1994, 90 (6), 837–841. 10.1039/FT9949000837. [DOI] [Google Scholar]
- Zoschke K.; Bornick H.; Worch E. Vacuum-UV Radiation at 185 nm in Water Treatment-A Review. Water Res. 2014, 52, 131–45. 10.1016/j.watres.2013.12.034. [DOI] [PubMed] [Google Scholar]
- Rao U.; Su Y.; Khor C. M.; Jung B.; Ma S.; Cwiertny D. M.; Wong B. M.; Jassby D. Structural Dependence of Reductive Defluorination of Linear PFAS Compounds in a UV/Electrochemical System. Environ. Sci. Technol. 2020, 54 (17), 10668–10677. 10.1021/acs.est.0c02773. [DOI] [PubMed] [Google Scholar]
- Rahn R. O. Potassium Iodide as a Chemical Actinometer for 254 nm Radiation: Use of Iodate as an Electron Scavenger. Photochem. Photobiol. 1997, 66, 450–455. 10.1111/j.1751-1097.1997.tb03172.x. [DOI] [Google Scholar]
- Backer L. C.; Esteban E.; Rubin C. H.; Kieszak S.; McGeehin M. A. Assessing Acute Diarrhea From Sulfate In Drinking Water. Journal - American Water Works Association 2001, 93 (9), 76–84. 10.1002/j.1551-8833.2001.tb09288.x. [DOI] [Google Scholar]
- Gu Y.; Liu T.; Zhang Q.; Dong W. Efficient Decomposition of Perfluorooctanoic Acid by a High Photon Flux UV/Sulfite Process: Kinetics and Associated Toxicity. Chemical Engineering Journal 2017, 326, 1125–1133. 10.1016/j.cej.2017.05.156. [DOI] [Google Scholar]
- Gu Y.; Dong W.; Luo C.; Liu T. Efficient Reductive Decomposition of Perfluorooctanesulfonate in a High Photon Flux UV/Sulfite System. Environ. Sci. Technol. 2016, 50 (19), 10554–10561. 10.1021/acs.est.6b03261. [DOI] [PubMed] [Google Scholar]
- Bentel M. J.; Liu Z.; Yu Y.; Gao J.; Men Y.; Liu J. Enhanced Degradation of Perfluorocarboxylic Acids (PFCAs) by UV/Sulfite Treatment: Reaction Mechanisms and System Efficiencies at pH 12. Environmental Science & Technology Letters 2020, 7 (5), 351–357. 10.1021/acs.estlett.0c00236. [DOI] [Google Scholar]
- Rosenfeldt E. J.; Linden K. G. Degradation of Endocrine Disrupting Chemicals Bisphenol A, Ethinyl Estradiol, and Estradiol during UV Photolysis and Advanced Oxidation Processes. Environmental science & technology 2004, 38 (20), 5476–5483. 10.1021/es035413p. [DOI] [PubMed] [Google Scholar]
- Rosenfeldt E. J.; Linden K. G.; Canonica S.; von Gunten U. Comparison of the Efficiency of ·OH Radical Formation During Ozonation and the Advanced Oxidation Processes O3/H2O2 and UV/H2O2. Water Res. 2006, 40 (20), 3695–704. 10.1016/j.watres.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Burns J. M.; Cooper W. J.; Ferry J. L.; King D. W.; DiMento B. P.; McNeill K.; Miller C. J.; Miller W. L.; Peake B. M.; Rusak S. A.; Rose A. L.; Waite T. D. Methods for Reactive Oxygen Species (ROS) Detection in Aqueous Environments. Aquatic Sciences 2012, 74 (4), 683–734. 10.1007/s00027-012-0251-x. [DOI] [Google Scholar]
- Pi Y.; Schumacher J.; Jekel M. The Use of para-Chlorobenzoic Acid (pCBA) as an Ozone/Hydroxyl Radical Probe Compound. Ozone: Science & Engineering 2005, 27 (6), 431–436. 10.1080/01919510500349309. [DOI] [Google Scholar]
- Ulliman S. L.; McKay G.; Rosario-Ortiz F. L.; Linden K. G. Low Levels of Iron Enhance UV/H2O2 Efficiency at Neutral pH. Water Res. 2018, 130, 234–242. 10.1016/j.watres.2017.11.041. [DOI] [PubMed] [Google Scholar]
- Ulliman S. L.; Miklos D. B.; Hübner U.; Drewes J. E.; Linden K. G. Improving UV/H2O2 Performance Following Tertiary Treatment of Municipal Wastewater. Environmental Science: Water Research & Technology 2018, 4 (9), 1321–1330. 10.1039/C8EW00233A. [DOI] [Google Scholar]
- Zepp R. G.; Braun A. M.; Hoigne J.; Leenheer J. A. Photoproduction of Hydrated Electrons From Natural Organic Solutes in Aquatic Environments. Environ. Sci. Technol. 1987, 21 (5), 485–490. 10.1021/es00159a010. [DOI] [PubMed] [Google Scholar]
- Thomas-Smith T. E.; Blough N. V. Photoproduction of Hydrated Electron from Constituents of Natural Waters. Environ. Sci. Technol. 2001, 35 (13), 2721–2726. 10.1021/es010552x. [DOI] [PubMed] [Google Scholar]
- Gu J.; Ma J.; Jiang J.; Yang L.; Yang J.; Zhang J.; Chi H.; Song Y.; Sun S.; Tian W. Q. Hydrated electron (eaq–) Generation from Phenol/UV: Efficiency, Influencing Factors, and Mechanism. Applied Catalysis B: Environmental 2017, 200, 585–593. 10.1016/j.apcatb.2016.07.034. [DOI] [Google Scholar]
- Li X.; Fang J.; Liu G.; Zhang S.; Pan B.; Ma J. Kinetics and Efficiency of the Hydrated Electron-induced Dehalogenation by the Sulfite/UV Process. Water Res. 2014, 62, 220–8. 10.1016/j.watres.2014.05.051. [DOI] [PubMed] [Google Scholar]
- Munch D. J.; Much J. W.; Pawlecki A. M.. Method 552.2 - Determination of Haloacetic Acids and Dalapon in Drinking Water by Liquid-Liquid Extraction, Derivatization and Gas Chromatography with Electron Capture Detection - Revision 1.0; U.S. Environmental Protection Agency: 1995. [Google Scholar]
- Anbar M.; Hart E. J. The Reaction of Haloaliphatic Compounds with Hydrated Electrons. J. Phys. Chem. 1965, 69 (1), 271–274. 10.1021/j100885a041. [DOI] [Google Scholar]
- Butler I. B.; Schoonen M. A.; Rickard D. T. Removal of Dissolved Oxygen From Water: A Comparison of Four Common Techniques. Talanta 1994, 41 (2), 211–215. 10.1016/0039-9140(94)80110-X. [DOI] [PubMed] [Google Scholar]
- Crittenden J. C.; Hu S.; Hand D. W.; Green S. A. A Kinetic Model for H2O2/UV Process In a Completely Mixed Batch Reactor. Water Res. 1999, 33 (10), 2315–2328. 10.1016/S0043-1354(98)00448-5. [DOI] [Google Scholar]
- Zhang R.; Sun P.; Boyer T. H.; Zhao L.; Huang C. H. Degradation of Pharmaceuticals and Metabolite in Synthetic Human Urine by UV, UV/H2O2, and UV/PDS. Environ. Sci. Technol. 2015, 49 (5), 3056–66. 10.1021/es504799n. [DOI] [PubMed] [Google Scholar]
- Ianni J. C.A Comparison of the Bader-Deuflhard and the Cash-Karp Runge-Kutta Integrators for the GRI-MECH 3.0 Model Based on the Chemical Kinetics Code Kintecus. In Computational Fluid and Solid Mechanics 2003; Bathe K. J., Ed.; Elsevier Science Ltd: Oxford, 2003; pp 1368–1372. [Google Scholar]
- Zhang T.; Huang C. H. Modeling the Kinetics of UV/Peracetic Acid Advanced Oxidation Process. Environ. Sci. Technol. 2020, 54 (12), 7579–7590. 10.1021/acs.est.9b06826. [DOI] [PubMed] [Google Scholar]
- Bulman D. M.; Mezyk S. P.; Remucal C. K. The Impact of pH and Irradiation Wavelength on the Production of Reactive Oxidants during Chlorine Photolysis. Environ. Sci. Technol. 2019, 53 (8), 4450–4459. 10.1021/acs.est.8b07225. [DOI] [PubMed] [Google Scholar]
- Li H.; McKay G. Relationships between the Physicochemical Properties of Dissolved Organic Matter and Its Reaction with Sodium Borohydride. Environ. Sci. Technol. 2021, 55 (15), 10843–10851. 10.1021/acs.est.1c01973. [DOI] [PubMed] [Google Scholar]
- Maurer F.; Christl I.; Kretzschmar R. Reduction and Reoxidation of Humic Acid: Influence on Spectroscopic Properties and Proton Binding. Environ. Sci. Technol. 2010, 44 (15), 5787–5792. 10.1021/es100594t. [DOI] [PubMed] [Google Scholar]
- Li C.; Zheng S.; Li T.; Chen J.; Zhou J.; Su L.; Zhang Y. N.; Crittenden J. C.; Zhu S.; Zhao Y. Quantitative Structure-activity Relationship Models for Predicting Reaction Rate Constants of Organic Contaminants with Hydrated Electrons and Their Mechanistic Pathways. Water Res. 2019, 151, 468–477. 10.1016/j.watres.2018.12.010. [DOI] [PubMed] [Google Scholar]
- Zheng S.; Li C.; Wei G. QSAR Modeling for Reaction Rate Constants of eaq– With Diverse Organic Compounds in Water. Environmental Science: Water Research & Technology 2020, 6 (7), 1931–1938. 10.1039/D0EW00244E. [DOI] [Google Scholar]
- Liu X.; Zhong J.; Fang L.; Wang L.; Ye M.; Shao Y.; Li J.; Zhang T. Trichloroacetic Acid Reduction by an Advanced Reduction Process Based on Carboxyl Anion Radical. Chemical Engineering Journal 2016, 303, 56–63. 10.1016/j.cej.2016.05.130. [DOI] [Google Scholar]
- Liu X.; Vellanki B. P.; Batchelor B.; Abdel-Wahab A. Degradation of 1,2-Dichloroethane with Advanced Reduction Processes (ARPs): Effects of Process Variables and Mechanisms. Chemical Engineering Journal 2014, 237, 300–307. 10.1016/j.cej.2013.10.037. [DOI] [Google Scholar]
- Sun C.; Chang W.; Ma W.; Chen C.; Zhao J. Photoreductive Debromination of Decabromodiphenyl Ethers in the Presence of Carboxylates Under Visible Light Irradiation. Environ. Sci. Technol. 2013, 47 (5), 2370–7. 10.1021/es3045604. [DOI] [PubMed] [Google Scholar]
- Wang L.; Liu X. Fast Degradation of Monochloroacetic Acid by BiOI-Enhanced UV/S(IV) Process: Efficiency and Mechanism. Catalysts 2019, 9 (5), 460. 10.3390/catal9050460. [DOI] [Google Scholar]
- Yu X.; Cabooter D.; Dewil R. Efficiency and Mechanism of Diclofenac Degradation by Sulfite/UV Advanced Reduction Processes (ARPs). Sci. Total Environ. 2019, 688, 65–74. 10.1016/j.scitotenv.2019.06.210. [DOI] [PubMed] [Google Scholar]
- Yu X.; Cabooter D.; Dewil R. Effects of Process Variables and Kinetics on the Degradation of 2,4-Dichlorophenol Using Advanced Reduction Processes (ARP). J. Hazard Mater. 2018, 357, 81–88. 10.1016/j.jhazmat.2018.05.049. [DOI] [PubMed] [Google Scholar]
- Liu X.; Yoon S.; Batchelor B.; Abdel-Wahab A. Degradation of Vinyl Chloride (VC) by the Sulfite/UV Advanced Reduction Process (ARP): Effects of Process Variables and a Kinetic Model. Sci. Total Environ. 2013, 454–455, 578–83. 10.1016/j.scitotenv.2013.03.060. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Zhang Q.; Chen B.; Long L.; Zhang G. Toward Better Understanding Vacuum Ultraviolet—Iodide Induced Photolysis via Hydrogen Peroxide Formation, Iodine Species Change, and Difluoroacetic Acid Degradation. Front. Environ. Sci. Eng. 2022, 16 (5), 55. 10.1007/s11783-021-1489-0. [DOI] [Google Scholar]
- Qiwen M.; Kim K. H.; Han I. Correlation Analysis of Pollutant Factors Influencing the Sulfite/UV-L Advanced Reduction Process. KSCE Journal of Civil Engineering 2018, 22 (2), 475–481. 10.1007/s12205-017-1536-3. [DOI] [Google Scholar]
- Yazdanbakhsh A.; Eslami A.; Mahdipour F.; Ghanbari F.; Ghasemi S. M.; Atamaleki A.; Maleksari H. S.; Lin K.-Y. A. Dye Degradation in Aqueous Solution by Dithionite/UV-C Advanced Reduction Process (ARP): Kinetic Study, Dechlorination, Degradation Pathway and Mechanism. J. Photochem. Photobiol., A 2021, 407, 112995. 10.1016/j.jphotochem.2020.112995. [DOI] [Google Scholar]
- Wang Z.; Liu W.; Chen H.; Zhang Z.; Yang Z.; Yang Q. Photoreductive Degradation of CCl4 by UV-Na2SO3: Influence of Various Factors, Mechanism and Application. Environ. Technol. 2021, 42 (2), 217–226. 10.1080/09593330.2019.1625957. [DOI] [PubMed] [Google Scholar]
- Asgari G.; Seidmohammadi A.; Rahmani A. R.; Samarghandi M. R.; Faraji H. Application of the UV/Sulfoxylate/Phenol Process in the Simultaneous Removal of Nitrate and Pentachlorophenol from the Aqueous Solution. J. Mol. Liq. 2020, 314, 113581. 10.1016/j.molliq.2020.113581. [DOI] [Google Scholar]
- Cao Y.; Qiu W.; Li J.; Zhao Y.; Jiang J.; Pang S. Sulfite Enhanced Transformation of Iopamidol by UV Photolysis in the Presence of Oxygen: Role of Oxysulfur Radicals. Water Res. 2021, 189, 116625. 10.1016/j.watres.2020.116625. [DOI] [PubMed] [Google Scholar]
- Yu X.; Gocze Z.; Cabooter D.; Dewil R. Efficient Reduction of Carbamazepine Using UV-activated Sulfite: Assessment of Critical Process Parameters and Elucidation of Radicals Involved. Chemical Engineering Journal 2021, 404, 126403. 10.1016/j.cej.2020.126403. [DOI] [Google Scholar]
- Akbari A.; Sadani M.; Sedighizadeh A.; Sedghi A.; Khajeh M.; Mahmoudi M.; Bagheri A.; Teimouri F.; Behin J. Reduction of Dinitrotoluene byHydrated Electrons Generated from UV Irradiation of Toluene in Wastewater: Towards Cleaner Production. Journal of Cleaner Production 2019, 238, 117857. 10.1016/j.jclepro.2019.117857. [DOI] [Google Scholar]
- Zhang T.; Wang J.; Yan D.; Wang L.; Liu X. Efficient Reduction of Bromate by Iodide-Assisted UV/Sulfite Process. Catalysts 2018, 8 (12), 652. 10.3390/catal8120652. [DOI] [Google Scholar]
- Liu X.; Zhang T.; Shao Y. Aqueous Bromate Reduction by UV Activation of Sulfite. CLEAN - Soil, Air, Water 2014, 42 (10), 1370–1375. 10.1002/clen.201300646. [DOI] [Google Scholar]
- Vellanki B. P.; Batchelor B. Nitrate Reduction by the Ultraviolet-Sulfite Advanced Reduction Process. Environ. Eng. Sci. 2021, 38, 927. 10.1089/ees.2021.0054. [DOI] [Google Scholar]
- Tan L.; Mao R.; Su P.; Gu J.; Chen H.; Jiang F.; Zhao X. Efficient Photochemical Denitrification by UV/Sulfite System: Mechanism and Applications. J. Hazard Mater. 2021, 418, 126448. 10.1016/j.jhazmat.2021.126448. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Qu Y.; Zhao X.; Zhou Q. Photoinduced Reductive Decomposition of Perflurooctanoic Acid in Water: Effect of Temperature and Ionic Strength. CLEAN - Soil, Air, Water 2015, 43 (2), 223–228. 10.1002/clen.201300869. [DOI] [Google Scholar]
- Qu Y.; Zhang C. J.; Chen P.; Zhou Q.; Zhang W. X. Effect of Initial Solution pH on Photo-induced Reductive Decomposition of Perfluorooctanoic Acid. Chemosphere 2014, 107, 218–223. 10.1016/j.chemosphere.2013.12.046. [DOI] [PubMed] [Google Scholar]
- Bentel M. J.; Liu Z.; Yu Y.; Gao J.; Men Y.; Liu J. Enhanced Degradation of Perfluorocarboxylic Acids (PFCAs) by UV/Sulfite Treatment: Reaction Mechanisms and System Efficiencies at pH 12. Environmental Science & Technology Letters 2020, 7 (5), 351–357. 10.1021/acs.estlett.0c00236. [DOI] [Google Scholar]
- Lyu X. J.; Li W. W.; Lam P. K.; Yu H. Q. Insights into Perfluorooctane Sulfonate Photodegradation in a Catalyst-Free Aqueous Solution. Sci. Rep 2015, 5, 9353. 10.1038/srep09353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C.; Zhang C.; Sun Z.; Zhao X.; Zhou Q.; Hoffmann M. R. Synergistic Impact of Humic Acid on the Photo-reductive Decomposition of Perfluorooctanoic Acid. Chemical Engineering Journal 2019, 360, 1101–1110. 10.1016/j.cej.2018.10.204. [DOI] [Google Scholar]
- Anbar M.; Hart E. J. On the Reactivity of Hydrated Electrons Toward Inorganic Compounds. In. Radiation Chemistry 1968, 81, 79–94. 10.1021/ba-1968-0081.ch006. [DOI] [Google Scholar]
- Anbar M.; Hart E. J. Reactions of Hydrated Electron. Solvated Electron 1965, 50, 55–81. 10.1021/ba-1965-0050.ch006. [DOI] [Google Scholar]
- Elliot A. J.; McCracken D. R.; Buxton G. V.; Wood N. D. Estimation of Rate Constants for Near-Diffusion-Controlled Reactions in Water at High Temperatures. J. Chem. Soc., Faraday Trans. 1990, 86 (9), 1539–1547. 10.1039/ft9908601539. [DOI] [Google Scholar]
- Thomas J. K.; Gordon S.; Hart E. J. The Rates of Reactions of the Hydrated Electron in Aqueous Inorganic Solutions. J. Phys. Chem. 1964, 68 (6), 1524–1527. 10.1021/j100788a043. [DOI] [Google Scholar]
- Atkins P.; Paula J. d.. Physical Chemistry, 7th ed.; Oxford University Press: 2003. [Google Scholar]
- Anbar M. The Reactions of Hydrated Electrons with Organic Compounds. Adv. Phys. Org. Chem. 1969, 7, 115–151. 10.1016/S0065-3160(08)60264-6. [DOI] [Google Scholar]
- Maza W. A.; Breslin V. M.; Owrutsky J. C.; Pate B. B.; Epshteyn A. Nanosecond Transient Absorption of Hydrated Electrons and Reduction of Linear Perfluoroalkyl Acids and Sulfonates. Environmental Science & Technology Letters 2021, 8 (7), 525–530. 10.1021/acs.estlett.1c00383. [DOI] [Google Scholar]
- Wentworth W. E.; Becker R. S.; Tung R. Thermal Electron Attachment to Some Aliphatic and Aromatic Chloro, Bromo, and Iodo Derivatives. J. Phys. Chem. 1967, 71, 1652–1665. 10.1021/j100865a017. [DOI] [Google Scholar]
- Shoute L. C. T.; Mittal J. P.; Neta P. Reduction and Defluorination of Pentafluorophenol in Aqueous Solutions. J. Phys. Chem. 1996, 100, 3016–3019. 10.1021/jp9513374. [DOI] [Google Scholar]
- Getoff N.; Solar S. Radiolysis and Pulse Radiolysis of Chlorinated Phenols in Aqueous Solutions. International Journal of Radiation Applications and Instrumentation 1986, 28 (5–6), 443–450. 10.1016/1359-0197(86)90165-7. [DOI] [Google Scholar]
- Ye M.Radiolytic Studies of Reduction and Oxidation of Aromatic Systems. Ph.D. Thesis, Notre Dame University, 1989. [Google Scholar]
- Rosario-Ortiz F. L.; Canonica S. Probe Compounds to Assess the Photochemical Activity of Dissolved Organic Matter. Environ. Sci. Technol. 2016, 50 (23), 12532–12547. 10.1021/acs.est.6b02776. [DOI] [PubMed] [Google Scholar]
- Lichtscheidl J.; Getoff N. Radiolysis of Halogenated Aromatic Compounds in Aqueous Solutions-I Conductometric Pulse Radiolysis and Steady-State Studies of the Reaction of eaq–. International Journal for Radiation Physics and Chemistry 1976, 8 (6), 661–665. 10.1016/0020-7055(76)90037-1. [DOI] [Google Scholar]
- Buck R. C.; Franklin J.; Berger U.; Conder J. M.; Cousins I. T.; de Voogt P.; Jensen A. A.; Kannan K.; Mabury S. A.; van Leeuwen S. P. Perfluoroalkyl and Polyfluoroalkyl Substances in the Environment: Terminology, Classification, and Origins. Integr Environ. Assess Manag 2011, 7 (4), 513–41. 10.1002/ieam.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino N.; Qu Y.; Deeb R. A.; Hawley E. L.; Hoffmann M. R.; Mahendra S. Degradation and Removal Methods for Perfluoroalkyl and Polyfluoroalkyl Substances in Water. Environmental Engineering Science 2016, 33 (9), 615–649. 10.1089/ees.2016.0233. [DOI] [Google Scholar]
- Van Hoomissen D. J.; Vyas S. Early Events in the Reductive Dehalogenation of Linear Perfluoroalkyl Substances. Environmental Science & Technology Letters 2019, 6 (6), 365–371. 10.1021/acs.estlett.9b00116. [DOI] [Google Scholar]
- Vione D.; Minella M.; Maurino V.; Minero C. Indirect Photochemistry in Sunlit Surface Waters: Photoinduced Production of Reactive Transient Species. Chemistry 2014, 20 (34), 10590–606. 10.1002/chem.201400413. [DOI] [PubMed] [Google Scholar]
- Wang W.; Zafiriou O. C.; Chan I.; Zepp R. C.; Blough N. V. Production of Hydrated Electrons from Photoionization of Dissolved Organic Matter in Natural Waters. Environ. Sci. Technol. 2007, 41 (5), 1601–1607. 10.1021/es061069v. [DOI] [PubMed] [Google Scholar]
- Dixit F.; Barbeau B.; Mostafavi S. G.; Mohseni M. Removal of Legacy PFAS and Other Fluorotelomers: Optimized Regeneration Strategies in DOM-rich Waters. Water Res. 2020, 183, 116098. 10.1016/j.watres.2020.116098. [DOI] [PubMed] [Google Scholar]
- Smith B. A.; Teel A. L.; Watts R. J. Identification of the Reactive Oxygen Species Responsible for Carbon Tetrachloride Degradation in Modified Fenton’s System. Environ. Sci. Technol. 2004, 38 (20), 5465–5469. 10.1021/es0352754. [DOI] [PubMed] [Google Scholar]
- da Silva-Rackov C. K. O.; Lawal W. A.; Nfodzo P. A.; Vianna M. M. G. R.; do Nascimento C. A. O.; Choi H. Degradation of PFOA by Hydrogen Peroxide and Persulfate Activated by Iron-modified Diatomite. Applied Catalysis B: Environmental 2016, 192, 253–259. 10.1016/j.apcatb.2016.03.067. [DOI] [Google Scholar]
- Panchangam S. C.; Lin A. Y.; Tsai J. H.; Lin C. F. Sonication-assisted Photocatalytic Decomposition of Perfluorooctanoic Acid. Chemosphere 2009, 75 (5), 654–660. 10.1016/j.chemosphere.2008.12.065. [DOI] [PubMed] [Google Scholar]
- He X.; Sun B.; He M.; Chi H.; Wang Z.; Zhang W.; Ma J. Highly Efficient Simultaneous Catalytic Degradation and Defluorination of Perfluorooctanoic Acid by the H2O2–carbon/MnO2 System Generating O2– and OH Synchronously. Applied Catalysis B: Environmental 2020, 277, 119219. 10.1016/j.apcatb.2020.119219. [DOI] [Google Scholar]
- Javed H.; Metz J.; Eraslan T. C.; Mathieu J.; Wang B.; Wu G.; Tsai A.-L.; Wong M. S.; Alvarez P. J. J. Discerning the Relevance of Superoxide in PFOA Degradation. Environmental Science & Technology Letters 2020, 7 (9), 653–658. 10.1021/acs.estlett.0c00505. [DOI] [Google Scholar]
- Neta P.; Huie R. E. Free-Radical Chemistry of Sulfite. Environ. Health Perspect. 1985, 64, 209–217. 10.1289/ehp.8564209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer D. T.; Valentine J. S. How Super is Superoxide?. Acc. Chem. Res. 1981, 14 (12), 393–400. 10.1021/ar00072a005. [DOI] [Google Scholar]
- Huang L.; Dong W.; Hou H. Investigation of the Reactivity of Hydrated Electron Toward Perfluorinated Carboxylates by Laser Flash Photolysis. Chem. Phys. Lett. 2007, 436 (1–3), 124–128. 10.1016/j.cplett.2007.01.037. [DOI] [Google Scholar]
- Szajdzinska-Pietek E.; Gebicki J. L. Pulse Radiolytic Investigation of Perfluorinated Surfactants in Aqueous Solutions. Res. Chem. Intermed. 2000, 26, 897–912. 10.1163/156856700X00381. [DOI] [Google Scholar]
- Benskin J. P.; De Silva A. O.; Martin J. W.. Isomer Profiling of Perfluorinated Substances as a Tool for Source Tracking: A Review of Early Findings and Future Applications; Springer: New York, 2010; Vol. 208. [DOI] [PubMed] [Google Scholar]
- Minakata D.; Mezyk S. P.; Jones J. W.; Daws B. R.; Crittenden J. C. Development of Linear Free Energy Relationships for Aqueous Phase Radical-involved Chemical Reactions. Environ. Sci. Technol. 2014, 48 (23), 13925–32. 10.1021/es504491z. [DOI] [PubMed] [Google Scholar]
- Le T. X. H.; Haflich H.; Shah A. D.; Chaplin B. P. Energy-Efficient Electrochemical Oxidation of Perfluoroalkyl Substances Using a Ti4O7 Reactive Electrochemical Membrane Anode. Environmental Science & Technology Letters 2019, 6 (8), 504–510. 10.1021/acs.estlett.9b00397. [DOI] [Google Scholar]
- Keen O.; Bolton J.; Litter M.; Bircher K.; Oppenländer T. Standard Reporting of Electrical Energy per Order (EEO) for UV/H2O2 Reactors (IUPAC Technical Report). Pure Appl. Chem. 2018, 90 (9), 1487–1499. 10.1515/pac-2017-0603. [DOI] [Google Scholar]
- Liu C. J.; Strathmann T. J.; Bellona C. Rejection of Per- and Polyfluoroalkyl Substances (PFASs) in Aqueous Film-Forming Foam by High-Pressure Membranes. Water Res. 2021, 188, 116546. 10.1016/j.watres.2020.116546. [DOI] [PubMed] [Google Scholar]
- Naumann R.; Lehmann F.; Goez M. Generating Hydrated Electrons for Chemical Syntheses by Using a Green Light-Emitting Diode (LED). Angew. Chem., Int. Ed. Engl. 2018, 57 (4), 1078–1081. 10.1002/anie.201711692. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- NDRL/NIST Solution Kinetics Database on the Web, 2002 ed.; National Institute of Standards and Technology: Notre Dame Radiation Laboratory, 2021. https://kinetics.nist.gov/solution/ (accessed October 5, 2021).