

ABSTRACT
The clinical successes of immune checkpoint blockade (ICB) in advanced cancer patients have recently spurred the clinical implementation of ICB in the neoadjuvant and perioperative setting. However, how neoadjuvant ICB therapy affects the systemic immune landscape and metastatic spread remains to be established. Tumors promote both local and systemic expansion of regulatory T cells (Tregs), which are key orchestrators of tumor-induced immunosuppression, contributing to immune evasion, tumor progression and metastasis. Tregs express inhibitory immune checkpoint molecules and thus may be unintended targets for ICB therapy counteracting its efficacy. Using ICB-refractory models of spontaneous primary and metastatic breast cancer that recapitulate the poor ICB response of breast cancer patients, we observed that combined anti-PD-1 and anti-CTLA-4 therapy inadvertently promotes proliferation and activation of Tregs in the tumor, tumor-draining lymph node and circulation. Also in breast cancer patients, Treg levels were elevated upon ICB. Depletion of Tregs during neoadjuvant ICB in tumor-bearing mice not only reshaped the intratumoral immune landscape into a state favorable for ICB response but also induced profound and persistent alterations in systemic immunity, characterized by elevated CD8+ T cells and NK cells and durable T cell activation that was maintained after treatment cessation. While depletion of Tregs in combination with neoadjuvant ICB did not inhibit primary tumor growth, it prolonged metastasis-related survival driven predominantly by CD8+ T cells. This study demonstrates that neoadjuvant ICB therapy of breast cancer can be empowered by simultaneous targeting of Tregs, extending metastasis-related survival, independent of a primary tumor response.
KEYWORDS: Breast cancer metastasis, myeloid cells, neoadjuvant immune checkpoint blockade, regulatory T cells, resistance mechanisms
Introduction
Encouraged by the clinical successes of immune checkpoint blockade (ICB) in late-stage cancer patients1–3, neoadjuvant ICB has now gained momentum4, demonstrating remarkable responses in, amongst others, patients with melanoma and mismatch-repair deficient colorectal cancer5,6. The first clinical trials with neoadjuvant ICB in combination with chemotherapy are showing promise in early-stage triple-negative breast cancer (TNBC) patients as well7,8. While the short-term goal of neoadjuvant ICB is to induce a pathological complete response (pCR), the long-term goal is the prevention of metastatic disease, which remains the biggest challenge in breast cancer patient care. Intriguingly, a recent clinical study demonstrated that neoadjuvant anti-PD-L1 plus chemotherapy significantly improved survival of TNBC patients despite showing only a modest increase in pathological complete response9, emphasizing that the absence of a therapeutic benefit in the primary tumor does not exclude therapeutic benefit against metastatic spread upon neoadjuvant immunotherapy-based treatment regimens. There is a growing appreciation that systemic immunity is required for effective cancer immunotherapy and the prevention of metastasis10,11. However, our understanding of how neoadjuvant ICB therapy affects the systemic immune landscape and metastatic spread, remains incomplete.
The immune system plays a dual role in metastasis formation. While optimally primed and activated cytotoxic immune cells can kill cancer cells, tumor-induced immunosuppression facilitates immune evasion, tumor progression, and metastasis12. Regulatory T cells (Tregs) are essential regulators of immune homeostasis by safeguarding self-tolerance and promoting the resolution of inflammation13, but Tregs also frequently infiltrate into tumor tissues, where they are key orchestrators of cancer-associated immunosuppression and contribute to immune evasion of cancer13,14. High intratumoral levels of Tregs correlate with tumor grade and poor survival in breast cancer patients15,16. In breast cancer mouse models, Tregs negatively impact anti-tumor immunity through inhibition of both innate and adaptive immune cell function17,18. It is becoming increasingly evident that tumors affect Tregs beyond the tumor microenvironment (TME). Elevated Treg levels have been reported in the circulation of breast cancer patients19,20, and their ex vivo immunosuppressive potential was predictive of tumor relapse21. Increased Treg frequencies were also found in tumor-invaded sentinel lymph nodes and correlated with metastatic spread to those lymph nodes22–24. Using a mouse model of spontaneous multi-organ breast cancer metastasis, we have previously demonstrated that mammary tumors induce the systemic accumulation of activated, immunosuppressive Tregs .that promote metastasis formation in the lymph nodes but not the lungs via local suppression of NK cell activation25, emphasizing that immune evasion of metastasis is a systemic, context-dependent process that is instigated by the primary tumor.
The main rationale of ICB is improving the priming, expansion and effector functions of tumor-specific CD8+ T cells26. However, the expression of immune checkpoint molecules such as PD-1 and CTLA-4 is not limited to CD8+ T cells but is also found on intratumoral Tregs in mouse models as well as cancer patients27–29. Recent data suggest that both anti-PD-1 and anti-CTLA-4-based antibody therapies may inadvertently lead to the activation and proliferation of Tregs30–32, and this has been associated with non-responsiveness to anti-PD-1 in NSCLC, gastric cancer and melanoma patients29,32, and hyperprogression upon anti-PD-1 in gastric cancer patients33. Moreover, several experimental mouse studies have demonstrated that Treg-targeting improves ICB response in immunogenic primary tumor models intrinsically sensitive to ICB34–37. However, none of these studies have investigated how Treg-targeting during ICB therapy in the neoadjuvant setting affects systemic immunity and metastatic spread. Moreover, the role of Tregs during ICB treatment in less immunogenic cancer models that are intrinsically unresponsive to ICB, remains to be elucidated.
Here, we set out to study how neoadjuvant anti-PD-1 and anti-CTLA-4 blockade affects Treg phenotype and function, and whether the potential activation of Tregs by ICB forms an obstacle for local and systemic anti-tumor immunity in ICB-unresponsive mouse models of primary and metastatic breast cancer. We make use of the transgenic K14cre;Cdh1F/F;Trp53F/F (KEP) mouse model of invasive lobular carcinoma (ILC)38 and the KEP-based mastectomy model for spontaneous multi-organ metastatic disease39, allowing side-by-side comparison of ICB responses during primary tumor growth and metastasis formation. We demonstrate that Tregs are inadvertently activated by ICB, locally in the TME as well as systemically in the tumor-draining lymph node (TDLN) and circulation, posing a barrier for anti-tumor immunity. Enhanced Treg frequencies in the circulation and increased FOXP3 expression in metastatic lesions were observed in breast cancer patients upon blockade of PD-1/PD-L1-axis. Although Treg-depletion during neoadjuvant ICB does not affect primary tumor control despite changing the TME into a state favorable for ICB response, it induces a robust and persistent systemic T cell activation which promotes a synergistic anti-metastatic response. Our data demonstrate that neoadjuvant ICB can be empowered by simultaneous targeting of Tregs extending metastasis-related survival independent of a primary tumor response.
Results
ICB inadvertently drives Treg accumulation in mammary tumor models and breast cancer patients
We first determined the expression pattern of PD-1 and CTLA-4 on intratumoral T cell populations in tumor-bearing KEP mice38. The proportion of Tregs expressing PD-1, CTLA-4, or co-expressing PD-1 and CTLA-4 was higher compared to CD8+ and CD4+ T cells (Figure 1A). As we have previously reported40, dual anti-PD-1/anti-CTLA-4 therapy is ineffective in controlling tumor growth in KEP mice bearing spontaneous mammary tumors (Figure 1B,C), consistent with poor response to ICB as monotherapy in breast cancer patients1,2. ICB did not alter the intratumoral infiltration of CD8+ and CD4+ T cells, but instead increased the accumulation of FOXP3+ cells (Figure 1D,E). As a result, the intratumoral ratio of CD8/FOXP3 cells decreased upon ICB (Figure 1F), whereas a high CD8/FOXP3 ratio has been associated with improved survival in breast cancer patients41. In line with this observation, we found increased expression of the proliferation marker Ki-67 in Tregs upon ICB (Figure 1G). In addition, Tregs, but not CD8+ T cells, were increased in blood of tumor-bearing KEP mice receiving ICB (Figure 1H,I). In the TDLN, Treg frequency was not significantly altered, but these Tregs did express higher levels of Ki-67 (Figure 1J).
Figure 1.
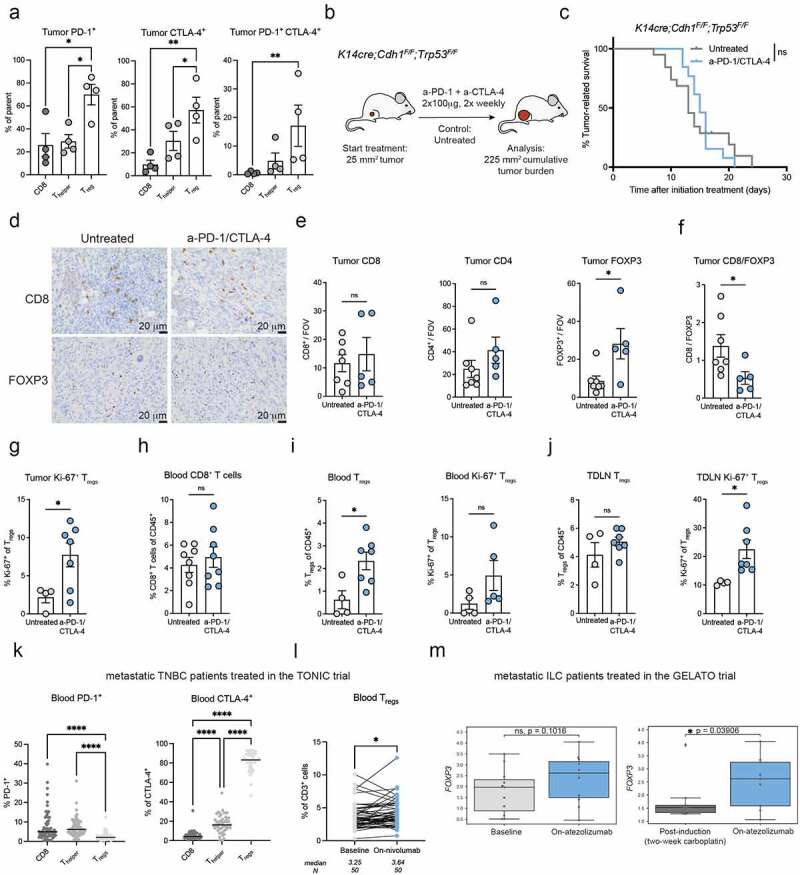
ICB fails to inhibit mammary tumor outgrowth and induces intratumoral and systemic Treg accumulation in transgenic KEP mice.
(A) Frequency of surface PD-1+, intracellular CTLA-4+, and surface PD-1+CTLA-4+ T cells subsets (% of parent) in untreated end-stage KEP tumors (mm2), determined by flow cytometry (n = 4-5). (B) Schematic overview of intervention study in KEP mice. (C) Kaplan-Meier survival curves of KEP mice left untreated (n = 15) or treated with a combination of anti-PD-1 and anti-CTLA-4 (ICB) (n = 12). (D) Representative images of immunohistochemical staining of CD8 and FOXP3 in tumors of KEP mice, treated as indicated. 40× magnifications, scale bar represents 20 m. (E) CD8, CD4 and FOXP3 counts in tumors of KEP mice treated as indicated, determined by immunohistochemical analysis (counts per 40× field of view, average of five randomly selected areas, n = 5-7 mice/group). (F) Ratio of CD8 and FOXP3 counts shown in Figure 1E. (G) Frequency of Ki-67 expression in Tregs (CD4+CD25+) in tumors of KEP mice treated as indicated, analyzed by flow cytometry (n = 4-7). (H) Frequency of CD8+ T cells as % of CD45+ cells in blood of tumor-bearing KEP mice, treated as indicated, determined by flow cytometry (n = 8). (I) Quantification of Tregs as % of CD45+ cells (left) and frequency of Ki-67 expression on Tregs (right) in blood of tumor-bearing KEP mice, treated as indicated, as determined by flow cytometry (n = 4-7). (J) Quantification of Tregs as % of CD45+ cells (left) and frequency of Ki-67 expression on Tregs (right), in tumor-draining lymph nodes (TDLN) of tumor-bearing KEP mice, treated as indicated, determined by flow cytometry (n = 4-7). (K) Frequency of PD-1 and CTLA-4 expression on T cell subsets (% of parent population) in baseline blood samples of metastatic TNBC patients treated in the TONIC-trial, analyzed by flow cytometry (n = 49-69 patients). (L) Frequency of circulating Tregs as % of CD3+ cells in blood samples taken at baseline and after 3 cycles of nivolumab/aPD-1 (on-nivolumab) in metastatic TNBC patients treated in the TONIC trial2, analyzed by flow cytometry (n = 50 patients). (M) FOXP3 gene expression in sequential tumor biopsies taken from a metastatic lesion at baseline, after 2 weeks of low-dose carboplatin treatment (post-induction), or after two cycles of atezolizumab/aPD-L1 in combination with carboplatin (on-atezolizumab) in metastatic ILC patients treated in the GELATO trial44 (n = 9-11 patients). Box plots display median with range. Data in A,E-J show mean ± SEM. P-values are calculated by One-way ANOVA with Sidak’s correction (A,K), Log-rank (Mantel-Cox) test (C), Unpaired Student’s T-test (E-J), Wilcoxon (L-M). ns, not significant, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
We set out to validate our findings in the orthotopic KEP transplantation model (figure S1A)39. Similar to the spontaneous KEP model and as described previously40, ICB did not affect tumor growth in this setting (figure S1B). Furthermore, Tregs, but not conventional T cells, were increased in frequency in blood and tumor and showed enhanced Ki-67 expression in tumors of ICB-treated mice (figure S1C-F), in line with our observations in the spontaneous KEP model. To evaluate whether ICB also influences Treg functionality, an in vitro T cell suppression assay was performed by co-culturing Tregs isolated from TDLNs of KEP tumor-bearing mice with in vitro activated splenic T cells. Tregs from Ctrl-treated or ICB-treated mice similarly inhibited the proliferation of responder T cells, demonstrating that ICB does not further enhance the suppressive capacity of Tregs in this setting (figure S1G).
We validated our findings in the Wap-cre;Cdh1F/F;AktE17K;Trp53KO (WEAP) tumor cell inoculation model of invasive lobular carcinoma42,43. Like in KEP tumors, the frequency of PD-1 and CTLA-4 expression was higher on intratumoral Tregs compared to CD8+ T cells (figure S1H). aPD-1/CTLA-4 treatment led to a modest survival benefit in mice bearing WEAP tumors (figure S1I). We found an increase in FOXP3 counts in WEAP tumors upon ICB, but interestingly also an increase in CD8 counts (figure S1J), potentially explaining the response to ICB in WEAP tumor-bearing mice which was not observed in KEP mice. Together, these data show that increased Treg infiltration by ICB is observed in two independent breast cancer models.
To assess the clinical relevance of our data, we obtained flow cytometry data on fresh blood samples from the TONIC trial, which evaluated the efficacy of nivolumab/anti-PD-1 after a two-week induction therapy with low-dose chemotherapy or irradiation in patients with metastatic TNBC (mTNBC)2,40. Blood was drawn before start of treatment (baseline) and after three cycles of anti-PD-1 (on-nivolumab). While only a very small proportion of circulating Tregs displayed PD-1 expression (Figure 1K), the vast majority of circulating Tregs displayed CTLA-4 expression. CTLA-4 expression was much lower on circulating CD8+ and CD4+ Thelper cells (Figure 1K). Strikingly, Treg frequency in the circulation increased from baseline to the on-nivolumab timepoint (Figure 1L). To assess whether intratumoral Treg infiltration may also be affected by immunotherapy in breast cancer patients, we examined FOXP3 gene expression in sequential tumor biopsies obtained in the GELATO trial44. Sequential tumor biopsies were taken from metastatic lesions before start of treatment (baseline), after two cycles of low-dose carboplatin as induction treatment (post-induction), and after two cycles of atezolizumab/anti-PD-L1 and continued carboplatin (on-atezolizumab) in patients with metastatic ILC (mILC). We found a trend in increased FOXP3 expression in metastatic lesions from baseline to the on-atezolizumab timepoint (p = 0.10) and a significant increase from the post-induction to on-atezolizumab timepoint (Figure 1M), suggesting that intratumoral Treg levels increase upon aPD-L1 in mILC patients. Our data are in line with previous observations showing an increase in circulating and intratumoral Tregs upon neoadjuvant/adjuvant aPD-1 in melanoma patients32.
Collectively, our preclinical findings in Keratin14-cre;Cdh1F/F;Trp53F/F and Wap-cre;Cdh1F/F;AktE17K;Trp53KO breast cancer models show that both circulating and intratumoral Tregs inadvertently respond to ICB, in line with our observations in breast cancer patients.
Depletion of Tregs in the context of neoadjuvant ICB induces the systemic expansion and activation of effector T cells and NK cells
To assess whether ICB-activated Tregs limit the efficacy of ICB, we utilized Foxp3DTR-GFP mice in which Tregs can be transiently depleted upon short-term diphtheria toxin (DT) treatment25,45. KEP tumor-bearing Foxp3DTR-GFP mice were treated with combinations of ICB or control antibody (Ctrl), and DT or PBS, until mastectomy was performed when tumors reached a size of ~120 mm2 (Figure 2A). Tregs were efficiently depleted upon Ctrl+DT or ICB+DT treatment in blood samples collected 1–2 days before mastectomy (“pre-mastectomy”) as well as in resected tumors (figure S2A-C).
Figure 2.
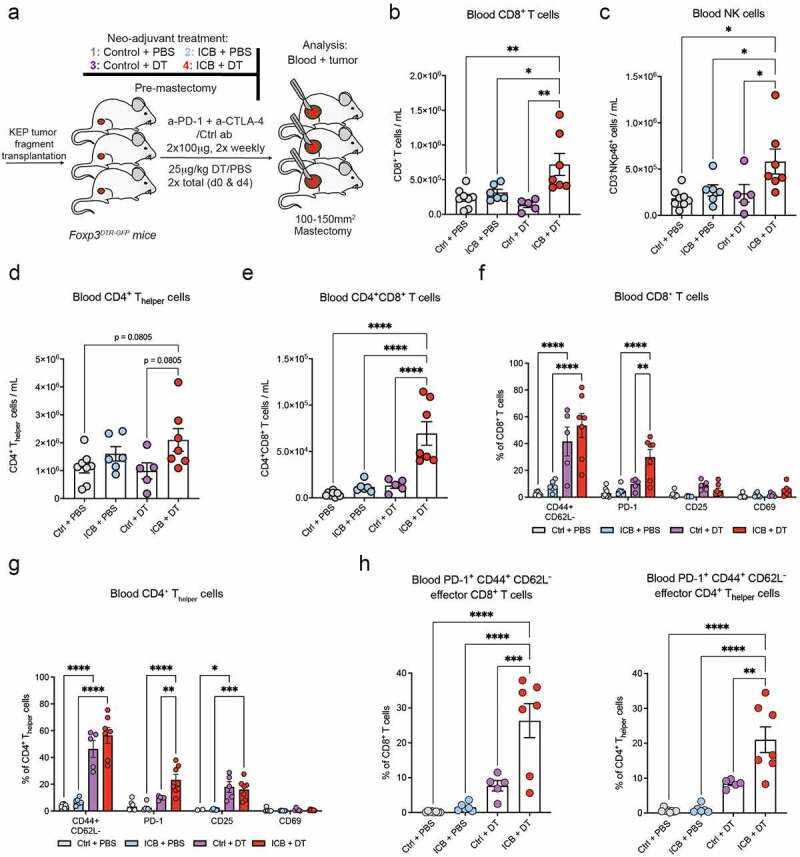
Depletion of Tregs during neoadjuvant ICB induces the systemic expansion and activation of T cells and NK cells.
(A) Schematic overview of intervention study using orthotopically transplanted KEP tumors. (B-E) Absolute cell counts of CD8+ T cells (B), NK cells (C), CD4+ Thelper cells (D), and CD4+CD8+ double-positive T cells (E) in blood of mice treated as indicated, determined by flow cytometry pre-mastectomy (n = 5-8). (F-G) Frequency of indicated markers (% of positive cells) gated on CD8+ T cells (F) or CD4+ Thelper cells (G) in blood of mice treated as indicated, determined by flow cytometry pre-mastectomy (n = 5-8). (H) Frequency of PD-1+ CD44+ CD62L− effector CD8+ and CD4+ T cells in the blood of mice treated as indicated, analyzed by flow cytometry pre-mastectomy (n = 5-8). Data in B-H show mean ± SEM. P-values were calculated by One-way ANOVA with Sidak’s correction (B-E,H) or Two-way ANOVA with Sidak’s correction (F,G). ns, not significant, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
In the pre-mastectomy blood samples, we found that combination of ICB+DT specifically induced a significant increase of both CD8+ T cells and NK cells (Figure 2B,C and S2D). A tendency toward increased number of CD4+ Thelper cells was also observed (Figure 2D). Notably, upon ICB+DT we also observed a strong increase in CD4+CD8+ T cells (Figure 2E), which have been described to be enriched in patients with auto-immune disease46 and cancer47, and have been shown to display reactivity toward autologous melanoma cell lines in vitro47,48. Treg-depletion was sufficient to strongly increase the frequency of CD44+CD62L− effector CD8+ T cells in the blood, and this was not further enhanced upon ICB+DT (Figure 2F). Similarly, Treg-depletion alone induced activation of circulating CD4+ Thelper cells (Figure 2G). Noteworthy, PD-1 expression on CD8+ and CD4+ T cells was further increased upon ICB+DT, compared to DT (Figure 2F,G). Further characterizing the phenotype of effector T cells induced upon ICB+DT, we found strong expression of PD-1 on effector CD8+ and CD4+ T cells specifically in mice treated with ICB+DT (Figure 2I), indicative of the antigen-experienced properties of these effector T cells49–51. These data demonstrate that combined ICB + Treg-depletion increases the frequency of circulating NK cells and activated PD-1+ effector CD8+ T cells.
Neoadjuvant ICB and Treg-depletion remodels the intratumoral immune landscape
Analysis of the resected tumors showed that ICB + Treg-depletion led to strong remodeling of tumor immune landscape, shifting the balance toward increased frequency of infiltrating T cells (figure S2E). Specifically, we found that ICB+DT induced a clear trend toward increased frequency of CD8+ T cells in the tumor and a statistically significant increase in the frequency of CD4+ Thelper cells and CD4+CD8+ T cells (Figure 3A-C). Similar to the blood, Treg-depletion was sufficient to promote the intratumoral activation of CD8+ and CD4+ T cells (Figure 3D,E). Interestingly, the frequency of CD69+CD8+ T cells in tumors was specifically increased upon ICB+DT compared to DT alone (Figure 3D), suggesting a more robust ICB-induced CD8+ T cell activation in the absence of Tregs. This is further indicated by the enhanced frequency of PD-1+ effector T cells and CD69+ effector T cells in the tumor upon ICB+DT compared to DT alone (Figure 3F-I).
Figure 3.
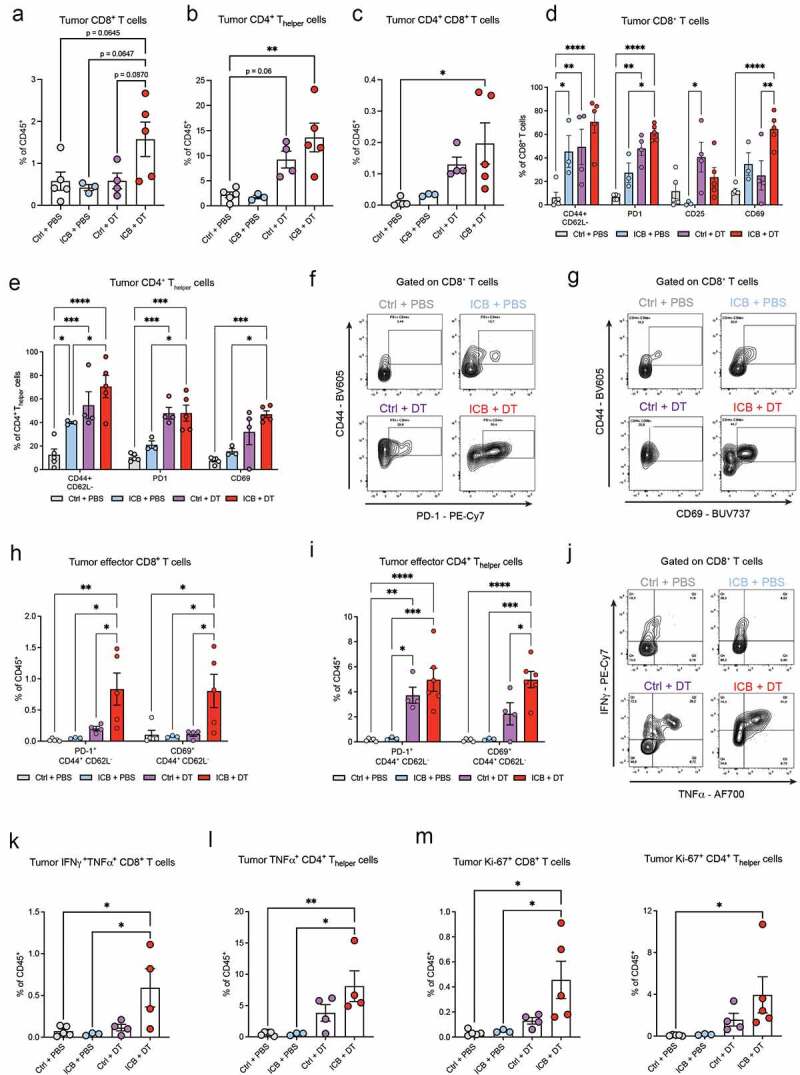
Increased tumor infiltration and activation of T cells upon depletion of Tregs during neoadjuvant ICB.
(A-C) Frequency of CD8+ T cells (A), CD4+ Thelper cells (B), and CD4+CD8+ double-positive T cells (C) in resected tumors of mice treated as indicated, determined by flow cytometry (n=3-5). (D-E) Frequency of indicated activation markers (% of positive cells) gated on CD8+ T cells (D) or CD4+ Thelper cells (E) in resected tumor of mice treated as indicated, determined by flow cytometry (n=5-8). (F-G) Representative contour plots depicting co-expression of PD-1 and CD44 (F) or CD69 and CD44 (G) on CD8+ T cells in resected tumors of mice treated as indicated. (H-I) Frequency of CD44+CD62L− effector CD8+ (H) and CD4+ (I) T cells co-expressing PD-1 or CD69 in the tumors of mice treated as indicated, analyzed by flow cytometry (n=3-5). (J) Representative contour plots depicting IFNγ- and TNFα-production by tumor CD8+ T cells in resected tumors of mice treated as indicated. (K-L) Frequency of IFNγ+TNFα+ CD8+ T cells (K) and TNFα+ CD4+ T cells (L) as % of CD45+ cells in the tumor of mice treated as indicated, measured by intracellular flow cytometry after ex vivo PMA/ionomycin stimulation (n=3-5). (M) Frequency of Ki-67+ CD8+ and CD4+ T cells as % of CD45+ cells in the tumors of mice treated as indicated, analyzed by intracellular flow cytometry (n=3-5). Data in A-E,H-I,K-M show mean ± SEM. P-values were calculated by One-way ANOVA with Sidak’s correction (A-C,K-M) or Two-way ANOVA with Sidak’s correction (D-E,H-I). ns, not significant, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
To characterize the functionality of the effector T cell populations induced by ICB+DT, we analyzed their proliferative state and ability to produce cytokines. This analysis revealed elevated levels of IFNγ– and TNFα–producing CD8+ T cells as well as TNFα-producing CD4+ Thelper cells in mice treated with ICB+DT (Figure 3J-L), providing strong indication of their cytotoxic functionally. Lastly, we found increased frequency of Ki-67+ CD8+ and CD4+ T cells in the tumor (Figure 3M), indicative of their increased activation in the absence of suppressive signals provided by Tregs, potentially explaining the increased frequency of circulating T cells we observed in the tumor and circulation.
Collectively, these data show that Treg-depletion is sufficient to induce some intratumoral T cell activation, but the addition of ICB further increases the frequency and activation of intratumoral CD8+ T cells, leading to a robust infiltration of highly proliferative, polyfunctional and antigen-experienced T cells.
Depletion of Tregs reshapes the systemic and tumor myeloid immune compartment toward a favorable anti-tumor immune compartment
We next assessed the impact of Treg-depletion on the myeloid immune cell compartment. In the blood, depletion of Tregs, independently from ICB, induced an increase in eosinophil numbers, but did not affect the abundance of other myeloid cells (Figure 4A & S2D). Of note, a high eosinophil count in the blood of cancer patients treated with ICB is often associated with a better outcome40,52 and eosinophils have been recently shown to promote T cell infiltration and anti-tumor responses in mouse models53,54, including the KEP mouse model40. Treg-depletion, either with or without ICB, induced strong changes in the myeloid compartment in resected tumors. In line with previous research53,55, we observed that Treg-depletion promoted an increase in eosinophils and a decrease in conventional dendritic cells type 2 (cDC2) infiltration in the tumor (Figure 4B,C). The latter observation is in line with a previous study in melanoma model describing how Treg-depletion enhances the migration of cDC2s from the tumor to the TDLN, where they gained increased ability to prime and activate CD4+ T cells in absence of Tregs55. Lastly, we found a decrease in neutrophil frequency upon Treg-depletion (Figure 4D), suggestive of reduced immunosuppression in the TME, as we have previously demonstrated that neutrophils promote metastasis formation via CD8+ T cell suppression in KEP mice56.
Figure 4.

Neoadjuvant depletion of Tregs leads to a favorable systemic and intratumoral immune landscape.
(A) Absolute eosinophil counts (CD11b+Ly6GlowSiglecF+SSC-Ahigh) in blood of mice treated as indicated, determined by flow cytometry 1-2 days before mastectomy (“pre-mastectomy”, n = 5-8 mice/group). (B-D) Frequency of eosinophils (CD11b+Ly6GlowSiglecF+F4/80int) (B), classical dendritic cells type 2 (cDC2; CD11c+F4/80−MHC-IIhighCD11b+) (C), and neutrophils (CD11b+Ly6G+) (D) in resected tumors of mice treated as indicated, as determined by flow cytometry (n = 3-5). (E) Representative contour plots depicting PD-L1 expression on macrophages (CD11b+Ly6G−SiglecF−Ly6C−F4/80+) in resected tumors of mice treated as indicated. (F) Frequency of PD-L1+ cells within the indicated immune cell subset in resected tumors of mice treated as indicated, determined by flow cytometry (n = 5-8). (G) Representative contour plots depicting MHC-II expression on macrophages in resected tumors of mice treated as indicated. (H) Frequency of MCH-II+ macrophages in resected tumors of mice treated as indicated, determined by flow cytometry (n = 5-8). (I-J) Representative images and quantification of immunohistochemical staining of PD-L1 (I) and MHC-II (J) in resected tumors of mice treated as indicated. 10× magnifications, scale bar represents 100 µm. Whole tumor slides were evaluated and scored blindly on a scale from 0 to 5 (n = 6-10 mice/group). Data in A-D,F,H-J show mean ± SEM. P-values were calculated by One-way ANOVA with Sidak’s correction (A-D,H-J) or Two-way ANOVA with Sidak’s correction (F). ns, not significant, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
In addition to changes in myeloid cell infiltration, we found that Treg-depletion resulted in an upregulation of PD-L1 on various myeloid cells (Figure 4E,F). Baseline PD-L1 expression in the TME is a predictive biomarker for response to ICB57,58. We also found a strong increase in MHC-II-expressing tumor-associated macrophages (TAMs) (Figure 4G,H), reflective of M1-like polarization to which anti-tumoral functions such as direct cytotoxicity and improved antigen-presentation have been attributed59. The increased expression of PD-L1 and MHC-II in tumors upon Treg-depletion was confirmed by immunohistochemical staining (Figure 4I,J). Importantly, these changes were not further enhanced when Treg-depletion was combined with ICB.
Together, these data show that depletion of Tregs, independently from ICB, drives broad pro-inflammatory changes in intratumoral myeloid cells, potentially reshaping the TME into a favorable anti-tumor immune compartment.
Treg-depletion during neoadjuvant ICB induces durable systemic T cell activation and inhibits metastasis formation
To assess whether the favorable changes in the TME observed upon ICB and Treg-depletion influence tumor development, we monitored the growth of the orthotopically transplanted mammary tumors upon the different treatments (Figure 5A). We observed that none of the treatments significantly delayed tumor growth (Figure 5B & S3A). These data indicate that Treg depletion during ICB does not drive effective anti-tumor responses against primary mammary tumors, despite the major changes in the tumor immune landscape.
Figure 5.
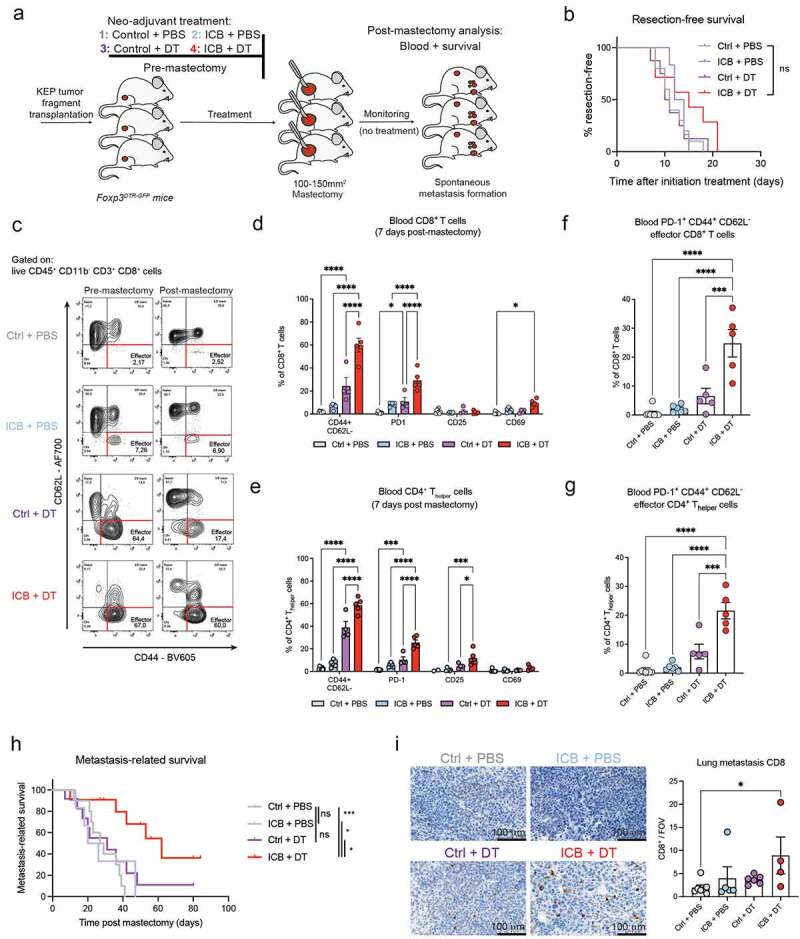
Neoadjuvant ICB combined with Treg-depletion induces durable systemic T cell activation and extends metastasis-related survival.
(A) Schematic overview of intervention study. Mice were treated as described in Figure 2A. After mastectomy, treatments were discontinued, and mice were monitored for the development of metastatic disease. (B) Kaplan-Meier curve displaying resection-free survival of mice treated as indicated, until the time they underwent mastectomy at tumor size ~120 mm2 (n = 7-10). (C) Representative contour plots depicting CD44 and CD62L expression on CD8+ T cells in blood of mice receiving indicated neoadjuvant treatments, analyzed 1-2 days before mastectomy (pre-mastectomy) and 7 days after mastectomy (post-mastectomy). (D-E) Frequency of indicated markers (% of positive cells) gated on CD8+ T cells (D) and CD4+ Thelper cells (E) in blood of mice post-mastectomy, previously treated as indicated (n = 6-8). (F-G) Frequency of PD-1+ CD44+ CD62L− effector CD8+ (F) and CD4+ (G) T cells in the blood of mice treated as indicated, analyzed by flow cytometry at post-mastectomy (n = 5-7). (H) Kaplan-Meier curve showing metastasis-related survival post mastectomy of mice treated with Ctrl+PBS (n = 10), ICB+PBS (n = 6, 1 censored), Ctrl+DT (n = 12, 2 censored), ICB+DT (n = 11, 4 censored). Censored cases indicate mice that were sacrificed due to metastasis-unrelated causes. (I) Representative image and quantification of immunohistochemical staining for CD8 in metastatic lesions in the lungs of mice previously treated in the neoadjuvant setting as indicated, analyzed at metastasis-related endpoint. Counts represent the average of 20-25 randomly selected 32× fields of views across metastatic lesions (n = 4-7 mice/group; scale bar 100 µm). Data in D-G,I show mean ± SEM. P-values were calculated by Log-rank (Mantel-Cox) test (B,H), Two-way ANOVA with Sidak’s correction (D,E), One-way ANOVA with Tukey’s correction (F,G), Kruskal-Wallis with Dunn’s correction (I). ns, not significant, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Ablation of Tregs during ICB mobilized both CD8+ T cells and NK cells in blood (Figure 2B,C), raising the question whether Tregs functionally impair systemic immune activation necessary to combat metastasis. To investigate this, we monitored the mice for development of overt metastatic disease after resection of primary tumors and cessation of neoadjuvant treatment (Figure 5A). In addition, T cell activation was analyzed in blood 7 days after mastectomy (“post-mastectomy”). Despite the discontinuation of the treatment at mastectomy, Tregs were still found to be increased in blood post-mastectomy in mice previously with ICB (figure S3B). These Tregs displayed increased expression of CD44 and PD-1, which was not observed in the initial pre-mastectomy characterization (figure S3C,D). Interestingly, CD44+ Tregs have been described to have strong immunosuppressive potential and play an important role in curbing autoimmunity60. In addition, a recent study showed that PD-1+ Tregs gain increased proliferative and immunosuppressive capacity upon PD-1 blockade in vitro 29,33. Thus, our data indicate that neoadjuvant ICB does not only drive the expansion of Tregs in blood, but also induces long-term phenotypical changes that are associated with Treg activation.
Our post-mastectomy analysis further demonstrated that both the CD8+ and CD4+ T cell compartments in the blood of mice previously treated with ICB+DT harbored a significantly increased frequency of CD44+CD62L− effector cells (Figure 5C-E). In addition, both CD8+ and CD4+ T cells in mice previously treated with ICB+DT showed increased expression of different activation markers (Figure 5D,E). Of note, CD69 expression in the circulation was negligible compared to the tumor (Figure 3D,E), likely due to the rapid up- and down-regulation of this marker after T cell activation. Interestingly, the T cell activation data post-mastectomy differs with what we observed at the pre-mastectomy time-point, where Treg-depletion alone was sufficient to promote a marked increase in activated effector T cells, independently from ICB (Figure 2F,G and 5C-E). These data suggest that neoadjuvant ICB in combination with Treg-depletion induces a more durable effect on circulating T cells, compared to Treg-depletion alone. This is further supported by the observation that the elevated levels of PD-1+ effector T cells we observed upon ICB+DT pre-mastectomy (Figure 2H), remain equally high post-mastectomy, whereas their levels are much lower upon Treg-depletion alone (Figure 5F,G). Thus, systemic depletion of Tregs in the context of neoadjuvant ICB leads to a more durable T cell activation, and CD8+ T cells in particular, raising the question whether these systemic pro-inflammatory conditions may lead to enhanced anti-metastatic effects of combined treatment.
Analysis of metastasis-related survival showed that Ctrl-treated mice developed metastatic disease, characterized by respiratory distress and/or end-stage metastatic tumor burden in axillary or caudal lymph nodes (Figure 5H & S3E). Strikingly, whereas neoadjuvant ICB or Treg-depletion alone did not improve survival, combined ICB and Treg-depletion significantly prolonged metastasis-related survival (Figure 5H & S3E). In the end, most mice succumbed to metastatic disease, thus we assessed whether there is a shift in location where metastases arise upon the different treatments. In line with previously published results describing an important role for Tregs in the development of lymph node metastasis25, we did not detect any lymph node metastasis in Treg-depleted mice, either with or without addition of ICB (figure S3F). The majority of mice in all treatment groups eventually developed lung metastases. Strikingly, immunohistochemical assessment of different immune cell parameters in the lung metastatic lesions, revealed a specific increase in CD8 counts in mice treated with neoadjuvant ICB+DT (Figure 5I & S3G-J). Considering that, on average, these mice developed respiratory distress around 7 weeks after mastectomy and cessation of treatment, this finding emphasizes the durability of the effects of this therapy combination on CD8+ T cells at the metastatic site.
As Treg deficiency and ICB are respectively associated with the development of autoimmune-related pathology45,61 and immune-related adverse events (irAEs) in patients, we investigated whether combining ICB with Treg-depletion exacerbates inflammation-related pathology compared to either ICB-treated or Treg-depleted mice. As expected, we observed an evident increase in autoimmunity-related pathology upon Treg-depletion, but no clear differences were observed between mice treated with Ctrl+DT and ICB+DT (figure S4A,B and supplementary table 1). In addition, no differences were observed in sizes of spleen and small intestine (figure S4C).
Altogether, these data show that the combination of neoadjuvant ICB and Treg-depletion promotes a synergistic anti-metastatic response without exacerbating irAEs induced by the depletion of Tregs, suggesting that neoadjuvant ICB can be empowered by simultaneous Treg-targeting, extending metastasis-related survival, independent of primary tumor response.
Therapeutic benefit of Treg-depletion during neoadjuvant ICB predominantly depends on CD8+ T cells
Since the combination of ICB and Treg-depletion specifically increased numbers of NK cells and durably activated, antigen-experienced CD8+ T cells in the circulation as well as increased CD8 counts in lung metastatic lesions, we assessed whether these two cell types are involved in the anti-metastatic response. Mice receiving anti-CD8 or anti-NK1.1 antibody during ICB+DT therapy displayed efficient CD8 or NK cell-depletion in blood pre-mastectomy and the reduction in cell frequencies remained apparent at least up to 1 month after discontinuation of treatment (figure S5A,B). CD8+ T cell-depletion showed a trend in reverting the therapeutic benefit of neoadjuvant ICB+DT (figure S5C, p = 0.12). In contrast, NK cell-depletion did not affect therapeutic benefit of treatment (figure S5C, p = 0.64). The pattern in which organ metastases arose was unaltered upon either CD8 or NK cell-depletion during ICB+DT (figure S5D). Of note, the discrepancy with our previous work where we showed that NK cell-depletion in context of anti-CD25-mediated Treg-depletion restores metastasis to the lymph nodes25, may be explained by the Treg-depletion strategy and addition of ICB to the treatment regimen. Collectively, these data suggest that CD8+ T cells contribute to the anti-metastatic response elicited by neoadjuvant ICB and Treg-depletion, but they are not the sole driver. Interestingly, neither CD8 or NK cell-depletion affected the changes in intratumoral myeloid compartment observed upon Treg-depletion (figure S5E-H). Collectively, these data suggest that the mechanisms driving the therapeutic benefit of ICB + Treg-depletion are multifactorial, with durably activated CD8+ T cells being an important, but not unique, driver of anti-metastatic immune response.
Discussion
Here, we used the clinically relevant KEP-based mastectomy model for spontaneous multi-organ breast cancer metastasis39, which is unresponsive to anti-PD-1/anti-CTLA4 therapy, to demonstrate that systemic immunosuppression driven by Tregs prevents an effective anti-metastatic immune response upon neoadjuvant immunotherapy. Moreover, our findings demonstrate that absence of therapeutic benefit in the primary tumor upon neoadjuvant therapy does not exclude therapeutic benefit against the future development of metastatic disease, indicative of the initiation of long-term beneficial systemic effects of neoadjuvant immunotherapy-based strategies. Our findings demonstrate that there is a window of opportunity for improving neoadjuvant ICB by simultaneously targeting of Tregs, inducing systemic anti-tumor immunity that delays metastatic outgrowth.
The ICB-induced accumulation and activation of Tregs we observed in mammary tumor-bearing mice and corroborated in breast cancer patients, affected both intratumoral and systemic immunosuppression, antagonizing anti-tumor immunity and regulation of metastasis. Based on our findings, we propose several mechanisms that may contribute to improved control of metastases observed upon Treg-depletion during neoadjuvant ICB. In tumors, Treg-depletion resulted in the upregulation of PD-1 on CD8+ T cells and PD-L1 on myeloid cells, which are both linked to response to anti-PD-129,57,58. PD-1 expression is a bona fide marker to identify tumor-specific CD8+ T cells49, as tumor-reactive T cells are identified within the PD-1+ CD8+ T cell population in human cancers50,51. In addition, we found increased frequency of IFNγ– and TNFα-producing CD8+ T cells in the tumor, indicative of their cytotoxic functionality. However, our CD8 depletion experiment demonstrated only a partial reversion of the therapeutic benefit of ICB + Treg-depletion, suggesting that the anti-metastatic response is multifactorial. In line with previous studies18,53, we find that Treg-depletion additionally reshaped the TME into a more pro-inflammatory, anti-tumorigenic environment, most notably characterized by increased infiltration of eosinophils and anti-tumor macrophage polarization. These populations have been described to have direct tumor-killing capacities62,63 as well as to promote CD8+ T cell activation, via amongst others, expression of T cell-recruiting and activating chemokines such as CXCL9 and increased antigen presentation capacity54,64,65. Interestingly, increased systemic and intratumoral eosinophil accumulation has previously been associated with ICB response in melanoma and TNBC patients40,52 and linked to anti-tumorigenic activity in response to ICB in mouse models of primary and metastatic breast cancer, including the KEP model40,54. We hypothesize that these pro-inflammatory conditions induced intratumorally by Treg-depletion, may have contributed in combination with ICB to the development of a robust anti-metastatic immune response.
In line with this hypothesis, we found that the combination of Treg-depletion and ICB synergistically increased the number of circulating CD8+ and CD4+CD8+ T cells, NK cells, and induced durable CD8+ T cell activation. Noticeably, increased CD8 counts were observed in metastatic lesions of mice treated with ICB+DT until time of mastectomy, which was on average 7 weeks before development of lung metastasis, indicative of the durability of the CD8+ T cell response induced by this neoadjuvant immunotherapeutic strategy. The persistent activation of T cells could be the product of improved priming of T cells in absence of Tregs and the consequent increased availability of IL-2 for CD8+ T cells. Whether the higher number of activated T cells is caused by an increased lifespan of optimally primed T cells or by the enhanced output or proliferation of activated T cells remains to be investigated. We speculate this persistent CD8+ T cell activation may confer protection against circulating cancer cells or metastasis formation, leading to improved survival. We demonstrate that CD8+ T cells are one of the main drivers of anti-metastatic immune response elicited by ICB and Treg-depletion, but other mechanisms likely contribute as well. Interesting, the favorable changes in the TME induced by ICB+DT were not affected by CD8+ T cell depletion. Which of these anti-cancer mechanisms are most important to curb metastasis, and by which underlying immune cell crosstalk Tregs suppress anti-metastatic immunity, remains a topic of future research.
In contrast to previous studies using more immunogenic tumor cell lines34–37, we observed no therapeutic benefit of Treg-targeting alone and the anti-tumoral effects of ICB + Treg-depletion were only observed in the metastatic context, and not the primary tumor. This suggests that additional hurdles for anti-tumor immunity are in place in primary KEP tumors, that are unrelated to Tregs. Previous research using the KEP model has shown that besides Tregs, primary tumors are infiltrated by immunosuppressive neutrophils56 and macrophages66, recapitulating the tumor immune landscape of breast cancer patients67,68. We speculate that these cells prevent effective anti-tumor immunity in the TME also in the absence of Tregs. Moreover, we cannot exclude contribution of other factors that were not included in our analysis such as IL-17-producing Th17 cells, whose crosstalk with Tregs is emerging as playing a role in both immunotherapy efficacy and toxicity69. Our findings are in line with a clinical study in TNBC patients that observed significantly improved survival after neoadjuvant anti-PD-L1 plus chemotherapy, despite showing only a modest increase in pathological complete response9, emphasizing that the absence of a pathological complete response does not exclude therapeutic benefit against metastatic spread upon neoadjuvant immunotherapy-based treatment regimens. Moreover, our data support further investigation into the unresolved clinical question whether continued immunotherapy after surgery adds benefit to neoadjuvant immunotherapy alone70.
We found that blockade of the PD-1/PD-L1-axis induces systemic and intratumoral Treg expansion in the blood and tumors of patients with metastatic TNBC or ILC, respectively, corroborating our findings in the mammary tumor models. Our results concerning the adverse role of Tregs in the response to immunotherapy are consistent with clinical data which have revealed correlations between PD-1+ Tregs and therapy response, relapse, and hyperprogressive disease in NCSLC, melanoma, and gastric cancer, respectively29,32,33, as well as an association between Treg proliferation and recurrence in melanoma patients32. Preclinical studies using inoculated B16 and MC38 cell line tumor models have shown that PD-1 blockade reactivates the proliferative and immunosuppressive capacity of PD-1+ Tregs, thereby promoting tumor growth29,33. Furthermore, the efficacy of PD-1 blockade was shown to be dependent on high PD-1 expression on CD8+ T cells, but low PD-1 expression on Tregs in the tumor29. Some mouse studies report that the therapeutic benefit of anti-CTLA-4 depends, at least in part, on Fcγ–receptor-dependent depletion of intratumoral Tregs71, while other studies found that anti-CTLA4 induces proliferation of tumor-associated Tregs in MC38 tumor-bearing mice72. In line with this latter study, and clinical observations showing that anti-CTLA-4 treatment expands immunosuppressive Tregs in blood and tumors of prostate, melanoma, and bladder cancer patients30,31, we did not observe Treg-depletion upon anti-PD-1/anti-CTLA-4 therapy but rather found strong accumulation and increased proliferation of Tregs. Of note, our study was not designed to tease apart the individual contributions of anti-PD-1 and anti-CTLA-4 on Tregs, since anti-CTLA-4 is rarely used for the treatment of breast cancer patients without the addition of anti-PD-1. We found that after discontinuation of treatment, the frequency of CD44+ and PD-1+ Tregs further increased in blood of mice that received neoadjuvant ICB, suggesting that ICB induces long-lasting systemic Treg activation. Whether Tregs respond directly to ICB by enhancing their proliferation and immunosuppressive activity33,72, or whether ICB-induced Treg expansion is due to upregulation of immunoregulatory feedback mechanisms upon an ongoing CD8+ T cell response32, remains to be investigated. Nevertheless, molecular understanding of how ICB induces Tregs may support the development of immunotherapeutic strategies that selectively activate conventional T cells, but not Tregs.
Finally, our data suggest that combining neoadjuvant ICB with Treg-targeting strategies is a potential avenue to improve ICB responses and combat metastasis. Due to the critical role of Tregs in prevention of auto-immune-related diseases, approaches that specifically deplete intratumoral Tregs or that only partially deplete Tregs, by targeting for example CCR8, OX-40, CCR4 and CD2513,33,35,37,73, may be more feasible for use in cancer patients. As this study provides proof-of-principle that Tregs impair anti-tumor immunity in the context neoadjuvant ICB, it will be crucial to identify how the variety of immunomodulatory drugs that are in clinical development will affect Treg activation beyond anti-PD-1 and anti-CTLA-4. These future studies may contribute to improved clinical decision making regarding the use of Treg-activating immunomodulatory drugs in cancer patients with abundant intratumoral accumulation of Tregs. Collectively, our data suggest that there is a window of opportunity for improving neoadjuvant ICB therapy to attenuate metastatic spread by simultaneously targeting of Tregs.
Material and methods
Mice
This study used wild-type FVB/N mice obtained from Janvier Labs and Keratin14(K14)-cre;Cdh1F/F;Trp53F/F (KEP)22 and Cdh1F/F;Trp53F/F;Foxp3GFP-DTR mice (referred to as Foxp3GFP-DTR) on FVB/N background generated and bred in the Netherlands Cancer Institute. Of note, Foxp3GFP-DTR mice have been generated on the KEP genetic background to ensure full match in genetic background. Starting at 6–7 weeks of age, female KEP mice were monitored twice weekly for the development of spontaneous mammary tumors. Upon mammary tumor formation, perpendicular tumor diameters were measured twice weekly using a caliper. Tumor-related endpoint was defined as cumulative tumor burden of 225 mm2. Mice were kept in individually ventilated cages at the animal laboratory facility of the Netherlands Cancer Institute under specific pathogen-free conditions. Food and water were provided ad libitum. All animal experiments were approved by the Netherlands Cancer Institute Animal Ethics Committee (license numbers AVD30100202215835 and AVD3010020172688), and performed in accordance with institutional, national, and European guidelines for Animal Care and Use.
WEAP tumor model
The Wap-cre;Cdh1F/F;AktE17K (WEA) tumor cell line was derived from a spontaneous tumor of a genetically engineered Wap-cre;Cdh1F/F;AktE17K (WEA) mouse as previously described42,43. Endogenous p53 was deleted from the WEA cell line to generate Wap-cre;Cdh1F/F;AktE17K ;Trp53KO (WEAP) as previously described42. To ensure relatedness to the parental tumor, a low passage of the generated polyclonal cell line was used for the intervention study. Cell suspensions of 5 × 105 WEAP tumor cells were injected into the mammary fat pad of female recipient 8–10-week-old wild-type FVB/N mice.
KEP metastasis model
The KEP metastasis model has been applied as previously described39. In short, KEP tumor fragments were orthotopically transplanted into the mammary fat pad of female recipient 8–16-week-old Foxp3DTR-GFP mice. Upon tumor outgrowth to a size of 100-150 mm2, tumors were surgically resected. Following mastectomy, mice were monitored for development of overt metastatic disease by daily palpation and observation of physical health, appearance, and behavior. Metastasis-related endpoint was defined as mice displaying signs of respiratory distress caused by metastatic disease or when lymph node metastasis reached the size of 225 mm2. Censored events are mice sacrificed for tumor- or metastasis-unrelated events including weight loss or local recurrence of the mastectomized tumor. At metastasis-related endpoint, lungs and axillary lymph nodes were collected and analyzed microscopically for the presence of metastatic foci by immunohistochemical cytokeratin 8 staining.
Intervention studies
Antibody treatments in KEP mice and transplanted KEP or WEAP tumor models in wild-type FVB/N mice were initiated at a tumor size of 25 mm2. Antibody treatments were initiated at 4 mm2 in orthotopic KEP tumor transplantation experiments in Foxp3GFP-DTR mice. Mice were intraperitoneally injected twice weekly with ICB; 100 µg of anti-PD-1 (clone RMP1–14, BioXCell) and 100 µg of anti-CTLA-4 (clone 9D9, BioXCell) or control; 100 µg rat IgG2a (clone 2A3, BioXCell). For cell depletion studies, mice were treated with 200 µg of anti-CD8 (clone 2.43, BioXCell) once a week with maximum of 3 injections or with an initial 400 µg, followed by 200 µg of anti-NK1.1 (clone PK136, BioXCell) once a week with a maximum of 4 injections. For depletion of Tregs, mice were treated with two doses of 25 µg/kg diphtheria toxin (Sigma) or PBS, starting at tumor size of 6-9 mm2 and again on day 4. All treatments were discontinued at cumulative tumor burden of 225 mm2 in the KEP and WEAP model, or upon mastectomy for KEP transplantation and metastasis experiments.
Flow cytometry analysis and cell sorting
Draining lymph nodes and tumors were collected in ice-cold PBS, and blood was collected via cardiac or tail vein puncture in heparin-containing tubes. Tissues were processed as previously described74. Blood erythrocyte lysis was performed in NH4Cl buffer for 5 min. For intracellular cytokine assessment, single cell suspensions were stimulated ex vivo with 50 ng/ml PMA, 1 μM ionomycin and Golgi-Plug (1:1000; BD) for 3 h at 37°C in IMDM medium supplemented with 8% FCS, 100 IU/ml Penicillin-Streptomycin (Roche) and 0.5% β-mercaptoethanol. For surface antigen staining, cells were incubated for 20 min with anti-CD16/32 (2.4G2, BD Biosciences), to block unspecific Fc receptor binding, and fluorochrome-conjugated antibodies diluted in FACS buffer (2.5% FBS, 2 mM EDTA in PBS). For analysis of intracellular proteins, cells were fixed and permeabilized after surface and live/dead staining using the FOXP3 Transcription buffer set (Thermofisher), according to manufacturer’s instruction. Fixation, permeabilization and intracellular staining was performed for 30 min. Data was analyzed on BD Symphony SORP or sorted on a FACS ARIA II (4 lasers). Absolute cell counts were determined using 123count eBeads (ThermoFisher) according to manufacturer’s instructions. The antibodies and viability-detection reagents used in this study are listed in supplemental table 2.
Clinical data
The clinical data used in the study were kindly provided by the investigators of the GELATO and TONIC trials in the Netherlands Cancer Institute. Clinical trial procedures were performed as described in their respective publications2,44. Briefly, the TONIC trial evaluated the efficacy of nivolumab (anti-PD-1) after a two-week induction therapy with low-dose chemotherapy or irradiation in patients with metastatic triple-negative breast cancer. Blood was drawn before start of treatment (baseline) and after three cycles of anti-PD-1 (on-nivolumab). The GELATO trial evaluated the efficacy of induction therapy with two cycles of low-dose carboplatin followed by combined atezolizumab (anti-PD-L1) and carboplatin treatment in patients with metastatic invasive lobular carcinoma. Sequential tumor biopsies from a metastatic lesion were taken before start treatment (baseline), after two weeks of carboplatin treatment (post-induction), and after two cycles of anti-PD-L1 (on-atezolizumab). Flow cytometry analysis of fresh blood samples was performed as previously described2,44. The clinical studies were approved by the medical-ethical committee of the NKI and conducted in accordance with ICH Harmonized Tripartite Guideline for Good Clinical Practice and the principles of the Declaration of Helsinki. All patients provided written informed consent to participate.
RNA-Seq data analysis
The RNA-Seq data was aligned to the reference genome GRCh38 with STAR version 2.7.1a75 with two-pass mode option set to “Basic” and gene counts were obtained using STAR-quantMode = GeneCounts option. FPKM were then transformed to TPM and log2 transformed. Data was analyzed with Python 3.7.6 and R 4.1.1. Pandas 1.3.376,77 was used for data handling. Seaborn 0.10.078, Matplotlib 3.1.379 and statannotations 0.4.380 have been used for plotting.
Treg suppression assays
Treg-T cell suppression assays were performed as previously described74. Tregs (Live CD45+CD3+CD8−CD4+CD25high) sorted from freshly isolated lymph nodes were activated overnight in IMDM containing 8% FCS, 100 IU/ml penicillin, 100 μg/ml streptomycin, 0.5% β-mercapto-ethanol, 300 U/mL IL-2, 1:5 bead:cell ratio CD3/CD28 coated beads (Thermofisher). Per condition, 2.5 × 104 cells were seeded in 96-wells plate, which were further diluted to appropriate ratios (1:2–1:16). Responder cells isolated from spleen (Live, CD45+CD3+CD4+CD25− and Live CD45+CD3+CD8+) were rested overnight. Next, responder cells were labeled with CellTraceViolet, and co-cultured with Tregs in cIMDM supplemented with CD3/CD28 beads (1:5 bead cell ratio) for 96 hours (without exogenous IL-2).
Immunohistochemistry
Immunohistochemical analyses were performed by the Animal Pathology facility at the Netherlands Cancer Institute. Formalin-fixed tissues were processed, sectioned and stained as described39.
Statistical analysis
Data analyses were performed using GraphPad Prism (version 9). Data show means ± SEM, unless stated otherwise. The statistical tests used are described in figure legends. P-values<0.05 were considered statistically significant. In vivo interventions were performed once with indicated sample sizes, unless otherwise indicated. In vitro experiments were repeated independently as indicated.
Supplementary Material
Acknowledgments
We thank the members of the Tumor Biology & Immunology Department at the Netherlands Cancer Institute for their insightful input. We thank the flow cytometry facility, animal laboratory facility, transgenesis facility and animal pathology facility of the Netherlands Cancer Institute for technical assistance.
Funding Statement
Research in the De Visser lab is funded by the Netherlands Organization for Scientific Research (NWO-VICI91819616), Dutch Cancer Society (KWF10083; KWF10623; KWF13191), Oncode Institute, and the KWF/Oncode consortium grant 14339. Additional funding was granted by NWO Oncology Graduate School Amsterdam Diamond Program to K.Ko. and by the Swiss National Science Foundation (P2FRP3_171794 and P400PM_18318/1) to L.S. The Dutch Cancer Society (10653ALPE) and A Sister’s Hope contributed to the blood immunophenotyping of the TNBC patients. The TONIC study was funded by BMS-International Immuno-Oncology Network (BMS/II-ON) and the Dutch Cancer Society (NKI2015-7710). The GELATO study was funded by F. Hoffmann-La Roche Ltd, Basel, Switzerland. This research was further supported by an institutional grant to the NKI of the Dutch Cancer Society and of the Dutch Ministry of Health, Welfare and Sport.
Disclosure statement
K.E.d.V. reports research funding from Roche and is consultant for Macomics, outside the scope of this work. M.K. reports funding to the institute from BMS, Roche/Genentech, AZ and an advisory role for BMS, Roche, MSD and Daiichi Sankyo, outside the submitted work.
Data availability statement
Data are available from the authors on reasonable request. The RNAseq data of the GELATO trial is available as previously described. Voorwerk L, Isaeva OI, Horlings HM, Balduzzi S, Chelushkin M, Bakker NAM, Champanhet E, Garner H, Sikorska K, Loo CE, Kemper I, Mandjes IAM, de Maaker M, van Geel JJL, Boers J, de Boer M, Salgado R, van Dongen MGJ, Sonke GS, de Visser KE, Schumacher TN, Blank CU, Wessels LFA, Jager A, Tjan-Heijnen VCG, Schröder CP, Linn SC, Kok M. PD-L1 blockade in combination with carboplatin as immune induction in metastatic lobular breast cancer: the GELATO trial. Nat Cancer. 2023 Apr 10. doi: 10.1038/s43018-023-00542-x. Epub ahead of print. PMID: 37038006.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2023.2201147
Authors’ contributions
O.S.B., K.Ko., L.S., and K.E.d.V. conceived the ideas and designed the experiments. O.S.B., K.Ko., L.S., M.D.W., D.E.M.D., and K.Ke. performed experiments and data analysis. C-S.H., D.K., E.A.M.R., and K.V. performed animal experiments. S.K. did the pathological assessments. Clinical data acquisition and analysis was done by O.I.I., H.G., and N.B. M.K. is the principal investigator of the TONIC and GELATO trials. K.E.d.V. supervised the study. K.E.d.V, K.Ko., and L.S. acquired funding. O.S.B., K.Ko, L.S., and K.E.d.V. wrote the paper and prepared the figures with input from all authors.
References
- 1.Adams S, Loi S, Toppmeyer D, Cescon DW, De Laurentiis M, Nanda R, Winer EP, Mukai H, Tamura K, Armstrong A, et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: cohort B of the phase II KEYNOTE-086 study. Annal Oncol. 2019;30(3):405–16. published Online First: 2018/11/27. doi: 10.1093/annonc/mdy518. [DOI] [PubMed] [Google Scholar]
- 2.Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, Nederlof I, Kluin RJC, Warren S, Ong S, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med. 2019;25(6):920–928. published Online First: 2019/05/16. doi: 10.1038/s41591-019-0432-4. [DOI] [PubMed] [Google Scholar]
- 3.Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im S-A, Yusof MM, Gallardo C, Lipatov O, Barrios CH, Holgado E, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet. 2020;396(10265):1817–1828. published Online First: 2020/12/07. doi: 10.1016/s0140-6736(20)32531-9. [DOI] [PubMed] [Google Scholar]
- 4.Maio M, Blank C, Necchi A, Di Giacomo AM, Ibrahim R, Lahn M, Fox BA, Bell RB, Tortora G, Eggermont AMM.. Neoadjuvant immunotherapy is reshaping cancer management across multiple tumour types: the future is now! Eur J Cancer. 2021;152:155–164. doi: 10.1016/j.ejca.2021.04.035. published Online First: 20210606. [DOI] [PubMed] [Google Scholar]
- 5.Chalabi M, Fanchi LF, Dijkstra KK, Van den Berg JG, Aalbers AG, Sikorska K, Lopez-Yurda M, Grootscholten C, Beets GL, Snaebjornsson P, et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med. 2020;26(4):566–576. published Online First: 2020/04/07. doi: 10.1038/s41591-020-0805-8. [DOI] [PubMed] [Google Scholar]
- 6.Blank CU, Rozeman EA, Fanchi LF, Sikorska K, van de Wiel B, Kvistborg P, Krijgsman O, van den Braber M, Philips D, Broeks A, et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat Med. 2018;24(11):1655–1661. published Online First: 2018/10/10. doi: 10.1038/s41591-018-0198-0. [DOI] [PubMed] [Google Scholar]
- 7.Schmid P, Cortes J, Dent R, Pusztai L, McArthur H, Kümmel S, Bergh J, Denkert C, Park YH, Hui R, et al. Event-free survival with pembrolizumab in early triple-negative breast cancer. N Engl J Med. 2022;386(6):556–567. doi: 10.1056/NEJMoa2112651. [DOI] [PubMed] [Google Scholar]
- 8.Mittendorf EA, Zhang H, Barrios CH, Saji S, Jung KH, Hegg R, Koehler A, Sohn J, Iwata H, Telli ML, et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet. 2020;396(10257):1090–1100. published Online First: 20200920. doi: 10.1016/S0140-6736(20)31953-X. [DOI] [PubMed] [Google Scholar]
- 9.Loibl S, Schneeweiss A, Huober J, Braun M, Rey J, Blohmer J-U, Furlanetto J, Zahm D-M, Hanusch C, Thomalla J, et al. Neoadjuvant durvalumab improves survival in early triple-negative breast cancer independent of pathological complete response. Ann Oncol. 2022;33(11):1149–1158. published Online First: 20220809. doi: 10.1016/j.annonc.2022.07.1940. [DOI] [PubMed] [Google Scholar]
- 10.Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. Systemic immunity is required for effective cancer immunotherapy. Cell. 2017;168(3):487–502.e15. published Online First: 20170119. doi: 10.1016/j.cell.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fransen MF, Schoonderwoerd M, Knopf P, Camps MGM, Hawinkels LJAC, Kneilling M, van Hall T, Ossendorp F. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight. 2018;3(23). published Online First: 20181206. doi: 10.1172/jci.insight.124507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garner H, de Visser KE. Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat Rev Immunol. 2020;20(8):483–497. doi: 10.1038/s41577-019-0271-z. published Online First: 20200205. [DOI] [PubMed] [Google Scholar]
- 13.Plitas GY, Rudensky AY. Regulatory T cells in cancer. Annu Rev Cancer Biol. 2020;4(1):459–477. doi: 10.1146/annurev-cancerbio-030419-033428. [DOI] [Google Scholar]
- 14.Kos K, de Visser KE. The multifaceted role of regulatory T cells in breast cancer. Annu Rev Cancer Biol. 2021. published Online First: 2021/10/12;5(1):291–310. doi: 10.1146/annurev-cancerbio-042920-104912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva E, Chudakov D, Rudensky A. Regulatory T cells exhibit distinct features in Human Breast Cancer. Immunity. 2016;45(5):1122–1134. doi: 10.1016/j.immuni.2016.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu S, Foulkes WD, Leung S, Gao D, Lau S, Kos Z, Nielsen TO. Prognostic significance of FOXP3+ tumor-infiltrating lymphocytes in breast cancer depends on estrogen receptor and human epidermal growth factor receptor-2 expression status and concurrent cytotoxic T-cell infiltration. Breast Cancer Res. 2014. published Online First: 20140906;16(5):432. doi: 10.1186/s13058-014-0432-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med. 2013. published Online First: 2013/10/16;210(11):2435–2466. doi: 10.1084/jem.20130762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark NM, Martinez LM, Murdock S, deLigio JT, Olex AL, Effi C, Dozmorov MG, Bos PD. Regulatory T cells support breast cancer progression by opposing IFN-γ-dependent functional reprogramming of Myeloid Cells. Cell Rep. 2020. published Online First: 2020/12/10;33(10):108482. doi: 10.1016/j.celrep.2020.108482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Decker T, Fischer G, Bücke W, Bücke P, Stotz F, Grüneberger A, Gropp-Meier M, Wiedemann G, Pfeiffer C, Peschel C, et al. Increased number of regulatory T cells (T-regs) in the peripheral blood of patients with Her-2/neu-positive early breast cancer. J Cancer Res Clin Oncol. 2012;138(11):1945–1950. published Online First: 20120704. doi: 10.1007/s00432-012-1258-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez SA, Karamouzis MV, Skarlos DV, Ardavanis A, Sotiriadou NN, Iliopoulou EG, Salagianni ML, Orphanos G, Baxevanis CN, Rigatos G, et al. CD4+CD25+ regulatory T-cell frequency in HER-2/neu (HER)-positive and HER-negative advanced-stage breast cancer patients. Clin Cancer Res. 2007;13(9):2714–2721. doi: 10.1158/1078-0432.ccr-06-2347. [DOI] [PubMed] [Google Scholar]
- 21.Wang L, Simons DL, Lu X, Tu TY, Solomon S, Wang R, Rosario A, Avalos C, Schmolze D, Yim J, et al. Connecting blood and intratumoral T(reg) cell activity in predicting future relapse in breast cancer. Nat Immunol. 2019;20(9):1220–1230. published Online First: 20190708. doi: 10.1038/s41590-019-0429-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang D, Gao Z, Cai Z, Wang M, He J. Clinicopathological and prognostic significance of FOXP3+ tumor infiltrating lymphocytes in patients with breast cancer: a meta-analysis. BMC Cancer. 2015. published Online First: 20151017;15(1):727. doi: 10.1186/s12885-015-1742-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Núñez NG, Tosello Boari J, Ramos RN, Richer W, Cagnard N, Anderfuhren CD, Niborski LL, Bigot J, Meseure D, De La Rochere P, et al. Tumor invasion in draining lymph nodes is associated with Treg accumulation in breast cancer patients. Nat Commun. 2020;11(1):3272. published Online First: 20200629. doi: 10.1038/s41467-020-17046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Pul KM, Vuylsteke R, van de Ven R, van Pul KM, van de Ven R, Te Velde EA, Rutgers EJT, van den Tol PM, Stockmann HBAC, de Gruijl TD. Selectively hampered activation of lymph node-resident dendritic cells precedes profound T cell suppression and metastatic spread in the breast cancer sentinel lymph node. J ImmunoTher Cancer. 2019. published Online First: 20190522;7(1):133. doi: 10.1186/s40425-019-0605-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kos K, Aslam MA, van de Ven R, Wellenstein MD, Pieters W, van Weverwijk A, Duits DEM, van Pul K, Hau C-S, Vrijland K, et al. Tumor-educated T(regs) drive organ-specific metastasis in breast cancer by impairing NK cells in the lymph node niche. Cell Rep. 2022;38(9):110447. doi: 10.1016/j.celrep.2022.110447. [DOI] [PubMed] [Google Scholar]
- 26.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013. published Online First: 2013/07/31;39(1):1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 27.Montler R, Bell RB, Thalhofer C, Leidner R, Feng Z, Fox BA, Cheng AC, Bui TG, Tucker C, Hoen H, et al. OX40, PD-1 and CTLA-4 are selectively expressed on tumor-infiltrating T cells in head and neck cancer. Clin Transl Immunol. 2016;5(4):e70. published Online First: 20160415. doi: 10.1038/cti.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin Y-T, Togashi Y, Kamada T, Irie T, Okumura G, Kono H, et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. 2022;40(2):201–18.e9. published Online First: 20220128. doi: 10.1016/j.ccell.2022.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, Vitaly K, Itahashi K, Maeda Y, Matsui S, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. 2020;21(11):1346–1358. published Online First: 2020/09/02. doi: 10.1038/s41590-020-0769-3. [DOI] [PubMed] [Google Scholar]
- 30.Kavanagh B, O’Brien S, Lee D, et al. CTLA4 blockade expands FoxP3+ regulatory and activated effector CD4+ T cells in a dose-dependent fashion. Blood. 2008;112(4):1175–1183. published Online First: 2008/06/05. doi: 10.1182/blood-2007-11-125435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, Allison JP, Ribas A, Sharma P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3(+) Regulatory T Cells (Tregs) in Human Cancers. Clin Cancer Res. 2019. published Online First: 2018/07/29;25(4):1233–1238. doi: 10.1158/1078-0432.CCR-18-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, Manne S, Kraya AA, Wubbenhorst B, Dorfman L, et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med. 2019;25(3):454–461. published Online First: 2019/02/26. doi: 10.1038/s41591-019-0357-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, Sato E, Fukuoka S, Tada Y, Tanaka A, et al. PD-1 + regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A. 2019;116(20):9999–10008. published Online First: 2019/04/28. doi: 10.1073/pnas.1822001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor NA, Vick SC, Iglesia MD, Brickey WJ, Midkiff BR, McKinnon KP, Reisdorf S, Anders CK, Carey LA, Parker JS, et al. Treg depletion potentiates checkpoint inhibition in claudin-low breast cancer. J Clin Invest. 2017;127(9):3472–3483. published Online First: 20170821. doi: 10.1172/jci90499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solomon I, Amann M, Goubier A, Arce Vargas F, Zervas D, Qing C, Henry JY, Ghorani E, Akarca AU, Marafioti T, et al. CD25-T(reg)-depleting antibodies preserving IL-2 signaling on effector T cells enhance effector activation and antitumor immunity. Nat Cancer. 2020;1(12):1153–1166. published Online First: 20201109. doi: 10.1038/s43018-020-00133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin Y, An X, Mao B, Sun R, Kumari R, Chen X, Shan Y, Zang M, Xu L, Muntel J, et al. Different syngeneic tumors show distinctive intrinsic tumor-immunity and mechanisms of actions (MOA) of anti-PD-1 treatment. Sci Rep. 2022;12(1):3278. published Online First: 20220228. doi: 10.1038/s41598-022-07153-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Damme H, Dombrecht B, Kiss M, Roose H, Allen E, Van Overmeire E, Kancheva D, Martens L, Murgaski A, Bardet PMR, et al. Therapeutic depletion of CCR8 + tumor-infiltrating regulatory T cells elicits antitumor immunity and synergizes with anti-PD-1 therapy. J ImmunoTher Cancer. 2021;9(2):e001749. doi: 10.1136/jitc-2020-001749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Derksen PW, Liu X, Saridin F, van der Gulden H, Zevenhoven J, Evers B, van Beijnum JR, Griffioen AW, Vink J, Krimpenfort P, et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell. 2006;10(5):437–449. published Online First: 2006/11/14. doi: 10.1016/j.ccr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 39.Doornebal CW, Klarenbeek S, Braumuller TM, Klijn CN, Ciampricotti M, Hau C-S, Hollmann MW, Jonkers J, de Visser KE. A preclinical mouse model of invasive lobular breast cancer metastasis. Cancer Res. 2013. published Online First: 2012/11/16;73(1):353–363. doi: 10.1158/0008-5472.CAN-11-4208. [DOI] [PubMed] [Google Scholar]
- 40.Blomberg OS, Spagnuolo L, Garner H. IL-5-producing CD4(+) T cells and eosinophils cooperate to enhance response to immune checkpoint blockade in breast cancer. Cancer Cell. 2023;41(1):106–23.e10. published Online First: 20221215. doi: 10.1016/j.ccell.2022.11.014. [DOI] [PubMed] [Google Scholar]
- 41.Tavares MC, Sampaio CD, Lima GE, Andrade VP, Gonçalves DG, Macedo MP, Cordeiro de Lima VC. A high CD8 to FOXP3 ratio in the tumor stroma and expression of PTEN in tumor cells are associated with improved survival in non-metastatic triple-negative breast carcinoma. BMC Cancer. 2021. published Online First: 2021/08/08;21(1):901. doi: 10.1186/s12885-021-08636-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wellenstein MD, Coffelt SB, Duits DEM, van Miltenburg MH, Slagter M, de Rink I, Henneman L, Kas SM, Prekovic S, Hau C-S, et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature. 2019;572(7770):538–542. published Online First: 20190731. doi: 10.1038/s41586-019-1450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Annunziato S, Kas SM, Nethe M, Yücel H, Del Bravo J, Pritchard C, Bin Ali R, van Gerwen B, Siteur B, Drenth AP, et al. Modeling invasive lobular breast carcinoma by CRISPR/Cas9-mediated somatic genome editing of the mammary gland. Gen Devel. 2016;30(12):1470–1480. doi: 10.1101/gad.279190.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Voorwerk LIsaeva OI, Horlings HM, Balduzzi S, Chelushkin M, Bakker NAM, Champanhet E, Garner H, Sikorska K, Loo CE, et al. PD-L1 blockade in combination with carboplatin as immune induction in metastatic lobular breast cancer: the GELATO trial. Nat Cancer. 2023. Apr 10. doi: 10.1038/s43018-023-00542-x. Epub ahead of print. PMID: 37038006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8(2):191–197. doi: 10.1038/ni1428. published Online First: 20061130. [DOI] [PubMed] [Google Scholar]
- 46.Wang S, Shen H, Bai B, Wu J, Wang J. Increased CD4+CD8+ Double-Positive T Cell in Patients with Primary Sjögren’s Syndrome Correlated with Disease Activity. J Immunol Res. 2021;2021:6658324. doi: 10.1155/2021/6658324. published Online First: 2021/06/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Desfrancois J, Moreau-Aubry A, Vignard V, Godet Y, Khammari A, Dréno B, Jotereau F, Gervois N. Double Positive CD4CD8 αβ T Cells: a New Tumor-Reactive Population in Human Melanomas. Plos One. 2010. published Online First: 2010/01/07;5(1):e8437. doi: 10.1371/journal.pone.0008437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schad SE, Chow A, Mangarin L, Pan H, Zhang J, Ceglia N, Caushi JX, Malandro N, Zappasodi R, Gigoux M, et al. Tumor-induced double positive T cells display distinct lineage commitment mechanisms and functions. J Exp Med. 2022;219(6):published Online First: 2022/05/24. doi: 10.1084/jem.20212169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inozume T, Hanada K, Wang QJ, Ahmadzadeh M, Wunderlich JR, Rosenberg SA, Yang JC. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J Immunother (1991). 2010;33(9):956–964. doi: 10.1097/CJI.0b013e3181fad2b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246–2259. published Online First: 20140325. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simon SCS, Hu X, Panten J, Grees M, Renders S, Thomas D, Weber R, Schulze TJ, Utikal J, Umansky V. Eosinophil accumulation predicts response to melanoma treatment with immune checkpoint inhibitors. Oncoimmunology. 2020. published Online First: 2020/03/03;9(1):1727116. doi: 10.1080/2162402X.2020.1727116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carretero R, Sektioglu IM, Garbi N, Salgado OC, Beckhove P, Hämmerling GJ. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8(+) T cells. Nat Immunol. 2015. published Online First: 2015/04/29;16(6):609–617. doi: 10.1038/ni.3159. [DOI] [PubMed] [Google Scholar]
- 54.Zheng X, Zhang N, Qian L, Wang X, Fan P, Kuai J, Lin S, Liu C, Jiang W, Qin S, et al. CTLA4 blockade promotes vessel normalization in breast tumors via the accumulation of eosinophils. Int J Cancer. 2020;146(6):1730–1740. published Online First: 2019/12/17. doi: 10.1002/ijc.32829. [DOI] [PubMed] [Google Scholar]
- 55.Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, Tsui J, Ruhland MK, Kersten K, Abushawish MA, et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell. 2019;177(3):556–71 e16. published Online First: 2019/04/09. doi: 10.1016/j.cell.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau C-S, Verstegen NJM, Ciampricotti M, Hawinkels LJAC, Jonkers J, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345–348. published Online First: 20150330. doi: 10.1038/nature14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, van der Merwe PA, Sivakumar S, van der Merwe PA. Biomarkers of response to PD-1 pathway blockade. Br J Cancer. 2022. published Online First: 2022/03/02;126(12):1663–1675. doi: 10.1038/s41416-022-01743-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davis AA, Patel VG. The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J ImmunoTher Cancer. 2019;7(1):278. doi: 10.1186/s40425-019-0768-9. published Online First: 2019/10/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol. 2020;11:583084. doi: 10.3389/fimmu.2020.583084. published Online First: 2020/12/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. 2014. published Online First: 2014/09/30;15(11):1070–1078. doi: 10.1038/ni.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu J, Blake SJ, Harjunpaa H, Fairfax KA, Yong MCR, Allen S, Kohrt HE, Takeda K, Smyth MJ, Teng MWL. Assessing Immune-Related Adverse Events of Efficacious Combination Immunotherapies in Preclinical Models of Cancer. Cancer Res. 2016. published Online First: 2016/08/10;76(18):5288–5301. doi: 10.1158/0008-5472.CAN-16-0194. [DOI] [PubMed] [Google Scholar]
- 62.Hollande C, Boussier J, Ziai J, Nozawa T, Bondet V, Phung W, Lu B, Duffy D, Paradis V, Mallet V, et al. Inhibition of the dipeptidyl peptidase DPP4 (CD26) reveals IL-33-dependent eosinophil-mediated control of tumor growth. Nat Immunol. 2019;20(3):257–264. published Online First: 2019/02/20. doi: 10.1038/s41590-019-0321-5. [DOI] [PubMed] [Google Scholar]
- 63.Zhang F, Parayath NN, Ene CI, Stephan SB, Koehne AL, Coon ME, Holland EC, Stephan MT. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat Commun. 2019. published Online First: 2019/09/05;10(1):3974. doi: 10.1038/s41467-019-11911-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akuthota P, Wang H, Weller PF. Eosinophils as antigen-presenting cells in allergic upper airway disease. Curr Opin Allergy Clin Immunol. 2010;10(1):14–19. doi: 10.1097/ACI.0b013e328334f693. published Online First: 2009/12/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.House IG, Savas P, Lai J, Chen AXY, Oliver AJ, Teo ZL, Todd KL, Henderson MA, Giuffrida L, Petley EV, et al. Macrophage-Derived CXCL9 and CXCL10 are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin Cancer Res. 2020;26(2):487–504. published Online First: 2019/10/23. doi: 10.1158/1078-0432.CCR-19-1868. [DOI] [PubMed] [Google Scholar]
- 66.Salvagno C, Ciampricotti M, Tuit S, Hau C-S, van Weverwijk A, Coffelt SB, Kersten K, Vrijland K, Kos K, Ulas T, et al. Therapeutic targeting of macrophages enhances chemotherapy efficacy by unleashing type I interferon response. Nat Cell Biol. 2019;21(4):511–521. published Online First: 20190318. doi: 10.1038/s41556-019-0298-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–945. published Online First: 20150720. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, Sheng K, Niu Y, Goldstein A, Zhao N, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol. 2019;21(9):1113–1126. published Online First: 20190826. doi: 10.1038/s41556-019-0373-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM, Paulos CM. When worlds collide: th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol. 2018. published Online First: 20180321;15(5):458–469. doi: 10.1038/s41423-018-0004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tarantino P, Corti C, Schmid P, Cortes J, Mittendorf EA, Rugo H, Tolaney SM, Bianchini G, Andrè F, Curigliano G. Immunotherapy for early triple negative breast cancer: research agenda for the next decade. NPJ Breast Cancer. 2022. published Online First: 20220218;8(1):23. doi: 10.1038/s41523-022-00386-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yofe I, Landsberger T, Yalin A, Solomon I, Costoya C, Demane DF, Shah M, David E, Borenstein C, Barboy O, et al. Anti-CTLA-4 antibodies drive myeloid activation and reprogram the tumor microenvironment through FcγR engagement and type I interferon signaling. Nat Cancer. 2022;3(11):1336–1350. published Online First: 20221027. doi: 10.1038/s43018-022-00447-1. [DOI] [PubMed] [Google Scholar]
- 72.Marangoni F, Zhakyp A, Corsini M, Geels SN, Carrizosa E, Thelen M, Mani V, Prüßmann JN, Warner RD, Ozga AJ, et al. Expansion of tumor-associated Treg cells upon disruption of a CTLA-4-dependent feedback loop. Cell. 2021;184(15):3998–4015 e19. published Online First: 2021/06/23. doi: 10.1016/j.cell.2021.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kidani Y, Nogami W, Yasumizu Y, Kawashima A, Tanaka A, Sonoda Y, Tona Y, Nashiki K, Matsumoto R, Hagiwara M, et al. CCR8-targeted specific depletion of clonally expanded Treg cells in tumor tissues evokes potent tumor immunity with long-lasting memory. Proc Natl Acad Sci U S A. 2022;119(7):published Online First: 2022/02/11. doi: 10.1073/pnas.2114282119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kos K, van Baalen M, Meijer DA, et al. Flow cytometry-based isolation of tumor-associated regulatory T cells and assessment of their suppressive potential. Methods Enzymol. 2020;632:259–281. doi: 10.1016/bs.mie.2019.07.035. published Online First: 2020/02/01. [DOI] [PubMed] [Google Scholar]
- 75.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. [DOI] [PMC free article] [PubMed]
- 76.pandas-dev/pandas: Pandas (v2.0.0rc1) . Zenodo. [program], 2023.
- 77.McKinney W Data structure for statistical computing in python. Proceedings of the 9th python in science conference, Austin, Texas, USA, 2010. [Google Scholar]
- 78.Waskom ML. Seaborn: statistical data visualization. J Open Source Softw. 2021;6(60):3021. doi: 10.21105/joss.03021. [DOI] [Google Scholar]
- 79.Hunter JD. Matplotlib: a 2D Graphics Environment. Comput Sci Eng. 2007;9(3):90–95. doi: 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 80.trevismd/statannotations: v0.5 (v0.5) . Zenodo. [program], 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the authors on reasonable request. The RNAseq data of the GELATO trial is available as previously described. Voorwerk L, Isaeva OI, Horlings HM, Balduzzi S, Chelushkin M, Bakker NAM, Champanhet E, Garner H, Sikorska K, Loo CE, Kemper I, Mandjes IAM, de Maaker M, van Geel JJL, Boers J, de Boer M, Salgado R, van Dongen MGJ, Sonke GS, de Visser KE, Schumacher TN, Blank CU, Wessels LFA, Jager A, Tjan-Heijnen VCG, Schröder CP, Linn SC, Kok M. PD-L1 blockade in combination with carboplatin as immune induction in metastatic lobular breast cancer: the GELATO trial. Nat Cancer. 2023 Apr 10. doi: 10.1038/s43018-023-00542-x. Epub ahead of print. PMID: 37038006.