

Abstract
Adult T-cell leukemia (ATL) is a peripheral T-cell malignancy caused by human T-cell leukemia virus type 1 (HTLV-1). Microsatellite instability (MSI) has been observed in ATL cells. Although MSI results from impaired mismatch repair (MMR) pathway, no null mutations in the genes encoding MMR factors are detectable in ATL cells. Thus, it is unclear whether or not impairment of MMR causes the MSI in ATL cells. HTLV-1 bZIP factor (HBZ) protein interacts with numerous host transcription factors and significantly contributes to disease pathogenesis and progression. Here we investigated the effect of HBZ on MMR in normal cells. The ectopic expression of HBZ in MMR-proficient cells induced MSI, and also suppressed the expression of several MMR factors. We then hypothesized that the HBZ compromises MMR by interfering with a transcription factor, nuclear respiratory factor 1 (NRF-1), and identified the consensus NRF-1 binding site at the promoter of the gene encoding MutS homologue 2 (MSH2), an essential MMR factor. The luciferase reporter assay revealed that NRF-1 overexpression enhanced MSH2 promoter activity, while co-expression of HBZ reversed this enhancement. These results supported the idea that HBZ suppresses the transcription of MSH2 by inhibiting NRF-1. Our data demonstrate that HBZ causes impaired MMR, and may imply a novel oncogenesis driven by HTLV-1.
Keywords: HTLV-1, HBZ, MSI, DNA mismatch Repair, NRF-1, ATL
1. Introduction
Human T-cell leukemia virus type 1 (HTLV-1) is the causative agent of adult T-cell leukemia (ATL), a distinct mature T-cell malignancy [1,2]. Only 3–5% of HTLV-1 infected individuals develop ATL decades after infection, suggesting a multistep process for ATL oncogenesis. Previous studies have shown that ATL cells have the instability of microsatellites, repetitive DNA regions that contain multiple repeats of one to six or more base pair motifs [3–7]. Microsatellites are unstable and prone to changes of repeat numbers with a frequency as high as 10−4 – 10−3 per generation, which is far higher than existing estimates of point mutation rate (around 10−8) [5]. The repeat number change is called microsatellite instability (MSI) and results from the slippage of replicative DNA polymerases during replication [8]. Multiple mechanisms prevent the microsatellite repeat number change during DNA replication, including the proofreading domain of the replicative DNA polymerase [9], Werner helicase [10], and mismatch repair (MMR). The repeat number of microsatellites changes independently of DNA replication, and the underlying molecular mechanism remains unclear [11]. Although a defect in MMR causes the most prominent MSI [12], ATL cells do not have null mutations in the genes encoding MMR factors [13]. Thus, the molecular mechanism underlying the MSI of ATL cells remains unclear.
MMR is a highly conserved biological process responsible for correcting the single-base mismatches and insertion-deletion loops that arise during DNA replication. Proteins involved in MMR system include MutS homologue 2 (MSH2), MSH3, MSH6, MutL homologue 1 (MLH1), MLH3, postmeiotic segregation increased 1 (PMS1) and PMS2 [12]. MMR system is very complex; the most abundant mismatch-binding factor is MutSα, the MSH2-MSH6 heterodimeric complex, which recognizes mismatched bases, leading to an ATP-dependent conformational change and subsequent recruitment of MutLα, the MLH1-PMS2 heterodimer [12]. MMR-deficient cells develop MSI, which is associated with elevated accumulation of spontaneous mutations and predisposition to cancers [12].
Two HTLV-1 proteins, Tax and HTLV-1 bZIP factor (HBZ), interact with numerous transcription factors and cellular signaling components, contributing to disease progression [2]. HBZ is expressed ubiquitously in ATL, while Tax expression is frequently lost [13,14]. HBZ impairs DNA damage response [15,16]. We also previously reported that HBZ downregulates the expression of tyrosyl-DNA phosphodiesterase 1 (TDP1), a DNA repair enzyme, by interfering with nuclear respiratory factor 1 (NRF-1) [17,18]. In this study, we investigate the effect of HBZ on MMR system and show that HBZ impairs MMR.
2. Methods
2.1. Cells
TK6 cells stably expressing a spliced form of HBZ were established by electroporation using the Neon transfection system (Invitrogen) followed by G418 (Nacalai Tesque) drug selection. MSH2- and MLH1-knockout TK6 cells were described previously [19]. These cells derived from TK6 were maintained in RPMI 1640 (Nacalai Tesque) supplemented with 5% horse serum (Gibco), 2 mM sodium pyruvate (Nacalai Tesque) and penicillin (100 U/ml), streptomycin (100 mg/ml), and L-glutamine (0.292 mg/ml) (PSG, Invitrogen). HEK293T cells were maintained in DMEM (Nacalai Tesque) containing 10% fetal bovine serum (Gibco) and PSG.
2.2. Plasmid constructs
The vector encoding myc-His-tagged HBZ has been described previously [18]. The MSH2 promoter sequences were generated by PCR amplification using genomic DNA from a healthy donor, and then subcloned into the pGL3-basic luciferase reporter vector (Promega). The vector encoding FLAG-tagged NRF-1 was gifted from Dr. H. Izumi [20].
2.3. Reverse transcription PCR
RNA was extracted using High Pure RNA Isolation Kit (Roche). cDNA was generated using ReverTra Ace qPCR RT Master Mix with gDNA Remover (TOYOBO). The following primers were used: HBZ (forward 5′-ATGGCGGCCTCAGGGCTGTT-3′/reverse 5′-GCGGCTTTCCTCTTCTAAGG-3′), ACTB (5′-CTGGAACGGTGAAGGTGACA-3′/5′- AAGGGACTTCCTGTAACAATGCA-3′)
2.4. Microsatellite analysis
DNA was extracted using QuickGene DNA whole blood kit (KURABO). Eight microsatellite loci, D10S190, D10S191, D18S21, BAT40, BAT25, BAT26, D2S123, and D5S346, were amplified by PCR from genomic DNA. For each microsatellite locus, the forward primer was fluorescently labeled with FAM, VIC or NED (Applied Biosystems, Foster City, CA). PCR reaction and analysis by capillary electrophoresis using ABI 3100 xl Genetic Analyzer (Applied Biosystems) were performed by SystemBiotics Inc. (Kanagawa, Japan). Data were processed using the Peak Scanner Software (Applied Biosystems). The primer sequences for PCR amplification of each microsatellite marker were: D10S190 (forward 5′-FAM-GTGTTTGGGTCATGGAGATG-3’/reverse 5′-AGGCAAAGCAGGAGCA-3′), D10S191 (5′-VIC-CTTTAATTGCCCTGTCTTC-3’/5′-TTAATTCGACCACTTCCC-3′), D18S21 (5′-VIC-GTGGTTATTGCCTTGAAAAG-3’/5′-GATGACATTTTCCCTCTAG-3′), BAT40 (5′-NED-ACAACCCTGCTTTTGTTCCT-3’/5′-GTAGAGCAAGACCACCTTG-3′), BAT25 (5′-VIC-TCGCCTCCAAGAATGTAAGT-3’/5′-TCTGGATTTTAACTATGGCTC-3′), BAT26 (5′-FAM-TGACTACTTTTGACTTCAGCC-3’/5′-GTTTCTAACCATTCAACATTTTTATCCC-3′), D2S123 (5′-NED-AAACAGGATGCCTGCCTTTA-3’/5′-GGACTTTCCACCTATGGGAC-3′), D5S346 (50-FAM-ACTCACTCTAGTGATAAATCGGG-3’/5′-AGCAGATAAGACAGTATTACTAGTT-3′).
2.5. Cell proliferation assay
Cell proliferation assay was performed as described previously [17]. 1-(2-chloroethyl)-3-cyclohexyl-nitrosourea (CCNU, L0251) was purchased from Tokyo Chemical Industry, and Mitomycin C (20898-21) from Nacalai Tesque. These were dissolved with dimethyl sulfoxide (Nacalai Tesque).
2.6. Quantitative real-time RT-PCR
Real-time RT-PCR was performed as described previously [17]. The following primers were used: MSH2 (5′-GGATCAAATGAAGGTTTTACAAGG-3′/5′-TGGCAGTTTTTGTGACTCCT-3′), MSH3 (5′-CGAATTCTGTCATCCTGCAC-3′/5′-CCTGCAGCCAGTAGAGCTG-3′), MSH6 (5′-CTTTGAGAGCTTTCTCTGCC-3′/5′-CTGCAACAACTTCCTCATCCC-3′), MLH1 (5′-GTGCTGGCAATCAAGGGACCC-3′/5′-CACGGTTGAGGCATTGGGTAG-3′), MLH3 (5′-CCTCCGGTGGTCTGGAGTAG-3′/5′-GAGAAGCCAATGAACTTCGG-3′), PMS1 (5′-GCGGCAACAGTTCGACTCCTT-3′/5′-AGCCTTGATACCCTCCCCGTT-3′), PMS2 (5′-TGACTGGAGCATTTTCATCG-3′/5′-CAGGGGCAGAGGCTCATA-3′), HPRT1 (5′-GCTGAGGATTTGGAAAGGGTG-3′/5′-TGAGCACACAGAGGGCTACAATG-3′). Data were normalized to the expression of HPRT1.
2.7. Western blotting
Immunoblotting was performed as described previously [17]. Primary antibodies used were: MSH3 (B-4, sc-271080), PMS1 (E-3, sc-515302), PMS2 (B-3, sc-25315), all from Santa Cruz Biotechnology; MSH2 (556349) from BD Pharmingen; MLH1 (ab92312) from Abcam; MSH6 (#VMA00250) from Bio-Rad; β-actin (A5441) and FLAG (M2, F3165) from Sigma-Aldrich. All Western blotting experiments were repeated independently at least three times, and representative results are shown.
2.8. Human microarray data processing
Microarray data processing was performed as described previously [18].
2.9. Luciferase reporter assay
Luciferase reporter assay was performed as previously described [18]. For Fig. 4C, HEK293T cells were transfected with 0.5 μg luciferase (Firefly) reporter plasmid driven by truncated MSH2 promoter and 0.5 ng control Renilla luciferase reporter plasmid. For Fig. 4D and E, 0.2 μg luciferase reporter driven by MSH2 promoter truncated at 278 bp upstream of the transcription start site and 0.5 ng control luciferase reporter were transfected together with a vector encoding FLAG-NRF-1 and/or myc-His-HBZ. Each transfection mixture was equalized with empty vector where necessary. MSH2-promoter driven Firefly luciferase activity was normalized by Renilla luciferase activity.
Fig. 4.
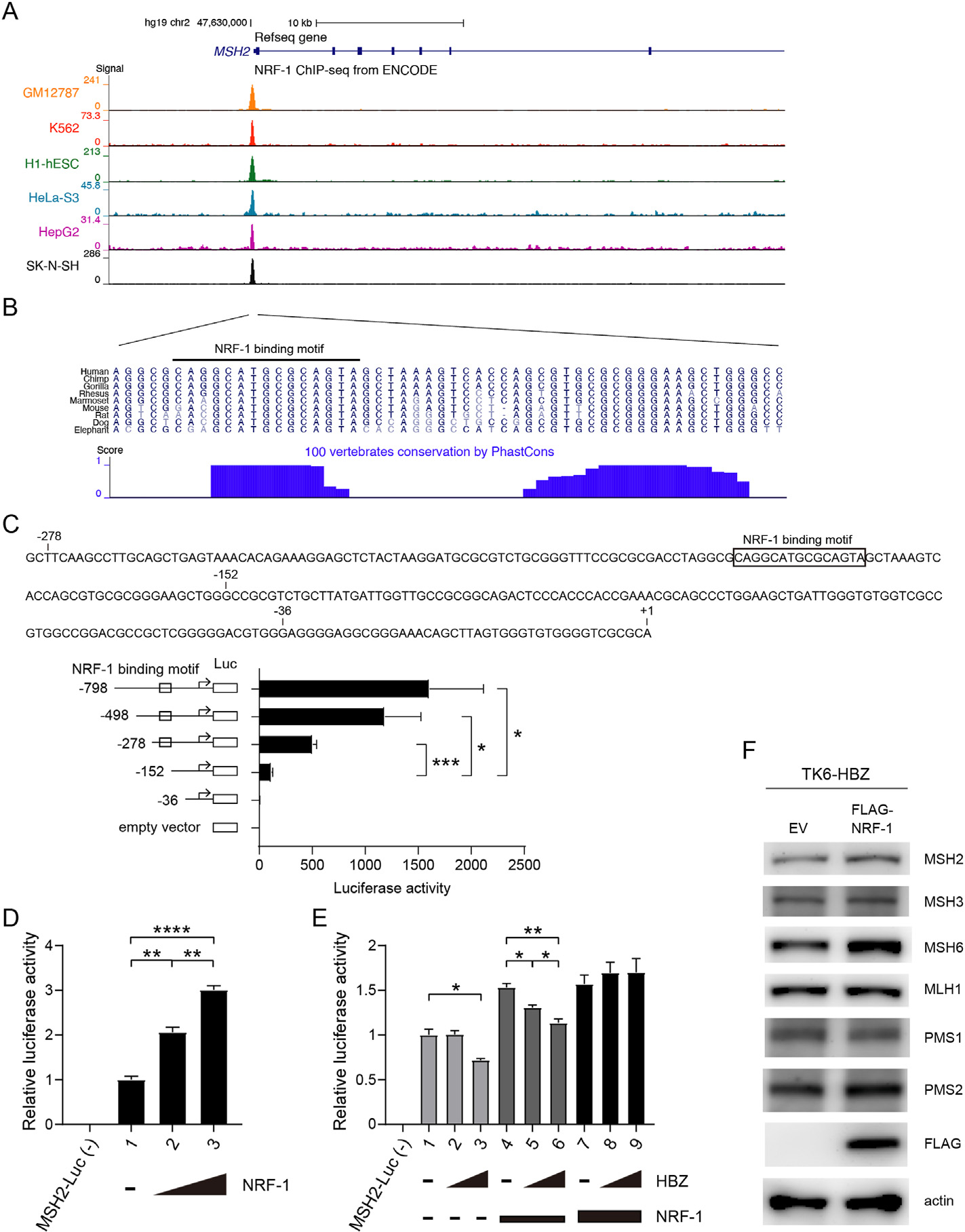
HBZ suppresses transcription of MSH2 via the transcription factor NRF-1 (A, B) UCSC genome browser view of the NRF-1 binding site in the MSH2 promoter. (A) Tracks show signals of NRF-1 ChIP-seq in the promoter region of MSH2 from the ENCODE project. (B) Sequences of the signal peak are shown. Blue bars represent the degree of sequence conservation across different vertebrates from the phastCons program. The black-lined region represents the NRF-1 binding motif predicted using Factorbook. (C) Activity of MSH2 promoter constructs truncated at different positions upstream of the transcription start site (TSS). Sequence upstream of the TSS of MSH2, and the NRF-1 binding motif predicted using Factorbook are shown. Each MSH2-Luc was transfected into HEK293T cells together with Renilla-Luc. MSH2-luciferase activity was assayed as relative to Renilla-Luc (n = 3, mean ± SEM). (D) Effects of NRF-1 expression on MSH2 promoter activity. MSH2-Luc and Renilla-Luc were transfected into HEK293T cells, with or without vectors expressing NRF-1 (0.05 μg or 0.2 μg) (n = 3, mean ± SEM). (E) Effects of HBZ on MSH2 promoter activity. MSH2-Luc and Renilla-Luc were transfected into HEK293T cells, with or without vectors expressing HBZ (0.4 μg or 0.6 μg) and NRF-1 (0.0125 or 0.025 μg) (n = 3, mean ± SEM). (F) Immunoblotting of the indicated proteins in TK6-HBZ cells infected with lentivirus expressing FLAG-tagged NRF-1 or the corresponding empty vector (EV).
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (unpaired Student’s t-test).
2.10. Transduction of NRF-1 using lentiviral vector
The FLAG-tagged NRF-1 was subcloned into lentiviral vector CSII-CMV-MCS-IRES-hrGFP, as described previously [18]. TK6-HBZ cells were infected with lentivirus expressing FLAG-NRF-1 or the corresponding empty vector, and confirmation of infectivity was based on hrGFP, as previously described [21]. Three days after infection, GFP+ cells were collected using a Cell Sorter SH800S instrument (Sony), and lysed for Western blotting.
3. Results
3.1. Transduction of HBZ into MMR-proficient cells induces microsatellite alterations
To investigate the effect of HBZ on MMR, we transduced HBZ or empty vector into MMR-proficient human TSCER2 B-lymphoblastoid cells [22], a TK6 subline for measuring heteroallelic homologous recombination, generating TK6-HBZ and TK6-EV (empty vector) cells (Fig. 1A). To analyze MSI, we analyzed eight microsatellite loci, including BAT25, BAT26, D2S123, and D5S346 from the National Cancer Institute panel, which are commonly used to assess MSI for solid cancers [23], and D10S190, D10S191, and D18S21, which were frequently altered in ATL in an earlier study testing many microsatellite loci [3]. We found that HBZ expression induced alterations in four microsatellite loci, D10S190, D18S21, BAT25, and D5S346 (Fig. 1B). To compare the extent of length changes of microsatellites, we also examined MSH2- or MLH1-deficient TK6 cells [19], and found that deficiency of MSH2 or MLH1 induced alterations similar to those induced by HBZ expression (Fig. S1). Regarding dinucleotide microsatellites, two distinct types of alterations have been described: relatively small length changes (≤6 bp, Type A) and drastic changes involving ≥8 bp (Type B) [24], and it was reported that ATL cells invariably showed Type A dinucleotide microsatellite changes [7]. Together, the ectopic expression of HBZ in MMR-proficient cells induced MSI Type A similar to those observed in human primary ATL samples.
Fig. 1.
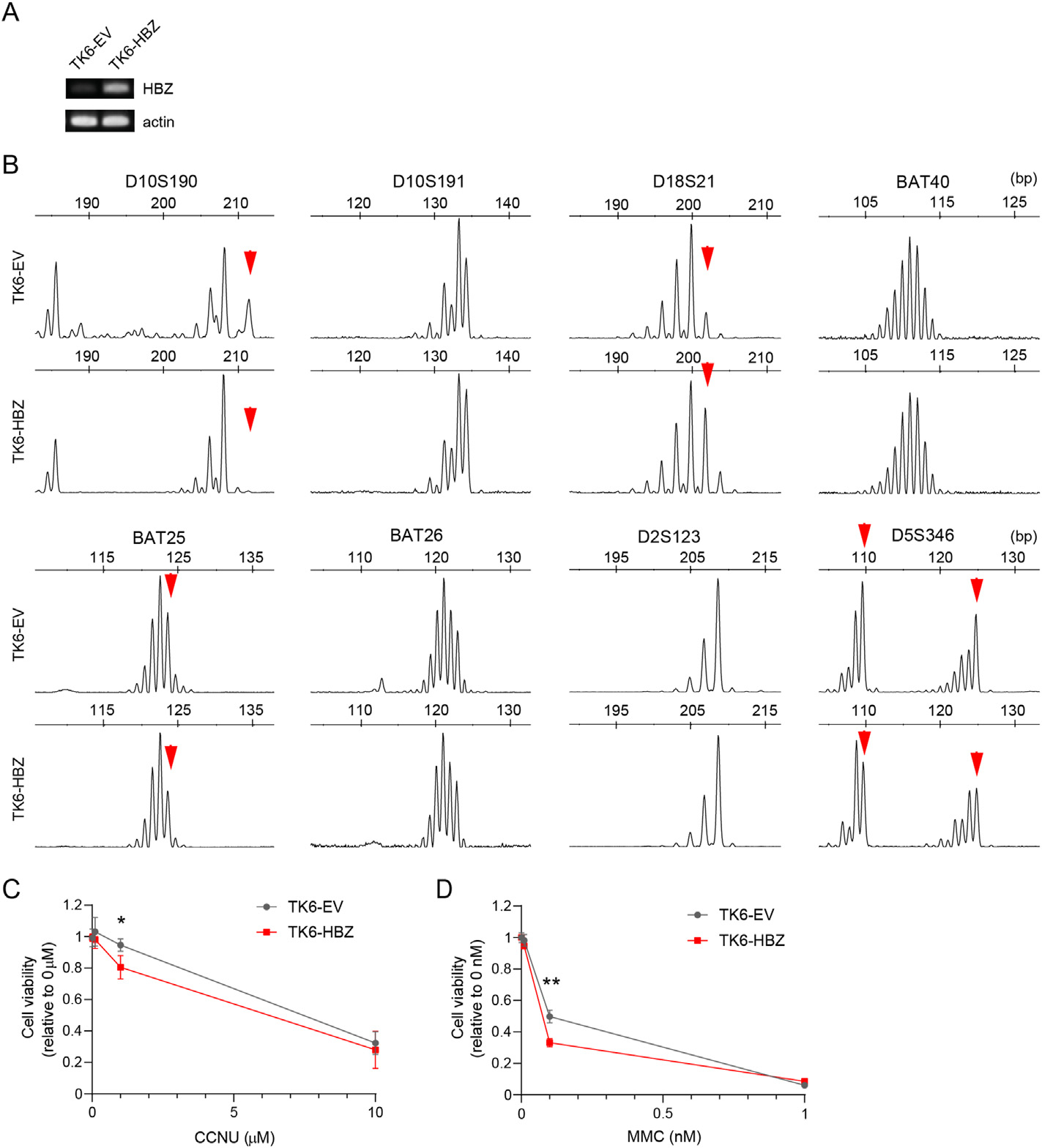
Transduction of HBZ into MMR-proficient cells induces microsatellite alterations (A) RT-PCR of the indicated genes in TK6 cells stably transduced with HBZ (TK6-HBZ) or empty vector (TK6-EV). (B) Electropherograms for eight loci of microsatellites (D10S190, D10S191, D18S21, BAT40, BAT25, BAT26, D2S123 and D5S346) in TK6-EV and TK6-HBZ cells. The arrows indicate microsatellite alterations. (C, D) TK6-EV and TK6-HBZ cells were treated with the indicated concentration of CCNU (C) or MMC (D) for 48h. Cell viability was determined by MTS assays and expressed as relative to vehicle-treated controls (n = 3, mean ± SD). *p < 0.05, **p < 0.01 (unpaired Student’s t-test).
Since previous studies suggested that MMR-deficient cells are more sensitive to 1-(2-chloroethyl)-3-cyclohexyl-nitrosourea (CCNU) and mitomycin C (MMC) than MMR-proficient cells [12,25], we tested the sensitivity to these drugs for TK6-HBZ and TK6-EV cells. Consistently, we found that TK6-HBZ cells were significantly more sensitive to CCNU and MMC compared with TK6-EV cells (Fig. 1C and D).
3.2. Transduction of HBZ into MMR-proficient cells suppresses the expression of several MMR factors
Genome-wide sequencing studies have never identified mutations in the genes encoding MMR factors in primary ATL samples [13]. Instead, a previous study pointed the reduction in expression levels of various MMR factors in ATL cells [26]. To unveil the mechanism of MSI observed in TK6-HBZ cells, we examined the expression levels of molecules involved in MMR. In TK6-HBZ cells, mRNA levels of MSH2, MLH1, MLH3 and PMS1 were suppressed compared with TK6-EV cells (Fig. 2A). In terms of protein levels, all of the factors that we tested were reduced in TK6-HBZ cells compared with TK6-EV cells (Fig. 2B). MLH3 was not assessed due to lack of an appropriate antibody for Western blotting. MMR proteins form functional heterodimeric complexes including MSH2-MSH6 (MutSα), MSH2-MSH3 (MutSβ) and MLH1-PMS2 (MutLα), in which MSH2 and MLH1 are essential to stabilize their binding partners [27–29]. Therefore, in TK6-HBZ cells, the reduced expression of MSH2 and MLH1 might consequently disrupt stability of their binding partners. Thus, HBZ repressed the expression of many of MMR components in protein levels.
Fig. 2.
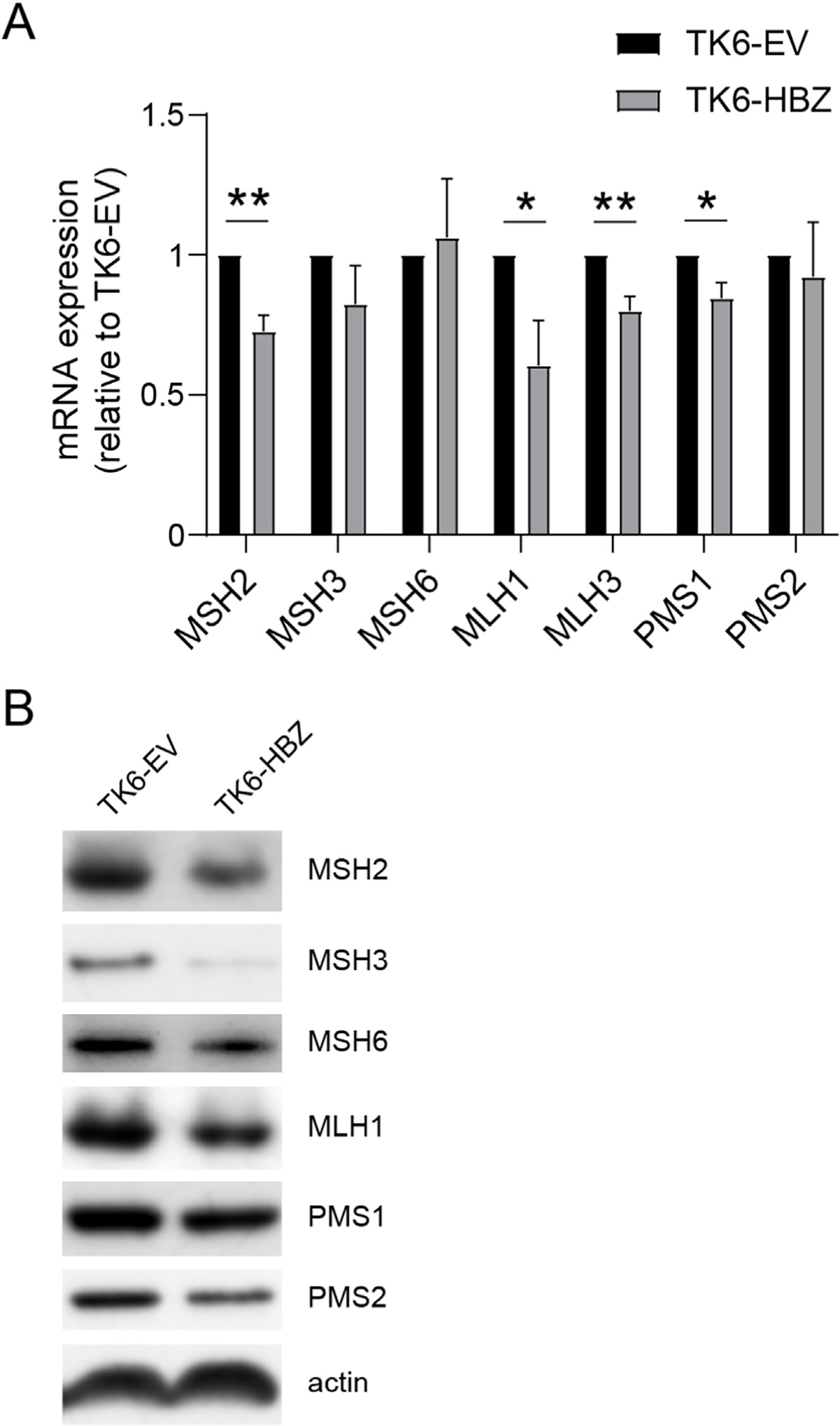
Transduction of HBZ into MMR-proficient cells suppresses the expression of several MMR factors (A) mRNA expression of indicated genes measured by qRT-PCR in TK6-EV and TK6-HBZ cells (n = 4, mean ± SEM). *p < 0.05, **p < 0.01 (unpaired Student’s t-test). (B) Immunoblotting of the indicated proteins in TK6-EV and TK6-HBZ cells.
3.3. HBZ suppresses transcription of MSH2 via the transcription factor NRF-1
To see whether the reduction in expression of MMR factors is recurrently present in ATL patients, we analyzed the publicly available microarray dataset (GSE43017) [30]. We found that MSH2 expression was significantly suppressed in acute type ATL cells compared with CD4+ T lymphocytes from healthy volunteers, while MLH1 expression was significantly upregulated and the other MMR genes were comparable (Fig. 3). According to the early study [26], MSH2 and PMS1 were most frequently suppressed in the tested eleven patients. These data suggest that MSH2 expression is recurrently downregulated in human acute type ATL.
Fig. 3.
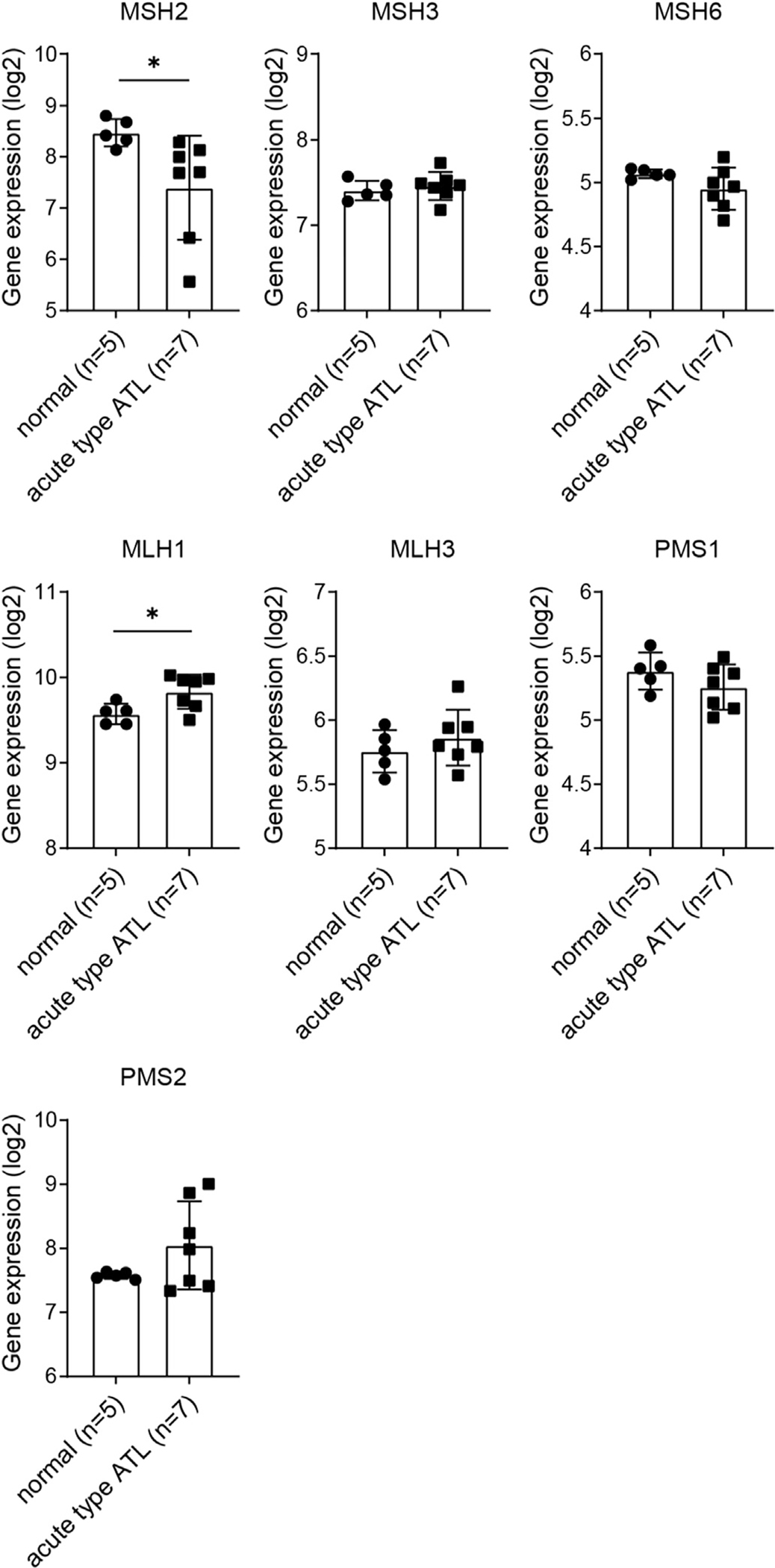
Expression of MMR genes in primary acute type ATL samples. mRNA expression levels of the indicated genes in human CD4+ T lymphocytes from the peripheral blood of healthy volunteers (n = 5) and primary acute type ATL cells (n = 7), obtained from the microarray dataset in the Gene Expression Omnibus database, accession number GSE43017 (mean ± SD). *p < 0.05 (unpaired Welch’s t-test).
Following this, we sought to investigate the mechanism through which HBZ represses MSH2 expression. Based on our previous data showing that HBZ binds directly to and functionally suppresses a transcription factor NRF-1 [18], we hypothesized that the HBZ/NRF-1 axis could contribute to the repression of MSH2. Analysis using the web-based resource CellMinerCDB (https://discover.nci.nih.gov/cellminercdb/) [31] revealed that MSH2 expression was significantly correlated with NRF-1 expression in the human NCI-60 cancer cell line database (Fig. S2A) and also in the Cancer Cell Line Encyclopedia (Fig. S2B). Likewise, the FANTOM gene expression atlas showed the significant correlation in the expression levels between MSH2 and NRF-1, for both human and mouse cells (Figs. S2C and S2D) [32]. Using Factorbook, we predicted a NRF-1 binding motif in the MSH2 promoter [33] and ENCODE ChIP-seq data showed that NRF-1 binds to the predicted motif [34] (Fig. 4A and B). We further found that the sequence of the NRF-1 binding motif is conserved in vertebrates [35] (Fig. 4B). Next, we transfected HEK293T cells with luciferase reporter constructs driven by MSH2 promoters (MSH2-Luc) truncated at various positions upstream of the transcription start site (TSS) and assessed their activity (Fig. 4C). We observed that MSH2-Luc without the NRF-1 binding motif (by truncation 152 bp upstream of the TSS) lost its promoter activity, compared with MSH2-Luc harboring the NRF-1 binding motif or longer upstream of the motif (truncated at −278 bp, −498 bp, and −798 bp) (Fig. 4C). Together, these data suggest that NRF-1 positively regulates the transcription of MSH2.
Next, we assessed MSH2 promoter activity when NRF-1 and/or HBZ are overexpressed, using MSH2-Luc truncated at 278 bp upstream of TSS. NRF-1 overexpression increased MSH2-Luc activity in a dose-dependent manner (Fig. 4D). HBZ expression inhibited MSH2-Luc activity in the absence of NRF-1 co-transfection (Fig. 4E, lanes 1–3) and when co-transfected with a small amount of NRF-1 (Fig. 4E, lanes 4–6). Notably, this inhibitory effect of HBZ was cancelled when cells were co-transfected with a larger amount of NRF-1 (Fig. 4E, lanes 7–9). Finally, to see if this effect of overexpressed NRF-1 is recapitulated also in TK6 cells, we measured the expression of MMR factors using the TK6-HBZ cells infected with lentivirus expressing NRF-1 or corresponding empty vector. The results showed that NRF-1 overexpression led to restoration of MSH2 expression level (Fig. 4F). Along with this MSH2 restoration, the amount of MSH6 was also clearly upregulated. In contrast, the expression levels of MSH3, MLH1, PMS1 or PMS2 were not robustly affected by NRF-1 overexpression. These findings reinforce our hypothesis that the suppressed expression of MSH2 could destabilize MSH6, which is a binding partner in functional heterodimeric complex MutSα (MSH2-MSH6). On the other hand, the mechanism of HBZ-driven MSH3 suppression is not likely due to destabilization of MutSβ (MSH2-MSH3) and remains unclear. Altogether, in combination with our previous report showing that HBZ interacts with and functionally interferes with NRF-1 [18], these data suggest that HBZ suppresses transcription of MSH2 through interfering with NRF-1.
4. Discussion
In this study, we show that HTLV-1-encoded protein HBZ impairs MMR system. We suggest that HBZ represses expression of multiple MMR components, which could associate with functional impairment of MMR and MSI. Further, in line with our previous results demonstrating the inhibitory effect of HBZ against NRF-1 [18], we suggest that HBZ suppresses MSH2 transcription through inhibiting NRF-1. Because MSH2 expression could be recurrently suppressed in acute type ATL, the HBZ/NRF-1/MSH2 axis could be a mechanism underlying the impaired MMR in HTLV-1-infected cells.
Two distinct types of dinucleotide microsatellite alterations have been described: relatively small length changes (≤6 bp, Type A) and drastic changes involving ≥8 bp (Type B) [24]. Intriguingly, MEF cells or tumors from MSH2- or MLH1-deficient mice, which are susceptible to cancer, invariably show Type A MSI, implying that Type A MSI is a direct consequence of defective MMR [24]. Notably, microsatellite changes observed in ATL cells are also invariably Type A [7]. A recent whole-genome sequencing study described that MSI is rarely observed in ATL [36], but this sequencing-based tool has been explored to detect high instability shown frequently in solid tumors [37] and might not be able to reflect Type A MSI. The HBZ-expressing TK6 cells, in which multiple MMR factors were reduced in expression, and the MSH2 or MLH1-knockout TK6 cells show similar extent of Type A MSI. This may suggest that, when many molecules of MMR are simultaneously suppressed, it could induce MMR disturbance equivalent to deficiency of the single essential MMR factor. Collectively, our data suggest that HBZ plays a crucial role in repressing the expression of multiple MMR factors, which could lead to MSI.
While it is suggested that MSH2 is recurrently reduced in acute type ATL, our results using HBZ-expressing TK6 cells suggest that HBZ suppresses various MMR factors. The aggressive subtypes (acute/lymphoma type) of ATL develop through multistep tumorigenesis during the long latent period of asymptomatic carriers or the precedent indolent subtypes of ATL [2]. Therefore, not only MSH2 but also other MMR molecules might be suppressed tentatively in HTLV-1-infected cells during the long period before the onset of aggressive ATL. However, it remains to be elucidated whether this suppression of multiple MMR components contributes to accumulation of gene mutations accelerating ATL tumorigenesis.
Regarding the mechanisms of reduced expression of MMR factors in ATL cells, it was suggested that reduced PMS1 expression could be attributed to DNA methylation [26], but it has never been described what represses MSH2 expression. In this study, we focused on MSH2 among MMR molecules and suggested a novel mechanism of MSH2 downregulation. In combination with our previous data showing that HBZ binds directly to and interferes with the DNA-binding ability of the transcription factor NRF-1 [18], we suggest that HBZ impedes NRF-1 from regulating the MSH2 transcription. This HBZ/NRF-1/MSH2 axis could be part of the mechanisms underlying the HBZ-driven MMR impairment.
From a therapeutic perspective, defective MMR and the resultant high mutation burden are correlated with sensitivity to immune checkpoint blockade therapies [38,39]. Given that ATL has relatively higher mutation rates than other hematological malignancies [13], a clinical trial of nivolumab (anti-PD-1 antibody) was conducted in ATL patients [40]. Rapid disease progression was observed in three patients of one study, warranting further studies on the efficiency of targeting PD-1 in the treatment of ATL [40,41]. A possible reason is that the somatic point mutation rate in ATL (2.3 mutations/Mb) appears to be much lower than in solid tumors such as colorectal cancers, lung cancers and melanoma [13,42]. Further studies will be necessary to utilize therapeutic vulnerabilities associated with MSI for ATL treatment.
In conclusion, we show that HBZ bears the potential to induce MMR abnormalities. We suggest that the suppression in amounts of multiple MMR molecules and the HBZ/NRF-1/MSH2 axis could be part of the mechanisms of HBZ-driven defective MMR. Our data have raised the possibility that HBZ-driven defective MMR may play crucial roles in the long process of HTLV-1-induced tumorigenesis.
Supplementary Material
Acknowledgements
This research was supported by JSPS KAKENHI Grant Number 16K09848, 16H06306, 19H04267 and by AMED Grant Numbers JP19fk0108113, JP20fk0410011, JP20fk0410014, JP21fk0410034; by the Center for Cancer Research, the Intramural Program of the National Cancer Institute, NIH (Z01-BC006150) (to Y.P.); the Japan Society for the Promotion of Science Core-to-Core Program, Advanced Research Networks (JPJSCCA20160007) (to S.T.); the RIKEN Junior Research Associate Program (to S.H.). We thank Dr. Atsushi Yamada for scientific advice; Kayoko Nagata for technical support; Dr. Anamaria Daniela Sarca for writing assistance.
Footnotes
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Masao Matsuoka reports a relationship with Kyowa Kirin Co Ltd that includes: speaking and lecture fees. Masao Matsuoka reports a relationship with Meiji Seika Pharma Co Ltd that includes: speaking and lecture fees. Masao Matsuoka reports a relationship with Bristol Myers Squibb Co that includes: speaking and lecture fees. Masao Matsuoka reports a relationship with Chugai Pharmaceutical Co Ltd that includes: speaking and lecture fees. Akifumi Takaori-Kondo reports a relationship with Ono Pharmaceutical Co Ltd that includes: funding grants.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbrc.2023.03.049.
References
- [1].Uchiyama T, Yodoi J, Sagawa K, et al. , Adult T-cell leukemia: clinical and hematologic features of 16 cases, Blood 50 (1977) 481–492. [PubMed] [Google Scholar]
- [2].Watanabe T, Adult T-cell leukemia: molecular basis for clonal expansion and transformation of HTLV-1-infected T cells, Blood 129 (2017) 1071–1081, 10.1182/blood-2016-09-692574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hatta Y, Yamada Y, Tomonaga M, et al. , Microsatellite instability in adult T-cell leukaemia, Br. J. Haematol. 101 (1998) 341–344, 10.1046/j.1365-2141.1998.00710.x. [DOI] [PubMed] [Google Scholar]
- [4].Hayami Y, Komatsu H, Iida S, et al. , Microsatellite instability as a potential marker for poor prognosis in adult T cell leukemia/lymphoma, Leuk. Lymphoma 32 (1999) 345–349, 10.3109/10428199909167395. [DOI] [PubMed] [Google Scholar]
- [5].Ellegren H, Microsatellites: simple sequences with complex evolution, Nature reviews, Genetics 5 (2004) 435–445, 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- [6].Magalhaes M, Oliveira PD, Bittencourt AL, et al. , Microsatellite alterations are also present in the less aggressive types of adult T-cell leukemia-lymphoma, PLoS Neglected Trop. Dis. 9 (2015), e0003403, 10.1371/journal.pntd.0003403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Miyashita K, Fujii K, Taguchi K, et al. , A specific mode of microsatellite instability is a crucial biomarker in adult T-cell leukaemia/lymphoma patients, J. Cancer Res. Clin. Oncol. 143 (2017) 399–408, 10.1007/s00432-016-2294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Viguera E, Canceill D, Ehrlich SD, Replication slippage involves DNA polymerase pausing and dissociation, EMBO J. 20 (2001) 2587–2595, 10.1093/emboj/20.10.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shioi S, Shimamoto A, Song Y, et al. , DNA polymerase delta Exo domain stabilizes mononucleotide microsatellites in human cells, DNA Repair 108 (2021), 103216, 10.1016/j.dnarep.2021.103216. [DOI] [PubMed] [Google Scholar]
- [10].van Wietmarschen N, Sridharan S, Nathan WJ, et al. , Repeat expansions confer WRN dependence in microsatellite-unstable cancers, Nature 586 (2020) 292–298, 10.1038/s41586-020-2769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Polyzos AA, McMurray CT, Close encounters: moving along bumps, breaks, and bubbles on expanded trinucleotide tracts, DNA Repair 56 (2017) 144–155, 10.1016/j.dnarep.2017.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jiricny J, The multifaceted mismatch-repair system, Nature reviews, Molecul. Cell Biol. 7 (2006) 335–346, 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- [13].Kataoka K, Nagata Y, Kitanaka A, et al. , Integrated molecular analysis of adult T cell leukemia/lymphoma, Nat. Genet. 47 (2015) 1304–1315, 10.1038/ng.3415. [DOI] [PubMed] [Google Scholar]
- [14].Satou Y, Yasunaga J, Yoshida M, et al. , HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells, Proc. Natl. Acad. Sci. U.S.A. 103 (2006) 720–725, 10.1073/pnas.0507631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Vernin C, Thenoz M, Pinatel C, et al. , HTLV-1 bZIP factor HBZ promotes cell proliferation and genetic instability by activating OncomiRs, Cancer Res. 74 (2014) 6082–6093, 10.1158/0008-5472.CAN-13-3564. [DOI] [PubMed] [Google Scholar]
- [16].Rushing AW, Hoang K, Polakowski N, et al. , The human T-cell leukemia virus type 1 basic leucine zipper factor attenuates repair of double-stranded DNA breaks via nonhomologous end joining, J. Virol. 92 (2018), 10.1128/JVI.00672-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tada K, Kobayashi M, Takiuchi Y, et al. , Abacavir, an anti-HIV-1 drug, targets TDP1-deficient adult T cell leukemia, Sci. Adv. 1 (2015), e1400203, 10.1126/sciadv.1400203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Takiuchi Y, Kobayashi M, Tada K, et al. , HTLV-1 bZIP factor suppresses TDP1 expression through inhibition of NRF-1 in adult T-cell leukemia, Sci. Rep. 7 (2017), 12849, 10.1038/s41598-017-12924-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rahman MM, Mohiuddin M, Shamima Keka I, et al. , Genetic evidence for the involvement of mismatch repair proteins, PMS2 and MLH3, in a late step of homologous recombination, J. Biol. Chem. (2020), 10.1074/jbc.RA120.013521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Izumi H, Ohta R, Nagatani G, et al. , p300/CBP-associated factor (P/CAF) interacts with nuclear respiratory factor-1 to regulate the UDP-N-acetyl-alpha-d-galactosamine: polypeptide N-acetylgalactosaminyltransferase-3 gene, Biochem. J. 373 (2003) 713–722, 10.1042/BJ20021902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sakamoto T, Kobayashi M, Tada K, et al. , CKIP-1 is an intrinsic negative regulator of T-cell activation through an interaction with CARMA1, PLoS One 9 (2014), e85762, 10.1371/journal.pone.0085762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Honma M, Izumi M, Sakuraba M, et al. , Deletion, rearrangement, and gene conversion; genetic consequences of chromosomal double-strand breaks in human cells, Environ. Mol. Mutagen. 42 (2003) 288–298, 10.1002/em.10201. [DOI] [PubMed] [Google Scholar]
- [23].Boland CR, Thibodeau SN, Hamilton SR, et al. , A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer, Cancer Res. 58 (1998) 5248–5257. [PubMed] [Google Scholar]
- [24].Oda S, Maehara Y, Ikeda Y, et al. , Two modes of microsatellite instability in human cancer: differential connection of defective DNA mismatch repair to dinucleotide repeat instability, Nucleic Acids Res. 33 (2005) 1628–1636, 10.1093/nar/gki303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fiumicino S, Martinelli S, Colussi C, et al. , Sensitivity to DNA cross-linking chemotherapeutic agents in mismatch repair-defective cells in vitro and in xenografts, Int. J. Cancer 85 (2000) 590–596, . [DOI] [PubMed] [Google Scholar]
- [26].Morimoto H, Tsukada J, Kominato Y, et al. , Reduced expression of human mismatch repair genes in adult T-cell leukemia, Am. J. Hematol. 78 (2005) 100–107, 10.1002/ajh.20259. [DOI] [PubMed] [Google Scholar]
- [27].Chang DK, Ricciardiello L, Goel A, et al. , Steady-state regulation of the human DNA mismatch repair system, J. Biol. Chem. 275 (2000) 18424–18431, 10.1074/jbc.M001140200. [DOI] [PubMed] [Google Scholar]
- [28].Drummond JT, Genschel J, Wolf E, et al. , DHFR/MSH3 amplification in methotrexate-resistant cells alters the hMutSalpha/hMutSbeta ratio and reduces the efficiency of base-base mismatch repair, Proc. Natl. Acad. Sci. U.S.A. 94 (1997) 10144–10149, 10.1073/pnas.94.19.10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Genschel J, Littman SJ, Drummond JT, et al. , Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha, J. Biol. Chem. 273 (1998) 19895–19901, 10.1074/jbc.273.31.19895. [DOI] [PubMed] [Google Scholar]
- [30].Nakahata S, Ichikawa T, Maneesaay P, et al. , Loss of NDRG2 expression activates PI3K-AKT signalling via PTEN phosphorylation in ATLL and other cancers, Nat. Commun. 5 (2014) 3393, 10.1038/ncomms4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Luna A, Elloumi F, Varma S, et al. , CellMiner Cross-Database (CellMinerCDB) version 1.2: exploration of patient-derived cancer cell line pharmacogenomics, Nucleic Acids Res. 49 (2021) D1083–D1093, 10.1093/nar/gkaa968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Consortium F, the Clst RP, et al. , A promoter-level mammalian expression atlas, Nature 507 (2014) 462–470, 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang J, Zhuang J, Iyer S, et al. , Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium, Nucleic Acids Res. 41 (2013) D171–D176, 10.1093/nar/gks1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Consortium EP, An integrated encyclopedia of DNA elements in the human genome, Nature 489 (2012) 57–74, 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Siepel A, Bejerano G, Pedersen JS, et al. , Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes, Genome Res. 15 (2005) 1034–1050, 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kogure Y, Kameda T, Koya J, et al. , Whole-genome landscape of adult T-cell leukemia/lymphoma, Blood (2021), 10.1182/blood.2021013568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bailey MH, Tokheim C, Porta-Pardo E, et al. , Comprehensive characterization of cancer driver genes and mutations, Cell 173 (2018) 371–385 e318, 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Le DT, Uram JN, Wang H, et al. , PD-1 blockade in tumors with mismatch-repair deficiency, N. Engl. J. Med. 372 (2015) 2509–2520, 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mandal R, Samstein RM, Lee KW, et al. , Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response, Science 364 (2019) 485–491, 10.1126/science.aau0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ratner L, Waldmann TA, Janakiram M, et al. , Rapid progression of adult T-cell leukemia-lymphoma after PD-1 inhibitor therapy, N. Engl. J. Med. 378 (2018) 1947–1948, 10.1056/NEJMc1803181. [DOI] [PubMed] [Google Scholar]
- [41].Ishitsuka K, Utsunomiya A, Ishida T, PD-1 inhibitor therapy in adult T-cell leukemia-lymphoma, N. Engl. J. Med. 379 (2018) 695, 10.1056/NEJMc1807852. [DOI] [PubMed] [Google Scholar]
- [42].Alexandrov LB, Nik-Zainal S, Wedge DC, et al. , Signatures of mutational processes in human cancer, Nature 500 (2013) 415–421, 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.