

Abstract
Introduction
Helicobacter pylori is a prevalent pathogenic bacterium that resides in the human stomach. Outer membrane vesicles (OMVs) are known as nanosized cargos released by H. pylori, which have been proposed to have a key role in disease progression, pathogenesis, and modulation of the immune system. There are multiple evidences for the role of H. pylori in extragastroduodenal illnesses especially liver-related disorders. However, the precise mechanism of H. pylori extragastroduodenal pathogenesis still remains unclear. In the current study, we aimed to determine the impact of H. pylori-isolated OMVs on hepatic stellate cell (HSC) activation and expression of liver fibrosis markers.
Materials and Methods
Five H. pylori clinical strains with different genotype profiles were used. Helicobacter pylori OMVs were isolated using ultracentrifugation and were analyzed by scanning electron microscopy (SEM) and dynamic light scattering (DLS). Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) analysis was applied to determine protein components of H. pylori-derived OMVs. Cell viability of LX-2 human hepatic stellate cell line exposed to OMVs was measured by MTT assay. LX-2 cells were treated with OMVs for 24 h. The gene expression of α-SMA, E-cadherin, vimentin, snail, and β-catenin was analyzed using quantitative real-time PCR. The protein expression of α-SMA, as a well-studied profibrotic marker, was evaluated with immunocytochemistry.
Results
Our results showed that H. pylori strains released round shape nanovesicles ranging from 50 to 500 nm. Totally, 112 various proteins were identified in OMVs by proteomic analysis. The isolated OMVs were negative for both CagA and VacA virulence factors. Treatment of HSCs with H. pylori-derived OMVs significantly increased the expression of fibrosis markers.
Conclusions
In conclusion, the present study demonstrated that H. pylori-derived OMVs could promote HSC activation and induce the expression of hepatic fibrosis markers. Further research is required to elucidate the definite role of H. pylori-derived OMVs in liver fibrosis and liver-associated disorders.
1. Introduction
Helicobacter pylori is a well-studied Gram-negative, microaerophilic, and motile pathogenic microorganism that is able to colonize the human stomach [1]. Helicobacter pylori strains can express various virulence factors such as urease, flagellum, BabA, SabA, OipA, HopQ, CagA, VacA, and HtrA, which facilitate its attachment, colonization, and disease development. Recently, H. pylori-derived outer membrane vesicles (OMVs) have been proposed as a potent virulence marker involved in the pathogenesis of this bacterium [2]. OMVs are nanosized structures released from live bacteria in a logarithmic phase of growth and carry a lot of factors such as enzymes, phospholipids, nucleic acids, toxins, and any different proteins rooted from cytoplasm, membrane, and outer membrane [3]. These extracellular vesicles (EVs) can be engulfed by different eukaryote cells via different endocytosis strategies and transfer several virulence factors directly into the host cell cytoplasm [4, 5]. Previous investigations have demonstrated that H. pylori-derived OMVs can contribute to disease development, and it is documented that OMVs promote local and systemic inflammatory responses and cause gastric barrier dysfunction [2, 3].
Traditionally, H. pylori is known as the major causative agent for developing various gastroduodenal pathologies including peptic ulcer disease, gastritis, gastric adenocarcinoma, and mucosal-associated lymphoid tissue (MALT) cancer [6]. Nowadays, there are several reports for the role of H. pylori in extragastroduodenal diseases such as neurodegenerative disease, inflammatory bowel disease (IBD), celiac disease, and also liver-related disorders [7]. Also, several studies have documented a positive association between nonalcoholic fatty liver disease (NAFLD) and H. pylori infection [8, 9]. However, any attempt targeting H. pylori detection or culture from liver and hepatic tissues has failed so far [10].
Liver fibrosis is a cell response in liver chronic inflammation that may lead to hepatocellular carcinoma (HCC) under uncontrolled conditions. Viral or autoimmune hepatitis, alcohol abuse, NAFLD, and microbial infections are the main causes of liver inflammation [11]. Liver inflammation can result in activation of hepatic stellate cells (HSCs), which are the key players mediating liver fibrosis [12]. Activation of HSCs requires the change from a quiescent to a proliferative, fibrogenic, and migratory phenotype (i.e., myofibroblast), which is characteristic of hepatic fibrogenesis process [13].
So far, little is known regarding the role of OMVs released from H. pylori clinical strains and their cargo in development of extragastroduodenal diseases in particular liver-associated disorders. In the present study, we aimed to investigate the direct effects of H. pylori-derived OMVs on HSCs activation, and their capability to induce liver fibrosis markers using LX-2 cell line.
2. Materials and Methods
2.1. H. pylori Strains and Culture
A collection of five different clinical strains of H. pylori obtained from nonrepetitive patients in our previous works were used in this study [14, 15]. The clinical and genotypic characteristics of selected H. pylori strains are shown in Table 1. Brucella agar (Merck, Darmstadt, Germany) enriched with 10% fetal calf serum (FCS), 7% (v/v) horse blood, and Skirrow supplement (polymyxin 0.05 mg/L, vancomycin 2.0 mg/L, and trimethoprim 1.0 mg/L) was used to culture the strains. The culture plates were kept at 37°C in microaerophilic conditions for 3-5 days. Helicobacter pylori strains were confirmed by Gram stain technique, colony morphology, and positive reactions to biochemical tests such as catalase, oxidase, and urease. Pure cultures from the selected strains were stored in brain heart infusion medium (Merck, Darmstadt, Germany) enriched with 20% FCS and 15% glycerol at -80°C.
Table 1.
Clinical H. pylori strains used in this study.
| Strain no. | CagA | CagL | VacA | BabA2 | SabA | OipA | CagPAI | Disease outcome | Age | Gender |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | + | + | s1m2 | + | + | On | Partial | PUD | 49 | M |
| 2 | + | + | s1m1 | + | — | On | Intact | CG | 54 | M |
| 3 | — | — | s2m2 | + | + | Off | Completely deleted | CG | 43 | F |
| 4 | + | + | s1m1 | + | + | On | Intact | PUD | 25 | F |
| 5 | + | + | s1m2 | + | + | On | Intact | PUD | 60 | F |
PUD: peptic ulcer disease; CG: chronic gastritis; male (M), female (F). Strain number 3 was used as a less virulent strain.
2.2. OMV Isolation and Characterization
To isolate OMVs, H. pylori strains were cultured in Brucella broth medium (Merck, Darmstadt, Germany) enriched with 10% FCS for 72 h at 37°C under microaerobic conditions with continuous rotation (120 rpm). Then, supernatants were concentrated using centrifugations (10,000 × g, 20 min and 4°C).
After centrifugations, supernatants were filtered through a 0.45 μm cellulose filter membrane (MCE, USA), and OMVs were extracted using ultracentrifugation (Optima XE-100; Beckman Coulter, USA) at 200,000 × g, for 3 h at 4°C as previously described with slight modifications [16]. The isolated OMVs were resuspended in sterile PBS and frozen at -80°C until use. Dynamic light scattering (DLS) and scanning electron microscopy (SEM) were performed to identify the OMVs size and morphology. BCA assay and sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) were used to determine the protein concentration and content of OMVs.
2.3. OMVs Proteome
For OMV proteomics, in the first step, samples were heated at 99°C for 5 min in SDS sample buffer to stabilize them and eliminate any protease activity. For SDS-PAGE, samples were subjected to electrophoresis in SDS-10% polyacrylamide gel electrophoresis based on the previous published method [17]. The gels were stained with Coomassie blue for visualizing the proteins bands. For each sample, the entire lane of stained protein was cut from the gel and digested with trypsin overnight as previously published [18]. Resulting peptides were desalted with STAGE tips based on previously described protocols [19]. After peptide preparation, 2 μg of each sample was injected into quadrupole-time of flight mass spectrometry (Impact II, Bruker Daltonics) coupled to an EASY-nLC 1000 (Thermo Fisher Scientific, USA) with previously described parameters [20]. Final results were analyzed by MaxQuant version 1.6.7.0 [21] and searched against H. pylori databases from the Uniprot database.
2.4. Cell Culture and Viability Assay
To examine the effects of OMVs on HSC activation, LX-2 cells were treated with the representative OMVs obtained from H. pylori clinical strain no. 1 as indicated in Table 1. The LX-2 human hepatic stellate cell line was kindly provided by Professor Friedman (Mount Sinai School of Medicine, New York, USA). LX-2 cells were cultured in complete Dulbecco's modified Eagle's medium (DMEM) enriched with 2% (v/v) heat-inactivated fetal bovine serum (FBS) (Gibco/Invitrogen, Carlsbad, CA, USA), 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 2 mM of L-glutamine. To determine the LX-2 cells viability, MTT assay was carried out with the commercial Cell Proliferation Kit I (Sigma-Aldrich, St. Louis, Missouri, USA) based on the producer's protocol. In brief, LX-2 cells at a density of 5 × 103 cells per each well were seeded in 96-well plates and then separately contaminated and incubated with varying concentrations (1, 5, 10, 15, 20, and 25 μm/mL) of H. pylori OMVs for 24 h. At the indicated time point, 10 μL of MTT reagent (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was added to each well, and the cells were kept for 4 h at 37°C. The process was stopped with dimethyl sulfoxide (DMSO), color developing substance, and absorbance at 560 nm was documented using a microplate reader (ELx808, BioTek Instruments, Winooski, Vermont, USA). The percentage of LX-2 cell viability was determined using the formula: cell viability (%) = (absorbance of treated cells × 100%)/absorbance of untreated cells.
2.5. LX-2 Cell Treatments
LX-2 cells were cultured in 6-well plates at a density of 5 × 105 cells in each well and kept at 37°C for 24 h in a CO2 incubator. Before treatments, the seeded cells were washed with PBS (pH 7.2), and the culture media were changed with DMEM medium. After that, the LX-2 cells were contaminated with 5 μg/mL of H. pylori-derived OMVs for 24 h. In addition, the untreated LX-2 cells were used as the control group. The experiments were performed in triplicate and repeated at least three times. After 24 h, the cell pellets were used for RNA extraction and gene expression assays.
2.6. Immunocytochemistry
The protein expression of α-SMA, as a well-studied profibrotic marker for the evaluation of HSC activation and fibrosis, was determined in the OMV-treated LX-2 cells by using immunocytochemistry (ICC) technique. Briefly, cells were seeded into six-well plates at a density of 5 × 105 cells/well, exposed to H. pylori OMVs at concentration of 5 μg/mL for 24 h. Treated cells were washed with sterile PBS (pH 7.2) for three times, fixed with 4% paraformaldehyde in sterile PBS (pH 7.5), and their membrane permeabilized using 0.5% Tween-20 in 5% bovine serum albumin (BSA) blocking solution for 30 min. For analysis of α-SMA, cells were incubated with rabbit polyclonal α-SMA antibody (1 : 100, Cell Signaling, Cat#: GTX10034) 24 h at 4°C. After 24 h, the treated cells were incubated with a goat anti-rabbit IgG Alexa Fluor 488 (Life Technologies, Cat# BA1054-2) for 1 h at ambient temperature. Also, DAPI staining was used for 15 min to see the fixed cells nuclei. Fluorescent images were documented by the Cytell-TM Cell Imaging System (GE, Healthcare, UK). The protein expression level was reported as the fold change comparing to the untreated control cells as negative control.
2.7. RNA Extraction and cDNA Synthesis
Total RNA was extracted from OMV-treated cells and untreated LX-2 cells, with the FavorPrep™ blood and cell total RNA mini kit (Favorgen® Biotech Corp., Pingtung, Taiwan) based on the developer's instruction. The RNA samples were stored at -80°C for further gene expression analysis. The quality and concentration of extracted RNAs were determined by measuring absorbance at 260 nm with a NanoDrop® ND-1000 spectrophotometer (Thermo Scientific, USA). RNAs were reverse transcribed to cDNA using the commercial cDNA synthesis kit (Favorgen® Biotech Corp., Pingtung, Taiwan) based on the manufacturer's instruction.
2.8. Quantitative Real-Time PCR
The quantitative real-time PCR assay was performed to investigate β-catenin, E-cadherin, snail, vimentin, and α-SMA genes expression level using the Rotor-Gene® Q real-time PCR system (Qiagen, Germany) and BioFACT™ 2X Real-Time PCR master mix (Biofact, Daejeon, South Korea). The primers and the size of each product used to examine the expression of β-catenin, E-cadherin, snail, vimentin, and α-SMA genes are presented in Table S1. The GAPDH housekeeping gene was used as the endogenous control. To evaluate the amplification process, melting curve analysis was used after each run. All reactions were run for three times. The relative gene expression was determined by applying the 2-ΔΔCt method, and the mRNA expression level was given as the fold change comparing to the untreated control cells.
2.9. Statistical Analysis
The obtained data were analyzed using the GraphPad Prism 8 (GraphPad Software, Inc., USA). One-way ANOVA was applied to calculate variations among different groups, and comparisons between each group were analyzed using t-test. Results were shown as the average ± standard error of the mean (SEM) of at least triplicate experiments, unless otherwise stated. Differences among the study groups were considered statistically significant when P < 0.05, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, and ∗∗∗∗P < 0.0001.
3. Results
3.1. Characterization of H. pylori OMVs and Their Protein Contents
The cell-free OMVs extracted from H. pylori clinical strains were purified by ultracentrifugation. The phenotypic characterization of OMVs was evaluated using DLS and SEM, respectively. Obtained data revealed that OMVs isolated from H. pylori clinical strains are round shape with a bilayer membrane and were ∼50-450 nm in diameter (Figure 1). This showed that OMVs were purified successfully without contamination with other bacterial components for subsequent cell experiments. The SDS-PAGE revealed protein bands with varying molecular weights, from 11 to 245 kDa, which shows the protein content of OMVs. To explore the protein contents of H. pylori-derived OMVs, liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) analysis was applied, and 112 different proteins were identified. Proteins are categorized in different classes including the following: 9 (8%) outer membrane proteins, 7 (6.2%) protein with unknown functions, 2 (1.7%) antibiotic resistance-associated proteins, 10 (8.9%) colonization factors, 5 (4.5%) acid resistance-related proteins, 12 (10.7%) potent virulence factors, 50 (44.6%) metabolism and vital functions, 3 (2.6%) motility, 3 (2.6%) ribosomal proteins, 2 (1.7%) iron-related proteins, and 11 (9.8%) others, ranked by abundance in Table 2. Among these different proteins that were detected, 47 (41.9%) proteins were common in the OMVs derived from H. pylori strains numbers 1, 2, 4, and 5 (OMV 1 had 8 (7.1%) unique proteins, OMV 2 had 13 (11.6%) unique proteins, OMV 4 had 30 (26.7%) unique proteins, and OMV 5 had 17 (15.1%) unique proteins) as indicated in Table S2. In addition, there were no unique proteins identified in OMV 3, which was obtained from a less virulent H. pylori strain. Among various proteins, urease and catalase enzymes were abundant, and metabolism plus proteins with vital functions was more condense. Also, all extracted OMVs were negative for both CagA and VacA virulence proteins.
Figure 1.
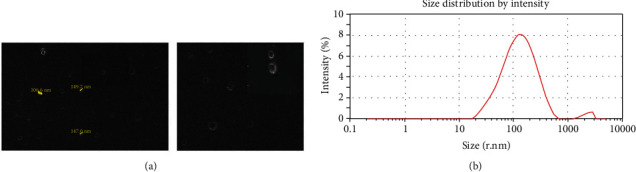
Morphological characterization of H. pylori-derived OMVs. (a) Scanning electron microscopic (SEM) image of OMVs indicated the round-shaped and double-layered vesicles in various sizes (range: 50-200 nm). (b) Size distribution by intensity of H. pylori-derived OMVs based on DLS. Size distribution is determined based on the intensity of OMVs in the ultracentrifugation technique. DLS confirmed nanosized OMVs in a range of about 50 to 450 nm, peaked at 100-150 nm.
Table 2.
Quantification proteomics analysis of OMVs derived from pathogenic clinical strains of H. pylori.
| Protein ID | Protein description | LFQ intensity OMV 1 | LFQ intensity OMV 2 | LFQ intensity OMV 4 | LFQ intensity OMV 5 | Possible function |
|---|---|---|---|---|---|---|
| Outer membrane proteins (OMP) | ||||||
| WP_000595790.1 | Outer membrane beta-barrel protein | 504540 | 3604860 | 963000 | 4892000 | OMP/colonization/adhesion |
| WP_000768579.1 | Outer membrane beta-barrel protein HofC | 0 | 203850 | 0 | 184520 | OMP/colonization/adhesion |
| WP_000395375.1 | Hop family outer membrane protein (OMP) HopE | 0 | 245410 | 693600 | 304390 | OMP/colonization/adhesion/type 4 secretion system assembly |
| WP_000753173.1 | Hop family adhesion BabB (Omp19) | 0 | 0 | 0 | 73925 | Blood group antigen binding adhesin B (BabB) |
| WP_000592437.1 | Hop family adhesion AlpA (Omp20) | 0 | 0 | 0 | 1082800 | Adherence-associated lipoprotein A (AlpA) |
| WP_000812546.1 | Hop family adhesion AlpB (Omp21) | 0 | 0 | 0 | 277750 | Adherence-associated lipoprotein B (AlpB) |
| WP_000831156.1 | Outer membrane protein Omp18 | 0 | 1459100 | 0 | 429570 | OMP/immune stimulator |
| WP_000751514.1 | Hop family adhesion HopQ (Omp27) | 0 | 362580 | 0 | 0 | OMP27/CagA transporter |
| WP_000716249.1 | Hop family adhesion BabA (Omp28) | 0 | 0 | 1279040 | 1267000 | Blood group antigen binding adhesin A (BabA) |
|
| ||||||
| Protein with unknown functions | ||||||
| WP_001268661.1 | Hypothetical protein | 0 | 0 | 0 | 50133 | Unknown |
| WP_000725112.1 | Hypothetical protein | 0 | 249580 | 0 | 196290 | Unknown |
| WP_000446639.1 | DUF874 family protein | 0 | 29293 | 0 | 0 | Unknown |
| WP_001225999.1 | Hypothetical protein | 23272 | 0 | 0 | 0 | Unknown |
| WP_001228436.1 | Hypothetical protein | 0 | 0 | 0 | 189012 | Unknown |
| WP_001202825.1 | Hypothetical protein | 0 | 0 | 0 | 80665 | Unknown |
| WP_000323695.1 | DUF3944 domain-containing protein | 0 | 450110 | 154830 | 561010 | Unknown |
|
| ||||||
| Antibiotic resistance | ||||||
| WP_000912892.1 | Leucyl aminopeptidase | 24719300 | 39469000 | 40399000 | 40792000 | Colonization/metronidazole resistant |
| WP_000670110.1 | NAD(P)H-dependent oxidoreductase | 0 | 0 | 194590 | 0 | Metronidazole resistant |
|
| ||||||
| Colonization factors | ||||||
| WP_000738964.1 | Polyisoprenoid-binding protein | 0 | 0 | 0 | 1887810 | Colonization |
| WP_001174648.1 | Thiol peroxidase | 0 | 0 | 0 | 147870 | Stress resistance and host colonization |
| WP_001285084.1 | Acetone carboxylase subunit alpha | 10039200 | 2686780 | 8418600 | 17540800 | Colonization |
| WP_001862402.1 | Acetone carboxylase subunit gamma | 0 | 0 | 1948520 | 0 | Colonization |
| WP_000731297.1 | Fibronectin type III domain-containing protein | 0 | 229900 | 0 | 175690 | Colonization/type 3 secretion system |
| WP_001269995.1 | Tol/Pal (colicin-tolerant/peptidoglycan-associated lipoprotein) system protein TolB | 0 | 0 | 0 | 53991 | TolB |
| WP_000795978.1 | LPP20 lipoprotein | 2583170 | 3121670 | 4231600 | 5133700 | Colonization |
| WP_000961643.1 | Peroxiredoxin | 21375000 | 20724500 | 16015000 | 24932200 | Colonization |
| WP_001215729.1 | Aliphatic amidase | 1222630 | 176590 | 4558450 | 7925500 | Ammonia production/increase colonization |
| WP_001254471.1 | Gamma-glutamyltransferase | 0 | 0 | 0 | 48692 | Gastric colonization |
|
| ||||||
| Acid resistance | ||||||
| WP_000038087.1 | Nickel-dependent hydrogenase large subunit | 0 | 422490 | 0 | 311320 | Urease maturation protein |
| WP_000709479.1 | Polyisoprenoid-binding protein | 0 | 0 | 353520 | 0 | Acid response protein |
| WP_000779223.1 | Urease subunit alpha | 48115000 | 112113000 | 321814000 | 107958000 | Urease A |
| WP_000724295.1 | Urease subunit beta | 87129000 | 68324000 | 274879000 | 157816000 | Urease B |
| WP_001099471.1 | Urease accessory protein UreD | 0 | 761510 | 988360 | 334590 | Urease D |
|
| ||||||
| Potent virulence factors | ||||||
| WP_000846523.1 | YbhB/YbcL family Raf kinase inhibitor-like protein | 0 | 0 | 199250 | 0 | Induce tumorigenesis |
| WP_001040419.1 | Polysaccharide deacetylase | 685340 | 1377940 | 0 | 2623860 | PG amidase/immune response inducer |
| WP_001053815.1 | Acetyl-CoA carboxylase biotin carboxyl carrier protein | 0 | 0 | 0 | 44002 | Adenocarcinoma progress |
| WP_000461024.1 | ATP-dependent protease subunit HslV | 0 | 0 | 0 | 32755 | Protease activity |
| WP_000890837.1 | Tumor necrosis factor alpha-inducing protein | 0 | 1332180 | 0 | 319850 | Accelerate inflammation and cancer development |
| WP_000467802.1 | Methyl-accepting chemotaxis protein | 2200500 | 4482860 | 3225630 | 3047290 | Chemotaxis |
| Periplasmic serine endoprotease DegP-like | 0 | 657760 | 909990 | 770250 | Protease high temperature requirement A (HtrA) | |
| WP_000034151.1 | Acyl-ACP--UDP-N-acetylglucosamine O-acyltransferase | 0 | 93066 | 0 | 0 | LPS assembly |
| WP_000846461.1 | DNA starvation/stationary phase protection protein | 7560800 | 6572800 | 16870200 | 11009900 | Accumulation of neutrophils and monocytes at the site of infection |
| WP_000858181.1 | 3-Deoxy-8-phosphooctulonate synthase | 0 | 50028 | 0 | 0 | 3-Deoxy-d-manno-octulosonic acid (KDO) |
| WP_000739470.1 | Peptidylprolyl isomerase CBF2 | 0 | 0 | 0 | 358410 | TH17 inflammation inducer |
| WP_000540573.1 | ATP-dependent Clp endopeptidase proteolytic subunit ClpP | 0 | 264590 | 0 | 563180 | ATP-dependent caseinolytic proteases (ClpP) |
| Metabolism and vital functions | ||||||
| WP_000506216.1 | Enoyl-ACP reductase FabI | 112930 | 0 | 0 | 0 | Lipid metabolism |
| WP_000866624.1 | Peptide-methionine (R)-S-oxide reductase MsrB | 0 | 0 | 1182160 | 0 | Repair enzyme for proteins that have been inactivated by oxidation |
| WP_000422563.1 | DEAD/DEAH box helicase | 0 | 0 | 0 | 45347 | Helicase |
| WP_001207734.1 | Ketol-acid reductoisomerase | 0 | 1747020 | 1312860 | 186410 | Amino acid synthesis enzyme |
| WP_001204347.1 | Menaquinone biosynthesis decarboxylase | 0 | 0 | 0 | 33451 | Respiratory mechanism |
| WP_000662799.1 | M3 family oligoendopeptidase | 214060 | 52823 | 759950 | 0 | Peptidoglycan endopeptidase |
| WP_000524601.1 | Redox-regulated ATPase YchF | 0 | 84152 | 0 | 0 | Regulator protein |
| WP_000608576.1 | 2,3,4,5-Tetrahydropyridine-2,6-dicarboxylate N-succinyltransferase | 0 | 173380 | 0 | 0 | Amino acid synthesis enzyme/unknown function |
| WP_001006979.1 | Acetyl-CoA C-acetyltransferase | 463830 | 0 | 0 | 0 | Metabolic enzyme |
| WP_000650637.1 | Hydantoinase/oxoprolinase family protein | 9456000 | 3187340 | 7925600 | 17515000 | Metabolic enzyme |
| WP_000862222.1 | GMP reductase | 0 | 0 | 32786 | 0 | Nucleic acid catabolic enzyme |
| WP_001268933.1 | ADP-glyceromanno-heptose 6-epimerase | 0 | 0 | 0 | 227002 | Catabolic enzyme |
| WP_001126584.1 | Carbamoyl-phosphate synthase large subunit | 0 | 0 | 402270 | 0 | Nitrogen metabolism |
| WP_001159546.1 | 4-Hydroxy-tetrahydrodipicolinate synthase | 0 | 0 | 0 | 370800 | Metabolic enzyme |
| WP_000060246.1 | AAA family ATPase | 0 | 0 | 0 | 612130 | ATPases |
| WP_000062669.1 | 3-Methyl-2-oxobutanoate hydroxymethyltransferase | 246880 | 1145910 | 0 | 559400 | Metabolic enzyme |
| WP_000037869.1 | DNA-directed RNA polymerase subunit beta/beta | 464440 | 1495780 | 0 | 1015630 | RNA polymerase |
| WP_000534771.1 | Formamidase | 0 | 0 | 0 | 856430 | Nitrogen metabolism |
| WP_001160526.1 | Class II fumarate hydratase | 0 | 0 | 1094120 | 0 | Nitrogen metabolism |
| WP_001862586.1 | S41 family peptidase | 0 | 0 | 565030 | 0 | Endopeptidase |
| WP_000438056.1 | 3-Hydroxyacyl-ACP dehydratase FabZ | 0 | 0 | 97338 | 0 | Catabolic enzyme |
| WP_000780126.1 | UbiX family flavin prenyltransferase | 0 | 127650 | 0 | 256960 | Catabolic enzyme |
| WP_000616334.1 | Transcription-repair coupling factor | 0 | 0 | 0 | 310300 | Repair enzyme |
| WP_000963128.1 | Recombinase RecA | 170880 | 0 | 0 | 0 | RecA |
| Superoxide dismutase [Fe] | 0 | 0 | 0 | 749400 | Superoxide dismutase | |
| WP_000955673.1 | Phosphopyruvate hydratase | 0 | 56407 | 0 | 0 | Enolase |
| WP_000412947.1 | Peroxiredoxin | 1152900 | 0 | 0 | 0 | Catabolic enzyme |
| WP_000080506.1 | F0F1 ATP synthase subunit alpha | 6236520 | 12373400 | 3033330 | 3668760 | ATPase |
| WP_001863096.1 | F0F1 ATP synthase subunit beta | 1873140 | 2158280 | 2582800 | 1605730 | ATPase |
| WP_000289187.1 | NADP-specific glutamate dehydrogenase | 0 | 496220 | 8149800 | 2938760 | Metabolic enzyme |
| WP_000864547.1 | DNA-directed RNA polymerase subunit alpha | 0 | 0 | 0 | 22302 | RNA polymerase |
| WP_001040579.1 | Elongation factor Tu | 2547840 | 2607030 | 1626200 | 1537060 | Elongation factor |
| WP_000117507.1 | Citrate synthase | 2217840 | 4356700 | 3076740 | 2120940 | Metabolic enzyme |
| WP_000323988.1 | Isocitrate dehydrogenase (NADP(+)) | 0 | 555280 | 0 | 331560 | Metabolic enzyme |
| WP_001242837.1 | Cystathionine gamma-synthase | 0 | 0 | 873810 | 0 | Metabolic enzyme |
| WP_000476591.1 | Porphobilinogen synthase | 0 | 0 | 0 | 74893 | Metabolic enzyme |
| WP_000002194.1 | F0F1 ATP synthase subunit gamma | 0 | 0 | 65433 | 0 | ATPase |
| WP_001221712.1 | IMP dehydrogenase | 806320 | 1544980 | 0 | 1451060 | Metabolic enzyme |
| WP_001124018.1 | Phosphogluconate dehydratase | 158780 | 161430 | 197580 | 608640 | Metabolic enzyme |
| WP_001217520.1 | Aspartate ammonia-lyase | 1172670 | 826180 | 924680 | 303884 | Nitrogen metabolism |
| WP_000131629.1 | Ribonuclease J | 0 | 0 | 0 | 127260 | Ribonuclease |
| WP_000774319.1 | Excinuclease ABC subunit (UvrC) | 0 | 158820 | 0 | 0 | UvrC |
| WP_000564420.1 | Thioredoxin-disulfide reductase | 0 | 0 | 304110 | 0 | Metabolic enzyme |
| WP_000187711.1 | Purine-nucleoside phosphorylase | 0 | 0 | 0 | 233580 | Metabolic enzyme |
| WP_010875529.1 | Excinuclease ABC subunit UvrA | 0 | 121980 | 0 | 0 | UvrA |
| WP_000020199.1 | Thioredoxin | 0 | 0 | 918110 | 1172960 | Metabolic enzyme |
| WP_000247370.1 | Catalase | 39582000 | 69503000 | 262561000 | 94226000 | Catalase |
| WP_000637150.1 | Type I glutamate--ammonia ligase | 0 | 0 | 3351300 | 0 | Metabolic enzyme |
| WP_000053246.1 | Glutamate--tRNA ligase | 0 | 0 | 0 | 154360 | Metabolic enzyme |
| WP_000699284.1 | Type II 3-dehydroquinate dehydratase | 0 | 4075540 | 0 | 8030600 | Catabolic enzyme |
| Motility | ||||||
| WP_000885496.1 | Flagellin A | 798030 | 3071800 | 50265700 | 2449870 | Flagellin A |
| WP_000010001.1 | Flagellin B | 0 | 132380 | 0 | 0 | Flagellin B |
| WP_000646667.1 | Flagellar sheath lipoprotein HpaA | 0 | 766340 | 0 | 1061070 | N-Acetylneuraminyllactose-binding hemagglutinin |
|
| ||||||
| Ribosomal proteins | ||||||
| WP_001018245.1 | 50S ribosomal protein L7/L12 | 3165000 | 0 | 0 | 0 | L7/L12 |
| WP_000529962.1 | 30S ribosomal protein S3 | 0 | 0 | 134180 | 0 | S3 |
| WP_001862443.1 | 50S ribosomal protein L9 | 0 | 0 | 111940 | 0 | L9 |
|
| ||||||
| Iron-related proteins | ||||||
| WP_000934548.1 | HugZ family heme oxygenase | 0 | 0 | 3413400 | 0 | Iron acquisition |
| WP_000949202.1 | Nonheme ferritin | 5908500 | 9860700 | 9396400 | 14801600 | Ferritin like protein |
|
| ||||||
| Others | ||||||
| WP_000342347.1 | Hybrid sensor histidine kinase/response regulator | 0 | 192370 | 0 | 0 | Response protein |
| WP_000785752.1 | Cag pathogenicity island protein Cag1 | 0 | 203798 | 0 | 0 | Membrane apparatus of T4SS |
| WP_000714010.1 | Insulinase family protein | 0 | 0 | 0 | 105420 | Insulinase/unknown activity |
| WP_001265981.1 | NAD(P)-dependent alcohol dehydrogenase | 0 | 0 | 0 | 439940 | Sensor protein |
| WP_000855958.1 | Transporter substrate-binding domain-containing protein | 2392480 | 3797000 | 4192600 | 2420660 | Transporter |
| WP_029671489.1 | Outer membrane beta-barrel protein | 0 | 0 | 0 | 64626 | Transporter |
| WP_001862559.1 | ABC transporter substrate-binding protein | 0 | 0 | 0 | 948250 | Transporter |
| WP_001210849.1 | Pyridoxine 5-phosphate synthase | 3573670 | 8087900 | 1476140 | 2574090 | Transferase |
| WP_000671934.1 | Cochaperone GroES | 2343300 | 3182200 | 2711900 | 3589100 | Chaperone |
| WP_001040293.1 | Chaperonin GroEL | 64544000 | 136783000 | 55262000 | 109558000 | Chaperone |
| WP_000664941.1 | Copper-translocating P-type ATPase CopA | 781570 | 0 | 0 | 0 | Chemotaxis/heat-shock response |
OMV1: OMVs derived from H. pylori strain 1; OMV2: OMVs derived from H. pylori strain 2; OMV4: OMVs derived from H. pylori strain 4, OMV5: OMVs derived from H. pylori strain 5. Abbreviations: H. pylori outer membrane protein family C: HofC, Helicobacter outer membrane proteins: Hop, nicotinamide adenine dinucleotide: NAD, Lpp20: lipoprotein 20, heat-shock locus V: hslV, lipopolysaccharides: LPS, protease high temperature requirement A: HtrA, deoxyribonucleic acid: DNA, 3-deoxy-d-manno-octulosonic acid: KDO, cell binding factor 2: CBF2, ATP-dependent caseinolytic proteases: ClpP, fatty acid biosynthesis I: FabI, methionine sulfoxide reductase B: MsrB, guanosine monophosphate: GMP, adenosine diphosphate: ADP, ATPases associated with diverse cellular activities: AAA, adenosine triphosphatase: ATPases, recombinase A: RecA, nicotinamide adenine dinucleotide phosphate: NADP, thermal unstable: Tu, inosine monophosphate: IMP, ultraviolet radiation: Uvr, Helicobacter pylori adhesion A: HpaA, heme utilization gene: Hug, cytotoxic associated gene: cag, copper-translocating P-type ATPase: CopA.
3.2. Effects of H. pylori-Derived OMVs on LX-2 Cell Viability
To assess cytotoxic effects of OMVs on HSCs, direct microscopic examination and cell viability assay were performed. Compared with the control cells after 24 h, OMVs derived from H. pylori induced significant increase in the number of viable LX-2 cells with 1 and 5 μg/mL concentrations during the coculture time. Furthermore, MTT assay indicated that H. pylori OMVs at concentrations of 10, 15, 20, and 25 μg/mL significantly affected the number of LX-2 cells in contrast to untreated control cells (Figure 2). Accordingly, OMVs with 5 μg/mL concentration was used in further coculture assays with LX-2 cells.
Figure 2.
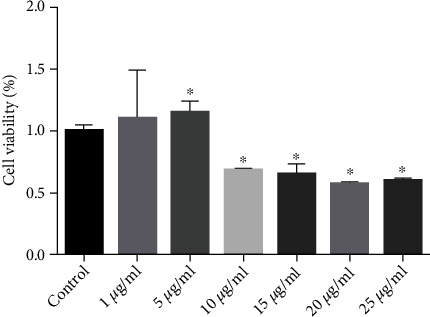
Effects of varying concentrations of the representative OMVs obtained from H. pylori clinical strain no. 1 (1, 5, 10, 15, 20, and 25 μg/mL) on cell viability of the LX-2 cells. Data are shown as the mean ± SEM. ∗P < 0.05.
3.3. OMVs Promoted Gene Expression of Fibrogenesis Markers in LX-2 Cells
To evaluate the expression of fibrotic markers in stimulated LX-2 cells, qRT-PCR was performed to detect the mRNA expression levels of E-cadherin, vimentin, β-catenin, α-SMA, and snail. The GAPDH gene was used as a reference gene. As shown in Figure 3, OMVs isolated from H. pylori strains numbers 1, 2, 4, and 5 significantly elevated the gene expression of α-SMA, β-catenin, and vimentin expression level in comparison to control cells. Moreover, OMVs derived from different H. pylori strains applied in this work downregulated the expression level of E-cadherin as compared to control cells. Regarding the snail expression, OMVs isolated from H. pylori strains numbers 1, 2, and 4 upregulated the expression of this marker, while OMVs derived from strains numbers 3 and 5 decreased its expression in comparison with untreated cells. These results demonstrate the capability of H. pylori OMVs to activate HSCs and stimulate the expression of liver fibrosis markers. Furthermore, there was no significant difference in the ability of OMVs originated from H. pylori strain number 3 (as less virulent strain according to genotyping results) to modulate the gene expression level of fibrosis markers, except for β-catenin, in comparison to OMVs derived from other H. pylori strains.
Figure 3.
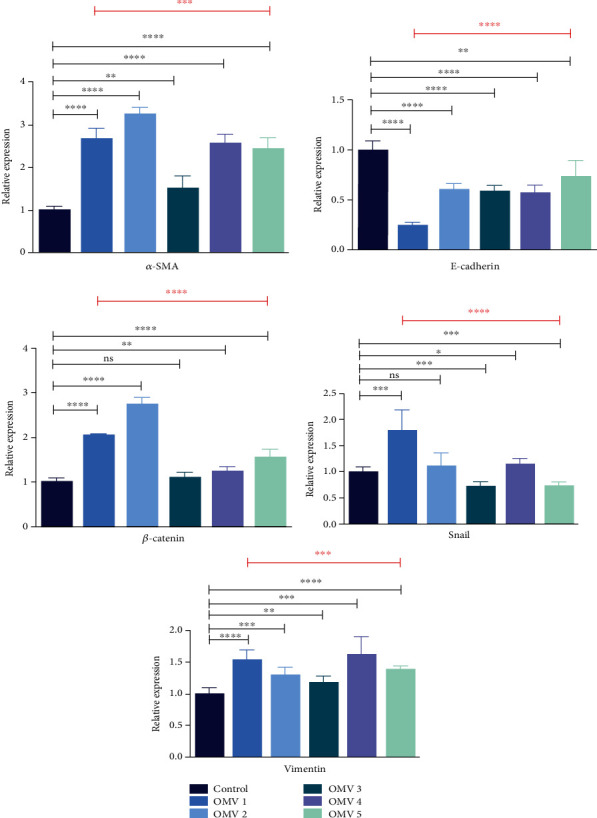
Gene expression of liver fibrosis markers in LX-2 cells upon treatment with 5 μg/mL of H. pylori OMVs. Relative gene expression of fibrosis markers (α-SMA, β-catenin, and vimentin) was markedly increased after treatment of LX-2 cells with OMVs derived from different H. pylori strains. The mRNA level of E-cadherin was decreased in LX-2 cells treated with OMVs isolated from different H. pylori strains. However, OMVs isolated from H. pylori strains numbers 1, 2, and 4 upregulated the expression of snail, while OMVs derived from strains numbers 3 and 5 decreased its expression in comparison with untreated cells. Data presented as means ± standard error (SEM) for three independent experiments. Data are shown as the mean ± SEM. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, and ∗∗∗∗P < 0.0001 by post hoc one-way ANOVA statistical analysis.
3.4. OMVs Increased the Protein Expression of α-SMA in LX-2 Cells
Immunocytochemistry was used to measure the protein expression level of α-SMA, as an important marker, which is overexpressed during HSC activation. Protein expression of α-SMA mostly confirmed the gene expression in the different studied groups. Our findings suggest that H. pylori OMVs have noticeable potential to increase more than twofold of the expression of HSC fibrotic markers as compared with untreated HSCs that served as control cells (Figure 4).
Figure 4.
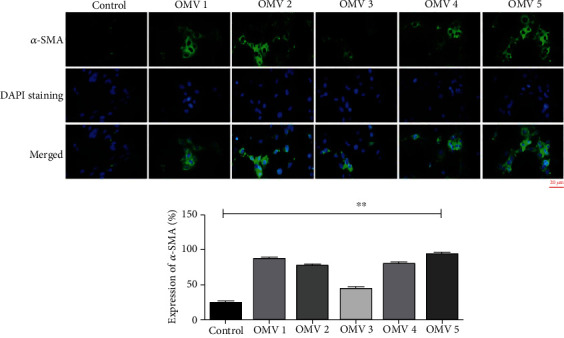
Protein expression of α-SMA in LX-2 cells upon treatment with 5 μg/mL of H. pylori OMVs. Relative protein expression of α-SMA was markedly increased after treatment of LX-2 cells with 5 μg/mL of H. pylori OMVs. Data presented as means ± standard error (SEM) for three independent experiments. Statistical significance was determined using one-way ANOVA with ∗∗P < 0.01. Scale bar: 20 μm, magnification 200X.
4. Discussion
Helicobacter pylori is a well-known and important pathogen involved in different gastric diseases such as acute and chronic gastritis, gastric atrophy, and gastric malignancy [22]. It is clear that H. pylori infection can lead to serious diseases in the stomach and duodenum, but there has also been growing documents of its clinical consequences in extragastroduodenal diseases. According to recent studies, H. pylori infection has been related to different disorders in the gastrointestinal tract, and there has been hypothesis suggesting possible association of H. pylori infection with NAFLD development, which is the well-studied hepatic syndrome associated with chronic disorders such as obesity, diabetes, and cardiovascular disease [23]. The evidence for H. pylori and NAFLD association focused on the detection of anti-H. pylori IgG in patients suffering from NAFLD [24–27]. Liver fibrosis is a multifactorial and complex process especially because of HSC activation and high overexpression of liver fibrosis markers which can cause hepatocellular dysfunction [26]. The principal extracellular causative agents that cause liver fibrosis include chronic infections and epithelial cell damage. To the author's knowledge, until now, H. pylori culture has been negative among liver biopsies. However, in some studies H. pylori DNA was detected in liver, which could be likely due to the presence of DNA in H. pylori-derived OMVs [28, 29]. These observations highlighted the possible effects of novel virulence factors such as OMVs in development of H. pylori-associated extragastroduodenal diseases.
Helicobacter pylori-released OMVs are abundant from outer membrane proteins (OMPs), toxins, several adhesins, and different molecules known to stimuli cell proliferation, chemokine and cytokine production, and other host cellular responses [30]. In our previous work, we demonstrated that H. pylori-derived OMVs may alter the content of hepatocyte-originated exosomes, and such modified exosomes could possibly activate HSCs and promote liver fibrosis progression. We reported that hepatocytes extracted exosomes treated with H. pylori-derived OMVs can cause HSC activation and elevate the expression of hepatic fibrosis markers (β-catenin, vimentin, α-SMA, and TIMP-1) [31].
The current in vitro study showed that treatment of H. pylori OMVs regardless to isolate genotypes can act as a virulence attribute to active HSCs and induce liver fibrosis markers. Similar to our findings, there are several reports supporting the putative role of H. pylori-isolated OMVs in bacterial pathogenesis and disease progression. For instance, data obtained from an in vitro study by Hock et al. [32] indicated that H. pylori OMVs can strongly induce expression of COX-2 by the peripheral blood mononuclear cells (PBMC) and significantly elevate levels of PGE2 and IL-10, leading to suppression of human T cell responses. They also demonstrated that these effects were independent of VacA cytotoxin expression. Additionally, Choi et al. [33] investigated the clinical importance of H. pylori OMVs on the development of gastric cancer. They purified H. pylori OMVs from gastric secretion samples of patients with different gastric diseases including patients suffering from gastric and duodenal ulcer and gastric cancer. Their study revealed that H. pylori-derived EVs, which are rich in the gastric secretions of GC patients, are involved in the induction of inflammatory response and cancer progression in the stomach. Another study conducted by Zhang et al. [34], demonstrated that H. pylori OMV packages can provoke the production of IL-8 in AGS cell line due to the presence of small noncoding RNAs (sR-2509025 and sR-989262) resulting in evasion of host immune surveillance and responses. These findings imply that H. pylori-derived OMVs are crucial factors, which can carry various pathogenic cargos, and play a key role in H. pylori-related diseases and malignancy, in particular extragastroduodenal disorders.
In the current study, according to proteomic data, we found 112 different proteins within the OMVs derived from four H. pylori virulent strains during the exponential phase of growth, which were abundant in OMPs and enzymes like catalase and urease. However, interestingly, our proteomics results confirmed that no CagA and VacA proteins were present in the extracted OMVs. Comparing to our findings, Mullaney et al. [30] showed the existence of VacA and CagA in H. pylori OMVs. They identified 162 OMV-associated proteins in the H. pylori strains J99 and 91 in the strain 11637. These reports showed that CagA and VacA proteins could be present or absent in OMVs derived from different H. pylori strains. Despite the fact that the CagA and VacA are known as major virulence factors in H. pylori disease development, presence of other potent pathogenic factors in OMVs may result in activation of HSCs. Furthermore, a previous study that was conducted by Carlsohn et al. [35] reported more than 60 unique membrane or membrane-associated proteins in different clinical isolates of H. pylori. Some membrane proteins such as BabA and Omp11 were detected to be expressed by 15 different clinical isolates of H. pylori. Also, they established a mass spectrometry-based method that allows detection of OMPs in clinical isolates of H. pylori. However, recently, it was also indicated that OMV contents are highly associated with the H. pylori OMV size range. The proteomic studies showed that adhesins (Hop, SabA, and BabA) are more abundant among larger OMVs compared to smaller OMVs, while smaller OMVs contain significantly more metabolic proteins. Interestingly, H. pylori survival- or virulence-related proteins such as neutrophil activating protein, VacA, urease A and B subunits, and the porin HopA were common in both small and large OMVs [5]. Moreover, the study conducted by Turkina et al. [36] identified that H. pylori OMVs containing CagA could interact with gastric epithelia and influence host gene transcription and potentially cause serious clinical outcomes as a consequence of H. pylori infection.
HSCs play an important role in the liver fibrosis process. Activated HSCs are identified by the overexpression of α-SMA and extreme expression of extra cellular matrix proteins (vimentin, β-catenin, and snail) through inflammatory cytokines such as TNF-α, IL-1β, and IL6, which increase the development of liver fibrosis [37, 38]. A study conducted by Camara et al. revealed that TNF-α markedly elevated the effects of TGF-β1 cytokine, a well-known profibrotic cytokine, on fibrosis [39]. Also, results obtained from Fielding et al. study demonstrated strictly the dependence of fibrosis on interleukin-6 (IL-6). Also, their study showed that repetitive inflammation process induced Th1 cell effector activation via IL-6 cytokine and increased STAT-1 (signal transducer and activator of transcription-1) pathway activation, which can cause fibrosis [40]. In this study, we showed overexpressing of α-SMA, vimentin, and β-catenin and downregulation of E-cadherin in HSCs treated with CagA and VacA-negative H. pylori OMVs. Our findings revealed the presence of 112 different proteins in the proteome of OMVs that potentially may engage in HSCs activation. Similar to our findings, Goo et al. [41] revealed that orogastric H. pylori infection could elevate the α-SMA mRNA expression in a liver fibrosis-induced murine model. It was also shown that the adverse effects of H. pylori on liver are not limited to cytotoxin producer strains. They demonstrated that not only the strain with no vacuolating cytotoxic activity such as SS1 strain with VacA genotype s2m2 but also the ATCC 43504 with VacA genotype s1m1 can induce liver fibrosis highlighting the role of other H. pylori-associated virulence attributes in the pathogenesis of liver fibrosis. However, Krzysiek-Maczka et al. [42] showed that VacA- and CagA-positive H. pylori strains could elevate the vimentin and α-SMA mRNA expression and cause differentiation of normal fibroblasts to cancer-associated fibroblasts (CAFs) using the normal rat gastric epithelial cells (RGM-1). Moreover, Yang et al. [43] in in vivo study demonstrated that H. pylori infection promotes gastric epithelial cells apoptosis through downregulation of E-cadherin and overexpression of cleaved caspase-3. Also, Yu et al. [44] showed that CagA is able to activate vimentin and TWIST1 expression and downregulates programmed cell death factor 4 (PDCD4), which inhibits E-cadherin expression. They concluded that H. pylori CagA protein may induce epithelial-mesenchymal transition (EMT) process in cancerous gastric cells. Their results provide new findings about H. pylori infection effects on molecular network of gastric cancer and presents a new signaling cascade of EMT induction in gastric cancer cell lines.
5. Conclusions
In recent years, the impact of H. pylori infection and its OMVs in development of extragastroduodenal diseases especially liver diseases have attracted great attention. However, because of insufficient documents in this regard, the virulence attributes and mechanisms behind H. pylori extragastroduodenal pathogenesis are not clear yet. In summary, the current study revealed the potential role of H. pylori-derived OMVs in HSC activation. Also, we showed that CagA- and VacA-negative OMVs could trigger activation of HSCs. Interestingly, we found that OMVs from more virulent H. pylori strains (OMVs extracted from strains 1, 2, 4, and 5) could have more potent impacts on induction of liver fibrosis compared to less virulent strains. Further research and in vivo studies are required to elucidate the precise role of H. pylori-derived OMVs in stimulation of liver fibrosis and to provide further understanding of the putative mechanisms engaged in H. pylori liver-associated disorders.
Acknowledgments
This work was funded by the National Institute for Medical Research Development of Iran (Grant No: 982958, Project No. IR.NIMAD.REC.1398.179). The authors wish to thank all laboratory staff of Foodborne and Waterborne Diseases Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran. We also are grateful to the Institute of Biophysics and Biochemistry of Tehran University, Tehran, Iran, and Michael Smith Laboratories, University of British Colombia, BC, Canada.
Contributor Information
Abbas Yadegar, Email: a.yadegar@sbmu.ac.ir.
Hossein Dabiri, Email: hodabiri@gmail.com.
Data Availability
Data generated during the study are available from the corresponding authors by request.
Conflicts of Interest
The authors declare that they have no competing interests.
Authors' Contributions
SB performed the microbiological experiments, proteomics, cell culture, molecular tests, immunocytochemistry, and data analysis, contributed to study design and methodology, and wrote the manuscript draft; SS participated in molecular testing, cell culture, and data analysis; AY, KB, and HD contributed to study design, methodology, conceptualization, and project administration; LJF and KMM contributed to LC/MS-MS experiments and proteomics data analysis; AY critically revised the manuscript; MJN participated in manuscript revision. All authors have seen and approved the final version of the manuscript and the author list.
Supplementary Materials
Primers used for real-time PCR assay in this study.
Proteomics analysis of each OMV derived from H. pylori strains used in this study.
References
- 1.Kusters J. G., Van Vliet A. H., Kuipers E. J. Pathogenesis of Helicobacter pylori infection. Clinical Microbiology Reviews . 2006;19(3):449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ansari S., Yamaoka Y. Helicobacter pylori virulence factors exploiting gastric colonization and its pathogenicity. Toxins . 2019;11(11):p. 677. doi: 10.3390/toxins11110677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chmiela M., Walczak N., Rudnicka K. Helicobacter pylori outer membrane vesicles involvement in the infection development and Helicobacter pylori-related diseases. Journal of Biomedical Science . 2018;25(1):p. 78. doi: 10.1186/s12929-018-0480-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chitcholtan K., Hampton M. B., Keenan J. I. Outer membrane vesicles enhance the carcinogenic potential of Helicobacter pylori. Carcinogenesis . 2008;29(12):2400–2405. doi: 10.1093/carcin/bgn218. [DOI] [PubMed] [Google Scholar]
- 5.Turner L., Bitto N. J., Steer D. L., et al. Helicobacter pylori outer membrane vesicle size determines their mechanisms of host cell entry and protein content. Frontiers in Immunology . 2018;9:p. 1466. doi: 10.3389/fimmu.2018.01466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Brito B. B., da Silva F. A. F., Soares A. S., et al. Pathogenesis and clinical management of Helicobacter pylori gastric infection. World Journal of Gastroenterology . 2019;25(37):5578–5589. doi: 10.3748/wjg.v25.i37.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bravo D., Hoare A., Soto C., Valenzuela M. A., Quest A. F. Helicobacter pylori in human health and disease: mechanisms for local gastric and systemic effects. World Journal of Gastroenterology . 2018;24(28):3071–3089. doi: 10.3748/wjg.v24.i28.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng D., He C., Hong Hui A., Huang Y., Nong-hua L. The possible role of Helicobacter pylori infection in non-alcoholic fatty liver disease. Frontiers in Microbiology . 2017;8:p. 743. doi: 10.3389/fmicb.2017.00743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sumida Y., Kanemasa K., Imai S., et al. Helicobacter pylori infection might have a potential role in hepatocyte ballooning in nonalcoholic fatty liver disease. Journal of Gastroenterology . 2015;50(9):996–1004. doi: 10.1007/s00535-015-1039-2. [DOI] [PubMed] [Google Scholar]
- 10.Okushin K., Tsutsumi T., Ikeuchi K., et al. Helicobacter pylori infection and liver diseases: epidemiology and insights into pathogenesis. World Journal of Gastroenterology . 2018;24(32):3617–3625. doi: 10.3748/wjg.v24.i32.3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seki E., Brenner D. A. Recent advancement of molecular mechanisms of liver fibrosis. Journal of Hepato-Biliary-Pancreatic Sciences . 2015;22(7):512–518. doi: 10.1002/jhbp.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moreira R. K. Hepatic stellate cells and liver fibrosis. Archives of Pathology & Laboratory Medicine . 2007;131(11):1728–1734. doi: 10.5858/2007-131-1728-HSCALF. [DOI] [PubMed] [Google Scholar]
- 13.Luedde T., Schwabe R. F. NF-κB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nature Reviews Gastroenterology & Hepatology . 2011;8(2):108–118. doi: 10.1038/nrgastro.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yarmohammadi M., Yadegar A., Ebrahimi M. T., Zali M. R. Effects of a potential probiotic strain Lactobacillus gasseri ATCC 33323 on Helicobacter pylori-induced inflammatory response and gene expression in coinfected gastric epithelial cells. Probiotics and Antimicrobial Proteins . 2021;13(3):751–764. doi: 10.1007/s12602-020-09721-z. [DOI] [PubMed] [Google Scholar]
- 15.Yadegar A., Mohabati Mobarez A., Zali M. R. Genetic diversity and amino acid sequence polymorphism in Helicobacter pylori CagL hypervariable motif and its association with virulence markers and gastroduodenal diseases. Cancer Medicine . 2019;8(4):1619–1632. doi: 10.1002/cam4.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Melo J., Pinto V., Fernandes T., et al. Isolation method and characterization of outer membranes vesicles of Helicobacter pylori grown in a chemically defined medium. Frontiers in Microbiology . 2021;12:p. 1253. doi: 10.3389/fmicb.2021.654193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Candiano G., Bruschi M., Musante L., et al. Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis . 2004;25(9):1327–1333. doi: 10.1002/elps.200305844. [DOI] [PubMed] [Google Scholar]
- 18.Foster L. J., De Hoog C. L., Mann M. Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proceedings of the National Academy of Sciences . 2003;100(10):5813–5818. doi: 10.1073/pnas.0631608100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishihama Y., Rappsilber J., Andersen J. S., Mann M. Microcolumns with self-assembled particle frits for proteomics. Journal of Chromatography A . 2002;979(1-2):233–239. doi: 10.1016/S0021-9673(02)01402-4. [DOI] [PubMed] [Google Scholar]
- 20.Kerr C. H., Skinnider M. A., Andrews D. D., et al. Dynamic rewiring of the human interactome by interferon signaling. Genome Biology . 2020;21(1):1–36. doi: 10.1186/s13059-020-02050-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tyanova S., Temu T., Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols . 2016;11(12):2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 22.Bauer B., Meyer T. F. The Human Gastric Pathogen Helicobacter pylori and Its Association with Gastric Cancer and Ulcer Disease. Ulcers . 2011;2011:23. doi: 10.1155/2011/340157.340157 [DOI] [Google Scholar]
- 23.Cindoruk M., Cirak M. Y., Unal S., et al. Identification of Helicobacter species by 16S rDNA PCR and sequence analysis in human liver samples from patients with various etiologies of benign liver diseases. European Journal of Gastroenterology & Hepatology . 2008;20(1):33–36. doi: 10.1097/MEG.0b013e3282efa4f2. [DOI] [PubMed] [Google Scholar]
- 24.He C., Cheng D., Wang H., Wu K., Zhu Y., Lu N. Helicobacter pylori infection aggravates diet-induced nonalcoholic fatty liver in mice. Clinics and Research in Hepatology and Gastroenterology . 2018;42(4):360–367. doi: 10.1016/j.clinre.2017.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Shapira Y., Agmon-Levin N., Renaudineau Y., et al. Serum markers of infections in patients with primary biliary cirrhosis: evidence of infection burden. Experimental and Molecular Pathology . 2012;93(3):386–390. doi: 10.1016/j.yexmp.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 26.Polyzos S. A., Kountouras J., Papatheodorou A., et al. Helicobacter pylori infection in patients with nonalcoholic fatty liver disease. Metabolism . 2013;62(1):121–126. doi: 10.1016/j.metabol.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 27.Dogan Z., Filik L., Ergül B., Sarikaya M., Akbal E. Association between Helicobacter pylori and liver-to-spleen ratio: a randomized-controlled single-blind study. European Journal of Gastroenterology & Hepatology . 2013;25(1):107–110. doi: 10.1097/MEG.0b013e3283590c10. [DOI] [PubMed] [Google Scholar]
- 28.Nilsson H. O., Mulchandani R., Tranberg K. G., Stenram U., Wadström T. Helicobacter species identified in liver from patients with cholangiocarcinoma and hepatocellular carcinoma. Gastroenterology . 2001;120(1):323–324. doi: 10.1053/gast.2001.21382. [DOI] [PubMed] [Google Scholar]
- 29.Nilsson H. O., Taneera J., Castedal M., Glatz E., Olsson R., Wadström T. Identification of Helicobacter pylori and other helicobacter species by PCR, hybridization, and partial DNA sequencing in human liver samples from patients with primary Sclerosing cholangitis or primary biliary cirrhosis. Journal of Clinical Microbiology . 2000;38(3):1072–1076. doi: 10.1128/JCM.38.3.1072-1076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mullaney E., Brown P. A., Smith S. M., et al. Proteomic and functional characterization of the outer membrane vesicles from the gastric pathogen Helicobacter pylori. Proteomics Clinical Applications . 2009;3(7):785–796. doi: 10.1002/prca.200800192. [DOI] [PubMed] [Google Scholar]
- 31.Zahmatkesh M. E., Jahanbakhsh M., Hoseini N., et al. Effects of exosomes derived from Helicobacter pylori outer membrane vesicle-infected hepatocytes on hepatic stellate cell activation and liver fibrosis induction. Frontiers in Cellular and Infection Microbiology . 2022;12, article 857570 doi: 10.3389/fcimb.2022.857570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hock B. D., McKenzie J. L., Keenan J. I. Helicobacter pylori outer membrane vesicles inhibit human T cell responses via induction of monocyte COX-2 expression. Pathogens and Disease . 2017;75(4) doi: 10.1093/femspd/ftx034. [DOI] [PubMed] [Google Scholar]
- 33.Choi H. I., Choi J. P., Seo J., et al. Helicobacter pylori -derived extracellular vesicles increased in the gastric juices of gastric adenocarcinoma patients and induced inflammation mainly via specific targeting of gastric epithelial cells. Experimental & Molecular Medicine . 2017;49(5):p. e330. doi: 10.1038/emm.2017.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang H., Zhang Y., Song Z., et al. sncRNAs packaged by Helicobacter pylori outer membrane vesicles attenuate IL-8 secretion in human cells. International Journal of Medical Microbiology . 2020;310(1, article 151356) doi: 10.1016/j.ijmm.2019.151356. [DOI] [PubMed] [Google Scholar]
- 35.Carlsohn E., Nyström J., Karlsson H., Svennerholm A. M., Nilsson C. L. Characterization of the outer membrane protein profile from disease-related Helicobacter pylori isolates by subcellular fractionation and nano-LC FT-ICR MS analysis. Journal of Proteome Research . 2006;5(11):3197–3204. doi: 10.1021/pr060181p. [DOI] [PubMed] [Google Scholar]
- 36.Turkina M. V., Olofsson A., Magnusson K. E., Arnqvist A., Vikström E. Helicobacter pylori vesicles carrying CagA localize in the vicinity of cell-cell contacts and induce histone H1 binding to ATP in epithelial cells. FEMS Microbiology Letters . 2015;362(11) doi: 10.1093/femsle/fnv076. [DOI] [PubMed] [Google Scholar]
- 37.Khurana A., Sayed N., Allawadhi P., Weiskirchen R. It's all about the spaces between cells: role of extracellular matrix in liver fibrosis. Annals of Translational Medicine . 2021;9(8):p. 728. doi: 10.21037/atm-20-2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee S. J., Kim K. H., Park K. K. Mechanisms of fibrogenesis in liver cirrhosis: the molecular aspects of epithelial-mesenchymal transition. World Journal of Hepatology . 2014;6(4):207–216. doi: 10.4254/wjh.v6.i4.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Câmara J., Jarai G. Epithelial-mesenchymal transition in primary human bronchial epithelial cells is Smad-dependent and enhanced by fibronectin and TNF-alpha. Fibrogenesis & Tissue Repair . 2010;3(1) doi: 10.1186/1755-1536-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fielding C. A., Jones G. W., McLoughlin R. M., et al. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity . 2014;40(1):40–50. doi: 10.1016/j.immuni.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goo M. J., Ki M. R., Lee H. R., et al. Helicobacter pylori promotes hepatic fibrosis in the animal model. Laboratory Investigation . 2009;89(11):1291–1303. doi: 10.1038/labinvest.2009.90. [DOI] [PubMed] [Google Scholar]
- 42.Krzysiek-Maczka G., Targosz A., Szczyrk U., et al. Role of Helicobacter pyloriinfection in cancer-associated fibroblast-induced epithelial-mesenchymal transition in vitro. Helicobacter . 2018;23(6, article e12538) doi: 10.1111/hel.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Y., Du J., Liu F., Wang X., Li X., Li Y. Role of caspase-3/E-cadherin in Helicobacter pylori-induced apoptosis of gastric epithelial cells. Oncotarget . 2017;8(35):59204–59216. doi: 10.18632/oncotarget.19471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu H., Zeng J., Liang X., et al. Helicobacter pylori promotes epithelial–mesenchymal transition in gastric cancer by downregulating programmed cell death protein 4 (PDCD4) PLoS One . 2014;9(8, article e105306) doi: 10.1371/journal.pone.0105306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used for real-time PCR assay in this study.
Proteomics analysis of each OMV derived from H. pylori strains used in this study.
Data Availability Statement
Data generated during the study are available from the corresponding authors by request.