

Abstract
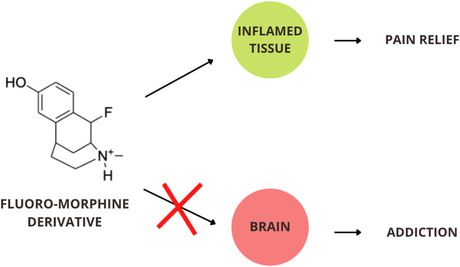
The opioid epidemic has impacted over 10 million Americans in 2019. Opioids, like morphine, bind non‐selectively in both peripheral tissue, leading to effective pain relief, and central tissue, resulting in dangerous side effects and addiction. The inflamed conditions of injured tissues have a lower pH (pH = 6–6.5) environment than healthy tissue (pH = 7.4). We aim to design a morphine derivative that binds selectively within inflamed tissue using molecular extension and dissection techniques. Morphine binds to the μ‐opioid receptor (MOR) when the biochemically active amine group is protonated. Fluorination of a β‐carbon from the tertiary amine group led to a reduced pKa of the derivative through induction. Through a decrease in the pKa, protonation is still statistically favored in lower pH environments of inflamed tissue but primarily deprotonated in healthy tissue. The cyclohexenol and N‐methyl‐piperidine rings of morphine are removed to increase conformational flexibility when binding while still maintaining the interactions required for analgesia. Electronic structure calculations were performed with Gaussian16 using the Keck Computational Research Cluster at Chapman University to determine the pKa. The theoretical pKa values are determined at the M06‐2X(SMD)/aug‐cc‐pVDZ level of theory to calculate the ΔG°aq values for the amine deprotonation reactions. Fluoromorphine β‐C2 was designed computationally and modeled within the MOR using Maestro: Schrödinger. This derivative exhibits a pKa reduction and enhanced ligand‐protein interactions within the MOR. β‐fluorination decreased the overall pKa values of the morphine derivatives (pKa: 6.1–7.83) relative to morphine, reducing binding within healthy, central tissue.
Keywords: addiction, amine, computational, fluorination, opioid, pH, pKa
The fluorinated morphine derivatives target binding prefentially with inflamed tissue, while being deterred from binding within central tissues such as the brain. Binding within the brain results in the addictive and dangerous side effects associated with opioid use; however, targetted binding in inflamed tissue will result in pain relief without the addictive component.
Abbreviations
- GPCR
G protein‐coupled receptor
- MOR
μ‐opioid receptor
1. INTRODUCTION
The opioid epidemic affected over 10 million Americans in 2019 with over 50 000 opioid‐related deaths reported in the United States. 1 Opioids are prescribed for a wide variety of reasons to provide relief for pathological pain, and these patients compose the majority of individuals who become addicted to opioids. Morphine, a benzylisoquinoline alkaloid, is a common opioid prescribed to treat pain, yet it is coupled with potentially deadly side effects that are enhanced by the addictive nature of morphine. Currently, there is no opioid medication that can provide pain relief without the inherent risk of addiction.
Pain relief results from the opioid molecule binding within peripheral opioid receptors in sites of pathological pain. Addiction and other dangerous side effects occur due to binding in healthy tissue within the central nervous system. Central binding in healthy tissue is responsible for activating the mesolimbic pathway in the brain that leads to addiction. 2 However, current opioids—including morphine—do not selectively target sites of pain, but instead bind indiscriminately in both peripheral and central opioid receptors. 3 While it was previously thought that opioids relieve pain centrally, in vivo studies show that there is minimal pain relief associated with central binding. 4 We aim to create a non‐addictive opioid by altering morphine's structure, causing it to selectively bind in peripheral inflamed tissue.
Morphine primarily binds to the μ‐opioid receptor (MOR), where binding is dependent on protonation of the biochemically active tertiary amino group of the ligand. 5 The protonated amino group forms an ion pair bond with the negatively charged carboxylate of Aspartate147 within the MOR binding site. 6 The majority of morphine molecules are protonated at physiological pH (pH = 7.4); therefore, binding occurs nonspecifically within all tissue. After injury, inflammation reduces the pH environment due to acidosis (pH = 6–6.5). 7 The discrepancy in pH conditions between healthy (central) and inflamed (peripheral) tissue provides an opportunity for alternative opioids that can bind preferentially in lower pH environments. Selectively binding within inflamed tissue is the target for opioid‐related pain relief with a lowered addictive risk. Decreasing the pKa of the biochemically active amine group promotes selective binding in peripheral opioid receptors within inflamed tissue. The aim of this study is to target opioid‐related pain relief by decreasing the pKa of the biochemically active amine group of morphine to promote selective protonation and therefore binding in peripheral opioid receptors within inflamed tissue.
Herein, molecular modeling and computational techniques are utilized to modify the structure of morphine to create and evaluate several morphine derivatives. Structural changes to morphine are proposed to enhance opioid ligand fit within the MOR binding pocket and to induce pH selective binding. The morphine derivative must bind efficiently within the MOR in order to activate pain‐relieving effects. Selectivity is induced by changing the conditions where the morphine derivative can protonate and therefore bind. The structural modifications include the addition of a fluorine atom to a β‐carbon site relative to the tertiary amine to decrease the pKa of the structure. Reducing the pKa of fentanyl has shown promising results in inducing peripherally selective binding in rats. 8 , 9 In morphine derivatives, the addition of a fluorine atom to a position beta of the amine decreases the pKa of the ligand. 10 The cyclohexenol (C) and N‐methyl‐piperidine rings (D) are dissected to increase conformational flexibility when binding to the MOR. Removal of these rings does not inhibit the biological function of morphine. 11
The novel morphine derivatives in this project were designed to have lowered pKa values that will bind specifically within peripheral opioid receptors of inflamed tissue, while avoiding central activation. This will address the needs of chronic and acute pain patients that require potent medication to relieve their pain, without facing the danger of addiction.
2. MATERIALS AND METHODS
2.1. Morphine derivative structures and modifications
Morphine structures were built within the graphical interface Gaussview6, and electronic structure calculations were done with Gaussian16 using the Keck Computational Research Cluster at Chapman University. Structural modifications were made to morphine computationally using molecular extension and dissection techniques. Structures were optimized using the M06‐2X hybrid functional, Dunning‐type cc‐pVDZ basis set, and the SMD solvent continuum model. 10
In previous studies, structural changes related to beta fluorination have been shown to reduce pKa. 10 , 12 In addition to this, dissection of the non‐biochemically active C and D rings is a new structural change made to morphine. Fluoromorphine β‐C1, fluoromorphine β‐C2, and fluoromorphine β‐C3 were fluorinated at β‐C1, β‐C2, β‐C3, respectively (Figure 1). The OH group of the A ring was removed in Dehydroxy‐fluoromorphine β‐C2 (Figure 1).
FIGURE 1.
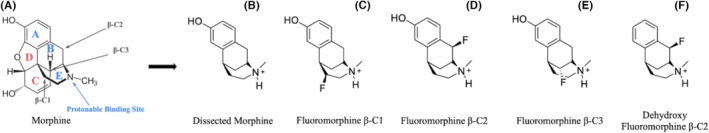
(A) Line drawing of morphine with rings A–E labeled. Sites of fluorination for derivatives are indicated at three β positions in the axial position relative to the proton of the tertiary amine. Blue indicates biochemically active components of morphine. Red indicates groups that were removed. (B) depicts dissected morphine, with C and D ring dissection and no β‐fluorination. (C–E) depict fluoromorphine derivatives with C and D ring dissection. (F) depicts a dehydroxy fluoromorphine derivative, which consists of OH group removal from the A ring, C and D ring dissection and β‐fluorination.
2.2. pKa
The direct method was utilized to calculate the pKa of each derivative from the electronic structures, which is supported when calculating the pKa of an amine group at the selected level of theory. 13 This method is based on the change in Gibbs free energy of the aqueous deprotonation equilibrium reaction (ΔGaq):
| (1) |
Overall Gibbs‐free energy of this deprotonation reaction was found using the equation:
| (2) |
After finding the overall Gibbs free energy, pKas were calculated using the equation:
| (3) |
This procedure is consistent with theoretical pKa calculations done in previous work. 10
2.3. Percent protonation
The percent protonation values were calculated using a derived Henderson‐Hasselbalch equation for morphine and theoretical derivatives modeled in physiological (7.4) and inflamed (6.5) pH conditions:
| (4) |
The power of 10 expression of the equation represents the ratio of the protonated to deprotonated derivative species (Equation 4). This was necessary in order to find the percent protonation, which is a ratio of the protonated morphine vs to the total morphine species concentration, of each morphine derivative in a specific pH environment:
| (5) |
2.4. Molecular modeling
Schrödinger: Maestro was used to model ligands within the MOR (PDB ID: 4DKL) from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). This crystallized structure has already been optimized from the RCSB PDB with a morphinan ligand docked within the binding pocket of 4DKL. 14 The analysis of the binding interactions of the derivatives was completed by superimposing each ligand upon the already optimized morphinan ligand (BFO 601). This method of superposition is commonly used when building ligands within a crystallized protein structure that contains a pre‐oriented ligand. 5
Ligand interaction diagrams were generated for each derivative within the MOR. Here, the interactions between the morphine derivatives and the amino acid residues within the entire binding pocket were visualized. Conformational searches using MacroModel were conducted for the protonated and deprotonated species of the derivatives to obtain the lowest energy conformer of each. The molecular geometries were optimized using molecular mechanics. All components of the crystallized protein were removed except for the Aspartate147 (Asp147) and Tyrosine148 (Tyr148) residues. These residues form the important ligand interactions within the binding pocket and thus are the residues of focus during modeling. All conformational searches were done in a water solvent using the OPLS4 force field. 15
3. RESULTS
3.1. Theoretical pKa and percent protonation
The pKa value of the dissected morphine derivatives increases with C and D ring removal (9.54), relative to morphine (8.00). Fluorination at any of the three potential beta positions available in morphine resulted in reductions of pKa. The largest reduction in pKa was in fluoromorphine β‐C3 which had a pKa value of 5.66, below the target for inflamed tissue. Fluoromorphine β‐C1 and β‐C2 have theoretical pKas of 7.83 and 7.04, respectively (Table 1). Removing the hydroxyl group from the A ring further impacts the pKa, as seen in the dehydroxy‐fluoromorphine derivative. Fluorination at positions β‐C1 and β‐C2 saw an increased reduction in pKa in comparison to the derivative with the hydroxyl group (Table 1).
TABLE 1.
The theoretical pKa values and percent protonation values in healthy (pH = 7.4) and inflamed (pH = 6.5, 6.0) tissues for the theoretical derivatives.
| Structure | pKa | % Protonation in healthy tissue (pH = 7.4) | % Protonation in inflamed tissue (pH = 6.5) | % Protonation in inflamed tissue (pH = 6.0) |
|---|---|---|---|---|
| Morphine | 8.00 | 86.3 | 98.0 | 99.0 |
| Dissected morphine | 9.54 | 99.3 | 99.9 | 99.9 |
| Fluoromorphine β‐C1 | 7.83 | 73.1 | 95.6 | 98.6 |
| Fluoromorphine β‐C2 | 7.04 | 30.8 | 77.9 | 91.8 |
| Fluoromorphine β‐C3 | 5.66 | 1.79 | 12.6 | 31.4 |
| Dehydroxy‐fluoromorphine β‐C2 | 6.88 | 23.2 | 70.5 | 88.3 |
Note: The experimentally determined pKa value of morphine is 8.21. 21 The computationally calculated pKa value of morphine obtained using the methodology outlined in this work is 8.00. The computationally derived pKa value obtained is within 2.6% of the experimental value. The bold values in Table 1 signify the pKa and percent protonation values in pH environments of 7.4, 6.5, and 6.0 of morphine. These values are bolded to distinguish them as benchmark values that the experimental morphine derivatives listed in Table 1 are compared to.
Percent protonation calculations model how reductions in pKa affect morphine binding in both healthy and inflamed tissue pH conditions. A higher protonation state indicates a statistically greater likelihood for binding. Morphine is 86.3% protonated in healthy tissue conditions and 98% protonated within inflamed tissue conditions (Table 1). Structural changes to morphine change the protonation states of the morphine derivative. When both C and D rings are removed in the dissected morphine derivative, protonation is higher in both tissue types, relative to morphine, at 99.3% and 99.9% in healthy and inflamed tissue, respectively (Table 1). Additionally, the removal of the hydroxyl group of the A ring increases the percent protonation of the structure further to 99.96% in inflamed conditions and 100% in healthy conditions. In comparison to morphine, fluoromorphine derivatives had a 13.2%–84.5% reduction in protonation in healthy pH conditions. Fluorination in both dissected and dehydroxy derivative at β‐C2 or β‐C3 resulted in greater than a 50% reduction in protonation in healthy pH conditions in comparison to morphine while still maintaining 59%–78% protonated in inflamed conditions (Table 2).
TABLE 2.
The changes in pKa and percent protonation values, in healthy (pH = 7.4) and inflamed (pH = 6.5, 6.0) tissue pH environments, of the theoretical derivatives relative to morphine.
| Structure | ΔpKa | Δ% Protonation in healthy tissue (pH = 7.4) | Δ% Protonation in inflamed tissue (pH = 6.5) | Δ% Protonation in inflamed tissue (pH = 6.0) |
|---|---|---|---|---|
| Morphine | 8.00 | 86.3 | 98.0 | 99.0 |
| Dissected Morphine | (+1.54) | (+13.0%) | (+1.9%) | (+0.9%) |
| Fluoromorphine β‐C1 | (−0.17) | (−13.2%) | (−2.4%) | (−0.4%) |
| Fluoromorphine β‐C2 | (−0.94) | (−55.5%) | (−20.1%) | (−7.2%) |
| Fluoromorphine β‐C3 | (−2.44) | (−84.5%) | (−85.4%) | (−67.6%) |
| Dehydroxy‐fluoromorphine β‐C2 | (−1.12) | (−63.1%) | (−27.5%) | (−10.7%) |
The bold values in Table 2 signify the pKa and percent protonation values in pH environments of 7.4, 6.5, and 6.0 of morphine. These values are bolded to distinguish them as benchmark values that the experimental morphine derivatives listed in Table 2 are compared to. However, the difference in the comparison of the experimental derivatives to morphine for this table is that the values in Table 2 represent the change in pKa units and change in percent protonation (%) relative to the benchmark values that are bolded.
3.2. Molecular modeling and computational simulation
Morphine and the theoretical derivatives are modeled to visualize their interactions within the MOR binding site. The model correctly described the primary ion pair bond that occurs between the protonated tertiary amino group of morphine and the negatively charged carboxylate of Asp147 which is recognized as essential for MOR activation. 6 , 16 The hydrogen of the protonated amino group of morphine also engaged in a hydrogen bond with the negatively charged oxygen of Asp147, which was an expected interaction in opioid binding. 16 Molecular modeling studies indicated a novel pi cation bond between the phenol group of Tyr148 and the protonated amino group of morphine. As expected, the theoretical derivatives maintain the same interactions as morphine when bound to the MOR (Figure 2).
FIGURE 2.
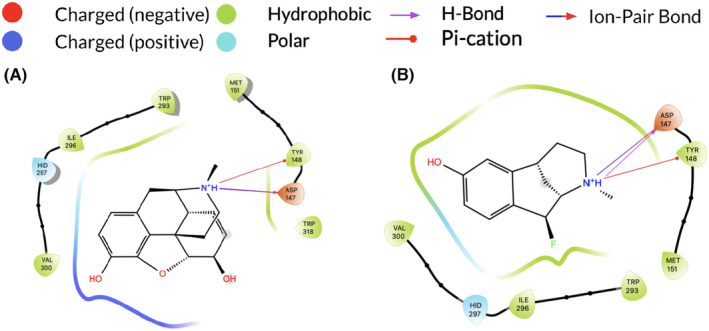
Intermolecular interactions between fluoromorphine derivatives within the 4KDL MOR are shown in 2D. (A) Morphine (B) Fluoromorphine β‐C2. Only one derivative is shown for clarity; however, the same interactions were maintained in all derivatives. Both morphine and fluoromorphine β‐C2 exhibit an ion pair bond and H bond between the protonated amino group and Asp147 and a pi cation bond between the protonated amino group and Tyr148.
Each derivative maintained the same binding interactions as morphine with a variation in distances between interactions. The substitution of hydrogen with fluorine resulted in a change of bond length from 1.09 Å (C–H bond) to 1.38 Å (C–F bond). The ion pair bond between the negatively charged oxygen of Asp147 and positively charged nitrogen of the derivatives was 2.69 ± 0.06 Å in length in comparison to 2.69 Å with morphine. Hydrogen bonds between protons of the amino groups and negatively charged oxygen of Asp147 were each 1.70 Å in length in morphine and all derivatives. The pi cation bond between the protonated amino group of the derivatives and phenyl group of Tyr148 was 5.92 ± 0.04 Å in length in comparison to 5.94 Å with morphine. In unbound states, all interactions were lost (Figure 3B). Interactions were dependent on protonation of the amino group of the morphine ligand and when the ligand was deprotonated, interactions were lost.
FIGURE 3.
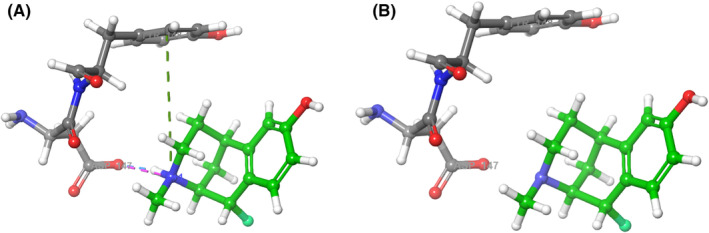
(A) The bound state of protonated morphine interacting with Asp147 and Tyr148 residues in the 4DKL MOR. The green dashed line indicates a pi‐cation bond between the aromatic ring on Tyr148 and the protonated tertiary amine. The pink‐dashed line indicates an ion pair bond between the protonated tertiary amine and carboxyl group on Asp147. The blue‐dashed line indicates a hydrogen bond between the protonated tertiary amine and carboxyl group of Asp147. (B) The unbound state of deprotonated morphine within the binding site of the 4DKL MOR. No binding interactions are present when morphine is deprotonated.
4. DISCUSSION
We propose multiple novel opioid derivatives that are capable of selectively binding within peripheral nerves of inflamed tissue. Of the theoretical morphine derivatives, fluoromorphine β‐C2 is deemed as a potential drug candidate for a non‐addictive opioid. Fluoromorphine β‐C2 exhibits selectivity to peripheral MORs due to increased binding in inflamed conditions than physiological conditions.
In order to effectively provide pain relief in inflamed tissue while avoiding central activation, there needs to be a reduction in protonation within healthy tissue pH while maintaining high protonation within inflamed tissue environments. Fluoromorphine β‐C2 reduces protonation in healthy tissue by 55%–70% relative to morphine (Table 2). While remaining dominantly deprotonated in central tissue, the fluoromorphine derivatives maintain high protonation in inflamed conditions (59%–77%; Table 2). Due to the discrepancy between healthy and inflamed tissue pH, the pKa values of the derivatives should be nearer to the pH range of inflamed tissue. In vivo studies have tested fentanyl derivatives designed for pH‐specific binding and those with pKa ranges between 6.73 and 6.93 provided effective pain relief, without eliciting central side effects in rats. 8 Of their fluorinated fentanyl derivatives, those within this pKa range exhibited the highest MOR binding and GPCR activation in rat models. 8 The theoretical pKa values of the morphine derivatives are reduced relative to morphine (Table 2). Derivatives with pKa values significantly below 6.73 (fluoromorphine β‐C3) will not remain dominantly protonated within any tissue in any environment and will therefore not elicit a pain‐relieving response. Derivatives with pKa ranges significantly above 6.93 will remain dominantly protonated in all tissues and will elicit the same dangerous side effects as morphine. Of the morphine derivatives evaluated, fluoromorphine β‐C2 will selectively bind in inflamed conditions because the pKa values are within the desired range.
β‐fluorination successfully reduces pKa to target protonation in inflamed conditions in all morphine derivatives. Fluorine is highly electronegative and its electron‐withdrawing properties lower the pKa via induction, destabilizing the tertiary amine. 17 A C–H to C–F bond replacement does not impose a large structural change (less than +0.3 Å change in C–X bond length) in comparison to morphine, thus not causing any steric hindrance upon morphine derivatives binding to the MOR. Other in vivo studies in fentanyl have also employed β‐fluorination to successfully lower pKa. 8 In previous computational studies, fluorination has shown reductions in pKa in morphine. 10
In conjunction with fluorination, structural changes made to morphine induce efficacious binding. While pH‐specific binding will combat the addictive properties, optimizing binding ensures that pain relief will occur in injured tissue. The dissection of the C and D rings increases the fit of the derivatives within the MOR and reduces steric hindrance upon binding, as seen in the structure of dissected morphine (Figure 1). This structural modification is inspired by benzomorphans, a popular opioid drug class with a C and D ring dissection. The benzomorphans are referred to as the universal opiate due to their high binding affinity and activity within δ, 𝜅, and μ‐opioid receptors, making them potent pain relievers. 11 , 18 Dissected morphine and dehydroxy dissected morphine display an increase in percent protonation in all tissues, suggesting an increase in binding capabilities but non‐specifically (Table 2). β‐fluorination ensures specificity while C and D ring removal maximizes MOR binding in inflamed tissue.
A final structural modification was considered that involves the removal of the hydroxyl group on the A ring of morphine, seen in Dehydroxy‐fluoromorphine β‐C2, to optimize binding in lower pH environments (Figure 1). An in vitro study investigated MOR activation in acidic pH conditions and found that opioids without this specific hydroxyl group had increased MOR activation and subsequent cAMP reduction. 8 Particularly important to MOR activation, a histidine residue (His297) is protonated in pH conditions of inflamed tissue. In its protonated form, opioid ligands with this hydroxyl group can form a hydrogen bond with the protonated His297. The in vitro study found that opioid ligands with the ability to form this hydrogen bond had decreased GPCR activation in acidic conditions. MOR activation of opioids that are incapable of forming this hydrogen bond was not affected. 8 However, other studies have shown that an electron‐donating or electron‐withdrawing group, such as a hydroxyl or acetal group, on the A ring are necessary components of the pharmacophore of morphine. 19
Specifically, analgesia was reduced when the aromatic hydroxyl group was modified or adversely affected by its complete removal. Our molecular modeling analysis found that all binding interactions were maintained when the hydroxyl group was removed, yet in vivo studies show this may negatively impact analgesic activity. This finding requires further computational investigation as to the occurrence and significance of interactions made between the hydroxyl group of the A ring and the analgesic receptor.
Molecular modeling studies confirmed that all fluoromorphine derivatives maintain the hydrogen bond and ion pair bond between Asp147 and protonated amino group (Figure 3). Maintaining this bond is crucial because the interaction initiates GPCR activation and subsequent cAMP reduction necessary for pain relief. 16 When the morphine ligand is deprotonated, all interactions are lost (Figure 3). In the deprotonated form, binding does not occur. The presence of a pi cation bond between Tyr148 and the protonated amino group occurred is an interesting finding as pi cation bonds are involved in many physiological binding processes and have been investigated for their role in binding specificity in GPCR binding. 20 Activation of the acetylcholine receptor and downstream K+ channels is vitally dependent on a pi cation bond between an aromatic amino acid and a positively charged ligand. 20 In the case of opioid binding, the pi cation bond between the positively charged amino group of the morphine ligand and Tyr148 of the MOR could influence GPCR activation and downstream signaling of K+ channels that act in morphine's mechanism of action to provide pain relief. This discovery requires further investigation to clarify the pi cation role within the MOR activation and signaling; however, it may be a significant discovery in understanding MOR activation.
The structural modifications made to morphine results in one ideal drug candidate that can provide pain relief by binding selectively within the periphery in inflamed tissue, while avoiding central activation. Fluoromorphine β‐C2 exhibits pH‐specific MOR binding within inflamed tissue as a result of reduced pKa since they maintain high percent protonation in inflamed tissues with reduced percent protonation within healthy tissue. While nullifying the addictive side effects and maintaining analgesia, this will address the needs of both chronic and acute pain patients. These results raise exciting possibilities for medication that is capable of providing pain relief, without the addiction risk of current opioids.
AUTHOR CONTRIBUTIONS
Makena Augenstein: Conceptualization, Visualization, Investigation, Writing‐original draft & review and editing. Nayiri Alexander: Conceptualization, Visualization, Investigation, Writing‐original draft & review and editing. Matthew Gartner: Conceptualization, Visualization, Investigation, Writing‐original draft & review and editing.
FUNDING INFORMATION
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Schmid College of Science and Technology at Chapman University.
DISCLOSURE
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
ETHICS STATEMENT
This article does not contain any studies with humans or animal participants. There are no human participants in this article and informed consent is not applicable.
Supporting information
Table S1
{kind=link}
ACKNOWLEDGMENTS
The authors would like to acknowledge the Schmid College of Science and Technology at Chapman University for funding this work.
Augenstein M, Alexander N, Gartner M. Computational design and molecular modeling of morphine derivatives for preferential binding in inflamed tissue. Pharmacol Res Perspect. 2023;11:e01075. doi: 10.1002/prp2.1075
Makena Augenstein and Nayiri Alexander should be considered joint first author.
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
REFERENCES
- 1. Jannetto PJ. The North American opioid epidemic. Ther Drug Monit. 2021;43:1‐5. [DOI] [PubMed] [Google Scholar]
- 2. Kosten T, George T. The neurobiology of opioid dependence: implications for treatment. Sci Pract Perspect. 2002;1(1):13‐20. doi: 10.1151/spp021113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zöllner C, Shaqura MA, Bopaiah CP, Mousa S, Stein C, Schäfer M. Painful inflammation‐induced increase in μ‐opioid receptor binding and G‐protein coupling in primary afferent neurons. Mol Pharmacol. 2003;64(2):202‐210. doi: 10.1124/mol.64.2.202 [DOI] [PubMed] [Google Scholar]
- 4. Jagla C, Martus P, Stein C. Peripheral opioid receptor blockade increases postoperative morphine demands—a randomized, double‐blind, placebo‐controlled trial. Pain. 2014;155(10):2056‐2062. doi: 10.1016/j.pain.2014.07.011 [DOI] [PubMed] [Google Scholar]
- 5. Lipiński PFJ, Jarończyk M, Dobrowolski JC, Sadlej J. Molecular dynamics of fentanyl bound to μ‐opioid receptor. J Mol Model. 2019;25(5):144. doi: 10.1007/s00894-019-3999-2 [DOI] [PubMed] [Google Scholar]
- 6. Coffman BL, Kearney WR, Green MD, Lowery RG, Tephly TR. Analysis of opioid binding to UDP‐glucuronosyltransferase 2B7 fusion proteins using nuclear magnetic resonance spectroscopy. Mol Pharmacol. 2001;59(6):1464‐1469. doi: 10.1124/mol.59.6.1464 [DOI] [PubMed] [Google Scholar]
- 7. Rajamäki K, Nordström T, Nurmi K, et al. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J Biol Chem. 2013;288(19):13410‐13419. doi: 10.1074/jbc.M112.426254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spahn V, Del Vecchio G, Labuz D, et al. A nontoxic pain killer designed by modeling of pathological receptor conformations. Science. 2017;355(6328):966‐969. [DOI] [PubMed] [Google Scholar]
- 9. Del Vecchio G, Labuz D, Temp J, et al. pKa of opioid ligands as a discriminating factor for side effects. Sci Rep. 2019;9:19344. doi: 10.1038/s41598-019-55886-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alexander N, Augenstein M, Sorensen AM, Garcia C, Greene A, Harrison AW. Computational design of β‐fluorinated morphine derivatives for ph‐specific binding. Chem Phys Lett. 2021;777:138723. doi: 10.1016/j.cplett.2021.138723 [DOI] [Google Scholar]
- 11. Chang KJ, Hazum E, Cuatrecasas P. Novel opiate binding sites selective for benzomorphan drugs. Proc Natl Acad Sci. 1981;78(7):4141‐4145. doi: 10.1073/pnas.78.7.4141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spahn V, Del Vecchio G, Rodriguez‐Gaztelumendi A, et al. Opioid receptor signaling, analgesic and side effects induced by a computationally designed ph‐dependent agonist. Sci Rep. 2018;8(1):8965. doi: 10.1038/s41598-018-27313-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dutra FR, Silva CS, Custodio R. On the accuracy of the direct method to calculate pKa from electronic structure calculations. J Phys Chem A. 2021;125(1):65‐73. doi: 10.1021/acs.jpca.0c08283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Manglik A, Kruse AC, Kobilka TS, et al. Crystal structure of the μ‐opioid receptor bound to a morphinan antagonist. Nature. 2012;485(7398):321‐326. doi: 10.1038/nature10954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chao L, Chuanjie W, Ghoreishi D, et al. OPLS4: improving force field accuracy on challenging regimes of chemical space. J Chem Theory Comput. 2021;17(7):4291‐4300. doi: 10.1021/acs.jctc.1c00302 [DOI] [PubMed] [Google Scholar]
- 16. Vo QN, Mahinthichaichan P, Shen J, Ellis CR. How μ‐opioid receptor recognizes fentanyl. Nat Commun. 2021;12(1):984. doi: 10.1038/s41467-021-21262-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shah P, Westwell AD. The role of fluorine in medicinal chemistry. J Enzyme Inhib Med Chem. 2007;22(5):527‐540. doi: 10.1080/14756360701425014 [DOI] [PubMed] [Google Scholar]
- 18. McLawhon RW, West RE, Miller RJ, Dawson G. Distinct high‐affinity binding sites for benzomorphan drugs and enkephalin in a neuroblastoma—brain hybrid cell line. Proc Natl Acad Sci. 1981;78(7):4309‐4313. doi: 10.1073/pnas.78.7.4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lešnik S, Bertalan É, Bren U, Bondar A. Opioid receptors and protonation‐coupled binding of opioid drugs. Int J Mol Sci. 2021;22(24):13353. doi: 10.3390/ijms222413353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kumar K, Woo SM, Siu T, Cortopassi WA, Duarte F, Paton RS. Cation–π interactions in protein–ligand binding: theory and data‐mining reveal different roles for lysine and arginine. Chem Sci. 2018;9(10):2655‐2665. doi: 10.1039/c7sc04905f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. PubChem . Morphine. NIH National Library of Medicine. https://pubchem.ncbi.nlm.nih.gov/compound/5288826 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its supplementary information files).