

Abstract
Background
In many countries tuberculosis (TB) remains a highly prevalent disease and a major contributor to infectious disease mortality. The fight against TB requires surveillance of the population of strains circulating worldwide and the analysis of the prevalence of certain strains in populations. Nowadays, whole genome sequencing (WGS) allows for accurate tracking of TB transmission. Currently, there is a lack of a comprehensive summary of the characteristics of TB outbreaks.
Methods
We systematically analyzed studies reporting TB outbreaks worldwide, monitored through WGS of Mycobacterium tuberculosis. We 1) mapped the reported outbreaks from 2011- 2020, 2) estimated the average size of the outbreaks, 3) indicated genetic lineages causing the outbreaks, and 4) determined drug-resistance patterns of M. tuberculosis strains involved in the outbreaks.
Results
Most data originated from Europe, Asia, and North America. We found that TB outbreaks were reported throughout the globe, on all continents, and in countries with both high and low incidences. The detected outbreaks contained a median of five M. tuberculosis isolates. Most strains causing the outbreaks belonged to lineage four, more rarely to lineage two. Reported outbreak isolates were often drug resistant.
Conclusions
We conclude that more WGS surveillance of M. tuberculosis outbreaks is needed. Globally standardized procedures might improve the control of M. tuberculosis infections.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12879-023-08197-w.
Keywords: Tuberculosis, Tuberculosis transmission, Local outbreaks, Whole genome sequencing, Recent transmission
Background
Tuberculosis (TB) is a communicable infectious disease caused by mycobacterium species belonging to the Mycobacterium tuberculosis complex. In most cases, the disease affects the respiratory system of the lung. However, TB can also involve lymph nodes, the central nervous system, bones, and joints and take a disseminated form [1, 2]. For several years, a decrease in TB cases can be observed worldwide, but the number of people contracting TB each year is still very high. In 2019, 10 million people contracted TB, and 1.2 million died from it, keeping TB among the top causes of death from infectious diseases alongside HIV/AIDS and, nowadays, COVID-19 [3]. The COVID-19 pandemic has slowed and even halted progress against TB. Complete eradication of TB worldwide seems difficult for various reasons. Currently there is no known therapy to completely eliminate latent TB. To date, there is no efficient vaccine giving adults herd immunity. The treatment of TB is difficult and consists of at least 6 months of drug administration. Drug-resistant strains are increasing – there were 160 684 drug-resistant TB cases in 2017, 186 772 in 2018, and 206 030 in 2019 [4–6].
Since so many people get affected by TB, often carrying high costs of treatment and facing poverty because of it, various agencies like World Health Organization (WHO), Centers for Disease Control and Prevention (CDC) and European Centre for Disease Prevention and Control (ECDC) implemented special programs to help combat TB. Breaking the chain of transmission is one of the most critical targets in TB eradication [7].
Before applying genetic methods in epidemiological investigations, M. tuberculosis transmission was monitored through the differentiation of strains based on phenotype. However, this method is not very practical for slow-growing organisms and is used only for drug resistance testing. Thanks to developments in molecular biology, genetic typing through repetitive or non-repetitive sequences and SNPs can be used. The first methods include IS6110, spoligotyping, and MIRU-VNTR, of which the last two are the most used in laboratory practice, especially in developing countries. IS6110 is based on RFLP analysis coupled with identification of the site of insertion. MIRU-VNTR is based on the identification of variation in polymorphic tandem repeats in specific chromosome regions. Spoligotyping is a PCR-based method analyzing the structure of the CRISPR-Cas locus. Discrimination occurs by analyzing the presence or absence of spacers in the 43-spacer set [8]. It is generally accepted that methods like spoligotyping or MIRU-VNTR can exclude transmission but are not sufficiently discriminative to confirm transmission chains. This is because genetic markers indicated by each method may show identical results for distinct strains due to convergent evolution and the lack of resolution needed to detect recent transmission [9]. Transmission chain confirmation is also done through whole-genome sequencing (WGS) and/or epidemiological/contact investigations. The advantage of WGS is its high discriminatory power since it analyzes vast genome regions. Furthermore, homoplasy and backward mutations are rare, and WGS mainly relies on SNPs (28). Additionally, the precision of data obtained through WGS allows for advancements in the mathematical modeling of transmission and machine learning [10]. Importantly, conventional genotyping is critical in current epidemiology, it is widely used, and it often precedes whole genome sequencing, especially for outbreak investigation.
Based on single nucleotide polymorphisms (SNPs) identification in other than drug resistance-associated genes, the global population of M. tuberculosis can be phylogenetically divided into nine major lineages, distributed in a way associated with specific geographic regions [11] due to tight association with historical human migration patterns [12]. Based on the presence of the TbD1 region (MmpS6/MmpL6-encoding Mtb-specific deletion region), M. tuberculosis lineages are divided into ancient and modern [13]. The modern lineages are mostly Eurasian lineages like lineage two (L2) (East Asian), L3 (East-African-Indian), and L4 (Euro-American). The ancient lineages are L1 (Indo-Oceanic) and the most restricted, only to specific regions of Africa, lineages L5 (West African 1), L6 (West African 2), and L7 (Ethiopia) [12]. Lineages 5 and 6 are also referred to as M. africanum [14]. Besides these seven lineages, two new lineages, L8 and L9, were separated from M. africanum based on phylogenomic analyses, drug resistance mutations and geographical distribution [15, 16]. Modern lineages are generally regarded as more worldwide spread than ancestral ones, which are considered more endemic [17, 18]. L2 is found mainly in East and Central Asia, while L4 is identified mainly in Europe, America, and Africa; both are found worldwide. L1 is found mainly in East Africa, the Philippines, and L7 in Ethiopia [12]. L4 has very high genetic diversity compared to other lineages and includes different sublineages with different geographic distribution e.g. X (4.1.1), Haarlem (4.1.2), Ghana (4.1.3), Cameroon (L4.6.2), Uganda (L4.6.1), LAM (4.3) [19]. L2 is compromised out of 2 sublineages, proto-Beijing or ancient Beijing (2.1) and modern Beijing (2.2), which is much more diverse than the ancient Beijing sublineage [20].
Currently, there is a lack of a comprehensive summary of the characteristics of TB outbreaks. The objective of this study was to synthesize available information on M. tuberculosis strains involved in the outbreaks to 1) map the reported outbreaks from 2011- 2020, 2) estimate the size of the outbreaks, 3) indicate genetic lineages causing the outbreaks, and 4) determine drug-resistance patterns of M. tuberculosis strains involved in the outbreaks. We focused on studies where the WGS of strains confirmed local outbreaks.
Materials and methods
The research aimed to summarize four characteristics of tuberculosis outbreaks: 1) the location of the outbreaks, 2) the size of the outbreaks, 3) the phylogenetic background of outbreak strains, and 4) the drug resistance pattern of outbreak strains. We systematically screened peer-reviewed reports deposited in the Pubmed database and Web of Science database for TB outbreaks confirmed by the currently most reliable epidemiological method, WGS.
Definitions and constraints
A cluster
A cluster was defined as two or more strains linked genetically through WGS.
An outbreak
An outbreak was defined as at least three cases of infection with evidence of serial transmission or a cluster consisting of at least three strains, as used previously [21–23].
Differentiation of population-based, drug-resistant population-based and outbreak investigation studies
Population-based studies were characterized by the use of large numbers of strains usually collected in National Reference Centers or Research Facilities to indicate strain population structure from more extensive areas. Drug-resistant population-based studies included strains from large collections but focused on drug-resistant strains population. Outbreak investigations concerned strains collected and spread in a specific place, sharing the same genotyping pattern obtained by classical methods of differentiation- spoligotyping and/or MIRU typing.
Time constraints on the indication of transmission
Since M. tuberculosis may enter a state of latency for decades, local transmission of clonal strains may be detected over long periods if the outbreak contains many isolates. All analyzed strains in this study were isolated within the last 30 years, which is within the lifespan of a single host. We did not impose time-range criteria on local outbreak studies included in this review. Of note, following the assumption that the mutation rate of M. tuberculosis is 0.3 SNP per year [24], the restriction of ≤ 12 SNPs to define recent transmission restricts transmission prior to approximately 20 years.
Location constraints on the indication of transmission
We classified the studies with five distinct levels of local outbreak extent: exact location (for example, a particular building), city, region, country, or international. We included international surveillance studies due to the current significant level of globalization and international travel.
Strain-relatedness constraints on the indication of transmission
We focused on studies where transmission cluster data based on WGS followed the rule, where two isolates involved in an outbreak differed no more than by 12 SNPs, indicating recent transmission. If there were no such criteria, but it was possible to extract data, we used the ≤ 12 SNPs rule to identify outbreak clusters.
Drug resistance
We reported data on drug resistance where available. A drug-resistant variant was defined when it was resistant to at least one first-line antibiotic used in the treatment of TB. There was no restriction on the identification method of drug resistance, whether it was reported through the in silico analysis of mutations or phenotypic testing.
Search strategy and inclusion/exclusion of articles
We conducted a systematic literature search of TB outbreaks worldwide identified by WGS. The search strategy was based on searching two major electronic databases, the PubMed and the Web of Science. The timeline restriction was 25 Nov 2020. We searched for phrases "whole genome sequencing tuberculosis" and "tuberculosis outbreak". We used the COVIDENCE tool (Covidence, Australia), the standard production platform for Cochrane Reviews, for screening and data collection.
Three authors, LZ, DZ, and AM, screened the articles. The articles obtained from the database search were screened in two steps: abstract and title screen, followed by a full-text screen and data extraction. At least two authors screened each manuscript, and in case of discrepancies, the consensus was reached by at least three authors. We considered English written manuscripts referencing human tuberculosis, where clustering of strains was determined by WGS (Table 1).
Table 1.
Criteria for including and excluding articles in this study
| Including | Excluding |
|---|---|
| M. tuberculosis studies | Other species of Mycobacteria |
| Human TB | Animal TB or zoonotic transmission of TB |
| Whole genome sequencing of at least three M. tuberculosis isolates from distinct people | Less than three isolates from distinct people sequenced and studies designed to investigate isolates from less than three possibly epidemiologically-linked people |
| Clustering analysis based on 12 or less SNP rule for clustering or possibility to identify clusters of 12 or less SNP of difference | Lack of clustering analysis or clustering analysis based on strains differing by more than 12 SNP, impossible to impose 12 or less rule for clustering |
| Cluster after sequencing containing three or more isolates | Largest cluster after sequencing containing less than three isolates or impossible to determine clusters of three and more isolates |
| English language | Any other language than English |
| Original analysis | Repurposed data or review manuscripts |
| Study not available in full text |
Statistics
The descriptive statistics of median and interquartile range used in this manuscript were calculated with Excel Microsoft Office (Microsoft, USA).
Results
Literature screening
One thousand three hundred sixty-five studies, excluding duplicates, went through title and abstract screening by at least two authors. In case of conflict between the authors, a consensus was reached. The vast majority of articles were found irrelevant to the topic of this review because they were either review articles or did not meet inclusion/exclusion criteria. Eighty-eight articles were chosen for data extraction through COVIDENCE (Fig. 1. Table S1).
Fig. 1.

The PRISMA overview of the process of obtaining data for analysis. We used the COVIDENCE tool to analyze data efficiently. We searched PubMed and Web of Science databases for the phrases "whole genome sequencing tuberculosis" and "tuberculosis outbreak" restricted to 25 Nov 2020. Each article was screened by at least two authors based on the title and abstract. Subsequently, articles that seemed to match the criteria were screened in full. We excluded studies written in languages other than English, describing other species of mycobacteria, and repeating data from previous years. We restricted articles that referred to human TB. The inclusion criteria were confirmation of transmission thru WGS
Data was dominated with reports mainly concerning Europe and Asia with 37% (n = 34) and 21% (n = 19) for each continent, respectively (Fig. 2A). For other continents, the number of reports was slightly smaller- North America 19% (n = 17) and Africa 13% (n = 12). The least represented were South America, 6% (n = 5), and Australia, 4% (n = 4).
Fig. 2.

The charts summarizing article data. A Geographic location of studies included in our dataset. Most studies that matched our criteria concerned Asia, North America, and Europe. Africa, Australia, and South America were less represented in our dataset. B Types of studies included in our dataset. Half of the studies were written in the form of the outbreak investigation, where strain relatedness was suspected or detected by traditional genotyping methods, like spoligotyping and MIRU-VNTR
The dataset included three types of studies (Fig. 2B). Two kinds of population-based studies assessed all strains in the population and drug-resistant strains in the population. The third type of study was "outbreak investigations," representing half of the reports included in our analysis. The outbreak investigations spanned over median 5 years (IQR 2–9.75).
The geographic location of outbreaks
Outbreaks were reported from countries located on all six continents included in our analysis (Fig. 3). Most outbreaks were reported in Malawi (n = 118), China (n = 58), and Ghana (n = 31). The notable number of outbreaks was also reported in European countries- United Kingdom, and Spain (27, and 25, respectively).
Fig. 3.

The geographic location of outbreaks. We screened the set of articles and excluded data regarding strain relatedness based on the assumption that no more than 12 SNPs separate strains involved in outbreaks. We counted the number of outbreaks reported for each country. We found outbreaks were reported by high and low-incidence countries
Size of the outbreaks
Across the 477 outbreaks for which we were able to extract data, the median number of M. tuberculosis isolates involved in an outbreak was five (IQR 3–8). Outbreak investigation studies revealed slightly bigger size outbreaks than the other two types of studies (Fig. 4). There were two reports of massive outbreaks which included more than a hundred isolates- in Greenland [25], and London, United Kingdom [26].
Fig. 4.

The number of isolates per each reported outbreak. We retrieved information regarding the size for 477 outbreaks. A Outbreak size for all analyzed studies. Five outbreaks, exceeding 65 isolates are not shown on the graph for better median and interquartile range visibility. The median number of M. tuberculosis isolates involved in an outbreak was 5 (IQR 3–8). B, C, D Outbreak size for population based studies, drug-resistant population based studies and outbreak investigations. Outbreak investigations revealed slightly bigger size outbreaks than the other two types of studies
Greenland is one of the world's least populated and relatively isolated regions. The authors sequenced M. tuberculosis isolates from all culture-positive cases gathered in East Greenland over 21 years (n = 182). The authors clustered isolates into four major clusters. The biggest cluster was estimated to evolve around 1972 in Tasiilaq and surrounding locations. The vast majority of isolates, 122, differed with ≤ 12 SNPs. The other two notable clusters were genetically similar and centered in different locations. The authors speculated that the most recent common ancestor of the three clusters was introduced in Greenland in 1894 during the foundation of a Danish colony in Tasiilaq. In summary, the study reported a very low diversity of circulating M. tuberculosis strains in East Greenland, with the outbreak possibly linked to "the founder effect" event over a hundred years ago.
In London, the authors sequenced 344 isolates collected over 14 years that were previously linked to an outbreak with traditional typing methods- IS6110 typing, spoligotyping, and MIRu-VNTR typing. The maximum number of SNPs between any pair of isolates was nine. The epidemiological investigation showed the outbreak spread in prison and squat frequented by intravenous drug abusers.
The lineage of strains involved in outbreaks
Data about lineage was available for 385 outbreaks. The most frequently reported lineage in outbreaks strains was L4 (57%, n = 220) (Fig. 5). The second most frequent lineage was L2, (27%, n = 106). We also observed minor numbers of outbreaks caused by lineages 1, 3, and 6, with L6 represented by one outbreak. The transmission of L6 took place in Ghana. It was not confirmed the transmission was not human to human [27].
Fig. 5.
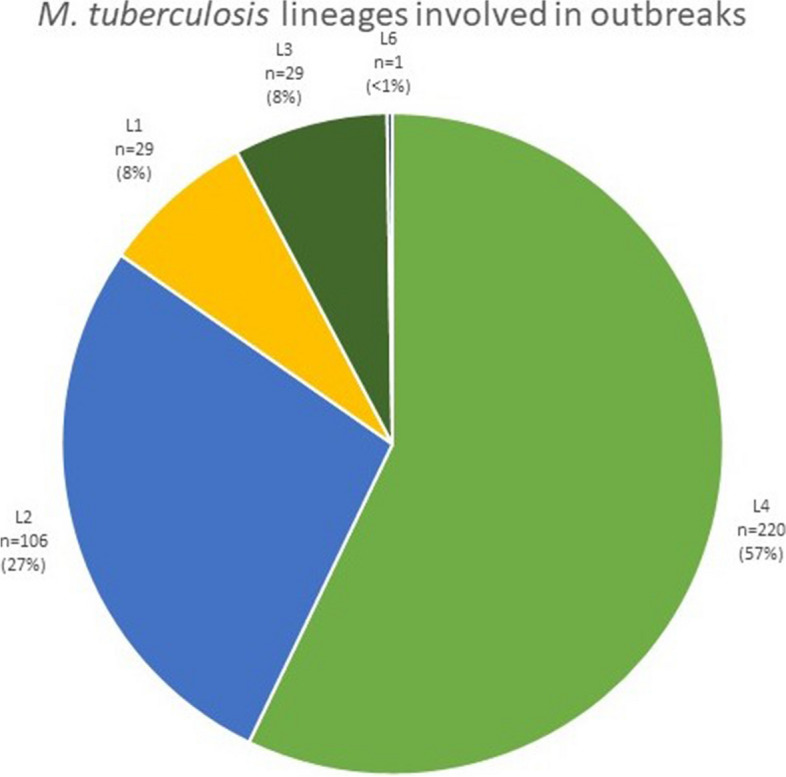
The phylogenetic lineage of M. tuberculosis strains involved in outbreaks. Data about lineage was available for 385 outbreaks. We did not find reports of outbreaks of L5 and L7-9
Drug resistance of outbreak-associated strains
Information on drug resistance in particular outbreaks was available for 163 outbreaks (Fig. 6). Pansusceptible outbreaks were the most prevalent ones, constituting 64% (n = 105) of the total. Drug-resistant outbreaks appeared in 28% of cases (n = 45). Mixed outbreaks, where drug resistance was acquired over time, were the smallest fraction of the total (8%, n = 13). Drug resistance was notably more often reported in outbreak investigations (52%) than in population based studies (10%) (Fig. 6B, and C).
Fig. 6.

The overview of drug resistance patterns of isolates reported in outbreaks. A overview of M. tuberculosis outbreak strains drug resistance B) isolates in population-based studies, and C) isolates in outbreak investigations. The outbreak was considered drug resistant if at least any strain in the cluster was drug resistant
Discussion
Study overview
We found that TB outbreaks are reported globally, on all continents, in both high and low-incidence countries. The detected outbreaks usually contain around five M. tuberculosis isolates. The strains causing the outbreaks typically belong to L4, more rarely to L2. Reported outbreak isolates often are or become drug resistant.
Many factors contribute to the transmission of TB. The immune competence of the host is an important facet because it affects whether TB develops into a primary infection or turns into a latent TB [28]. The rate at which a person produces droplets containing bacteria is also important [29]. For unclear reasons, gender is also an important determinant of acquiring TB: TB seems to affect men two times more than women worldwide [30]. In 2019, 56% of people who contracted TB were men (aged ≥ 15 years) [4]. There is also rising evidence that some people are superspreaders, which means they are more infectious than others [31, 32]. Other significant factors are co-infections and coexisting diseases impacting immunity, like diabetes, alcoholism, and smoking. Apart from host-related factors, different environmental factors also play important roles in TB transmission, e.g., duration of time spent in close proximity of the infected person or country's medical infrastructure, signifying faster treatment and detection of infected people [29].
It is not well known to what extent pathogen factors contribute to the development and spread of TB since mycobacteria lack typical virulence factors [17]. The virulence level does not necessarily go along with the increased transmission. Our results support that certain genetic lineages of M. tuberculosis are more adapted to spread and cause disease in humans. This review's results agree with observation reporting widespread lineages two and four [33]. It is unclear whether these particular lineages are more adapted to the infection process. However, L2 and L4 seem to spread more and are often associated with increased virulence or high transmission [34]. There is possibility that certain lineages, sublineages, or strains within the sublineage, may have intrinsic genes increasing their resistance and subsequently transmission, like the Cameroon genotype [35]. The Beijing lineage shows significant strain characteristics variability, including transmissible and low-transmissible strains [36]. Compared with L4 and L2, L1 and L3 are much more geographically restricted [], which is reflected in this review. L5 seems less transmissible, possibly causing outbreaks thru infection from the external source [37]. Similar observations were made for L6 [38]. L8 is highly restricted and found only in the African Great Lakes region, but it was so far identified only in 2 patients [16]. Similarly, L9 was identified only in a few patients [15].
We observed the highest number of outbreaks in high-burden countries- Malawi, Ghana, and China. However, the high number of reported outbreaks is also likely a result of the technological availability of WGS technology, for a significant number of outbreaks was also reported in low-incidence countries of the European region.
The summarized results show that 36% of outbreak reports involved at least one drug-resistant isolate. According to WHO data, rifampin-resistant strains constitute less than 7% of the global population [5]. It seems reasonable to assume that drug-resistant outbreaks gather more attention than drug-susceptible ones, probably resulting in more frequent sequencing of such strains. Indeed, drug resistance was notably lower in population based studies than in outbreak investigations. However, based on the results summarized in this review, it should be monitored carefully whether drug-resistant strains are more likely to transmit and cause disease. In early studies, MDR/XDR strains were thought to be less virulent in a guinea pig model, which meant they were less adaptable to the environment of a new host (not likely to spread); therefore, they were less evolutionary fit [39]. For that reason, there was less concern than should have been for MDR strains causing outbreaks. While some drug-resistant strains may be less fit, they constitute a significant percentage of all circulating M. tuberculosis strains.
Study limitations
It needs to be stressed that the data obtained in this review is possibly biased. Strains in which drug resistance is detected tend to receive more attention than the susceptible strains. Often, laboratories genotype only certain strains due to money restrictions, which usually means sequencing only MDR or XDR strains. Therefore outbreak strains reported in this review do not necessarily represent the M. tuberculosis population. Another possible problem is that there were reports that included longitudinal samples together with isolates obtained from distinct patients. Such an approach may have skewed the results regarding the outbreak frequency. Finally the actual size of the outbreaks, especially for outbreak investigations, may be skewed. Conventional genotyping methods usually detect larger groups of possibly linked cases, yet only a fraction of isolates undergoes WGS.
Recommendations for reporting outbreak studies
A very small fraction of isolates is currently being sequenced. If we are to control the TB epidemic, we need more information regarding transmission chains. Identification of exact routes of transmission could facilitate the identification of superspreaders and asymptomatic patients. Even though it is not currently achievable to introduce WGS to the routine diagnosis of TB on a global scale, we should encourage those types of studies to increase our control over the epidemic. The more strains we sequence, the more we will know about the characteristics of the global population of M. tuberculosis and its ability to transmit and cause disease.
Collecting data about outbreaks should be precise and standardized to facilitate data extraction. We recommend that for efficient reporting of outbreaks, authors should catalog analyzed strains in DNA sequence repositories, for example European Nucleotide Archive or NCBI Genome Database. The information about strains should include information about the date and place of collection and the drug resistance of the strain. Preferably, patient data should also be included. The information about outbreaks should include the starting dates in which the outbreak is observed, the number of patients affected by the outbreak, and the area where the outbreak was detected. Further, the authors should state the rules conferring genetic relatedness between the two strains and precise data about strain lineage.
Conclusions
We conclude that TB outbreaks are reported globally, contain median of five isolates that typically belong to L4, and often are or become drug resistant. Our study summarizes data available up to date and indicates areas of research that require organization and future development.
Supplementary Information
Acknowledgements
Not applicable.
Abbreviations
- CDC
Centers for Disease Control and Prevention
- ECDC
European Centre for Disease Prevention and Control
- L1–9
Lineage one – Lineage nine
- MDR
Multidrug – resistant
- R
Resistant
- RR
Rifampicin monoresistant
- SNPs
Single nucleotide polymorphisms
- TB
Tuberculosis
- WGS
Whole genome sequencing
- WHO
World Health Organization
- XDR
Extensively drug – resistant strains
Authors’ contributions
LZ, AM, DZ, KS collected the data. LZ, AM wrote the manuscript and conducted analysis. AM was main originator of the manuscript. AZ, MK, EAK, JD, AM supervised and corrected the manuscript. EAK funded the study. All authors contributed to the article and approved the submitted version.
Funding
This work is a part of the research project financed by the National Science Center of Poland, grant number 2019/35/B/NZ7/00942.
Availability of data and materials
All data was collected from publically published manuscripts. All datasets were referenced in the supplementary material of this manuscript.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Footnotes
This article has been updated to correct the funding number.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
11/2/2023
A Correction to this paper has been published: 10.1186/s12879-023-08751-6
References
- 1.Sia JK, Rengarajan J. Immunology of Mycobacterium tuberculosis Infections. Microbiol Spectr. 2019;7. https://journals.asm.org/doi/full/10.1128/microbiolspec.GPP3-0022-2018. [DOI] [PMC free article] [PubMed]
- 2.Suárez I, Fünger SM, Kröger S, Rademacher J, Fätkenheuer G, Rybniker J. The Diagnosis and Treatment of Tuberculosis. Dtsch Arztebl Int. 2019;116:729–735. doi: 10.3238/arztebl.2019.0729. [DOI] [PubMed] [Google Scholar]
- 3.Global tuberculosis report 2021. https://www.who.int/publications-detail-redirect/9789240037021. Accessed 20 Oct 2021.
- 4.Global tuberculosis report 2020. https://www.who.int/publications-detail-redirect/9789240013131. Accessed 17 Nov 2021.
- 5.World Health Organization . Global tuberculosis report 2019. 2019. [Google Scholar]
- 6.World Health Organization. Global tuberculosis report 2018. Geneva: World Health Organization; 2018. https://www.who.int/publications/i/item/9789241565646.
- 7.Matteelli A, Rendon A, Tiberi S, Al-Abri S, Voniatis C, Carvalho ACC, et al. Tuberculosis elimination: where are we now? Eur Respir Rev. 2018;27:180035. doi: 10.1183/16000617.0035-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jagielski T, van Ingen J, Rastogi N, Dziadek J, Mazur PK, Bielecki J. Current Methods in the Molecular Typing of Mycobacterium tuberculosis and Other Mycobacteria. Biomed Res Int. 2014;2014:645802. doi: 10.1155/2014/645802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mokrousov I. Current topics of molecular mycobacteriology. Infect Genet Evol. 2019;73:132–138. doi: 10.1016/j.meegid.2019.04.027. [DOI] [PubMed] [Google Scholar]
- 10.Xu Y, Stockdale JE, Naidu V, Hatherell H, Stimson J, Stagg HR, et al. Transmission analysis of a large tuberculosis outbreak in London: a mathematical modelling study using genomic data. Microb Genom. 2020 doi: 10.1099/mgen.0.000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJM. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science. 2006;312:1944–1946. doi: 10.1126/science.1124410. [DOI] [PubMed] [Google Scholar]
- 12.Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45:1176–1182. doi: 10.1038/ng.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci USA. 2002;99:3684–3689. doi: 10.1073/pnas.052548299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gagneux S, Small PM. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis. 2007;7:328–337. doi: 10.1016/S1473-3099(07)70108-1. [DOI] [PubMed] [Google Scholar]
- 15.Coscolla M, Gagneux S, Menardo F, Loiseau C, Ruiz-Rodriguez P, Borrell S, et al. Phylogenomics of Mycobacterium africanum reveals a new lineage and a complex evolutionary history. Microb Genom. 2021;7:000477. doi: 10.1099/mgen.0.000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ngabonziza JCS, Loiseau C, Marceau M, Jouet A, Menardo F, Tzfadia O, et al. A sister lineage of the Mycobacterium tuberculosis complex discovered in the African Great Lakes region. Nat Commun. 2020;11:2917. doi: 10.1038/s41467-020-16626-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coscolla M, Gagneux S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin Immunol. 2014;26:431–444. doi: 10.1016/j.smim.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dinkele R, Gessner S, McKerry A, Leonard B, Leukes J, Seldon R, et al. Aerosolization of Mycobacterium tuberculosis by Tidal Breathing. Am J Respir Crit Care Med. 2022 doi: 10.1164/rccm.202110-2378OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stucki D, Brites D, Jeljeli L, Coscolla M, Liu Q, Trauner A, et al. Mycobacterium tuberculosis Lineage 4 comprises globally distributed and geographically restricted sublineages. Nat Genet. 2016;48:1535–1543. doi: 10.1038/ng.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ajawatanawong P, Yanai H, Smittipat N, Disratthakit A, Yamada N, Miyahara R, et al. A novel Ancestral Beijing sublineage of Mycobacterium tuberculosis suggests the transition site to Modern Beijing sublineages. Sci Rep. 2019;9:13718. doi: 10.1038/s41598-019-50078-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bao H, Liu K, Wu Z, Wang X, Chai C, He T, et al. Tuberculosis outbreaks among students in mainland China: a systematic review and meta-analysis. BMC Infect Dis. 2019;19:972. doi: 10.1186/s12879-019-4573-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitruka K, Oeltmann JE, Ijaz K, Haddad MB. Tuberculosis Outbreak Investigations in the United States, 2002–2008. Emerg Infect Dis. 2011;17:425–431. doi: 10.3201/eid1703.101550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denholm J, Coulter C, Bastian I, and the National Tuberculosis Advisory Committee Defining a tuberculosis cluster or outbreak. Commun Dis Intell Q Rep. 2016;40:E356–9. [PubMed] [Google Scholar]
- 24.Menardo F, Duchêne S, Brites D, Gagneux S. The molecular clock of Mycobacterium tuberculosis. PLoS Pathog. 2019;15:e1008067. doi: 10.1371/journal.ppat.1008067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bjorn-Mortensen K, Soborg B, Koch A, Ladefoged K, Merker M, Lillebaek T, et al. Tracing Mycobacterium tuberculosis transmission by whole genome sequencing in a high incidence setting: a retrospective population-based study in East Greenland. Sci Rep. 2016;6:33180. doi: 10.1038/srep33180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Casali N, Broda A, Harris SR, Parkhill J, Brown T, Drobniewski F. Whole genome sequence analysis of a large isoniazid-resistant tuberculosis outbreak in london: a retrospective observational study. PLoS Med. 2016;13:e1002137. doi: 10.1371/journal.pmed.1002137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asare P, Otchere ID, Bedeley E, Brites D, Loiseau C, Baddoo NA, et al. Whole genome sequencing and spatial analysis identifies recent tuberculosis transmission hotspots in Ghana. Front Med (Lausanne) 2020;7:161. doi: 10.3389/fmed.2020.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Correa-Macedo W, Cambri G, Schurr E. The Interplay of Human and Mycobacterium Tuberculosis Genomic Variability. Front Genet. 2019;10:865. doi: 10.3389/fgene.2019.00865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mathema B, Andrews JR, Cohen T, Borgdorff MW, Behr M, Glynn JR, et al. Drivers of Tuberculosis Transmission. J Infect Dis. 2017;216(suppl_6):S644–53. doi: 10.1093/infdis/jix354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadikot RT. Host Factors in Tuberculosis. In: Hasnain SE, Ehtesham NZ, Grover S, editors. Mycobacterium Tuberculosis: Molecular Infection Biology, Pathogenesis, Diagnostics and New Interventions. Singapore: Springer; 2019. pp. 27–36. [Google Scholar]
- 31.Guthrie JL, Strudwick L, Roberts B, Allen M, McFadzen J, Roth D, et al. Whole genome sequencing for improved understanding of Mycobacterium tuberculosis transmission in a remote circumpolar region. Epidemiol Infect. 2019;147:e188. doi: 10.1017/S0950268819000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melsew YA, Gambhir M, Cheng AC, McBryde ES, Denholm JT, Tay EL, et al. The role of super-spreading events in Mycobacterium tuberculosis transmission: evidence from contact tracing. BMC Infect Dis. 2019;19:244. doi: 10.1186/s12879-019-3870-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coscolla M, Gagneux S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin Immunol. 2014;26(6):431–44. 10.1016/j.smim.2014.09.012. [DOI] [PMC free article] [PubMed]
- 34.Guerra-Assunção JA, Houben RMGJ, Crampin AC, Mzembe T, Mallard K, Coll F, et al. Recurrence due to relapse or reinfection with Mycobacterium tuberculosis: a whole-genome sequencing approach in a large, population-based cohort with a high HIV infection prevalence and active follow-up. J Infect Dis. 2015;211:1154–1163. doi: 10.1093/infdis/jiu574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merker M, Nikolaevskaya E, Kohl TA, Molina-Moya B, Pavlovska O, Brännberg P, et al. Multidrug- and Extensively Drug-Resistant Mycobacterium tuberculosis Beijing Clades, Ukraine, 2015. Emerg Infect Dis. 2020;26:481–490. doi: 10.3201/eid2603.190525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aguilar D, Hanekom M, Mata D, Gey van Pittius NC, van Helden PD, Warren RM, et al. Mycobacterium tuberculosis strains with the Beijing genotype demonstrate variability in virulence associated with transmission. Tuberculosis (Edinb). 2010;90:319–25. doi: 10.1016/j.tube.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 37.Asare P, Otchere ID, Bedeley E, Brites D, Loiseau C, Baddoo NA, Asante-Poku A, Osei-Wusu S, Ahu Prah D, Borrell S, Reinhard M, Forson A, Koram KA, Gagneux S, Yeboah-Manu D. Whole Genome Sequencing and Spatial Analysis Identifies Recent Tuberculosis Transmission Hotspots in Ghana. Front Med (Lausanne). 2020;7:161. 10.3389/fmed.2020.00161. [DOI] [PMC free article] [PubMed]
- 38.Asare P, Asante-Poku A, Prah DA, Borrell S, Osei-Wusu S, Otchere ID, et al. Reduced transmission of Mycobacterium africanum compared to Mycobacterium tuberculosis in urban West Africa. Int J Infect Dis. 2018;73:30–42. doi: 10.1016/j.ijid.2018.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Middlebrook G. Isoniazid-resistance and catalase activity of tubercle bacilli; a preliminary report. Am Rev Tuberc. 1954;69:471–472. doi: 10.1164/art.1954.69.3.471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data was collected from publically published manuscripts. All datasets were referenced in the supplementary material of this manuscript.