

Abstract
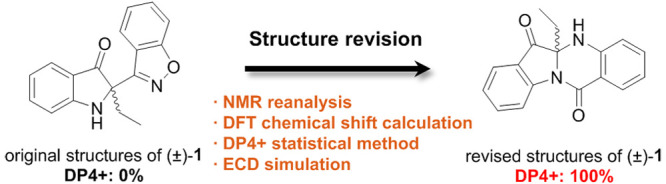
(R)- and (S)-2-(benzo[d]isoxazol-3-yl)-2-ethylindolin-3-one [(±)-1] were previously isolated from NIRAM, a natural blue dye from Polygonum tinctorium, and their structures were initially proposed to possess a 1,2-benzisoxazole ring. In this study, the structures of (±)-1 were revised to have an indole–anthranilic acid fused tetracyclic ring rather than the 1,2-benzisoxazole ring by reanalysis of one-dimensional (1D) and two-dimensional (2D) NMR followed by density functional theory (DFT) chemical shift calculation, DP4+ technique, and ECD simulation.
1. Introduction
NMR technique is the most powerful tool to determine the structure of small organic molecules for natural product chemists and synthetic chemists. However, even if the NMR data are completely interpreted, the full structural characterization is sometimes not possible due to missing information on connectivity among substructures and/or stereochemistry. This issue is exacerbated when isolated/synthesized compounds have a low H/C ratio (e.g., flavonoids, anthraquinones, tetracyclines).1−3 One of the alternative methods to propose the structure of compounds is the computational chemistry approach, and it has been successful in the structural determination and revision of organic molecules for the last few decades.4−8
(R)- and (S)-2-(benzo[d]isoxazol-3-yl)-2-ethylindolin-3-one [(±)-1] are indole alkaloids isolated from NIRAM, a natural blue dye from Polygonum tinctorium, and showed anti-inflammatory activity from our previous research in 2019 (Figure 1A, left).9 Their chemical structures were initially proposed to possess a 1,2-benzisoxazole ring. However, we later found that the 1,2-benzisoxazole functionality is extremely rare in natural products except for fusavenin, the only fungal metabolite (Figure 1A, right),10 and moreover, most of the other co-isolated compounds from NIRAM contained an anthranilic acid moiety, which is not present in (±)-1 (Figure 1B).9 These chemical features led us to reinvestigate the structures of (±)-1 and their NMR data. Here, we report the structure revision of (±)-1 through density functional theory (DFT) calculation of NMR chemical shifts followed by the DP4+ statistical method and ECD simulation.
Figure 1.

Structures of reported natural products containing 1,2-benzisoxazole ((A), red color) or anthranilic acid ((B), blue color). All of the compounds but fusavenin were isolated from NIRAM by Kim et al.9
2. Results and Discussion
We began intensive reanalysis of one-dimensional (1D) and two-dimensional (2D) NMR data of (±)-1. We found that the 3-bond coupling of H-8 and C-3′ was not observed in the heteronuclear multiple bond correlation (HMBC) (Figure 2) and that the chemical shift of the mononitrogenated carbon C2 was relatively large (80.1 ppm, Table 1).
Figure 2.

Structure revision of (±)-1. Reanalysis of the HMBC correlation and DP4+ result of original and revised (±)-1.
Table 1. Experimental (Exp.) and Calculated (Cal.) 13C NMR Chemical Shift Values of Original and Revised (±)-1 Used for DP4+ Analysis.
| exp. | cal. (|exp. – cal.|) |
||
|---|---|---|---|
| carbon | (±)-1 | original (±)-1 | revised (±)-1 |
| 2 | 80.1 | 74.3 (5.8) | 80.7 (0.6) |
| 3 | 198.4 | 198.6 (0.2) | 198.6 (0.2) |
| 3a | 124.0 | 120.8 (3.2) | 122.5 (1.5) |
| 4 | 125.5 | 123.9 (1.6) | 124.8 (0.7) |
| 5 | 126.1 | 126.1 (0) | 126.8 (0.7) |
| 6 | 139.4 | 138.8 (0.6) | 140.5 (1.1) |
| 7 | 118.4 | 119.3 (0.9) | 117.5 (0.9) |
| 7a | 153.1 | 162.5 (9.4) | 155.7 (2.6) |
| 8 | 30.2 | 35.7 (5.5) | 31.1 (0.9) |
| 9 | 7.7 | 10.5 (2.8) | 7.5 (0.2) |
| 3′ | 162.1 | 164.0 (1.9) | 161.0 (1.1) |
| 3′a | 117.3 | 114.0 (3.3) | 117.1 (0.2) |
| 4′ | 129.6 | 126.1 (3.5) | 130.7 (1.1) |
| 5′ | 120.4 | 119.7 (0.7) | 119.5 (0.9) |
| 6′ | 136.0 | 131.2 (4.8) | 136.7 (0.7) |
| 7′ | 117.3 | 110.2 (7.1) | 115.7 (1.6) |
| 7′a | 149.0 | 159.0 (10.0) | 148.3 (0.7) |
| MAE | 3.6 | 0.9 | |
| DP4+ | 0% | 100% | |
Therefore, we assumed that H-8 and C-3′ were more than 4-bond away, and therefore, the C2–C3′ bond should be replaced with a new C2–N bond. The nitrogen atom attached to C3′ was further proposed to belong to an anthranilic acid moiety. The presence of a carbonyl group was proposed based on the degree of unsaturation of the molecule, and finally, we were able to suggest tetracyclic structures of (±)-1 as shown in Figure 2. In these revised structures of (±)-1, H-8 and C-3′ were 4-bond away, which was consistent with the absence of HMBC correlation between these two nuclei (Figure 2). Then, we revisited and reanalyzed the previous HMBC spectrum of (±)-1, and the results supported the revised structures (Figure S1). To further confirm the revised structures of (±)-1, DFT chemical shift calculations were performed on both the original and revised structures of (±)-1. As shown in Table 1, the mean absolute error (MAE) of the revised (±)-1 (0.9 ppm) was much smaller than that of the original (±)-1 (3.6 ppm). Further, the calculated and experimental 1H and 13C NMR chemical shift values were subjected to DP4+ analysis, and the results also indicated revised (±)-1 to be the more likely structure with 100% probability (Figures 2 and S2, Table 1). Collectively, the planar structures of (±)-1 were revised from the 1,2-benzisoxazole-bearing structures to the indole-anthranilic acid fused tetracyclic ring-bearing structures.
In our previous research, (±)-1 were initially isolated as a racemic mixture and each enantiomer was further obtained by the chiral separation technique.9 The fact that the purified (+)-1 and (−)-1 showed the exact mirror images in their experimental ECD spectra (Figure 3A) confirmed that they are enantiomerically pure. There is only one stereogenic center at C-2 in (±)-1, and to determine its absolute configuration, we simulated ECD spectra of (±)-1 at B3LYP/6-31G*, CAM-B3LYP/TZVP, and CAM-B3LYP/SVP levels and compared them to the experimental ECD spectra. As shown in Figure 3A, the simulated ECD spectra of 2R- and 2S-1 matched with the experimental ECD spectra of (+)-1 and (−)-1, respectively. Therefore, we elucidated the structures of (+)-1 and (−)-1 as (R)- and (S)-5a-ethyl-5,5a-dihydroindolo[2,1-b]quinazoline-6,12-dione, respectively.
Figure 3.

Determination of absolute configuration of (±)-1. (A) Experimental ECD spectra of (±)-1 and simulated ECD spectra of 2R- and 2S-1. (B) Comparison of specific rotation values of (±)-1 and the previously reported (+)-indigodole B.
An extensive literature search revealed (+)-1 to have the same chemical structure as indigodole B (Figure 3B).11 However, intriguingly, it showed no significant Cotton effect in the ECD spectrum, indicating that indigodole B should be a racemic mixture. While the R configuration at C-2 in indigodole B was assigned by its positive specific optical rotation value, [α]D22 = +9.9, we proposed that this small specific optical rotation value would be attributable to impurity. Consequently, we newly named (+)- and (−)-1 as (+)- and (−)-tetrapolygonine, respectively, to distinguish them from indigodole B, a probable racemic mixture.
Despite continuing development of modern sophisticated analytical tools, there are still a large number of incorrect natural product structures being reported.12 Our present work is emphasizing again that a careful and thorough analysis of NMR data is needed when natural product chemists solve the structure of isolated compounds with these data.
3. Experimental Section
3.1. Computational Analysis
All conformers used in this study were found using the Macromodel (version 2022-1, Schrödinger LLC) module with “Mixed torsional/Low-mode sampling” in the MMFF force field. The searches were implemented in the gas phase with a 10 kJ mol–1 energy window limit and 10 000 maximum number of steps to explore all potential conformers. The Polak–Ribiere conjugate gradient (PRCG) method was utilized to minimize conformers with 10 000 iterations and a 0.001 kJ (mol Å)−1 convergence threshold on the root mean square (RMS) gradient. All of the conformers found were subjected to geometry optimization using the Gaussian 16 package (Gaussian Inc.) in the gas phase at the B3LYP/6-31G(d) level and proceeded to the calculation of the gauge-independent atomic orbital (GIAO) nuclear magnetic shielding tensor at the mPW1PW91/6-311G(d) level in the PCM (methanol). The 1H and 13C NMR chemical shift values were calculated from the Boltzmann-averaged shielding tensor using the equation below.
The calculated NMR properties were used for calculations of DP4+ probability analysis facilitated by the Excel sheet (DP4+) provided by Grimblat et al.13
All of the conformers from revised 2S-1 were proceeded to the calculation of excitation energies, oscillator strength, and rotatory strength at B3LYP/6-31G*, CAM-B3LYP/TZVP, and CAM-B3LYP/SVP levels in the polarizable continuum model (PCM, methanol). The ECD spectra were Boltzmann-averaged and visualized with SpecDis software (version 1.71, https://specdis-software.jimdofree.com) with a σ/γ value of 0.25 eV (default value), and the resulting spectra were corrected based on the UV spectra (Figure S3).14
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (Nos. 2021R1C1C1011045 and 2022R1A6A1A03054419), by the Sungkyunkwan University, by the BK21 FOUR (Graduate School Innovation) funded by the Ministry of Education (MOE, Korea) and National Research Foundation of Korea (NRF) and by the National Supercomputing Center with supercomputing resources including technical support (KSC-2021-CRE-0273).
Data Availability Statement
Data will be made available on request.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c00408.
Computational data, revised HMBC correlations, and the results of DP4+ analysis (PDF)
Author Contributions
∥ J.R.L. and K.J.P. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Kim C. S.; Bae M.; Oh J.; Subedi L.; Suh W. S.; Choi S. Z.; Son M. W.; Kim S. Y.; Choi S. U.; Oh D. C.; Lee K. R. Anti-neurodegenerative biflavonoid glycosides from Impatiens balsamina. J. Nat. Prod. 2017, 80, 471–478. 10.1021/acs.jnatprod.6b00981. [DOI] [PubMed] [Google Scholar]
- Lee T. H.; Ham S. L.; Lee D. Y.; Lee J. R.; Kim J.; Kim C. S. Structure revision of balsamisides A–D and establishment of an empirical rule for distinguishing four classes of biflavonoids. J. Nat. Prod. 2022, 85, 2461–2467. 10.1021/acs.jnatprod.2c00694. [DOI] [PubMed] [Google Scholar]
- Kim H. R.; Kim J.; Yu J. S.; Lee B. S.; Kim K. H.; Kim C. S. Isolation, structure elucidation, total synthesis, and biosynthesis of dermazolium A, an antibacterial imidazolium metabolite of a vaginal bacterium Dermabacter vaginalis. Arch. Pharm. Res. 2023, 46, 35–43. 10.1007/s12272-022-01424-z. [DOI] [PubMed] [Google Scholar]
- Kim C. S.; Oh J.; Lee T. H. Structure elucidation of small organic molecules by contemporary computational chemistry methods. Arch. Pharm. Res. 2020, 43, 1114–1127. 10.1007/s12272-020-01277-4. [DOI] [PubMed] [Google Scholar]
- Nugroho A. E.; Morita H. Circular dichroism calculation for natural products. J. Nat. Med. 2014, 68, 1–10. 10.1007/s11418-013-0768-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grauso L.; Teta R.; Esposito G.; Menna M.; Mangoni A. Computational prediction of chiroptical properties in structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 1005–1030. 10.1039/C9NP00018F. [DOI] [PubMed] [Google Scholar]
- Mándi A.; Kurtan T. Applications of OR/ECD/VCD to the structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 889–918. 10.1039/C9NP00002J. [DOI] [PubMed] [Google Scholar]
- Menna M.; Imperatore C.; Mangoni A.; Della Sala G.; Taglialatela-Scafati O. Challenges in the configuration assignment of natural products. A case-selective perspective. Nat. Prod. Rep. 2019, 36, 476–489. 10.1039/C8NP00053K. [DOI] [PubMed] [Google Scholar]
- Kim D. H.; Kim C. S.; Subedi L.; Kim S. Y.; Lee K. R. Alkaloids of NIRAM, natural dye from Polygonum tinctorium, and their anti-inflammatory activities. Tetrahedron Lett. 2019, 60, 151130 10.1016/j.tetlet.2019.151130. [DOI] [Google Scholar]
- Jiang C. X.; Li J.; Zhang J. M.; Jin X. J.; Yu B.; Fang J. G.; Wu Q. X. Isolation, identification, and activity evaluation of chemical constituents from soil fungus Fusarium avenaceum SF-1502 and endophytic fungus Fusarium proliferatum AF-04. J. Agric. Food Chem. 2019, 67, 1839–1846. 10.1021/acs.jafc.8b05576. [DOI] [PubMed] [Google Scholar]
- Lee C. L.; Wang C. M.; Hu H. C.; Yen H. R.; Song Y. C.; Yu S. J.; Chen C. J.; Li W. C.; Wu Y. C. Indole alkaloids indigodoles A–C from aerial parts of Strobilanthes cusia in the traditional Chinese medicine Qing Dai have anti-IL-17 properties. Phytochemistry 2019, 162, 39–46. 10.1016/j.phytochem.2019.02.016. [DOI] [PubMed] [Google Scholar]
- Burns D. C.; Reynolds W. F. Minimizing the risk of deducing wrong natural product structures from NMR data. Magn. Reson. Chem. 2021, 59, 500–533. 10.1002/mrc.4933. [DOI] [PubMed] [Google Scholar]
- Grimblat N.; Zanardi M. M.; Sarotti A. M. Beyond DP4: an improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. 10.1021/acs.joc.5b02396. [DOI] [PubMed] [Google Scholar]
- Bruhn T.; Schaumloffel A.; Hemberger Y.; Bringmann G. SpecDis: quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. 10.1002/chir.22138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.