

Abstract
Acute myeloid leukemia (AML) is an aggressive disease of clonal hematopoiesis with a high rate of relapse and refractory disease despite intensive therapy. Traditionally, relapsed or refractory AML has increased therapeutic resistance and poor long-term survival. In recent years, advancements in the mechanistic understanding of leukemogenesis has allowed for the development of targeted therapies. These therapies offer novel alternatives to intensive chemotherapy and have prolonged survival in relapsed or refractory AML. Unfortunately, a significant portion of patients do not respond to these therapies and relapse occurs in most patients who initially responded. This review will focus on the mechanisms of resistance to targeted therapies in relapsed or refractory AML.
Graphical Abstract
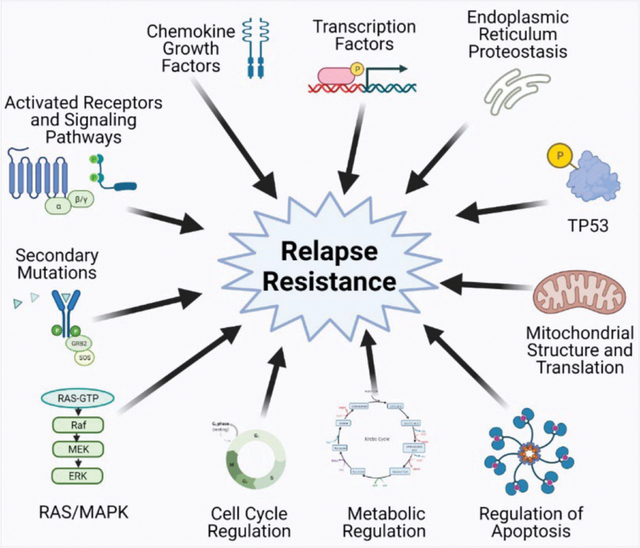
Proposed mechanisms of relapse and resistance following targeted therapies in AML. Figure created with biorender.com.
Introduction
Acute myeloid leukemia (AML) is an aggressive clonal disease of hematopoietic stem and progenitor cells. Historically, treatment has focused on induction with intensive chemotherapy followed by consolidation with chemotherapy or allogeneic hematopoietic cell transplant. Despite intensive therapy, approximately 10–40% of patients have primary refractory disease and relapse remains the primary cause of long term treatment failure1,2. In patients who relapse, the five-year overall survival (OS) rates are estimated around 10%2,3. Relapsed and refractory AML therefore remains a central problem for improving outcomes in the treatment of AML.
The mechanisms of leukemogenesis and disease relapse have been an intense area of study over the last several decades. Leukemic stem cells (LSC) are a population of cells that retain transcriptional and molecular features of hematopoietic stem and progenitor cells with the ability to regenerate leukemic cells4–7. Historically, these cells have been associated with increased therapeutic resistance and relapse4,5,8,9. Mechanistic studies suggest that clonal evolution of pre-leukemic stem cells or a dominant LSC population can occur through acquisition of additional mutations or dysregulated cellular processes, which provide a survival advantage and molecular basis for relapse4–6,10.
Advancements in mechanistic studies of leukemogenesis have allowed for the development of targeted therapies, which has improved survival in relapsed and refractory AML. Unfortunately, despite these advancements, long-term survival after targeted therapy remains poor with refractory disease in at least one fourth of patients and eventual relapse in most cases. This review will discuss our current understanding of the mechanisms of resistance to targeted therapies in relapsed or refractory AML.
FMS-Like Tyrosine Kinase 3
FMS-like tyrosine kinase 3 (FLT3) is a receptor tyrosine kinase that has been implicated in signaling pathways for hematopoietic stem cell (HSC) differentiation, cell survival and proliferation11. FLT3 mutations arise from internal tandem duplications (ITD) or point mutations in the tyrosine kinase domain (TKD)12. FLT3 mutations are estimated to occur in approximately 30% of de novo AML cases, but at relapse, can be acquired or lost in approximately 20% of cases11,13,14. FLT3 ITD and TKD mutations lead to increased FLT3 tyrosine kinase activity and activation of downstream signaling pathways mediated by phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) as well as direct phosphorylation of transcription factor STAT5, which provides a survival advantage and contributes to leukemogenesis12,15 (figure 1a). Several tyrosine kinase inhibitors (TKI) have been developed and tested in clinical trials as induction, consolidation, and maintenance therapy12,16,17. As monotherapy in relapsed or refractory AML, first generation inhibitors midostaurin and sorafenib have limited response11,12 while second generation inhibitors gilteritinib, crenolanib, and quizartinib have greater activity11,18,19(table 1 and figure 1b). Gilteritinib increased median overall survival to 9.3 months compared to 5.6 months with salvage chemotherapy in a randomized phase 3 trial in patients with relapsed or refractory FLT3-mutated AML and received FDA approval in this setting18. Despite these advances, approximately one fourth of patients had no response to gilteritinib treatment and the median duration of response remains less than one year in responders.
Figure 1:
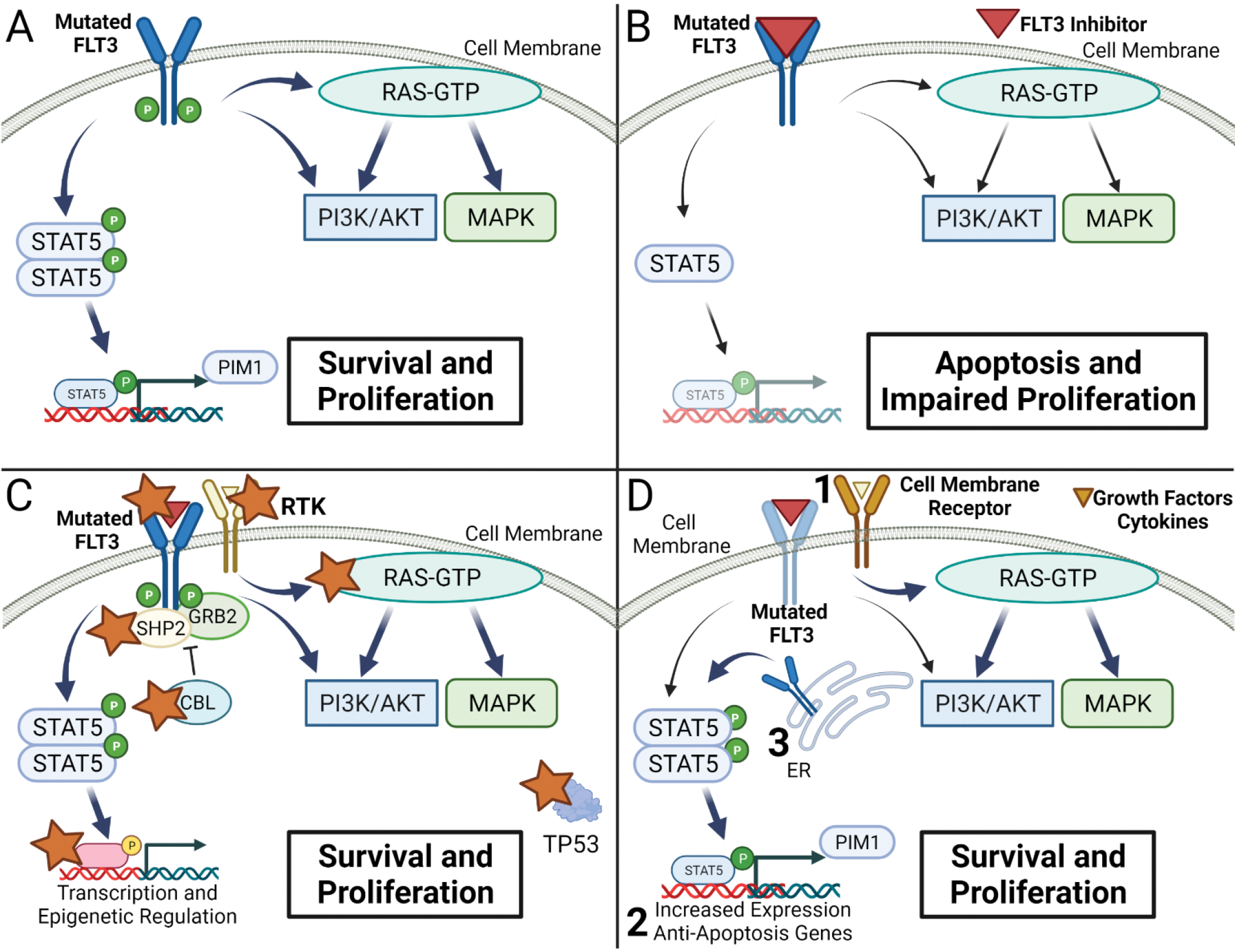
Mechanisms of Resistance to tyrosine kinase inhibitors in FLT3 mutated AML. A. Altered signaling in previously untreated newly diagnosed or relapsed AML with mutated FLT3. B. Decreased FLT3 mediated signaling and induction of apoptosis with tyrosine kinase inhibitor treatment in sensitive cells carrying FLT3 mutations. C. Mutations associated with resistance in relapsed refractory FLT3 mutated AML treated with tyrosine kinase inhibitors. Star indicates reported mutations. D. Altered cellular regulatory processes that allow for improved survival in treatment resistant cell lines or relapsed or refractory AML samples. Increased survival can be mediated through upregulation of alternative cell surface receptors or ligand-mediated signaling (1), expression of anti-apoptotic proteins (2), and STAT5 activation by mutated FLT3 in the endoplasmic reticulum (ER) (3).
Table 1:
Summary of targeted therapies and clinical trials in relapsed or refractory AML.
| Inhibitor Type | Target | FDA Approval | Clinical Trials | Trial Overview | Outcome | |
|---|---|---|---|---|---|---|
| FLT3-Tyrosine Kinase Inhibitors | ||||||
| Midostaurin | Type 1 1st generation |
FLT3, PKC, SYK, FLK-1, AKT, KIT, FGR, PDGFR, VEGFR 1/212 | Yes* 2017 | 125,126 | • Phase 2: 17/20 with FLT3-ITD or FLT3-TKD RR AML125 • Phase 2: 85/89 RR AML; 35/95 FLT3-ITD or FLT3-TKD126 |
• CR 0/20, PR 1/20 (5%)125 • CR or CRi 0/92 (0%), 1/92 patients PR (1%)126 |
| Gilteritinib | Type 1 2nd generation |
FLT3, LTK, ALK, AXL12 | Yes† 2018 | 18 | Phase 3: FLT3-ITD or FLT3-TKD RR AML Control-Chemotherapy |
OS 9.3 vs 5.6 months CR/CRi: 84/247 (34%) vs 19/124 |
| Lestaurtinib | Type 1 1st generation |
FLT3, JAK, TRK12 | No | 127 | Phase 1/2: FLT-3-ITD or FLT3-TKD mutated RR AML | 1/17 blast reduction to <5% |
| Crenolanib | Type 1 2nd generation |
FLT3, PDGFRB12 |
No | 128,129 | Phase 2: FLT3-ITD or FLT3-TKD RR AML | CRi: 4/34 (12%)129 and 7/18 (39%)128 |
| Sorafenib | Type 2 1st generation | FLT3, RAF, VEGFR, PDGFRB, KIT, RET12 | No | 21,130–132 | Monotherapy: • Retrospective: FLT3-ITD RR AML130 • Phase 2: FLT3-ITD RR AML21 Combination: FLT3-ITD RR AML • Phase 2: AZA131 • Case review-Decitabine132 |
• CR or CRi in 15/65 (23%)130 • CRi 6/13 (46%)21 Combination • AZA: CR or CRi 16/37 (43.2%)131 • Decitabine: 4/5 CRi (80%)132 |
| Quizartinib | Type 2 2nd generation |
FLT3, KIT, PDGFR12 | No | 19 | Phase 3: FLT3-ITD mutated RR AML. Control-Chemotherapy | OS 6.2 vs 4.7 months |
| Isocitrate Dehydrogenase 1/2 | ||||||
| Ivosidenib | IDH 1 | IDH1 | Yes‡ 2018 | 58 | Phase 1: IDH1-mutated RR AML | CR or CRi in 54/179 (30%) |
| Enasidenib | IDH 2 | IDH2 | Yes† 2017 | 59 | Phase 1/2: IDH2-mutated RR AML | CR or CRi in 29/109 (27%) Median OS 9.3 months |
| BCL-2 | ||||||
| Venetoclax | BH-3 Mimetic | BCL-2 | Yes* 2020 | 70 | Phase 1/2: RR AML in 94% | CR or CRi 6/32 (19%) |
| 73–79 | Retrospective: LDAC/HMA in RR AML§ | CR or CRi 21–51%; OS 3–7.8 months§ | ||||
Food and Drug Administration (FDA). RR: Relapsed or Refractory. CR: Complete Remission. CRi: Complete remission with incomplete hematologic recovery, PR: partial response, OS: Overall Survival. LDAC: low dose cytarabine, HMA: hypomethylating agent, AZA: Azacitidine
FDA Approval is in combination therapy for newly diagnosed AML.
Approved as monotherapy in relapsed or refractory AML.
Approved as monotherapy in relapsed or refractory disease or newly diagnosed AML >75 years old with IDH1 mutation meeting treatment criteria58,133.
Data combined from series of reports with dynamic range listed for response rate and survival as available.
Mutational Evolution
The acquisition of clones with additional mutations has been proposed as a mechanism of resistance in targeted therapy. Initial studies found that TKI monotherapy is more effective in relapsed AML or in samples with a high allelic burden, suggesting that selection of clones with a FLT3 driver mutation and total clonal heterogeneity plays a role in responsiveness to TKI monotherapy20. Initially, there was concern that acquisition of secondary FLT3-TKD mutations could drive relapse, especially in patients treated with type II inhibitors which can have decreased activity against constitutively active TKD mutations21–25. A retrospective analysis comparing targeted next generation sequencing (NGS) panels at relapse in patients treated with type I versus type II TKIs identified the emergence of secondary FLT3D835 mutations in approximately 30% of patients treated with type II inhibitors, but not patients treated with type I inhibitors26. Additional secondary mutations at FLT3N676, FLT3F691, or FLT3N841 have been reported in approximately 1–12% patients following treatment with both type I and type II inhibitors26–29. While the acquisition of secondary FLT3 mutations represents a possible mechanism of resistance to TKI therapy, they appear less common with type I inhibitors and in some cases may be present but may not act as the driver mutation associated with relapse27.
Selection of clones with activating mutations of RAS/MAPK signaling pathway is frequently observed at disease progression in patients who received frontline TKI combination therapy27 or monotherapy for relapsed/refractory AML26,28,30. A recent study comparing NGS panels pre- and post-gilteritinib treatment in relapsed/refractory AML found that activation of the RAS/MAPK pathway was present in 15/41 (36.6%) patients at disease progression28. Mutations in NRAS, KRAS, PTPN11, CBL and BRAF have been reported in resistant cells in patients with relapsed or refractory AML treated with either type I or type II inhibitors26,28,31 and following induction therapy with midostaurin in combination with chemotherapy in de novo AML (figure 1c)27. These studies suggest that the RAS/MAPK pathway provides a survival advantage in the presence of TKI therapy and RAS-driven clonal evolution at relapse or progressive disease can occur independent of TKI type or initial mutational status.
While secondary FLT3-TKD and RAS/MAPK pathway mutations account for about 39–49% of mutations in resistant samples to FLT3 inhibitors26,28, alternative mutations have been identified in retrospective NGS analysis of paired patient samples at relapse. Mutations in WT1, CEBPA, IDH1/2, RUNX1, TET2, GATA2, TP53, chromatin-cohesion/splicing have been identified in a smaller percentage of relapsed or refractory diseases (figure 1c)26–28. Rare mutations resulting in BCR-ABL1 fusion have been identified in resistant cases of relapsed/refractory disease treated with TKI28,32. These analyses highlight the heterogeneity of mutations that may lead to treatment resistance.
Dysregulation of Signaling Pathways and Gene Expression
In addition to somatic mutations that activate downstream signaling of FLT3, mechanistic studies revealed that dysregulation of MAPK and STAT5 can contribute to cell survival and TKI resistance (figure 1d). An in vitro CRISPR mutagenesis screen observed that loss of function mutations in SPRY3 and GSK3 led to drug resistance through downstream activation of RAS/MAPK and increased WNT signaling33. Activation of alternative signaling pathways mediated by AXL34 and SYK35 are associated with resistance to TKI in FLT3-ITD mutated cells. Increased cyclin D3 expression was identified in a subset of FLT3-ITD mutated AML patient samples which is associated with enhanced proliferation in the presence of TKI36. Additionally, upregulation of anti-apoptotic genes MCL-137, BCL-xL38, BCL2A139 and PIM-140,41 has been associated with resistance in pre-clinical models. Upregulation of BCL-xL and RAD51 are associated with hyperactivation of STAT5 and TKI resistance in FLT3 dual ITD-TKD mutated in vitro cell models38. Resistance from increased anti-apoptotic protein expression can be reversed by co-treatment with rapamycin or BH3 mimetic inhibitors38,42. Alternatively, combination therapy with inhibitors of the MAPK pathway39 or targeting STAT5 activation43,44 have been proposed in pre-clinical studies to overcome activation of downstream pathways that provide a survival advantage and therapeutic resistance to TKIs. These studies suggest a complex relationship between dysregulated gene expression and modulation of downstream signaling pathways that control cell proliferation, apoptosis, and differentiation, which ultimately can induce resistance to TKI therapy.
FLT3-ITD mutated protein may alter protein homeostasis in leukemic cell lines as it is retained in the endoplasmic reticulum in a hypoglycosylated form where it associates with chaperones including HSP90 and calnexin41. This process is associated with aberrant STAT5 activation and increased PIM-1 expression. Mechanistic studies suggest that PIM-1 phosphorylates FLT3-ITD, which stabilizes the protein and induces a positive feedback loop. Inhibition of glycosylation or PIM-1 alters downstream signaling pathways and increases sensitivity to TKI treatment40. These studies raise the interesting hypothesis that protein quality control systems in endoplasmic reticulum may play an important role in modulating the activity of mutant oncoproteins and impact the response to targeted therapies.
Chemokine and cytokine signaling mediated by the leukemic microenvironment have also been implicated in TKI resistance. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin-3 (IL-3) restore colony formation, viability and proliferation in FLT3 mutated cells treated with crenolanib, an effect dependent on the activation of STAT5 and PIM-145. Exogenous FLT3 ligand and stromal cells have been shown to decrease sensitivity to TKI in vitro through increased cytokine signaling and persistent activation of ERK46. Increased levels of fibroblast growth factor (FGF) are observed in samples of AML patients following initiation of therapy47 and addition of FGF1 induces resistance to TKI therapy in vitro while FGF inhibition sensitizes cells33. Enhanced CXCL12-CXCR4 has been shown to induce leukemic cell migration with TKI resistance with aberrant expression of ROCK1 and altered chemotaxis48.
While metabolic alterations have been extensively studied in AML and chemotherapy response49–51, there are limited studies in relapsed or refractory disease after TKI treatment. Sorafenib resistant AML cell lines have decreased oxidative phosphorylation and increased expression of glycolytic enzymes with enhanced glucose uptake52. Treatment with glycolytic inhibitors increases sensitivity to sorafenib and induces cell death in resistant cells, suggesting that metabolic regulation may represent another target for novel therapies in treatment resistance49,52,53.
Many of these mechanistic findings have been described with in vitro cell models or cultured patient samples, which requires further validation with in vivo models. However, these studies begin to suggest a complex network in which resistance to FLT3 inhibition may be a consequence of altered gene expression, activated signaling pathways, dysregulated proteostasis, microenvironmental factors and metabolic reprogramming. Modulation of these pathways in concert with TKI treatment may increase sensitivity in vitro but further study is required to evaluate their therapeutic potential.
Isocitrate Dehydrogenase 1/2
IDH1 and IDH2 mutations were identified with DNA sequencing in AML samples and are estimated to occur in up to 15–30% of cases54,55. Mechanistic studies in IDH1/2 mutated leukemic cells identified the pathogenesis occurs through the production of oncometabolite 2-hydroxyglutarate (2-HG), which leads to TET inhibition, histone hypermethylation, and impaired hematopoietic differentiation55–57. IDH1 inhibitor ivosidenib received FDA approval based on phase 1/dose expansion study in 125 patients with IDH1 mutated relapsed or refractory AML which reported a composite response with complete remission with full (CR) or partial hematologic response (CRi) of 30.4% and median duration of remission lasting 8.2 months58 (table 1). IDH2 inhibitor enasidenib received FDA approval for use in relapsed IDH2 mutated AML based on a phase 1/2 study which reported an overall response rate (ORR) of 40.3% (CI 29.4–48.3) with median response duration of 5.8 months59 (table 1). Resistance to both IDH1 and IDH2 inhibitors have been reported.
Resistance to IDH1 Inhibitors
Mutational analysis by NGS of samples from 101 patients with IDH1-mutated relapsed or refractory AML treated with ivosidenib found that baseline mutations in receptor tyrosine kinases, NRAS, KRAS, PTPN11, KIT are associated with a lower likelihood of achieving a complete response60,61. De novo RAS/MAPK and FLT3 mutations were identified at relapse, which suggests acquisition of additional mutations or expansion of a rare sub-clone that was below the limits of detection at diagnosis60,61. Secondary IDH1/2 mutations were another common driver mutation detected at relapse. IDH1S280F was initially identified in a case study of ivosidenib resistance and is predicted to sterically hinder ivosidenib binding to IDH162. Since this study, 5 additional IDH1 mutations have been identified, all of which are predicted to alter drug/cofactor binding or lead to conformational change in active sites60. Second site IDH1 mutations were present in 17/74 and de novo IDH2 mutations were present in 9/74 samples60. The presence of these mutations was associated with an increase in 2-HG in 15/16 available samples60. While activation of RAS/MAPK pathway or secondary IDH1/2 mutations have been implicated in the development of resistance with ivosidenib, single cell DNA sequencing revealed significant clonal heterogeneity at relapse with multiple clonotypes carrying different mutations60. These findings suggest again that relapse may be attributed to expansion of heterogenous clones.
Resistance to IDH2 Inhibitors
Mutational analysis with NGS in patients treated with enasidenib for relapsed and refractory AML identified FLT3 and NRAS mutations in primary refractory disease. Capture-based NGS studies found that co-occurring mutations known to activate RAS signaling led to decreased response rate and were associated with a higher mutational burden61,63. In addition, SRSF2, DNMT3A, ASLX1, RUNX1 and BCOR were also associated with non-response61,63,64. At relapse, a retrospective cohort analysis found 2-HG remained suppressed in 14/16 patients65. NGS mutational analysis found clonal evolution with new mutations including CSF3R, FLT3, U2AF1, NFKB1, RUNX1, BCROL1, BCL11A and GATA2 in these patients. In the two patients with rising 2-HG levels, an IDH1 mutation was detected65. Furthermore, secondary IDH2 mutations IDH2Q316E and IDH2I319M have been reported at hematologic progression65,66. In these cases, second site mutations in IDH2 allele emerged in a clone without the initial IDH2R140Q mutations66. Structural modeling predicted decreased binding of enasidenib to these mutant IDH2 proteins. Similar to IDH1, resistance to IDH2 may be mediated through secondary IDH1/2 mutations in a subset of patients, although a majority may occur through clonal evolution and activation of alternative signaling pathways.
Evaluation of cytosine methylation profiling and RNA sequencing in a longitudinal cohort analysis from 60 IDH1 or IDH2 mutated patient samples reports that differential regulation of genes associated with hematopoietic differentiation and increased stemness are present in relapsed or refractory disease61. This suggests that acquisition of stem cell features may drive resistance to IDH1/2 directed therapies. Stuni et al report increased oxidative phosphorylation and fatty acid beta oxidation in IDH1/2 mutated leukemic cells67, suggesting that metabolic compensation and altered mitochondrial regulation can mediate resistance, which may be abrogated by cotreatment with mitochondrial inhibitors. These studies provide a strong foundation for further mechanistic studies to understand how relapse after targeted therapy modulates downstream cellular process and provides a basis for potential clinical trials with novel targets or therapeutic combinations to overcome resistance.
B-Cell Lymphoma 2 (BCL-2)
Apoptosis is a carefully regulated process with a balance between pro-apoptotic proteins (BAX, BAK,) and anti-apoptotic proteins (BCL-2, BCl-xL, MCL-1). These proteins form a tight regulatory network which ultimately control cytochrome C release and oligomeric pore formation, two essential processes for initiating apoptosis. BCL-2 overexpression has been implicated in impaired apoptosis and increased survival of LSCs68,69. An BCL-2 inhibitor, venetoclax which tips the balance toward pro-apoptotic proteins (figure 2A), was recently FDA approved for treatment of newly diagnosed AML in patient ineligible for intensive induction or over 75 years old. It was initially tested as a single agent in a phase 2 study in relapsed/refractory AML which reported a composite response rate (CR or CRi) of 19%70. Improved composite responses were noted when combined with a hypomethylating agent (HMA) (CR or CRi 64.7–84%) or low dose cytarabine (LDAC) (CR or CRi 48%) for frontline induction therapy in patients not eligible for intensive chemotherapy71–73, which has since been applied in relapsed or refractory disease. Survival data from several retrospective analysis for venetoclax with HMA or LDAC in relapsed or refractory AML report median overall survival of 3–7.8 months with composite response rate of approximately 21–51%, which suggests worse outcomes when used in the relapsed or refractory setting73–79 (table 1). Retrospective analysis of 41 patients found median overall survival was limited to 2.4 months in patients with primary refractory or relapsed disease following frontline venetoclax and HMA therapy80. While combination therapy with venetoclax offers an effective frontline therapy in patients not eligible for intensive therapy, poor outcomes after progression and decreased efficacy in relapsed and refractory disease remain a significant concern.
Figure 2:
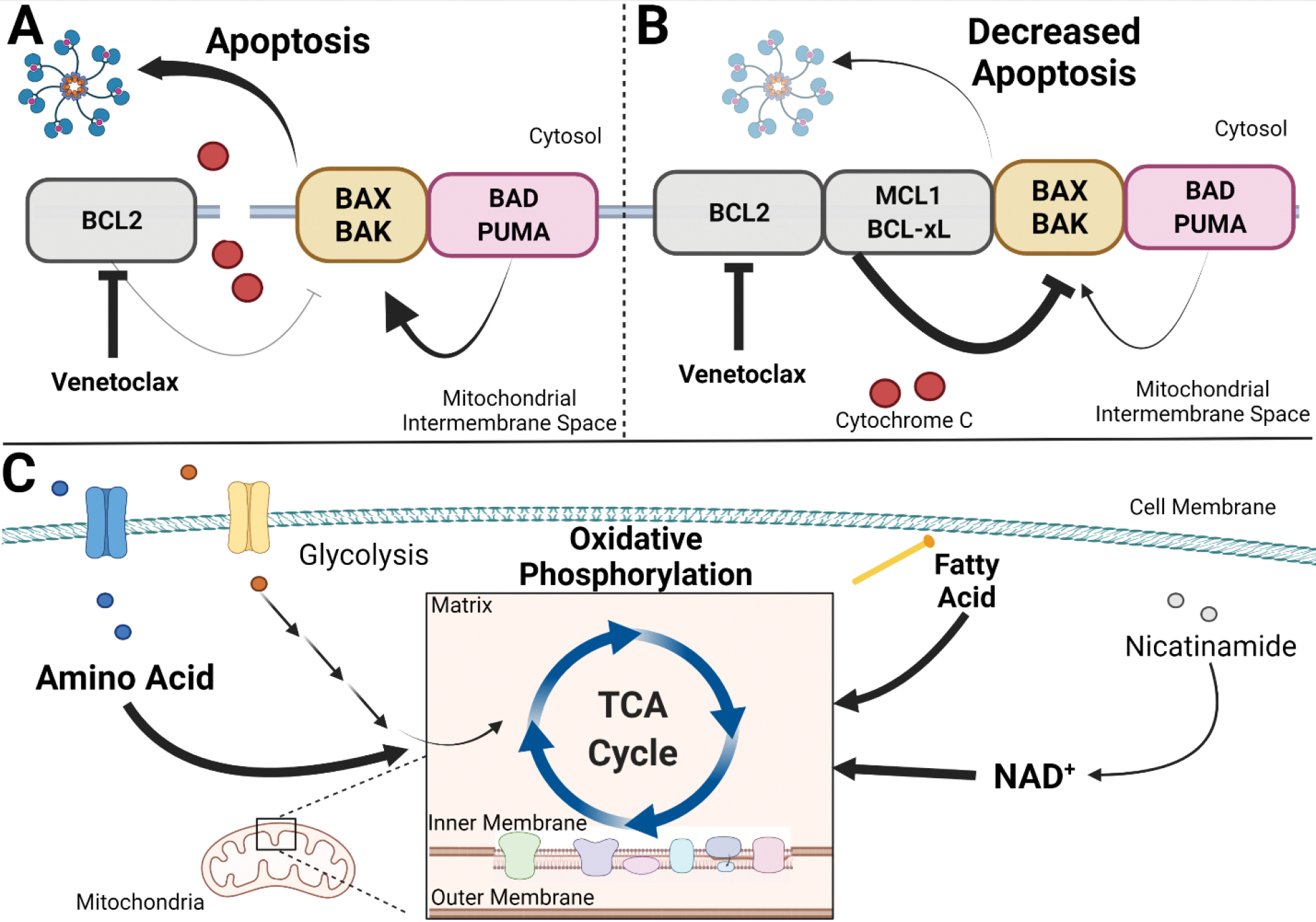
Mitochondrial regulation relapsed or refractory AML cells. A. Venetoclax inhibits anti-apoptotic protein BCL-2, which leads to increased cytochrome C release and induction of intrinsic apoptosis in sensitive cells. B. In resistant relapsed or refractory AML cells, increased expression of anti-apoptotic proteins (MCL1, BCL-xL) leads to a decrease in intrinsic apoptosis. C. In relapsed or refractory AML cells resistant to venetoclax, increased oxidative phosphorylation can be supported by amino acid uptake, beta oxidation of fatty acids, and nicotinamide adenine dinucleotide (NAD+) synthesis.
Mutational Evolution
Retrospective analysis of paired samples from 81 patients treated with venetoclax and HMA or LDAC as front line therapies identified mutations associated with durable remission or refractory/relapsed disease81. Durable remission is associated with baseline NPM1, DNMT3A, IDH1, and IDH2 mutations. Primary resistance correlates with TP53, RUNX1 and signaling mutations including FLT3, RAS, MPL and PTPN11. At relapse, FLT3-ITD and TP53 mutations are most frequently identified. A retrospective analysis in 86 patients with relapsed or refractory AML treated with venetoclax plus HMA or LDAC reports similar associations with treatment response75. At relapse, novel mutations in NRAS, FLT3, ASXL1, BCOR, TET2 and DNMT3A were identified. These findings support the notion that treatment resistance and relapse is associated with mutations that activate alternative signaling pathways, regulate differentiation, or overcome pro-apoptotic signaling.
Dysregulation of Gene Expression
Several studies have reported that venetoclax sensitivity can be hindered by utilization of other anti-apoptotic proteins (figure 2B). Retrospective review of patients with relapsed and refractory AML treated with venetoclax therapy found that BCL-2 sensitive protein index correlates with a longer duration of therapy, whereas dependence on other anti-apoptotic proteins such as BCL-xL or MCL1 had a negative correlation with therapy duration70. Studies from in vivo PDX models from samples resistant to venetoclax showed that resistance can emerge by displacing BIM to MCL-1 leading to survival dependency on MCL-1 instead of BCL-282. High levels of MCL-1 or phosphorylated BCL-2 can result in BAX displacement from pBCL-2 and BCL-xL leading to treatment resistance83. A genome wide CRISPR/Cas9 in vitro screen reported that deletion of pro-apoptotic BAX, TP53 and MAIP1 proteins led to resistance whereas knockout of anti-apoptotic proteins MDM2 and MCL1 sensitized cells to venetoclax84,85. Resistance mediated by TP53 knockout correlated with protection from mitochondrial stress and altered metabolic properties85. Similarly, inhibiting BCL-2 phosphorylation or decreasing MCL-1 and BCL-xL levels sensitizes cells to venetoclax treatment69. While studies have shown modulating activity or expression of regulatory proteins for apoptosis can lead to venetoclax resistance, others report variable expression of these proteins in resistant cells raising the possibility for other mechanisms of escape independent of apoptosis regulatory pathways84. Clinical trials with novel combination therapies including MCL-1 inhibitors are ongoing, which may mitigate resistance and relapse with venetoclax therapies87.
Metabolic and Mitochondrial Regulation
Preliminary studies reported metabolism is dysregulated in LSCs with a high dependence of oxidative phosphorylation but have relatively lower basal metabolic rate as compared to the leukemic bulk88. Venetoclax and azacitidine treatment decreases amino acid uptake and oxidative phosphorylation in patient derived LSCs, suggesting downstream effects on metabolism contribute to cellular toxicity88,89. Decreased oxidative phosphorylation is circumvented in resistant LSCs by increasing fatty acid metabolism, which provides alternative substrate for the TCA cycle89–91. Knockdown of acyl-CoA dehydrogenase restores venetoclax sensitivity in resistant LSCs92. In addition to altered amino acid metabolism, metabolomic analysis of 6 paired patient samples with venetoclax resistance found increased nicotinamide in resistant LSCs93. Treatment of cells with nicotinamide, a precursor required for nicotinamide adenine dinucleotide (NAD+) synthesis, negated the cytotoxic effect of venetoclax plus azacitidine. These effects could be reversed by limiting NAD+ synthesis with an inhibitor of nicotinamide phosphoribosyltransferase (NAMPT), which decreased LSC engraftment and oxidative phosphorylation. Similarly, TP53 insufficiency induces venetoclax resistance and increases mitochondrial oxidation with altered levels of amino acids and intermediates in glycolysis, pentose phosphate pathway, nucleotide synthesis and the urea cycle85. These studies suggest that restoration of oxidative phosphorylation either through increasing fatty acid metabolism, altered NAD+ synthesis, or utilization of alternative metabolic pathways can mediate resistance to venetoclax therapy (figure 2C).
Venetoclax has been reported to cause abnormal mitochondrial ultrastructure with lower numbers of cristae, increased cristae lumen width and loss of TMRM staining84. There is also a loss of long optic atrophy 1 (OPA1) forms suggestive of increased proteolysis, which allows for opening of cristae junctions and cytochrome c redistribution for caspase activation and induction of apoptosis. Resistant clones have a higher number of cristae with tighter cristae morphology and increase in OPA1 expression, all of which can be protective against apoptosis. RNA sequencing analysis in resistant cells identified differential expression of genes involved in mitochondrial membrane organization, potential and depolarization84. Single guide RNA targeting identified genes involved in mitochondrial transcription, such as DAP3, MRPL54, MRPL17, RBFA are associated with venetoclax resistance89. Cotreatment with tedizolid, an inhibitor of mitochondrial translation, increases sensitivity to venetoclax. These studies indicate modulation of mitochondrial structure and function may provide a survival advantage and lead to resistance to venetoclax therapies.
Cellular Heterogeneity: A Challenge to Targeted Therapies
There is growing evidence that treatment resistance and relapse in AML can occur through heterogenous mechanisms of clonal evolution94–97. Recent large scale single cell DNA sequencing on 123 AML patients found that branching clonal evolution occurs in approximately 45% of cases and in some cases convergent evolution can lead to multiple subclones with leukemia initiating capabilities95. This likely reflects recent reports in which several scenarios for clonal expansion at disease progression have been observed. In refractory disease, the primary clone with a driver mutation may respond to targeted therapy, but disease progression is associated with the outgrowth of subclones carrying treatment resistant mutations (RAS/MAPK/FLT3)27,95. However, in other cases, persistent and continued outgrowth of the primary clone with or without resistant co-mutations is thought to cause disease progression27. These instances cannot always be identified with bulk NGS alone, highlighting the need for single cell analysis to dissect the clonal architecture and evolution during disease progression.
At relapse, several scenarios of clonal expansion have been reported. Analysis of clonal evolution at relapse in FLT3-ITD AML patients treated with frontline midostaurin found persistence and expansion of the original clone, loss of the original FLT3-ITD clone with expansion of subclones with different mutational profiles, or persistence of FLT3-ITD clone with novel co-occurring mutations27. Multiple instances of polyclonal expansion at relapse have been reported in venetoclax combination therapy, FLT3, and IDH1 directed therapies25,27,60,61,65,75,81. In one case, polyclonal relapse after frontline venetoclax therapy was associated with clones carrying 6 different activating receptor kinase or RAS mutations (FLT3-ITD, FLT3N676K, FLT3D835H, NRASG12A, NRASQ61K, NRASG13R)81. Additional genetic alterations have been noted with single cell sequencing following quizartinib treatment in FLT3-ITD disease, which reported a high degree of heterogeneity within the FLT3 locus, approximately 3–7 new coding mutations by whole exome sequencing and cytogenetic changes in one out of four patients25. Alternatively, loss of heterozygosity of FLT3-ITD was associated with amplification of FLT3-ITD signaling, which may provide a survival advantage for clonal expansion95. This suggests that genomic alterations may be more complex within individual clones and highlights the importance of single cell analysis and cytogenetic microarray at relapse94,98. Expanding these genomic studies to a broader patient population may allow for a better understanding of clonal hierarchy. Further studies are required to test whether defining clonal hierarchy in AML patients can predict treatment response or provide prognostication at relapse. Given the concern for polyclonal expansion and heterogenous mutations at relapse, developing therapies directed towards commonly shared downstream signaling pathways or unique features of LSCs may have a broader therapeutic potential.
RAS Mutations: A Common Mechanism of Treatment Resistance
RAS genes encode a group of signaling GTPase proteins that regulate pathways implicated in cell survival, proliferation, and differentiation. RAS proteins are recruited to activated receptor tyrosine kinases and serve to transduce signals to downstream mediators including the MAPK and PI3K/AKT/mTOR pathways99. RAS mutations at codon 12, 13, and 61 lead to decreased b inactivation due to defective intrinsic GTPase activity and impaired responsiveness to GTPase activating proteins (GAP), which results in activation of downstream signaling pathways99. Although recent meta-analysis and cohort studies suggest limited prognostic value of RAS mutations for de novo AML in adults, the frequent identification of RAS/MAPK mutations in relapsed/refractory AML highlights the need to better understand the mechanisms by which RAS mediate leukemogenesis and treatment resistance100–102.
As described above, despite different cellular targets for FLT3, IDH1/2, and BCL-2 directed therapies, RAS/MAPK mutations represent a common pathway of resistance. The mechanism by which RAS/MAPK mutations provide a survival advantage and treatment resistance remains unclear and is an active area of study. Initial studies characterizing NRAS in treatment resistance to targeted therapies implicate activation of MAPK or PI3K, the canonical RAS effector pathways103,104. NRAS mutated cell lines display gilteritinib resistance with sustained ERK phosphorylation, cell growth and decreased apoptosis despite continued STAT5 and AKT suppression28. Trametinib, a MEK inhibitor, abrogated resistance to gilteritinib in vitro28. In vitro screens in cell lines identified that RAF1, SOC2 and PREX1 are required for MAPK activation in RAS mutated cells105. Given implications of MAPK pathway mediated signaling in RAS activating mutations, several studies have tested RAF and MEK inhibitors alone or as combination therapy in both pre-clinical models and phase 1/2 clinical trials103,106–109.
While a clear association of RAS activating mutations with disease resistance and relapse has been reported; it is unclear if this association will result in shared mechanism by which RAS promotes a survival advantage and clonal expansion in LSCs. Since evidence suggests FLT3 mutations transform cells by activating RAS/MAPK signaling, RAS activating mutations are believed to render resistance by activating downstream signaling to bypass FLT3 inhibition. Other reports, however, suggest alterative mechanisms by activation of non-canonical signaling or alteration of oxidative stress and mitochondrial programs. AML cell lines harboring a PTPN11 mutation were resistant to venetoclax and azacitidine treatment In vitro, which was in part mediated through increased oxidative phosphorlyation92. These protective effects can be ameliorated through MCL1 inhibition, suggesting that hyperactive RAS signaling may promote survival benefit and treatment resistance via modulating oxidative stress and mitochondrial programs.
The importance of hyperactive RAS signaling in leukemogenesis and treatment response has been recapitulated in mouse models. Hyperactive Nras or Kras mutations have been shown to induce development of myeloproliferative neoplasms, HSC proliferation and competitive advantage in mouse models109–113. Mice harboring Ras mutations alone typically develop myeloproliferative disorders reminiscent of human juvenile or chronic myelomonocytic leukemia (JMML or CMML), however, the presence of co-mutations promoted the development of AML114–120, which suggests Ras mutations may act cooperatively with other mutations in AML development.
Using these mouse models, our recent studies showed that NrasG12D confers a survival benefit to HSCs and progenitors following metabolic and genotoxic stress121. This effect was not affected by inhibition of the canonical RAS effectors, such as MEK and PI3K. Inhibition of the non-canonical RAS effector pathway protein kinase C (PKC) however, ameliorated the protective effects of NRASG12D. Mechanistically, N-RasG12D lowers levels of reactive oxygen species (ROS), mitochondrial membrane potential and ATP levels. Inhibition of PKC, importantly, restored the levels of ROS and abrogated the protective effects granted by N-RasG12D. Interestingly, a recent study showed that hyperactive Ras signaling promotes resistance to JAK inhibitors by suppressing BAD-mediated apoptosis122. Studies in KrasG12D knock-in mouse models have implicated NOTCH signaling and increased oxidative phosphorylation in development of MPN, which was abrogated by induction of DUSP, a phosphatase that inactivates MEK/ERK pathway123. Collectively, these studies suggest resistance may be mediated by activation of non-canonical Ras signaling pathways, altered mitochondrial regulation and dysregulated cellular metabolism. Further studies are required to establish the pathogenic and mechanistic role of RAS in refractory and relapsed AML treated with targeted therapy, which may be instrumental in identifying novel therapeutic targets to overcome treatment resistance.
Conclusion
The development of targeted therapies has increased therapeutic options and offers survival benefit in relapsed/refractory AML. Despite these advances, primary resistance and relapse remain a major barrier to long-term survival. Mutational analysis has provided insight into common pathways associated with resistance and mechanistic studies have started to characterize how modulation of signaling pathways, metabolism, proteostasis, and mitochondrial regulation contribute to treatment resistance and provide a survival advantage in leukemic stem cells. Among these, RAS activating mutations have emerged as a commonly shared mechanism of resistance to targeted therapies. Although it remains unclear how hyperactive RAS signaling provides resistance and whether common downstream pathways are induced in refractory cases after therapies targeting FLT3, IDH and BCL-2, both canonical and non-canonical RAS pathways are likely involved. Future studies are required to characterize the signaling and cellular processes altered by the RAS pathways and to guide strategies to overcome the diverse mechanisms of resistance. Given the clonal diversity of relapsed refractory AML, combination therapies targeting downstream signaling pathways, anti-apoptotic proteins, or metabolic regulation in LSCs may have a broader impact on treating the disease rather than targeting driver mutations individually. Several clinical trials have started to test this approach, which may help to provide improved response and survival in relapsed and refractory AML12,55,124.
Acknowledgements:
We thank the patients who participated in clinical trials and provided samples for mechanistic studies that contribute to our current understanding in this disease. Funding sources include E. Kropp (T32CA9357-39) and Q. Li (NIH/NHLBI R01HL132392 and NIH/NHLBI 1R01HL150707). The authors have no relevant conflicts of interest to report.
References
- 1.Thol F, Schlenk RF, Heuser M, Ganser A. How I treat refractory and early relapsed acute myeloid leukemia. Blood. 2015;126(3):319–327. doi: 10.1182/BLOOD-2014-10-551911 [DOI] [PubMed] [Google Scholar]
- 2.DeWolf S, Tallman MS. How I treat relapsed or refractory AML. Blood. 2020;136(9):1023. doi: 10.1182/BLOOD.2019001982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forman SJ, Rowe JM. The myth of the second remission of acute leukemia in the adult. Blood. 2013;121(7):1077–1082. doi: 10.1182/BLOOD-2012-08-234492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Gils N, Denkers F, Smit L. Escape From Treatment; the Different Faces of Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia. doi: 10.3389/fonc.2021.659253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shlush LI, Mitchell A, Heisler L, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nat Publ Gr. 2017;547. doi: 10.1038/nature22993 [DOI] [PubMed] [Google Scholar]
- 6.Yamashita M, Dellorusso PV., Olson OC, Passegué E. Dysregulated haematopoietic stem cell behaviour in myeloid leukaemogenesis. Nat Rev Cancer. 2020;20(7):365. doi: 10.1038/S41568-020-0260-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol 2004 57. 2004;5(7):738–743. doi: 10.1038/ni1080 [DOI] [PubMed] [Google Scholar]
- 8.Ishikawa F, Yoshida S, Saito Y, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol 2007 2511. 2007;25(11):1315–1321. doi: 10.1038/nbt1350 [DOI] [PubMed] [Google Scholar]
- 9.Duy C, Li M, Teater M, et al. Chemotherapy induces senescence-like resilient cells capable of initiating AML recurrence. Cancer Discov. 2021;11(6):1542. doi: 10.1158/2159-8290.CD-20-1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miles LA, Bowman RL, Merlinsky TR, et al. Single cell mutation analysis of clonal evolution in myeloid malignancies. Nature. 2020;587(7834):477. doi: 10.1038/S41586-020-2864-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33(2):299–312. doi: 10.1038/S41375-018-0357-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scholl S, Fleischmann M, Schnetzke U, Heidel FH. Molecular Mechanisms of Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia: Ongoing Challenges and Future Treatments. Cells. 2020;9(11). doi: 10.3390/CELLS9112493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thol F, Ganser A. Treatment of Relapsed Acute Myeloid Leukemia. Curr Treat Options Oncol. 2020;21(8):66. doi: 10.1007/S11864-020-00765-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCormick SR, McCormick MJ, Grutkoski PS, et al. FLT3 Mutations at Diagnosis and Relapse in Acute Myeloid Leukemia: Cytogenetic and Pathologic Correlations, Including Cuplike Blast Morphology. Arch Pathol Lab Med. 2010;134(8):1143–1151. doi: 10.5858/2009-0292-OA.1 [DOI] [PubMed] [Google Scholar]
- 15.Moser B, Edtmayer S, Witalisz-Siepracka A, Stoiber D. The Ups and Downs of STAT Inhibition in Acute Myeloid Leukemia. Biomedicines. 2021;9(8). doi: 10.3390/BIOMEDICINES9081051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larrosa-Garcia M, Baer MR. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Published online 2017. doi: 10.1158/1535-7163.MCT-16-0876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bazinet A, Assouline S. A review of FDA-approved acute myeloid leukemia therapies beyond ‘7 + 3.’ Expert Rev Hematol. 2021;14(2):185–197. doi: 10.1080/17474086.2021.1875814 [DOI] [PubMed] [Google Scholar]
- 18.Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3 - Mutated AML. N Engl J Med. 2019;381(18):1728–1740. doi: 10.1056/NEJMoa1902688 [DOI] [PubMed] [Google Scholar]
- 19.Cortes JE, Khaled S, Martinelli G, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019;20(7):984–997. doi: 10.1016/S1470-2045(19)30150-0 [DOI] [PubMed] [Google Scholar]
- 20.Pratz KW, Sato T, Murphy KM, Stine A, Rajkhowa T, Levis M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood. 2010;115(7):1425–1432. doi: 10.1182/BLOOD-2009-09-242859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Man CH, Fung TK, Ho C, et al. Sorafenib treatment of FLT3-ITD+ acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood. 2012;119(22):5133–5143. doi: 10.1182/BLOOD-2011-06-363960 [DOI] [PubMed] [Google Scholar]
- 22.Smith CC, Wang Q, Chin CS, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260. doi: 10.1038/NATURE11016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heidel F, Solem FK, Breitenbuecher F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107(1):293–300. doi: 10.1182/BLOOD-2005-06-2469 [DOI] [PubMed] [Google Scholar]
- 24.Smith CC, Lin K, Stecula A, Sali A, Shah NP. FLT3 D835 Mutations Confer Differential Resistance to Type II FLT3 Inhibitors. Leukemia. 2015;29(12):2390. doi: 10.1038/LEU.2015.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith CC, Paguirigan A, Jeschke GR, et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood. 2017;130(1):48. doi: 10.1182/BLOOD-2016-04-711820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alotaibi AS, Yilmaz M, Kanagal-Shamanna R, et al. Patterns of Resistance Differ in Patients with Acute Myeloid Leukemia Treated with Type I versus Type II FLT3 Inhibitors. Blood Cancer Discov. 2021;2(2):125. doi: 10.1158/2643-3230.BCD-20-0143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmalbrock LK, Dolnik A, Cocciardi S, et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood. 2021;137(22):3093. doi: 10.1182/BLOOD.2020007626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mcmahon CM, Ferng T, Canaani J, et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia Secondary Resistance to Selective FLT3 Inhibition in AML. Cancer Discov. 2019;9:1050–1063. doi: 10.1158/2159-8290.CD-18-1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarver TC, Hill JE, Rahmat L, et al. Gilteritinib is a clinically active FLT3 inhibitor with broad activity against FLT3 kinase domain mutations. Published online 2020. doi: 10.1182/bloodadvances.2019000919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McMahon CM, Canaani J, Rea B, et al. Gilteritinib induces differentiation in relapsed and refractory FLT3-mutated acute myeloid leukemia. Blood Adv. 2019;3(10):1581. doi: 10.1182/BLOODADVANCES.2018029496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H, Savage S, Schultz AR, et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat Commun. 2019;10(1). doi: 10.1038/S41467-018-08263-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alotaibi AS, Yilmaz M, Loghavi S, et al. Emergence of BCR–ABL1 Fusion in AML Post–FLT3 Inhibitor-Based Therapy: A Potentially Targetable Mechanism of Resistance – A Case Series. Front Oncol. 2020;10. doi: 10.3389/FONC.2020.588876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hou P, Wu C, Wang Y, et al. A genome-wide CRISPR screen identifies genes critical for resistance to FLT3 inhibitor AC220. Cancer Res. 2017;77(16):4402. doi: 10.1158/0008-5472.CAN-16-1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park IK, Mundy-Bosse B, Whitman SP, et al. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia. 2015;29(12):2382. doi: 10.1038/LEU.2015.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puissant A, Fenouille N, Alexe G, et al. SYK Is a Critical Regulator of FLT3 In Acute Myeloid Leukemia. Cancer Cell. 2014;25(2):226. doi: 10.1016/J.CCR.2014.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith CC, Viny AD, Massi E, et al. Recurrent Mutations in Cyclin D3 Confer Clinical Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Clin Cancer Res. 2021;27(14):4003–4011. doi: 10.1158/1078-0432.CCR-20-3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Breitenbuecher F, Markova B, Kasper S, et al. A novel molecular mechanism of primary resistance to FLT3-kinase inhibitors in AML. Blood. 2009;113(17):4063–4073. doi: 10.1182/BLOOD-2007-11-126664 [DOI] [PubMed] [Google Scholar]
- 38.Bagrintseva K, Geisenhof S, Kern R, et al. FLT3-ITD-TKD dual mutants associated with AML confer resistance to FLT3 PTK inhibitors and cytotoxic agents by overexpression of Bcl-x(L). Blood. 2005;105(9):3679–3685. doi: 10.1182/BLOOD-2004-06-2459 [DOI] [PubMed] [Google Scholar]
- 39.Yamatani K, Ai T, Saito K, et al. Inhibition of BCL2A1 by STAT5 inactivation overcomes resistance to targeted therapies of FLT3-ITD/D835 mutant AML. Transl Oncol. 2022;18:101354. doi: 10.1016/J.TRANON.2022.101354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Green AS, Maciel TT, Hospital MA, et al. Pim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemia. Sci Adv. 2015;1(8). doi: 10.1126/SCIADV.1500221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Natarajan K, Xie Y, Burcu M, Linn DE, Qiu Y, Baer MR. Pim-1 kinase phosphorylates and stabilizes 130 kDa FLT3 and promotes aberrant STAT5 signaling in acute myeloid leukemia with FLT3 internal tandem duplication. PLoS One. 2013;8(9). doi: 10.1371/JOURNAL.PONE.0074653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kohl TM, Hellinger C, Ahmed F, et al. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia. 2007;21(8):1763–1772. doi: 10.1038/SJ.LEU.2404776 [DOI] [PubMed] [Google Scholar]
- 43.Wingelhofer B, Maurer B, Heyes EC, et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leuk 2018 325. 2018;32(5):1135–1146. doi: 10.1038/s41375-017-0005-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Page BDG, Khoury H, Laister RC, et al. Small Molecule STAT5-SH2 Domain Inhibitors Exhibit Potent Antileukemia Activity. J Med Chem. 2012;55(3):1047–1055. doi: 10.1021/jm200720n [DOI] [PubMed] [Google Scholar]
- 45.Sung PJ, Sugita M, Koblish H, Perl AE, Carroll M. Hematopoietic cytokines mediate resistance to targeted therapy in FLT3-ITD acute myeloid leukemia. Blood Adv. 2019;3(7):1061. doi: 10.1182/BLOODADVANCES.2018029850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang X, Sexauer A, Levis M. Bone marrow stroma-mediated resistance to FLT3 inhibitors in FLT3-ITD AML is mediated by persistent activation of extracellular regulated kinase. Br J Haematol. 2014;164(1):61. doi: 10.1111/BJH.12599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Traer E, Martinez J, Javidi-Sharifi N, et al. FGF2 from Marrow Microenvironment Promotes Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Cancer Res. 2016;76(22):6471–6482. doi: 10.1158/0008-5472.CAN-15-3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Onish C, Mori-Kimachi S, Hirade T, et al. Internal Tandem Duplication Mutations in FLT3 Gene Augment Chemotaxis to Cxcl12 Protein by Blocking the Down-regulation of the Rho-associated Kinase via the Cxcl12/Cxcr4 Signaling Axis. J Biol Chem. 2014;289(45):31053. doi: 10.1074/JBC.M114.568287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stuani L, Sabatier M, Sarry J-E. Exploiting metabolic vulnerabilities for personalized therapy in acute myeloid leukemia. BMC Biol. 2019;17(1):57. doi: 10.1186/s12915-019-0670-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen W-L, Wang J-H, Zhao A-H, et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood. 2014;124(10):1645–1654. doi: 10.1182/blood-2014-02-554204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Åbacka Hannah, Hansen Jesper S., Huang Peng, et al. Targeting GLUT1 in acute myeloid leukemia to overcome cytarabine resistance. Haematologica. 2020;106(4):1163–1166. doi: 10.3324/haematol.2020.246843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang A, Ju H-Q, Liu K, et al. Metabolic alterations and drug sensitivity of tyrosine kinase inhibitor resistant leukemia cells with a FLT3/ITD mutation. Cancer Lett. 2016;377(2):149–157. doi: 10.1016/j.canlet.2016.04.040 [DOI] [PubMed] [Google Scholar]
- 53.Ju H-Q, Zhan G, Huang A, et al. ITD mutation in FLT3 tyrosine kinase promotes Warburg effect and renders therapeutic sensitivity to glycolytic inhibition. Leukemia. 2017;31:2143–2150. doi: 10.1038/leu.2017.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mardis ER, Ding L, Dooling DJ, et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N Engl J Med. 2009;361(11):1058–1066. doi: 10.1056/NEJMoa0903840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McMurry H, Fletcher L, Traer E. IDH Inhibitors in AML—Promise and Pitfalls. Curr Hematol Malig Rep. 2021;16(2):207–217. doi: 10.1007/s11899-021-00619-3 [DOI] [PubMed] [Google Scholar]
- 56.Ward PS, Patel J, Wise DR, et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. doi: 10.1016/J.CCR.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474. doi: 10.1038/NATURE10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DiNardo CD, Stein EM, de Botton S, et al. Durable Remissions with Ivosidenib in IDH1 -Mutated Relapsed or Refractory AML. N Engl J Med. 2018;378(25):2386–2398. doi: 10.1056/NEJMoa1716984 [DOI] [PubMed] [Google Scholar]
- 59.Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722. doi: 10.1182/BLOOD-2017-04-779405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choe S, Wang H, DiNardo CD, et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020;4(9):1894. doi: 10.1182/BLOODADVANCES.2020001503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang F, Morita K, DiNardo CD, et al. Leukemia stemness and co-occurring mutations drive resistance to IDH inhibitors in acute myeloid leukemia. Nat Commun. 2021;12(1). doi: 10.1038/S41467-021-22874-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oltvai ZN, Harley SE, Koes D, et al. Assessing acquired resistance to IDH1 inhibitor therapy by full-exon IDH1 sequencing and structural modeling. Mol Case Stud. 2021;7(2):a006007. doi: 10.1101/mcs.a006007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amatangelo MD, Quek L, Shih A, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017;130(6):732–741. doi: 10.1182/BLOOD-2017-04-779447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stein EM, DiNardo CD, Fathi AT, et al. Molecular remission and response patterns in patients with mutant- IDH2 acute myeloid leukemia treated with enasidenib. Blood. 2019;133(7):676–687. doi: 10.1182/BLOOD-2018-08-869008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quek L, David MD, De Botton S, et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat Med. 2018;24(8):1167–1177. doi: 10.1038/s41591-018-0115-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Intlekofer AM, Shih AH, Wang B, et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature. 2018;559(7712):125. doi: 10.1038/S41586-018-0251-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stuani L, Sabatier M, Saland E, et al. Mitochondrial metabolism supports resistance to IDH mutant inhibitors in acute myeloid leukemia. J Exp Med. 2021;218(5). doi: 10.1084/JEM.20200924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilde L, Ramanathan S, Kasner M. B-cell lymphoma-2 inhibition and resistance in acute myeloid leukemia. World J Clin Oncol. 2020;11(8):528. doi: 10.5306/WJCO.V11.I8.528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Andreeff M, Jiang S, Zhang X, et al. Expression of Bcl-2-related genes in normal and AML progenitors: changes induced by chemotherapy and retinoic acid. Leukemia. 1999;13(11):1881–1892. doi: 10.1038/SJ.LEU.2401573 [DOI] [PubMed] [Google Scholar]
- 70.Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and Biological Correlates of Response in a Phase 2 Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016;6(10):1106. doi: 10.1158/2159-8290.CD-16-0313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med. 2020;383(7):617–629. doi: 10.1056/NEJMoa2012971 [DOI] [PubMed] [Google Scholar]
- 72.Wei AH, Montesinos P, Ivanov V, et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo-controlled trial. Blood. 2020;135(24):2137–2145. doi: 10.1182/BLOOD.2020004856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DiNardo CD, Maiti A, Rausch CR, et al. Ten-day Decitabine with Venetoclax in Acute Myeloid Leukemia: A Single-arm Phase 2 Trial. Lancet Haematol. 2020;7(10):e724. doi: 10.1016/S2352-3026(20)30210-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aldoss I, Yang D, Aribi A, et al. Efficacy of the combination of venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Haematologica. 2018;103(9):e404–e407. doi: 10.3324/HAEMATOL.2018.188094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stahl M, Menghrajani K, Derkach A, et al. Clinical and molecular predictors of response and survival following venetoclax therapy in relapsed/refractory AML. Blood Adv. 2021;5(5). doi: 10.1182/BLOODADVANCES.2020003734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.DiNardo CD, Rausch CR, Benton C, et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am J Hematol. 2018;93(3):401–407. doi: 10.1002/AJH.25000 [DOI] [PubMed] [Google Scholar]
- 77.Gaut D, Burkenroad A, Duong T, Feammelli J, Sasine J, Schiller G. Venetoclax combination therapy in relapsed/refractory acute myeloid leukemia: A single institution experience. Leuk Res. 2020;90:106314. doi: 10.1016/J.LEUKRES.2020.106314 [DOI] [PubMed] [Google Scholar]
- 78.Aldoss I, Yang D, Pillai R, et al. Association of leukemia genetics with response to venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Am J Hematol. 2019;94(10):E253–E255. doi: 10.1002/AJH.25567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang YW, Tsai CH, Lin CC, et al. Cytogenetics and mutations could predict outcome in relapsed and refractory acute myeloid leukemia patients receiving BCL-2 inhibitor venetoclax. Ann Hematol 2020 993. 2020;99(3):501–511. doi: 10.1007/S00277-020-03911-Z [DOI] [PubMed] [Google Scholar]
- 80.Maiti A, Rausch CR, Cortes JE, et al. Outcomes of relapsed or refractory acute myeloid leukemia after front-line hypomethylating agent and venetoclax regimens. Haematologica. 2021;106(3):894. doi: 10.3324/HAEMATOL.2020.252569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.DiNardo CD, Tiong IS, Quaglieri A, et al. Myeloid Neoplasia: Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135(11):791. doi: 10.1182/BLOOD.2019003988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bhatt S, Pioso MS, Olesinski EA, et al. Reduced Mitochondrial Apoptotic Priming Drives Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Cell. 2020;38(6):872–890.e6. doi: 10.1016/J.CCELL.2020.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10(5):375–388. doi: 10.1016/j.ccr.2006.10.006 [DOI] [PubMed] [Google Scholar]
- 84.Chen X, Glytsou C, Zhou H, et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to Venetoclax treatment. Cancer Discov. 2019;9(7):890. doi: 10.1158/2159-8290.CD-19-0117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nechiporuk T, Kurtz SE, Nikolova O, et al. The TP53 Apoptotic Network is a Primary Mediator of Resistance to BCL2 inhibition in AML Cells. Cancer Discov. 2019;9(7):910. doi: 10.1158/2159-8290.CD-19-0125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med. 2020;383(7):617–629. doi: 10.1056/NEJMOA2012971 [DOI] [PubMed] [Google Scholar]
- 87.Wang H, Guo M, Wei H, Chen Y. Targeting MCL-1 in cancer: current status and perspectives. J Hematol Oncol 2021 141. 2021;14(1):1–18. doi: 10.1186/S13045-021-01079-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lagadinou ED, Sach A, Callahan K, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12(3):329. doi: 10.1016/J.STEM.2012.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sharon D, Cathelin S, Mirali S, et al. Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Sci Transl Med. 2019;11(516):2863. doi: 10.1126/scitranslmed.aax2863 [DOI] [PubMed] [Google Scholar]
- 90.CL J BM, S AD, et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell. 2018;34(5):724–740.e4. doi: 10.1016/J.CCELL.2018.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pollyea DA, Stevens BM, Jones CL, et al. Venetoclax with Azacitidine Disrupts Energy Metabolism and Targets Leukemia Stem Cells in Acute Myeloid Leukemia Patients. Nat Med. 2018;24(12):1859. doi: 10.1038/S41591-018-0233-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.BM S CL J, DA P, et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat cancer. 2020;1(12):1176–1187. doi: 10.1038/S43018-020-00126-Z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jones CL, Stevens BM, Pollyea DA, et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Published online 2020. doi: 10.1016/j.stem.2020.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hackl H, Astanina K, Wieser R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J Hematol Oncol. 2017;10(1). doi: 10.1186/S13045-017-0416-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Morita K, Wang F, Jahn K, et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat Commun. 2020;11(1):5327. doi: 10.1038/s41467-020-19119-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Potter N, Miraki-Moud F, Ermini L, et al. Single cell analysis of clonal architecture in acute myeloid leukaemia. Leuk 2018 335. 2018;33(5):1113–1123. doi: 10.1038/s41375-018-0319-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shouval R, Shlush LI, Yehudai-Resheff S, et al. Single cell analysis exposes intratumor heterogeneity and suggests that FLT3-ITD is a late event in leukemogenesis. Exp Hematol. 2014;42(6):457–463. doi: 10.1016/J.EXPHEM.2014.01.010 [DOI] [PubMed] [Google Scholar]
- 98.Xu X, Bryke C, Sukhanova M, et al. Assessing copy number abnormalities and copy-neutral loss-of-heterozygosity across the genome as best practice in diagnostic evaluation of acute myeloid leukemia: An evidence-based review from the cancer genomics consortium (CGC) myeloid neoplasms working. Cancer Genet. 2018;228–229:218–235. doi: 10.1016/j.cancergen.2018.07.005 [DOI] [PubMed] [Google Scholar]
- 99.Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012;120(17):3397–3406. doi: 10.1182/BLOOD-2012-05-378596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu X, Ye Q, Zhao XP, et al. RAS mutations in acute myeloid leukaemia patients: A review and meta-analysis. Clin Chim Acta. 2019;489:254–260. doi: 10.1016/J.CCA.2018.08.040 [DOI] [PubMed] [Google Scholar]
- 101.Bacher U, Haferlach T, Schoch C, Kern W, Schnittger S. Implications of NRAS mutations in AML: a study of 2502 patients. Blood. 2006;107(10):3847–3853. doi: 10.1182/BLOOD-2005-08-3522 [DOI] [PubMed] [Google Scholar]
- 102.Bowen DT, Frew ME, Hills R, et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood. 2005;106(6):2113–2119. doi: 10.1182/BLOOD-2005-03-0867 [DOI] [PubMed] [Google Scholar]
- 103.Gritsman K, Yuzugullu H, Von T, et al. Hematopoiesis and RAS-driven myeloid leukemia differentially require PI3K isoform p110α. J Clin Invest. 2014;124(4):1794. doi: 10.1172/JCI69927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kadia TM, Kantarjian H, Kornblau S, et al. Clinical and Proteomic Characterization of Acute Myeloid Leukemia with Mutated RAS. Cancer. 2012;118(22):5550. doi: 10.1002/CNCR.27596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang T, Yu H, Hughes NW, et al. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell. 2017;168(5):890. doi: 10.1016/J.CELL.2017.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Khoury JD, Tashakori M, Yang H, et al. Pan-RAF Inhibition Shows Anti-Leukemic Activity in RAS-Mutant Acute Myeloid Leukemia Cells and Potentiates the Effect of Sorafenib in Cells with FLT3 Mutation. Cancers (Basel). 2020;12(12):1–13. doi: 10.3390/CANCERS12123511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Maiti A, Naqvi K, Kadia TM, et al. Phase II Trial of MEK Inhibitor Binimetinib (MEK162) in RAS-mutant Acute Myeloid Leukemia. Clin Lymphoma Myeloma Leuk. 2019;19(3):142–148.e1. doi: 10.1016/J.CLML.2018.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tambe M, Karjalainen E, Vähä-Koskela M, et al. Pan-RAF inhibition induces apoptosis in acute myeloid leukemia cells and synergizes with BCL2 inhibition. Leukemia. 2020;34(12):3186–3196. doi: 10.1038/S41375-020-0972-0 [DOI] [PubMed] [Google Scholar]
- 109.Burgess MR, Hwang E, Mroue R, et al. KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence In Cancer. Cell. 2017;168(5):817. doi: 10.1016/J.CELL.2017.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sabnis AJ, Cheung LS, Dail M, et al. Oncogenic Kras Initiates Leukemia in Hematopoietic Stem Cells. Jordan CT, ed. PLoS Biol. 2009;7(3):e1000059. doi: 10.1371/JOURNAL.PBIO.1000059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li Q, Bohin N, Wen T, et al. Oncogenic Nras has bimodal effects on stem cells that sustainably increase competitiveness. Nat 2013 5047478. 2013;504(7478):143–147. doi: 10.1038/nature12830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang J, Kong G, Liu Y, et al. NrasG12D/+ promotes leukemogenesis by aberrantly regulating hematopoietic stem cell functions. Blood. 2013;121(26):5203. doi: 10.1182/BLOOD-2012-12-475863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Braun BS, Tuveson DA, Kong N, et al. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci U S A. 2004;101(2):597. doi: 10.1073/PNAS.0307203101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li Q, Haigis KM, McDaniel A, et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood. 2011;117(6):2022–2032. doi: 10.1182/BLOOD-2010-04-280750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Carratt SA, Braun TP, Coblentz C, et al. Mutant SETBP1 enhances NRAS-driven MAPK pathway activation to promote aggressive leukemia. Leuk 2021 3512. 2021;35(12):3594–3599. doi: 10.1038/s41375-021-01278-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shi X, Yang Y, Shang S, et al. Cooperation of Dnmt3a R878H with Nras G12D promotes leukemogenesis in knock-in mice: a pilot study. BMC Cancer. 2019;19(1):1072. doi: 10.1186/s12885-019-6207-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jin X, Qin T, Zhao M, et al. Oncogenic N-Ras and Tet2 haploinsufficiency collaborate to dysregulate hematopoietic stem and progenitor cells. Blood Adv. 2018;2(11):1259–1271. doi: 10.1182/BLOODADVANCES.2018017400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kunimoto H, Meydan C, Nazir A, et al. Cooperative Epigenetic Remodeling by TET2 Loss and NRAS Mutation Drives Myeloid Transformation and MEK Inhibitor Sensitivity. Cancer Cell. 2018;33(1):44–59.e8. doi: 10.1016/j.ccell.2017.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang J, Kong G, Rajagopalan A, et al. p53−/− synergizes with enhanced NrasG12D signaling to transform megakaryocyte-erythroid progenitors in acute myeloid leukemia. Blood. 2017;129(3):358–370. doi: 10.1182/BLOOD-2016-06-719237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zuber J, Radtke I, Pardee TS, et al. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009;23(7):877–889. doi: 10.1101/GAD.1771409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ney GM, Yang KB, Ng V, et al. Oncogenic N-Ras Mitigates Oxidative Stress–Induced Apoptosis of Hematopoietic Stem Cells. Cancer Res. 2021;81(5):1240–1251. doi: 10.1158/0008-5472.CAN-20-0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Winter PS, Sarosiek KA, Lin KH, et al. RAS signaling promotes resistance to JAK inhibitors by suppressing BAD-mediated apoptosis. Sci Signal. 2014;7(357). doi: 10.1126/scisignal.2005301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kong G, You X, Wen Z, et al. Downregulating Notch counteracts KrasG12D-induced ERK activation and oxidative phosphorylation in myeloproliferative neoplasm. Leukemia. 2019;33(3):671. doi: 10.1038/S41375-018-0248-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Smith CC. The growing landscape of FLT3 inhibition in AML. Hematol (United States). 2019;2019(1):539–547. doi: 10.1182/hematology.2019000058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stone RM, DeAngelo DJ, Klimek V, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105(1):54–60. doi: 10.1182/BLOOD-2004-03-0891 [DOI] [PubMed] [Google Scholar]
- 126.Fischer T, Stone RM, DeAngelo DJ, et al. Phase IIB Trial of Oral Midostaurin (PKC412), the FMS-Like Tyrosine Kinase 3 Receptor (FLT3) and Multi-Targeted Kinase Inhibitor, in Patients With Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome With Either Wild-Type or Mutated FLT3. J Clin Oncol. 2010;28(28):4339. doi: 10.1200/JCO.2010.28.9678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Smith BD, Levis M, Beran M, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103(10):3669–3676. doi: 10.1182/BLOOD-2003-11-3775 [DOI] [PubMed] [Google Scholar]
- 128.Cortes JE, Kantarjian HM, Kadia TM, et al. Crenolanib besylate, a type I pan-FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3-ITD and D835 AML. J Clin Oncol. 2016;34(15_suppl):7008–7008. doi: 10.1200/JCO.2016.34.15_suppl.7008 [DOI] [Google Scholar]
- 129.Randhawa JK, Kantarjian HM, Borthakur G, et al. Results of a Phase II Study of Crenolanib in Relapsed/Refractory Acute Myeloid Leukemia Patients (Pts) with Activating FLT3 Mutations. Blood. 2014;124(21):389–389. doi: 10.1182/BLOOD.V124.21.389.389 [DOI] [Google Scholar]
- 130.MetzelDer SK, SchroeDer T, Finck A, et al. High activity of sorafenib in FLT3-ITD-positive acute myeloid leukemia synergizes with allo-immune effects to induce sustained responses. Leuk 2012 2611. 2012;26(11):2353–2359. doi: 10.1038/leu.2012.105 [DOI] [PubMed] [Google Scholar]
- 131.Ravandi F, Alattar ML, Grunwald MR, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121(23):4655–4662. doi: 10.1182/blood-2013-01-480228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Muppidi MR, Portwood S, Griffiths EA, et al. Decitabine and Sorafenib Therapy in FLT-3 ITD-Mutant Acute Myeloid Leukemia. Clin Lymphoma, Myeloma Leuk. 2015;15(S):S73–S79. doi: 10.1016/J.CLML.2015.02.033 [DOI] [PubMed] [Google Scholar]
- 133.DiNardo CD, Stein AS, Stein EM, et al. Mutant Isocitrate Dehydrogenase 1 Inhibitor Ivosidenib in Combination With Azacitidine for Newly Diagnosed Acute Myeloid Leukemia. J Clin Oncol. 2021;39(1):57–65. doi: 10.1200/JCO.20.01632 [DOI] [PMC free article] [PubMed] [Google Scholar]