

Abstract
Pathogenic variants in ANKRD11 or microdeletions at 16q24.3 are the cause of KBG syndrome (KBGS), a neurodevelopmental syndrome characterized by intellectual disability, dental and skeletal anomalies, and characteristic facies. The ANKRD11 gene encodes the ankyrin repeat-containing protein 11A transcriptional regulator, which is expressed in the brain and implicated in neural development. Syndromic conditions caused by pathogenic variants in epigenetic regulatory genes show unique patterns of DNA methylation (DNAm) in peripheral blood, termed DNAm signatures. Given ANKRD11’s role in chromatin modification, we tested whether pathogenic ANKRD11 variants underlying KBGS are associated with a DNAm signature. We profiled whole-blood DNAm in 21 individuals with ANKRD11 variants, 2 individuals with microdeletions at 16q24.3 and 28 typically developing individuals, using Illumina’s Infinium EPIC array. We identified 95 differentially methylated CpG sites that distinguished individuals with KBGS and pathogenic variants in ANKRD11 (n = 14) from typically developing controls (n = 28). This DNAm signature was then validated in an independent cohort of seven individuals with KBGS and pathogenic ANKRD11 variants. We generated a machine learning model from the KBGS DNAm signature and classified the DNAm profiles of four individuals with variants of uncertain significance (VUS) in ANKRD11. We identified an intermediate classification score for an inherited missense variant transmitted from a clinically unaffected mother to her affected child. In conclusion, we show that the DNAm profiles of two individuals with 16q24.3 microdeletions were indistinguishable from the DNAm profiles of individuals with pathogenic variants in ANKRD11, and we demonstrate the diagnostic utility of the new KBGS signature by classifying the DNAm profiles of individuals with VUS in ANKRD11.
Introduction
ANKRD11 (previously known as ANCO-1) encodes the ankyrin repeat-containing protein 11, a nuclear protein belonging to a family of ankyrin repeat-containing coactivators known to regulate transcriptional activity (1,2). Truncating variants in ANKRD11 or microdeletions at 16q24.3 encompassing ANKRD11 cause KBG syndrome (KBGS; MIM# 148050), a rare autosomal dominant neurodevelopmental disorder (NDD) (3–5). KBGS is characterized by specific facial dysmorphia (e.g. triangular face, brachycephaly, synophrys, widely spaced eyes), dental and palatal abnormalities, most commonly macrodontia of permanent upper central incisors and dental pits, skeletal anomalies such as postnatal short stature and brachydactyly, learning difficulty and developmental delay of variable severity (4–6). No sex differences in the frequency of this syndrome have been reported. The initials ‘KBG’ are the first letters of the surnames of the first families in which this disorder was diagnosed in 1975 by Hermann et al. Today, there are more than 200 individuals reported in the medical literature associated with ANKRD11 variants or 16q24.3 microdeletions (3,6).
The majority (>75%) of variants in ANKRD11 causing KBGS are frameshift and nonsense variants, and most are de novo; approximately 30% are inherited (6). However, a small proportion (~5%) of missense or splice site variants have also been reported. A recent survey of all published cases (n = 253) of KBG syndrome noted both genotype–phenotype correlation and variable expressivity. A higher frequency of developmental delay and intellectual disability is seen in individuals with truncating variants compared to those with missense variants (7). Of those patients with missense variants and a clinical diagnosis of KBGS (n = 13; ~5%), six individuals inherited the variant from a parent who did not meet the clinical diagnostic criteria for KBGS, and in four of those individuals, transmission is from mother to son (7). Based on published reports, a highly variable clinical presentation of inherited missense variants appears to be a hallmark of ANKRD11 (5,8,9).
The ANKRD11 protein is composed of four protein domains: ankyrin domain with five ankyrin repeats (amino acids 162–284), two repression domains (amino acids 318–611 and 2369–2663) and one activation domain (amino acids 1851–2145) (2). This protein acts as a chromatin regulator with a unique dual function, allowing it to interact with coactivators and corepressors of nuclear receptors (NRs). Studies have shown that ANKRD11 interacts with p160 NR coactivators (RAC3, NCOA1, NCOA2), components of the P/CAF (p300/CBP-associated factor) acetyltransferase complex, and histone deacetylases, particularly HDAC3 (1,10,11). The role of ANKRD11 in epigenetic regulation is not fully characterized; however, evidence of ANKRD11’s interactions with writers and eraser of histone acetylation suggest it as an epigenetic regulator. Modification of chromatin structure through histone acetylation is important for nervous system and bone development (12,13). The ANKRD11 protein is highly expressed in the adult human brain and localizes to nuclei of neurons and glial cells (11,14). Using ANKRD11-deficient murine and human cell models, studies have shown that ANKRD11 is a vital co-regulator of neurogenesis, including neural positioning and dendritic differentiation (11,14).
Our research group and others have reported unique genome-wide changes in DNA methylation (DNAm) caused by pathogenic variants in genes encoding epigenetic regulators, termed ‘DNAm signatures’ (15–21). DNAm signatures are gene- and disorder-specific; to date, DNAm signatures for >50 disorders have been defined, most of which are caused by variants in genes encoding epigenetic regulators. These DNAm signatures are likely established via crosstalk between histone modifications and DNAm.
Although the exact molecular mechanisms underpinning DNAm signatures are not yet fully elucidated, a rapidly expanding body of work has emerged demonstrating that DNAm signatures have diagnostic utility in classifying variants of uncertain significance (VUS). For VUS classification, the DNAm profile for a single case is compared to a gene-specific DNAm signature, derived from analysis of samples from patients with pathogenic variants within the gene in question. Here, we generated a unique DNAm signature for pathogenic ANKRD11 variants in a cohort of individuals with KBGS. We demonstrate the diagnostic utility of the signature and classify four ANKRD11 VUS. Gene targets overlapping the KBGS-specific DNAm signature sites also demonstrate the functional relevance of DNAm signatures, as we found some of those genes to be directly implicated in bone and skeletal development.
Results
Molecular genetics
In this study, we report 21 individuals with ANKRD11 (NM_013275.5) variants that are predicted to adversely impact protein function and are classified as pathogenic using the ACMG variant classification guidelines (22) (Table 1 and Supplementary Material, Table S1). These are frameshift or nonsense variants located within the largest exon (9), an observed mutational hotspot in ANKRD11. In addition, there is one individual with a splice site variant between exons 2 and 3, and two individuals with microdeletions at 16q24.3, which result in full deletion of the ANKRD11 gene. Variants in all these individuals are associated with KBG syndrome phenotypes (43% females). The remaining variants (n = 4) in ANKRD11 are missense VUS, two of which are located within exon 9, and an additional two from a parent–child duo located in exon 7 overlapping ankyrin repeats. Figure 1 provides a schematic of the ANKRD11 gene structure and variant location generated using ProteinPaint (23).
Table 1.
Demographic and mutation information for individuals with ANKRD11 variants used for signature discovery and validation
| Sample_ID | Sex | Age at blood collection (years) | cDNA change | Protein change | Group |
|---|---|---|---|---|---|
| EX1 | M | 9 | c.3770_3771del | p. (Lys1257Argfs*25) | Discovery |
| EX2 | F | 13 | c.2329_2332del | p. (Glu777Argfs*5) | Discovery |
| EX3 | M | 8 | c.2344_2345dup | p. (Leu782Phefs*2) | Discovery |
| EX4 | F | 18 | c.2175_2178del | p. (Asn725Lysfs*23) | Discovery |
| EX5 | M | 18 | c.3309del | p. (Asp1104Metfs*214) | Discovery |
| EX6 | F | 14 | c.1920_1921insT | p. (Lys641*) | Discovery |
| EX7 | F | 9 | c.1903_1907del | p. (Lys635Glnfs*26) | Discovery |
| EX8 | M | 10 | c.3786_3789del | p. (Lys1262Asnfs*55) | Discovery |
| EX9 | F | 13 | c.3201dupA | p. (Asp1068Argfs*34) | Discovery |
| EX10 | F | 11 | c.1903_1907del | p. (Lys635Glnfs*26) | Discovery |
| EX11 | F | 4 | c.1902_1905del | p. (Lys635Thrfs*17) | Discovery |
| EX12 | F | 2 | c.2398_2401del | p. (Glu800Asnfs*62) | Discovery |
| EX13 | M | 15.5 | c.4374delG | p. (Lys1459Argfs*72) | Discovery |
| EX14 | M | 3.4 | c.5174dupC | p. (Ser1726Valfs*6) | Discovery |
| EX15 | M | 43 | c.1902_1905del | p. (Lys635Thrfs*17) | Validation |
| EX16 | F | 41 | c.3786_3789del | p. (Lys1262Asnfs*55) | Validation |
| EX17 | M | 8 | - | Protein Del 507 kb (ACSF3, CDH15, ANKRD11) | Validation |
| EX18 | M | 2 | - | Protein Del 635 kb (GALNS, ACSF3, CDH15, ANKRD11) | Validation |
| EX19 | F | 16 | c.226+1G>T | c.(226+1G>T) | Validation |
| EX20 | M | 1 | c.3860_3861delAG | p. (Glu1287Glyfs*4) | Validation |
| EX21 | M | 13 | c.6738delA | p. (Glu2247Asnfs*90) | Validation |
| EX22 | M | 17 | c.2178del | p. (Asp727Thrfs*22) | Validation |
| EX23 | F | 14 | c.1903_1907del | p. (Lys635Glnfs*26) | Validation |
Figure 1.

Genomic location of ANKRD11 variants. Schematic representation of the ANKRD11 protein (GenBank: ANKRD11; NM_013275; GRCh37), its functional domains and variants used in this study. The number in each lollipop represents the number of individuals with that variant. Exon structure, based on GenBank: NM_013275.5, is provided by dashed lines. ANK, ankyrin tandem repeats (167–292); repression domain I (318–611); repression domain II (2369–2663); one activation domain (amino acids 1851–2145).
KBG syndrome DNAm signature generation
To generate a KBG-syndrome-specific DNAm signature, we profiled genome wide DNAm in blood from individuals with a clinical diagnosis of KBG syndrome and a truncating variant in ANKRD11 (Table 1 and Supplementary Material, Table S1). The KBG syndrome discovery cohort (n = 14) included eight females and six males with mean age at sample collection of 10.6 ± 5.1 years (range 2–18 years). The 28 sex- and age-matched control subjects included 14 females and 14 males with mean ages at sample collection of 9.5 ± 4.7 years (range 2–18 years) (Supplementary Material, Table S2).
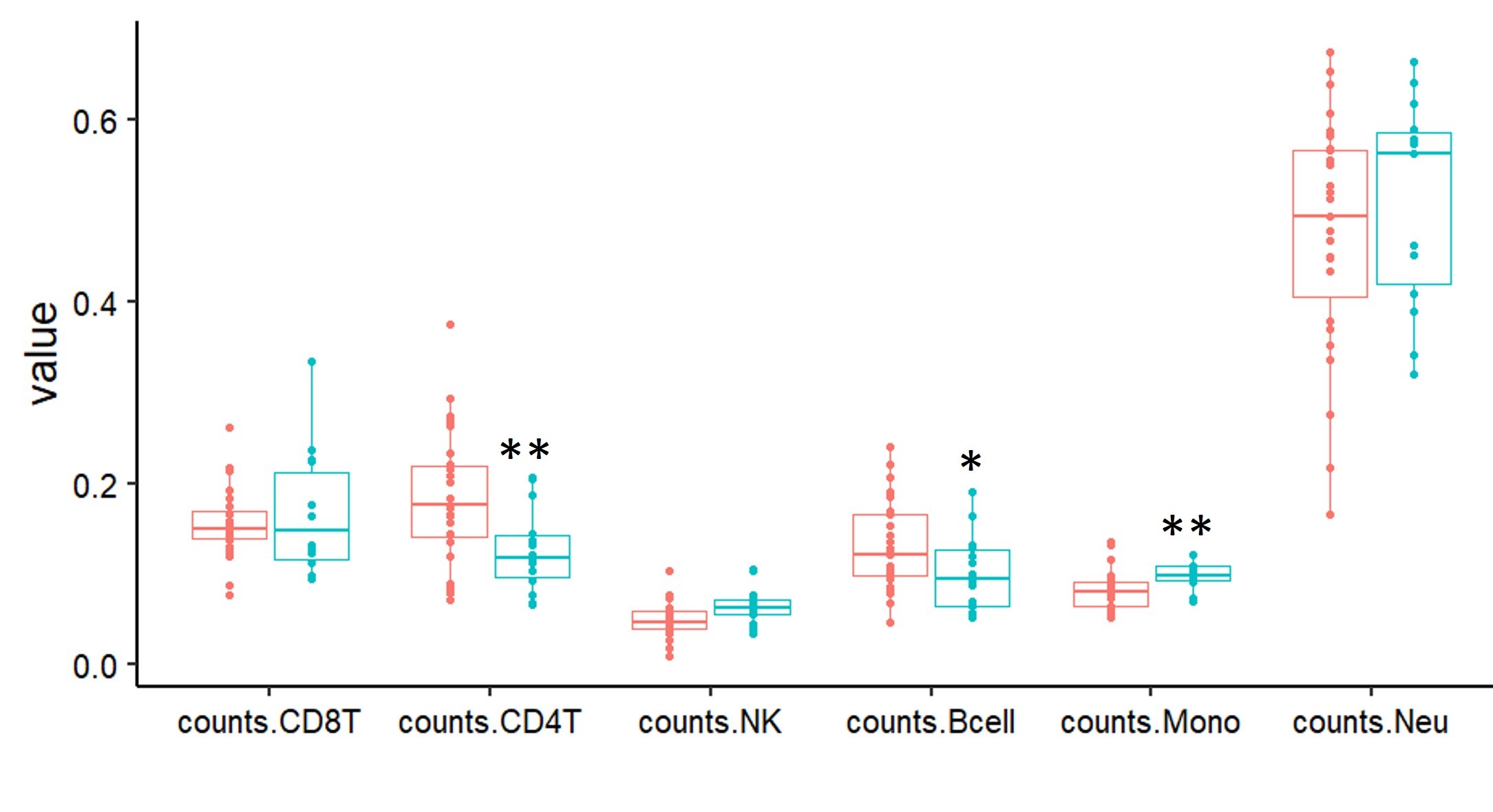
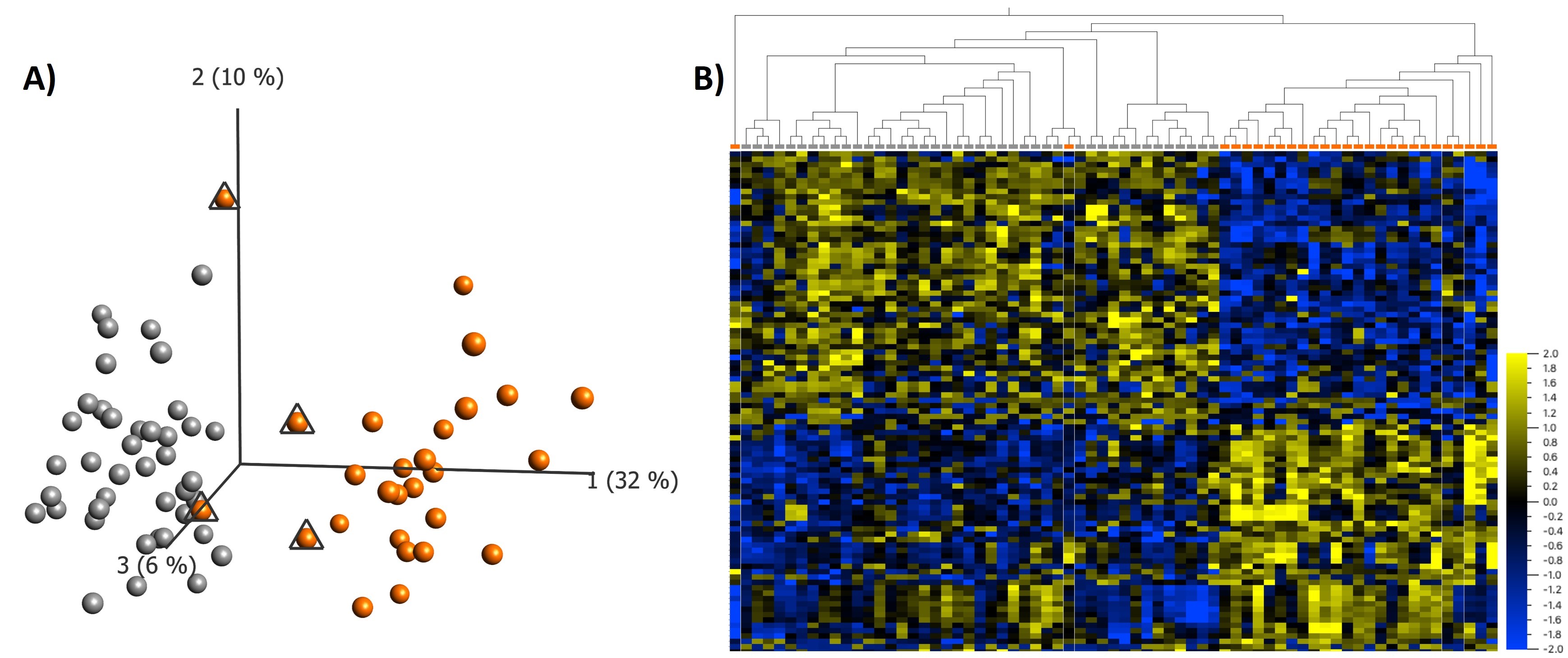
We identified 95 differentially methylated CpG sites that meet thresholds of false discovery rate (FDR) < 0.05 and |Δβ| > 0.10 (10% DNAm difference; Supplementary Material, Table S3), using linear regression modeling. We accounted for cell types as covariates in the linear regression model (Supplementary Material, Fig. S1). We visualized DNAm data at signature sites using principal component analysis (PCA) and hierarchal clustering (Fig. 2). DNAm at 95 signature sites clearly clusters individuals with KBG syndrome from typically developing controls; 45% of the signature CpG sites were hypermethylated and 55% were hypomethylated. Approximately 56% of CpG signature sites overlapped CpG islands or shores (within 2 kb of islands). This was significantly higher than the percentage of total probes on the array mapping to islands and shores (37%) (P-value = 4.9E−3; hypergeometric test). There were no significant DNAm differences based on sex at the identified KBGS-specific signature sites.
Figure 2.

Loss-of-function variants in ANKRD11 are associated with a distinct DNAm signature. (A) PCA and (B) heatmap showing clustering of the KBGS discovery subjects (n = 14) and control discovery subjects (n = 28) using DNAm values at the 95 CpG sites identified in the KBGS DNAm signature. The heatmap color gradient indicates the normalized DNAm value ranging from −2.0 (blue) to 2.0 (yellow). Euclidean distance metric is used in the heatmap clustering dendrograms.
Independent validation of KBG syndrome DNAm signature
Using the KBG syndrome DNAm signature, we used a machine learning classification model to robustly categorize variants from independent samples as ‘KBG-like’ or ‘control-like’ based on DNAm levels at signature sites. We trained a support vector machine (SVM) model on data from the discovery cohort used to generate the signature. The model generated a probability of pathogenicity score ranging from 0 ‘control-like’ to 1 ‘KBGS-like’ for each sample (Supplementary Material, Table S4). We classified a validation cohort of nine individuals with pathogenic sequence variants (n = 7) and 16q24.3 microdeletions (n = 2) involving ANKRD11 (Table 1). The SVM model generated high pathogenicity scores (75–98%) for all samples in the validation cohort demonstrating high sensitivity of the signature (Fig. 3 and Supplementary Material, Table S4). We found that individuals with 16q24.3 microdeletions have a similar DNAm profile to individuals with single variants in ANKRD11 (pathogenicity scores >88%). To test the specificity of the KBGS DNAm signature, we included DNAm data for an additional 150 typically developing controls (46% females, and ages 1–42 years), all of which had low SVM scores (0–25%) demonstrating high specificity of the signature (Fig. 3). To further validate the specificity of the KBG syndrome DNAm signature generated, we classified cohorts of individuals with Kabuki (n = 9) and Weaver (n = 17) syndromes, caused by pathogenic variants in genes encoding the epigenetic regulators, KMT2D and EZH2, respectively. Both cohorts had pathogenicity scores within the control range (1–22%) (Fig. 3).
Figure 3.
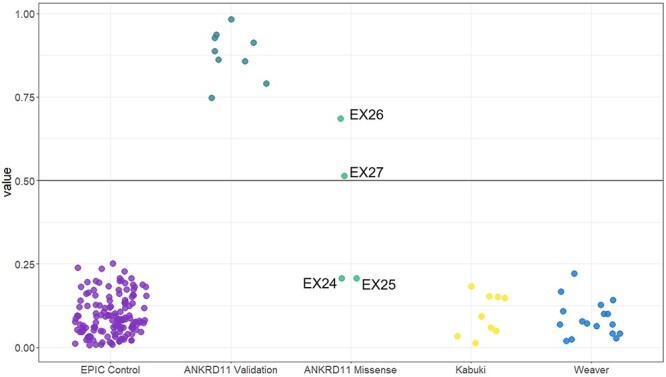
Classification of samples using SVM machine learning models based on the KBG DNAm signature. Sample groups were scored using the KBGS SVM model. KBGS validation subject (n = 7) classified as KBG-like demonstrating 100% sensitivity of the model. Whereas validation control subjects (n = 150) classified with low probabilities demonstrating 100% specificity of the model. ANKRD11 missense variants (n = 4), two of which belong to a child–parent duo and have KBG syndrome-like probabilities, whereas the remaining two had control-like probabilities.
Classification of VUS in ANKRD11
Having illustrated the efficacy of the KBG syndrome DNAm signature in classifying individuals with pathogenic ANKRD11 variants, we next classified four individuals with VUS in ANKRD11. Two of the individuals with VUS in ANKRD11 are a parent–child duo (EX26, EX27), whereas the VUS in EX24 is de novo, and inheritance for EX25 is unknown (Supplementary Material, Table S1). The SVM model based on the KBG syndrome DNAm signature sites generated pathogenicity scores within the control range for EX24 [p. (Ile2427Thr), 20%] and EX25 [p. (Lys459Asn), 20%] (Fig. 3 and Supplementary Material, Table S4). For the parent–child duo EX26 and EX27 [p. (Thr236Met)], the SVM model generated intermediate pathogenicity scores, the child’s score was 68% and the mother’s was 51% (Fig. 3). To investigate the possibility of somatic mosaicism in the mother, we used quantitative pyrosequencing to genotype the ANKRD11 variant and found the percentage of the variant allele in blood to be 49%, suggesting no mosaicism was present. We clustered all classification and validation samples on signature sites using heatmap and PCA approaches (Supplementary Material, Fig. S2). We assessed predicted pathogenicity of these VUS using Alamut variant annotation software, which applies multiple prediction algorithms for comparison (Table 2). All in silico predictions indicate that these variants are deleterious; however, the parent–child duo has a higher CADD score compared to the other two VUS (Table 2). We also used the MetaDome web server (24) to assess ANKRD11’s protein tolerance of the three reported missense VUS (Fig. 4); the lower the score, the more intolerant the protein is to variation. All three missense variants are intolerant; however, p. (Thr236Met) in the duo was ‘highly intolerant’ with a score of 0.12, whereas p. (Ile2427Thr) and p. (Lys459Asn) were ‘intolerant’ with scores of 0.2 and 0.38, respectively. No formal clinical genetic assessments were available for these individuals. The limited clinical data available are as follows: Individual EX24 with a de novo ANKRD11 VUS p. (Ile2427Thr) had microcephaly, short stature and developmental delay but did not have macrodontia or other dental abnormalities, one of the more frequently observed dysmorphic features associated with KBGS. Phenotypic information for individual EX25 p.(Lys459Asn) was even more limited; the individual is reported to have autism spectrum disorder. Individual EX26 who had a maternally inherited ANKRD11 VUS had microcephaly, developmental delay, and borderline intellectual disability, but no KBGS-specific facial dysmorphic features. Her mother, individual EX27, was assessed by a clinical geneticist who deemed her to be unaffected, although specific information was not available.
Table 2.
Predicted pathogenicity of missense ANKRD11 variants generated using Alamut
| Sample | Protein change (ANKRD11 NM013275.5) | Predicted pathogenicity | ||||
|---|---|---|---|---|---|---|
| SIFT (score) | PolyPhen-2 | MutationTaster | CADD | SVM score (%) | ||
| EX24 | p.Ile2427Thr | Deleterious | Probably damaging | Disease causing | 25.3 | 21.5 |
| EX25 | p.Lys459Asn | Deleterious | Probably damaging | Disease causing | 25.7 | 21.0 |
| EX26 | p.Thr236Met | Deleterious | Probably damaging | Disease causing | 29.4 | 68.0 |
| EX27a | p.Thr236Met | Deleterious | Probably damaging | Disease causing | 29.4 | 51.0 |
aMother of EX26
Figure 4.
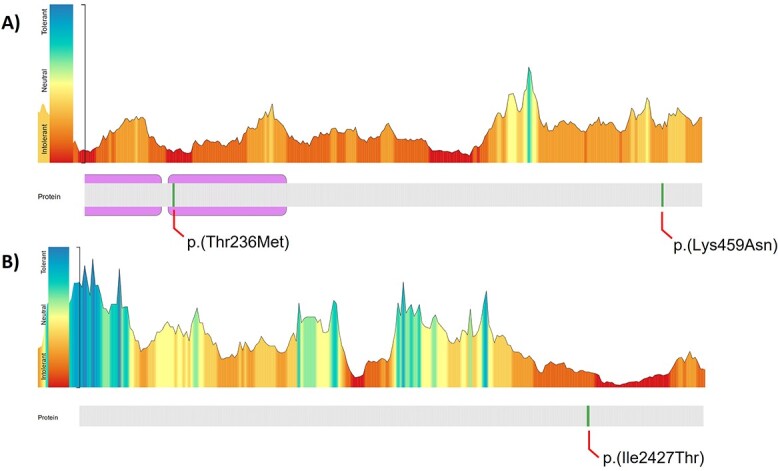
Predicting ANKRD11 protein tolerance to missense variants. The diagram on the top illustrates ANKRD11’s tolerance to missense changes landscape according to MetaDome web server. The protein structure is depicted on the bottom with ankyrin repeat domains shown. Missense variants analyzed in the present work are indicated in green. (A) Zoomed view for variants p.(Thr236Met) and p.(lys459Asn). (B) Zoomed view for p.(Ile2427Thr).
Ontology of KBG syndrome DNAm signature sites
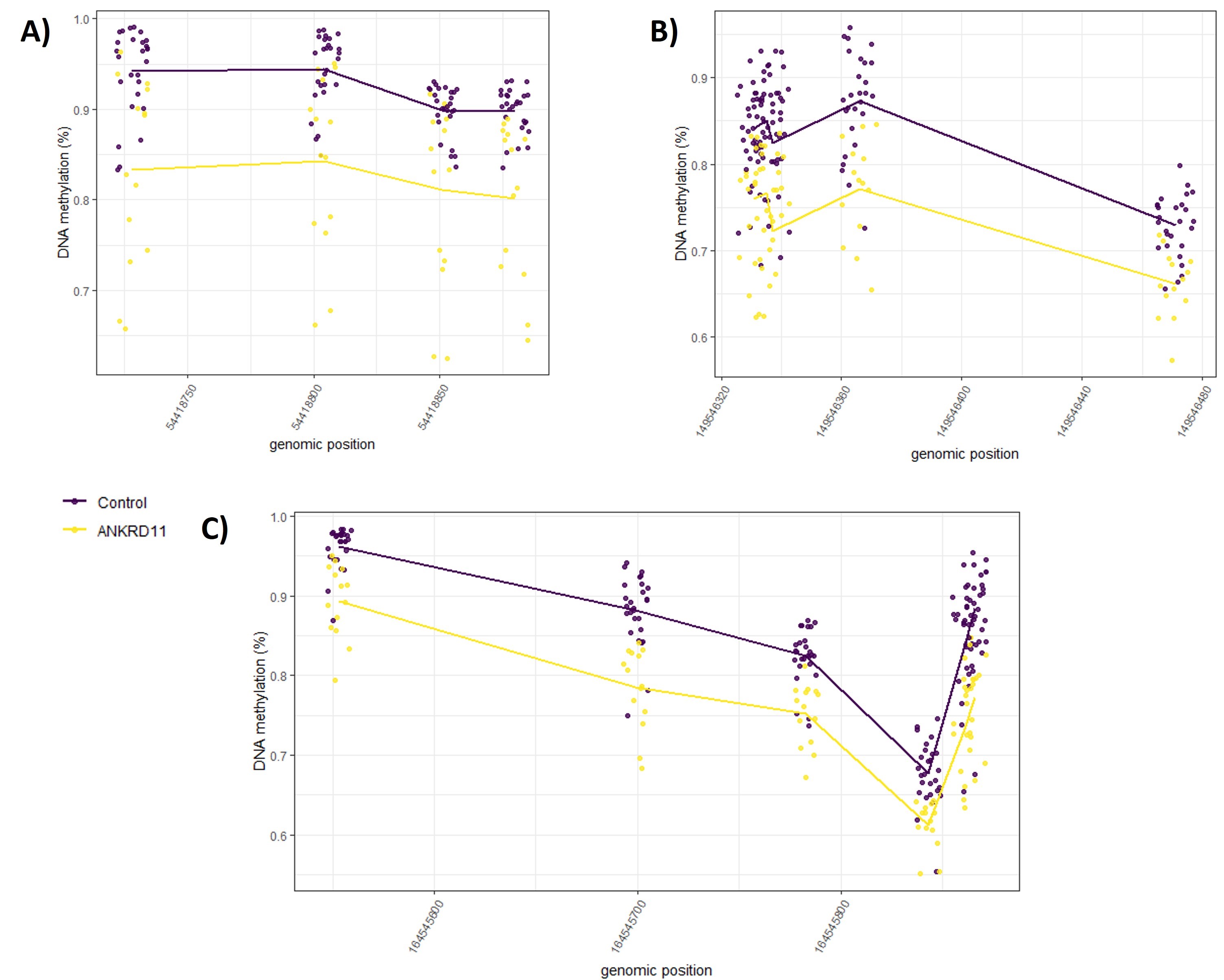
We assessed the ontology of genes overlapping CpG sites in the KBG syndrome DNAm signature using GREAT (25). Gene ontology analyses can be used to describe the role of gene targets in two biological domains: larger processes accomplished by multiple molecular functions (biological processes) and phenotypic abnormalities of human disorders that gene targets are predicted to contribute to (human phenotypes). There were 41 unique genes that overlapped 54 of the 95 signature CpG sites, with 9 genes overlapping 2 or more CpG sites (BAHCC1, EBF4, SH3BP2, C13orf26, BMP4, CDX1, PBX1, SNED1, TET1). We identified significant enrichment (FDR < 0.05, gene hits ≥2) for 28 biological processes and 27 human phenotypes (Supplementary Material, Tables S5 and S6). The top biological processes, ranked based on the number of gene hits, were related to skeletal and bone development. Key genes in these biological processes were BMP4, TBX1 and CDX1. Human phenotypes were related to skeletal abnormalities, also enriched for BMP4 and TBX1, in addition to SH3BP2 and NFIX. We plotted signature CpG sites overlapping BMP4 in addition to two CpG sites located within the same island. All four CpGs are hypomethylated in individuals with KBGS compared to controls (Supplementary Material, Fig. S3). The two additional CpGs did not meet our statistical thresholds to be included in the signature but clearly show the same methylation trends. We also plotted CpGs within CDX1 and PBX1, each overlapping two CpG sites, located within CpG islands. In the CDX1 gene, the CpGs were located within the transcriptional start site of the gene, whereas in PBX1 the CpG sites were located within the gene body. We assessed DNAm at CpG sites adjacent to the signature CpG sites located within the same islands overlapping CDX1 and PBX1. Although adjacent CpG sites did not meet statistical thresholds to be included in the signature, these sites follow the observed trend in signature CpGs and are hypomethylated in individuals with KBGS compared to typically developing controls (Supplementary Material, Fig. S3).
Discussion
We identified a unique genome-wide DNAm signature associated with truncating pathogenic variants in the ANKRD11 gene in peripheral blood of individuals with a clinical diagnosis of KBGS. The signature discovery cohort (n = 14) included individuals with truncating variants in exon 9 (the largest exon), the reported mutational hotspot for ANKRD11 (6). Variants in exon 9 are predicted to cause ANKRD11 haploinsufficiency via nonsense-mediated decay (NMD); however, a dominant negative effect has not been ruled out for variants in ANKRD11’s C-terminal repression domain 2 (26). Exon 9 encodes the first repression domain (318–611), the activation domain (1851–2145) and the beginning of the second repression domain (2369–2663), which extends to the final exon (exon 13) (2). Although there are no truncating variants in the C-terminal region encompassing repression domain 2 in our study cohort, the validation cohort (n = 9) included two individuals with 16q24.3 microdeletions encompassing ANKRD11 and one individuals with a splice site variant between exons 2 and 3. The KBGS-specific DNAm signature classified these three individuals with high pathogenicity scores, resembling DNAm profiles of individuals with truncating variants in exon 9. We have previously observed the same phenomenon in our DNAm studies of Kleefstra syndrome, where the DNAm pattern of individuals with 9q34.3 microdeletions was similar to that of individuals with single variants in the EHMT1 gene (27). These data support the hypothesis that the DNAm signatures for these copy number variants are driven by loss of function of a gene involved in epigenetic regulation. The KBGS-specific signature demonstrated >99% specificity when it was used to classify two cohorts of individuals with pathogenic variants in other chromatin modifying genes (KMT2D and EZH2). These individuals with Kabuki (n = 9) and Weaver (n = 17) syndromes, respectively, all classified as control-like.
Using a machine learning model based on the unique KBG syndrome DNAm signature, we classified the DNAm profiles of a mother–child duo who share a missense variant overlapping the ankyrin repeats domain p. (Thr236Met). The DNAm profiles of the clinically affected child had a predicted pathogenicity score of 68%, just below the range of KBGS individuals, whereas the clinically unaffected mother had a pathogenicity score of 51% and did not classify clearly with either KBGS individuals or controls. Given this intermediate score, we tested the mother for somatic mosaicism for the ANKRD11 variant and found that her two alleles were equally expressed. This does not exclude the possibility of mosaicism in the mother in tissues other than blood. This type of variable intrafamilial expression of KBGS has been reported in multiple families in which an ANKRD11 missense variant is vertically transmitted (5,7–9). Another report investigating de novo missense variants within ANKRD11’s repression domain 2 concluded that the phenotype of individuals with these variants was in line with KBGS (28). The report also confirmed these variants have a loss-of-function effect on the ANKRD11 protein. While the transcript can escape NMD, the protein is unstable and unable to carry out trans-repression activities (28). That is, missense variants in ANKRD11 have a variable impact on the protein expression and function, which may contribute to the variable phenotype observed.
It will be important to evaluate additional parent–child duos to determine whether the consistent finding of a milder phenotype in the parent versus the child represents an ascertainment bias. If so, we would predict that studies of larger cohorts of missense duos should uncover some parent–child duos in which the parent is equally or more severely affected than the child. Nonetheless, these reports on ANKRD11 missense variants suggest that caution is warranted in the interpretation and classification of these variants, particularly inherited variants. The typical pipeline for trio exome analysis, which excludes variants, found in unaffected parents should be reconsidered for ANKRD11 missense variants as the potential pathogenicity of such variants may be missed. The impact of inherited missense variants may need to be considered more generally in genome sequencing pipelines, with a recent report demonstrating phenotypic variation in the general adult population is associated with genomic variants in genes known to cause rare monogenic developmental disorders (29). Lastly, it may be informative to determine whether the severity of the KBGS phenotype is consistently reflected by the DNAm data. We have demonstrated this relationship for missense variants and DNAm in the HNRNPK gene associated with Au-Kline syndrome (30). In the future, DNAm profiling could be helpful in predicting pathogenicity and severity of the KBGS phenotype as we have shown for a missense ANKRD11 variant inherited from a clinically unaffected parent.
While the role of ANKRD11 in epigenetic regulation has not been fully characterized, studies have identified a dual function for ANKRD11 in transcriptional regulation, through protein–protein interactions with co-activators and co-repressors of NRs (1,10). NRs are DNA-binding transcription factors that control hormone-dependent gene expression in many important biological processes (31,32). Coactivators and corepressors are cofactors that mediate activation and repression functions for NRs, respectively (32). The p160 NR coactivator binds liganded NR to enhance transcriptional activation through recruitment of histone acetyltransferases (HAT), i.e. CBP and p300 (31). Conversely, co-repressors bind unliganded NR and recruit histone deacetylates (HDAC) to neutralize histone acetyltransferase activity and attenuate transcriptional activation (31). The ANKRD11 protein was initially identified to interact with RAC3 (1), a p-160 coactivator protein (33), where it can recruit and interact with HDACs to neutralize transactivation activity (1). Subsequent studies have characterized additional ANKRD11-interacting proteins such as ADA3 and p53 (10,34). The human alteration/deficiency in activation 3 protein, ADA3, is a NR coactivator and a component of the P/CAF complex (p300/CBP [CREB (cAMP-response-element binding protein)-binding protein]-associated factor), which functions to link co-activators to histone acetylation and basal transcription machinery (35). Collectively, these studies demonstrate an integral role for ANKRD11 in transcriptional regulation and chromatin modification via histone acetylation.
Chromatin modification via histone acetylation is required for development and function of the nervous, bone and skeletal systems (12,36). Therefore, more recent studies have investigated the role of ANKRD11 during the development of these systems. A study by Gallagher et al. (11) confirmed the expression of ANKRD11 in neurons and their precursors in the developing mouse cortex and human forebrain. In addition, targeted knockdown of ANKRD11 in cultured murine and human cortical precursors disrupted proliferation and neurogenesis (11). Using in vivo techniques, targeted knockdown of Ankrd11 in the developing embryonic murine cortex caused mislocalization of cortical neurons and decreased neurogenesis (11). Histone acetylation and p53 acetylation were markedly altered in Ankrd11-deficient murine embryonic cortical precursors and postnatal neurons (11,14). These studies emphasize ANKRD11’s role in neural development and histone acetylation.
To investigate ANKRD11’s role in craniofacial bone and palate development, a study by Roth et al. (37) used conditional ablation of Ankrd11 in developing murine neural crest cells, which contribute to the development of the anterior craniofacial complex. Mice with heterozygous Ankrd11 deletions displayed reduction in ossification of midfacial bone, hypoplastic palatal shelves, and delayed bone maturation and remodeling during development (37). These mice had KBGS-like craniofacial manifestations such as retrognathia and midfacial hypoplasia (37). Ontology analysis for the KBGS DNAm signature sites identified relevant biological processes to the KBG syndrome phenotypes such as bone development, skeletal development, odontogenesis of dentin-containing tooth, and endochondral ossification. A number of genes overlapping the signature are driving the enrichment of these biological processes including BMP4, CDX1 and PBX1. Studies have shown that bone morphogenetic protein 4 (BMP4) has a role in bone and skeletal development and limb patterning (38). The loss of BMP4 in murine models resulted in severe impairment in osteogenesis and differentiation of cells to mature osteoblasts (38). In the KBGS signature, there are two hypomethylated CpG sites in a CpG island located in BMP4’s gene body (Supplementary Material, Fig. S3). Homeobox transcription factors CDX1 and PBX1 also have critical functions in bone and skeletal development, particularly the proliferation and differentiation of osteoblasts and chondrocytes, respectively (39,40). A study in osteoblast cell culture showed that PBX1 is associated with histone deacetylases and attenuates activations of osteoblast-related genes important for osteogenesis (41). In the KBGS DNAm signature, CpG sites overlapping CDX1 and PBX1 are also located within CpG islands, and all were hypomethylated (Supplementary Material, Fig. S3).
While the exact mechanisms by which ANKRD11 haploinsufficiency triggers DNAm alterations in these genes are not fully understood, many of the signature genes have a direct role in skeletal patterning and bone development, and in some instances, their expression is regulated by histone acetylation, e.g. histone 3 lysine 9 acetylation of the BMP4 promotor increases expression (42). In addition to DNAm signature sites overlapping genes implicated in bone and skeletal development, there are sites that also overlap genes implicated in autism, including CNOT3, GAS7 and TET1. While we do not yet understand how differential methylation of these genes impacts their function and expression in KBGS, all three are highly expressed in brain tissues, implicated in neuronal development and identified as candidate autism genes (43–46).
In conclusion, we report a unique DNAm signature that is highly sensitive and specific for ANKRD11 haploinsufficiency and KBG syndrome. Gene targets in the signature have a direct role in bone development and transcriptional regulation. The DNAm signature classified a mother–child duo with a VUS in the ankyrin repeats domain as KBGS-like. The different levels of DNAm disruption identified in the child with KBGS and the unaffected mother provide further insights into the mechanisms underlying the clinical variability observed in multiple families. For future studies, additional functional studies of missense variants in ANKRD11 paired with DNAm studies will likely contribute to our understanding of molecular mechanisms underpinning this disease. In addition, in vitro models of induced pluripotent stem cells derived from patient fibroblast, combined with multi-omics approaches to measure histone acetylation and gene expression, will further elucidate molecular and epigenetic changes associated with ANKRD11 variants causing KBG syndrome.
Materials and Methods
Research participants
Informed consent was obtained from all research participants and/or their guardian(s) according to the protocol approved by the Research Ethics Board of the Hospital for Sick Children (REB# 1000038847). Individuals were recruited through our International Epigenetic Consortium (IEC), which includes both local and international collaborators. Individuals with ANKRD11 variants were also identified through MSSNG (46), one of the largest whole-genome sequencing project for Autism Spectrum Disorder and SFARI (Simons Foundation Autism Research Initiative) (47) and the Simons Simplex Collection using the Genotypes and Phenotypes in Families (GPF) tool (https://gpf.sfari.org/). Following recruitment, our cohort consisted of 25 individuals with ANKRD11 variants and 2 individuals with 16q24.3 microdeletions. The patient demographics, clinical phenotype and variant information are presented in Supplementary Material, Table S1. We divided individuals with classic features of KBG and pathogenic truncating variants in ANKRD11 (n = 23) into discovery (n = 14) and validation (n = 9) (including 2 microdeletion samples) cohorts. The remaining ANKRD11 cohort included four (n = 4) individuals with a VUS that were included in order to classify them as KBGS- or control-like using the generated KBG DNAm signature. Banked DNA samples from age- and sex-matched typically developing individuals (n = 150) were included as control subjects (Supplementary Material, Table S2). These individuals were recruited from the Hospital for Sick Children and the Province of Ontario Neurodevelopmental Disorders (POND) Network and were deemed typically developing (Dr Gregory Hanna). ‘Typically developing’ was defined as healthy and developmentally normal by using formal cognitive/behavioral assessments (POND) or via physician/parental screening questionnaires (SickKids).
DNAm profiling and data processing
Genomic DNA was extracted from peripheral blood and bisulfite converted using the EpiTect PLUS Bisulfite Kit (QIAGEN, Germany). Sodium bisulfite converted DNA was then hybridized to the Illumina Infinium Human Methylation EPIC BeadChip to interrogate more than 850 000 CpG sites in the human genome at The Center for Applied Genomics (TCAG), Hospital for Sick Children Research Institute, Toronto, Ontario, Canada. Samples were run in two batches. On each microarray chip, cases and controls were randomly assigned a chip position. The minfi Bioconductor package in R was used to preprocess data including quality control, Illumina normalization and background subtraction, followed by extraction of beta (β) values as previously described (48). Standard quality control metrics in minfi were used, including median intensity QC plots, density plots and control probe plots. Probes with detection flaws (n = 816), probes near SNPs with minor allele frequencies above 1% (n = 29 958), cross-reactive probes (n = 41 975) (49), probes with raw beta of 0 or 1 in >0.25% of samples (n = 247), non-CpG probes (n = 2925) and X and Y chromosome probes (n = 19 627) were removed, resulting in a total of n = 774 245 probes remained for differential methylation analysis.
DNAm signature derivation
To assess DNAm patterns, we identified differentially methylated sites in whole blood derived DNA from n = 14 individuals with truncating variants in ANKRD11 and a clinical diagnosis of KBGS compared to 28 sex- and age-matched typically developing controls (Supplementary Material, Tables S1 and S2). For all samples, we applied the blood cell-type proportion estimation tool in minfi based on Illumina EPIC array data (50). We identified differentially methylated CpG sites using Limma (51) regression modelling with age, sex, cell type proportions, and one surrogate variable, to account for latent variables outside of our known and measured covariates. The surrogate variable was identified using the ‘sva’ package in R (52). The thresholds for differentially methylated CpG sites were Benjamini–Hochberg adjusted P-value <0.05 and a |Δβ| > 0.10. Δβ represents the difference in average DNAm (β) between groups. PCA and hierarchical clustering were generated using Qlucore Omics Explorer V3.7 (QOE, www.qlucore.com).
Machine learning classification models
We developed a machine learning model using the KBGS DNAm signature. Using the R package ‘caret’, CpG sites with correlations equal to or greater than 90% to other signature CpGs were removed as previously described (16). A SVM model, trained on signature CpG sites, was set to ‘probability’ mode to generate SVM scores ranging between 0 and 1 (0–100%), classifying variants as ‘KBGS-like’ (score >0.5) or ‘control-like’ (score <0.5). To test model specificity, EPIC array data from additional typically developing controls (n = 150) were scored. To test model sensitivity, we classified n = 9 validation samples, from individuals with a confirmed truncating variants in ANKRD11 (n = 7) or a 16q24.3 microdeletion that includes the ANKRD11 gene (n = 2) and a clinical diagnosis of KBGS. Lastly, we classified individuals with Kabuki (n = 9) and Weaver (n = 17) syndromes, carrying pathogenic variants in KMT2D and EZH2, respectively, two genes encoding epigenetic regulators and implicated in NDDs.
Gene Ontology analysis
The list of CpG positions comprising the DNAm signature was submitted to GREAT (25) (Genomic Regions Enrichment of Annotations Tool) for Gene Ontology (GO) enrichment analysis. Enrichment of each GO term within the gene list was calculated using a foreground/background hypergeometric test over genomic regions, using the set of CpG sites after minfi probe quality control (n = 774 245) as a background set. Overlapping genes were mapped using default GREAT settings with the following exceptions: the cut-off to annotate a CpG as overlapping with a gene (‘distal gene mapping’ setting) was set to 10 kb, and only enriched terms with two or more gene hits and FDR < 0.05 were reported.
Pyrosequencing
Genotyping was performed using quantitative pyrosequencing for ANKRD11 variant: GenBank: NM_013275.5, c.707G>A (p.Thr236Met) in a mother–child pair (EX26 and EX27). Targeted assay was designed using the PyroMark Assay Design Software 2.0 (QIAGEN). Primer set sequences consisted of forward primer 50-GGGATGCCAACCTTGTAGTGC-30; reverse primer 50-CGCCAGGGTTTTCCCAGTCACGACGCAGGTGCGGAGGTGAAC-30 and sequencing primer 50-AGCGTCGTGCAAAGG-30. The amplification protocol was developed using a biotinylated universal primer approach. Regions of interest were amplified by PCR and pyrosequencing was carried out using the PyroMark Q24 pyrosequencer (QIAGEN) according to the manufacturer’s protocol. Output data were analyzed using PyroMark Q24 Software (QIAGEN), which calculates the allelic percentage for each allele, allowing quantitative comparisons.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We are grateful to all the study participants and their families and the many clinicians who recruited them into this study. We acknowledge the administrative assistance of Khadine Wiltshire, and the technical assistance of Youliang Lou and Chunhua Zhao. Thank you as well to Dr Greg Hanna for contributing blood DNA samples from typically developing control individuals who had undergone cognitive/behavioral assessments. The authors also wish to acknowledge the resources of MSSNG (www.mss.ng), Autism Speaks and The Centre for Applied Genomics at The Hospital for Sick Children, Toronto, Canada. We thank the participating families for their time and contributions to this database, as well as the generosity of the donors who supported this program.
Conflicts of Interest statement: The authors have no conflicts of interest to declare.
Contributor Information
Zain Awamleh, Genetics and Genome Biology Program, Research Institute, The Hospital for Sick Children, Toronto, ON M5G 1X8, Canada.
Sanaa Choufani, Genetics and Genome Biology Program, Research Institute, The Hospital for Sick Children, Toronto, ON M5G 1X8, Canada.
Cheryl Cytrynbaum, Division of Clinical and Metabolic Genetics, The Hospital for Sick Children, Toronto, ON M5G 1X8, Canada.
Fowzan S Alkuraya, Department of Translational Genomics, Center for Genomic Medicine, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia.
Stephen Scherer, Genetics and Genome Biology Program, Research Institute, The Hospital for Sick Children, Toronto, ON M5G 1X8, Canada; Department of Molecular Genetics, University of Toronto, Toronto, ON M5S 1A8, Canada; Institute of Medical Sciences, University of Toronto, Toronto, ON M5S 1A8, Canada.
Sofia Fernandes, Medical Genetics Unit, Hospital Pediátrico, Centro Hospitalar e Universitário de Coimbra, EPE, Coimbra, Portugal; Familial Risk Clinic, Instituto Português de Oncologia de Lisboa Francisco Gentil, Lisbon, Portugal.
Catarina Rosas, Medical Genetics Unit, Hospital Pediátrico, Centro Hospitalar e Universitário de Coimbra, EPE, Coimbra, Portugal.
Pedro Louro, Medical Genetics Unit, Hospital Pediátrico, Centro Hospitalar e Universitário de Coimbra, EPE, Coimbra, Portugal.
Patricia Dias, Serviço de Genética Médica, Departamento de Pediatria, Centro Hospitalar Lisboa Norte, Hospital de Santa Maria, EPE, Lisbon, Portugal.
Mariana Tomásio Neves, Serviço de Genética Médica, Departamento de Pediatria, Centro Hospitalar Lisboa Norte, Hospital de Santa Maria, EPE, Lisbon, Portugal.
Sérgio B Sousa, Medical Genetics Unit, Hospital Pediátrico, Centro Hospitalar e Universitário de Coimbra, EPE, Coimbra, Portugal.
Rosanna Weksberg, Genetics and Genome Biology Program, Research Institute, The Hospital for Sick Children, Toronto, ON M5G 1X8, Canada; Division of Clinical and Metabolic Genetics, The Hospital for Sick Children, Toronto, ON M5G 1X8, Canada; Department of Molecular Genetics, University of Toronto, Toronto, ON M5S 1A8, Canada; Institute of Medical Sciences, University of Toronto, Toronto, ON M5S 1A8, Canada; Department of Paediatrics, University of Toronto, Toronto, ON M5S 1A8, Canada.
Data availability
The datasets generated during the current study are not publicly available due to institutional ethical restrictions but are available from the corresponding author on reasonable request.
Funding
Canadian Institutes of Health Research (CIHR) (grant PJT-178315 to R.W.); Ontario Brain Institute (Province of Ontario Neurodevelopmental Disorders (POND) network (grant IDS11-02 to R.W.); Simons Foundation Autism Research Initiative (887172 for R.W.).
Ethical approval
Informed consent was obtained from all research participants and/or their guardian(s) according to the protocol approved by the Research Ethics Board of the Hospital for Sick Children (REB# 1000038847).
References
- 1. Zhang, A., Yeung, P.L., Li, C.W., Tsai, S.C., Dinh, G.K., Wu, X., Li, H. and Chen, J.D. (2004) Identification of a novel family of ankyrin repeats containing cofactors for p160 nuclear receptor coactivators. J. Biol. Chem., 279, 33799–33805. [DOI] [PubMed] [Google Scholar]
- 2. Zhang, A., Li, C.W. and Chen, J.D. (2007) Characterization of transcriptional regulatory domains of ankyrin repeat cofactor-1. Biochem. Biophys. Res. Commun., 358, 1034–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Herrmann, J., Pallister, P.D., Tiddy, W. and Opitz, J.M. (1975) The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig. Artic. Ser., 11, 7–18. [PubMed] [Google Scholar]
- 4. Sirmaci, A., Spiliopoulos, M., Brancati, F., Powell, E., Duman, D., Abrams, A., Bademci, G., Agolini, E., Guo, S., Konuk, B.et al. (2011) Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet., 89, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Low, K., Ashraf, T., Canham, N., Clayton-Smith, J., Deshpande, C., Donaldson, A., Fisher, R., Flinter, F., Foulds, N., Fryer, A.et al. (2016) Clinical and genetic aspects of KBG syndrome. Am. J. Med. Genet. A, 170, 2835–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Morel Swols, D. and Tekin, M. (2018) Adam MP, Ardinger H., Pagon RA (ed.). KBG Syndrome. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022. GeneReviews, in press. [PubMed] [Google Scholar]
- 7. Gao, F., Zhao, X., Cao, B., Fan, X., Li, X., Li, L., Sui, S., Su, Z. and Gong, C. (2022) Genetic and phenotypic Spectrum of KBG syndrome: a report of 13 new Chinese cases and a review of the literature. J. Pers. Med., 12, 407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parenti, I., Mallozzi, M.B., Hüning, I., Gervasini, C., Kuechler, A., Agolini, E., Albrecht, B., Baquero-Montoya, C., Bohring, A., Bramswig, N.C.et al. (2021) ANKRD11 variants: KBG syndrome and beyond. Clin. Genet., 100, 187–200. [DOI] [PubMed] [Google Scholar]
- 9. Bestetti, I., Crippa, M., Sironi, A., Tumiatti, F., Masciadri, M., Smeland, M.F., Naik, S., Murch, O., Bonati, M.T., Spano, A.et al. (2022) Expanding the molecular spectrum of ANKRD11 gene defects in 33 patients with a clinical presentation of KBG syndrome. Int. J. Mol. Sci., 23, 5912–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li, C.W., Dinh, G.K., Zhang, A. and Chen, J.D. (2008) Ankyrin repeats-containing cofactors interact with ADA3 and modulate its co-activator function. Biochem. J., 413, 349–357. [DOI] [PubMed] [Google Scholar]
- 11. Gallagher, D., Voronova, A., Zander, M.A., Cancino, G.I., Bramall, A., Krause, M.P., Abad, C., Tekin, M., Neilsen, P.M., Callen, D.F.et al. (2015) Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Dev. Cell, 32, 31–42. [DOI] [PubMed] [Google Scholar]
- 12. Cho, B., Kim, H.J., Kim, H. and Sun, W. (2011) Changes in the histone acetylation patterns during the development of the nervous system. Exp. Neurobiol., 20, 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mossink, B., Negwer, M., Schubert, D. and Nadif Kasri, N. (2021) The emerging role of chromatin remodelers in neurodevelopmental disorders: a developmental perspective. Cell. Mol. Life Sci., 78, 2517–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ka, M. and Kim, W.Y. (2018) ANKRD11 associated with intellectual disability and autism regulates dendrite differentiation via the BDNF/TrkB signaling pathway. Neurobiol. Dis., 111, 138–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choufani, S., Cytrynbaum, C., Chung, B.H., Turinsky, A.L., Grafodatskaya, D., Chen, Y.A., Cohen, A.S., Dupuis, L., Butcher, D.T., Siu, M.T.et al. (2015) NSD1 mutations generate a genome-wide DNA methylation signature. Nat. Commun., 6, 10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Butcher, D.T., Cytrynbaum, C., Turinsky, A.L., Siu, M.T., Inbar-Feigenberg, M., Mendoza-Londono, R., Chitayat, D., Walker, S., Machado, J., Caluseriu, O.et al. (2017) CHARGE and kabuki syndromes: gene-specific DNA methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. Am. J. Hum. Genet., 100, 773–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chater-Diehl, E., Ejaz, R., Cytrynbaum, C., Siu, M.T., Turinsky, A., Choufani, S., Goodman, S.J., Abdul-Rahman, O., Bedford, M., Dorrani, N.et al. (2019) New insights into DNA methylation signatures: SMARCA2 variants in Nicolaides-Baraitser syndrome. BMC Med. Genet., 12, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Siu, M.T., Butcher, D.T., Turinsky, A.L., Cytrynbaum, C., Stavropoulos, D.J., Walker, S., Caluseriu, O., Carter, M., Lou, Y., Nicolson, R.et al. (2019) Functional DNA methylation signatures for autism spectrum disorder genomic risk loci: 16p11.2 deletions and CHD8 variants. Epigenetics, 11, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Choufani, S., Gibson, W.T., Turinsky, A.L., Chung, B.H.Y., Wang, T., Garg, K., Vitriolo, A., Cohen, A.S.A., Cyrus, S., Goodman, S.et al. (2020) DNA methylation signature for EZH2 functionally classifies sequence variants in three PRC2 complex genes. Am. J. Hum. Genet., 106, 596–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Courraud, J., Chater-Diehl, E., Durand, B., Vincent, M., Del Mar Muniz Moreno, M., Boujelbene, I., Drouot, N., Genschik, L., Schaefer, E., Nizon, M.et al. (2021) Integrative approach to interpret DYRK1A variants, leading to a frequent neurodevelopmental disorder. Genet. Med., 23, 2150–2159. [DOI] [PubMed] [Google Scholar]
- 21. Rots, D., Chater-Diehl, E., Dingemans, A.J.M., Goodman, S.J., Siu, M.T., Cytrynbaum, C., Choufani, S., Hoang, N., Walker, S., Awamleh, Z.et al. (2021) Truncating SRCAP variants outside the Floating-Harbor syndrome locus cause a distinct neurodevelopmental disorder with a specific DNA methylation signature. Am. J. Hum. Genet., 108, 1053–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., Grody, W.W., Hegde, M., Lyon, E., Spector, E.et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med., 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou, X., Edmonson, M.N., Wilkinson, M.R., Patel, A., Wu, G., Liu, Y., Li, Y., Zhang, Z., Rusch, M.C., Parker, M.et al. (2016) Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet., 48, 4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wiel, L., Baakman, C., Gilissen, D., Veltman, J.A., Vriend, G. and Gilissen, C. (2019) MetaDome: pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum. Mutat., 40, 1030–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McLean, C.Y., Bristor, D., Hiller, M., Clarke, S.L., Schaar, B.T., Lowe, C.B., Wenger, A.M. and Bejerano, G. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol., 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walz, K., Cohen, D., Neilsen, P.M., Foster, J., 2nd, Brancati, F., Demir, K., Fisher, R., Moffat, M., Verbeek, N.E., Bjørgo, K.et al. (2015) Characterization of ANKRD11 mutations in humans and mice related to KBG syndrome. Hum. Genet., 134, 181–190. [DOI] [PubMed] [Google Scholar]
- 27. Goodman, S.J., Cytrynbaum, C., Chung, B.H.-Y., Chater-Diehl, E., Aziz, C., Turinsky, A.L., Kellam, B., Keller, M., Ko, J.M., Caluseriu, O.et al. (2020) EHMT1 pathogenic variants and 9q34.3 microdeletions share altered DNA methylation patterns in patients with Kleefstra syndrome. J. Transl. Genet. Genom., 4, 144–158. [Google Scholar]
- 28. de Boer, E., Ockeloen, C.W., Kampen, R.A., Hampstead, J.E., Dingemans, A.J.M., Rots, D., Lütje, L., Ashraf, T., Baker, R., Barat-Houari, M.et al. (2022) Missense variants in ANKRD11 cause KBG syndrome by impairment of stability or transcriptional activity of the encoded protein. Genet. Med., 24, 2051–2064. [DOI] [PubMed] [Google Scholar]
- 29. Kingdom, R., Tuke, M., Wood, A., Beaumont, R.N., Frayling, T.M., Weedon, M.N. and Wright, C.F. (2022) Rare genetic variants in genes and loci linked to dominant monogenic developmental disorders cause milder related phenotypes in the general population. Am. J. Hum. Genet., 109, 1308–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Choufani, S., McNiven, V., Cytrynbaum, C., Jangjoo, M., Adam, M.P., Bjornsson, H.T., Harris, J., Dyment, D.A., Graham, G.E., Nezarati, M.M.et al. (2022) An HNRNPK-specific DNA methylation signature makes sense of missense variants and expands the phenotypic spectrum of au-Kline syndrome. Am. J. Hum. Genet., 109, 1867–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Millard, C.J., Watson, P.J., Fairall, L. and Schwabe, J.W. (2013) An evolving understanding of nuclear receptor coregulator proteins. J. Mol. Endocrinol., 51, T23–T36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McKenna, N.J., Lanz, R.B. and O'Malley, B.W. (1999) Nuclear receptor coregulators: cellular and molecular biology. Endocr. Rev., 20, 321–344. [DOI] [PubMed] [Google Scholar]
- 33. Li, H., Gomes, P.J. and Chen, J.D. (1997) RAC3, a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF2. Proc. Natl. Acad. Sci. U. S. A., 94, 8479–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Neilsen, P.M., Cheney, K.M., Li, C.W., Chen, J.D., Cawrse, J.E., Schulz, R.B., Powell, J.A., Kumar, R. and Callen, D.F. (2008) Identification of ANKRD11 as a p53 coactivator. J. Cell Sci., 121, 3541–3552. [DOI] [PubMed] [Google Scholar]
- 35. Balasubramanian, R., Pray-Grant, M.G., Selleck, W., Grant, P.A. and Tan, S. (2002) Role of the Ada2 and Ada3 transcriptional coactivators in histone acetylation. J. Biol. Chem., 277, 7989–7995. [DOI] [PubMed] [Google Scholar]
- 36. Li, K., Han, J. and Wang, Z. (2021) Histone modifications centric-regulation in osteogenic differentiation. Cell Death Dis., 7, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roth, D.M., Baddam, P., Lin, H., Vidal-García, M., Aponte, J.D., De Souza, S.T., Godziuk, D., Watson, A.E.S., Footz, T., Schachter, N.F.et al. (2021) The chromatin regulator Ankrd11 controls palate and cranial bone development. Front. Cell Dev. Biol., 9, 645386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bandyopadhyay, A., Tsuji, K., Cox, K., Harfe, B.D., Rosen, V. and Tabin, C.J. (2006) Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet., 2, e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Subramanian, V., Meyer, B.I. and Gruss, P. (1995) Disruption of the murine homeobox gene Cdx1 affects axial skeletal identities by altering the mesodermal expression domains of Hox genes. Cell, 83, 641–653. [DOI] [PubMed] [Google Scholar]
- 40. Selleri, L., Depew, M.J., Jacobs, Y., Chanda, S.K., Tsang, K.Y., Cheah, K.S., Rubenstein, J.L., O'Gorman, S. and Cleary, M.L. (2001) Requirement for Pbx1 in skeletal patterning and programming chondrocyte proliferation and differentiation. Development, 128, 3543–3557. [DOI] [PubMed] [Google Scholar]
- 41. Gordon, J.A., Hassan, M.Q., Saini, S., Montecino, M., vanWijnen, A.J., Stein, G.S., Stein, J.L. and Lian, J.B. (2010) Pbx1 represses osteoblastogenesis by blocking Hoxa10-mediated recruitment of chromatin remodeling factors. Mol. Cell. Biol., 30, 3531–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang, P., Liu, Y., Jin, C., Zhang, M., Lv, L., Zhang, X., Liu, H. and Zhou, Y. (2016) Histone H3K9 acetyltransferase PCAF is essential for osteogenic differentiation through bone morphogenetic protein signaling and may be involved in osteoporosis. Stem Cells, 34, 2332–2341. [DOI] [PubMed] [Google Scholar]
- 43. Deciphering Developmental Disorders Study (2017) Prevalence and architecture of de novo mutations in developmental disorders. Nature, 542, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Garbett, K., Ebert, P.J., Mitchell, A., Lintas, C., Manzi, B., Mirnics, K. and Persico, A.M. (2008) Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol. Dis., 30, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vogel Ciernia, A. and LaSalle, J. (2016) The landscape of DNA methylation amid a perfect storm of autism aetiologies. Nat. Rev. Neurosci., 17, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yuen, R.K.C., Merico, D., Bookman, M., Howe, J.L., Thiruvahindrapuram, B., Patel, R.V., Whitney, J., Deflaux, N., Bingham, J., Wang, Z.et al. (2017) Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci., 20, 602–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fischbach, G.D. and Lord, C. (2010) The Simons simplex collection: a resource for identification of autism genetic risk factors. Neuron, 68, 192–195. [DOI] [PubMed] [Google Scholar]
- 48. Aryee, M.J., Jaffe, A.E., Corrada-Bravo, H., Ladd-Acosta, C., Feinberg, A.P., Hansen, K.D. and Irizarry, R.A. (2014) Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics, 30, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen, Y.A., Choufani, S., Grafodatskaya, D., Butcher, D.T., Ferreira, J.C. and Weksberg, R. (2012) Cross-reactive DNA microarray probes lead to false discovery of autosomal sex-associated DNA methylation. Am. J. Hum. Genet., 91, 762–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Horvath, S. (2013) DNA methylation age of human tissues and cell types. Genome Biol., 14, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ritchie, M.E., Phipson, B., Wu, D., Hu, Y., Law, C.W., Shi, W. and Smyth, G.K. (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res., 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leek, J.T., Johnson, W.E., Parker, H.S., Jaffe, A.E. and Storey, J.D. (2012) The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics, 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are not publicly available due to institutional ethical restrictions but are available from the corresponding author on reasonable request.