

Abstract
Myotonic dystrophy type 1 (DM1) is a multi-systemic disorder caused by expansion of CTG microsatellite repeats within DMPK. The most severe form, congenital myotonic dystrophy (CDM), has symptom onset at birth due to large intergenerational repeat expansions. Despite a common mutation, CDM individuals present with a distinct clinical phenotype and absence of common DM1 symptoms. Given the clinical divergence, it is unknown if the hallmark of DM1 pathology, dysregulation of alternative splicing (AS) due to sequestration of MBNL proteins within toxic CUG repeat RNAs, contributes to disease throughout pediatric development. To evaluate global transcriptomic dysregulation, RNA-seq was performed on 36 CDM skeletal muscle biopsies ages 2 weeks to 16 years, including two longitudinal samples. Fifty DM1 and adult/pediatric controls were also sequenced as comparative groups. Despite a large CTG expansion and shared age of onset, CDM individuals presented with a heterogenous, MBNL-dependent mis-splicing signature. Estimation of intracellular MBNL concentrations from splicing responses of select events correlated with total spliceopathy and revealed a distinct, triphasic pattern of AS dysregulation across pediatric development. CDM infants (< 2 years) possess severe mis-splicing that significantly improves in early childhood (2–8 years) independent of sex or CTG repeat load. Adolescent individuals (8–16 years) stratified into two populations with a full range of global splicing dysregulation. DMPK expression changes correlated with alterations in splicing severity during development. This study reveals the complex dynamics of the CDM muscle transcriptome and provides insights into new therapeutic strategies, timing of therapeutic intervention, and biomarker development.
Introduction
Myotonic dystrophy type 1 (DM1) is a progressive, multi-systemic disorder caused by the expansion of CTG trinucleotide repeats within the 3′ untranslated region (UTR) of the dystrophia myotonia protein kinase (DMPK) gene (1–3). It is an autosomal dominantly inherited disorder and the most common form of adult-onset muscular dystrophy, with a prevalence of 1 in 2100 (4). While affected individuals present with a highly dynamic phenotypic profile, common symptoms include myotonia, progressive distal muscle weakness, cardiac conduction defects, cataracts, and central nervous system impairment (5). Adult-onset DM1 is defined as the onset of symptoms after the age of 10 years (6). In approximately 10–20% of individuals with DM1, symptomatic onset begins at birth, a classification known as congenital myotonic dystrophy (CDM) (7). The occurrence of CDM is commonly associated with intergenerational, large-scale repeat expansions from the maternal allele (8–11). Despite a shared genetic cause of disease, infants with CDM present with a distinct clinical phenotype characterized by respiratory failure, clubfoot, feeding difficulties, and hypotonia in the first 72 hours of life (12–14). In contrast to adult-onset DM1 where symptoms are persistently progressive over years, children with CDM have a period of natural improvement in their motor function and rarely develop myotonia until adolescence (13,15). These divergent phenotypes require an investigation of similarities into the underlying pathophysiology.
There is strong evidence to support that a major pathogenic event in DM1 is widespread alterations in RNA metabolism due to sequestration of the muscleblind-like (MBNL) family of RNA-binding proteins (RBP) with toxic CUG repeat RNAs transcribed from the disease locus (16,17). As a critical regulator of fetal to adult mRNA isoform transitions, depletion of MBNL from the nucleoplasm has been shown to lead to global perturbations in alternative splicing (AS) with reversion of transcripts to the fetal isoform in affected individuals (18–22). Mis-splicing of specific events has been directly linked to DM1 symptoms, including CLCN1 and myotonia (23,24), SCN5A and cardiac arrhythmias (25), and BIN1 and muscle weakness (26). Although event dependent, the severity of MBNL-dependent mis-splicing correlates with muscle strength and has been utilized for biomarker development to assess disease severity (27–30).
CDM phenotypic presentation is thought to, in part, be caused by severe global spliceopathy occurring early in tissue development (31,32). Previous work has provided evidence that MBNL-dependent RNA splicing is disrupted in CDM. Mis-splicing of disease-associated events, including BIN1, has been observed in myoblasts derived from tissues with CTG repeat lengths consistent with the development of CDM (26,33). Severe mis-splicing of disease-associated events has also been observed in CDM skeletal muscle autopsies under 15 months of age or from aborted fetuses characterized by high repeat lengths (31,32). The use of postmortem tissue may skew data to reflect those children most severely affected and precludes longitudinal assessment of transcriptomic changes through the dynamic phenotypic progression observed in CDM.
To investigate transcriptomic dysregulation in children with CDM, we conducted RNA sequencing (RNA-seq) on skeletal muscle biopsies collected from individuals ranging from 2 weeks to 16 years of age. Using this approach, we identified a MBNL-dependent splicing signature shared by CDM and DM1. Additionally, we captured and described the complex heterogeneity of transcriptomic dysregulation in CDM individuals across pediatric development and defined distinct populations of individuals with CDM based upon age and overall splicing dysregulation. This collective analysis redefines our previous understanding of molecular perturbations in these individuals and provides a critical foundation for future biomarker and therapeutic discovery efforts targeted to the CDM population.
Results
Skeletal muscle biopsies from a large collection of CDM and adult-onset DM1 individuals were subjected to RNA-seq
To gain insight on the extent of transcriptomic dysregulation in CDM, we performed total RNA-seq on 36 skeletal muscle biopsies collected from CDM individuals, the largest group of samples sequenced to date. This sample set consists of an even distribution of sex and age ranging from 2 weeks to 16 years (Fig. 1A, Supplementary Material, Table S1). In addition, 22 adult biopsies from individuals with adult-onset DM1, 7 unaffected adult control (AdCo) and 21 pediatric control (PeCo) biopsies were subjected to RNA-seq as comparative groups (Fig. 1A). This dataset also contains two individuals with CDM that provided repeat biopsies at two distinct ages: the first at 2 weeks and 8 years (CDM-1), the second at 12 and 16 years (CDM-32). These sample pairs provided a unique opportunity to assess potential changes in the CDM transcriptome and related disease progression across pediatric development. Total RNA from all samples (n = 86) was sequenced to a depth of at least 50 M reads, with most samples possessing >70 M reads, facilitating in-depth quantitative analysis of both AS variation and differential gene expression within and between sample groups.
Figure 1.

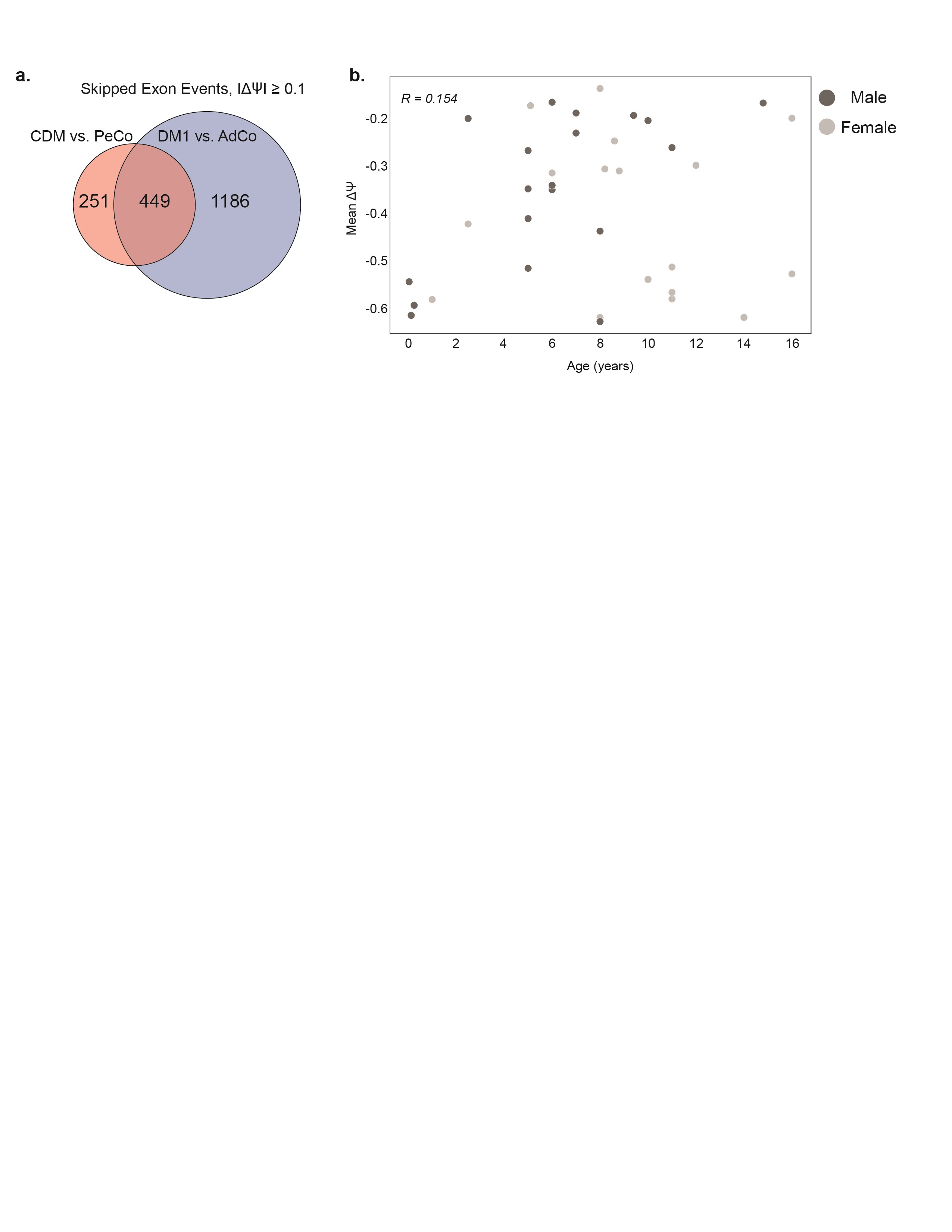
Analysis of alternative splicing dysregulation in both adult-onset and congenital myotonic dystrophy muscle biopsy transcriptomes compared to unaffected control samples. (A) List of muscle biopsy samples subjected to RNA-seq, including CDM, DM1, AdCo, and PeCo sample size, range of age at biopsy, and sex distribution. (B) Principal component analysis of skipped exon (SE) events significantly mis-spliced between affected (CDM and DM1) and unaffected (PeCo and AdCo) groups where |ΔΨ| ≥ 0.1, FDR ≤ 0.05. (C) Heatmap displaying estimated percent spliced in (PSI, Ψ) of top 50 significantly dysregulated SE events between affected and unaffected groups. Both rows (SE events) and columns (individual samples) were subjected to hierarchical clustering. Sample group and sex are annotated above the heatmap and sample age in years is listed below. CDM repeat biopsies are annotated as * (CDM-1) and ** (CDM-32). (D) Ψ values for specific skipped exon events in all sample groups. (E) Heatmap showing enrichment of k-mers (k = 4) around the 274 most significantly dysregulated skipped exons (Affected versus Unaffected, |ΔΨ| ≥ 0.2, FDR ≤ 0.05). Columns from left to right denote the intronic region from +1 to +300 bp and −300 bp to −1 bp of the upstream intron, the skipped exon itself, and the intronic region from +1 to +300 bp and −300 to −1 bp of the downstream intron.
CDM and DM1 patients share a heterogenous, MBNL-dependent mis-splicing signature
Given extensive research defining global, MBNL-dependent AS dysregulation, specifically in adult-onset DM1 (28,29,34), we first sought to investigate the extent to which this disease-specific splicing signature is shared by individuals with CDM. We quantified percent spliced in (PSI or Ψ) values for skipped exon (SE) events between affected (CDM and DM1) and unaffected controls (AdCo and PeCo). Significantly mis-spliced events (|ΔΨ| ≥ 0.1, false discovery rate (FDR) ≤ 0.05, n = 967, Supplementary Material, Table S2) were used to perform principal component analysis, and all samples were plotted against the first two principal components (Fig. 1B). As expected, both AdCo and PeCo samples clustered tightly together. In contrast, both CDM and DM1 samples were distributed across the first principal component which accounted for a significant majority of the sample variance (44.9%). The observed distribution pattern for adults with DM1 was consistent to that observed for a separate DM1 cohort (29). CDM individuals were similarly distributed across the sample population with a distinct group clustering near controls, while others were dispersed amongst the DM1 samples.
This unexpected heterogeneity of AS dysregulation in CDM was further demonstrated by visualization of a panel of the 50 SE events most differentially spliced between affected and control groups (Fig. 1C, Supplementary Material, Table S3). While AdCo and PeCo showed minimal variability in Ψ, individuals with DM1 possessed a range of splicing dysregulation, whereby a subset of SE events were only moderately dysregulated in some individuals, while others were mis-spliced across nearly all samples. Surprisingly, the same was not true of CDM; while some individuals showed similar patterns of AS dysregulation to DM1, a large cohort of CDM samples possessed minimal splicing dysregulation. This included common disease-associated, MBNL-dependent events such as INSR e11 (35–37), CLCN1 e7a (38), NFIX e7 (39),and ATP2A1 e22 (40,41) (Fig. 1C and D). Despite a range of sensitivity to MBNL sequestration (28), none of these classically associated DM events were always mis-spliced in all CDM individuals. CDM biopsies often displayed a wider distribution of observed Ψ values compared with DM1. In combination, these results indicate a previously underappreciated variability in the range of AS dysregulation within CDM despite severe symptomatic onset at birth and association with higher mean CTG repeat length (8).
To identify cis-elements associated with mis-splicing in this comparative analysis, we enumerated 4-mers in the regions flanking and within the most significantly mis-spliced SE events (|ΔΨ| ≥ 0.2, FDR ≤ 0.05, n = 274) compared with a set of control SEs expressed in skeletal muscle. We observed a strong enrichment for MBNL binding motifs (YGCY where Y = C or U), including TGCT and CGCT in regions consistent with positional-dependent MBNL splicing regulation (i.e. aberrantly excluded exons show an enrichment in the downstream intronic region while aberrantly included exons contained motifs within the upstream intron and exon) (19,42,43) (Fig. 1E). These results are consistent with a core pattern of MBNL-dependent mis-splicing in affected individuals similar to that observed in other DM1 affected tissues (29,34). Despite differences in disease onset and symptomatic presentation, individuals with CDM and DM1 share a heterogenous range of AS dysregulation and a MBNL-dependent, mis-splicing signature.
CDM patient cohort displays variable and dynamic presentation of mis-splicing compared with age-matched controls
To further discern the range of AS dysregulation within our CDM cohort, we next characterized global spliceopathy of CDM individuals when compared only to pediatric controls. As expected, this analysis revealed extensive global splicing dysregulation, including SEs, mutually exclusive exons (MXE), alternative 5′ splice sites (A5SS) and alternative 3′ splice sites (A3SS) (|ΔΨ| ≥ 0.1, FDR ≤ 0.05) (Fig. 2A, Supplementary Material, Table S4). Hundreds of AS events had substantial changes in Ψ with the largest differences detected in the SE category (n = 700) (Fig. 2A). Enumeration of 4-mers in the regions flanking the most-significantly mis-spliced SE events (|ΔΨ| ≥ 0.2, FDR ≤ 0.05, n = 194) compared with non-dysregulated exons revealed a strong enrichment of YGCY motifs consistent with positional-dependent splicing regulation by, and depletion of, MBNL proteins (Fig. 2B) (19,42,43). Consistent with a shared MBNL-dependent spliceopathy in all affected populations, 64% of all dysregulated exons within CDM biopsies were also mis-spliced in adult-onset DM1 when compared to their associated, age-matched unaffected population (Supplementary Material, Fig. S1A, Supplementary Material, Table S5).
Figure 2.

Analysis of alternative splicing dysregulation in CDM individuals compared with age-matched pediatric controls. (A) Violin plot displaying distribution of splicing dysregulation based on ΔΨ compared with all PeCo samples. Events significantly dysregulated (ΔΨ ≥ 0.1, FDR ≤ 0.05) within each AS category, including skipped exons (SE), mutually exclusive exons (MXE), alternative 5′ splice sites (A5SS) and alternative 3′ splice sites (A3SS), are annotated as colored circles. The number of each AS event type is listed above. (B) Heatmap showing enrichment of k-mers (k = 4) around the 194 most significantly dysregulated skipped exons (CDM versus PeCo, |ΔΨ| ≥ 0.2, FDR ≤ 0.05). Columns from left to right denote the intronic region from +1 to +300 bp and −300 to −1 bp of the upstream intron, the skipped exon itself, and the intronic region from +1 to +300 bp and −300 to −1 bp of the downstream intron. (C) Heatmap displaying estimated Ψ of top 50 significantly dysregulated SE events between CDM and PeCo groups (CDM versus PeCo, |ΔΨ| ≥ 0.1, FDR ≤ 0.05). Rows (SE events) were subjected to hierarchical clustering, while columns (individual samples) were ranked by mean ΔΨ of these 50 events as shown above. Sample group, sex, and age at biopsy are also listed. CDM repeat biopsies are annotated as * (CDM-1) and ** (CDM-32). (D) Ψ values of specific skipped exon events for CDM and PeCo samples.
By visualizing a panel of the 50 most dysregulated SEs between CDM and PeCo we observed that while some exons displayed widespread dysregulation (i.e. SUN1 e7), most exons showed a range of dysregulation whereby Ψ was only moderately dysregulated in some individuals and largely dysregulated in others (i.e. MBNL1 e5, CLCN1 e7a, CLASP1 e19) (28,38,44) (Fig. 2C and D, Supplementary Material, Table S6). This range of AS heterogeneity within the full CDM cohort is further emphasized by calculation of mean ΔΨ which was used to rank samples by overall observed mis-splicing (Fig. 2C). Visualization of AS dysregulation in this manner revealed that a significant proportion of individuals with CDM possessed moderate splicing dysregulation compared with unaffected PeCos. Conversely, a subset of CDM individuals displayed widespread AS dysregulation consistent with severe response to disease state; this pattern closely matched those of previously published skeletal muscle transcriptomes derived from CDM postmortem tissue (Supplementary Material, Fig. S2, Supplementary Material, Table S7) (32). No correlation was observed between overall splicing dysregulation as represented by mean ΔΨ and sex or age to explain this observed variability (Supplementary Material, Fig. S1B). Using this analysis, we also observed a previously unreported occurrence whereby AS dysregulation was reduced for CDM-1, but relatively stable for CDM-32 upon secondary biopsy (Fig. 2C). Although limited to two individuals, these observations are consistent with a dynamic variability of AS dysregulation in CDM.
Inferred [MBNL] captures global changes in CDM transcriptomic dysregulation across pediatric development
Due to the diverse presentation of global splicing dysregulation across the full CDM cohort we sought to condense these observations into an aggregate metric. Given the MBNL-dependent spliceopathy observed within the CDM cohort (Fig. 2B–D), we chose to utilize a previously published methodology of calculating intracellular concentrations of free MBNL ([MBNL]inferred) using Ψ values from a small collection of SE events with high predictive power of overall global mis-splicing in DM1 TA muscle (28). Plotting of Ψ for the nine SE events used to quantify [MBNL]inferred exhibited the expected sigmoidal, dose-dependent splicing response whereby PeCo and AdCo samples showed consistent exon inclusion patterns with high estimations of intracellular [MBNL] (Fig. 3A and Supplementary Material, Fig. S3A). While the majority of adult-onset DM1 individuals possessed relatively low [MBNL]inferred values indicative of high MBNL depletion and consistent with the significant mis-splicing dysregulation observed (Fig. 1C and D), individuals with CDM were divided into two distinct groups with approximately two-thirds of patients possessing elevated estimates of intracellular [MBNL] ([MBNL]inferred > 0.5) (Fig. 3B). Importantly, a strong correlation was observed between [MBNL]inferred and total splicing dysregulation as assessed by mean ΔΨ within the CDM cohort (Fig. 3C and Supplementary Material, Fig. S3B). The high proportion of CDM individuals with high [MBNL]inferred, especially as compared with DM1, are concordant with both the widespread variability in measured Ψ for CDM (Figs 1 and 2) and the overall reduced number of mis-spliced events detected compared with DM1 (Supplementary Material, Fig. S1B).
Figure 3.

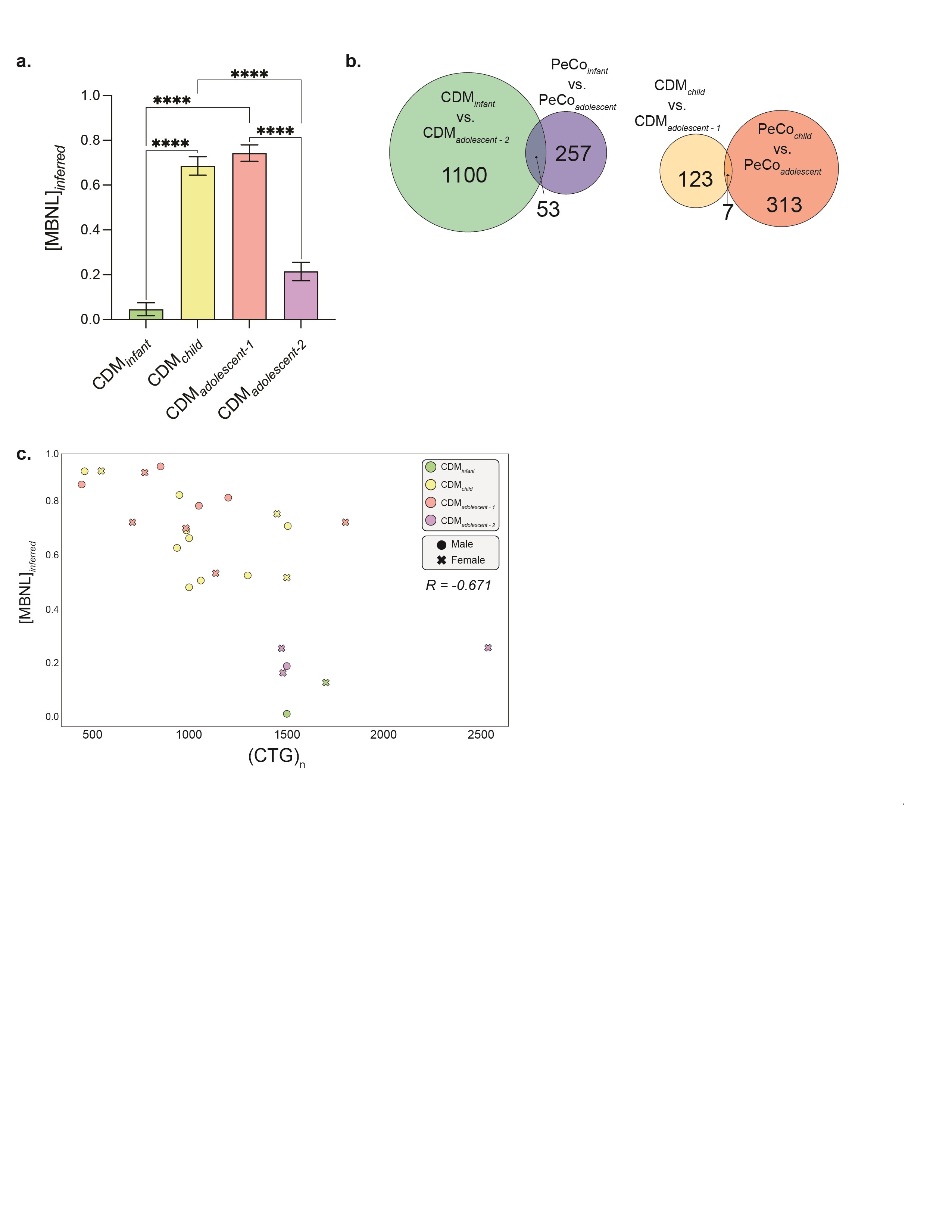
[MBNL]inferred captures dynamic variability of splicing dysregulation within CDM skeletal muscle transcriptomes during pediatric development. (A) Percent spliced in (PSI, Ψ) values derived from Affected (CDM and DM1) versus Unaffected (PeCo and AdCo) comparison for MBNL1 e5 and CLASP1 e19 plotted against [MBNL]inferred. Ψ values for individuals in each sample group are differentiated by color, and 95% confidence of estimated Ψ value at a given [MBNL]inferred is shown in gray. (B) Bar graph displaying distribution of calculated [MBNL]inferred for CDM and DM1 cohorts. (C) [MBNL]inferred of CDM and PeCo samples correlates with total splicing dysregulation as estimated by mean ΔΨ of top 50 most significantly dysregulated SE events (Affected versus Unaffected |ΔΨ| ≥ 0.1, FDR ≤ 0.05). R2 value is reported. (D) Scatter plot of [MBNL]inferred for CDM patients versus age at biopsy. Individual sample’s sex and classification within outlined CDM groups is displayed as described in associated table legend. Table outlines age and [MBNL]inferred inclusion criteria for four CDM subcohorts: CDMinfant, CDMchild, CDMadolescent-1 and CDMadolescent-2. These inclusion criteria are also visually annotated on scatter plot with boundaries of circled groupings. CDM repeat biopsies are annotated as * (CDM-1) and ** (CDM-32).
Plotting of [MBNL]inferred against age at biopsy revealed a distinct pattern of changes in MBNL depletion and associated spliceopathy across early muscle development in CDM (Fig. 3D). Consistent with severe symptomatic presentation of infants with CDM in the first years of life (12–14), all individuals below 2 years had low [MBNL]inferred values indicative of significant MBNL depletion and severe, global mis-splicing (n = 4, [MBNL]inferred < 0.2). A significant increase in [MBNL]inferred was observed for all samples collected in early childhood (n = 13, [MBNL]inferred > 0.48). In fact, many individuals within this age range possessed [MBNL]inferred values equivalent to those of controls ([MBNL]inferred = 0.75–1.0). Variability of [MBNL]inferred was only observed in individuals > 8 years. While 37% of patients were assigned low [MBNL]inferred values (n = 7, [MBNL]inferred < 0.4), 63% of individuals were estimated to have high intracellular [MBNL] (n = 12, [MBNL]inferred > 0.4). Given these distinct groupings of samples, we subdivided the overall CDM cohort into four groups based on both age and [MBNL]inferred: CDMinfant, CDMchild, CDMadolescent-1 and CDMadolescent-2 (Fig. 3D).
These results point to a dynamic progression of transcriptomic alterations in skeletal muscle across childhood development whereby CDM patients exhibit severe spliceopathy and MBNL depletion at birth (CDMinfant) that improves in early childhood (CDMchild). This projected pathway of alterations in CDM spliceopathy is supported by the increase in [MBNL]inferred for CDM-1 between 2 weeks and repeat sampling at 8 years (Δ[MBNL]inferred = 0.56). Post 8 years, individuals with CDM segregate into two distinct groups: individuals within adolescent-1 maintain high [MBNL]inferred levels while those within adolescent-2 revert to low estimates of [MBNL]inferred like that of their infant counterparts.
Changes in CDM spliceopathy across pediatric development underscore complexity of transcriptomic dysregulation in the presence of large CTG repeat expansions
To further evaluate the dynamics of CDM AS dysregulation, we identified shared and independently dysregulated SEs within our four CDM subcohorts when compared only to age-matched, unaffected PeCos (|ΔΨ| ≥ 0.1, FDR ≤ 0.05). The number of overlapping events shared between each possible combination of CDM subcohorts was visualized via an UpSet plot (Fig. 4A). Consistent with low [MBNL]inferred values quantified for the infant and adolescent-2 groups, these two populations possessed significant mis-splicing of over 1600 SE events (Fig. 4A, Supplementary Material, Table S8). Conversely, we observed that individuals assigned to child or adolescent-1 cohorts shared a reduced number of dysregulated SE events (Fig. 4A). Congruent with mean [MBNL]inferred of each CDM subcohort and the associated severity of global mis-splicing, increase of the ΔΨ threshold to 0.2 and 0.4 led to a substantial reduction in the number of significantly mis-spliced SE events within CDMchild and CDMadolescent-1. Conversely, many events continued to be dysregulated in the two most affected subcohorts (Fig. 4B and Supplementary Material, Fig. S4A).
Figure 4.

CDM subcohorts display variable alternative splicing dysregulation compared with age-matched pediatric controls in the presence of large CTG repeat expansions. (A) UpSet plot displaying number of shared skipped exon (SE) events shared between all four CDM subcohorts (CDMGroup versus age-matched PeCo |ΔΨ| ≥ 0.1, FDR ≤ 0.05) as outlined in associated table. Each column corresponds to a CDM group or set of CDM groups (dots connected by lines below x-axis) sharing the same events. The first four columns represent the number of SE events dysregulated in each subcohort compared with age-matched controls and are colored accordingly. All other columns highlighting shared events are shown in gray. The number of SE events in each set is shown above the column. The total number of dysregulated SEs within each CDM subcohort is displayed in a separate bar plot in the bottom left. (B) Number of SE events identified as dysregulated within each CDM subcohort when compared with age-matched PeCos as outlined in (A) upon increase in threshold for IΔΨI. (C) Area-proportional Venn diagram displaying overlapping SE events mis-spliced in select CDM subcohorts (CDMGroup versus age-matched PeCo |ΔΨ| ≥ 0.1, FDR ≤ 0.05). (D) Scatter plot of Ψ for 392 SE events only dysregulated in CDMadolescent-2 and CDMinfantgroups. Events differentially spliced (|ΔΨ| ≥ 0.2) are colored in red. (E) Scatter plot of CDM individuals’ CTG repeat expansion size versus age at biopsy. Pearson correlation is shown. Individuals with repeat biopsies were only plotted once and colored according to the assigned CDM cohort at first biopsy. (F) Mean CTG repeat length for CDM subcohorts. (CTG)n was only available for some individuals. Repeat length for individuals with repeat biopsies was only utilized for age and CDM group assigned at first biopsy. Data represented as mean ± standard deviation, *P < 0.05, one-way ANOVA, Tukey’s post-hoc test. All unannotated comparisons were not significant.
Although a similar number of SE events were quantified as dysregulated within CDMinfant and CDMadolescent-2, approximately 60% of events in either population were uniquely mis-spliced only within their associated group (Fig. 4C). Of those events mis-spliced only in these two subcohorts (n = 392), the directionality and magnitude of Ψ were generally preserved, although the overall ΔΨ for select events was increased for CDMinfant (Fig. 4D). This may be due to overall increased spliceopathy in this subcohort as exemplified by lower mean [MBNL]inferred (Supplementary Material, Fig. S4A). While we initially hypothesized these distinct differences in AS dysregulation to be a consequence of skeletal muscle development, only 4.6% of all events identified as differentially spliced between CDMinfant and CDMadolescent-2 were also found to have a significant change in Ψ when compared to their associated age-matched PeCos (Supplementary Material, Fig. S4B). These results suggest that while both CDM cohorts share low estimated [MBNL]inferred values and widespread splicing dysregulation, individuals in both groups are characterized by distinct patterns of severe spliceopathy with age-specific variability in affected events unrelated to muscle development.
In contrast to the diversity of AS dysregulation observed between infant and adolescent-2 populations, greater than 55% of SE events identified as mis-spliced in either the CDMchild or CDMadolescent-1 groups were also dysregulated in the other; the remaining variability was not explained by simple differences between age-matched controls used to quantify Ψ (Fig. 4C, Supplementary Material, Fig. S4B). These observations reveal minimal transcriptomic variability independent of age within these two CDM subcohorts.
Consistent with significant differences in mean [MBNL]inferred between CDM adolescent subcohorts (Supplementary Material, Fig. S4A), a substantially greater number of SE events were mis-spliced at an increased magnitude in adolescent-2 compared with adolescent-1 (Fig. 4A–C). While select SE events were dysregulated in both populations when compared with their shared age-matched control group (PeCoadolescent, n = 284) (Fig. 4C), direct transcriptomic comparison revealed that 1449 SE events were differentially spliced between the two adolescent subcohorts (Supplementary Material, Fig. S4B). This is in sharp contrast to CDMadolescent-1 and CDMchild whereby only 130 events were differentially spliced (Supplementary Material, Fig. S4B). These results indicate that CDMadolescent-2 has a distinct transcriptomic signature from all other CDM subcohorts, including their age-matched affected counterparts in CDMadolescent-1.
Prior work in adult DM1 has demonstrated a modest relationship between the size of the CTG repeat expansion and disease severity (27,45). While we also observed a modest correlation between CTG repeat length and severity of disease associated mis-splicing across CDM samples (Supplementary Material, Fig. S4C), the overall dynamics of splicing dysregulation across pediatric development were not explained by repeat size alone. Individuals within each CDM subcohort possessed similar large-scale repeat expansions (Fig. 4F) and no correlation was observed between CTG repeat size and age at biopsy to explain the variability in splicing dysregulation observed (Fig. 4E). The only significant difference in (CTG)n was for CDMadolescent-2 which possessed a higher mean repeat length than either their adolescent-1 or child counterparts (Fig. 4F). This higher repeat load may contribute to the segregation of individuals into either adolescent subcohort post 8 years of age. However, this assessment is potentially skewed by the low number of samples within CDMadolescent-2 with available repeat information.
Overall, this analysis revealed a triphasic pattern of transcriptomic dysregulation across childhood development in CDM. This consists of an early infantile pattern of mis-splicing whereby a substantial proportion of events unique only to these affected individuals are mis-spliced (CDMinfant). Post 2 years of age all patients transition to a pattern of reduced global spliceopathy independent of sex or CTG repeat expansion size (Fig. 4E). This conclusion is supported by the significant improvement in AS dysregulation observed from CDM-1 longitudinal biopsies sampled while the individual resided in two distinct CDM subcohorts at 2 weeks and 8 years of age (Fig. 3D). This overall improvement in spliceopathy in early childhood represents a secondary stage of disease progression and is consistent with clinical observations of motor function improvement during this stage of development (13,15). Finally, a third phase occurs post 8 years of age whereby individuals segregate based on observed levels of AS dysregulation. Patients within CDMadolescent-1 maintain an overall reduced spliceopathy like CDMchild. Inclusion of muscle transcriptomes derived from a secondary longitudinal biopsy set from CDM-32 at 12 and 16 years of age highlights the stability of this reduced spliceopathy within this subcohort (Δ[MBNL]inferred = 0.02). Conversely, the CDMadolescent-2 population displays a transcriptomic regression and enhanced spliceopathy, possibly due to increased size of the repeat expansion (Fig. 4F). Interestingly, we identified a greater proportion of females within adolescent-2 compared with adolescent-1 (86 versus 58%, respectively). While female CDM patients may regress within this tertiary phase of disease progression earlier than their male counterparts due to an earlier onset of pubescence, the small proportion of males >8 years in this sample cohort did not allow us to evaluate this hypothesis. Regardless, since disease onset was matched, the divergent transcriptomic signatures observed indicate that other genetic or environmental factors yet unknown may modify the disease progression.
Subset of adolescent CDM individuals (CDM adolescent-2) possess a transcriptomic signature similar to adult-onset DM1
Previous clinical observations suggest that CDM individuals display progressive impairment associated with adult DM1 features during adolescence, including myotonia and distal weakness (13). The divergence of splicing dysregulation within the two CDM adolescent cohorts led us to investigate if global patterns of mis-splicing within CDMadolescent-2 aligned with those observed in adult-onset DM1. To assess this hypothesis, we performed direct transcriptomic comparison of each CDM subcohort to adult-onset DM1 populations with a matched distribution of [MBNL]inferred (CDMadolescent-1 versus DM1, [MBNL]inferred > 0.4 and CDMadolescent-2 versus DM1, [MBNL]inferred ≤ 0.4). While a distinct population of SE events were differentially spliced in CDMadolescent-2 when compared to DM1 (n = 372, |ΔΨ| ≥ 0.1, FDR ≤ 0.05), a higher number of exons (n=586) were differentially spliced within CDMadolescent-1. This value is even higher than that mis-spliced when individuals were compared to age-matched controls (PeCoadolescent (n = 480, Fig. 4A). Consistent with the distinct AS profile of infantile CDM patients, 1293 events were differentially spliced compared with DM1 (CDMinfant versus DM1, [MBNL]inferred ≤ 0.4). Together, these observations suggest that CDMadolescent-2 individuals begin to possess AS patterns of adult-onset DM1 individuals.
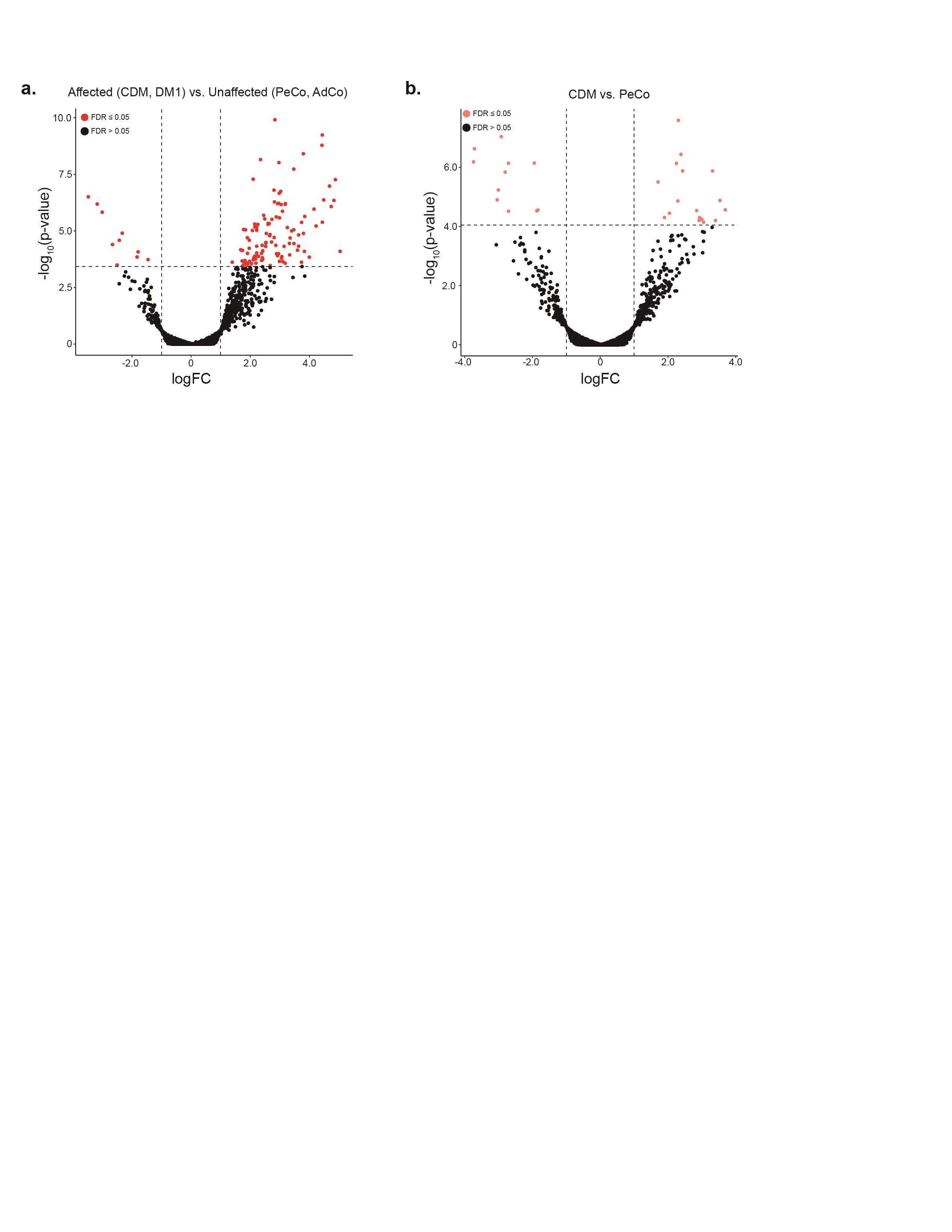
To further evaluate DM1 and CDM transcriptomic variation, gene expression values from biopsies were estimated and differential expression quantified for all affected individuals compared with their associated age-matched controls. Using this approach, we observed a distinct pattern of dysregulation within the DM1 cohort whereby over 90% of the 180 genes identified were significantly upregulated (Fig. 5A, Supplementary Material, Table S9). Importantly, only 10 of these genes were also differentially spliced. Few genes (n = 28) were found to be dysregulated within CDM individuals when analyzed as a complete cohort (Supplementary Material, Fig. S5A). When divided into the previously described subcohorts, gene expression changes were only detected within individuals categorized as CDMadolescent-2 (Fig. 5B). While fewer genes were differentially expressed compared with DM1 (n = 65), the evident distribution of gene expression upregulation was preserved. Of these genes, 46% were also dysregulated within the DM1 cohort (Fig. 5C). While gene expression changes have not been previously associated with disease-specific presentation (46), this shared gene expression signature further supports our hypothesis of a triphasic disease progression whereby individuals segregated into the CDMadolescent-2 group possess a transcriptomic signature most like DM1.
Figure 5.

Adult-onset DM1 and CDMadolescent-2 individuals share a differential gene expression signature. (A, B) Volcano plot of differentially expressed genes in adult-onset DM1 and CDMadolescent-2 cohorts compared with age-matched unaffected controls. Genes significantly differentially expressed are colored accordingly (logFC ≥1, FDR ≤ 0.05). (C) Venn diagram displaying number of differentially expressed genes shared between DM1 and CDMadolescent-2 cohorts when compared to age-matched controls.
Changes in DMPK expression contribute to unique pattern of transcriptomic dysregulation observed across CDM disease progression
Given the strong mechanistic connection between expression of toxic, CUG repeat containing RNAs from the DMPK locus, sequestration of MBNL, and downstream dysregulation of RNA metabolism, we assessed DMPK gene expression levels for all biopsies collected. When plotted against [MBNL]inferred a strong negative correlation was observed (Fig. 6A). Consistent with low [MBNL]inferred and enhanced global spliceopathy quantified in CDMinfant, CDMadolescent-2 and DM1 individuals, all three groups possessed qualitatively higher levels of DMPK compared with unaffected controls, which had similar expression patterns independent of age (Fig. 6A). No correlation was observed between MBNL1 and MBNL2 levels for all individuals when plotted against [MBNL]inferred or upon qPCR analysis of select samples within each subcohort (Supplementary Material, Fig. S6A–C).
Figure 6.

Variable DMPK expression within CDM subcohorts correlates with MBNL-dependent mis-splicing signature conserved between all adult-onset DM1 and CDM individuals. (A) Scatter plot of DMPK expression levels within all sample groups as detected by RNA-seq (logCPM) plotted against [MBNL]inferred. Pearson correlation is reported and CDM repeat biopsies are annotated. (B) Aligned dot plot of DMPK relative expression normalized to GAPDH as measured by qPCR of all CDM subcohorts, DM1, and AdCo. Results are expressed as mean ± standard deviation; one-way ANOVA with Tukey’s post hoc analysis; *P < 0.05; **P < 0.005. Unless annotated, relative expression from all comparisons was not significant. (C) Relative fold-change in expression of DMPK in CDM-1 and CDM-32 repeat biopsies at the first and second collection compared with AdCo as measured by qPCR. Age at collection for each sample is listed in brackets (years) and bars are colored according to assigned CDM subcohort. Results are expressed as mean ± standard deviation; one-way ANOVA with Tukey’s post-hoc analysis; **P < 0.005; ****P < 0.0001. Unless annotated, relative expression from all comparisons was not significant (ns). (D) Percent spliced in (PSI, Ψ) values of three skipped exon events mis-spliced in DM1 and all CDM subcohorts. Ψ values derived from comparisons between each affected group and associated age-matched controls (|ΔΨ| ≥ 0.1, FDR ≤ 0.05).
Quantification of relative gene expression via qPCR for representative samples within each affected subcohort identified a significant increase in relative DMPK expression for infant CDM individuals compared with nearly all other populations (Fig. 6B). While DMPK transcript levels as evaluated by RNA-seq appeared increased within DM1 and CDMadolescent-2, no significant increase compared with AdCo was quantified within either subcohort. We assert this is due to the variability of quantified transcript levels due to selection of individuals across the full [MBNL]inferred spectrum within each group. The overall significance of expression differences, especially for DM1, appears diluted due to the correlation between DMPK levels and [MBNL]inferred (Supplementary Material, Fig. S6C).
CDM individuals categorized into child and adolescent-1 cohorts presented with relative DMPK expression patterns similar to unaffected controls (Fig. 6B). These differences in DMPK levels across CDM subcohorts were further exemplified by the notable reduction in DMPK expression observed within CDM-1 upon transition from the CDMinfant to CDMadolescent-1 cohort (Fig. 6A and C). Consistent with minimal changes in [MBNL]inferred for CDM-32, relative levels of DMPK remained below that of controls upon repeat biopsy (Fig. 6A and C). Overall, these analyses indicate that dynamic changes in DMPK expression over the course of CDM disease progression may contribute to the variability of transcriptomic dysregulation described within each CDM subcohort.
Given the variability of DMPK expression, estimations of intracellular [MBNL], and global AS perturbations observed throughout our CDM cohort, we sought to identify universally dysregulated AS events that may serve as biomarkers of total muscle spliceopathy. A total of 93 SE events were found to be dysregulated in all CDM subcohorts, 90 of which were also mis-spliced in adult-onset DM1 (Fig. 4A). This distilled population of events included commonly mis-spliced, MBNL-dependent cassette exons such as CLCN1 e7a (38) and CLASP1 e19 (28), along with several events used to derive [MBNL]inferred (28) (Fig. 6D and Supplementary Material, Fig. S7, Supplementary Material, Table S10). Consistent with a shared MBNL-dependent spliceopathy, mean ΔΨ of these 90 events strongly correlated with [MBNL]inferred (Supplementary Material, Fig. S8). While most mis-spliced exons exhibited a sigmoidal, dose-dependent response to changes in inferred [MBNL] concentrations, select events such as MYH11 e6 displayed uniform dysregulation of Ψ independent of [MBNL]inferred (Fig. 6D and Supplementary Material, Fig. S7). We propose that these events represent the most universal pool of potential biomarkers for assessment of AS dysregulation within all CDM and DM1 individuals.
Discussion
AS dysregulation of CDM skeletal muscle is highly dynamic across pediatric development
Through generation and analysis of transcriptomic data derived from a large collection of CDM skeletal muscle biopsies, we identified a dynamic, variable, and previously unreported pattern of AS dysregulation during pediatric development. Utilization of biopsies sampled from children from 2 weeks to 16 years of age and select longitudinal samples (CDM-1 and CDM-32) revealed a widespread range of AS dysregulation greater than previously described (31,32) with many individuals displaying Ψ values consistent with that measured for unaffected controls (Figs 1B–D and2C–D). Analysis of cis-elements flanking significantly dysregulated exons in CDM revealed an enrichment of MBNL motifs consistent with functional depletion of this RBP being a major driver of mis-splicing (Figs 1E and2B). Via utilization of a Bayesian estimation methodology that employs Ψ values for select SE events previously shown to respond to changes in intracellular [MBNL] in a dose-dependent manner (28), [MBNL]inferred was derived for all muscle transcriptomes (Fig. 3B). As [MBNL]inferred correlated strongly with total splicing dysregulation as estimated by mean ΔΨ in our CDM cohort (Fig. 3C and Supplementary Material, Fig. S3B), this metric was used to condense and subsequently visualize AS dysregulation as a function of age at biopsy. Using this approach, we identified four subcohorts of CDM patients segregated by age and [MBNL]inferred that revealed a triphasic modality of disease progression (Fig. 3D).
All CDM individuals under the age of 2 years (CDMinfant) presented with severe global spliceopathy followed by a significant reversal of splicing dysregulation in early childhood (CDMchild). The complete lack of observation of individuals under 8 years of age with intermediate [MBNL]inferred ([MBNL]inferred = 0.15–0.45) and the dramatic increase in [MBNL]inferred observed for CDM-1 between 2 weeks and 8 years highlights previously unobserved, partial reversal in global spliceopathy for CDM in early childhood (Fig. 3D). Another phase of disease progression emerges in adolescence where individuals separate into two distinct groups. Individuals categorized within CDMadolescent-2 saw a return to enhanced transcriptomic dysregulation that resembled adult-onset DM1 (Figs 4A–C and5). Conversely, individuals within CDMadolescent-1 maintained an AS pattern like that of their CDMchild counterparts (Figs 3D and4A–C). Inclusion of a secondary repeat biopsy from CDM-32 at 12 and 16 years of age also revealed that individuals can naturally maintain these high inferred levels of [MBNL] into late adolescence. Though cross-sectional, the transition from a childhood phenotype to one that more closely matches adult-onset DM1 is divergent amongst individuals. Additional research is required to identify the mechanism by which the adult DM1 splicing and gene expression profile emerges.
Overall, these combined analyses of a large cohort of CDM patients expand previous descriptions of overall transcriptomic dysregulation in CDM, especially past early infantile development. Our observations of severe, MBNL-dependent splicing dysregulation within the CDMinfant group are consistent with that previously described for CDM patients <15 months (31,32). These studies also reported significant gene expression dysregulation in CDM, most notably increased expression of genes related to hypoxia and inflammation, including components of the interlukin-6 (IL6) pathway (31,32). Increased expression of these genes was also correlated with histological assessment of muscle immaturity (31). No genes were found to be significantly dysregulated in age-matched CDM individuals (CDMinfant) when compared to unaffected samples, and where expression differences were identified (CDMadolescent-2) genes associated with these pathways were not affected (Fig. 5B, Supplementary Material, Fig. S5B, Supplementary Material, Table S9). These inconsistent findings may be related to the use of autopsy tissue where increases in expression of hypoxia and signal-transduction-related genes postmortem have been reported (47). Further evaluation of gene expression changes over pediatric development in CDM is required to further define its contribution to disease pathology.
While our categorization of each CDM subcohort’s age of inclusion was based on transcriptomes derived from available biopsy materials, the addition of more individuals may refine the defined inclusion criteria. Additionally, the PeCo biopsies gifted for use in this study, while graded as histopathologically normal, were obtained from children referred clinically for a muscle biopsy. Use of transcriptomes from these PeCo individuals as part of the unaffected cohort may have modified baseline Ψ values used to quantify overall mis-splicing in CDM individuals. Finally, because some biopsies were obtained ad hoc (e.g. controls) or different muscles were sampled for safety reasons (vastus lateralis in CDM versus tibialis anterior in DM1), it is possible that there may be transcriptomic differences obscured by the selection of muscles. Fortunately, since muscle selection was consistent within the DM1 and CDM cohorts, it is unlikely these differences would vary significantly. Overall, these data describe the highly dynamic nature of the CDM skeletal muscle transcriptome during pediatric development that correlates with improvement of musculoskeletal phenotypic presentation in early childhood. However, this transcriptome profile may not reflect changes in the brain or other organ systems that are severely affected in CDM (12,48). This is consistent with previous reports using DM1 RNA-seq datasets from different tissues, including the frontal cortex, heart and tibialis anterior, where AS events were only dysregulated in some tissues or differentially mis-spliced across tissues (29,34).
Variation in CUG repeat RNA toxicity through CDM disease progression
While a minimum CTG repeat copy number required for CDM disease presentation remains unclear (7,8,49–51), the shared age of onset within this assembled CDM cohort allowed for assessment of repeat length dependency on the trajectory of disease progression. Such analyses have proved difficult in adult-onset DM1 patients due to assessment of repeat length in unaffected tissues and confounding variability from differences in age of onset and variant repeat interruptions (52,53). Both the size of our CDM sample set and inclusion of longitudinal biopsies support a repeat-independent improvement in global spliceopathy post 2 years of age that is maintained through early childhood. This is followed by a possible repeat size dependent regression of AS dysregulation once individuals enter adolescence (Fig. 4F and Supplementary Material, Fig. S4C). However, due to lack of repeat sizing available for a proportion of CDM individuals, acquisition of additional biopsies with matched CTG repeat length will be vital to effectively evaluate the effects of the expansion on segregation of patients into each adolescent subcohort. Additionally, while repeat sizing was defined from blood, somatic mosaicism of the CTG expansion across affected patient tissues has been previously observed in adult-onset DM1 individuals, although the impacts have never been evaluated in CDM (27,54). As such, determination of CTG repeat size within CDM muscle tissue would further define how overall repeat length contributes to the dynamic changes in disease-associated mis-splicing across pediatric development.
The occurrence of CUG repeat RNA toxicity during prenatal tissue development and into childhood is predicted to contribute to overall muscle immaturity and reduced myogenic potential in CDM muscle (31,32,55,56). Low estimates of free [MBNL] and severe mis-splicing of muscle-specific events within CDMinfant support this pathogenic mechanism. However, the concurrent observed partial restoration of splicing dysregulation with a period of motor function improvement in early childhood (15) suggests that various trans-acting, possibly protective modifiers contribute to this observed change in toxicity of CUG repeat RNAs and associated mis-splicing. This may include developmental dynamics of tissue-specific RBP expression (57) or altering contributions from other DM-associated disease mechanisms, such as repeat-associated non-AUG translation (RAN) (58,59), which have never been assessed in CDM tissues. The effects of trans-acting modifiers of disease severity are further exemplified by the unexpected observation of mis-splicing of events linked to specific symptoms in the absence of such phenotypic presentation. For example, aberrant splicing of CLCN1 is linked to clinical presentation of myotonia in DM1 patients and in mouse models of the disease (23,24). Dysregualtion of this event was observed in individuals as young as 2 weeks old, an observation inconsistent with a lack of clinical myotonia in this population.
Enhanced DMPK expression and shared MBNL-dependent mis-splicing signature in CDM highlight common molecular perturbations amenable to therapeutic intervention
Expression analysis of disease relevant genes revealed a strong correlation of DMPK expression with variability in CDM spliceopathy during pediatric development (Fig. 6A). More specifically, high DMPK levels were detected in the youngest CDM individuals (CDMinfant) whereas reduced expression similar to unaffected controls was observed in CDMchild and CDMadolescent-1 groups (Fig. 6A and B). This observed increase in DMPK expression within the youngest CDM samples (CDMinfant) is consistent with that previously reported in fetuses or CDM autopsy samples (31,32). Current models of CDM pathogenesis link this observed increase in DMPK expression to CpG hypermethylation of CTCF sites flanking the expanded CTG repeat and subsequent increase in sense DMPK transcription (11,31,60). This enhanced production of CUG repeat containing RNA species is hypothesized to be responsible for severe mis-splicing occurring due to accumulation of RNA foci and disruption of MBNL intracellular distribution early in muscle development (32,61,62).
While the severity of mis-splicing and DMPK expression in our youngest CDM samples support this early model of disease pathogenesis, the reversion of DMPK expression to unaffected levels in early childhood and its maintenance into adolescence for select individuals (CDMadolescent-1) indicate that this model does not encompass the full variability of CDM disease progression. Developmental transitions connected to transcriptional regulation of the disease locus, including alterations in the epigenetic landscape, may contribute to the overall transcriptomic transitions identified in our cohort for CDM individuals over 2 years of age. While increased CpG methylation flanking the expanded CTG repeat have been detected in peripheral blood leukocytes sampled from CDM patients up to 26 years of age (63), this does not preclude tissue-specific epigenetic modifications in skeletal muscle tissue during CDM disease progression. Additionally, trans-acting modifiers of toxic RNA turnover, including ribonuclear foci aggregation and retention, may contribute to observed reduction of detectable DMPK transcripts. Previous efforts to model CDM have focused on expression of a large CTG repeat or skeletal muscle specific knockout of MBNL isoforms (32,64). While these resources have been vital in understanding the effects of both the CTG repeat expansion size and MBNL depletion on global transcriptomic dysregulation, our observations of variable DMPK expression and dynamic influence of the CTG repeat expansion over pediatric development indicate that these models may only be partially representative of the entire CDM cohort.
Consistent with decreased [MBNL]inferred and overall splicing dysregulation observed in DM1 and CDMadolescent-2 cohorts, a qualitative increase in DMPK expression was observed within these populations (Fig. 6A). However, no significant upregulation was quantified upon qPCR confirmatory analysis for select samples due to overall variability in these cohorts (Fig. 6B). Further research, including acquisition of additional muscle biopsies from CDM patients within this subcohort, is required to evaluate if and how DMPK expression may contribute to disease pathology in this CDM group.
Despite these observations, DMPK was not identified as differentially expressed within any CDM or DM1 population when compared with age-matched, unaffected individuals. However, given the normalized age of onset for all CDM patients and the ability to map global transcriptomic changes across the course of aging, the observable shifts in DMPK expression assert that minimal alterations in DMPK transcript levels, already defined to be a low-abundance RNA species (65), may have significant impacts on skeletal muscle disease pathology. Overall, these observations indicate that RNA-modifying therapies targeting toxic, CUG-repeat containing transcripts for degradation (66,67) are likely to be impactful in CDM patients, especially early in disease progression, to assist in advancement or maintenance of transcriptomic improvement. These data also indicate that therapeutic interventions designed to halt production of DMPK transcripts (68–70), specifically the CTG repeat-containing allele, may be an effective alternative therapeutic approach, especially given reports of minimal effects of DMPK knockout or knockdown in mouse, non-human primate, and cell culture models (71,72).
Finally, through comparative analysis of all CDM and DM1 biopsies, we identified a MBNL-dependent mis-splicing signature shared between all affected individuals. A total of 90 SE events were found to be mis-spliced in all CDM subcohorts and DM1 individuals when compared with age-matched controls (Fig. 6D, Supplementary Material, Fig. S7). This distilled subset of SE events, most of which displayed a dose-dependent relationship to [MBNL]inferred, included many MBNL-dependent, disease-associated events (NFIX, INSR, CLCN1). These findings are similar to other efforts in adult DM1 that have identified a subset of splicing events that may serve as biomarkers (27,28,30). These events represent the most comprehensive group of splicing events which may be utilized for development of a universal biomarker panel representative of disease severity in all DM patient populations.
Materials and Methods
Muscle biopsy collection
Children with CDM were enrolled for sample collections if they met the clinical criteria for diagnosis: (1) an onset of symptoms within the first month of life requiring > 72 h of hospitalization and (2) a history of hypotonia, respiratory failure, talipes equinovarus and/or feeding difficulty. Adults with DM1 were enrolled if they possessed: (1) a diagnosis of DM1 and (2) age of onset after 10 years. Individuals were excluded if they had a history of a bleeding disorder, on anticoagulation or had a platelet count less than 50 000. All samples were collected under an Institutional Review Board approved protocol. Adult DM1 biopsies were collected from the tibialis anterior using a Bergstrom needle as previously described (27). Given the small muscle mass, a 14-gauge SuperCore needle (Argon Medical Devices) was used to collect a biopsy from the vastus lateralis in children with CDM. Adult controls biopsies were collected from individuals with no neuromuscular diagnoses using either a Bergstrom of the vastus lateralis (courtesy of Dr. M Drummond) or 14-gauge needle of the tibialis anterior. All biopsies, regardless of collection method, were flash frozen in liquid nitrogen immediately post collection. Pediatric control biopsies were gifted from the University of Iowa (courtesy of Dr. Steve Moore) after being evaluated as histopathologically normal. Biopsy location, if available, is provided in Supplementary Material, Table S1, along with sex and age at biopsy for all samples. CTG repeat sizes from peripheral blood for each individual, where available, were obtained from clinical reports (Supplementary Material, Table S1).
RNA extraction and RNA-seq library preparation
RNA was extracted from biopsies via a TRIzol-chloroform extraction supplemented with a bead-based tissue homogenization (Benchmark Scientific) and purified using the Zymo Clean and Concentrator—5 kit following the standard protocol for on-column DNase treatment (Zymo Research). RNA quality and abundance were assessed using fragment analysis (Agilent Fragment Analyzer 5200, DNF-472 (15 nt) RNA kit) and use of the Qubit 4 Fluorometer (Invitrogen). Samples with an assigned RNA integrity number (RIN) > 4 were sequenced. Total RNA-seq libraries were prepared using the NEBNext rRNA Depletion Kit in conjunction with the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs). Deviations from the manufacturer’s protocol include modifications of the recommended 5-fold adaptor dilution to a 40-fold adaptor dilution for RNA inputs between 250 and 100 ng and RNA fragmentation times based on RIN and 28S:18S ratios. Samples with an RIN ≥7.9 were fragmented for 15 min, but if the 28S:18S ratio was ≤0.5, then the samples were fragmented for 12 min. Additionally, samples with a RIN between 7.0 and 7.8 were fragmented for 12 min. Samples with an RIN ≤ 6.9 were fragmented for 8 min unless they had 28S:18S ratio ≥ 0.6, in which case they were fragmented for 12 min. Libraries were paired-end sequenced using the Illuminia NextSeq 550 v2.5 high output kit (150 cycles). All RNA-seq libraries are available in GEO (accession number GSE201255).
RNA-seq library quality assessment and read mapping
Adapters were trimmed from reads using Trim Galore. FastQC, RSeQC, and MultiQC were used to assess overall quality of the RNA-seq libraries produced and to obtain summary information including read distribution, splicing junction saturation and annotation (73,74). RNA-seq reads were aligned to hg38 using STAR in basic two-pass while being supplied a SJDB file generated using a GTF file from Ensembl release 98 (75).
Alternative splicing analysis
Splicing analysis was performed using rMATS to obtain differential splicing between any two cohort comparisons (76). Isoform percent spliced in (PSI, Ψ) for each event was calculated and were considered significantly mis-spliced with a threshold of |ΔΨ| ≥ 0.1, FDR ≤ 0.05, unless otherwise stated. Principal component analysis was performed on significant SE events using scikit-learn decomposition python package (https://scikit-learn.org/stable/about.html#citing-scikit-learn) and plotted using seaborn (https://seaborn.pydata.org/citing.html) and matplotlib (https://matplotlib.org/stable/users/project/citing.html) python packages.
Motif enrichment analysis
Motif enrichment was performed for both inclusion and exclusion events using a custom python script. Motifs of k-mer = 4 were enumerated across five different regions, including 300 bp downstream of the upstream exon, 300 bp upstream and downstream of the SE, the entire SE itself, and 300 bp upstream of the downstream exon. Motif occurrences were counted and compared against the total number of motifs in that region for both significant and non-significant exon events identified by rMATS (76). Motifs were considered enriched by performing a binomial test of the observed ratio of a given motif in significantly mis-spliced events against the ratio of that motif in background splicing events. Significance was corrected for multiple testing using Bonferroni correction. Motif enrichment was plotted using the odds ratio (OR) for that given motif. If a motif was not considered significantly enriched in that region, but appears in another region, its OR was set to 0 for visualization purposes.
Derivation of [MBNL]inferred
[MBNL]inferred was calculated within this sample cohort as previously described (28). While originally the top 10 events previously identified were to be utilized (28), a singular event was not identified (PDLIM3) and was excluded from the analysis. Nine total splicing events were used to quantify [MBNL]inferred (CLASP1, PDLIM3.2, CACNA2D1, MAPT.1, CACNA1S, PHTF2, CAPZB, PHKA1, MBNL1 and MAPT). While original gene coordinates were provided using hg19, coordinates were transitioned to hg38 using UCSC liftOver tool. Ψ values utilized were derived from rMATs Affected (CDM, DM1) versus Unaffected (AdCo, PeCo) comparison.
Differential gene expression analysis
Gene level read counts were obtained using featureCounts (77). Gene information, including Entrez ID, gene symbol, gene name, biotype, and description, was obtained using biomaRt (78). An initial filtering threshold was performed requiring a logCPM > 1 in at least 10 individual samples. Differential gene expression was performed using edgeR and glmTreat function (79). Genes were considered differentially expressed with a logFC ≥1 and FDR ≤ 0.05.
Quantitative PCR
Samples from each cohort were chosen within a range of [MBNL]inferred scores to demonstrate the variability within each grouping and had an even distribution of sex and age. Due to lack of sample material, PeCo samples were not included in this analysis. Each reaction was run in triplicate with three biological replicates per cohort, except for CDMadolescent-1 (n = 4), which includes the CDM-32 repeat biopsy. The cDNA was synthesized using SuperScript IV Reverse Transcriptase (Invitrogen) as per the manufacturer’s protocol. The following probe-based PrimeTime assays (Integrated DNA Technologies) were utilized: GAPDH (Hs.PT.39a.22214836, NM_002046), DMPK (Hs.PT.58.20386668, NM_004409), MBNL1 (Hs.PT.58.24846081, NM_207297) and MBNL2 (Hs.PT.58.45708803, NM_207304). Quantitative PCR was performed using the PrimeTime Gene Expression Master Mix (Integrated DNA Technologies) as per the manufacturer’s protocol and the QuantStudio 3 (Applied Biosystems). Gene expression was normalized to GAPDH and the fold-change in relative expression was calculated by 2-ΔΔCt methodology against adult controls with statistical analysis performed via one-way ANOVA with Tukey’s post-hoc test using Prism9.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Thanks to Dr. Steve Moore for gifting pediatric muscle biopsies from the Iowa Wellstone Muscular Dystrophy Specialized Research Center, NS053672, National Institutes of Health. Thanks to Dr. Mike Drummond for the gift of select adult unaffected biopsies. Dr Johnson has provided consultation for AMO Pharma, AveXis, Fulcrum Therapeutics, Dyne, Avidity, Vertex and Entrada.Conflict of Interest statement. None.
Contributor Information
Melissa A Hale, Department of Neurology, Virginia Commonwealth University, Richmond, VA 23298, USA.
Kameron Bates, Department of Neurology, Virginia Commonwealth University, Richmond, VA 23298, USA.
Marina Provenzano, Department of Neurology, Virginia Commonwealth University, Richmond, VA 23298, USA.
Nicholas E Johnson, Department of Neurology, Virginia Commonwealth University, Richmond, VA 23298, USA.
Funding
National Institutes of Health (R01NS104010; R21TR003184 to N.E.J.); Centers for Disease Control and Prevention (U01DD001242 to N.E.J.); Food and Drug Administration (R01FD006071 to N.E.J.); Dyne; AveXis; Vertex Pharmaceuticals; Fulcrum Therapeutics; ML Bio; Sarepta; Triplet Therapeutics; Avidity Biosciences; AMO Pharma; University of Rochester for the CCMDHI and CMTHI (to N.E.J.).
References
- 1. Mahadevan, M., Tsilfidis, C., Sabourin, L., Shutler, G., Amemiya, C., Jansen, G., Neville, C., Narang, M., Barceló, J., O’Hoy, K.et al. (1992) Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science (80-. ), 255, 1253–1255. [DOI] [PubMed] [Google Scholar]
- 2. Fu, Y.H., Pizzuti, A., Fenwick, R.G., King, J., Rajnarayan, S., Dunne, P.W., Dubel, J., Nasser, G.A., Ashizawa, T., De Jong, P.et al. (1992) An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science (80-. ), 255, 1256–1258. [DOI] [PubMed] [Google Scholar]
- 3. Brook, J.D., McCurrach, M.E., Harley, H.G., Buckler, A.J., Church, D., Aburatani, H., Hunter, K., Stanton, V.P., Thirion, J.P., Hudson, T.et al. (1992) Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell, 68, 799–808. [DOI] [PubMed] [Google Scholar]
- 4. Johnson, N.E., Butterfield, R.J., Mayne, K., Newcomb, T., Imburgia, C., Dunn, D., Duval, B., Feldkamp, M.L. and Weiss, R.B. (2021) Population-based prevalence of myotonic dystrophy type 1 using genetic analysis of statewide blood screening program. Neurology, 96, e1045–e1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harper, P.S. (2001) Myotonic Dystrophy, 3rd edn. W.B. Saunders, London. [Google Scholar]
- 6. Johnson, N.E., Aldana, E.Z., Angeard, N., Ashizawa, T., Berggren, K.N., Marini-Bettolo, C., Duong, T., Ekström, A.B., Sansone, V., Tian, C.et al. (2019) Consensus-based care recommendations for congenital and childhood-onset myotonic dystrophy type 1. Neurol. Clin. Pract., 9, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Campbell, C., Levin, S., Siu, V.M., Venance, S. and Jacob, P. (2013) Congenital myotonic dystrophy: Canadian population-based surveillance study. J. Pediatr., 163, 120–125.e3. [DOI] [PubMed] [Google Scholar]
- 8. Tsilfidis, C., MacKenzie, A.E., Mettler, G., Barceló, J. and Korneluk, R.G. (1992) Correlation between CTG trinucleotide repeat length and frequency of severe congenital myotonic dystrophy. Nat. Genet., 1, 192–195. [DOI] [PubMed] [Google Scholar]
- 9. Duthel, S., Bost, M., Ollagnon, E., Vial, C., Petiot, P., Chazot, G. and Vandenberghe, A. (1999) CTG instability in myotonic dystrophy: molecular genetic analysis of families from south-eastern France with characteristics of intergenerational variation in CGT repeat numbers. Ann. Genet., 42, 151–159. [PubMed] [Google Scholar]
- 10. Harley, H.G., Rundle, S.A., MacMillan, J.C., Myring, J., David Brook, J., Crow, S., Reardon, W., Fenton, L., Shaw, D.J. and Harper, P.S. (1993) Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am. J. Hum. Genet., 52, 1164–1174. [PMC free article] [PubMed] [Google Scholar]
- 11. Barbé, L., Lanni, S., López-Castel, A., Franck, S., Spits, C., Keymolen, K., Seneca, S., Tomé, S., Miron, I., Letourneau, J.et al. (2017) CpG methylation, a parent-of-origin effect for maternal-biased transmission of congenital myotonic dystrophy. Am. J. Hum. Genet., 100, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson, N.E., Butterfield, R., Berggren, K., Hung, M., Chen, W., Dibella, D., Dixon, M., Hayes, H., Pucillo, E., Bounsanga, J.et al. (2016) Disease burden and functional outcomes in congenital myotonic dystrophy. Neurology, 87, 160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johnson, N.E., Ekstrom, A.B., Campbell, C., Hung, M., Adams, H.R., Chen, W., Luebbe, E., Hilbert, J., Moxley, R.T. and Heatwole, C.R. (2016) Parent-reported multi-national study of the impact of congenital and childhood onset myotonic dystrophy. Dev. Med. Child Neurol., 58, 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pucillo, E.M., Dibella, D.L., Hung, M., Bounsanga, J., Crockett, B., Dixon, M., Butterfield, R.J., Campbell, C. and Johnson, N.E. (2017) Physical function and mobility in children with congenital myotonic dystrophy. Muscle Nerve, 56, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Quigg, K.H., Berggren, K.N., McIntyre, M., Bates, K., Salmin, F., Casiraghi, J.L., DʼAmico, A., Astrea, G., Ricci, F., McKay, M.J.et al. (2021) 12-Month progression of motor and functional outcomes in congenital myotonic dystrophy. Muscle Nerve, 63, 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miller, J.W., Urbinati, C.R., Teng-Umnuay, P., Stenberg, M.G., Byrne, B.J., Thornton, C.A. and Swanson, M.S. (2000) Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J., 19, 4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee, K.Y., Li, M., Manchanda, M., Batra, R., Charizanis, K., Mohan, A., Warren, S.A., Chamberlain, C.M., Finn, D., Hong, H.et al. (2013) Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med., 5, 1887–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin, X., Miller, J.W., Mankodi, A., Kanadia, R.N., Yuan, Y., Moxley, R.T., Swanson, M.S. and Thornton, C.A. (2006) Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet., 15, 2087–2097. [DOI] [PubMed] [Google Scholar]
- 19. Wang, E.T., Cody, N.A.L., Jog, S., Biancolella, M., Wang, T.T., Treacy, D.J., Luo, S., Schroth, G.P., Housman, D.E., Reddy, S.et al. (2012) Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell, 150, 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. López-Martínez, A., Soblechero-Martín, P., De-La-puente-ovejero, L., Nogales-Gadea, G. and Arechavala-Gomeza, V. (2020) An overview of alternative splicing defects implicated in myotonic dystrophy type I. Genes, 11, 1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Degener, M.J.F., vanCruchten, R.T.P., Otero, B.A., Wang, E.T., Wansink, D.G. and t Hoen, P.A.C. (2022) A comprehensive atlas of fetal splicing patterns in the brain of adult myotonic dystrophy type 1 patients. NAR Genom. Bioinform., 4, lqac016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Konieczny, P., Stepniak-Konieczna, E. and Sobczak, K. (2014) MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res., 42, 10873–10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Charlet-B, N., Savkur, R.S., Singh, G., Philips, A.V., Grice, E.A. and Cooper, T.A. (2002) Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell, 10, 45–53. [DOI] [PubMed] [Google Scholar]
- 24. Mankodi, A., Takahashi, M.P., Jiang, H., Beck, C.L., Bowers, W.J., Moxley, R.T., Cannon, S.C. and Thornton, C.A. (2002) Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell, 10, 35–44. [DOI] [PubMed] [Google Scholar]
- 25. Freyermuth, F., Rau, F., Kokunai, Y., Linke, T., Sellier, C., Nakamori, M., Kino, Y., Arandel, L., Jollet, A., Thibault, C.et al. (2016) (2016) Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat. Commun., 71, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fugier, C., Klein, A.F., Hammer, C., Vassilopoulos, S., Ivarsson, Y., Toussaint, A., Tosch, V., Vignaud, A., Ferry, A., Messaddeq, N.et al. (2011) Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med., 17, 720–725. [DOI] [PubMed] [Google Scholar]
- 27. Nakamori, M., Sobczak, K., Puwanant, A., Welle, S., Eichinger, K., Pandya, S., Dekdebrun, J., Heatwole, C.R., McDermott, M.P., Chen, T.et al. (2013) Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol., 74, 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wagner, S.D., Struck, A.J., Gupta, R., Farnsworth, D.R., Mahady, A.E., Eichinger, K., Thornton, C.A., Wang, E.T. and Berglund, J.A. (2016) Dose-dependent regulation of alternative splicing by MBNL proteins reveals biomarkers for myotonic dystrophy. PLoS Genet., 12, e1006316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang, E.T., Treacy, D., Eichinger, K., Struck, A., Estabrook, J., Olafson, H., Wang, T.T., Bhatt, K., Westbrook, T., Sedehizadeh, S.et al. (2019) Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet., 28, 1312–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wojciechowska, M., Sobczak, K., Kozlowski, P., Sedehizadeh, S., Wojtkowiak-Szlachcic, A., Czubak, K., Markus, R., Lusakowska, A., Kaminska, A. and Brook, J.D. (2018) Quantitative methods to monitor RNA biomarkers in myotonic dystrophy. Sci. Reports, 81, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakamori, M., Hamanaka, K., Thomas, J.D., Wang, E.T., Hayashi, Y.K., Takahashi, M.P., Swanson, M.S., Nishino, I. and Mochizuki, H. (2017) Aberrant myokine signaling in congenital myotonic dystrophy. Cell Rep., 21, 1240–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thomas, J.D., Sznajder, Ł.J., Bardhi, O., Aslam, F.N., Anastasiadis, Z.P., Scotti, M.M., Nishino, I., Nakamori, M., Wang, E.T. and Swanson, M.S. (2017) Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev., 31, 1122–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buj-Bello, A., Furling, D., Tronchère, H., Laporte, J., Lerouge, T., Butler-Browne, G.S. and Mandel, J.L. (2002) Muscle-specific alternative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells. Hum. Mol. Genet., 11, 2297–2307. [DOI] [PubMed] [Google Scholar]
- 34. Otero, B.A., Poukalov, K., Hildebrandt, R.P., Thornton, C.A., Jinnai, K., Fujimura, H., Kimura, T., Hagerman, K.A., Sampson, J.B., Day, J.W.et al. (2021) Transcriptome alterations in myotonic dystrophy frontal cortex. Cell Rep., 34, 108634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Savkur, R.S., Philips, A.V. and Cooper, T.A. (2001) (2001) Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet., 291, 40–47. [DOI] [PubMed] [Google Scholar]
- 36. Sen, S., Talukdar, I., Liu, Y., Tam, J., Reddy, S. and Webster, N.J.G. (2010) Muscleblind-like 1 (Mbnl1) promotes insulin receptor exon 11 inclusion via binding to a downstream evolutionarily conserved intronic enhancer. J. Biol. Chem., 285, 25426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Echeverria, G.V. and Cooper, T.A. (2014) Muscleblind-like 1 activates insulin receptor exon 11 inclusion by enhancing U2AF65 binding and splicing of the upstream intron. Nucleic Acids Res., 42, 1893–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kino, Y., Washizu, C., Oma, Y., Onishi, H., Nezu, Y., Sasagawa, N., Nukina, N. and Ishiura, S. (2009) MBNL and CELF proteins regulate alternative splicing of the skeletal muscle chloride channel CLCN1. Nucleic Acids Res., 37, 6477–6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Du, H., Cline, M.S., Osborne, R.J., Tuttle, D.L., Clark, T.A., Donohue, J.P., Hall, M.P., Shiue, L., Swanson, M.S., Thornton, C.A.et al. (2010) Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol., 17, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kimura, T., Nakamori, M., Lueck, J.D., Pouliquin, P., Aoike, F., Fujimura, H., Dirksen, R.T., Takahashi, M.P., Dulhunty, A.F. and Sakoda, S. (2005) Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum. Mol. Genet., 14, 2189–2200. [DOI] [PubMed] [Google Scholar]
- 41. Hino, S.I., Kondo, S., Sekiya, H., Saito, A., Kanemoto, S., Murakami, T., Chihara, K., Aoki, Y., Nakamori, M., Takahashi, M.P.et al. (2007) Molecular mechanisms responsible for aberrant splicing of SERCA1 in myotonic dystrophy type 1. Hum. Mol. Genet., 16, 2834–2843. [DOI] [PubMed] [Google Scholar]
- 42. Goers, E.S., Purcell, J., Voelker, R.B., Gates, D.P. and Berglund, J.A. (2010) MBNL1 binds GC motifs embedded in pyrimidines to regulate alternative splicing. Nucleic Acids Res., 38, 2467–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lambert, N., Robertson, A., Jangi, M., McGeary, S., Sharp, P.A. and Burge, C.B. (2014) RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol. Cell, 54, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gates, D.P., Coonrod, L.A. and Berglund, J.A. (2011) Autoregulated splicing of muscleblind-like 1 (MBNL1) Pre-mRNA. J. Biol. Chem., 286, 34224–34233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cumming, S.A., Jimenez-Moreno, C., Okkersen, K., Wenninger, S., Daidj, F., Hogarth, F., Littleford, R., Gorman, G., Bassez, G., Schoser, B.et al. (2019) Genetic determinants of disease severity in the myotonic dystrophy type 1 OPTIMISTIC cohort. Neurology, 93, e995–e1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bachinski, L.L., Baggerly, K.A., Neubauer, V.L., Nixon, T.J., Raheem, O., Sirito, M., Unruh, A.K., Zhang, J., Nagarajan, L., Timchenko, L.T.et al. (2014) Most expression and splicing changes in myotonic dystrophy type 1 and type 2 skeletal muscle are shared with other muscular dystrophies. Neuromuscul. Disord., 24, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sanoudou, D., Kang, P.B., Haslett, J.N., Han, M., Kunkel, L.M. and Beggs, A.H. (2004) Transcriptional profile of postmortem skeletal muscle. Physiol. Genomics, 16, 222–228. [DOI] [PubMed] [Google Scholar]
- 48. Lindeblad, G., Kroksmark, A.K. and Ekström, A.B. (2019) Cognitive and adaptive functioning in congenital and childhood forms of myotonic dystrophy type 1: a longitudinal study. Dev. Med. Child Neurol., 61, 1214–1220. [DOI] [PubMed] [Google Scholar]
- 49. Barcelo, J.M., Pluscauskas, M., MacKenzie, A.E., Tsilfidis, C., Narang, M. and Korneluk, R.G. (1994) Additive influence of maternal and offspring DM-kinase gene CTG repeat lengths in the genesis of congenital myotonic dystrophy. Am. J. Hum. Genet., 54, 1124. [PMC free article] [PubMed] [Google Scholar]
- 50. Novelli, G., Gennarelli, M., Menegazzo, E., Angelini, C. and Dallapiccola, B. (1995) Discordant clinical outcome in myotonic dystrophy relatives showing (CTG)n > 700 repeats. Neuromuscul. Disord., 5, 157–159. [DOI] [PubMed] [Google Scholar]
- 51. Spranger, M., Janssen, B., Rating, D. and Spranger, S. (1999) Disease picture of myotonic muscular dystrophy in patients with large CTG triplet expansion. Nervenarzt, 70, 131–135. [DOI] [PubMed] [Google Scholar]
- 52. Hogrel, J.Y., Ollivier, G., Ledoux, I., Hébert, L.J., Eymard, B., Puymirat, J. and Bassez, G. (2017) Relationships between grip strength, myotonia, and CTG expansion in myotonic dystrophy type 1. Ann. Clin. Transl. Neurol., 4, 921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Overend, G., Legare, C., Mathieu, J., Bouchard, L., Gagnon, C. and Monckton, D.G. (2019) Allele length of the DMPK CTG repeat is a predictor of progressive myotonic dystrophy type 1 phenotypes. Hum. Mol. Genet., 28, 2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thornton, C.A., Johnson, K. and Moxley, R.T. (1994) Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann. Neurol., 35, 104–107. [DOI] [PubMed] [Google Scholar]
- 55. Farkas-Bargeton, E., Barbet, J.P., Dancea, S., Wehrle, R., Checouri, A. and Dulac, O. (1988) Immaturity of muscle fibers in the congenital form of myotonic dystrophy: its consequences and its origin. J. Neurol. Sci., 83, 145–159. [DOI] [PubMed] [Google Scholar]
- 56. Furling, D., Coiffier, L., Mouly, V., Barbet, J.P., Lacau, J., Taneja, K., Gourdon, G., Junien, C. and Butler-Browne, G.S. (2001) Defective satellite cells in congenital myotonic dystrophy. Hum. Mol. Genet., 10, 2079–2087. [DOI] [PubMed] [Google Scholar]
- 57. Shi, D.L. and Grifone, R. (2021) RNA-binding proteins in the post-transcriptional control of skeletal muscle development, regeneration and disease. Front. Cell Dev. Biol., 9, 2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zu, T., Gibbens, B., Doty, N.S., Gomes-Pereira, M., Huguet, A., Stone, M.D., Margolis, J., Peterson, M., Markowski, T.W., Ingram, M.A.C.et al. (2011) Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. U. S. A., 108, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Castelli, L.M., Huang, W.P., Lin, Y.H., Chang, K.Y. and Hautbergue, G.M. (2021) Mechanisms of repeat-associated non-AUG translation in neurological microsatellite expansion disorders. Biochem. Soc. Trans., 49, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lanni, S. and Pearson, C.E. (2019) Molecular genetics of congenital myotonic dystrophy. Neurobiol. Dis., 132, 104533. [DOI] [PubMed] [Google Scholar]
- 61. Michel, L., Huguet-Lachon, A. and Gourdon, G. (2015) Sense and antisense DMPK RNA foci accumulate in DM1 tissues during development. PLoS One, 10, e0137620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. André, L.M., Van Cruchten, R.T.P., Willemse, M. and Wansink, D.G. (2019) (CTG)n repeat-mediated dysregulation of MBNL1 and MBNL2 expression during myogenesis in DM1 occurs already at the myoblast stage. PLoS One, 14, e0217317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Morales, F., Corrales, E., Zhang, B., Vásquez, M., Santamaría-Ulloa, C., Quesada, H., Sirito, M., Estecio, M.R., Monckton, D.G. and Krahe, R. (2021) Myotonic dystrophy type 1 (DM1) clinical subtypes and CTCF site methylation status flanking the CTG expansion are mutant allele length-dependent. Hum. Mol. Genet., 31, 262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. André, L.M., vanCruchten, R.T.P., Willemse, M., Bezstarosti, K., Demmers, J.A.A., vanAgtmaal, E.L., Wansink, D.G. and Wieringa, B. (2019) Recovery in the myogenic program of congenital myotonic dystrophy myoblasts after excision of the expanded (CTG) n repeat. Int. J. Mol. Sci., 20, 5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gudde, A.E.E.G., González-Barriga, A., van denBroek, W.J.A.A., Wieringa, B. and Wansink, D.G. (2016) A low absolute number of expanded transcripts is involved in myotonic dystrophy type 1 manifestation in muscle. Hum. Mol. Genet., 25, 1648–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thornton, C.A., Wang, E. and Carrell, E.M. (2017) Myotonic dystrophy: approach to therapy. Curr. Opin. Genet. Dev., 44, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Overby, S.J., Cerro-Herreros, E., Llamusi, B. and Artero, R. (2018) RNA-mediated therapies in myotonic dystrophy. Drug Discov. Today, 23, 2013–2022. [DOI] [PubMed] [Google Scholar]
- 68. Coonrod, L.A., Nakamori, M., Wang, W., Carrell, S., Hilton, C.L., Bodner, M.J., Siboni, R.B., Docter, A.G., Haley, M.M., Thornton, C.A.et al. (2013) Reducing levels of toxic RNA with small molecules. ACS Chem. Biol., 8, 2528–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Siboni, R.B., Nakamori, M., Wagner, S.D., Struck, A.J., Coonrod, L.A., Harriott, S.A., Cass, D.M., Tanner, M.K. and Berglund, J.A. (2015) Actinomycin D specifically reduces expanded CUG repeat RNA in myotonic dystrophy models. Cell Rep., 13, 2386–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pinto, B.S., Saxena, T., Oliveira, R., Méndez-Gómez, H.R., Cleary, J.D., Denes, L.T., McConnell, O., Arboleda, J., Xia, G., Swanson, M.S.et al. (2017) Impeding transcription of expanded microsatellite repeats by deactivated Cas9. Mol. Cell, 68, 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pandey, S.K., Wheeler, T.M., Justice, S.L., Kim, A., Younis, H.S., Gattis, D., Jauvin, D., Puymirat, J., Swayze, E.E., Freier, S.M.et al. (2015) Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1s. J. Pharmacol. Exp. Ther., 355, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Carrell, S.T., Carrell, E.M., Auerbach, D., Pandey, S.K., Bennett, C.F., Dirksen, R.T. and Thornton, C.A. (2016) Dmpk gene deletion or antisense knockdown does not compromise cardiac or skeletal muscle function in mice. Hum. Mol. Genet., 25, 4328–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang, L., Wang, S. and Li, W. (2012) RSeQC: quality control of RNA-seq experiments. Bioinformatics, 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- 74. Ewels, P., Magnusson, M., Lundin, S. and Käller, M. (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shen, S., Park, J.W., Lu, Z.X., Lin, L., Henry, M.D., Wu, Y.N., Zhou, Q. and Xing, Y. (2014) rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U. S. A., 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Liao, Y., Smyth, G.K. and Shi, W. (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–930. [DOI] [PubMed] [Google Scholar]
- 78. Durinck, S., Spellman, P.T., Birney, E. and Huber, W. (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc., 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chen, Y., Lun, A.T.L. and Smyth, G.K. (2016) From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Research, 5, 1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.