

Abstract
Objectives
To report a novel likely pathogenic variant in the SERAC1 gene associated with early adult-onset parkinsonism and progressive dystonia.
Methods
Clinical, biochemical, and imaging assessments were performed on 2 affected adult brothers with a genetically unsolved progressive neurologic disorder followed by whole-genome sequencing.
Results
A homozygous likely pathogenic variant in the SERAC1 gene (c.[129-2A > C], p.[(?)];[(?)]) was discovered.
Discussion
We describe a novel homozygous variant in the serine active site-containing protein 1 gene (SERAC1) in 2 brothers with a progressive extrapyramidal movement disorder of early onset parkinsonism and dystonia. Previous variants have been associated with a severe 3-methylglutaconic aciduria with dystonia, deafness, hepatopathy, encephalopathy and Leigh-like syndrome, or juvenile onset complicated spastic paraparesis. Our cases expand the phenotype of SERAC1 variants, with an adult-onset presentation of dystonia-parkinsonism.
The serine active site-containing protein 1 gene (SERAC1) encodes a PGAP1-like protein responsible for phospholipid phosphatidylglycerol remodeling—essential for mitochondrial function and intracellular cholesterol trafficking.1 Previously described homozygous or compound heterozygous variants have been associated with severe 3-methylglutaconic aciduria with dystonia, deafness, hepatopathy, encephalopathy and Leigh-like syndrome (MEGDHEL),1-3 or juvenile onset complicated spastic paraparesis.4 We report a novel homozygous variant presenting in 2 brothers with a late-onset phenotype.
Methods
Ethics
This project was approved by the University of Western Australia Human Research Ethics Committee (RA/4/20/1008).
Patient Consent
Written informed consent for genetic analysis, data publication, and clinical material disclosure for publication was provided.
Genetics Investigations
Whole-genome sequencing (WGS) of both individuals was performed using the Illumina HiSeq platform as previously published5 and data analyzed in the GENESIS platform.6 The variant was confirmed by bidirectional Sanger sequencing of DNA from both affected individuals. DNA from other family members was not available.
Data Availability
All data are available from the corresponding author on reasonable request.
Case Description
Two brothers aged 53 and 59 years had been patients of our clinic for 25 years with a progressive extrapyramidal syndrome. Both had normal birth and development, playing football in school, and while not academically minded, attended school until age 15 years. The elder brother (patient 1) was a laborer on the family farm, and the younger (patient 2) worked in the farm shop. At the ages 23 and 17 years, the brothers were in a motor vehicle accident and afterward sought medical attention for tremor, stiffness, dysphonia, and difficulty with handwriting. Initial examination showed hypomimia, slurring dysarthria and hypophonia, increased tone with sustained clonus, brisk reflexes with spread, bradykinesia, and an abnormal gait with no arm swing and toe walking with a flexed truncal posture. There was no freezing, festination, or loss of postural control. General examination showed gum hypertrophy, a high-arched palate, normal eye movements, and no orthostatic hypotension.
The unaffected deceased parents came from the same Italian village. Consanguinity was suspected but not confirmed. A deceased elder brother presented at age 2.5 years with seizures, followed by functional deterioration with an extrapyramidal syndrome, cognitive decline, and dysphagia. A postmortem at age 27 years gave a pathologic diagnosis of striatonigral degeneration; further details were not available. An autosomal recessive mode of inheritance was suspected.
Various medications were trialled with no clinical benefit: levodopa, benzhexol, tetrabenazine, and benztropine. Both developed sensorineural hearing loss, worsening dysarthria, and deterioration in mobility over the following 25-year period, requiring electric wheelchairs and assistance with personal care.
The most recent examinations at ages 59 and 53 years (Video 1, Video 2), revealed generalized dystonia, resting, and postural upper limb tremors, dysarthria, and hypophonia. Patient 2 had prominent orofacial dyskinesia, blepharospasm, and cog-wheeling rigidity, while patient 1 had more lower limb spasticity. There were no bradykinesia or cerebellar signs. Eye movements were essentially normal.
Neurologic examination of Patient 1.Download Supplementary Video 1 (19MB, mp4) via http://dx.doi.org/10.1212/200067_Video_1
Neurologic examination of Patient 2.Download Supplementary Video 1 (18.9MB, mp4) via http://dx.doi.org/10.1212/200067_Video_2
Results
MRI of the brain showed bilateral symmetric atrophy of the caudate head and anterior putamen, with associated gliosis (Figure). Initial testing of urine spot organic acids was reported within normal limits, with a nonsignificant increase in 3-methylglutaconic acid.
Figure. Pedigree and MRI.

(A) Family pedigree, with unknown degree of consanguinity. Patient 1 = II.2, Patient 2 = II.3. Age corresponds to current age or age at death. (B) MRI showing putamen (white arrows) and caudate (yellow arrows) atrophy and gliosis.
Previous genetic testing on a customized Dystonia/Parkinson gene panel by massively parallel sequencing was negative (eAppendix 1, links.lww.com/NXG/A590). Duo WGS and analysis revealed a homozygous likely pathogenic essential splice site variant in SERAC1 (c.[129-2A > C], p.[(?)];[(?)]). No other likely pathogenic or pathogenic variants were identified in clinically concordant genes. This variant meets the American College of Medical Genetics and Genomics criteria for likely pathogenic: PVS1 and PM2.
Subsequent repeat testing of urine spot organic acids showed a significant increase in 3-methylglutaconate levels (3+) and a mild increase in 3-methylglutarate (2+). Liver function tests, coagulation profile, and lipid profile continued to be normal.
Discussion
We present a new likely pathogenic splice variant of SERAC1 (c.[129-2A > C], p.[(?)];[(?)]) with a phenotype of adult-onset parkinsonism and progressive dystonia. The c.[129-2A > C] variant is in intron 3 within the splice-acceptor site for exon 4 of SERAC1 and is not present in gnomAD.7 Splice prediction software predicts 2 possible outcomes: skipping of the out-of-frame exon 4 or activation of a cryptic splice site 2 bases into the exon resulting in a frameshift. Either of these would result in a null allele, and hence, no protein product. Functional studies would be required to determine this.
The previously documented MEGDHEL phenotype has a neonatal onset and early mortality, with a median lifespan of 9 years.1-3 Another variant with a milder phenotype of juvenile onset complicated spastic paraplegia has been documented, with the onset of cognitive delay and spasticity at 2–7 years, and thought to be secondary to a truncated protein product.4 There are no long-term data on the evolution of this subtype. Our cases demonstrate the evolution of this forme fruste MEGDHEL, with delayed onset and significantly slower progression (Table). They demonstrate the same cardinal features of dystonia, basal ganglia abnormalities on MRI, and eventual sensorineural hearing loss and 3-methylglutaconic aciduria in the third decade of disease. Cognitive involvement is mild, and there was no significant regression documented at times of metabolic stress to support a Leigh-like syndrome and no hepatopathy. The parkinsonian features may not be apparent in the earlier onset forms of disease because of earlier spasticity.
Table.
Clinical Comparison of Case Series in the Literature With Patient 1 and 2
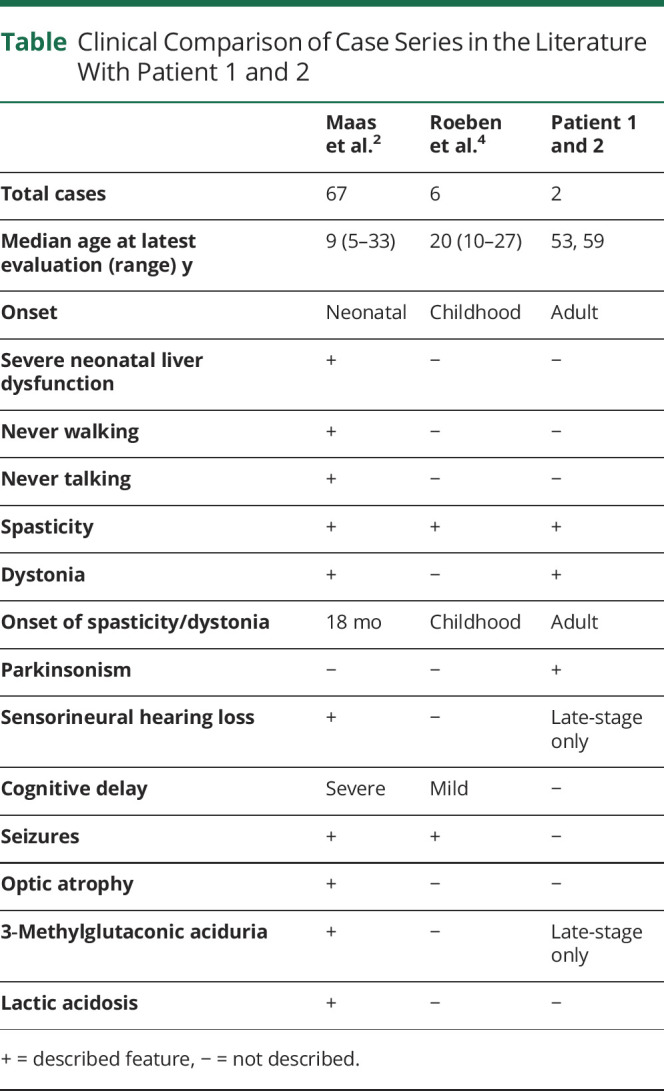
The milder phenotype of our patients, in comparison with their deceased brother, may be due to other yet unknown modifying factors, including earlier metabolic stress. These cases demonstrate the phenotypic variability of the secondary mitochondriopathies and the influence of phospholipid phosphatidylglycerol remodeling on neurodegeneration. The lack of biochemical abnormalities on initial metabolic screening supports the inclusion of SERAC1 genetic testing in patients with dystonia-parkinsonism.
Supplementary Material
Acknowledgment
The authors thank this family for agreeing to participate in the study.
Appendix. Authors
Study Funding
N.G. Laing was funded by an Australian National Health and Medical Research Council (NHMRC) Fellowship APP1117510. G. Ravenscroft is supported by a NHMRC EL2 Fellowship (APP2007769).
Disclosure
G. Ravenscroft and N. Laing are supported by NHMRC grant funding, as disclosed in the manuscript. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Wortmann SB, Vaz FM, Gardeitchik T, et al. . Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat Genet. 2012;44(7):797-802. doi: 10.1038/ng.2325 [DOI] [PubMed] [Google Scholar]
- 2.Maas RR, Iwanicka-Pronicka K, Kalkan Ucar S, et al. . Progressive deafness-dystonia due to SERAC1 mutations: a study of 67 cases. Ann Neurol. 2017;82(6):1004-1015. doi: 10.1002/ana.25110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radha Rama Devi A, Lingappa L. Novel mutations in SERAC1 gene in two Indian patients presenting with dystonia and intellectual disability. Eur J Med Genet. 2018;61(2):100-103. doi: 10.1016/j.ejmg.2017.07.013 [DOI] [PubMed] [Google Scholar]
- 4.Roeben B, Schüle R, Ruf S, et al. . SERAC1 deficiency causes complicated HSP: evidence from a novel splice mutation in a large family. J Med Genet. 2018;55(1):39-47. doi: 10.1136/jmedgenet-2017-104622 [DOI] [PubMed] [Google Scholar]
- 5.Martin PB, Kigoshi-Tansho Y, Sher RB, et al. . NEMF mutations that impair ribosome-associated quality control are associated with neuromuscular disease. Nat Commun. 2020;11(1):4625. doi: 10.1038/s41467-020-18327-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez M, Falk MJ, Gai X, Postrel R, Schüle R, Zuchner S. Innovative genomic collaboration using the GENESIS (GEM.app) platform. Hum Mutat. 2015;36(10):950-956. doi: 10.1002/humu.22836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karczewski KJ, Francioli LC, Tiao G, et al. . The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Neurologic examination of Patient 1.Download Supplementary Video 1 (19MB, mp4) via http://dx.doi.org/10.1212/200067_Video_1
Neurologic examination of Patient 2.Download Supplementary Video 1 (18.9MB, mp4) via http://dx.doi.org/10.1212/200067_Video_2
Data Availability Statement
All data are available from the corresponding author on reasonable request.