

Abstract
Secondary bacterial infections increase influenza-related morbidity and mortality, particularly if acquired after 5–7 d from the viral onset. Synergistic host responses and direct pathogen–pathogen interactions are thought to lead to a state of hyperinflammation, but the kinetics of the lung pathology have not yet been detailed, and identifying the contribution of different mechanisms to disease is difficult because these may change over time. To address this gap, we examined host–pathogen and lung pathology dynamics following a secondary bacterial infection initiated at different time points after influenza within a murine model. We then used a mathematical approach to quantify the increased virus dissemination in the lung, coinfection time-dependent bacterial kinetics, and virus-mediated and postbacterial depletion of alveolar macrophages. The data showed that viral loads increase regardless of coinfection timing, which our mathematical model predicted and histomorphometry data confirmed was due to a robust increase in the number of infected cells. Bacterial loads were dependent on the time of coinfection and corresponded to the level of IAV-induced alveolar macrophage depletion. Our mathematical model suggested that the additional depletion of these cells following the bacterial invasion was mediated primarily by the virus. Contrary to current belief, inflammation was not enhanced and did not correlate with neutrophilia. The enhanced disease severity was correlated to inflammation, but this was due to a nonlinearity in this correlation. This study highlights the importance of dissecting nonlinearities during complex infections and demonstrated the increased dissemination of virus within the lung during bacterial coinfection and simultaneous modulation of immune responses during influenza-associated bacterial pneumonia.
Keywords: influenza, pneumococcus, viral–bacterial coinfection, lung injury, inflammation, mathematical modeling
Histomorphometry of the IAV–Sp coinfected lung showing the areas of influenza NP-positive “active” lesions (red), previously infected “inactive” lesions with minimal antigen-positive debris (green), or mixed active and inactive regions (orange) during IAV infection or IAV–Sp coinfection.
Introduction
Influenza A virus (IAV) infections can be complicated by coinfecting bacterial pathogens, which has resulted in increased morbidity and mortality during seasonal epidemics and pandemics (Louria et al. 1959, Thompson 2004, Brundage and Shanks 2008, Morens et al. 2008, Chien et al. 2009, Klugman et al. 2009, Morens et al. 2009, Weinberger et al. 2012, Reed et al. 2015). Streptococcus pneumoniae (Sp; pneumococcus) is a common coinfecting pathogen that has accounted for up to 95% of influenza-related mortality in past pandemics (Louria et al. 1959, Morens et al. 2008, Chien et al. 2009, Weinberger et al. 2012). The viral infection deteriorates both the physiological and immunological protective barriers and renders hosts more susceptible to invading pathogens [reviewed in references McCullers (2006), Short et al. (2012), Metzger and Sun (2013), Smith and McCullers (2014), Kash and Taubenberger (2015), and Rynda-Apple et al. (2015)]. Viral and bacterial strain and dose, the timing and order of infections, and numerous host and pathogen factors seem to influence the susceptibility and pathogenicity of severe pneumonia. However, viral–bacterial coinfections are typically quite complex with multiple mechanisms concurrently affecting the dynamics, and current knowledge about how each pathogen contributes and affects lung pathology is limited.
Numerous studies have identified alveolar macrophages (AMΦs) as critical in defining bacterial susceptibility during influenza while neutrophils have a greater role in coinfection pathogenicity [reviewed in references McCullers (2006), Short et al. (2012), Metzger and Sun (2013), Smith and McCullers (2014), Kash and Taubenberger (2015), and Rynda-Apple et al. (2015)]. The dynamic depletion of AMΦs throughout an IAV infection is predictive of the initial trajectory and rate of bacterial growth during a secondary pneumococcal infection and one reason why lethality is maximal at 7 d postinfluenza, which is when AMΦ depletion is at its greatest (Ghoneim et al. 2013, Smith et al. 2013, Smith and Smith 2016). Our mathematical model was critical in defining this mechanism (Smith et al. 2013) and established a nonlinear threshold that governs whether bacteria exhibit a growth or clearance phenotype within the first 4 h of infection based on the extent of AMΦ depletion and the bacterial inoculum size (Smith and Smith 2016). However, bacterial loads rebound if they are not cleared within this time frame (Smith and Smith 2016). This may be a consequence of further AMΦ loss due to either the bacteria or virus, but currently no studies have defined the effect on these cells once the bacteria has invaded.
Another important feature of an IAV–Sp coinfection is the rebound of viral loads (Smith et al. 2013, 2021), which few studies have examined. Our previous studies quantified the rebound for a coinfection 7 d after influenza (Smith et al. 2013, 2021), but it is unknown whether this rebound occurs if the coinfection occurs at other times or how much this affects disease severity. In addition, the underlying mechanism(s) leading to this increase in virus have been more elusive. Our model hypothesized that it was the consequence of a bacteria-induced increase in the rate of virus production (Smith et al. 2013). While our model could not identify the exact mechanism, there have since been a few studies that identified mechanisms that could potentially lead to the viral rebound. One in vitro study showed that Staphylococcus aureus can enter a virus-infected cell and interfere with the antiviral interferon response, which led to an increase in viral loads (Warnking et al. 2015). Whether pneumococcus can also do this is unknown. Another study showed that CD8 T cells are depleted once the bacteria is introduced, which may contribute to an increase in viral loads (Blevins et al. 2014), but our recent study on these cells (Myers et al. 2021) suggested that their level would need to be quite low for this to lead to a significant and immediate increase in viral loads. More recently, it was shown that the influenza virus can attach to pneumococcus (Rowe et al. 2019). Although the in vivo consequences were not thoroughly explored, it could feasibly lead to an increase in virus dissemination in the lung. The current study addresses this possibility.
If the increase in viral loads during bacterial coinfection is a consequence of viral dissemination in the lung, we may expect this to contribute to lung damage and, thus, disease severity. However, our recent study on influenza suggested that virus-induced lung damage is nonlinearly related to disease severity (Myers et al. 2021). In addition, inflammation, which had distinct dynamics compared with the lung damage, was also nonlinearly related to the severity and log-linearly correlated to both inflammatory macrophages and neutrophils. While we may expect that the significant neutrophilia during influenza-associated bacterial infections would increase lung inflammation, these nonlinearities make it difficult to directly assess the consequences of host–pathogen interactions.
To better understand the host–pathogen dynamics during IAV–Sp coinfection and the contributions of the virus, bacteria, and host immune responses to lung pathologies, we combined a mathematical model with data from mice infected with influenza followed by pneumococcus at either 3, 5, or 7 d postvirus infection (pvi). The data showed that viral loads rebound regardless of coinfection timing, and the model suggested that this was primarily due to an increase in the number of infected cells. This finding was confirmed using quantitative histomorphometry, which showed that the number of infected cells increases and does so more robustly for earlier bacterial invasion. At all coinfection times, additional AMΦ loss was evident following bacterial establishment, and our model suggested that this was primarily virus-mediated. However, it was not sufficient to have a robust impact on bacterial dynamics. Unexpectedly, inflammation was generally not enhanced during the coinfection and was not correlated with neutrophilia. These results highlight important nonlinearities in host–pathogen dynamics and improve our knowledge of viral–bacterial coinfections.
Materials and methods
Ethics statement
All experimental procedures were performed under protocols O2A-020 or 17–096 approved by the Animal Care and Use Committees at St. Jude Children’s Research Hospital (SJCRH) or the University of Tennessee Health Science Center (UTHSC), respectively, under relevant institutional and American Veterinary Medical Association (AVMA) guidelines. All experimental procedures were performed in a biosafety level 2 facility, i.e. accredited by the American Association for Laboratory Animal Science (AALAS).
Mice
Adult (6 week old) female BALB/cJ mice were obtained from Jackson Laboratories (Bar Harbor, ME) or Charles River Laboratories (Wilmington, MA). Mice were housed in groups of five mice in high-temperature 31.2 cm × 23.5 cm × 15.2 cm polycarbonate cages with isolator lids (SJCRH) or in 38.2 cm × 19.4 cm × 13.0 cm solid-bottom polysulfone individually ventilated cages (UTHSC). Rooms used for housing mice were maintained on a 12:12-hour light:dark cycle at 22 ± 2°C with 50% humidity in the biosafety level 2 facility at SJCRH (Memphis, TN) or UTHSC (Memphis, TN). Before inclusion in the experiments, mice were allowed at least 7 d to acclimate to the animal facility such that they were 7 weeks old at the time of infection. Laboratory Autoclavable Rodent Diet (PMI Nutrition International, St. Louis, MO; SJCRH) or Teklad LM-485 Mouse/Rat Sterilizable Diet (Envigo, Indianapolis, IN; UTHSC) and autoclaved water were available ad libitum. All experiments were performed under an approved protocol and in accordance with the guidelines set forth by the Animal Care and Use Committee at SJCRH or UTHSC.
Infection experiments
The mouse-adapted influenza A/Puerto Rico/8/34 (H1N1; PR8) and type 2 pneumococcal strain D39 were used for all experiments. The viral infectious dose (TCID50) was determined by interpolation using the Reed and Muench method (Reed and Muench 1938) with serial dilutions of virus on Madin-Darby canine kidney (MDCK) cells and hemagglutination of chicken red blood cells (cRBCs). Bacterial infectious doses (CFU) were determined using serial dilutions on tryptic soy-agar plates supplemented with 3% sheep erythrocyte (TSA plates). Inocula were diluted in sterile PBS and administered intranasally (total volume of 100 µl, 50 µl per nostril) to groups of five mice lightly anesthetized with 2.5% inhaled isoflurane (Baxter, Deerfield, IL). For coinfection dynamics, mice were infected with either PBS or 75 TCID50 PR8 at day 0 then with 103 CFU of D39 at 3, 5, or 7 d later. Animals were weighed daily to monitor illness and mortality. Animals were weighed daily to monitor illness and euthanized if they became moribund or lost 30% of their starting body weight.
Lung harvesting for titering and flow cytometry
Mice were euthanized by CO2 asphyxiation (SJCRH) or 33% isoflurane inhalation (UTHSC). Lungs were digested with collagenase (1 mg/ml, Sigma C0130), and physically homogenized by syringe plunger against a 40 μm cell strainer. Cell suspensions were centrifuged at 4°C, 500 × g for 7 min. The supernatants were used to determine the viral and bacterial titers. Following red blood cell lysis, cells were washed in MACS buffer (PBS, 0.01 M HEPES, 5 mM EDTA, and 5% heat-inactivated FBS). Cells were then counted with trypan blue exclusion using a Cell Countess System (Invitrogen, Grand Island, NY) and prepared for flow cytometric analysis as indicated below.
Lung immunohistopathology and immunohistochemistry (IHC)
The lungs from IAV infected mice were fixed via intratracheal infusion and then immersion in 10% buffered formalin solution. Tissues were paraffin-embedded, sectioned, and stained for influenza virus using a primary goat polyclonal antibody (US Biological, Swampscott, MA) against influenza A, USSR (H1N1) at 1:1000 and a secondary biotinylated donkey antigoat antibody (sc-2042; Santa Cruz Biotechnology, Santa Cruz, CA) at 1:200 on tissue sections subjected to antigen retrieval for 30 min at 98°C. The extent of virus spread was quantified by capturing digital images of whole-lung sections (2D) stained for viral antigen using an Aperio ScanScope XT Slide Scanner (Aperio Technologies, Vista, CA) then manually outlining defined fields. Alveolar areas containing virus antigen-positive pneumocytes were highlighted in red (defined as “active” infection), whereas lesioned areas containing minimal or no virus antigen-positive debris were highlighted in green (defined as “inactive” infection). Lesions containing a mix of virus antigen-positive and antigen-negative pneumocytes were highlighted in orange (defined as “mixed” infection). The percentage of each defined lung field was calculated using the Aperio ImageScope software. Pulmonary lesions in HE-stained histologic sections were assigned scores based on their severity and extent. Pulmonary lesions were assigned severity scores on a scale from 0 to 5 by a veterinary pathologist, where 0 = normal: no tissue affected, 1 = minimal: rare or inconspicuous lesions, 2 = mild: multifocal or small, but prominent lesions, 3 = moderate: multifocal, prominent lesions, 4 = marked: extensive to coalescing lesions or areas of inflammation with some loss of structure, and 5 = severe: extensive or diffuse lesions with effacement of normal structure. Intermediate severity grades were assigned where necessary.
Flow cytometric analysis
Single cell suspensions were stained for flow cytometric analysis. Fc receptors were blocked using human-γ globulin, followed by surface marker staining with antimouse antibodies: CD11c (eFluor450, clone N418, eBioscience), CD11b (Alexa700, clone M1/70, BD Biosciences), Ly6G (PerCp-Cy5.5, clone 1A8, Biolegend), Ly6C (APC, clone HK1.4, eBioscience), F4/80 (PE, clone BM8, eBioscience), CD3e (PE-Cy7, clone 145–2C11, BD Biosciences or BV785 Biolegend), CD4 (PE-Cy5, clone RM4-5, BD Biosciences), CD8a (BV605, clone 53–6.7, BD Biosciences), CD49b (APC-Cy7, Biolegend or APC-e780, clone DX5, Affymetrix Inc), and MHC-II (FITC, clone M5/114.15.2, eBioscience). Samples were run on a LSRII Fortessa (Becton Dickinson, San Jose, CA) and data were analyzed using FlowJo 10.0.8 (Tree Star, Ashland, OR). Viable cells were gated from a forward scatter/side scatter plot and singlet inclusion. Following neutrophil exclusion (Ly6Ghi), macrophages (MΦ) were gated as CD11chiF4/80hi with alveolar macrophages (AMΦ) subgated as CD11b– and inflammatory macrophages (iMacs) as CD11b+. AMΦ were confirmed to have high side-scatter and be negative for MHC-II, CD11b, CD3e, CD4, CD8a, and DX5. The absolute numbers of cells were calculated based on the gating of viable events by flow cytometry and normalized to the total number of viable cells in the lung digest.
Mathematical model
We used differential equation models that describe IAV infection (Smith et al. 2018, Myers et al. 2021) and IAV–Sp coinfection (Smith et al. 2013). Briefly, target cells become infected with the virus at rate βV (β = 5.5e-5 per TCID50/day). These cells transition from an eclipse phase (I1) at rate k (4/d) before the cells (I2) produce virus (V) at rate p (3.2 TCID50/cell/d), which was assumed to be enhanced by the presence of bacteria (B) according to the function aBz (a = 3e-4 per CFU; z = 0.5). Virus is cleared at rate c (18.1/d), and virus-producing infected cells are cleared in a density-dependent manner at rate δ(I2) = δ/(Kδ+I2) (δ = 1.7e6 cells/day; Kδ = 1.4e5 cells). Bacteria are produced logistically at rate r (27 CFU/d) and carrying capacity KB (2.8e7 CFU) and cleared by alveolar macrophages (AM) at rate γ(B) = γKM2n2/(B2+ KMn2) (γ = 1.4e-4; KM = 7.5e5; n = 5), i.e. reduced with AMΦ loss (φ(AM) = 1-AM/AM(0) is the percentage of depletion). AMΦs were assumed to be constantly produced at rate sA (3.0e5 cells/day), cleared at rate dA (3.3 per day), and depleted by an unknown intermediate (C) at rate ρ (7.4e6 per C/d). This intermediate was assumed to be produced proportional to the infected cell classes at rates ν1 (6.1e-3 C/cell/d) and ν2 (6.1e-2 C/cell/d), respectively, and at rate ω (3.1e-7 per cell/d) after a delay τ (2.6 d) and cleared at rate dC (1.5 per day). Inflammation (LI) was modeled by assuming that virus-infected cells (I1 and I2) produce cytokines that result in the infiltration of other cells (e.g. macrophages and neutrophils). The parameters α1 (4.3e-7 score/cell/day) and α2 (8.7e-8 score/cell/day) define the contribution from each infected cell class.
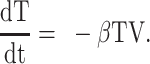 |
(1) |
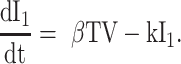 |
(2) |
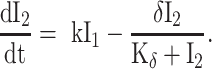 |
(3) |
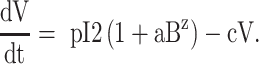 |
(4) |
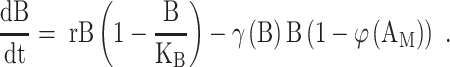 |
(5) |
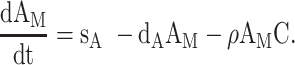 |
(6) |
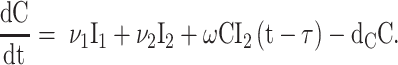 |
(7) |
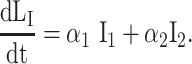 |
(8) |
The function cumtrapz in MATLAB was used to estimate the CAUC of the infected cells (I2).
Statistical analysis
Unpaired t-tests were used on the linear values of the data to assess the significance. P values of < 0.05 were considered significant. The function polyfit in MATLAB was used to perform linear regressions.
Results
IAV–Sp coinfection kinetics vary depending on time of bacterial initiation
To better assess the dynamics during IAV–Sp coinfection, BALB/cJ mice were infected with 75 TCID50 influenza A/Puerto Rico/8/34 (PR8) followed by either a mock infection or 103 CFU type 2 pneumococcus strain D39 at 3, 5, or 7 d pvi (Fig. 1). In the IAV-infected groups, the depletion of AMΦ was evident immediately after influenza where 17% were lost within 1 d pvi. Similar to our prior results, this continued to increase significantly and was maximal at 7 d pvi with an ∼83% loss (Ghoneim et al. 2013, Smith et al. 2013). The loss of these cells increased during IAV–Sp coinfected groups where 66%, 89%, and 94% of the cells were lost within 48 h after bacterial infection at 3, 5, or 7 d pvi, respectively, compared with 56%, 83%, and 62% at the same time points during IAV infection (Fig. 1A–C).
Figure 1.

IAV–Sp coinfection kinetics. Viral titers, bacterial titers, and alveolar macrophages (AMΦ) during IAV infection (75 TCID50 PR8; black squares) or IAV–Sp coinfection (colored circles), where animals were infected with 75 TCID50 PR8 followed by 103 CFU D39 at 3 d (A; magenta), 5 d (B; green), or 7 d (C; blue) pvi. Data are shown for five individual animals with virus, bacteria, and AMΦ simultaneously measured. Dynamics of the model in Equations (1–8) are shown (solid line) for each scenario, and two trajectories with differing values of KM = 7.5e5 (solid) or 4e5 (dashed) are shown for a coinfection at 3 d pvi. (D) Percentage of weight following IAV infection (black squares) or IAV–Sp coinfection (colored circles). Time indicates time postinfluenza infection.
Consistent with our prior study showing the importance of the ratio of bacteria to AMΦs in defining the initial trajectory and heterogeneity of bacterial loads, the kinetics of pulmonary bacterial loads depended on the day of coinfection (Fig. 1). When animals were infected with bacteria at 3 d pvi, which is below an initial dose threshold (Smith and Smith 2016), 30%–40% of the animals were able to clear the bacteria while others had their bacterial loads at higher levels [maximum of 5.38 log10 colony forming units (CFU)] at 24 h pbi (Fig. 1A). By 72 h pbi, the maximum reached 7.58 log10 CFU. Our prior study showed if clearance occurs, it would be within the first 4 h (Smith and Smith 2016). In addition, the heterogeneity was reproducible and decreased as the ratio of bacteria to AMΦs is closer to the initial dose threshold (Smith and Smith 2016). This was observed in the groups that were coinfected at 5 d pvi, where bacterial loads were higher at 24 h and 48 h pbi (maximums of 7.50 and 8.12 log10 CFU, respectively), on average, compared to the 3 d coinfected groups (average of 5.84 and 6.67 log10 CFU versus 2.90 and 3.35 log10 CFU; P = 0.08 and P = 0.04), and the heterogeneity was reduced (SD: 1.6 and 1.8 versus 2.2 and 3.1; Fig. 1B). However, some animals had bacterial levels that were below the value of the inoculum (minimum was 2.24 log10 CFU). The heterogeneity was relatively abolished during a coinfection at 7 d pvi (SD: 0.4 and 0.6), where all animals had high bacterial loads (average of 7.69 log10 CFU) that stayed relatively constant (Fig. 1C). Similar to prior studies (Ghoneim et al. 2013, Smith and Smith 2016), these coinfection time-varying dynamics correspond to the level of AMΦ depletion at the time of coinfection.
Viral loads, on the other hand, were not dependent on the time of coinfection. Rather, viral loads in the lungs of coinfected animals rebounded following coinfection for each group with significantly higher levels by 24 h pbi (P < 0.01; Fig. 1). Interestingly, the pattern was similar regardless of the coinfection timing and viral clearance was only visible during the 7 d coinfection, a time when robust CD8+ T cell-mediated clearance is occurring (Myers et al. 2021). On the time scale that we evaluated (24–72 h pbi), mortality only occurred in the 7 d coinfection group with 1/10 succumbing to the infection at 48 h pbi and 3/10 at 72 h pbi. Of the remaining animals at 72 h pbi, four were able to clear the virus. Weight loss significantly increased by > 10% within 72 h pbi during each of the coinfections [3% versus 17% (3 d coinfection); 7% versus 18.3% (5 d coinfection); and 13.4% versus 23.5% (7 d coinfection); all P < 0.01; Fig. 1D].
Modeling coinfection kinetics suggests contributions of different mechanisms
To better detail the dynamics of the viral rebound, bacterial growth, and AMΦ depletion, we simulated the model in Equations (1–8) for each coinfection timing (Fig. 1). As expected, the model produced time-varying bacterial load dynamics due to the depletion of AMΦs where the model predicted immediate clearance of bacteria if the coinfection was initiated at 3 d pvi but rapid growth if it was given at 5 or 7 d pvi. Small alterations to one parameter (KM) could reproduce variable dynamics (outgrowth versus clearance) in the 3 d coinfection group (Fig. 1A), confirming the tight regulation of the underlying processes. Interestingly, the dynamics of the intermediate that creates the decline in AMΦ within our model [C; Equation (7)] matched the dynamics of IFN-γ (not shown) during IAV infection, which aligns with prior reports suggesting this cytokine mediates their decline (Verma et al. 2020). In our prior work, modeling a bacterial coinfection at 7 d pvi (Smith et al. 2013), our mathematical model suggested that the rate of virus production was increased by the bacteria according to the nonlinear function aBz. However, including only this term in the model was insufficient to replicate the data for coinfections at 3 or 5 d pvi when bacteria were at lower levels. Because influenza virus can attach to pneumococci (Rowe et al. 2019), we hypothesized that this may transport the virus to new areas of the lung, which would increase the number of susceptible cells available for the virus to infect. Increasing the number of target cells by 1.8e7, 1.1e7, or 3.2e6 cells for coinfections at 3, 5, or 7 d pvi, respectively, was able to recover the viral load dynamics (Fig. 1). With the increased number of infected cells, the model predicted an additional loss of AMΦs, but it was not sufficient to capture the stark decline in these cells at 7 d pvi. However, assuming that interactions between AMΦs and bacteria could also lead to their decline produced dynamics consistent with the data (Fig. 1C), and supports the unaltered IFN-γ dynamics during coinfection (not shown).
Enhanced virus-induced lung injury during bacterial coinfection
To examine whether our model predicted the increase in infected cells accurately, we quantified these cells using whole lung histomorphometry (Fig. 2). The percentage of the lung that was infected by IAV significantly increased for coinfections at 3, 5, or 7 d pvi, respectively. The influenza nucleoprotein (NP)-positive areas of the lung (“active” lesions) significantly increased for coinfections at 3 or 5 d pvi and were only slightly higher for a coinfection at 7 d pvi (Fig. 2A–D). For a coinfection at 3 d pvi, the percentage of the lung that was infected by IAV rose from ∼20% (3 d pvi) to ∼74% within 48 h pbi versus 29.7% in the IAV-infected group at the same time point (5 d pvi; P < 1e-6). No cleared (“inactive”) lesions were visible in either the IAV-infected or IAV–Sp coinfected groups between 3 and 5 d pvi. For a coinfection at 5 d pvi, the % of the lung that was infected by IAV rose from ∼30% (5 d) to ∼65% within 48 h pbi versus ∼37% in the IAV-infected group at the same time point (7 d pvi; P < 0.001). No cleared (“inactive”) lesions were visible in the IAV–Sp coinfected groups while there was clearance (11.66% of the lung) beginning in the IAV-infected group (Fig. 2E). For a coinfection at 7 d pvi, the % of the lung that was infected by IAV increased slightly to ∼0.5% at 48 h pbi compared to 0% (P > 0.05) for IAV-infected groups at the same time point (9 d pvi). However, additional infection and subsequent clearance were more visible in the images and quantification of the inactive lesion where the damaged area was at an average of 53.8% in the coinfected group and 42.6% ( P = 0.2) in the IAV-infected group at 9 d pvi.
Figure 2.

Histomorphometry of the IAV–Sp coinfected lung. (A)–(C) Whole lung sections with histomorphometry showing the areas of influenza NP-positive “active” lesions (red), previously infected “inactive” lesions with minimal antigen-positive debris (green), or mixed active and inactive regions (orange) during IAV infection (Myers et al. 2021; 75 TCID50 PR8; top) or IAV–Sp coinfection (bottom), where animals were infected with 75 TCID50 PR8 followed by 103 CFU D39 at 3, 5, or 7 d pvi. Day pvi is indicated, and representative images from each group are shown. (D)–(E) Percent active lesion (D) and inactive lesion (E) for IAV (Myers et al. 2021; black) and IAV–Sp (magenta, 3 d coinfection; green, 5 d coinfection; and blue, 7 d coinfection). For the active lesion, the cumulative area under the curve (CAUC) of the predicted infected cell dynamics (I2) is plotted (solid line). To include the data and CAUC(I2) on the same scale, the CAUC(I2) was multiplied by a scaling factor of 14.2% per 1 × 107 cells. (F) Fit of a saturating function (black line) to the mean percental total lesion, which is the addition of the active and inactive lesions, and mean weight loss for all time points during IAV infection (Myers et al. 2021). All data are shown as mean ± standard deviation.
Our prior work established that the percentage of the active lesion could be estimated using the cumulative area under the curve (CAUC) of the productively infected cells (I 2) because it was a measure of the influenza-positive cells. Plotting the CAUC of I2 against the percentage of active lesion for each of the coinfections showed that our model-predicted increase in the number of infected cells was accurate (Fig. 2D). In addition, we previously found that, during an IAV infection, the percentage of total lesion nonlinearly correlates to weight loss and could be approximated using a Hill (S-shaped) function (Myers et al . 2021; Fig. 2F). However, this function saturated once the total lesion was at ∼40% and the weight loss was > 6%. Thus, alterations to this correlation were not needed due to the high levels of weight loss.
Inflammation is relatively unaffected by bacterial coinfection
In addition to quantifying the amount of the lung that was infected, the lungs were also scored for alveolar and interstitial inflammation. Unlike the IAV infection where alveolar and interstitial inflammation were similar in their scores (Myers et al. 2021; Fig. 3A), these two measurements had some differences during the coinfection. However, the scores showed that alveolar and interstitial inflammation were not enhanced after a coinfection at 3 (all P > 0.1), 5 ( P > 0.1 for 24 h pbi), or 7 (all P > 0.2) d coinfection with the exception of 48 h after a 5 d coinfection (Fig. 3A). At this time point, the alveolar inflammation score was, on average, 4.8 out of 5 compared with 3 out of 5 (P = 0.0008) during an IAV infection, while the interstitial inflammation was lower (average score of 3 for all groups), which was similar to the score during an IAV infection (3.4 out of 5; P > 0.1).
Figure 3.
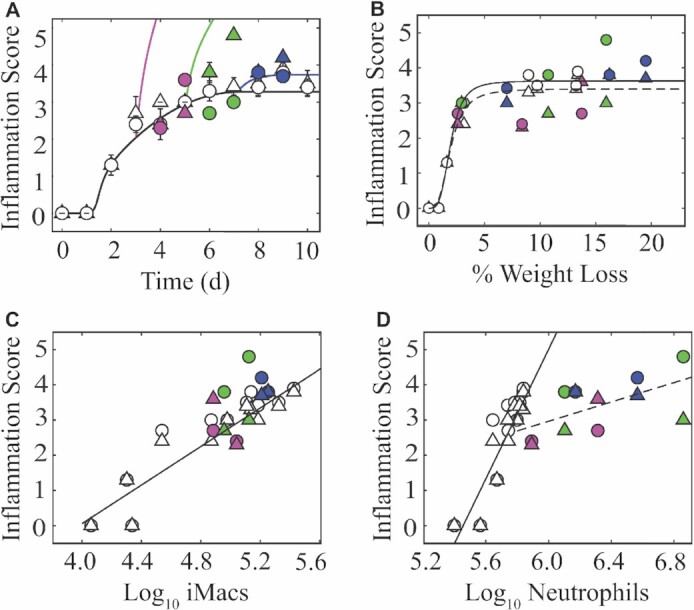
Lung inflammation during IAV–Sp coinfection. (A) Fit of the model to the alveolar (circles) and interstitial (triangles) inflammation scores from an IAV infection (Myers et al. 2021; 75 TCID50 PR8; white) or simulation of the equation for IAV–Sp coinfection [75 TCID50 PR8 followed by 103 CFU D39 at 3 d (magenta), 5 d (green), or 7 d (blue) d pvi]. Data are shown as mean ± standard deviation. (B) The average alveolar and interstitial inflammation scores plotted against the average percent weight loss. Fit of a saturating function (black line) to the mean alveolar (solid line) and interstitial (dashed line) inflammation scores and mean weight loss for all time points during IAV infection (Myers et al. 2021). (C)–(D) Regression analysis of the average alveolar inflammation and interstitial inflammation scores with the average (C) log10 inflammatory macrophages (iMacs; F480hiCD11chiCD11b+) and (D) log10 neutrophils (Ly6Ghi) for IAV infection (Myers et al. 2021; solid line). During IAV–Sp coinfection, the log-linear correlation between neutrophils and inflammation scores was not similar to IAV infection and the line was refit to these data (dashed line).
In our prior work, we approximated the inflammation dynamics with Equation (8) (Fig. 3A), which assumes inflammation can be approximated using the infected cell dynamics (Myers et al. 2021). Plotting this equation for the coinfections was accurate if the bacteria was given at 7 d pvi, but overestimated the amount of inflammation for the other two timings due to the heightened number of new infected cells (Fig. 3A), suggesting either that other cells are helping reduce the inflammation during the earlier time points (i.e. 3–5 d pvi) or that our equation is too simplistic.
Similar to the connection between lung lesions and disease severity, we also found inflammation tightly and nonlinearly correlated to disease severity during an IAV infection. This could also be approximated using a Hill (S-shaped) function (Myers et al. 2021; Fig. 3B), which saturated once inflammation reached a score of 3.4–3.6 and the weight loss was > 6%. Because the weight loss was greater than this amount and inflammation was not altered, the correlation between inflammation and weight loss was still accurate (Fig. 3B).
We previously found that inflammation was log-linearly correlated to macrophages and neutrophils (Myers et al. 2021; Fig. 3C and D). Because neutrophils are known to enhance disease during IAV–Sp coinfections, we plotted inflammation against these cells for the coinfections. This suggested that inflammatory macrophages were again correlated with inflammation. Comparatively, neutrophils were significantly exacerbated during the coinfection and did not support the same log-linear correlation as in the IAV-infected groups. Refitting a line to these data did not support a strong correlation between neutrophils and inflammation.
Discussion
Secondary bacterial infections increase influenza-associated disease and understanding their complex dynamics and impact on the lung is important to crafting better therapeutic strategies. Comparing the dynamics at different coinfection timings and using a mathematical approach allowed us to better evaluate time-dependencies and whether different effects were independent mechanisms or downstream consequences.
Both clinically and experimentally, bacterial complications are most pronounced after 5–7 d of influenza, and our data support reduced bacterial outgrowth at earlier time points. However, there was a larger impact on the lung when animals were infected with bacteria at 3 d pvi compared to 5 d pvi, which was also greater than 7 d pvi (Fig. 2). While one might expect increased virus dissemination in the lung to significantly impact disease severity, it seems to play a relatively minor role. This may be why antiviral administration has little impact on the outcome of influenza-associated bacterial coinfections (McCullers 2004, 2011, Smith 2016). Our model and data indicated that bacterial presence allowed the virus to infect new areas of the lung, which may be a consequence of direct attachment of the virus to the bacteria (Rowe et al. 2019). Interestingly, bacterial coinfection with other viruses, such as SARS-CoV-2, does not result in increased viral loads or new areas of the lung becoming infected (Smith et al. 2022). Here, the model also suggested that an increase in virus production was still playing a role, albeit smaller than the increase in the number of infected cells. Reduced IFN signaling was observed during influenza coinfection with S. aureus in vitro in primary bronchial epithelial cells (Warnking et al. 2015). This may be more difficult to tease apart in vivo given the robust increases in IFN-β during IAV–Sp coinfection (Li et al. 2012, Lee et al. 2015, Shepardson et al. 2016), which may be from a combination of virus-infected cells and immune cells.
The increased heterogeneity in bacterial loads during a coinfection at 3 d pvi was not reflected in the heterogeneity in viral loads or postbacterial AMΦ levels, which also did not depend on the time of bacterial initiation. This seems to support our model result suggesting that the additional AMΦ loss was due primarily to the increase in virus-infected cells, and our prior results showing that AMΦs were not significantly depleted during a high-dose primary bacterial infection (Smith et al. 2021). Curiously, additional mechanisms were needed in the model to capture the postbacterial decline in AMΦs at the 7 d coinfection (Fig. 1C), but this could be because these cells may be in a different state (e.g. engaged in wound healing and/or population restoration) at the time when the CD8+ T cells were arriving to rapidly clear infected cells (Myers et al. 2021). Although our current model excludes CD8+ T cells, we would not expect their depletion (Blevins et al. 2014) to strongly contribute to the viral rebound. This speculation is supported by the ability of some to clear virus during the 7 d coinfection (Fig. 1C). It is also based on our previous findings showing that the CD8+ T cell population would need to be quite low for a robust impact on virus clearance (Myers et al. 2021). However, evaluating their loss during IAV–Sp coinfection more definitively is the subject of our ongoing studies.
Numerous studies report that influenza–bacterial coinfection is hyperinflammatory and that severity is a result of robust immune cell infiltration and hyperproduction of proinflammatory immune cytokines (McCullers 2006, Short et al. 2012, Metzger and Sun 2013, Smith and McCullers 2014, Kash and Taubenberger 2015, Rynda-Apple et al. 2015). It is, thus intriguing that inflammation was not significantly enhanced in most of our coinfection groups, at least not within the first 24–48 h pbi. This is largely due to the nonlinearity of the relation (Myers et al. 2021; Fig. 3), which saturates for large weight losses. We found it especially interesting that the significant increases in the amount of the lung that became infected with the virus during the coinfections at 3 or 5 d pvi had no impact on inflammation. This may be due to these timings being at the height of the innate response, which could feasible reduce any ensuing inflammation. The log-linear correlation with macrophages may be supportive (Fig. 3C). Comparatively, a coinfection at 7 d pvi is at a point where the switch from innate to adaptive has occurred. Nevertheless, these data highlighted limitations in our mathematical approximation of inflammation, which was quite simplistic but the first detailing of the dynamics during influenza (Myers et al. 2021).
Although our model, which excluded neutrophils and inflammatory macrophages, was able to produce rebounding bacterial dynamics (e.g. Fig. 1B), it was not on the time scale that our prior study indicated this rebound occurs at (i.e. ∼3–4 h; Smith and Smith 2016) and the additional AMΦ loss was not profound enough to have a strong influence on the bacterial dynamics. We might anticipate that neutrophils arriving at this time would be able to clear the infection, particularly for the animals whose bacterial loads were less than 102-103 CFU, but this does not seem to be the case. Neutrophil dysfunction has been noted in numerous studies (Abramson et al. 1982, Abramson and Wheeler 1994, Colamussi et al. 1999, Engelich et al. 2001, McNamee and Harmsen 2006, Ishikawa et al. 2016), and our recent work showed that bacterial metabolism may affect the function of these cells particularly during viral–bacterial coinfection (Smith et al. 2021). In that study, postbacterial pathogenicity could be ameliorated with the deletion of select bacterial genes. This was also sufficient to decrease the type I IFN response, which has been proposed to result in defective neutrophil function and recruitment, high bacterial burden, and increased mortality (Shahangian et al. 2009, Li et al. 2012, Shepardson et al. 2016). Better detailing the dynamics of these responses using mathematical models should be interesting and fruitful.
In conclusion, the pairing of time-resolved data with a modeling approach stressed the importance of understanding nonlinearities in host–pathogen dynamics and the time dependency of different interactions between influenza and invading bacteria. Continuing to establish the predictive capabilities of models like the one here should improve our knowledge of viral–bacterial coinfections and our ability to forecast their impact on the lung. This should increase our chances of finding new ways to treat or prevent coinfections.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the NIH grants AI100946, AI125324, and AI139088.
Contributor Information
Amanda P Smith, Department of Pediatrics, University of Tennessee Health Science Center, Memphis, TN, United States.
Lindey C Lane, Department of Pediatrics, University of Tennessee Health Science Center, Memphis, TN, United States.
Ivan Ramirez Zuniga, Department of Pediatrics, University of Tennessee Health Science Center, Memphis, TN, United States.
David M Moquin, Department of Anesthesiology, Washington University School of Medicine, St. Louis, MO, United States.
Peter Vogel, Department of Pathology, St. Jude Children’s Research Hospital, Memphis, TN, United States.
Amber M Smith, Department of Pediatrics, University of Tennessee Health Science Center, Memphis, TN, United States.
Conflict of interest statement
None declared.
References
- Abramson JS, Lewis JC, Lyles DSet al. Inhibition of neutrophil lysosome-phagosome fusion associated with influenza virus infection in vitro. Role in depressed bactericidal activity. J Clin Invest. 1982;69:1393–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramson JS, Wheeler JG. Virus-induced neutrophil dysfunction: role in the pathogenesis of bacterial infections. Pediatr Infect Dis J. 1994;13:643–52. [PubMed] [Google Scholar]
- Blevins LK, Wren JT, Holbrook BCet al. Coinfection with Streptococcus pneumoniae negatively modulates the size and composition of the ongoing influenza-specific CD8+ T cell response. J Immunol. 2014;193:5076–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundage JF, Shanks GD. Deaths from bacterial pneumonia during 1918–19 influenza pandemic. Emerg Infect Dis. 2008;14:1193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y-W, Klugman KP, Morens DM. Bacterial pathogens and death during the 1918 influenza pandemic. N Engl J Med. 2009;361:2582–3. [DOI] [PubMed] [Google Scholar]
- Colamussi ML, White MR, Crouch E.et al. Influenza A virus accelerates neutrophil apoptosis and markedly potentiates apoptotic effects of bacteria. Blood. 1999;93:2395–403. [PubMed] [Google Scholar]
- Engelich G., White M., Hartshorn KL. Neutrophil survival is markedly reduced by incubation with influenza virus and Streptococcus pneumoniae: role of respiratory burst. J Leukoc Biol. 2001;69:50–56. [PubMed] [Google Scholar]
- Ghoneim HE, Thomas PG, McCullers JA. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J Immunol. 2013;191:1250–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Fukui T, Ino Set al. Influenza virus infection causes neutrophil dysfunction through reduced G-CSF production and an increased risk of secondary bacteria infection in the lung. Virology. 2016;499:23–29. [DOI] [PubMed] [Google Scholar]
- Kash JC, Taubenberger JK. The role of viral, host, and secondary bacterial factors in influenza pathogenesis. Am J Pathol. 2015;185:1528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugman KP, Chien Y.-W, Madhi SA. Pneumococcal pneumonia and influenza: a deadly combination. Vaccine. 2009;27:C9–C14. [DOI] [PubMed] [Google Scholar]
- Lee B, Robinson KM, McHugh KJet al. Influenza-induced type I interferon enhances susceptibility to Gram-negative and Gram-positive bacterial pneumonia in mice. Am J Physiol Lung Cell Mol Physiol. 2015;309:L158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Moltedo B, Moran T. M. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of γδ t cells. J Virol. 2012;86:12304–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louria DB, Blumenfeld HL, Ellis JT. Studies on influenza in the pandemic of 1957-1958. II. Pulmonary complications of influenza*†. J Clin Invest. 1959;38:213–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers JA. Effect of antiviral treatment on the outcome of secondary bacterial pneumonia after influenza. J Infect Dis. 2004;190:519–26. [DOI] [PubMed] [Google Scholar]
- McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev. 2006;19:571–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers JA. Preventing and treating secondary bacterial infections with antiviral agents. Antivir Ther. 2011;16:123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamee LA, Harmsen A. G. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect Immun. 2006;74:6707–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger DW, Sun K. Immune dysfunction and bacterial coinfections following influenza. J Immunol. 2013;191:2047–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198:962–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morens DM, Taubenberger JK, Fauci AS. The persistent legacy of the 1918 influenza virus. N Engl J Med. 2009;361:225–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MA, Smith AP, Lane LCet al. Dynamically linking influenza virus infection kinetics, lung injury, inflammation, and disease severity. Elife. 2021;10:e68864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed C, Chaves SS, Daily Kirley P. Estimating influenza disease burden from population-based surveillance data in the United States. PLoS ONE. 2015;10:e0118369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints12. Am J Epidemiol. 1938;27:493–7. [Google Scholar]
- Rowe HM, Meliopoulos VA, Iverson A. Direct interactions with influenza promote bacterial adherence during respiratory infections. Nat Microbiol. 2019;4:1328–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rynda-Apple A, Robinson KM, Alcorn JF. Influenza and bacterial superinfection: illuminating the immunologic mechanisms of disease. Infect Immun. 2015;83:3764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahangian A, Chow EK, Tian Xet al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest. 2009;119:1910–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepardson KM, Larson K, Morton RVet al. Differential type I interferon signaling is a master regulator of susceptibility to postinfluenza bacterial superinfection. Mbio. 2016;7:e00506–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short KR, Habets MN, Hermans PWet al. Interactions between Streptococcuspneumoniae and influenza virus: a mutually beneficial relationship?. Fut Microbiol. 2012;7:609–24. [DOI] [PubMed] [Google Scholar]
- Smith AM, Adler FR, Ribeiro RMet al. Kinetics of coinfection with influenza a virus and Streptococcus pneumoniae. PLoS Pathog. 2013;9:e1003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, McCullers JA. Secondary bacterial infections in influenza virus infection pathogenesis. In: Compans RW, Oldstone MBA (eds), Influenza Pathogenesis and Control. 1. Cham: Springer International Publishing, 327–56., 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Smith AP. A critical, nonlinear threshold dictates bacterial invasion and initial kinetics during influenza. Sci Rep. 2016;6:38703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM. Quantifying the therapeutic requirements and potential for combination therapy to prevent bacterial coinfection during influenza. J Pharmacokinet Pharmacodyn. 2016; 44:81–93. DOI:10.1007/s10928-016-9494-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AP, Lane LC, van Opijnen Tet al. Dynamic pneumococcal genetic adaptations support bacterial growth and inflammation during coinfection with influenza. Infect Immun. 2021;89:e0002321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AP, Moquin DJ, Bernhauerova V. Influenza virus infection model with density dependence supports biphasic viral decay. Front Microbiol. 2018;9:1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AP, Williams EP, Plunkett TRet al. Time-dependent increase in susceptibility and severity of secondary bacterial infections during SARS-CoV-2. Front Immunol. 2022;13:894534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WW. Influenza-associated hospitalizations in the United States. JAMA. 2004;292:1333. [DOI] [PubMed] [Google Scholar]
- Verma AK, Bansal S, Bauer Cet al. Influenza infection induces alveolar macrophage dysfunction and thereby enables noninvasive Streptococcuspneumoniae to cause deadly pneumonia. J Immunol. 2020;205:1601–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnking K, Klemm C, Löffler Bet al. Super-infection with Staphylococcusaureus inhibits influenza virus-induced type i IFN signalling through impaired STAT1-STAT2 dimerization: influenza virus- and S. aureus -mediated signalling. Cell Microbiol. 2015;17:303–17. [DOI] [PubMed] [Google Scholar]
- Weinberger DM, Simonsen L, Jordan Ret al. Impact of the 2009 influenza pandemic on pneumococcal pneumonia hospitalizations in the United States. J Infect Dis. 2012;205:458–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.