

Abstract
Bacterial membrane vesicles (MVs) have been reported to kill other bacteria. In the case of Pseudomonas aeruginosa the bactericidal activity has been attributed to an unidentified 26 kDa peptidoglycan (PG) hydrolase that is associated with MVs and gives rise to a lytic band on zymograms using murein sacculi as substrate. In this study, we employed a proteomics approach to show that this PG hydrolase is the AmphD3 amidase. The analysis of an amphD3 mutant as well as of an AmphD3 overexpression derivative revealed that this enzyme is not required for the bactericidal activity of P. aeruginosa MVs but is involved in cell wall recycling and thus protects the cell against PG damage. Another 23 kDa PG hydrolase, which we observed on zymograms of SOS-induced MVs, was identified as the endolysin Lys, which triggers explosive cell lysis but is shown to be dispensable for MV-mediated killing. We conclude that the lytic activities observed on zymograms do not correlate with the bactericidal potential of MVs. We demonstrate that P. aeruginosa MVs are enriched for several autolysins, suggesting that the predatory activity of MVs depends on the combined action of different murein hydrolases.
Keywords: membrane vesicles, explosive cell lysis, bacterial killing, endolysin, AmpDh3, peptidoglycan turnover
Peptidoglycan hydrolytic activities on zymograms do not correlate with the bacteriocidal potential of membrane vesicles.
Introduction
Extracellular vesicles are produced by species across all domains of life, suggesting that vesiculation represents a fundamental principle of living matter (Gill et al. 2019). Bacterial membrane vesicles (MVs), which are typically 40 to 350 nm in size, carry specific cargos, including periplasmic and cytosolic proteins, DNA and RNA, and are known to transport virulence factors (Wai et al. 2003, Altindis et al. 2014, Sjöström et al. 2015, Koeppen et al. 2016, Turnbull et al. 2016, Bitto et al. 2017). MVs can fuse with both eukaryotic and prokaryotic membranes and thereby can deliver their contents into target cells (Kimmitt et al. 2000, Kuehn and Kesty 2005, Chatterjee and Chaudhuri 2011). Bacterial MVs were first shown to originate from blebs of the outer membrane of Gram-negative bacteria and are therefore often referred to as outer membrane vesicles (OMVs). More recent work has provided evidence that Gram-negative bacteria can produce different types of vesicles and that many Gram-positive bacteria also release MVs (Toyofuku et al. 2019). Moreover, it has been demonstrated that MVs are not only produced by living cells but can also originate from endolysin-triggered cell lysis in both Gram-negative (Turnbull et al. 2016) and Gram-positive (Toyofuku et al. 2017) bacteria via mechanisms termed ‘explosive cell lysis’ and ‘bubbling cell death’, respectively (Toyofuku et al. 2019). Endolysins are typically used by double-stranded DNA phages to lyse their hosts, so that the phage progeny can be released. In P. aeruginosa, DNA-damaging stress induces the SOS response that leads to the expression of the endolysin Lys, which is part of a pyocin biosynthesis gene cluster. As Lys hydrolyses the peptidoglycan (PG) layer, the cells round up, explode and the remaining shattered membrane fragments self-assemble into MVs. Explosive cell lysis was shown to be stimulated in biofilms and under anoxic conditions (Toyofuku et al. 2014, Turnbull et al. 2016).
Various studies showed that MVs affect diverse biological processes, including virulence, export of metabolites, horizontal gene transfer, cell-to-cell communication, and protection against phages and antibiotics (Brown et al. 2015, Schwechheimer and Kuehn 2015, Orench-Rivera and Kuehn 2016). Moreover, in a seminal study, Kadurugamuwa and Beveridge demonstrated that MVs of P. aeruginosa PAO1 can kill Gram-negative as well as Gram-positive bacteria and suggested that MVs may represent a conceptual new group of antibiotics (Kadurugamuwa and Beveridge 1996). A subsequent study showed that naturally produced MVs isolated from 15 Gram-negative bacteria exhibited bactericidal effects against various bacteria, albeit to different degrees (Li et al. 1998). The toxicity of such ‘predatory’ MVs was proposed to be caused by the lytic action of peptidoglycan hydrolases that are associated with the MVs and which could be visualized by the aid of zymograms using murein sacculi as substrates (Li et al. 1996, Li et al. 1998). In the case of P. aeruginosa PAO1 it has been shown that the luminal content of the MVs contains a 26 kDa murein hydrolase, which was suggested to play an important role in bacterial killing. Although more recent proteomics studies identified various MV-associated autolysins, the identity of this 26 kDa autolysin has remained unidentified (Clarke 2018).
In this study we demonstrate the 26 kDa major autolysin of P. aeruginosa MVs is the AmpDh3 amidase. Furthermore, we show that a 23 kDa hydrolytic enzyme, which we observed in addition to the AmpDh3 band on zymograms of SOS-induced MVs, as the endolysin Lys, which triggers explosive cell lysis. We provide evidence that AmpDh3 is not involved in the bactericidal activity of P. aeruginosa MVs but rather plays a critical role in cell wall recycling, and thereby protects the cell against PG damage and counteracts explosive cell lysis. Our data show that the lytic activities observed on zymograms do not correlate with the bactericidal potential of the MVs. A proteomics approach revealed that MVs are enriched for several autolysins, which were not detectable on zymograms, and thus the predatory activity of MVs appears to depend on the combined action of different murein hydrolases.
Material and methods
Bacterial strains, plasmids, and growth conditions
Bacteria strains and plasmids used in this study are listed in Table S1. Gene deletion mutants of ampDh2 and ampDh3 were constructed using the I-SceI mutagenesis system as described previously (Flannagan et al. 2008). In short, the ampDh2 and ampDh3 deletion cassettes were inserted into plasmid pGPI using the restriction sites XbaI and EcoRI. The resulting plasmids were conjugated into PAO1, and mutants were selected on PIA supplemented with tetracycline. Rhamnose-inducible overexpression of ampDh3, pbpG, ftsI and ampR was achieved by cloning the respective genes into pIN299 using the restriction sites NdeI and XbaI. All bacteria were cultured in Luria-Bertani (LB) broth at 37°C. If required, antibiotics were added at the following final concentrations: for E. coli: 15 μg ml−1 gentamycin, 20 μg ml−1 chloramphenicol or 20 μg ml−1 tetracycline; for P. aeruginosa: 60 μg ml−1 chloramphenicol, 100 μg ml−1 tetracycline or 80 μg ml−1 gentamycin.
For rhamnose-inducible overexpression of target genes, overnight cultures were inoculated into fresh medium at an OD600 = 0.01. Following 4 h of incubation at 37°C, rhamnose was added to a final concentration of 2% and the cultures were incubated overnight. To test the effect of different growth conditions on MV productions, overnight cultures were inoculated at an OD600 = 0.01 and were grown for 4 h at 37°C before they were treated with 200 ng ml−1 mitomycin C (MMC, AG scientific), 100 ng ml−1 ciprofloxacin (CIP), 50 µg ml−1 ceftacidime (CAZ) or exposured to UV-C for 10 min. The cultures were grown for additional 4 h before they were harvested.
MV isolation, quantification and nanoparticle tracking analysis
Cell cultures were centrifuged at 10 000 g at 4°C for 20 min and the supernatants were filtered through a 0.45 µm Durapore Membrane Filter (hydrophilic PVDF; Merck). The filtrates were ultracentrifuged at 150 000 g at 4°C for 90 min, the pellets were resuspended in double distilled water (ddH2O) and MVs were quantified using three assays: (i) staining with the fluorescent dye FM1-43 (Life Technologies, USA), (ii) protein quantification using the BCA protein assay kit (Thermo Scientific), and (iii) nanoparticle tracking analysis (NTA) using the Nanosight NS300 (Malvern Panalytical) to determine particle concentrations and size distributions. For NTA, MV samples were diluted in HyClone HyPure Water (GE) to a final concentration of 20 to 80 particles per frame before they were measured in the light scattering mode for 1 min. Maximum camera detection sensitivity was set, and the processing threshold was set at 5. Data are mean values of 15 technical replicates. The amounts of MVs were normalized by OD600 of the culture.
Peptidoglycan extraction and zymography
Peptidoglycan (PG) was extracted using the protocol as described (Bernadsky et al. 1994). To this end, overnight cultures were centrifuged and the cell pellet was resuspended in 4% SDS and boiled for 1 h, followed by ultracentrifugation at 150 000 g at 25°C for 1 h. The pellet was washed twice with ddH2O and resuspended in 50 mM Tris buffer pH 8.0 containing 5 mM EDTA and 0.5 mg ml−1 trypsin (T7409, Sigma) at 25°C for overnight digestion. The samples were then ultracentrifuged, washed twice with ddH2O and lyophilized. Purified PG samples were store at −20°C. To cast zymogram gels, the PG was resuspended in ddH2O and sonicated with a SONOPULS HD 2200 homogeniser using a MS72 probe (BANDELIN) at 25% power, 40% pulsation for 2 min on ice. Zymographic analysis was performed as described by Moak and Molineux with 20 µg protein per lane (Moak and Molineux 2004).
Protein identification and proteomics
For protein identification the in-gel digestion method described by Liu et al. (Liu et al. 2018) was used with minor modifications. Hydrolytic bands were cut out of zymograms and de-stained in 50/50 (v/v) MeOH/ammonium bicarbonate (ABC) buffer (50 mM (NH4)HCO3). The gel pieces were washed with acetonitrile (ACN) and dried by vacuum evaporation. The gel pieces were soaked with ABC supplemented with 10 mM dithiothreitol (43819, Fluka) and incubated at 60°C for 1 h, followed by washing with ACN. The gel pieces were then incubated in ABC containing 50 mM iodoacetamide (I1149, Sigma) and incubated in dark for 30 min, washed twice with ABC, transferred to 80% ACN and dried by vacuum evaporation. The samples were digested with 1 µg trypsin (V5113, Promega) in 50 mM Triethylammonium bicarbonate (pH 8.5) at room temperature overnight. After acidification with trifluoroacetic acid (TFA) to a final concentration of 0.5% the samples were desalted with C18 stage tips (Rappsilber et al. 2003). For label-free quantitative proteomic, the filter-aided sample preparation method as described previously (Wisniewski et al. 2009) was used. iRT peptides (Biognosys) were added to the samples for the calibration of retention times.
Peptides were separated on an ACQUITY UPLC M-Class System (Waters) equipped with a HSS T3 C18 reverse-phase column (1.8 µm, 75 µm × 250 mm, Waters) and analysed by an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific). Thermo raw files were converted to the Mascot generic format (MGF) by the Proteome Discoverer, v2.0 (Thermo Fisher Scientific) using the automated rule-based converter control (Barkow-Oesterreicher et al. 2013). The significance threshold was set to P < 0.05 for protein identification. For quantitative proteomics, Progenesis QI software (Waters) was used. The data presented are based on two independent biological experiments.
Growth inhibition assays
Overnight cultures were diluted to an OD600 of 0.01 and inoculated into 96-well plates. PA14 or PAO1 MVs with protein concentrations of 25 and 40 µg, respectively, were added to the wells. Growth curves were measured over 24 h at 37°C in a microtiter plate reader (Synergy HT; Bio-Tek, Germany). Growth inhibition of B. subtilis 168 was determined after 6 h and of S. aureus DSM20235 and P. aeruginosa PAO1 were after 12 h of incubation.
Self-toxicity assays
Overnight cultures of overexpression derivatives were diluted into fresh medium to an OD600 of 0.01. Following 3 h of incubation at 37°C, L-rhamnose was added to a final concentration of 2% (w/v) to induce protein expression. After overnight incubation cells were harvested and washed twice with PBS. Viability was assessed by staining with 5 µM SYTO 9 (Thermo Fisher Scientific) and 1 µg ml−1 propidium iodide (Thermo Fisher Scientific). Microscopic image acquisition was performed using a confocal laser scanning microscope (DM5500Q, Leica) equipped with × 100/1.44 oil objective.
Metabolic labelling and quantification
For microscopic analysis cells were labelled with D-alkyne alanine as described previously (Siegrist et al. 2013) with some modifications. Bacterial overnight cultures were inoculated at an OD600 of 0.02 in fresh LB medium and grown for 3 h at 37°C before 2% rhamnose was added to induce protein overexpression. Following 16 h of incubation, either 1 mM D-alanine (ACROS) or 1 mM D-alkyne-alanine ((R)-alpha-propargylglycine, ACROS) was added to the cultures. After 90 min the cells were centrifuged and washed twice with PBS and fixed with 2% formaldehyde. The Cu(I)-catalyzed azide−alkyne cycloaddition (CuAAC) reaction was initiated with 128 µM TBTA, 1 mM CuSO4, 1.2 mM Sodium ascorbate, and 20 µM Azide-fluor 488 (Sigma) for 30 min at room temperature. Samples were washed and inspected using a DM5500Q microscope fitted with a TCS SPE confocal laser scanning unit (Leica), an HCX PL APO CS 100x oil-immersion objective (NA = 1.44, Leica) and a laser line set at 488 nm. Eight-bit TIFF images were acquired in the 490–575 nm fluorescence window with corresponding bright field signal using the LAS software (Leica). Azide-fluor 488 signal quantifications were performed with Fiji 1.53. Cells displaying mean fluorescence values greater than the calculated mean Azide-fluor unspecific binding signal were considered positively labelled.
For chemical quantification, bacterial cultures were inoculated at an OD600 of 0.02 in fresh LB medium and grown for 3 h at 37°C before 2% rhamnose was added. After 1 h of incubation 1 mM D-alanine or 1 mM D-alkyne-alanine were added. Following 1 h of incubation the cells were pelleted and the CuAAC reaction was initiated with Azide-fluor 488 (Sigma). PG was then extracted as described before and treated with 1 mg lysozyme (SERVA, Germany) in 1 ml 50 mM ABC buffer (pH 6.24) at 25°C overnight. The samples were analysed by the aid of a Dionex UltiMate 3000 UHPLC (Thermo Fisher Scientific) coupled to a Compact QTOF (Bruker). An ACQUITY UPLC BEH C18 (130Å, 1.7 µm, 2.1 mm × 100 mm, Waters) column was used in combination with solvents A (H2O + 0.1% formic acid) and B (ACN + 0.1% formic acid) buffer. The gradient started from 0% B for 2 min, followed by a ramp to 100% B for 10 min and was continued at 100% B for 4 min with a flow rate of 0.3 ml min−1. MSMS fragmentation was performed for 940.40 m/z and 964.40 m/z with CID 40 eV for PG containing D-alanine and alkyne-D-alanine, respectively. Alkyne-D-alanine labelled PG was quantified by integrating the peak of extracted ion mass at 482.70 m/z ([M+2H]2+) and 964.40 m/z ([M+H]+) at 5.91 min. D-alanine containing PG was quantified by integrating the peak of extracted ion mass at 470.70 m/z ([M+2H]2+) and 940.40 m/z ([M+H]+) at 5.82 min. Experiments were performed in biological triplicates.
Results
MMC treatment increases the amounts of cell wall-degrading enzymes in P. aeruginosa PAO1 MVs
When purified native P. aeruginosa PAO1 MVs were analysed for PG hydrolytic activities by zymography, a lytic band with a molecular weight (MW) of approximately 28 kDa was detected (Fig. 1A). This is in good agreement with the 26 kDa murein hydrolase first observed on zymograms of P. aeruginosa MVs by Li et al. (Li et al. 1996). As recent work has demonstrated that DNA damaging agents and certain antibiotics can stimulate MV formation and that depending on their formation routes they appear to carry different cargos (Toyofuku et al. 2019), we analysed the PG hydrolytic activities of MVs isolated from cultures treated with different antimicrobial compounds. When the cells were treated with the DNA damaging agent mitomycin C (MMC) or the fluoroquinolone antibiotic ciprofloxacin (CIP) the 28 kDa lytic band was enhanced and a very faint 23 kDa band became visible (Fig. 1B, data not shown). As both compounds are well characterized triggers of the SOS response, these results suggest that MVs that originate from explosive cells lysis are enriched for these two cell wall-degrading enzymes. Importantly, two hydrolytic bands (a strong band of 26 kDa and a barely visible band at 23 kDa) have also been observed by Kadurugamuwa and Beveridge (Kadurugamuwa and Beveridge 1996) with both native and gentamicin-induced PAO1 MVs. Lytic bands were only observed with PG of P. aeruginosa as substrate in the zymograms but not with PG of Bacillus subtilis or Burkholderia cenocepacia (Fig. S1), indicating that the MV-associated PG hydrolytic activity is specific for certain PG types.
Figure 1.
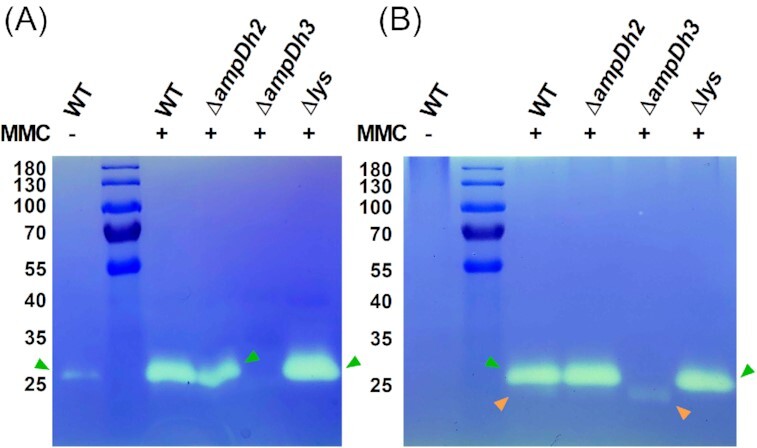
Electrophoretic profile (zymogram) of MV- and cell-associated autolysins. Bands with PG-hydrolysing activities were observed as clear zones in renatured SDS-PAGE gels containing 1% murein sacculi of P. aeruginosa PAO1. (A) Purified MVs of cultures of the PAO1 wild-type strain grown in the presence or absence of MMC were compared to MMC-induced MVs of the ampDh2, ampDh3 and lys deletion mutants. (B) MMC-induced cell fractions of the ampDh2, ampDh3, and lys deletion mutants. Orange and green arrows indicate the 23 and 28 kDa bands, respectively.
The AmpDh3 amidase and the endolysin Lys are responsible for the PG hydrolytic activities observed on zymograms
To ascertain the identity of the hydrolytic enzymes observed in the zymograms, the two bands were excised from the SDS gel and the eluted proteins were digested and subjected to LC-MS analysis. Among the proteins identified for the 28 kDa lytic band (Table S2), were the AmpDh2 (PA5485, predicted MW of 28.9 kDa) and AmpDh3 amidases (PA0807, predicted MW of 28.7 kDa) while the endolysin Lys (PA0629), which has a predicted MW of 23.0 kDa, was among the proteins identified for the 23 kDa band (Table S3). To validate our results, we analysed purified MVs isolated from MMC-induced cultures of defined mutants, in which either ampDh2, ampDh3 or lys have been inactivated, by zymography. We observed that the 28 kDa hydrolytic band was missing from samples isolated from cultures of the ampDh3 mutant but not the ampDh2 mutant, while the 23 kDa band was missing in samples of the lys mutant (Fig. 1A and B).
Collectively, our data show that the cell-wall hydrolytic activities of P. aeruginosa PAO1 MVs observed in zymograms are the result of the enzymatic actions of the amidase AmpDh3 and the endolysin Lys. Our data also suggest that production of both AmpDh3 and Lys are boosted by the induction of the SOS response.
Neither the AmpDh3 amidase nor the endolysin Lys is the killing agent of P. aeruginosa MVs
Previous studies have suggested that the bactericidal activity of P. aeruginosa MVs is dependent on the 26 kDa murein hydrolase observed in zymograms (Li et al. 1998). We therefore speculated that AmpDh3 or Lys, which showed cell-wall degrading activities in our zymograms, could be responsible for MV-mediated bacterial killing. To test this hypothesis, we determined the growth inhibitory effect of MVs isolated from the wild-type strain, an ampDh3 and a lys mutant. Both native and MMC-induced MVs inhibited the growth of the Gram-positive bacteria Bacillus subtilis and Staphylococcus aureus (Fig. 2A and 2B) but not of P. aeruginosa PAO1 (Fig. 2C). To exclude that the toxic effect is due to traces of MMC, which was used to stimulate MV formation, we induced explosive cell lysis by treating cultures with UV-C light (Fig. S2A-C). No significant difference in the toxicity of UV- and MMC-induced MVs was observed. Interestingly, native MVs showed a significantly stronger inhibitory effect than induced MVs, indicating that the toxic cargo of induced and native MVs is different. Neither the MVs of the ampDh3 nor of the lys mutant showed decreased growth inhibition when compared to wild-type MVs. We also tested MVs of a strain overexpressing AmpDh3. The inhibitory effect was similar to the one observed for MVs of the wild-type strain (Fig. S2D).
Figure 2.

Growth inhibitory effect of MVs on cultures of B. subtilis(A), S. aureus(B) and P. aeruginosa PAO1 (C). MVs were purified from supernatants of MMC-treated or untreated cultures of the wild-type strain and various deletion mutants. Following addition of MVs to cultures of the three target bacteria, growth was monitored spectrophotometrically. Data are presented as mean±SD from triplicate measurements and significant differences between groups were determined by one-way ANOVA followed by Tukey post-hoc test. *, P< 0.05; **, P< 0.01; ***, P< 0.001; ns, not significant.
In conclusion, our data suggest that the bactericidal effect of P. aeruginosa MVs cannot be correlated to the lytic activities observed in our zymograms. Hence, bacterial killing may not depend on a single autolysin but rather on the synergistic action of multiple enzymes. This may also explain why no hydrolytic activity could be detected on zymograms with PG from B. subtilis as substrate, although P. aeruginosa MVs showed strong bactericidal activity against this organism.
To further test this hypothesis, we determined the toxicity of MVs isolated from cultures of P. aeruginosa PA14 mutants, in which different genes encoding PG-modifying enzymes had been inactivated, for growth inhibition of B. subtilis, S. aureus, and P. aeruginosa PAO1. A strong inhibitory effect was seen with all tested MV preparations against the Gram-positive bacteria whereas no inhibitory effect was seen against P. aeruginosa PAO1 (Fig. S3). While none of the tested mutants showed a significant decrease of bactericidal activity, we observed an approximately 2-fold increase in growth inhibition of B. subtilis with MVs of the pbpG (PA14_53020), dacB (PA14_24690), sltB1 (PA14_12080) and pbpA (PA14_12060) mutants (Fig. S3A). Nanoparticle tracking analysis (NTA) revealed that the MVs of the pbpG, dacB, mltF, pbpA, and SltB1 mutants were larger than the ones of the wild-type strain (Fig. S3D). Moreover, we observed a great variation in the amounts of MVs produced by the different mutants (Fig. S3D). Although additional work will be required to unravel why some mutants show increased bactericidal activity, we speculate that the inactivation of some PG-modifying enzymes may disturb PG homeostasis and thus lead to an increased or unbalanced biosynthesis of PG hydrolases. It is also important to note that none of the mutants showed decreased activity, strengthening the idea that the bactericidal effect of MVs likely depends on the combined action of multiple enzymes.
MMC-induced MVs are enriched for PG-modifying enzymes
To validate that expression of Lys and AmpDh3 is induced by MMC and to investigate whether additional PG-modifying enzymes are regulated by the SOS response, we employed a quantitative proteomics approach. Treatment of a P. aeruginosa PAO1 culture with 200 ng ml−1 MMC resulted in a differential increase of various enzymes involved in PG metabolism both in cells and MVs (Table 1). The amounts of Lys, AmpDh3, PbpG, Tse3, and FtsI were more than 10-fold increased in MMC-treated cells relative to the untreated control. As with Lys and AmpDh3, expression of PbpG, Tse3 and FtsI appears to be upregulated by the SOS response. Given that these enzymes are involved in cell wall hydrolysis, it is tempting to speculate that they could assist Lys-triggered explosive cell lysis. We also observed that some PG-modifying enzymes were selectively enriched (>10 fold) in MMC-induced MVs, including MtgA, MltA, DacB, MltF, SltB1, and PbpA. It is interesting to note that many of these enzymes are predicted to be associated with the inner membrane, indicating that these MVs were formed as a consequence of cell lysis.
Table 1.
Differential abundance of cell wall-modifying enzymes in cells and MVs of P. aeruginosa PAO1 upon MMC treatment. Predicted localization of proteins: C, cytoplasm; IM, inner membrane associated; P, periplasm; OM, outer membrane associated; E, extracellular.
| Gene number | Gene name | Protein description | Size (kDa) | Possible localization | Fold increase | |
|---|---|---|---|---|---|---|
| Cells | MVs | |||||
| PA0378 | mtgA | Biosynthetic peptidoglycan transglycosylase | 26.5 | IM | < 2.0 | 11.2 |
| PA0629 | lys | Endolysin | 23.0 | P | 27.4 | 2.1 |
| PA0807 | ampDh3 | AmpDh3 | 28.7 | P | 98.0 | 2.9 |
| PA0869 | pbpG (PBP7) | D-alanyl-D-alanine endopeptidase | 34.0 | P | 30.5 | < 2.0 |
| PA1171 | sltB2 (sltG) | Soluble lytic transglycolase | 42.7 | IM | 6.8 | 3.7 |
| PA1222 | mltA | Membrane-bound lytic murein transglycosylase A | 41.9 | OM | 5.9 | 20.4 |
| PA2854 | erfK | Putative L,D-transpeptidase | 34.8 | P | 2.5 | < 2.0 |
| PA2963 | mltG | Endolytic murein transglycosylase | 39.6 | IM | < 2.0 | 5.5 |
| PA3020 | mltC | Probable soluble lytic transglycosylase | 73.4 | P | 5.7 | < 2.0 |
| PA3047 | dacB (PBP4) | Probable D-alanyl-D-alanine carboxypeptidase | 51.9 | P | < 2.0 | 14.1 |
| PA3484 | tse3 | Muramidase (toxin of Type VI secretion system) | 44.4 | E | 11.6 | < 2.0 |
| PA3764 | mltF | Membrane-bound lytic murein transglycosylase F | 55.2 | OM | 3.0 | 26.6 |
| PA3992 | sltB3 (sltH) | Soluble lytic (exolytic) transglycosylase | 47.8 | IM | 3.1 | 3.7 |
| PA4000 | rlpA | Endolytic peptidoglycan transglycosylase RlpA | 36.5 | IM | < 2.0 | 7.0 |
| PA4001 | sltB1 | Soluble lytic transglycosylase B | 37.9 | IM | < 2.0 | 16.1 |
| PA4003 | pbpA | Peptidoglycan D,D-transpeptidase MrdA | 72.2 | IM | 6.8 | 17.6 |
| PA4412 | murG | UDP-N-acetylglucosamine–N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase | 37.8 | IM | < 2.0 | 5.1 |
| PA4413 | ftsW | Probable peptidoglycan glycosyltransferase FtsW | 43.8 | IM | < 2.0 | 2.3 |
| PA4414 | murD | UDP-N-acetylmuramoylalanine–D-glutamate ligase | 48.1 | C | 2.3 | < 2.0 |
| PA4417 | murE | UDP-N-acetylmuramoyl-L-alanyl-D-glutamate–2,6-diaminopimelate ligase | 51.3 | C | < 2.0 | 4.2 |
| PA4418 | ftsI (PBP3) | Peptidoglycan D,D-transpeptidase FtsI | 62.9 | IM | 59.4 | 5.5 |
| PA4450 | murA | UDP-N-acetylglucosamine-1-carboxyvinyltransferase | 44.6 | C | 2.5 | <2.0 |
| PA4700 | mrcB (PBP1b) | Penicillin-binding protein 1B | 85.5 | IM | 7.7 | 3.6 |
| PA4749 | glmM | Phosphoglucosamine mutase | 47.8 | C | 4.2 | < 2.0 |
| PA4947 | amiB | N-acetylmuramoyl-L-alanine amidase | 50.7 | OM | 3.9 | < 2.0 |
| PA5045 | mrcA (PBP1a) | Penicillin-binding protein 1A | 91.2 | IM | 3.0 | 6.0 |
| PA5485 | ampDh2 | AmpDh2 | 28.9 | OM | 4.1 | 7.0 |
Despite the presence of various MMC-inducible PG hydrolytic enzymes in MVs, only two lytic bands were observed on our zymograms (Fig. 1), suggesting that the other enzymes identified by our proteomics analysis may be produced in low amounts, cannot be renatured in the zymograms or are inefficient to degrade high molecular weight PG. To test this hypothesis, we determined the hydrolytic activities of MVs isolated from P. aeruginosa PAO1 cultures overexpressing enzymes that were particularly abundant in MMC-induced cells, including AmpDh3, PbpG and FtsI. (Fig. 3). MVs from the AmpDh3 overexpression strain showed an enhanced activity of the 28 kDa band, whereas overexpression of PbpG or FtsI gave rise to a weak band at approximately 23 kDa corresponding to Lys. Overexpression of AmpR, a global transcriptional factor that controls expression of many PG-modifying enzymes (Kong et al. 2005, Balasubramanian et al. 2015), resulted in increased lytic activities of both bands, suggesting that AmpDh3 and Lys were upregulated by AmpR overexpression. These data confirm that the hydrolytic bands at 28 kDa and 23 kDa are caused by the activities of AmpDh3 and Lys, respectively, and show that neither PbpG nor FtsI can be visualized on zymograms under the conditions used in our experimental setup.
Figure 3.
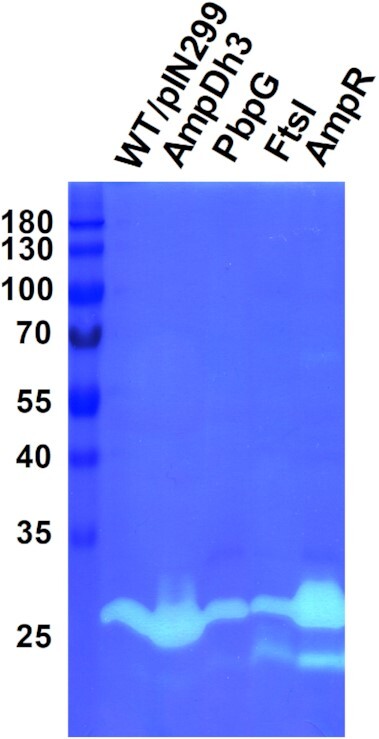
Zymographic analysis of MVs from P. aeruginosa PAO1 strains overexpressing AmpDh3, PbpG, FtsI, and AmpR. Production of PG modifying enzymes was induced with 2% rhamnose.
PG modifying enzymes affect MV formation in P. aeruginosa
As our data suggest that many PG hydrolases are upregulated under MMC treatment, they may contribute to Lys-triggered cell lysis. To test this hypothesis, we quantified the amount of MVs produced by ampDh3 and lys mutants in the presence of MMC by NTA. While the amount of MVs produced by the lys mutant was reduced as previously reported (Turnbull et al. 2016, Zhang et al. 2020), the ampDh3 mutants produced more MVs relative to the wild-type strain (Fig. 4A). We also observed that MMC-induced MVs were larger than native MVs, suggesting that MVs originating from cell lysis may be larger than those of MVs originating from classic blebbing mechanisms. Moreover, MVs of the lys mutant were found to be smaller compared to wild-type MVs, while MVs of the ampDh3 mutant showed a similar size distribution (Fig. 4A).
Figure 4.
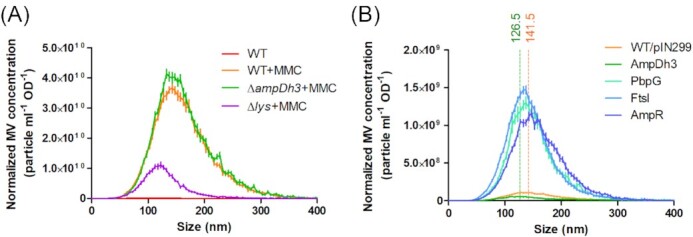
Normalized amounts and size distribution of MVs isolated from MMC-treated and untreated cultures of the PAO1 wild-type carrying pIN299 (A) and various overexpression mutants (B) as assessed by NTA. The mean sizes of MVs from the PAO1 wild-type and the AmpDh3 overexpression strain were determined to be 141.5 ± 0.29 nm (orange dotted lines, R2 = 0.8988) and 126.5 ± 0.27 nm (green dotted lines, R2 = 0.8991), respectively.
We also determined the amounts and sizes of MVs isolated from the culture supernatants of P. aeruginosa PAO1 strains overexpressing AmpDh3, PbpG, FtsI and AmpR. Overexpression of either PbpG, FtsI or AmpR was found to strongly increase MV production (Fig. 4B), indicating that these enzymes might play a role in explosive cell lysis. By contrast, AmpDh3 overexpression resulted in a reduction of MV production (Fig. 4B), which implies that AmpDh3 counteracts explosive cell lysis. In support of this idea, we found that the average size of MVs from the AmpDh3 overexpression strain was slightly decreased relative to wild-type MVs (Fig. 4B), which showed a size distribution similar to the lys mutant (Fig. 4A).
Overexpression of PbpG, FtsI and AmpR but not AmpDh3 affect cell viability
We noticed that overexpression of PbpG, FtsI and AmpR caused growth defects of P. aeruginosa PAO1, whereas the AmpDh3 overexpressing strain grew to a slightly higher optical density than the wild-type strain after 16 h of incubation (Fig. 5A). Inspection of the cultures by confocal laser scanning microscopy (CLSM) after staining with Syto 9 (green) and propidium iodide (PI, magenta) to visualize living and dead cells, respectively (Fig. 5B). In agreement with their impaired growth, we observed in cultures overexpressing PbpG, FtsI or AmpR high numbers of dead cells as well as extracellular DNA (eDNA). Moreover, the cells often exhibited an aberrant morphology or were ghost cells. Quantification of the eDNA showed increased amounts with the PbpG, FtsI and AmpR overexpressing strains compared to the wild type, while no significant effect was observed when AmpDh3 was overexpressed (Fig. 5C). These results suggest that overexpression of PbpG, FtsI and AmpR cause an imbalance of PG turnover with the consequence of cell lysis and concomitant MV formation. By contrast, overexpression of AmpDh3 did not affect cell viability (Fig. 5A). This is in agreement with our observation that this strain neither showed a growth defect nor increased cell lysis and suggests that induction of AmpDh3 expression upon MMC treatment rather represents the cells’ attempt to repair the damage caused by murein hydrolytic enzymes and thus dampens explosive cell lysis.
Figure 5.
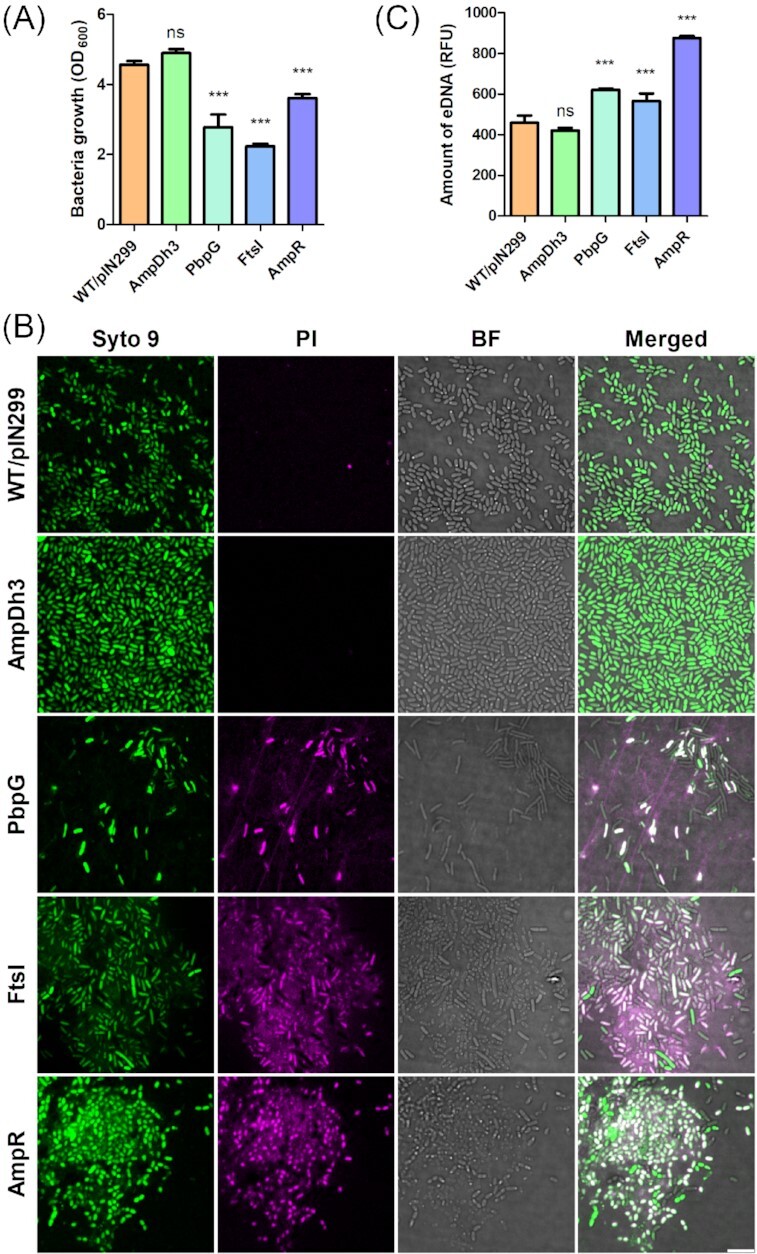
Self-toxicity of PAO1 derivatives overexpressing PG-modifying enzymes. Bacteria from an overnight culture were inoculated in LB at an initial OD600 of 0.01. Following 3 h of growth, 2% rhamnose was added. After 16 h of incubation, bacterial cell densities were determined (A) and samples were inspected by bright field microscopy (BF) and CLSM (B). Bacterial cells were stained with Syto 9 (green) and dead cells were visualized by staining with propidium iodide (PI, magenta). Bar indicates 5 µm. We also quantified the amounts of eDNA in the supernatants of the tested strains by staining with SYTOX (C). Data are presented as mean±SD from triplicate measurements and significant differences between groups were determined by one-way ANOVA followed by Dunnett's post-hoc test. *, P< 0.05; **, P< 0.01; ***, P< 0.001; ns, not significant.
AmpDh3 is involved in PG turnover
Despite the strong hydrolytic activity of AmpDh3 on zymograms it is neither involved in MV-mediated killing nor does its overexpression impair cell viability. In fact, our data rather suggest a role of this amidase in PG repair thereby counteracting explosive cell lysis. To test this possibility, we determined the rate of PG turnover in the wild-type and the AmpDh3 overexpression strain. To this end, we employed a metabolic labelling method that is based on the D-alanine (D-ala) analogue alkyne-D-alanine (alk-ala) that can readily be covalently linked to an azido-fluorophore using a copper-catalysed click chemistry (Siegrist et al. 2013) as detailed in the Material and Methods. Stationary phase cells were incubated with alk-ala or D-ala for 90 min prior to fluorescent labelling with Azide-fluor 488 to visualize the newly synthesized PG (Fig. S4). Quantification of the fluorescent signal of single cells by CLSM showed that AmpDh3 overexpression resulted in a significantly higher incorporation of alk-ala relative to the wild-type strain (Fig. 6A). Furthermore, we extracted the PG from the two exponential phase cultures and subjected them to quantitative LC-MS analysis (Fig. S5A). PG molecules with either D-ala or alk-ala incorporation were confirmed by MS/MS fragmentation (Fig. S5B). In support of the results of the microscopic investigation, we found that the PG of the overexpression strain incorporated much higher amounts of alk-ala, indicating an accelerated PG renewal in the AmpDh3 overexpression strain (Fig. 6B). In conclusion, our data suggest that AmpDh3 plays a critical role in cell wall recycling and thereby protects the cell against PG damage.
Figure 6.
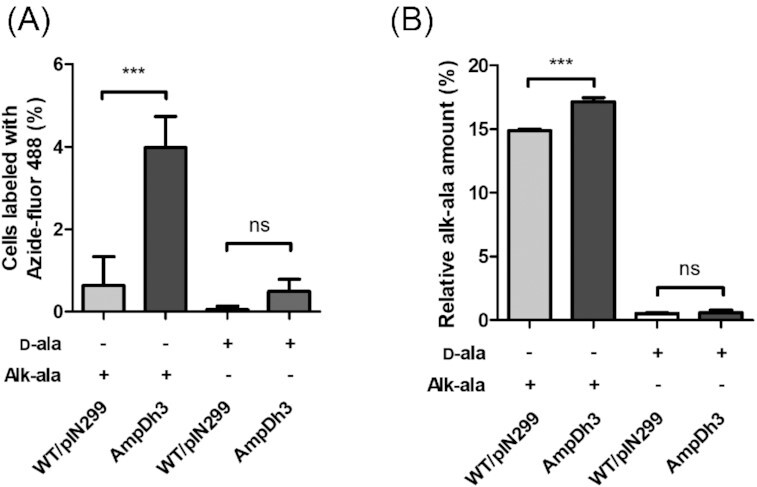
PG turnover rates of the P. aeruginosa PAO1 wild-type carrying pIN299 and an AmpDh3 overexpression strain. The incorporation of the D-alanine (D-ala) analogue alkyne-D-alanine (alk-ala) was determined. (A) Quantification of Azide-fluor 488 labelled cells from stationary phase cultures after CuAAC crosslinking. (B) LC-MS quantification of incorporated alk-ala in extracted PG from exponential phase cultures. Data are presented as mean±SD from triplicate measurements and significant differences between groups were determined by one-way ANOVA followed by Bonferroni's post-hoc test. *, P < 0.05; **, P< 0.01; ***, P< 0.001; ns, not significant.
Discussion
Work by Beveridge and co-workers showed that MVs contain PG hydrolases that lyse other bacteria, suggesting that MVs may represent a conceptually new group of antibiotics (Kadurugamuwa and Beveridge 1996). In their initial study, they demonstrated that MVs isolated from culture supernatants of P. aeruginosa PAO1 hydrolyse PG sacculi isolated from Gram-positive and Gram-negative bacteria (Kadurugamuwa and Beveridge 1996). The bacteriocidal effect was tested by adding MVs to viable cells of S. aureus D2C, E. coli DH5α, P. aeruginosa PAO1 and P. aeruginosa 8803 and followed growth by determining the CFUs. Only in the case of P. aeruginosa 8803 inhibition of growth was observed and it was therefore speculated that the limited bacteriocidal effect on intact cells of other bacteria was because actively growing cells were able to overcome and cope with the localized hydrolysis of PG after attack by MV-associated autolysins. To avoid growth of the target bacteria, a different killing assay was employed by the same group in a subsequent study, where, in contrast to their initial report, weak inhibition of P. aeruginosa PAO1 by its own MVs was observed (Li et al. 1998). Using this assay, in which the target bacteria were poured into agar plates without nutrients, and clearing zones caused by MV samples were measured after overnight incubation, the authors showed that MVs isolated from 15 Gram-negative bacteria exhibited bactericidal effects against various Gram-positive bacteria as well as E. coli K12 and P. aeruginosa PAO1. In agreement with the initial report, we show that in a liquid culture assay P. aeruginosa MVs exhibit no activity against the Gram-negative bacteria tested, while we did observe growth inhibition of the Gram-positive bacteria B. subtilis and S. aureus. In conclusion, while our work confirms that MVs are effective to inhibit growth of Gram-positive bacteria, additional work will be required to fully evaluate their potential against Gram-negative bacteria under different assay conditions.
On zymograms using P. aeruginosa PAO1 murein sacculi as substrate, Kadurugamuwa and Beveridge (1996) observed a major hydrolytic band with a MW of approximately 26 kDa and a very faint band at approximately 23 kDa (Kadurugamuwa and Beveridge 1996). However, the MWs of these bands are not entirely clear, as no size markers were included on the gels. In another study analysing P. aeruginosa PAO1 whole cells and supernatants, the Beveridge group observed two lytic bands with MWs of 26 and 29 kDa (with no size markers included on the gels) and concluded that the 26 kDa enzyme is the major autolysin of P. aeruginosa (Li et al. 1996). This 26 kDa enzyme was also proposed to be required for MV-mediated killing of other bacteria. Yet, the identity of this autolysin has remained unknown. In this study we used a proteomics approach to identify the proteins present within the two lytic bands observed in our zymograms. In good agreement with the estimated MW of 23 and 28 kDa for the lytic enzymes, we identified the 23 kDa endolysin Lys and the 28.7 kDa AmphD3 amidase within the bands, respectively. Further evidence that these enzymes are responsible for the hydrolytic activities of MVs observed on zymograms was obtained by the analysis of defined lys and ampDh3 mutants, each of which missed the respective hydrolytic band. Based on these results we hypothesize that the 26 kDa major autolysin that has been observed by the Beveridge group is most likely AmpDh3. Immunogold labeling of thin sections of P. aeruginosa PAO1 using antibodies raised against the 26 kDa autolysin has shown that this enzyme is mainly localized within the periplasm (Li et al. 1996). AmpDh3 is in fact a periplasmic enzyme that anchors to the PG in a multivalent manner (Lee et al. 2013, Zhang et al. 2013). We think that it is rather unlikely that the 26 kDa autolysin observed by the Beveridge laboratory represents Lys, as this endolysin causes explosive cell lysis when it enters the periplasmic space through pores that are formed by co-expressed holins. In contrast, the 26 kDa autolysin was shown to be associated with the cell envelopes of intact cells when cells are rapidly dividing (Li et al. 1996). Moreover, AmpDh3 was the major PG hydrolysing enzyme detected on zymograms of MVs isolated from untreated cultures, while Lys was barely detectable (Fig. 1) (Li et al. 1996).
AmpDh3 is a periplasmic zinc protease that together with the AmpDh2 amidase, which is sequestered on the inner leaflet of the outer membrane, is intimately involved in cell-wall recycling by removing the peptide stems from the PG (Martínez-Caballero et al. 2013, Lee et al. 2017). AmpDh3 was shown to have a higher activity than AmpDh2 and to interact preferentially with the polymeric and insoluble component of the cell wall (Lee et al. 2013). This may explain why this enzyme causes a pronounced clearing zone in zymograms. Yet, MVs of an ampDh3 mutant did not exhibit reduced antibacterial activity nor showed MVs of an AmpDh3 overexpressing strain increased biocidal activity, providing strong evidence that AmpDh3 is not the killing agent of MVs. Our data rather confirm previous reports that AmpDh3 is critical for PG turnover and repair.
The bacteriophage-derived endolysin lys is part of a gene cluster encoding the R- and F-type pyocins in P. aeruginosa (Nakayama et al. 2000). The release of these phage tail-like bacteriocins requires Lys-dependent cell lysis. To reach the periplasmic space, Lys is dependent on small hydrophobic proteins called holins that upon oligomerization in the cytoplasmic membrane form pores that allow the endolysin to assess the PG (Catalão et al. 2013). As a consequence of PG degradation cells undergo explosive cell lysis which not only releases the pyocines but also leads to the liberation of eDNA and the formation of MVs (Turnbull et al. 2016, Toyofuku et al. 2019). The production and release of the pyocins is induced through the RecA-mediated SOS response of P. aeruginosa (Nakayama et al. 2000, Cirz et al. 2006, Penterman et al. 2014), which is triggered by genotoxic stress. In agreement with this model, we show that the amount of Lys associated with MVs is greatly increased when cells are treated with the DNA-damaging antibiotics MMC and CIP. Interestingly, we observed that expression of AmpDh3 is also induced by MMC and CIP, indicating that the SOS response also stimulates expression of this amidase. We speculate that AmpDh3 counteracts explosive cell lysis, possibly by recycling damaged PG and thus reduces both MV and eDNA production.
While our data support the role of Lys in explosive cell lysis and the genesis of MVs we could not find evidence that Lys is required for MV-mediated bacterial growth inhibition. In fact, native MVs contained very low amounts of Lys, as under normal growth conditions only a small subpopulation (estimated to be > 1%) undergoes explosive cell lysis (Turnbull et al. 2016). While MMC, CIP and UV treatment increased the amounts of Lys associated with MVs (Fig. 1), their bactericidal activities were not enhanced nor showed MVs of a lys mutant reduced activity (Figs. 2 and S2). Collectively, our results suggest the two enzymes that showed activity on zymograms, the amidase AmpDh3 and the endolysin Lys, are not responsible for the antimicrobial effect of MVs. Our proteomics analysis confirmed previous reports that showed that several PG hydrolases are associated with MVs. However, apart from AmpDh3 and Lys we were unable to visualized these hydrolases on zymograms under the conditions used. Although additional work will be required to unravel their contribution to MV toxicity our data suggest that the bactericidal effect of MVs may not depend on a single autolysin but rather on the synergistic action of several enzymes. Moreover, secondary metabolites with antimicrobial activity that can be associated with MVs (Wettstadt 2020) could potentially also contribute to their toxicity. Work currently under progress aims at investigating this possibility.
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded by the Swiss National Science Foundation grants CRSII5_186410 and 310030_192800. We would like to thank Aurélien Bailly for help with confocal microscopy and image-analysis, Carlotta Fabbri for technical support and Bernd Roschitzki from the Functional Genomics Center Zurich for his advice with the MS analysis.
Contributor Information
Yi-Chi Chen, Department of Plant and Microbial Biology, University of Zürich, Switzerland.
Ratchara Kalawong, Department of Plant and Microbial Biology, University of Zürich, Switzerland.
Masanori Toyofuku, Faculty of Life and Environmental Sciences, University of Tsukuba, Japan.
Leo Eberl, Department of Plant and Microbial Biology, University of Zürich, Switzerland.
Conflict of interest statement
None declared.
References
- Altindis E, Fu Y, Mekalanos JJ. Proteomic analysis of Vibriocholerae outer membrane vesicles. Proc Natl Acad Sci. 2014;111:E1548–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D, Kumari H, Mathee K. Pseudomonas aeruginosa Ampr: an acute-chronic switch regulator. Pathog Dis. 2015;73:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkow-Oesterreicher S, Turker C, Panse C. FCC - An automated rule-based processing tool for life science data. Source Code Biol Med. 2013;8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernadsky G, Beveridge TJ, Clarke AJ. Analysis of the sodium dodecyl sulfate-stable peptidoglycan autolysins of select gram-negative pathogens by using renaturing polyacrylamide gel electrophoresis. J Bacteriol. 1994;176:5225–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitto NJ, Chapman R, Pidot Set al. . Bacterial membrane vesicles transport their DNA cargo into host cells. Sci Rep. 2017;7:7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown L, Wolf JM, Prados-Rosales Ret al. . Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat Rev Microbiol. 2015;13:620–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalão MJ, Gil F, Moniz-Pereira Jet al. . Diversity in bacterial lysis systems: bacteriophages show the way. FEMS Microbiol Rev. 2013;37:554–71. [DOI] [PubMed] [Google Scholar]
- Chatterjee D, Chaudhuri K. Association of cholera toxin with Vibriocholerae outer membrane vesicles which are internalized by human intestinal epithelial cells. FEBS Lett. 2011;585:1357–62. [DOI] [PubMed] [Google Scholar]
- Cirz RT, O'Neill BM, Hammond JAet al. . Defining the Pseudomonasaeruginosa SOS response and its role in the global response to the antibiotic ciprofloxacin. J Bacteriol. 2006;188:7101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AJ. The “hole” story of predatory outer-membrane vesicles. Can J Microbiol. 2018;64:589–99. [DOI] [PubMed] [Google Scholar]
- Flannagan RS, Linn T, Valvano MA. A system for the construction of targeted unmarked gene deletions in the genus Burkholderia. Environ Microbiol. 2008;10:1652–60. [DOI] [PubMed] [Google Scholar]
- Gill S, Catchpole R, Forterre P. Extracellular membrane vesicles in the three domains of life and beyond. FEMS Microbiol Rev. 2019;43:273–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadurugamuwa JL, Beveridge TJ. Bacteriolytic effect of membrane vesicles from Pseudomonasaeruginosa on other bacteria including pathogens: conceptually new antibiotics. J Bacteriol. 1996;178:2767–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmitt PT, Harwood CR, Barer MR. Toxin gene expression by shiga toxin-producing Escherichiacoli: the role of antibiotics and the bacterial SOS response. Emerg Infect Dis. 2000;6:458–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppen K, Hampton TH, Jarek Met al. . A novel mechanism of host-pathogen interaction through sRNA in bacterial outer membrane vesicles. PLoS Pathog. 2016;12:e1005672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong KF, Jayawardena SR, Indulkar SDet al. . Pseudomonas aeruginosa AmpR is a global transcriptional factor that regulates expression of AmpC and PoxB beta-lactamases, proteases, quorum sensing, and other virulence factors. Antimicrob Agents Chemother. 2005;49:4567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn MJ, Kesty NC. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 2005;19:2645–55. [DOI] [PubMed] [Google Scholar]
- Lee M, Artola-Recolons C, Carrasco-López Cet al. . Cell-wall remodeling by the zinc-protease Ampdh3 from Pseudomonasaeruginosa. J Am Chem Soc. 2013;135:12604–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Hesek D, Lastochkin Eet al. . Deciphering the nature of enzymatic modifications of bacterial cell walls. ChemBioChem. 2017;18:1696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Clarke AJ, Beveridge TJ. A major autolysin of Pseudomonasaeruginosa: subcellular distribution, potential role in cell growth and division and secretion in surface membrane vesicles. J Bacteriol. 1996;178:2479–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Clarke AJ, Beveridge TJ. Gram-negative bacteria produce membrane vesicles which are capable of killing other bacteria. J Bacteriol. 1998;180:5478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Pessi G, Riedel Ket al. . Identification of AHL- and BDSF-Controlled proteins in burkholderiacenocepacia by proteomics. Methods Mol Biol. 2018;1673:193–202. [DOI] [PubMed] [Google Scholar]
- Martínez-Caballero S, Lee M, Artola-Recolons Cet al. . Reaction products and the X-ray structure of Ampdh2, a virulence determinant of Pseudomonasaeruginosa. J Am Chem Soc. 2013;135:10318–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moak M, Molineux IJ. Peptidoglycan hydrolytic activities associated with bacteriophage virions. Mol Microbiol. 2004;51:1169–83. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Takashima K, Ishihara Het al. . The R-type pyocin of Pseudomonasaeruginosa is related to P2 phage, and the F-type is related to lambda phage. Mol Microbiol. 2000;38:213–31. [DOI] [PubMed] [Google Scholar]
- Orench-Rivera N, Kuehn MJ. Environmentally controlled bacterial vesicle-mediated export. Cell Microbiol. 2016;18:1525–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penterman J, Singh PK, Walker GC. Biological cost of pyocin production during the SOS response in Pseudomonasaeruginosa. J Bacteriol. 2014;196:3351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem. 2003;75:663–70. [DOI] [PubMed] [Google Scholar]
- Schwechheimer C, Kuehn MJ. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat Rev Microbiol. 2015;13:605–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegrist MS, Whiteside S, Jewett JCet al. . (D)-Amino acid chemical reporters reveal peptidoglycan dynamics of an intracellular pathogen. ACS Chem Biol. 2013;8:500–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström AE, Sandblad L, Uhlin BEet al. . Membrane vesicle-mediated release of bacterial RNA. Sci Rep. 2015;5:15329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku M, Cárcamo-Oyarce G, Yamamoto Tet al. . Prophage-triggered membrane vesicle formation through peptidoglycan damage in Bacillussubtilis. Nat Commun. 2017;8:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku M, Nomura N, Eberl L. Types and origins of bacterial membrane vesicles. Nat Rev Microbiol. 2019;17:13–24. [DOI] [PubMed] [Google Scholar]
- Toyofuku M, Zhou S, Sawada Iet al. . Membrane vesicle formation is associated with pyocin production under denitrifying conditions in Pseudomonasaeruginosa PAO1. Environ Microbiol. 2014;16:2927–38. [DOI] [PubMed] [Google Scholar]
- Turnbull L, Toyofuku M, Hynen ALet al. . Explosive cell lysis as a mechanism for the biogenesis of bacterial membrane vesicles and biofilms. Nat Commun. 2016;7:11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai SN, Lindmark B, Söderblom Tet al. . Vesicle-mediated export and assembly of pore-forming oligomers of the enterobacterial ClyA cytotoxin. Cell. 2003;115:25–35. [DOI] [PubMed] [Google Scholar]
- Wettstadt S. Death in a sphere: Chromobacterium violaceum secretes outer membrane vesicles filled with antibiotics. Environ Microbiol Rep. 2020;12:255–7. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj Net al. . Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–62. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhao SQ, Zhang Jet al. . Proteomic analysis of vesicle-producing pseudomonas aeruginosa PAO1 exposed to X-Ray irradiation. Front Microbiol. 2020;11:558233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Lee M, Hesek Det al. . Reactions of the three AmpD enzymes of Pseudomonasaeruginosa. J Am Chem Soc. 2013;135:4950–3..x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.