

Abstract
The “eye-of-the-tiger” sign in brain magnetic resonance imaging (MRI) is typically associated with neurodegeneration with brain iron accumulation disorders, especially pantothenate kinase-associated neurodegeneration. However, very similar neuroimaging findings may be seen in other neurodegenerative disorders involving the basal ganglia. We report here a patient with fucosidosis who had MRI brain findings closely resembling the “eye-of-the-tiger” sign.
Keywords: eye-of-the-tiger sign, fucosidosis, neurodegeneration with brain iron accumulation
Introduction
The “eye-of-the-tiger” sign, which refers to the hypointensity of the globi pallidi with a central region of hyperintensity in T2-weighted images on magnetic resonance imaging (MRI) of the brain, is typically associated with neurodegeneration with brain iron accumulation (NBIA) disorders, especially pantothenate kinase-associated neurodegeneration (PKAN). The hypointensity results from accumulation of iron (in the form of ferritin) and the central hyperintensity occurs due to gliosis. 1 However, certain other neurodegenerative disorders involving the basal ganglia may have very similar neuroimaging findings. 1 We report here a 2.5-year-old male patient with neuroregression and dystonia, who was presumed to have PKAN in view of his MRI brain findings closely resembling the “eye-of-the-tiger” sign, but following detailed clinical phenotyping and molecular genetic testing was identified to have fucosidosis.
Case Report
A 2.5-year-old male child was referred for clinical genetic evaluation to our center, with a referral diagnosis of PKAN, in view of the history of neuroregression and findings resembling “eye-of-the-tiger” sign in MRI brain. He was the first child of a third-degree consanguineous couple, and had a 10-month-old younger female sibling who was asymptomatic as per the given history. There were no adverse events in the perinatal or neonatal period. Global developmental delay was noted since early infancy. He attained stable head control by the age of 7 months, sitting without support by 14 months, independent walking by 18 months, and bisyllabic speech by 18 months. Gradual regression of all attained milestones was perceived after 18 months of age, along with abnormal body posturing and swallowing and feeding difficulties. Vision and hearing were not impaired, and there was no history of seizures.
On clinical evaluation, his growth parameters were within the normal centiles. He was noted to have coarse facial features with a prominent forehead, mild ocular hypertelorism, broad nasal bridge, smooth philtrum, and thick lips ( Fig. 1A ). There were no other significant external dysmorphic features. The eyes, skin, and joints were normal; and corneal clouding, angiokeratomas, and joint contractures were absent. Neurological examination revealed increased tone and exaggerated deep tendon reflexes in both upper and lower limbs, extensor plantar reflex bilaterally, and intermittent dystonic posturing. Choreoathetosis or other involuntary movements were absent. He was able to sit briefly with support and lacked even bisyllabic speech. There was no organomegaly, and other systems were clinically normal.
Fig. 1.
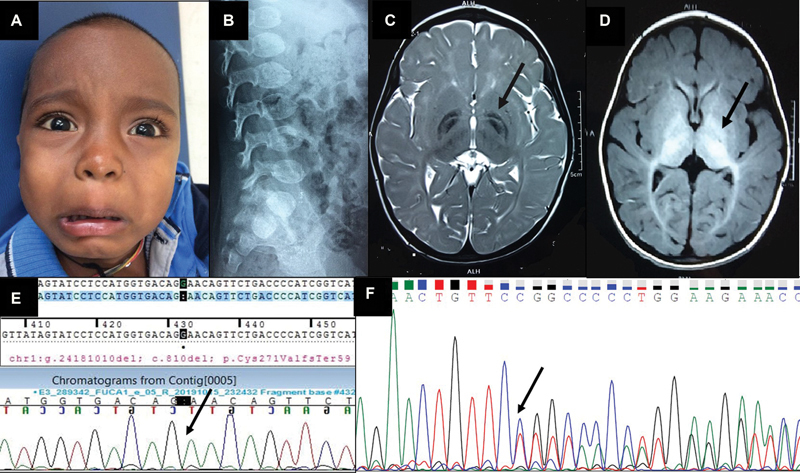
( A ) Close-up of the child's face showing the coarse facial features, mild ocular hypertelorism, and broad nasal bridge and thick lips. ( B ) Lateral radiograph of the lumbosacral spine showing the ovoid vertebrae and the anterior-inferior beaking indicative of dysostosis multiplex. ( C ) T2-weighted axial image of MRI brain showing bilaterally symmetrical hypointensity of both the globi pallidi with a central streak of hyperintensity between the medial and lateral segments of the globi pallidi, resembling the “eye-of-the-tiger” sign (pointed out by the arrow), along with symmetric hyperintensities in bilateral cerebral white matter. ( D ) T1-weighted axial image of MRI brain showing bilaterally symmetrical hyperintensity of globi pallidi, substantia nigra, and subthalamic nuclei (pointed out by the arrow). ( E ) Sanger sequence chromatogram of the proband showing the homozygous c.810del variant in the FUCA1 gene (the position of the single nucleotide deletion is marked with the arrow). ( F ) Sanger sequence chromatogram of the mother showing the heterozygous c.810del variant in the FUCA1 gene (the position of the single nucleotide deletion is marked with the arrow).
Lateral radiographs of the cervical and thoracolumbar spines showed cervical platyspondyly and ovoid vertebrae with anterior beaking ( Fig. 1B ). The available MRI brain images were reviewed. T2-weighted images showed bilaterally symmetrical hypointensity of both globi pallidi with a central streak of hyperintensity between the medial and lateral segments of the globi pallidi resembling the “eye-of-the-tiger” sign, along with symmetrical hypointensity in bilateral substantia nigra and subthalamic nuclei ( Fig. 1C ). T1-weighted images showed bilaterally symmetrical hyperintensity in the globi pallidi, substantia nigra, subthalamic nuclei, and optic radiations ( Fig. 1D ). In addition, symmetric hyperintensities in bilateral cerebral white matter sparing the peri-Rolandic white matter (in the T2-weighted and FLAIR images) and thinning of body of the corpus callosum were also noted.
The possibilities of type I fucosidosis and type II (late-infantile form) GM1 gangliosidosis were considered clinically. The referral diagnosis of PKAN appeared unlikely in view of the additional findings of coarse facies, dysostosis multiplex, and associated white matter changes in the brain MRI.
Molecular genetic testing through next generation sequencing-based clinical exome sequencing using the Illumina sequencing platform (Illumina Inc., San Diego, California, United States), showed a homozygous, single base-pair deletion c.810del in exon 5 of the FUCA1 gene (transcript id ENST00000374479.3), resulting in frameshift and premature termination of the protein, 59 amino acids downstream to codon 271 (p.Cys271ValfsTer59 ) . This is a known pathogenic variant listed in HGMD ( http://www.hgmd.cf.ac.uk/ac/ ) and ClinVar ( https://www.ncbi.nlm.nih.gov/clinvar/ ). It is not reported in the 1000 genomes ( https://www.internationalgenome.org/1,000-genomes-browsers/ ) or gnomAD ( https://gnomad.broadinstitute.org/ ) databases. It is classified as “pathogenic” as per the criteria outlined by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP variant classification guidelines 2015). 2 The in silico prediction of the variant is disease causing by MutationTaster2 ( http://www.mutationtaster.org/ ) and the reference base is conserved across mammals.
As the FUCA1 gene has a pseudogene in chromosome 2 with 80% sequence homology, targeted Sanger sequencing was done, which confirmed presence of the variant ( Fig. 1E ), thereby confirming the diagnosis of type I fucosidosis in the child. Further, Sanger sequencing-based targeted variant testing was also done in both parents and the asymptomatic younger sibling, and all three were found to be heterozygous carriers for this variant ( Fig. 1F ).
The parents were counseled about the course of the disorder, continuance of multidisciplinary supportive care and follow-up, the autosomal recessive pattern of inheritance with 25% risk of recurrence in each subsequent offspring, and the availability of prenatal and preimplantation genetic testing options for their subsequent pregnancies.
Discussion
Fucosidosis (OMIM 230000) is a rare, neurodegenerative, lysosomal storage disorder caused by deficiency of the lysosomal enzyme α-L-fucosidase, which is responsible for degradation of fucose-containing glycoproteins and glycolipids. 3 It occurs due to biallelic mutations in the FUCA1 gene. Only 36 mutations in the FUCA1 gene are listed in the Human Gene Mutation Database ( http://www.hgmd.cf.ac.uk/ac/index.php ) and very few mutation-confirmed and/or enzymatically-proven patients have been reported from India till date. 4 5 6 The homozygous single base deletion identified in this patient is expected to cause a shift in the reading frame and premature termination of the protein. We plan to perform further functional validation experiments using mammalian cell lines to confirm the pathogenicity of this variant, through site-directed mutagenesis and cell transfection of the mutant cDNA construct, followed by α-L-fucosidase assay and immunoblotting of the transfected cell-lysate.
Fucosidosis presents with a wide spectrum of manifestations and is classified based on the age of onset and progression of manifestations as type I, which presents in early infancy, is rapidly progressive and fatal within the first decade of life, and type-II, which has later onset, slower progression, and longer survival. Various manifestations of fucosidosis include intellectual disability (95%), neuroregression (88%), coarse facies (79%), growth retardation (78%), recurrent infections (78%), kyphoscoliosis (66%), dysostosis multiplex (58%), angiokeratomas (52%), joint contractures (48%), seizures (38%), visceromegaly (30%), hearing loss (12%), hernia (9%), and visual impairment (6%). 7 Our patient had type I fucosidosis with the typical features of early onset psychomotor retardation, neuroregression, coarse facies, and dysostosis multiplex. He also had generalized dystonia, which is a relatively uncommon neurological finding in fucosidosis. In a literature review of 75 cases of fucosidosis compiled from different scientific publications, Wali et al found that dystonia was reported in only nine (i.e., 12%) of patients and six of these had generalized dystonia, while one each had focal, segmental, and hemidystonia. 5
Clinical findings of fucosidosis overlap with those of other neuroregressive lysosomal storage disorders, especially GM1 gangliosidosis and mannosidosis, and the neuroimaging findings overlap with those of PKAN and GM1 gangliosidosis. As in our patient, findings closely resembling the “eye-of-the-tiger” sign, which is most commonly described in PKAN, have been previously reported in a few other patients with fucosidosis. 8 9 10 Galluzzi et al described findings very similar to those observed in our patient, that is, hypointensity of both globi pallidi with a central curvilinear streak of hyperintensity between the medial and lateral segments of the globi pallidi in T2-weighted images in the MRI brain of three patients with fucosidosis. This streak corresponds to the medial medullary lamina of each globus pallidus, and though the exact pathogenesis of the altered signal intensity is not clearly understood, it is hypothesized to be related to the pallidal accumulation of glycolipids in fucosidosis. 8 “Eye-of-the-tiger” like appearance in MRI brain has also been described in some children with Leigh disease and mitochondrial membrane protein-associated neurodegeneration (MPAN), as well as in adults with multiple system atrophy, neuroferritinopathy, and primary familial brain calcification. 1 11 12 The clinical phenotype and presence of additional findings in MRI brain, as outlined in Table 1 , help to differentiate between these conditions.
Table 1. Comparison of childhood-onset neurodegenerative conditions with neuroimaging findings similar to fucosidosis.
| Features | Fucosidosis | PKAN (NBIA) |
MPAN (NBIA) |
GM1 gangliosidosis | Proband |
|---|---|---|---|---|---|
| Clinical features | |||||
| Neuroregression | + | + | + | + | + |
| Pyramidal signs | + | + | + | + | + |
| Dystonia, dysarthria, and other extrapyramidal signs | +/− (less common) | + | + | +/− (in juvenile-onset form) | + |
| Seizures | +/− | − | − | + | − |
| Other neurological features | Peripheral neuropathy, hearing loss | − | Psychiatric disturbances, motor axonopathy | − | − |
| Coarse facies | + | − | − | + | + |
| Dysostosis multiplex | + | − | − | + | + |
| Other extra-neurological findings | Angiokeratoma, anhidrosis, cardiomegaly, hepatosplenomegaly, hernia | − | − | Hepatosplenomegaly, cardiomyopathy, gingival hyperplasia, hernia, angiokeratoma | − |
| Ocular findings | Hypertelorism, tortuosity of conjunctival vessels | Abnormal eye movements (including vertical saccades), Adie's pupil, pigmentary retinal degeneration | Optic atrophy | Corneal clouding, cherry red spot in fundus | Mild hypertelorism |
| Neuroimaging findings | |||||
| Basal ganglia | T2-weighted hypointensity of globus pallidus with streak of hyperintensity involving medial medullary lamina in some cases, resembling “eye-of-the-tiger” sign | “Eye-of-the-tiger” sign; calcification of globus pallidus in CT scan | T2-weighted hypointensity of globus pallidus with streak of hyperintensity involving medial medullary lamina that resembles “eye-of-the-tiger” sign | T2-weighted hypointensity of globus pallidus | T2-hypointensity of globus pallidus with central streak of hyperintensity involving medial medullary lamina resembling “eye-of-the-tiger” sign |
| Cerebral atrophy | +/− | − | + | + | − |
| Cerebellar atrophy | − | − | + | +/− | − |
| White matter changes | + | − | − | + | + |
| Genetic basis | |||||
| Gene | FUCA1 | PANK2 | C19orf12 | GLB1 | FUCA1 |
Abbreviations: MPAN, mitochondrial membrane protein-associated neurodegeneration; NBIA, neurodegeneration with brain iron accumulation; PKAN, pantothenate kinase-associated neurodegeneration.
Detailed clinical phenotyping and analysis of MRI brain findings in entirety are thus essential to obtain the correct diagnosis to ensure appropriate management and follow-up of the patient and accurate genetic counseling of the family to prevent recurrence.
Conflict of Interest None declared.
Note
The authors are grateful to the patient and his family, for their participation in this study. Molecular genetic testing of the patient was performed in MedGenome Labs Ltd., Bengaluru, India.
References
- 1.Hayflick S J, Hartman M, Coryell J, Gitschier J, Rowley H. Brain MRI in neurodegeneration with brain iron accumulation with and without PANK2 mutations. AJNR Am J Neuroradiol. 2006;27(06):1230–1233. [PMC free article] [PubMed] [Google Scholar]
- 2.ACMG Laboratory Quality Assurance Committee . Richards S, Aziz N, Bale S. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(05):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.George H T. 8th ed. New York: McGraw-Hill; 2001. Disorders of glycoprotein degradation: α-mannosidosis, β-mannosidosis, α-fucosidosis and sialidosis; pp. 3507–3533. [Google Scholar]
- 4.Sheth J, Mistri M, Bhavsar R. Lysosomal storage disorders in Indian children with Neuroregression attending a genetic center. Indian Pediatr. 2015;52(12):1029–1033. doi: 10.1007/s13312-015-0768-x. [DOI] [PubMed] [Google Scholar]
- 5.Wali G, Wali G M, Sue C M, Kumar K R. A novel homozygous mutation in the FUCA1 gene highlighting fucosidosis as a cause of dystonia: case report and literature review. Neuropediatrics. 2019;50(04):248–252. doi: 10.1055/s-0039-1684052. [DOI] [PubMed] [Google Scholar]
- 6.Gowda V K, Srinivasan V M, Vegda H, Bhat M. Fucosidosis with pathogenic variant in FUCA1 gene. Indian J Pediatr. 2020;87(10):867–868. doi: 10.1007/s12098-020-03246-7. [DOI] [PubMed] [Google Scholar]
- 7.Willems P J, Gatti R, Darby J K. Fucosidosis revisited: a review of 77 patients. Am J Med Genet. 1991;38:111–131. doi: 10.1002/ajmg.1320380125. [DOI] [PubMed] [Google Scholar]
- 8.Galluzzi P, Rufa A, Balestri P, Cerase A, Federico A. MR brain imaging of fucosidosis type I. AJNR Am J Neuroradiol. 2001;22(04):777–780. [PMC free article] [PubMed] [Google Scholar]
- 9.Gautschi M, Merlini L, Calza A M, Hayflick S, Nuoffer J M, Fluss J. Late diagnosis of fucosidosis in a child with progressive fixed dystonia, bilateral pallidal lesions and red spots on the skin. Eur J Paediatr Neurol. 2014;18(04):516–519. doi: 10.1016/j.ejpn.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Zubarioglu T, Kiykim E, Zeybek C A. Clinical and neuroradiological approach to fucosidosis in a child with atypical presentation. Ann Indian Acad Neurol. 2015;18(04):471–474. doi: 10.4103/0972-2327.160090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gregory A, Hayflick S J.Pantothenate kinase-associated neurodegeneration 2002Seattle, WA: University of Washington;1993–2020. Available at:https://www.ncbi.nlm.nih.gov/books/NBK1490/ [PubMed] [Google Scholar]
- 12.Yoganathan S, Sudhakar S V, Thomas M, Dutta A K, Danda S. “Eye of tiger sign” mimic in an adolescent boy with mitochondrial membrane protein associated neurodegeneration (MPAN) Brain Dev. 2016;38(05):516–519. doi: 10.1016/j.braindev.2015.10.017. [DOI] [PubMed] [Google Scholar]