

Abstract
Neurofibromatosis type 1 (NF1) is one of the most common inherited disorders. It is caused by mutations in the neurofibromin-1 gene ( NF1 ) and affects the formation and growth of nerve tissues. More than 3,600 pathogenic variants in the NF1 gene have been identified from patients with most of the germline variants are from the Western populations. We found 16 patients (15 Chinese and 1 Asian Indian) who had heterozygous variants in NF1 through targeted next-generation sequencing. There were 15 different variants: 4 frameshift, 4 nonsense, 5 missense, and 2 splice variants. One nonsense variant and three frameshift variants had never been reported in any population or patient database. Twelve of the 16 patients met the NF1 diagnostic criteria, and each was found to have a pathogenic or likely pathogenic variant. Three different missense variants of unknown significance were discovered in the other four patients who did not meet NF1 diagnostic criteria. Our findings add four novel variants to the list of genetic mutations linked to NF1's various clinical manifestations.
Keywords: neurofibromatosis type 1, NF1 variants , café-au-lait spots, Lisch nodules, optic pathway glioma
Introduction
Neurofibromatosis is an autosomal dominant disorder that affects the formation and growth of nerve tissues. There are three different types based on the age of onset and genes involved. The most common is the von Recklinghausen peripheral type, known as neurofibromatosis type 1 (NF1, MIM# 162200), which accounts for more than 90% of all neurofibromatosis. 1 2 It is also one of the most common inherited disorders with a prevalence between 1:2,000 and 1:4,500. 3 4 5 6 The phenotype for type 1 usually appears from childhood. In contrast, type 2 (MIM# 101000) has a later onset from late teens for schwannoma development and other presentations such as tinnitus, hearing loss, and focal neurologic problems. The third type, schwannomatosis (MIM# 162091), presents as an asymptomatic mass or diffuse or localized pain and usually affects adults older than 30 years.
The main presentations of NF1 include café-au-lait spots (CALSs) or cafe-au-lait macules (CALMs), Lisch nodules, freckling in the axillary or inguinal regions, neurofibromas, optic pathway gliomas, and skeletal abnormalities. NF1 patients are also at increased risk for vascular dysplasia, pheochromocytoma and neuronal benign/cancerous tumors, and neurocognitive difficulties that may include intellectual disabilities and social and behavioral issues. 7 8 9 While the penetrance is essentially complete by 20 years, 10 clinical presentations are highly variable. They may vary in patients of different ages and between individuals and family members who carry the same pathogenic variant. 11 In addition, there is poor genotype–phenotype correlation except for observed milder phenotype associated with a three-nucleotide deletion in exon 22. 12
Haploinsufficiency caused by loss-of-function mutations and microdeletions involving the neurofibromin gene ( NF1 , OMIM# 613113) has been identified in patients with NF1. The gene was first mapped to chromosome 17q11.2 13 with 58 exons spread over 287 kilobases. 14 15 16 17 18 It encodes neurofibromin, a GTPase-activating protein (GAP) which serves as a negative regulator of the RAS oncogene signal transduction pathway. Through its GAP-related domain (GRD), which is responsible for interaction with RAS proteins and GTP hydrolysis, neurofibromin downregulates the RAS pathway and plays an essential role in cellular processes such as proliferation and apoptosis. 19 20 21
Although NF1 can be diagnosed clinically, genetic testing may be useful for distinguishing between NF1 and related conditions such as Legius' syndrome (multiple CALS and freckling) and other rasopathies due to overlapping features such as short stature and developmental delay. 22 23 As clinical signs may evolve with age, genetic testing can lead to its early confirmation in children who may not fulfill clinical diagnostic criteria at the time of clinical evaluation. This will allow age-specific monitoring of specific clinical features to enable early treatment as they occur. A laboratory result could facilitate engagement with patients and their parents that could explain the phenotype and enhance the likelihood of regular return to the clinic for reviews. Here, we report the identification of 15 unique NF1 variants from 16 patients and present the phenotypic characteristics of these patients.
Materials and Methods
This study was conducted with approval of the SingHealth Central Institutional Review Board, which oversees all research activities in KK Women's and Children's Hospital. Written informed consent was obtained from the patients or their parents, or legal guardians. The respective clinical geneticists assessed the patients for features related to NF1.
According to the manufacturer's instructions, genomic DNA was extracted from blood samples using Gentra Puregene Blood Kit (Qiagen Inc., Germantown, United States). Next-generation sequencing was performed using a customized panel or the TruSight One panel (Illumina, San Diego, United States) and the MiSeq system (Illumina). Identified variants were confirmed by Sanger sequencing using BigDye v3.1 (Applied Biosystems, Waltham, United States) before testing with parental samples to determine inheritance status.
Output sequence reads were mapped and aligned to human reference sequence GRCh37/hg19. Variants data were generated using MiSeq Reporter and annotated using the online wANNOVAR program. 24 Candidate variants were further annotated with Alamut Visual v2.11 from Interactive Biosoftware, Rouen, France ( http://www.interactive-biosoftware.com/alamut-visual/ ). The pathogenicity of the variants was classified according to the published guidelines of the American College of Medical Genetics and Genomics (ACMG) and the Association of Molecular Pathology (AMP). 25
Results
Clinical Features
The demographic and clinical presentations of the patients found to have NF1 variants are shown in Table 1 . Apart from one Asian Indian, the rest were of Chinese ancestry. Three were females and the other 13 were males. The mean age at clinical diagnosis or recruitment for testing was 6.5 years, while the median age was 4.1 years.
Table 1. Demographic characteristics and clinical presentations of the 16 patients.
| Patient no. | Gender | Age at presentation or diagnosis (y) | Age at last visit (y) | NIH criteria | Other presentations | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ≥6 CALS with diameter of >5 mm (prepubertal) or >15 mm (postpubertal) | ≥2 neurofibromas or 1 plexiform neurofibroma | Freckling in the axillary or inguinal regions | ≥2 Lisch nodules and (iris hamartomas) | Optic glioma | Scoliosis, sphenoid dysplasia or thinning of long bone cortex | 1 deg relative to NF1 | Short stature | Developmental delay | Learning disability | Social and behavioral disorders | ||||
| 1 | F | 5 | 19 | + | + | + | + | − | − | − | + | − | − | − |
| 2 | M | 14 | 25 | + | + | + | NT | − | − | + | + | − | + | − |
| 3 | M | 1.2 | 14 | + | + | + | − | − | + | − | − | + | + | − |
| 4 | M | 10.9 | 16 | + | + | + | − | − | − | − | − | − | − | − |
| 5 | M | 1.3 | 8 | + | − | + | − | − | − | − | + | + | + | − |
| 6 | M | 5.9 | 11 | + | − | + | − | − | − | − | − | − | + | − |
| 7 | M | 2.4 | 11 | + | − | + | − | − | − | − | + | − | − | − |
| 8 | F | 3.5 | 5 | + | + | + | − | + | − | − | + | + | + | + |
| 9 | M | 4.8 | 14 | + | − | + | − | − | − | − | − | + | + | + |
| 10 | M | 1.2 | 13 | + | − | + | + | − | − | + | + | − | + | − |
| 11 | F | 4.6 | 10 | + | + | − | − | − | − | − | − | + | + | + |
| 12 | M | 17.2 | 19 | + | + | + | − | − | − | − | + | − | − | − |
| 13 | M | 0.4 | 8 | − | − | − | NT | − | − | − | − | − | − | − |
| 14 | M | 28.6 | 32 | − | + | − | NT | − | − | − | − | − | − | − |
| 15 | M | 0.2 | 15 | − | − | − | − | − | − | − | − | − | − | − |
| 16 | M | 2.9 | 8 | + | − | − | − | − | − | − | + | − | − | − |
Abbreviations: CALS, café-au-lait spot; NF1, neurofibromatosis type 1; NIH, National Institutes of Health; NT, not tested.
Patients 1 to 12 fulfilled clinical diagnostic criteria, that is, had at least two of the seven main features of the National Institutes of Health diagnostic criteria for NF1. 26 Their most common major phenotypic features were CALMs (12/12), skin freckling (11/12), and neurofibromas (7/12). Other commonly shared features were learning disability (8/12), short stature (7/12), and developmental delay (5/12) ( Table 1 ).
Some of the 12 patients also had additional features that are not major criteria for NF1. Patient 3 had thoracolumbar scoliosis, while Patient 8 developed an NF1-related glioma and had seizures. Other patients had clinical features not usually associated with NF1: Patient 6 presented with bilateral hearing loss and had a history of uteropelvic obstruction and adynamic ureter. Patient 7 had a history of chronic gastrointestinal symptoms, iron deficiency anemia, and right inguinal hernia. Patient 9 had autism spectrum disorder and allergic rhinitis.
Four of the 16 patients had fewer than two major features and did not meet the diagnostic criteria for NF1. They had additional features such as right hemihyperplasia involving the right arm and right leg for Patient 13 who also had naevus flammeus on the forehead and nape of the neck, and also generalized telangiectasia. Patient 14 had plexiform neurofibroma and a large CALS overlying the neurofibroma. Although he had multiple small CALMs on the neck and upper limbs, all of them were < 15 mm. His features presented only in adulthood, so he could be mosaic for a pathogenic variant that might not be detected in his germline DNA. Patient 15 demonstrated mainly eye-related abnormalities, including left microphthalmia, left iris coloboma, and left choanal atresia. He was suspected of CHARGE syndrome, but no variant in CHD7 was identified. He also had mild learning difficulties. Patient 16 and his father both had small CALSs, but only the father had axillary freckling, and both did not meet the criteria for NF1.
Molecular Analysis
Fifteen unique NF1 variants were identified from the 16 patients. Patients 13 and 14 had the same missense variant ( Table 2 ). There were four variants in the cysteine and serine-rich domain (CSRD), three near or within the tubulin-binding domain (TBD), two in the GRD, one at the boundary of the GRD and RAS regulatory domain (Sec14-PH), one in Sec14-PH, and two in the carboxyl-terminal domain. Two of the variants did not map to any protein domain.
Table 2. Characteristics of the NF1 variants identified; positions are based on NM_000267.3 a .
| Patient no. | Nucleotide change | Consequence | Exon | Domain | HGMD b | Known? | Inheritance | ACMG |
|---|---|---|---|---|---|---|---|---|
| 1 | c.1393_1394delAG | p.L466Yfs*3 | 13 | − | − | Known | De novo | P |
| 2 | c.1748A > G | p.K583R | 16 | CSRD | CM1111788 | Known | NT | LP |
| 3 | c.1924C > T | p.Q642* | 17 | CSRD | CM1815723 | Known | De novo | P |
| 4 | c.2681T > C | p.F894S | 21 | CSRD | − | Known | De novo | LP |
| 5 | c.3203delT | p.L1068Wfs*9 | 25 | TBD | − | Novel | De novo | P |
| 6 | c.3734_3735delCT | p.L1246Vfs*2 | 28 | GRD | − | Novel | De novo | P |
| 7 | c.4269G > A | p.K1423 =/splice | 32 | GRD | CS177447 | Known | De novo | P |
| 8 | c.4537C > T | p.R1513* | 35 | GRD/Sec14-PH | CM941093 | Known | De novo | P |
| 9 | c.5752_5753delAT | p.I1918* | 40 | Sec14-PH | − | Novel | De novo | P |
| 10 | c.5944–5A > G | Splice | Intron 40 | − | CS941516 | Known | Paternal | P |
| 11 | c.6711delA | p.V2238Cfs*6 | 45 | CTD | − | Novel | De novo | P |
| 12 | c.6792C > A | p.Y2264* | 46 | CTD | CM981382 | Known | De novo | P |
| 13 | c.1933A > G | p.M645V | 17 | CSRD | CM134541 | Known | Maternal | VUS |
| 14 | NT | |||||||
| 15 | c.3362A > G | p.E1121G | 26 | TBD | CM145714 | Known | Paternal | VUS |
| 16 | c.A3484G | p.M1162V | 26 | TBD | CM1815605 | Known | Paternal | VUS |
Abbreviations: ACMG, American College of Medical Genetics and Genomics classification; CSRD, cysteine and serine-rich domain; CTD, carboxyl-terminal domain; GRD, GTPase-activating protein–related domain; HGMD, Human Gene Mutation Database; LP, likely pathogenic; NF1, neurofibromatosis type 1; NT, not tested; P, pathogenic; TBD, tubulin-binding domain; VUS, variant of unknown significance—insufficient evidence.
ClinVar data are based on NM_001042492.2, which shares the identical genomic, cDNA, and amino acids sequences with NM_000267.3.
Human Gene Mutation Database entries associated with NF1 .
No variant in SPRED1 was identified for any of the patients. Our sequencing was based on short reads and targeted the exons only, so it would not be able to detect either copy number alterations or deep intronic variants in the associated genes.
Of the 12 variants found in the 12 patients who met NF1 diagnostic criteria, there were four frameshifts, four nonsense, two missense, and two splice variants (one exonic and one intronic) ( Table 2 ). Four of the 12 variants were novel: all 4 were deletions (2 were deletions of one nucleotide and 2 were deletions of two nucleotides) ( Table 2 ). Although another frameshift variant (c.1393_1394delAG) had not been reported in the published literature, it had two submissions (both classified as pathogenic) in ClinVar ( www.ncbi.nlm.nih.gov/clinvar/ ).
Parental testing showed that 10 of the 12 variants were de novo and 1 inherited from an affected father. The remaining variant did not have parental samples to test inheritance status, but it is a known mutation found in multiple patients with NF1. All 12 variants could be classified as either “pathogenic” or “likely pathogenic” based on the ACMG–AMP guidelines ( Table 2 ).
For the four patients (Patients 13–16) who did not meet the clinical criteria for NF1, each was heterozygous for a missense variant. Patients 13 and 14 had the same Met > Val variant in the CSRD, while the variants found in Patients 15 and 16 mapped to the TBD. Although all three missense variants are documented in Human Gene Mutation Database (HGMD) (as disease causing or likely disease causing) with additional submissions in ClinVar, targeted testing of parental samples found that they were inherited in three cases (Patients 13, 15, and 16), while parental samples were not available for Patient 14.
Discussion
In this study, we report 16 patients with variants in NF1 and summarized their clinical presentations. Among the 12 who met the NF1 diagnostic criteria, CALMs and axillary freckling were the most frequent clinical presentations, followed by neurofibromas. In contrast, only 2 of the 12 had Lisch nodules. At the last review, three patients were eight years old or younger and might not have Lisch nodules yet. 27 28 Only 2 of our 12 NF1 patients had a family member with NF1, and only 1 patient had optic pathway glioma.
In general, the 10 patients with truncating variants did not have a more severe phenotype compared with the two who carried missense variants. There was also no correlation between the location of the variant and specific phenotypic features. As with several previous reports, 29 30 a recent study of previously unreported variants in 108 patients did not find any correlation between the type of mutation and the clinical presentations. 31 Unlike a study from China, 32 we did not find an association of short stature with having variants at the N-terminal for our patients. The seven patients with short stature from our cohort had mutations spread through the gene ( Fig. 1 ).
Fig. 1.
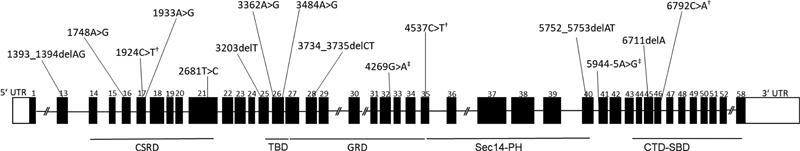
Distribution of the variants identified in the patient cohort. Schematic diagram (not drawn to scale) of the NF1 gene exons and the corresponding protein domains 34 35 with the approximate locations of the identified variants indicated above the respective exons. CSRD, cysteine and serine-rich domain; CTD, carboxyl-terminal domain; GRD, GTPase-activating protein–related domain; PH, pleckstrin homology; SBD, syndecan-binding domain; TBD, tubulin-binding domain. † Nonsense variant. ‡ Splice variant.
There were 15 unique NF1 variants, of which 4 were novel ( Table 2 ). Consistent with variants associated with NF1 being mostly truncating, 10 were small deletions or essential splice site variants that were predicted to affect the production of fully functional mRNA or protein. The remaining five were missense variants, two of which could be classified as likely pathogenic and documented in multiple patients with NF1 in HGMD.
The other three missense variants were found in four patients (Patients 13–16) who did not meet NF1 diagnostic criteria. However, all three could be found in HGMD with the associated phenotype of NF1. The c.1933A > G variant in exon 17, which was found in two of our patients (Patients 13 and 14), also had 11 submissions in ClinVar with classifications of “benign” or “unknown” and was deemed unlikely to be damaging. The c.3362A > G variant in exon 26 had two submissions in ClinVar, both classified as “unknown significance.” Yet, this variant has been reported in two related NF1 patients (father and son) whose peripheral blood showed a mixture of transcripts with the variant and shorter transcripts missing the exon containing the variant. 33 Also, within exon 26, our third missense variant c.3484A > G had two listings (“unknown significance” and “benign”) in ClinVar. It was also reported as benign in a patient with NF1 from China who had an additional variant (c.7189G > A) which was damaging. 34 Our patient inherited this variant from his father. Both of them had some pigmentary presentations, but neither met the clinical criteria for NF1. Besides, this patient was found to have a de novo pathogenic SOS1 variant, consistent with his features that met the Noonan syndrome criteria.
Conclusion
In summary, we reported 12 molecularly confirmed NF1 patients from Singapore who were heterozygous for a pathogenic/likely pathogenic NF1 variant and 4 other patients who had some features indicative of genetic disorders. Our study describes the phenotypic features of the patients. It adds four novel NF1 variants to the molecular spectrum of NF1 germline mutations from the Asian population, which has proportionally fewer reported variants than Western populations.
Acknowledgments
This research is supported by the National Research Foundation Singapore under its NMRC Centre Grant Program (NMRC Project No. NMRC/CG/M003/2017) and administered by the Singapore Ministry of Health's National Medical Research Council.
Conflict of Interest None declared.
These two authors are the first co-authors.
References
- 1.Kresak J L, Walsh M. Neurofibromatosis: a review of NF1, NF2, and schwannomatosis. J Pediatr Genet. 2016;5(02):98–104. doi: 10.1055/s-0036-1579766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson J L, Gutmann D H. Neurofibromatosis type 1. Handb Clin Neurol. 2015;132:75–86. doi: 10.1016/B978-0-444-62702-5.00004-4. [DOI] [PubMed] [Google Scholar]
- 3.Evans D G, Howard E, Giblin C. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. 2010;152A(02):327–332. doi: 10.1002/ajmg.a.33139. [DOI] [PubMed] [Google Scholar]
- 4.Lammert M, Friedman J M, Kluwe L, Mautner V F. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol. 2005;141(01):71–74. doi: 10.1001/archderm.141.1.71. [DOI] [PubMed] [Google Scholar]
- 5.Kallionpää R A, Uusitalo E, Leppävirta J, Pöyhönen M, Peltonen S, Peltonen J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet Med. 2018;20(09):1082–1086. doi: 10.1038/gim.2017.215. [DOI] [PubMed] [Google Scholar]
- 6.Uusitalo E, Leppävirta J, Koffert A. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol. 2015;135(03):904–906. doi: 10.1038/jid.2014.465. [DOI] [PubMed] [Google Scholar]
- 7.Gutmann D H, Ferner R E, Listernick R H, Korf B R, Wolters P L, Johnson K J. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017;3:17004. doi: 10.1038/nrdp.2017.4. [DOI] [PubMed] [Google Scholar]
- 8.NF France Network . Bergqvist C, Servy A, Valeyrie-Allanore L, Ferkal S, Combemale P, Wolkenstein P. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis. 2020;15(01):37. doi: 10.1186/s13023-020-1310-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams V C, Lucas J, Babcock M A, Gutmann D H, Korf B, Maria B L. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123(01):124–133. doi: 10.1542/peds.2007-3204. [DOI] [PubMed] [Google Scholar]
- 10.DeBella K, Szudek J, Friedman J M.Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children Pediatrics 2000105(3 Pt 1):608–614. [DOI] [PubMed] [Google Scholar]
- 11.Friedman J M. Seattle (WA): University of Washington, Seattle; 2019. Neurofibromatosis 1. [PubMed] [Google Scholar]
- 12.Upadhyaya M, Huson S M, Davies M. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation . Am J Hum Genet. 2007;80(01):140–151. doi: 10.1086/510781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ledbetter D H, Rich D C, O'Connell P, Leppert M, Carey J C. Precise localization of NF1 to 17q11.2 by balanced translocation. Am J Hum Genet. 1989;44(01):20–24. [PMC free article] [PubMed] [Google Scholar]
- 14.Wallace M R, Marchuk D A, Andersen L B.Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients Science 1990249(4965):181–186. [DOI] [PubMed] [Google Scholar]
- 15.Abramowicz A, Gos M. Neurofibromin in neurofibromatosis type 1 - mutations in NF1 gene as a cause of disease . Dev Period Med. 2014;18(03):297–306. [PubMed] [Google Scholar]
- 16.Cawthon R M, Weiss R, Xu G F. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62(01):193–201. doi: 10.1016/0092-8674(90)90253-b. [DOI] [PubMed] [Google Scholar]
- 17.White R, O'Connell P. Identification and characterization of the gene for neurofibromatosis type 1. Curr Opin Genet Dev. 1991;1(01):15–19. doi: 10.1016/0959-437x(91)80034-j. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, O'Connell P, Breidenbach H H. Genomic organization of the neurofibromatosis 1 gene ( NF1 ) . Genomics. 1995;25(01):9–18. doi: 10.1016/0888-7543(95)80104-t. [DOI] [PubMed] [Google Scholar]
- 19.Ballester R, Marchuk D, Boguski M. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63(04):851–859. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- 20.Scheffzek K, Ahmadian M R, Wiesmüller L. Structural analysis of the GAP-related domain from neurofibromin and its implications. EMBO J. 1998;17(15):4313–4327. doi: 10.1093/emboj/17.15.4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shapira S, Barkan B, Friedman E, Kloog Y, Stein R. The tumor suppressor neurofibromin confers sensitivity to apoptosis by Ras-dependent and Ras-independent pathways. Cell Death Differ. 2007;14(05):895–906. doi: 10.1038/sj.cdd.4402057. [DOI] [PubMed] [Google Scholar]
- 22.Brems H, Legius E. Legius syndrome, an update. Molecular pathology of mutations in SPRED1. Keio J Med. 2013;62(04):107–112. doi: 10.2302/kjm.2013-0002-re. [DOI] [PubMed] [Google Scholar]
- 23.Jouhilahti E M, Peltonen S, Heape A M, Peltonen J. The pathoetiology of neurofibromatosis 1. Am J Pathol. 2011;178(05):1932–1939. doi: 10.1016/j.ajpath.2010.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet. 2012;49(07):433–436. doi: 10.1136/jmedgenet-2012-100918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.ACMG Laboratory Quality Assurance Committee . Richards S, Aziz N, Bale S. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(05):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.National Institutes of Health Consensus Development Conference . Stumpf D, Alksne J, Annegers J. Neurofibromatosis. Conference statement. Arch Neurol. 1988;45(05):575–578. [PubMed] [Google Scholar]
- 27.Lubs M L, Bauer M S, Formas M E, Djokic B. Lisch nodules in neurofibromatosis type 1. N Engl J Med. 1991;324(18):1264–1266. doi: 10.1056/NEJM199105023241807. [DOI] [PubMed] [Google Scholar]
- 28.Nichols J C, Amato J E, Chung S M. Characteristics of Lisch nodules in patients with neurofibromatosis type 1. J Pediatr Ophthalmol Strabismus. 2003;40(05):293–296. doi: 10.3928/0191-3913-20030901-11. [DOI] [PubMed] [Google Scholar]
- 29.De Luca A, Schirinzi A, Buccino A. Novel and recurrent mutations in the NF1 gene in Italian patients with neurofibromatosis type 1 . Hum Mutat. 2004;23(06):629. doi: 10.1002/humu.9245. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J, Tong H, Fu X. Molecular characterization of NF1 and neurofibromatosis type 1 genotype-phenotype correlations in a Chinese population. Sci Rep. 2015;5:11291. doi: 10.1038/srep11291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonatti F, Adorni A, Matichecchia A. Patterns of novel alleles and genotype/phenotype correlations resulting from the analysis of 108 previously undetected mutations in patients affected by neurofibromatosis type I. Int J Mol Sci. 2017;18(10):E2071. doi: 10.3390/ijms18102071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao R, Yu T, Xu Y. Clinical presentation and novel pathogenic variants among 68 Chinese neurofibromatosis 1 children. Genes (Basel) 2019;10(11):E847. doi: 10.3390/genes10110847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu W, Yang X, Hu X, Li S. Fifty-four novel mutations in the NF1 gene and integrated analyses of the mutations that modulate splicing . Int J Mol Med. 2014;34(01):53–60. doi: 10.3892/ijmm.2014.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu-Chou Y H, Hung T C, Lin Y T. Genetic diagnosis of neurofibromatosis type 1: targeted next-generation sequencing with Multiple Ligation-Dependent Probe Amplification analysis. J Biomed Sci. 2018;25(01):72. doi: 10.1186/s12929-018-0474-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ratner N, Miller S J. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015;15(05):290–301. doi: 10.1038/nrc3911. [DOI] [PMC free article] [PubMed] [Google Scholar]