

Abstract
Cystine-dense peptides (CDPs) are a miniprotein class that can drug difficult targets with high affinity and low immunogenicity. Tools for their design, however, are not as developed as those for small molecule and antibody drugs. CDPs have diverse taxonomic origins but structural characterization is lacking. Here we adapted Iterative Threading ASSEmbly Refinement (I-TASSER) and Rosetta protein modeling software for structural prediction of 4298 CDP scaffolds and performed in silico pre-screening for CDP binders to targets of interest. Mammalian display screening of a library of docking-enriched, methionine and tyrosine scanned (DEMYS) CDPs against PD-L1 yielded binders from four distinct CDP scaffolds. One was affinity-matured and co-crystallography yielded a high-affinity (KD = 202 pM) PD-L1-binding CDP that competes with PD-1 for PD-L1 binding. Its subsequent incorporation into a CD3-binding bispecific T cell engager produced a molecule with pM-range in vitro T cell killing potency and which substantially extends survival in two different xenograft tumor-bearing mouse models. Both in vitro and in vivo, the CDP-incorporating bispecific molecule out-performed a comparator antibody-based molecule. This CDP modeling and DEMYS technique can accelerate CDP therapeutic development.
One Sentence Summary:
A PD-L1-binding cystine-dense peptide produced using in silico and in vitro screening outperforms an antibody in a bispecific T cell engager.
INTRODUCTION
As targeted drug discovery pipelines mature, drug discovery innovation has gradually shifted towards addressing “undruggable” targets with difficult interfaces (1). Alternative strategies beyond simple target inhibition through binding are also maturing, many of which rely on modular design for multiple functions (e.g., chimeric antigen receptors, bispecific antibodies, Proteolysis-targeting chimeras [PROTACs]) (1–3). Miniproteins are small (roughly <10 kDa in size) proteins that are expanding the toolkit of targeted therapeutics in ways that both small molecules and antibodies are naturally limited (4). Miniproteins represent a wide variety of structures and sources, but generally, they are self-contained, globular domains whose stable tertiary structures confer specificity and affinity with sufficient size to disrupt protein-protein interactions. Some are extraordinarily resistant to proteolysis and thermal denaturation (5–7). Smaller size also permits a greater degree of tissue penetration than larger protein drugs (8, 9) along with whole molecule structure-activity relationship studies using saturation mutagenesis (10). The choice of a recombinant or synthetic production strategy is another advantage. Their screening strategies are also often based on surface display (11–15), which permits tremendous chemical diversity without being limited by antigen presentation or host homology. Additionally, as stable, globular domains, they lend themselves to modular assembly (16–18).
One such miniprotein class is cystine-dense peptides (CDPs, also referred to as cystine-dense miniproteins). With folding and stability typically dominated by 3 to 5 cystines (Cys-Cys disulfide bridges), CDPs can be exceptionally thermostable and protease-resistant in their native forms (5, 6, 19). As efficient recognition by the adaptive immune system requires breakdown of proteins into short peptides for antigen presentation, this protease resistance may explain the low immunogenicity seen in patients (19). The best-studied CDPs are those whose pharmacology is suited to direct clinical utility, such as ion-channel modulators derived from venom proteins (5, 19). However, CDPs represent the most structurally and taxonomically diverse class of miniproteins (4, 6, 10). While this diversity permits a wide array of shapes and surfaces for high affinity target contact, such diversity is of limited utility when their structures are largely uncharacterized. At least 100,000 CDP-like domains exist in public sequence databases, but despite this, we can find fewer than 800 in structural form in the Research Collaboratory for Structural Bioinformatics (RCSB) protein data bank.
To address this structural knowledge limitation, we structurally modeled diverse CDP sequences, generating high-confidence models for 4,298 sequences from a protease-resistant, high-expression CDP library. We leveraged these high-confidence models using in silico docking to select scaffolds most likely to bind to targets of interest, focusing on PD-L1 as a representative cell surface target of the well-studied immunoglobulin structural domain. Combining docking enrichment with methionine and tyrosine scanning (DEMYS), we screened for PD-L1 binders, identifying four candidates and maturing one to ~200 pM KD affinity. To demonstrate the modularity of CDPs in biologic drug design, this high-affinity PD-L1-binding CDP was incorporated into a CD3-targeting bispecific T cell engager and was used alongside activated T cells (ATCs) to treat subcutaneous tumors from two unrelated human cell lines (prostate cancer PC3 and triple negative breast cancer MDA-MB-231) in immune compromised mice. Treatment with ATCs plus the CDP-based molecule completely eliminated PC3 tumors in 27 of 30 mice between two experiments (only 1 of 10 were eliminated by an antibody-based comparator) while substantially extending survival in mice carrying MDA-MB-231 tumors. With these ex silico modeling and screening tools, diverse CDP scaffolds can become an effective and efficient tool in modular drug design.
RESULTS
Constructing an improved CDP screening library
Selection of CDP scaffolds for modeling began with identifying homologs of CDPs that demonstrate high expression and/or protease resistance in mammalian display (10) (Fig. 1A). We re-scored CDPs from the taxonomically diverse CDP library NCL1 (Native CDP Library 1, N = 10,000) using a composite surface folding and expression (SFE) score incorporating surface expression and protease resistance. A subset of high-performing scaffolds (N = 953 that scored above −20 SFE units) was identified (Fig. 1B). A curated library of 96,000 natural CDPs (Swiss-Prot and TrEMBL) was searched for members sharing sequence homology or cysteine topology with high-SFE CDPs, identifying 7,940 additional CDPs. Combining these with the 953 high-scoring NCL1 members created NCL2 (Native CDP Library 2) (Fig. 1C). As a pool, NCL2 demonstrates higher expression and greater protease resistance than NCL1 (Fig. 1D).
Fig. 1. Deriving a second-generation CDP library with improved surface folding characteristics.
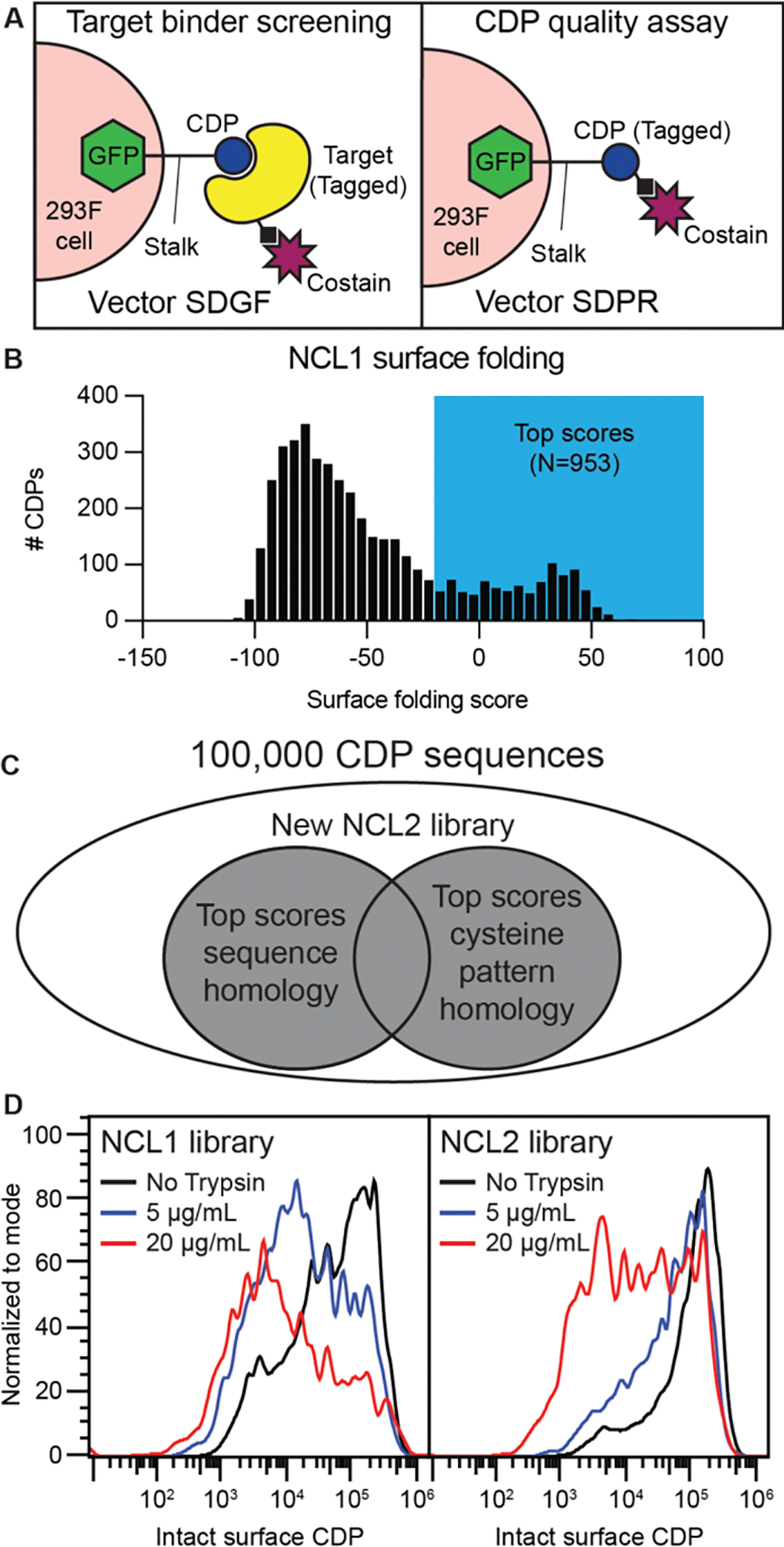
(A) Illustration of mammalian surface display principle. If the CDP is not tagged (vector SDGF), tagged (e.g., biotin) target along with a fluorescent co-stain (e.g., streptavidin) stains cells to enrich for target-binding CDPs of interest. When the CDP itself is tagged (e.g., 6xHis, vector SDPR), the fluorescent co-stain (e.g., anti-6xHis) will detect intact CDPs on the surface of the cell. (B and C) High surface folding scores (B) from pooled analysis of library NCL1 permitted homology-based selection from a large CDP library (C) to create a second-generation CDP library, NCL2. (D) Flow-based comparison of SDPR-cloned 6xHis-tagged NCL1 and NCL2 with or without trypsin (5 or 20 μg/mL, 5 mins) followed by DTT treatment (5 mins) and subsequent anti-6xHis antibody staining.
Modeling NCL2 CDP structures to assess structural diversity
High-quality models of CDP scaffolds would facilitate structure-activity relationship (SAR) studies and docking simulations. For structural modeling, we combined I-TASSER (20) and Rosetta (21) for our CDP modeling pipeline (Fig. 2A). Both can be run on local servers and consistently perform in the top 3 of the community-wide CASP (critical assessment of techniques for protein structure prediction) competition. I-TASSER’s structural predictions were fed into Rosetta’s “ForceDisulfides” program to convert close cysteine pairs into disulfides in the models. This pipeline was used for 19 CDPs (Fig. 2B), comparing models with determined crystal structures (6, 22), including six determined as part of this work (table S1). Backbone atom alignment (PyMol v2.4.0) yielded an average root-mean-square deviation (RMSD) of 0.924 Å. Asymmetric units of crystals aligned to each other with 0.333 Å average RMSD. An average RMSD of < 1 Å compares well with the ~1.0–2.3 Å RMSD values (model vs. crystal structure or nuclear magnetic resonance [NMR] ensemble) typically interpreted as evidence of successful modeling of de novo miniprotein designs (23, 24).
Fig. 2. NCL2 structural modeling using I-TASSER and Rosetta produces accurate, diverse models.
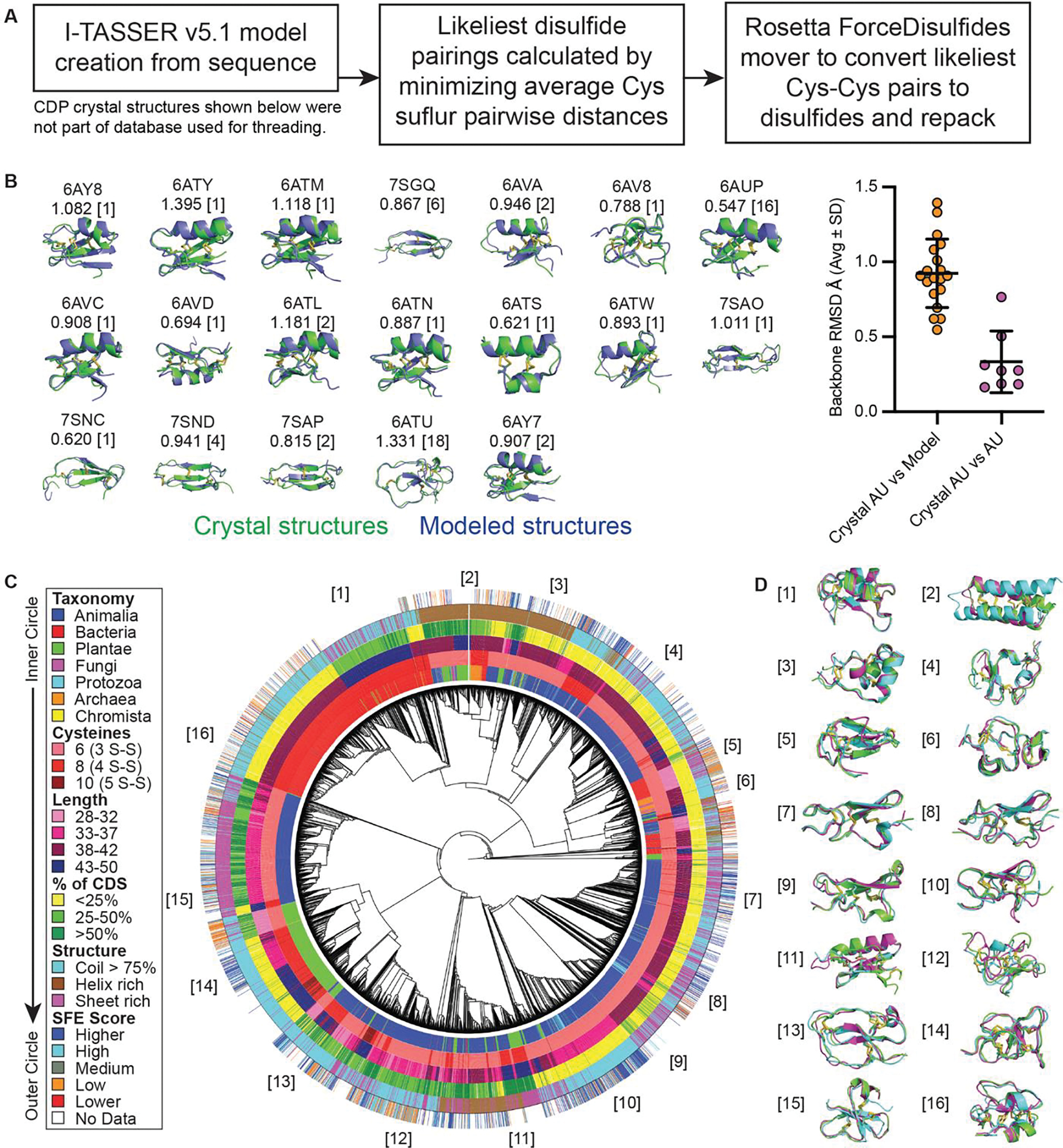
(A) Flow chart of modeling pipeline. (B) 19 CDP crystal structures were compared to their I-TASSER/Rosetta model for structural alignment. Above each model is the RCSB PDB identifier and RMSD (in Å) of the backbone alignment. The asymmetric unit (AU) count in the crystal is in brackets; where AU > 1, RMSD represents the average of the model aligned to each AU. Plotted on the right are the alignment values of crystal vs model (left) compared to the intra-crystal AU alignments of each CDP for which AU > 1. (C) The 8893 members of NCL2 yielded 4298 models of sufficient quality for reliable structural comparisons. The high quality NCL2 models were aligned with one another, shown as a circular cladogram phylogenetic tree. The CDPs’ taxonomy, cysteine count, length, coding sequence (CDS) proportion, dominant structural elements, and surface folding/expression (SFE) score are shown. (D) Example CDP models within a given subclade, identified by number in the cladogram.
Applying this pipeline to the 8893-member NCL2 library, 4298 structures passed our confidence thresholds (I-TASSER C-score > 0 and Rosetta energy score < 80). An all-vs-all pairwise structural homology matrix (TMalign) (25) was converted to a phylogenetic tree (Fig. 2C) that includes each sequence’s taxonomic source, cysteine count, length, percent of coding sequence (CDP length / coding sequence length), dominant secondary structural elements (structural identification [STRIDE] algorithm) (26, 27), and SFE score (as was assessed for NCL1 previously (10)). Overall, the NCL2 library models demonstrate high structural diversity (Fig. 2D), indicating that the CDPs within have a wide variety of shapes and surfaces to provide a complementary interface for target binding.
Low-resolution docking of NCL2 models for in silico pre-screening
We wished to use our miniprotein structural model library to predict favorable binding to targets. Because high-resolution docking simulations are computationally expensive at this scale, we performed low-resolution docking with RosettaDock scripts (28) for 2,000 iterations per scaffold to predict favorable docking regions where high-scoring docks clustered (DBSCAN) (29). An example of this docking pipeline for a transferrin receptor (TfR)-binding CDP and TfR (30) is illustrated in fig. S1A. Extending this method to four other miniprotein:target pairs [Insulin:InsR (31), Affitin H4:Lysozyme (32), Fynomer 4C-A4:Chymase (33), and Affibody ZHER2:HER2 (34)], using the miniprotein binder as the candidate ligand in low resolution docking (fig. S1B), identified plausible docking sites for all targets, including one near the center of mass of the ligand for all five (distances ranged from 1.9 Å to 14.4 Å [average 7.3 ± 4.6 Å] apart).
We used this pipeline to dock the full NCL2 model library against PD-L1 (PDB 4ZQK (35)). PD-L1 has several known binders of common modalities (e.g., antibodies), facilitating direct comparison of binder activities. After docking all 4298 scaffolds, common target regions for high-scoring CDP docks were identified (Fig. 3A). This clustering data can be used to generate sub-libraries of PD-L1 binders to specific sites. We identified a diverse set of 238 CDPs with high scores at the PD-1 binding site (Fig. 3, B and C). To leverage surface display as a high complexity screening platform, this limited sublibrary was further diversified by identifying surface-exposed, non-Cys, non-hydrophobic (i.e. not Tyr/Phe/Trp/Met/Ile/Leu) residues and substituting them with either Tyr or Met to create hydrophobic patches that can seed novel protein:protein interactions. Tyr and Met are aromatic and aliphatic, respectively, residues that contain a polar atom, avoiding the extreme hydrophobic character of similarly sized Phe and Leu residues (36) that could impact solubility. This strategy for combining sub-library selection of docking-capable scaffolds with Tyr and Met scanning is referred to as DEMYS (Docking-Enriched M-Y Scan) screening(Fig. 3D).
Fig. 3. NCL2 model library docking and DEMYS concept.
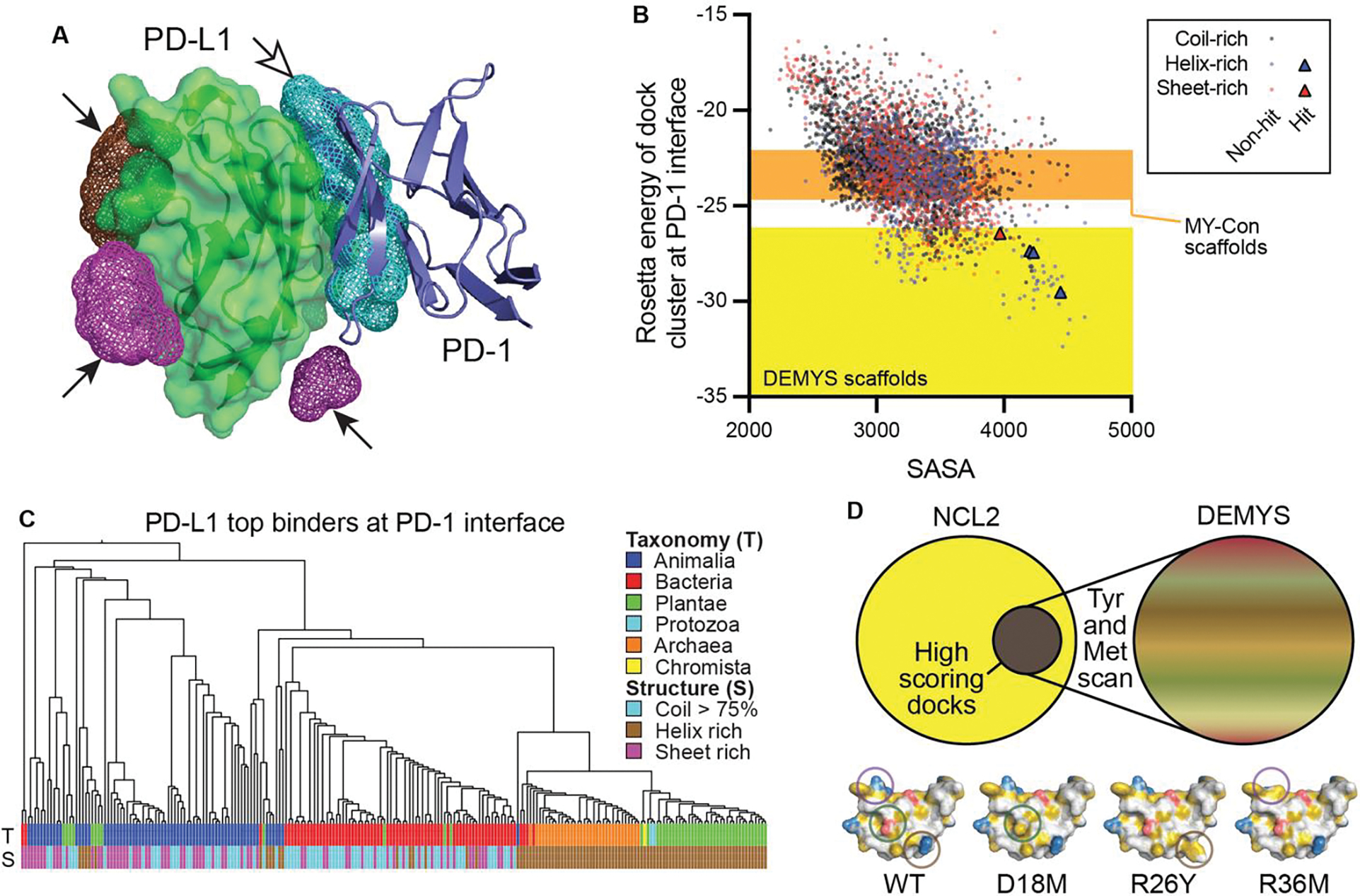
(A) CDP binder identification for PD-L1 as co-crystalized with PD-1 [PDB 4ZQK]. It was screened in silico against the NCL2 model library with low-resolution RosettaDock to identify possible docking sites. Mesh clouds with arrowheads: DBSCAN docking clusters (up to 200 NCL2 model docking sites); hollow arrowhead indicates cluster of interest for sublibrary production. (B) NCL2 low-resolution docking scores for the PD-L1:PD-1 interface vs solvent accessible surface area (SASA). Color-coded by dominant structural element in the scaffold. Orange and yellow shaded regions illustrate source range for MY-Con and DEMYS libraries, respectively. (C) All-vs-all structural alignment cladogram of PD-L1:PD-1 site DEMYS CDP library. (D) The DEMYS concept. A sample scaffold, color coded by hydrophobicity (yellow are carbon atoms not contacting a polar atom, red are acidic atoms, blue are basic atoms, and white are all others), is shown as WT and three example Met or Tyr mutations that seed or expand hydrophobic patches.
DEMYS screening yielded PD-L1 binders with predictable binding properties
We created a DEMYS library targeted to the PD-L1:PD-1 interface (PD-L1 DEMYS), as well as a control library of 300 scaffolds randomly selected from those scoring in the median 50% of docks but similarly M-Y scanned (MY-Con). Including NCL2 itself, all three libraries were cloned after error-prone PCR mutagenesis to create pools of ~107 unique CDP sequences. The three pools were screened for PD-L1 binders using the SDGF (Surface Display GFP FasL) vector system (10) and 100 nM biotinylated PD-L1 + streptavidin (Fig. 4, A to C). MY-Con yielded no hits (Fig. 4A), suggesting that M-Y scanning alone is insufficient when the library lacks scaffolds predicted to dock to PD-L1. NCL2 yielded one validated binding scaffold (Fig. 4B and fig. S2A), which we called PDL1B1G1 (PD-L1 binder 1, generation 1). The DEMYS library yielded hits representing four distinct parental CDP scaffolds (Fig. 4C). The highest staining of the four DEMYS hits was a variant of PDL1B1G1 while the other binders represented distinct scaffolds (PDL1B2G1, PDL1B3G1, and PDL1B4G1) (fig. S2B). The modeled scaffold for PDL1B1G1 is shown in Fig. 4D, while all four DEMYS hits’ sequences and parent scaffold models are shown in fig. S2C. The native parent scaffolds of PDL1B2G1 and PDL1B4G1 share a modeled fold with that of PDL1B1G1, though their relatively low pairwise homology (59% identical for B1 vs. B2, 44% for B1 vs. B4, and 63% for B2 vs. B4) suggests their modes of binding may differ. The native parent of PDL1B3G1 (a defensin) has a very different modeled fold from the others.
Fig. 4. PD-L1-binding CDP screening and binding behavior.
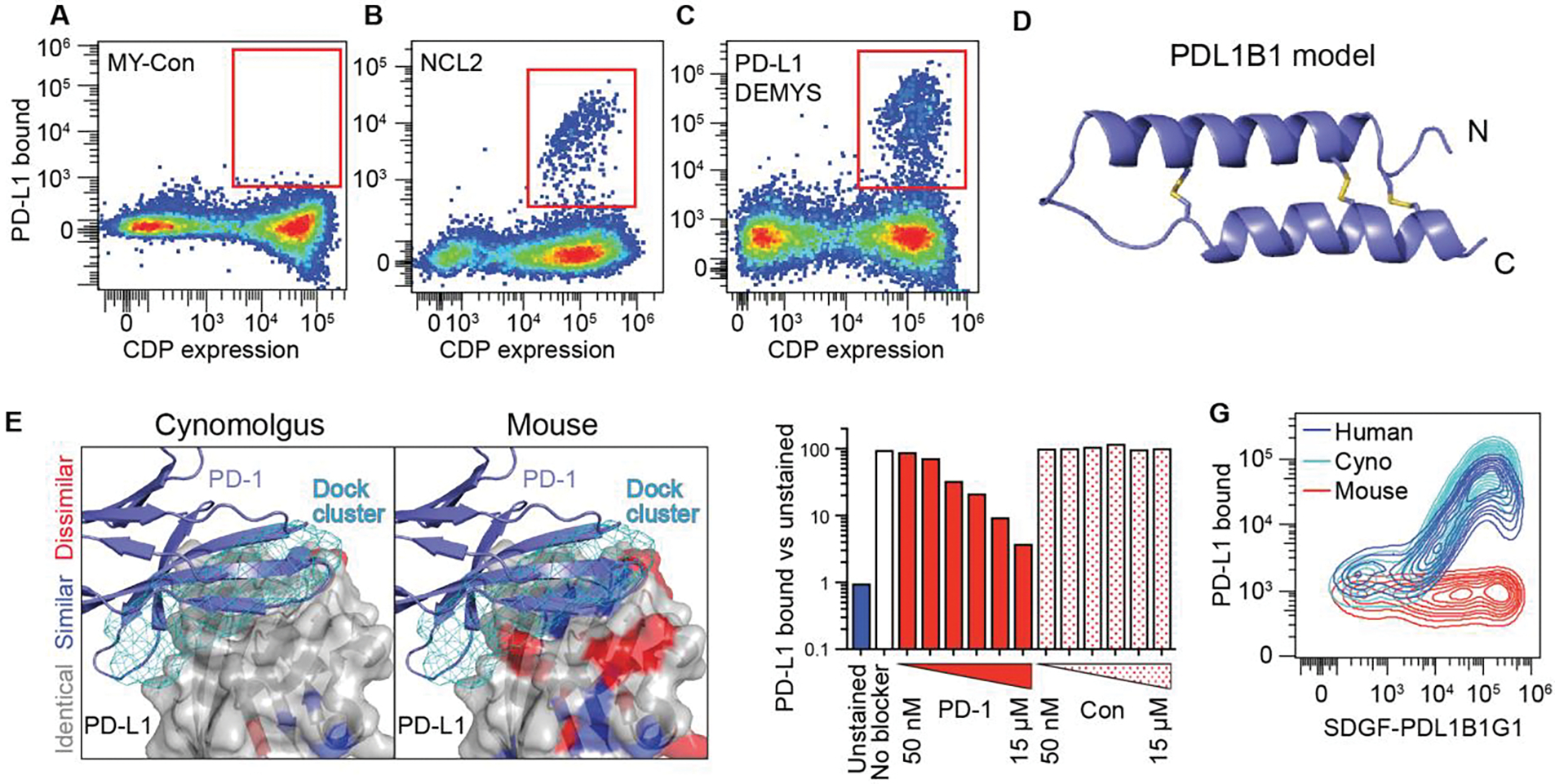
(A to C) Flow profiles after one magnetic sort and two flow sorts (100 nM PD-L1; flow included 100 nM streptavidin-AlexaFluor 647) to enrich for SDGF-displayed PD-L1 binding CDPs in MY-Con (A), NCL2 (B) and PD-L1 DEMYS (C). NCL2 yielded a single hit and DEMYS screening yielded four. (D) Ribbon model of the parental scaffold of PDL1B1 (PD-L1 binder 1). (E) PD-L1 DEMYS scaffold docking sites (top 200, cyan mesh) on human PD-L1, color-coded for cynomolgus monkey (left) and mouse (right) homology (white, identical residues; blue, similar; red, dissimilar). Ribbon structure is PD-1 bound to PD-L1 (PDB 4ZQK). (F) SDGF-PDL1B1G1:PD-L1 staining with or without PD-1-Fc competitor (“PD-1”) or control Fc fusion (“Con”) at 50 nM, 150 nM, 500 nM, 1.5 μM, 5 μM, or 15 μM. (G) SDGF-PDL1B1G1-expressing cells stained with human, cynomolgus, or mouse PD-L1.
The DEMYS library was derived from top scoring scaffolds at the PD-L1:PD-1 interface which has substantial cynomolgus monkey homology but poor murine homology (Fig. 4E). If PDL1B1G1 binds to this site, its binding should be competitive with PD-1, and its binding to cynomolgus PD-L1 should be stronger than seen with murine PD-L1. Both properties were demonstrated in PDL1B1G1-expressing cells (Fig. 4, F and G).
Affinity-maturation of PDL1B1G1 yielded well-folded, high-affinity CDPs
PDL1B1G1 was affinity-matured using site-saturation mutagenesis (SSM). After two rounds of staining, flow sorting, and regrowth (fig. S3, A to C), the resulting variant pool and the input pool were sequenced to identify enriched and depleted variants (Fig. 5A). Enriched variants represented mutations that directly (interface) or indirectly (folding/stability) improve target binding, while depletion suggested disruption of binding capability. Six highly enriched mutations (E11W, A13M, Y15G, I22N, Y36K, and W40F) were combined into a single “All 6” variant alongside six variants representing all-but-one enriched mutation (fig. S3D). Upon testing for PD-L1 binding, the highest-staining variant contained all except the E11W mutation, so PDL1B1G1-A13M-Y15G-I22N-Y36K-W40F became PDL1B1G2 (PD-L1 binder 1, generation 2). Furthermore, PDL1B1G1 differed from its parental scaffold by five mutations (D25Y, T27I, Q29R, F39V, and V43L), and the behavior of the corresponding reversion mutations suggested that F39V and V43L contribute to PD-L1 binding while D25Y, T27I, and Q29R are likely to be benign passenger mutations picked up during library generation.
Fig. 5. Affinity maturation of PD-L1-binding CDP and soluble CDP characterization.
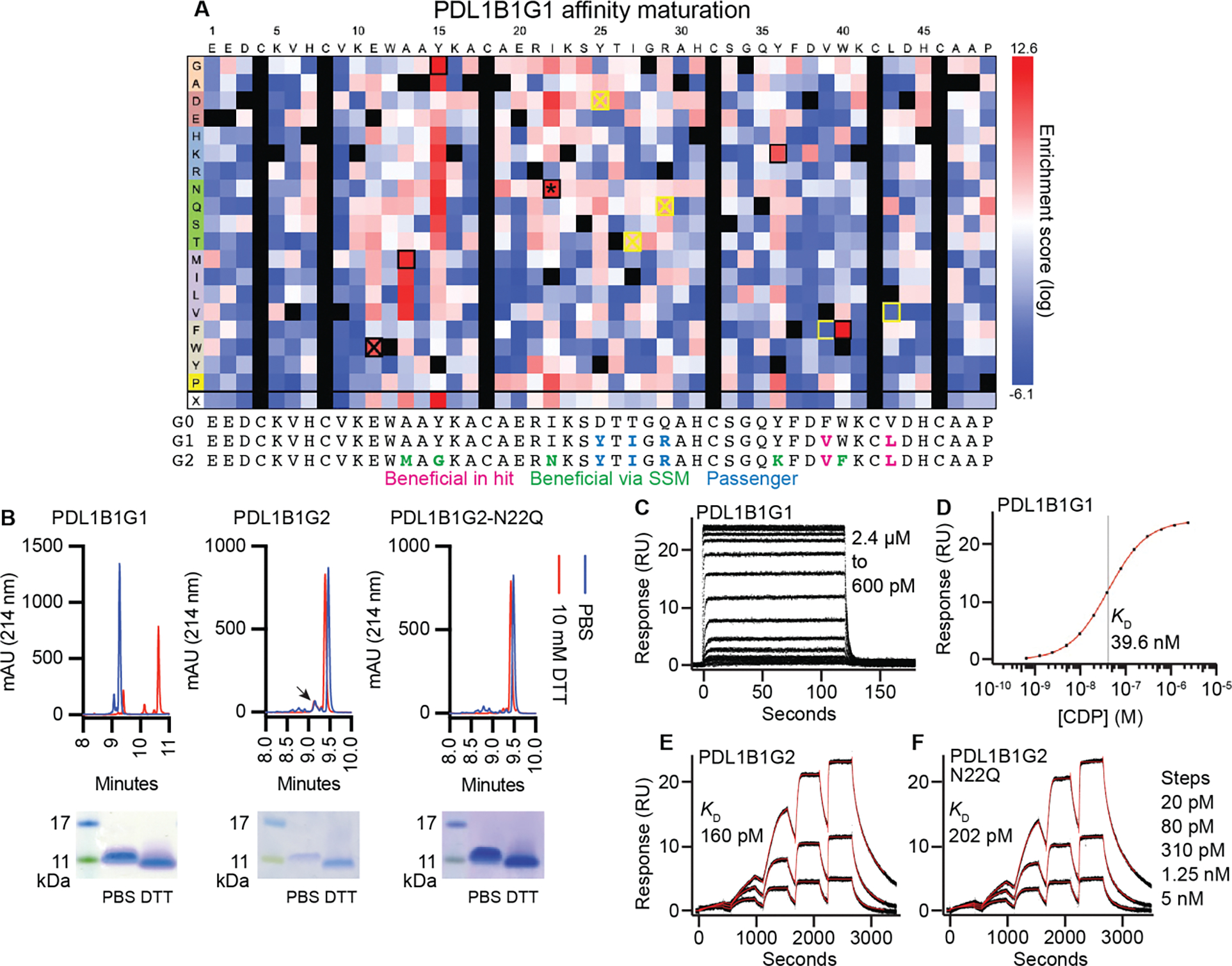
(A) Affinity maturation of PDL1B1G1. Heat map represents log2-transformed, normalized enrichment of each variant after two rounds of sorting (top ~7%) and regrowth. Positive enrichment scores represent increased relative abundance and therefore improved binding. Yellow box: detrimental reversion. Crossed out yellow box: Inconsequential reversion. Black box: beneficial and synergistic. Crossed out black box: beneficial but not synergistic. *: novel N-linked glycosite. Below the heatmap are sequences of the parental scaffold (here called G0 for PDL1B1G0), the primary hit (G1, PDL1B1G1), and the affinity-matured final variant (G2, PDL1B1G2), with beneficial (in original hit or via SSM) and passenger mutations indicated. (B) RP-HPLC and SDS-PAGE of recombinant PDL1B1G1, PDL1B1G2, and PDL1B1G2-N22Q. Arrowhead: possible minor species representing glycosylated PDL1B1G2. Note: mobility of CDPs in SDS-PAGE does not always correlate with size markers. For full SDS-PAGE gels, see fig. S12. (C-F) SPR traces of the three CDPs demonstrating a ~200-fold improvement in KD after affinity maturation. C: PDL1B1G1 steady-state traces. D: Steady-state curve fit. E: Single-cycle PDL1B1G2 traces. F: Single-cycle PDL1B1G2-N22Q traces. Black lines are data, red lines are the model fits.
PDL1B1G1, PDL1B1G2, and PDL1B1G2-N22Q (a variant that eliminates a canonical N-linked glycosite acquired during affinity maturation) were produced as soluble molecules in the Daedalus platform (37) (Fig. 5B). All three molecules were produced at high yields (13–23 mg/L) and high homogeneity (as evaluated by reversed-phase high-performance liquid chromatography [RP-HPLC]). While PDL1B1G2 contains a canonical N-linked glycosite, there is minimal evidence of glycosylation; a small peak (arrow in Fig. 5B, middle panel) could represent a minor product of glycosylated PDL1B1G2. Homogeneity of the products strongly suggests the presence of only a single primary fold, which is commonplace for soluble CDPs that demonstrate high SFE scores in surface display (6, 10) and bodes well for developability. Upon exposure of the CDPs to high concentrations of a strong reducing agent (10 mM DTT), PDL1B1G1 demonstrated a substantial mobility shift in RP-HPLC consistent with losing all cystine-stabilized tertiary structure. The affinity matured variants PDL1B1G2 and PDL1B1G2-N22Q, however, demonstrated only a subtle mobility shift in RP-HPLC upon reduction. Because mass spectrometry (MS) (fig. S4) confirmed that the DTT-treated samples were ~6 Da heavier than the PBS-treated samples, we concluded that both PDL1B1G2 variants lose three cystines to DTT reduction. Therefore, the minimal RP-HPLC mobility shift of the PDL1B1G2 variants upon reduction suggests that some or all of the mutations acquired during affinity maturation contribute to cystine-independent tertiary structure stabilization. Even so, because the interface involves both of the CDP’s primary structural elements (helices), any change in structure is expected to change the binding interface, so the altered RP-HPLC mobility upon reduction indicates that cystines are likely crucial for high-affinity PDL1B1G2-N22Q:PD-L1 binding. Binding to PD-L1 was evaluated by surface plasmon resonance (SPR) (Fig. 5, C to F). PDL1B1G1 bound PD-L1 with an equilibrium dissociation constant (KD) of 39.6 ± 0.3 nM, whereas affinity-matured PDL1B1G2 and PDL1B1G2-N22Q bound PD-L1 with KD = 160 ± 1 pM and 202 ± 2 pM, respectively.
PDL1B1G2:PD-L1 co-crystal structure is consistent with observed binding data
PDL1B1G2-N22Q was co-crystalized with PD-L1 (Fig. 6A and table S2). A portion of the CDP, from A19 through Q35, was unresolved in the 2.0 Å structure. This is consistent with the SSM data, as the average SSM enrichment scores of unresolved residues were less extreme (deviated less from 0) than seen with resolved residues (P = 0.0055, Fig. 6B), suggesting the specific side chain identities of unresolved residues were less important to high affinity binding. K36 and F37 resolved but were not part of the D38-A48 helix. The C4-C46 and C8-C42 cystines that resolved were the expected pairing from the model, leaving the third cystine (C18-C32, known by MS [fig. S4] to exist) unresolved. Although low-resolution docking models informed scaffold selection, only 3 of 34 docks between PDL1B1G0 (the parental scaffold of PDL1B1G2-N22Q) and PD-L1 are found at the PD-1 site in positions loosely resembling the co-crystal CDP (fig. S5), and the docking scores of these three were not particularly high.
Fig. 6. Co-crystallization of PDL1B1G2-N22Q with PD-L1.
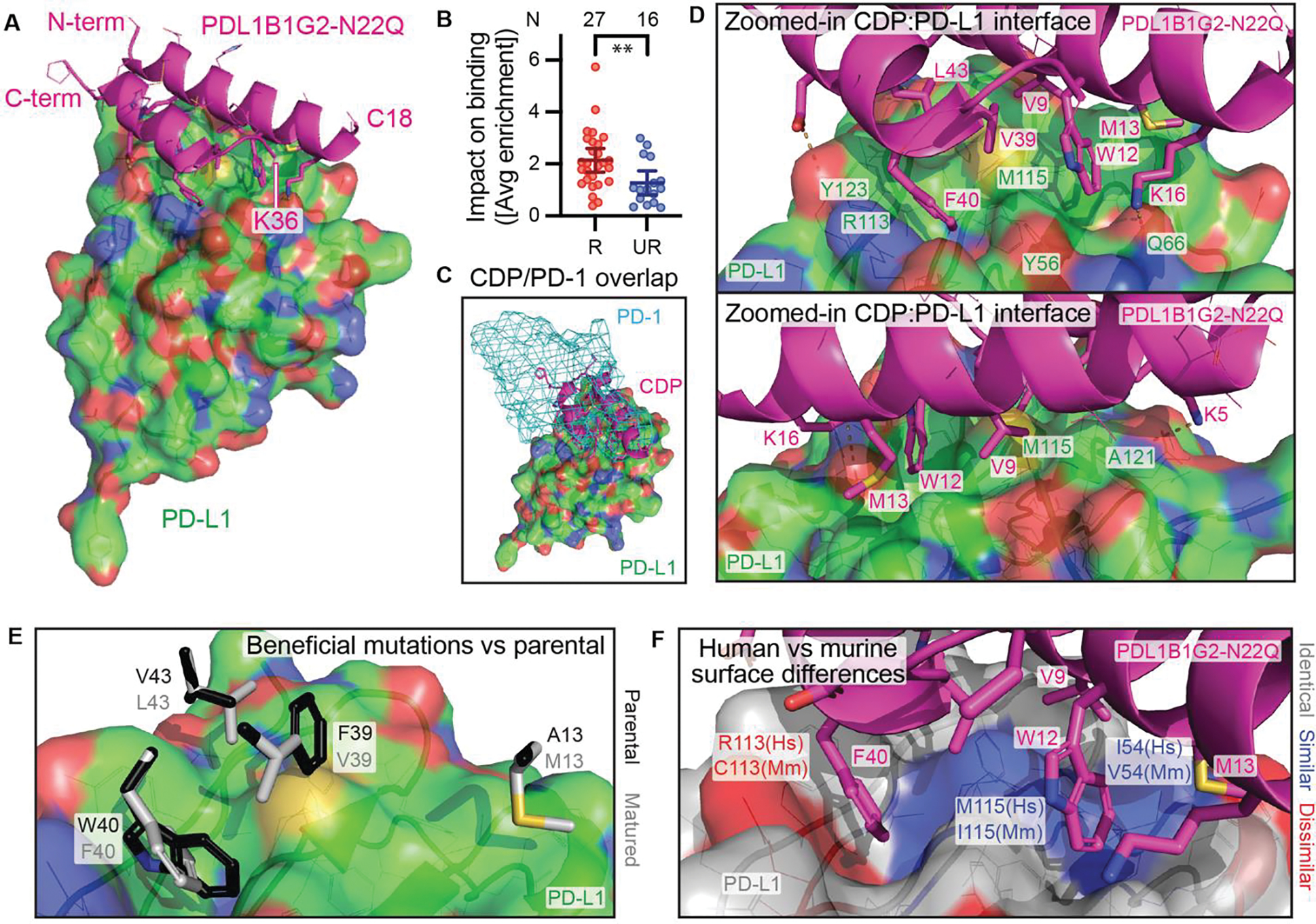
(A) Co-crystal structure of PDL1B1G2-N22Q (magenta cartoon) with PD-L1 (green surface). (B) Impact on binding (shown as absolute values of residues’ average SSM enrichment scores) of mutations to resolved (R; N = 27) residues compared to unresolved (UR; N = 16) residues, mean ± 95% CI. **P = 0.0055 by Mann-Whitney test. (C) PD-1 (cyan mesh surface, from PDB 4ZQK) binding site overlaps with CDP site. (D) From two different angles, a zoomed-in view of the CDP:PD-L1 interface. (E) Select side chains from PDL1B1G2-N22Q (white) at the PD-L1 interface are shown with parental residues (black, minimally clashing rotamers) superimposed. (F) CDP:PD-L1 interface where the PD-L1 surface is color-coded for human (Hs) vs murine (Mm) homology (white, identical residues; blue, similar; red, dissimilar). For surfaces in panels A, C, D, and E, and for side chain lines/sticks in all panels, non-C atoms are color-coded as follows: red, O; blue, N; yellow, S.
The resolved portion had an interface surface area of 620 Å2 as assessed by PISA (PDBe PISA v1.52), similar to the observed interface surface area of PD-L1 with PD-1 (622 Å2, PDB 4ZQK) and occupying the same space (Fig. 6C). The CDP makes use of many of the same interaction sites as PD-1 (Fig. 6D and fig. S6). Both PDL1B1G2-N22Q K5 and PD-1 K78 make a salt bridge with the PD-L1 A121 backbone oxygen, while both PDL1B1G2-N22Q D44 and PD-1 E136 form a salt bridge with PD-L1 Y123. PDL1B1G2-N22Q F40 sits in pocket formed by PD-L1 Y56, R113, M115, and Y123, making hydrophobic contacts (M115), herringbone ring stacking interactions (two Y’s), and a cation-pi interaction (R113). This pocket is also occupied by PD-1 I134. Furthermore, PDL1B1G2-N22Q V9, W12, and L43 also share sites of hydrophobic interactions used by PD-1 L128, A132, and I126 respectively. The interface-adjacent mutations that differentiate PDL1B1G2-N22Q from its parental scaffold would all be expected to disrupt binding if reverted (Fig. 6E): the hydrophobic interactions of both M13 and L43 with the surface of PD-L1 would be lost in the parental A13 and V43; the pocket occupied by F40 would have to distort to accommodate the parental W40, altering the interface elsewhere; and parental F39 does not neatly fit against the surface as V39 does. Finally, studying the human/mouse and human/cynomolgus monkey homology on the PD-L1 surface, it is apparent that the interaction site contains several non-homologous side chains between human and mouse (Fig. 6F). The human/cyno homology is perfect at this interface, consistent with the cross-reactivity data.
Incorporation of PDL1B1G2-N22D into a potent bispecific T cell engager
A PD-L1-binding CDP that disrupts the PD-L1:PD-1 interface could be an effective checkpoint inhibitor. However, using such a molecule in a bispecific T cell engager format, similar in mechanism to blinatumomab (38), could convert a protective signal into a liability for the cancer cells: high PD-L1 expression would induce, rather than suppress, T cell killing. For these studies, we used PDL1B1G2-N22D; D substitution also eliminates the N22 glycosite but was more enriched than Q at that site in SSM. PDL1B1G2-N22D was fused to an Fc construct containing T366S/L368A/Y407V “hole” mutations facilitating knob-and-hole heterodimerization (39) along with N297A/P329G mutations to blunt FcɣR-related effector function (40, 41) and improve T cell trafficking (42). This was paired with an Fc fusion (T366W “knob” with N297A/P329G) to a human CD3-engaging scFv (43) (Fig. 7A), producing CS-BTTC (CDP/scFv bispecific tumor/T cell crosslinker) (Fig. 7B). A comparator molecule using an anti-PD-L1 scFv (44) that lacks murine cross-reactivity (fig. S7) in place of the CDP, referred to as SS-BTTC, was also constructed (Fig. 7C).
Fig. 7. Incorporation of PDL1B1G2-N22D and comparator anti-PD-L1 scFv into CD3-engaging BTTCs.
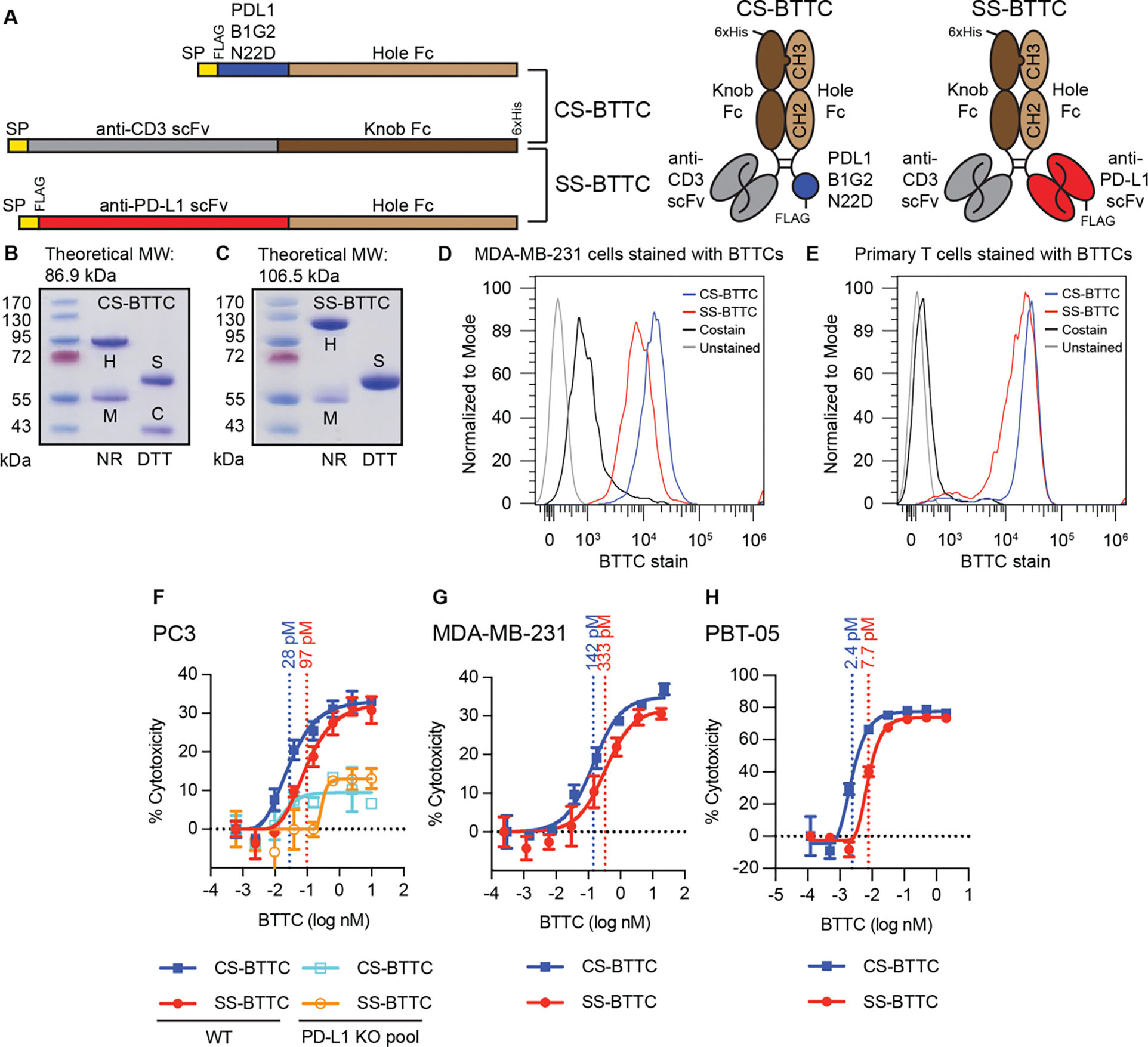
(A) Construct design. Both CS-BTTC and SS-BTTC share the anti-CD3-scFv-Fc arm; CS-BTTC was co-expressed with a PDL1B1G2-N22D-Fc, while SS-BTTC was co-expressed with anti-PD-L1-scFV-Fc. Knob-and-hole design enriches products for heterodimers via His-tag purification (nickel IMAC). SP = signal peptide. Primary sequence designs on the left, cartoons of purified heterodimers on the right. (B and C) SDS-PAGE of CS-BTTC (B) and SS-BTTC (C); heterodimer theoretical molecular weight (MW) above. Heterodimer [H] is present in non-reduced (NR) lanes, while in reduced (DTT) lanes, separate CDP-Fc [C] and scFv-Fc [S] species are visible. In both preps, anti-CD3-scFv-Fc monomer [M] is present as an impurity that did not affect TCK performance. (D and E) Flow staining of MDA-MB-231 cells (D) and primary T cells from PBMCs (E) using CS-BTTC and SS-BTTC along with fluorescent anti-6xHis antibody (costain). (F to H) In vitro TCK assays in three cancer cell lines (N = 3 per concentration): PC3 (F), MDA-MB-231 (G), and PBT-05 (H). Each BTTC’s EC50 concentration is shown. For PC3 (F), pooled PD-L1 knockout cells were tested in parallel. For full SDS-PAGE gels, see fig. S12. EC50 values determined using Graphpad Prism version 9.
Both CS-BTTC and SS-BTTC stained PD-L1+ human triple negative breast cancer line MDA-MB-231 (45) (Fig. 7D) and primary T cells purified from patient-derived peripheral blood mononuclear cells (PBMCs) (Fig. 7E). In vitro T cell killing (TCK) assays using cancer cells incubated with PBMCs or purified human T cells showed that the BTTCs potently induced T cell killing against three human cancer lines (prostate cancer cell line PC3, MDA-MB-231 cells, and patient-derived pediatric brain tumor line PBT-05 (46)) (Fig. 7, F to H). In all three cell lines, CS-BTTC appeared roughly 2–3x more potent than SS-BTTC. Two assays also demonstrated that PD-L1-engagement is required for maximal activity. First, pooled PD-L1 knockout PC3 cells demonstrated substantially less T cell killing upon BTTC incubation (Fig. 7F); the small activity observed is likely related to remaining PD-L1+ cells in the knockout pool or residual FcɣR-related effector function. Second, both BTTC preps contained impurities representing monomeric CD3-engaging scFv-Fc. Anti-FLAG-M1 affinity purification of BTTCs away from the anti-CD3 scFv-Fc monomers was performed (fig. S8A). Extra-purified BTTC preps were compared with the IMAC-purified preps in TCK assays with PBT-05 cells (fig. S8B). The presence of the CD3-engaging scFv-Fc monomer did not impact performance, suggesting the molecules’ activities are dependent on bispecific engagement of both targets and that the impurities found in the preps do not confer TCK activity.
CDP-containing BTTC drives ATC-mediated inhibition and elimination of tumors
The BTTCs were tested in mice bearing subcutaneous flank PC3 (N = 2 studies) or MDA-MB-231 (N = 1 study) tumors. Mice received either 1 or 0.1 nmol of CS-BTTC, SS-BTTC, or anti-PD-L1 durvalumab on days 1, 4, 8, and 11 as well as 7.5×106 activated human T cells (ATCs) on days 2 and 9 (Fig. 8A). In the first cohort of PC3-bearing mice (Fig. 8, B and C), the median survival of ATC-only mice (25 days) was longer than vehicle only mice (20 days), P = 0.004. This was further extended to 61.5 days by adding SS-BTTC (P = 0.002): 8 of 10 mice treated with ATC + SS-BTTC responded to treatment with a reduction in tumor volume beginning on study day 16 ± 3, though tumors re-grew in 7 of the 8 responding mice within 5 weeks. In contrast, ATC + CS-BTTC treatment resulted in 9 of 10 mice experiencing complete, sustained tumor elimination beginning on study day 16 ± 2 and having no palpable tumor at the conclusion of the study on day 95 (one mouse died on day 88). No overt signs of toxicity (weight loss or observed behavioral changes) were apparent in either BTTC treatment group (fig. S9).
Fig. 8. Immunocompromised mice with subcutaneous tumors treated with ATCs and BTTCs.
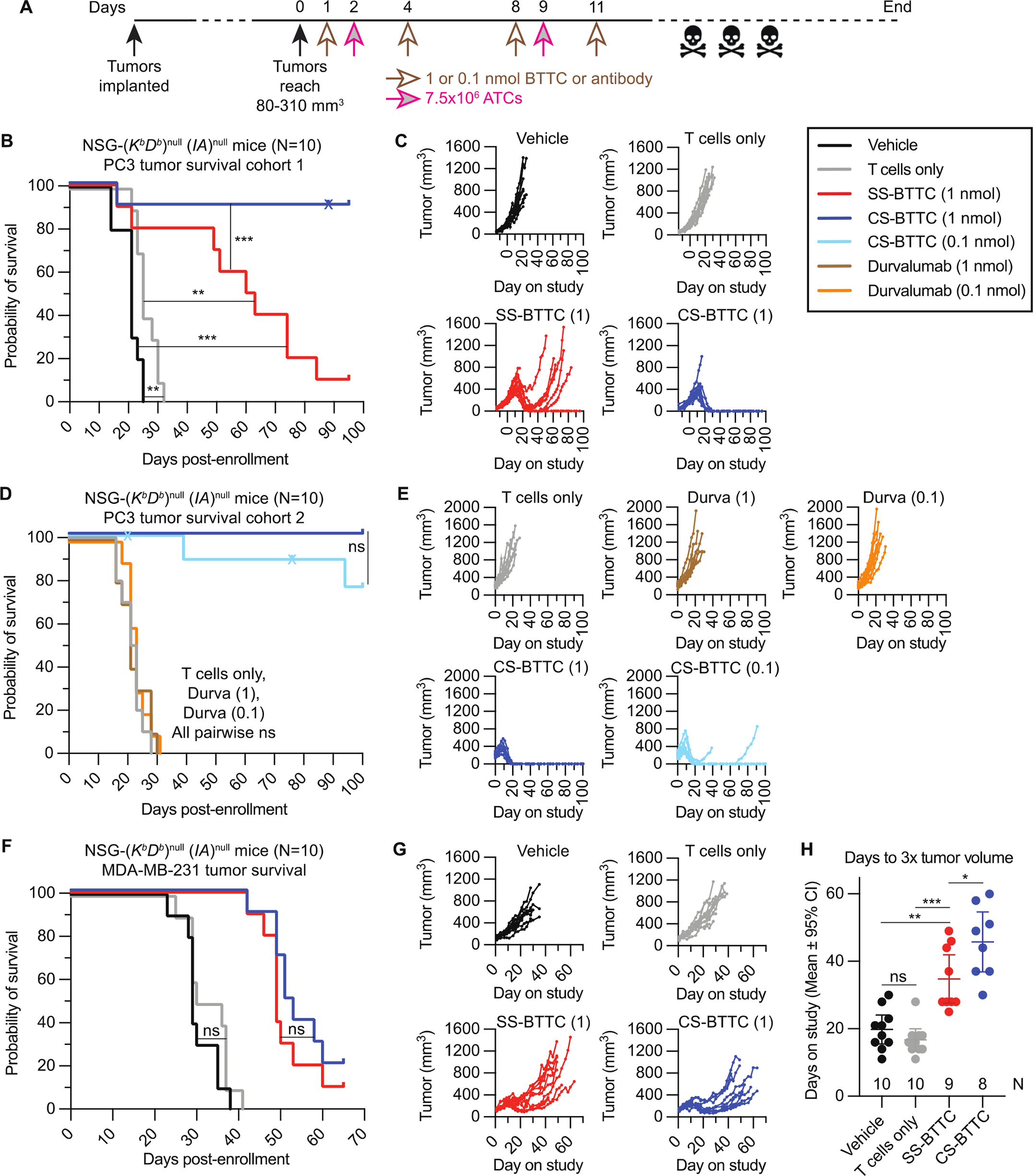
(A) Experimental design; n = 10 per arm. (B) PC3 tumor cohort 1 survival curve of four treatment arms: Vehicle (no T cells or BTTC), T cells only (no BTTC), SS-BTTC 1 nmol (T cells plus 1 nmol SS-BTTC), and CS-BTTC 1 nmol (T cells plus 1 nmol CS-BTTC). (C) PC3 tumor cohort 1 tumor growth curves. (D) PC3 tumor cohort 2 survival curve of five treatment arms: Vehicle, durvalumab 1 nmol (T cells plus 1 nmol durvalumab), durvalumab 0.1 nmol (T cells plus 0.1 nmol durvalumab), CS-BTTC 1 nmol, and CS-BTTC 0.1 nmol. (E) PC3 tumor cohort 2 tumor growth curves. (F) MDA-MB-231 tumor survival curves of four treatment arms; same design as (B). (G) MDA-MB-231 tumor growth curves. (H) Days on study for MDA-MB-231 tumors to triple in volume, mean ± 95% CI. Mice euthanized prior to tumor reaching 3x starting volume were censored (one from SS-BTTC group, two from CS-BTTC group). ns: not statistically significant. *: P < 0.05. **: P < 0.01. ***: P < 0.001. Unlabeled: P ≤ 0.0001. Kaplan-Meier curve P values are by Mantel-Cox test. x: Censored. Days to 3x tumor volume comparisons are by Mann-Whitney test.
A second cohort of PC3-bearing mice (Fig. 8, D and E) tested a lower dose of CS-BTTC as well as ATCs + durvalumab, a non-murine-cross-reactive (47) anti-PD-L1 antibody with similar clinical efficacy to atezolizumab (48) to evaluate whether the effects seen in cohort 1 were due to PD-L1 inhibition or bispecific activity. No tumor growth suppression was seen in ATC + durvalumab treatment, but both arms of ATC + CS-BTTC (1 nmol/dose and 0.1 nmol/dose) were highly potent at eliminating tumors; all 10 mice in the 1 nmol/dose repeat arm and 6 of 8 mice in the 0.1 nmol/dose arm (two mice in this arm died prior to tumor-related culling and were censored) showed complete tumor elimination, demonstrating that even a 1/10 dose of CS-BTTC out-performed SS-BTTC in this model. Histological examination (fig. S10) revealed extensive and intense ATC infiltration and activation (human CD8 and granzyme B staining) 14 days after treatment with 1 nmol CS-BTTC, whereas the only apparent response to durvalumab treatment was the recruitment of trace numbers of ATCs infiltrating the tumor.
In mice bearing MDA-MB-231 tumors (Fig. 8, F to H), for which the BTTCs demonstrated lower potency in vitro, CS-BTTC treatment did not extend survival compared to SS-BTTC treatment, but both BTTCs significantly (P < 0.0001) increased lifespan (Fig. 8F). Median survival of 33 days in ATC-only mice was extended to 49 and 52 days with ATC + SS-BTTC and ATC + CS-BTTC, respectively. Although no mice demonstrated complete MDA-MB-231 tumor clearance (Fig. 8G), histology confirmed extensive T cell infiltration and activation only in BTTC-treated tumors (fig. S11), though with less extensive infiltration and less intense granzyme B staining than seen in CS-BTTC-treated PC3 tumors (fig. S10). Comparing MDA-MB-231 tumor growth between the BTTCs (Fig. 8, G and H), CS-BTTC treatment generated slower tumor growth kinetics than SS-BTTC (P = 0.033) as measured by time for tumors to triple in volume after enrollment. In both tumor models and with both PD-L1-engaging modalities, a single two-week treatment of ATCs with PD-L1/CD3 bispecific molecules presented substantial tumor mass reduction and survival enhancement, with the CDP-containing CS-BTTC either subtly (MDA-MB-231 tumors) or substantially (PC3 tumors) out-performing the scFv-containing SS-BTTC comparator.
DISCUSSION
CDPs have great potential to address poorly served clinical needs, including difficult-to-drug targets or those within compartments poorly accessed by other biologic modalities (e.g., intracellular or central nervous system [CNS]). While the two clinically approved CDP drugs (49, 50) validate the therapeutic potential of this class, the modality lacks the existing discovery and development infrastructures for antibodies and small molecules. This work provides several new tools and examples for CDP therapeutics that can enable and motivate further development of powerful drug candidates from this class. First, we show that well-established protocols can yield CDPs and CDP libraries with high expressibility, stability, and affinity prior to soluble molecule production (synthetic or recombinant). Second, the structures of native CDPs are solvable in silico, permitting computational pre-screening like DEMYS to improve library designs. The structures also facilitate hypothesis-driven decisions on proposed improvements or liability reduction, such as identifying conjugation or fusion sites that do not interfere with activity or loops for binding epitope integration (51). Third, while a CDP in isolation can be an effective drug, many miniproteins, including CDPs, are capable of incorporation into tandem multimers as modular units (17, 18). CS-BTTC is an example of this utility; even though it would benefit from further lead optimization (e.g., improving manufacturability to eliminate the observed impurities), it demonstrates remarkable potency in vitro and in vivo.
We wanted to assess the potential and strategies for developing CDP drugs against well-established targets using in silico docking of diverse CDP scaffolds. PD-L1, being an extracellular target that relies on protein:protein interactions to perpetuate disease-associated signaling, is an appropriate test case for this approach. While PD-L1 disruption is commonly addressed with antibody-based drugs (52), it is a valuable target for CDP-binder screening for three reasons. First, by demonstrating viability against a target with known translational relevance in a head-to-head comparison with antibody-based technology, we have confidence in extending this approach to targets for which high-affinity antibodies are difficult to raise. Second, most antibody-based drugs have difficulty accessing the central nervous system (CNS), but CDPs with this capability have been identified either with (30) or without (53) selection for blood-brain barrier (BBB) transport, so a BBB-penetrant, PD-L1-disrupting CDP may have potential for CNS immuno-oncology applications. Third, both the dimensions and binding kinetics of CDPs differ from that of antibodies (10, 30), raising the possibility that CDP-based bispecific molecules could have improved pharmacology on targets for which antibody-based molecules already exist.
Given that the dimensions of a bispecific molecule may impact its activity, the difference in potency in vitro and efficacy in vivo of the two BTTCs (CS-BTTC and SS-BTTC) is notable. CS-BTTC stains MDA-MB-231 cells roughly twice as well as SS-BTTC, correlating with roughly double the potency (Fig. 7G). However, the relative sizes of an scFv vs a CDP could impact potency by creating differing BTTC-induced immune synapse dimensions. The PDL1B1G2-N22Q co-crystal structure places both termini near the surface of PD-L1. Meanwhile, an scFv is roughly 40 Å (4 nm) in length between the CDR loops and its C-terminus. Depending on where the scFv is binding, SS-BTTC and CS-BTTC could produce immunological synapses differing in dimensions by ~4 nm. Considering that a conventional TCR-driven immunological synapse can have an intermembrane distance of ~15 nm (54, 55), a 4 nm difference may prove substantial and could have implications on T cell killing activity (such as rate of degranulation), activation and exhaustion dynamics, and T cell mobility within the tumor microenvironment, among others. We will note that it is also possible that the improved in vivo potency of CS-BTTC over SS-BTTC could be influenced by differences in pharmacokinetic (PK) or other pharmacologic properties, which we did not test in these studies. Both molecules used the same Fc domain for half-life extension, but if PK differences are found to be in the CDP-based molecule’s favor, it adds another variable in addition to size and affinity for investigating the nature of CDP-based molecule advantages.
A PD-L1/CD3-engaging bispecific molecule is interesting for its ability to turn a cancer cell-protective mechanism into a liability (56, 57). However, while PD-L1 is overexpressed in many cancers, it is far from cancer-specific. Pancreatic β cells, for example, are enriched for PD-L1, and atezolizumab-linked Type 1 diabetes has been reported (58). Such a PD-L1/CD3 bispecific molecule could be de-risked several ways. The T cell engaging arm can bind costimulatory receptors CD28 or 4-1BB instead of CD3, which may result in a molecule that is preferentially active in the tumor microenvironment where T cell activation signals are more abundant. Alternatively, as the bispecific molecule is recombinant, it is possible to use it as a “genetic adjuvant” for chimeric antigen receptor (CAR) T cell therapies where the CAR T cell generates target-dependent T cell activation while also secreting the bispecific molecule within the tumor microenvironment. There is some preclinical evidence of this approach succeeding in EGFR-driven glioma cells (59). PD-L1 is only one among dozens of targets considered for bispecific molecule approaches, some of which could have greater tumor selectivity (60). It is possible that CDP-based bispecific molecules against other targets will show similar improvements in activity vs larger antibody-based bispecific molecules.
Future development of this methodology will address the limitations of this study. For example, the in silico docking in DEMYS identified scaffolds with promising target-binding properties, but it was performed in low-resolution mode for ease of use. As high-resolution docking becomes more accurate and less computationally expensive, DEMYS may benefit from its use. Recent advances in protein structural prediction like AlphaFold (61) may also improve scaffold models for DEMYS, whether CDP or other modalities. Given the promising efficacy of CS-BTTC in reducing tumor growth in PC3 and MDA-MB-231 lines was promising, and extending its testing to other cancer models to see how generalizable it is could push its development towards cancers for which it is especially efficacious. Potential safety liabilities will also need to be addressed, as PD-L1 expression is not unique to cancerous tissue, though a recent study provides some encouragement that this may be surmountable (56).
In conclusion, through extensive structural modeling of an improved CDP library, we have described a method for discovering CDPs of potential clinical interest. We have demonstrated that CDPs have potential both as independent drugs and through incorporation into other biologic drug scaffolds. Structure-driven DEMYS screening can yield hits of interest as well as provide information about candidate docking sites (and therefore mechanism of action), situating CDPs, and miniproteins at large, in a position to efficiently and effectively address clinical needs poorly served by other modalities.
MATERIALS AND METHODS
Study design
This work explored in silico screening capabilities with diverse CDP structural models for their ability to identify clinically relevant binders to targets of interest, the latter of which was evaluated in the context of BTTC molecules for T cell-driven tumor clearance. In vitro TCK experiments in the three cancer cell lines were performed once and utilized triplicates of each drug at eight concentrations to arrive at reported EC50 potencies, and all cross-BTTC comparisons (CS-BTTC vs SS-BTTC) were performed simultaneously. In vivo experiments were performed in adult male and female NSG-(KbDb)null (IA)null mice with 10 mice per treatment arm. Mice with tumors between 80 and 310 mm3 in volume were enrolled in study, normalizing for equal volume between groups. 2 studies were run with PC3 tumors, 1 study with MDA-MB-231 tumors. The research was blinded to treatment identity for the first PC3 experiment. In total, three mice died unrelated to either tumors or treatment (died after tumor response [tumors reduced to <100 mm3], without tumor burden, and having demonstrated no treatment-related toxicity); these were censored from survival statistical analysis.
CDP library construction and flow cytometry
CDP library cloning and surface display flow cytometry were based on previously described protocols (62), with details found in the Supplementary Methods. Each library screen for PD-L1 binder identification was conducted once.
Recombinant protein production and QC
The scFv used in SS-BTTC was derived from the VK4 and VH2 sequences from (63) in VKVH orientation with a [GGGGS]3 linker. Soluble protein production was as described in Bandaranayake et al. (37). In short, 293 FreeStyle cells (293F, ThermoFisher R79007) were transduced at MOI ≈ 10; for BTTC constructs, two separate lentivectors were used. Cultures were grown until terminal volume (typically 2–4 L) in FreeStyle 293 Expression Medium (Invitrogen 12338018). Media samples containing secreted proteins were harvested, 0.22 μm sterile filtered, and purified via FPLC IMAC Ni-Affinity Chromatography (Cytiva 17525501). Protein-specific details for purification and quality control are found in the Supplementary Methods.
Crystallization and crystallography
Crystallography conditions, phasing, and refinement notes are found in the Supplementary Methods. The PD-L1:CDP complex was deposited to the RCSB database as PDB 7SJQ. Please note that because the recombinant CDP used for these studies contains a leading “GS” dipeptide leftover from TEV cleavage, the N-terminus of the CDP (EEDC…) begins at amino acid number 3 of chain D in the deposited PDB file, resulting in the numbering being offset by 2. Structure 7SLT, not featured in Fig. 2, was discovered upon crystallization to contain a single amino acid deletion causing misfolding and improper dimerization. It was deposited but not included in alignment analysis.
In vitro TCK
Prior to incubation, the three cell lines (PC3 [Synthego control line for custom CD274 knockout pool], MDA-MB-231 [ATCC HTB-26], and PBT-05 [FHCRC BTRL line PBT-05FHTC]) were transduced with lentivirus delivering an infrared fluorescent protein (iRFP) for more robust cell killing quantitation. PBMCs (Bloodworks Northwest) in the PC3 and PBT-05 tests, or purified naïve T cells (EasySep 19555) in the MDA-MB-231 tests, were seeded with target cells at a 5:1 E:T ratio in 200 μL/well of complete T cell media (RPMI with 25 mM HEPES [Gibco], 10% human serum [Valley Biomedical Product], 4 mM GlutaMax [Gibco], 1x Pen/Strep [Gibco], and 1x β-mercaptoethanol [Sigma]) in 96-well plates. Experiments were conducted once per cell line. Test molecules were added in triplicate (biological replicates) and cells were incubated for 48 hrs (37°C, 5% CO2). Cells were imaged using ImageXpress Nano (Molecular Devices) to measure cytotoxicity.
In vivo subcutaneous flank xenograft studies
All mice were maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals with approval from the Fred Hutchinson Cancer Research Center Institutional Animal Care and Use Committee (protocol 51073). NSG-(KbDb)null (IA)null were purchased from Jackson laboratories (#025216) and bred on site. PC3 and MDA-MB-231 tumor models were purchased from ATCC (CRL-3471 and CRM-HTB-26) and verified human pathogen- and mycoplasma-free before implantation into mice. BTTCs were produced in house as described above and tested endotoxin free. Clinical grade durvalumab was purchased through the Seattle Cancer Care Alliance Pharmacy (NDC 0310-4500-12). ATC were purchased as PBMC from Bloodworks Northwest (donor 3126), stimulated with a human T Cell Activation/Expansion Kit (Miltenyi 130-091-441), expanded, verified human pathogen free, and cryo-preserved. Cells were thawed immediately before administration. Mice were implanted with tumor when 10–19 weeks of age. Male and female mice were used in all studies. Study enrollment was done en masse on day 0 when the average tumor volume was 130–220 mm3. Tumors smaller than 80 mm3 or larger than 310 mm3 were excluded from enrollment. 10 mice were randomly enrolled in each treatment group, normalizing for equal starting tumor volume. Vehicle, ATC, BTTC, and antibody treatments were administered as a 100 μl bolus in sterile PBS via tail vein injection; BTTC or antibody (1 or 0.1 nmol per mouse) on days 1, 4, 8, and 11, ATC (7.5×106 per mouse) on days 2 and 9, vehicle. 1 nmol = 100 μg SS-BTTC, 80 μg CS-BTTC, 150 μg durvalumab per dose. Doses were chosen for consistency with existing bispecific molecule literature (57, 64). Tumor volume and body weight were recorded every Monday, Wednesday, and Friday. Mice were removed from study and euthanized when tumors reached 1500 mm3, if tumors developed an open ulcer on their surface, or if body weight dropped to 80% of the starting value. A small cohort of extra mice were enrolled in the MDA-MB-231 and second PC3 studies for immunohistochemical analysis of T cell tumor infiltration and activity. 2 extra mice were enrolled in each group in the MDA-MB-231 study and 3 mice were enrolled in the ATC-only, 1 nmol CS-BTTC, and 1 nmol durvalumab groups in the PC3 study; tumors were collected on day 14. All mice were group housed with unrestricted mobility and free access to food and water for the duration of their study.
Statistical analysis
Statistics were performed using GraphPad Prism v9.1.2 software. In vitro TCK experiments were performed in triplicate (3 wells per condition), and EC50 calculations used asymmetric sigmoidal (5PL) least squares curve fitting with bottom set to 0. Significance of inter-group comparisons (enrichment scores’ effect on binding [Fig. 6B] and days to 3x tumor volume in MDA-MB-231 subcutaneous tumors [Fig. 8H]) were by Mann-Whitney test (two-tailed non-parametric rank comparison). Kaplan-Meier curve P values (Fig. 8B, 8D, and 8F) are by Mantel-Cox test (proportional hazards). For in vivo studies, each comparison used arms of ten mice; sample size was chosen for consistency with similar studies in-house and in literature rather than by power calculation. Subject-level murine data are provided in data file S1.
Supplementary Material
Acknowledgments:
We thank S. Turnbaugh and A. Williams from the Molecular Design and Therapeutics Core for producing and analyzing the CDPs that were integral to this work; G. Rocklin, J. Carter, K. Grabstein, P. Bradley, and J. Simon for intellectual input and scientific guidance.
Funding:
The project was supported by NIH grants R01CA223674, U01CA232490, and R01CA114567 (to J.M.O.), NIH grants S10OD020069 and S10OD028685 (Fred Hutch Scientific Computing Shared Resource), NIH grant P30CA015704 (Fred Hutch Flow Cytometry Shared Resource), Project Violet, Unravel Pediatric Cancer, Washington Research Foundation, and Blaze Bioscience.
Footnotes
Competing interests: Blaze Bioscience, Inc. retains intellectual property rights to the CDPs used in this manuscript. J.M.O. is a founder and shareholder of Blaze Bioscience Inc. and serves as a compensated Scientific Advisory Board Member. Z.R.C., G.P.S., T.-D.P., and N.W.N. are currently employees of Blaze Bioscience Inc. with stock options, and Z.R.C. and G.P.S. were paid consultants for Blaze Bioscience, Inc. prior to employment. Z.R.C., N.W.N., C.E.C., and J.M.O. are inventors on patent application PCT/US21/61039 (“PD-L1 Binding Peptides and Peptide Complexes and Methods of Use Thereof”) of which these molecules are a subject. E.J.G., M.-Y.B., P.B.R., D.J.F., M.M.G., M.C., I.L., R.R., F.P., A.B., C.E.C., A.J.M., and R.K.S. have no competing interests.
Supplementary Materials
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. Protein crystal structures have been deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (rcsb.org), accession numbers 7SGQ, 7SLT, 7SAO, 7SNC, 7SND, 7SAP, and 7SJQ. Requests for additional information and/or materials related to this study, including recombinant proteins and raw, non-proprietary datasets not found in the manuscript or its supplements, should be addressed to the corresponding author and will be made available on reasonable terms
References and Notes
- 1.Coleman N, Rodon J, Taking Aim at the Undruggable, Am. Soc. Clin. Oncol. Educ. B., e145–e152 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Yoo HJ, Harapan BN, Chimeric antigen receptor (CAR) immunotherapy: basic principles, current advances, and future prospects in neuro-oncology, Immunol. Res. (2021), doi: 10.1007/s12026-021-09236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antonarelli G, Giugliano F, Corti C, Repetto M, Tarantino P, Curigliano G, Research and Clinical Landscape of Bispecific Antibodies for the Treatment of Solid Malignancies, Pharmaceuticals 14, 884 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crook ZR, Nairn NW, Olson JM, Miniproteins as a Powerful Modality in Drug Development, Trends Biochem. Sci. 45, 332–346 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busby RW, Kessler MM, Bartolini WP, Bryant AP, Hannig G, Higgins CS, Solinga RM, Tobin JV, Wakefield JD, Kurtz CB, Currie MG, Pharmacologic Properties, Metabolism, and Disposition of Linaclotide, a Novel Therapeutic Peptide Approved for the Treatment of Irritable Bowel Syndrome with Constipation and Chronic Idiopathic Constipation, J. Pharmacol. Exp. Ther. 344, 196–206 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Correnti CE, Gewe MM, Mehlin C, Bandaranayake AD, Johnsen WA, Rupert PB, Brusniak M-Y, Clarke M, Burke SE, De Van Der Schueren W, Pilat K, Turnbaugh SM, May D, Watson A, Chan MK, Bahl CD, Olson JM, Strong RK, Screening, large-scale production and structure-based classification of cystine-dense peptides, Nat. Struct. Mol. Biol. 25, 270–278 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu X, Yang Y-P, Dikici E, Deo SK, Daunert S, Beyond Antibodies as Binding Partners: The Role of Antibody Mimetics in Bioanalysis, Annu. Rev. Anal. Chem. 10, 293–320 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cruz E, Kayser V, Monoclonal antibody therapy of solid tumors: clinical limitations and novel strategies to enhance treatment efficacy, Biol. Targets Ther. Volume 13, 33–51 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thurber GM, Schmidt MM, Wittrup KD, Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance, Adv. Drug Deliv. Rev. 60, 1421–1434 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crook ZR, Sevilla GP, Friend D, Brusniak M-Y, Bandaranayake AD, Clarke M, Gewe M, Mhyre AJ, Baker D, Strong RK, Bradley P, Olson JM, Mammalian display screening of diverse cystine-dense peptides for difficult to drug targets., Nat. Commun. 8, 2244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleetwood F, Klint S, Hanze M, Gunneriusson E, Frejd FY, Ståhl S, Löfblom J, Simultaneous targeting of two ligand-binding sites on VEGFR2 using biparatopic Affibody molecules results in dramatically improved affinity, Sci. Rep. 4, 7518 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hulme JT, D’Souza WN, McBride HJ, Yoon B-RP, Willee AM, Duguay A, Thomas M, Fan B, Dayao MR, Rottman JB, Merriam K, Xie J, Smith R, Alba BM, Case RB, Dang K, Montalvan A, Grinberg N, Sun H, Black RA, Gabel CA, Sims JE, Moore K, Bakker A, Li P, Novel protein therapeutic joint retention strategy based on collagen-binding Avimers, J. Orthop. Res. 36, 1238–1247 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Zhang D, Whitaker B, Derebe MG, Chiu ML, FcγRII-binding Centyrins mediate agonism and antibody-dependent cellular phagocytosis when fused to an anti-OX40 antibody, MAbs 10, 463–475 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goux M, Becker G, Gorré H, Dammicco S, Desselle A, Egrise D, Leroi N, Lallemand F, Bahri MA, Doumont G, Plenevaux A, Cinier M, Luxen A, Nanofitin as a New Molecular-Imaging Agent for the Diagnosis of Epidermal Growth Factor Receptor Over-Expressing Tumors, Bioconjug. Chem. 28, 2361–2371 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Wensel D, Sun Y, Li Z, Zhang S, Picarillo C, McDonagh T, Fabrizio D, Cockett M, Krystal M, Davis J, Discovery and Characterization of a Novel CD4-Binding Adnectin with Potent Anti-HIV Activity, Antimicrob. Agents Chemother. 61 (2017), doi: 10.1128/AAC.00508-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Löfblom J, Feldwisch J, Tolmachev V, Carlsson J, Ståhl S, Frejd FY, Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications, FEBS Lett. 584, 2670–2680 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Silverman J, Lu Q, Bakker A, To W, Duguay A, Alba BM, Smith R, Rivas A, Li P, Le H, Whitehorn E, Moore KW, Swimmer C, Perlroth V, Vogt M, Kolkman J, Stemmer WPC, Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains, Nat. Biotechnol. 23, 1556–1561 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Lipovšek D, Adnectins: engineered target-binding protein therapeutics, Protein Eng. Des. Sel. 24, 3–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarcha EJ, Olsen CM, Probst P, Peckham D, Muñoz-Elías EJ, Kruger JG, Iadonato SP, Safety and pharmacodynamics of dalazatide, a Kv1.3 channel inhibitor, in the treatment of plaque psoriasis: A randomized phase 1b trial, PLoS One 12, 1–19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y, The I-TASSER Suite: protein structure and function prediction, Nat. Methods 12, 7–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban YEA, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popović Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P, Rosetta3: An object-oriented software suite for the simulation and design of macromolecules, Methods Enzymol. 487, 545–574 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cook Sangar ML, Girard EJ, Hopping G, Yin C, Pakiam F, Brusniak M-Y, Nguyen E, Ruff R, Gewe MM, Byrnes-Blake K, Nairn NW, Miller DM, Mehlin C, Strand AD, Mhyre AJ, Correnti CE, Strong RK, Simon JA, Olson JM, A potent peptide-steroid conjugate accumulates in cartilage and reverses arthritis without evidence of systemic corticosteroid exposure, Sci. Transl. Med. 12, eaay1041 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Buchko GW, Pulavarti SVSRK, Ovchinnikov V, Shaw EA, Rettie SA, Myler PJ, Karplus M, Szyperski T, Baker D, Bahl CD, Cytosolic expression, solution structures, and molecular dynamics simulation of genetically encodable disulfide-rich de novo designed peptides, Protein Sci. 27, 1611–1623 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhardwaj G, Mulligan VK, Bahl CD, Gilmore JM, Harvey PJ, Cheneval O, Buchko GW, Pulavarti SVSRK, Kaas Q, Eletsky A, Huang P-S, Johnsen WA, Greisen PJ, Rocklin GJ, Song Y, Linsky TW, Watkins A, Rettie SA, Xu X, Carter LP, Bonneau R, Olson JM, Coutsias E, Correnti CE, Szyperski T, Craik DJ, Baker D, Accurate de novo design of hyperstable constrained peptides, Nature 538, 329–335 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, TM-align: a protein structure alignment algorithm based on the TM-score, Nucleic Acids Res. 33, 2302–2309 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frishman D, Argos P, Knowledge-based protein secondary structure assignment., Proteins 23, 566–79 (1995). [DOI] [PubMed] [Google Scholar]
- 27.Heinig M, Frishman D, STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins., Nucleic Acids Res. 32, W500–2 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marze NA, Roy Burman SS, Sheffler W, Gray JJ, Valencia A, Ed. Efficient flexible backbone protein–protein docking for challenging targets, Bioinformatics 34, 3461–3469 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ester M, Kriegel HP, Sander J, Xiaowei X, A density-based algorithm for discovering clusters in large spatial databases with noise, Proc. 2nd Int. Conf. Knowl. Discov. Data Min., 226–231 (1996). [Google Scholar]
- 30.Crook ZR, Girard E, Sevilla GP, Merrill M, Friend D, Rupert PB, Pakiam F, Nguyen E, Yin C, Ruff RO, Hopping G, Strand AD, Finton KAK, Coxon M, Mhyre AJ, Strong RK, Olson JM, A TfR-Binding Cystine-Dense Peptide Promotes Blood–Brain Barrier Penetration of Bioactive Molecules, J. Mol. Biol. 432, 3989–4009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Menting JG, Yang Y, Chan SJ, Phillips NB, Smith BJ, Whittaker J, Wickramasinghe NP, Whittaker LJ, Pandyarajan V, Wan Z, Yadav SP, Carroll JM, Strokes N, Roberts CT, Ismail-Beigi F, Milewski W, Steiner DF, Chauhan VS, Ward CW, Weiss MA, Lawrence MC, Protective hinge in insulin opens to enable its receptor engagement., Proc. Natl. Acad. Sci. U. S. A. 111, E3395–404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Correa A, Pacheco S, Mechaly AE, Obal G, Béhar G, Mouratou B, Oppezzo P, Alzari PM, Pecorari F, Potent and specific inhibition of glycosidases by small artificial binding proteins (affitins)., PLoS One 9, e97438 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlatter D, Brack S, Banner DW, Batey S, Benz J, Bertschinger J, Huber W, Joseph C, Rufer AC, van der Klooster A, Weber M, Grabulovski D, Hennig M, Generation, characterization and structural data of chymase binding proteins based on the human Fyn kinase SH3 domain, MAbs 4, 497–508 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eigenbrot C, Ultsch M, Dubnovitsky A, Abrahmsén L, Härd T, Structural basis for high-affinity HER2 receptor binding by an engineered protein, Proc. Natl. Acad. Sci. U. S. A. 107, 15039–15044 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, Dömling A, Dubin G, Holak TA, Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1., Structure 23, 2341–2348 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moon CP, Fleming KG, Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers, Proc. Natl. Acad. Sci. 108, 10174–10177 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bandaranayake AD, Correnti C, Ryu BY, Brault M, Strong RK, Rawlings DJ, Daedalus: a robust, turnkey platform for rapid production of decigram quantities of active recombinant proteins in human cell lines using novel lentiviral vectors., Nucleic Acids Res. 39, e143 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu J, Fu J, Zhang M, Liu D, Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia, J. Hematol. Oncol. 8, 104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Atwell S, Ridgway JB, Wells JA, Carter P, Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library, J. Mol. Biol. 270, 26–35 (1997). [DOI] [PubMed] [Google Scholar]
- 40.Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E, Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions, Protein Eng. Des. Sel. 29, 457–466 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Zhou Q, Jaworski J, Zhou Y, Valente D, Cotton J, Honey D, Boudanova E, Beninga J, Rao E, Wei R, Mauriac C, Pan C, Park A, Qiu H, Engineered Fc-glycosylation switch to eliminate antibody effector function, MAbs 12, 1814583 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, Hoseini SS, Xu H, Ponomarev V, Cheung N-K, Silencing Fc Domains in T cell–Engaging Bispecific Antibodies Improves T-cell Trafficking and Antitumor Potency, Cancer Immunol. Res. 7, 2013–2024 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Friedrich M, Henn A, Raum T, Bajtus M, Matthes K, Hendrich L, Wahl J, Hoffmann P, Kischel R, Kvesic M, Slootstra JW, Baeuerle PA, Kufer P, Rattel B, Preclinical characterization of AMG 330, a CD3/CD33- bispecific T-cell-engaging antibody with potential for treatment of acute myelogenous leukemia, Mol. Cancer Ther. 13, 1549–1557 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Freeman GJ, Ahmed R, Jones TD, Carr FJ, Gregson JP, ANTI-PD-L1 ANTIBODIES AND USES THEREFORUS 8,552,154 B2 (2013). [Google Scholar]
- 45.Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, Su X, Wang Y, Gonzalez-Angulo AM, Akcakanat A, Chawla A, Curran M, Hwu P, Sharma P, Litton JK, Molldrem JJ, Alatrash G, PD-L1 Expression in Triple-Negative Breast Cancer, Cancer Immunol. Res. 2, 361–370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brabetz S, Leary SES, Gröbner SN, Nakamoto MW, Şeker-Cin H, Girard EJ, Cole B, Strand AD, Bloom KL, Hovestadt V, Mack NL, Pakiam F, Schwalm B, Korshunov A, Balasubramanian GP, Northcott PA, Pedro KD, Dey J, Hansen S, Ditzler S, Lichter P, Chavez L, Jones DTW, Koster J, Pfister SM, Kool M, Olson JM, A biobank of patient-derived pediatric brain tumor models, Nat. Med. 24, 1752–1761 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Magiera-Mularz K, Kocik J, Musielak B, Plewka J, Sala D, Machula M, Grudnik P, Hajduk M, Czepiel M, Siedlar M, Holak TA, Skalniak L, Human and mouse PD-L1: similar molecular structure, but different druggability profiles, iScience 24, 101960 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J, Wang J, Xu H, Comparison of atezolizumab, durvalumab, pembrolizumab, and nivolumab as first-line treatment in patients with extensive-stage small cell lung cancer, Medicine (Baltimore). 100, e25180 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Corsetti M, Tack J, Linaclotide: A new drug for the treatment of chronic constipation and irritable bowel syndrome with constipation, United Eur. Gastroenterol. J. 1, 7–20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGivern JG, Ziconotide: a review of its pharmacology and use in the treatment of pain, Neuropsychiatr. Dis. Treat. 3, 69–85 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kimura RH, Levin AM, Cochran FV, Cochran JR, Engineered cystine knot peptides that bind αvβ3, αvβ5, and α5β1 integrins with low-nanomolar affinity, Proteins Struct. Funct. Bioinforma. 77, 359–369 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Akinleye A, Rasool Z, Immune checkpoint inhibitors of PD-L1 as cancer therapeutics, J. Hematol. Oncol. 12, 92 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patil CG, Walker DG, Miller DM, Butte P, Morrison B, Kittle DS, Hansen SJ, Nufer KL, Byrnes-Blake KA, Yamada M, Lin LL, Pham K, Perry J, Parrish-Novak J, Ishak L, Prow T, Black K, Mamelak AN, Phase 1 Safety, Pharmacokinetics, and Fluorescence Imaging Study of Tozuleristide (BLZ-100) in Adults With Newly Diagnosed or Recurrent Gliomas, Neurosurgery 85, E641–E649 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Dustin ML, The Immunological Synapse, Cancer Immunol. Res. 2, 1023–1033 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krummel MF, Cahalan MD, The immunological synapse: A dynamic platform for local signaling, J. Clin. Immunol. 30, 364–372 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu L, Chen J, Bae J, Li H, Sun Z, Moore C, Hsu E, Han C, Qiao J, Fu Y-X, Rejuvenation of tumour-specific T cells through bispecific antibodies targeting PD-L1 on dendritic cells, Nat. Biomed. Eng. 5, 1261–1273 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Horn LA, Ciavattone NG, Atkinson R, Woldergerima N, Wolf J, Clements VK, Sinha P, Poudel M, Ostrand-Rosenberg S, CD3xPDL1 bi-specific T cell engager (BiTE) simultaneously activates T cells and NKT cells, kills PDL1+ tumor cells, and extends the survival of tumor-bearing humanized mice, Oncotarget 8, 57964–57980 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sothornwit J, Phunmanee A, Pongchaiyakul C, Atezolizumab-Induced Autoimmune Diabetes in a Patient With Metastatic Lung Cancer, Front. Endocrinol. (Lausanne). 10 (2019), doi: 10.3389/fendo.2019.00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, Bailey SR, Boroughs AC, Frigault MJ, Leick MB, Scarfò I, Cetrulo CL, Demehri S, V Nahed B, Cahill DP, Wakimoto H, Curry WT, Carter BS, V Maus M, CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity, Nat. Biotechnol. 37, 1049–1058 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Zhou S, Liu M, Ren F, Meng X, Yu J, The landscape of bispecific T cell engager in cancer treatment, Biomark. Res. 9, 38 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Highly accurate protein structure prediction with AlphaFold, Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crook ZR, Sevilla GP, Mhyre AJ, Olson JM, Mammalian Surface Display Screening of Diverse Cystine-Dense Peptide Libraries for Difficult-to-Drug Targets., Methods Mol. Biol. 2070, 363–396 (2020). [DOI] [PubMed] [Google Scholar]
- 63.Freeman GJ, Ahmed R, Jones TD, Carr FJ, Gregson JP, Anti-PD-L1 antibodies and uses therefor (2013) (available at https://pdfpiw.uspto.gov/.piw?Docid=08552154&homeurl=http%3A%2F%2Fpatft.uspto.gov%2Fnetacgi%2Fnph-Parser%3FSect1%3DPTO2%2526Sect2%3DHITOFF%2526p%3D1%2526u%3D%25252Fnetahtml%25252FPTO%25252Fsearch-bool.html%2526r%3D1%2526f%3DG%2526l%3D50%2526co1%3DAND%2526).
- 64.Caraccio C, Krishna S, Phillips DJ, Schürch CM, Bispecific Antibodies for Multiple Myeloma: A Review of Targets, Drugs, Clinical Trials, and Future Directions, Front. Immunol. 11, 1–25 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Otwinowski Z, Minor W, Processing of X-ray diffraction data collected in oscillation mode., Methods Enzymol. 276, 307–26 (1997). [DOI] [PubMed] [Google Scholar]
- 66.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, Phaser crystallographic software J Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS, Overview of the CCP 4 suite and current developments, Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roussel A, Mathieu M, Dobbs A, Luu B, Cambillau C, Kellenberger C, Complexation of Two Proteic Insect Inhibitors to the Active Site of Chymotrypsin Suggests Decoupled Roles for Binding and Selectivity, J. Biol. Chem. 276, 38893–38898 (2001). [DOI] [PubMed] [Google Scholar]
- 69.Emsley P, Cowtan K, Coot : model-building tools for molecular graphics, Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Murshudov GN, Vagin AA, Dodson EJ, Refinement of Macromolecular Structures by the Maximum-Likelihood Method, Acta Crystallogr. Sect. D Biol. Crystallogr. 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- 71.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC, MolProbity: all-atom contacts and structure validation for proteins and nucleic acids., Nucleic Acids Res. 35, W375–83 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, The Protein Data Bank., Nucleic Acids Res. 28, 235–42 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. Protein crystal structures have been deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (rcsb.org), accession numbers 7SGQ, 7SLT, 7SAO, 7SNC, 7SND, 7SAP, and 7SJQ. Requests for additional information and/or materials related to this study, including recombinant proteins and raw, non-proprietary datasets not found in the manuscript or its supplements, should be addressed to the corresponding author and will be made available on reasonable terms