

Abstract
Proteomic analysis of limited samples and single cells requires specialized methods that prioritize high sensitivity and minimize sample loss. Consequently, sample preparation is one of the most important steps in limited sample analysis workflows to prevent sample loss. In this work, we have eliminated sample handling and transfer steps by processing intact cells directly in the separation capillary, online with capillary electrophoresis coupled to tandem mass spectrometry (CE-MS/MS) for top-down proteomic (TDP) analysis of low numbers of mammalian cancer cells (<10) and single cells. We assessed spray voltage injection of intact cells from a droplet of cell suspension (~1000 cells) and demonstrated 0–9 intact cells injected with a dependency on the duration of spray voltage application. Spray voltage applied for 2 min injected an average of 7 ± 2 cells and resulted in 33–57 protein and 40–88 proteoform identifications (N = 4). To analyze single cells, manual cell loading by hydrodynamic pressure was used. Replicates of single HeLa cells (N = 4) lysed on the capillary and analyzed by CE-MS/MS demonstrated a range of 17–40 proteins and 23–50 proteoforms identified. An additional cell line, THP-1, was analyzed at the single-cell level, and proteoform abundances were compared to show the capabilities of single-cell TDP (SC-TDP) for assessing cellular heterogeneity. This study demonstrates the initial application of TDP in single-cell proteome-level profiling. These results represent the highest reported identifications from TDP analysis of a single HeLa cell and prove the tremendous potential for CE-MS/MS on-capillary sample processing for high sensitivity analysis of single cells and limited samples.
Graphical Abstract
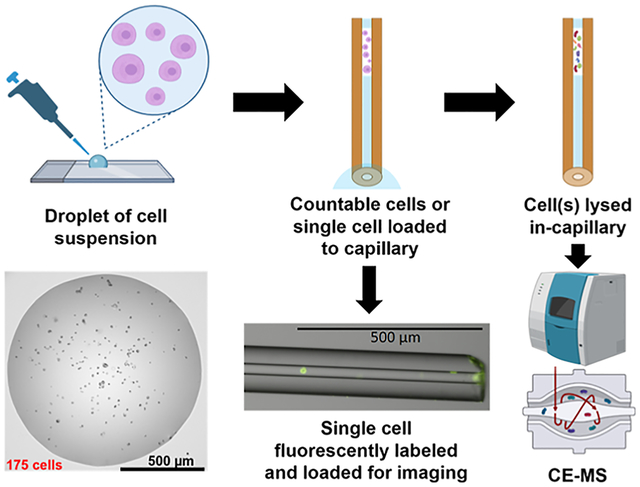
In recent years, the field of single-cell proteomics (SCP) has grown immensely,1 especially as MS technologies have improved to achieve superior levels of sensitivity. The rationale for investigating proteomes at the single-cell level is primarily based on moving away from the traditional bulk sample proteomic analysis to gain insights into cellular heterogeneity that would be lost with the bulk sampling approach that results in an averaging of protein levels from all cells. This averaging effect from bulk sampling is particularly detrimental to the profiling of rare populations of cells that are masked by the signal from the predominant cell types. This aim has driven many researchers to focus their efforts on improving the SCP workflow, including sample processing steps, separations, MS, and data analysis.2–4 Certain types of limited samples are inherently mass-limited due to their naturally low abundance or the application of minimally invasive sampling techniques, such as isolation of circulating tumor cells from small volume liquid biopsies and procurement of microbiopsies from disease-affected tissue loci. Molecular profiling of such samples can also benefit from these SCP workflows that emphasize high sensitivity and minimal sample loss.5
The vast majority of the progress in SCP workflows to date has centered around the bottom-up proteomic approach involving the digestion of proteins into peptides prior to analysis. With these workflows, >1000 protein groups can be profiled from a single mammalian cell,6–8 representing an impressive level of proteome coverage for such a low sample amount. However, top-down approaches for SCP analysis have not yet been as widely studied. Top-down proteomics (TDP) is the analysis of intact proteins and can more accurately represent protein identifications by distinguishing specific proteoforms that are present in the sample. The term proteoform was established to describe protein variants resulting from post-translational modifications (PTMs), alternative splicing, and genetic mutation.9 Understanding these protein variants has been demonstrated to reveal links to certain diseases and their underlying mechanisms.10,11 Proteoform information cannot be determined from the bottom-up approach, where the proteins are proteolytically digested prior to analysis, highlighting the necessity for top-down approaches. However, there are several additional challenges for separations, MS acquisition, and data analysis in TDP12 that limit the proteome coverage that can be achieved. MS analysis of intact proteins is complicated by the diversity of ionization products for a single proteoform, resulting in multiple charge states and adducts that cause signal dilution and contribute to spectral overlap. Data processing presents an additional challenge, as many top-down analysis software packages were not designed to handle complex “omics” data, and there is currently no universal data evaluation platform.
As evidenced by the establishment of the Consortium for Top-Down Proteomics13 in 2012, TDP analysis is gaining a lot of traction in the field due to an interest in developing a greater understanding of proteoform-specific information that is unobtainable from a bottom-up analysis. Several recent studies have shown deep proteome coverage at the top-down level, including the identification of 5700 unique proteoforms from Escherichia coli14 and 2778 unique proteoforms from HeLa cell lysate.15 A larger effort to identify proteoform-level differences across human tissues was able to map 11,466 proteoforms across five distinct tissues, including PTMs specific to certain tissue types.16 To achieve this level of coverage, microgram or even milligram amounts of sample and multidimensional separation approaches were required. High sensitivity TDP analysis has been conducted at the level of ~70 HeLa cells demonstrating ~170 proteoforms17 using the nanoPOTs3 approach for limited sample processing. Others have achieved measurements of a single intact protein (hemoglobin) from single erythrocytes with an integrated microfluidic device platform for single-cell injection and processing online with MS detection;18 however, TDP of limited samples and single cells remains a challenge.
In this work, we report a proof-of-concept capillary electrophoresis coupled to tandem mass spectrometry (CEMS/MS)-based pilot approach for proteome-level TDP analysis of <10 mammalian cancer cells, down to the single-cell level. Applying our novel method for single-cell proteomics (SC-TDP), which can also be referred to as top-down single-cell proteomics (TD-SCP), we were initially able to identify 40 proteins and 50 proteoforms from a single HeLa cell, marking an improvement in previous single-cell intact protein analyses that were able to characterize only 1–2 proteins.18–20 Since CE is an open-tubular separation, it is possible to inject intact cells and lyse them directly in the separation column, thereby eliminating any sample losses that occur during sample processing and transfer steps. In fact, a study that was conducted nearly two decades ago achieved the first MS measurements of hemoglobin from single erythrocytes using a similar approach.20 Here, we investigated two mechanisms of injection of intact cells directly onto CE separation capillary; the first relies on a flow rate generated by the application of electrospray at the outlet of the capillary to draw cells onto the capillary from a nanoliter-sized droplet of cell suspension at the inlet, similar to the injection mechanism described in the recently published report;21 the second utilizes the hydrodynamic pressure differential created by lowering the outlet end of the capillary and generating low flow to load a single cell. We demonstrated each of these modes of injection allowed for cell lysis directly within the separation capillary and could be successfully followed by CE-MS/MS analysis, resulting in the confident identification of intact proteins and proteoforms.
MATERIALS AND METHODS
Sample Preparation.
Cell pellets were harvested by centrifuging at 500g for 5 min. The harvested cells were resuspended in PBS to wash off the residual medium. The washing step was repeated another two times. Before the final centrifugation, the cell density was estimated by hemocytometer counting. The PBS wash buffer was removed, and cell pellets were then resuspended in a corresponding volume of 200 mM ammonium acetate (pH = 6.7) to get to a final cell density of 1.5 million HeLa cells per mL. The cell viability was typically around 80% in the experiments. The cell suspension was kept on ice and mixed thoroughly before each cell injection.
Spray Voltage Injection of Intact Cells.
Intact HeLa cells were injected into the separation capillary from a 0.5 μL droplet on a glass slide (Swiss Glass, Norwich, CT). The glass slide was pre-treated with Aquapel to provide a hydrophobic coating, which produces a higher contact angle with cell droplets to improve injection. A micropipette (Eppendorf Research, Hamburg, Germany) was used to deposit 0.5 μL of cell suspension (1.5 × 106 cells/mL, ~1000 cells) to the glass slide. The inlet end of the separation capillary was positioned in the droplet, and the emitter tip was positioned ~2 mm from the MS inlet. A +2 kV spray voltage was applied on the MS for 1 or 2 min to perform cell injections. Prior to injection, a plug of 75% formic acid (FA) was loaded into the capillary by applying 5 psi for 20 s. Following the injection, the capillary was dipped first into 75% FA and then into the water to clean the capillary and prevent contamination of the lysis buffer and BGE. A plug of 75% FA was injected after the cells to lyse cells on the capillary by applying 5 psi for 27 s. Following the second injection of 75% FA, a 3 min wait period was included before beginning the analysis to enable FA diffusion through the sample plug for cell lysis.
Hydrodynamic Loading of Single HeLa Cells.
Single cells were loaded onto the capillary manually by applying a low flow generated by lowering the sprayer tip end of the capillary by ~45 cm relative to the capillary inlet. The injection was monitored under the microscope at 10× magnification. To avoid the introduction of air bubbles into the inlet of the capillary before sample loading, the outlet of the capillary was lifted ~35 cm above the capillary inlet. The capillary was filled first with BGE before the injection, and the inlet end was immobilized on a glass slide and immersed in a droplet of 30 μL of 200 mM ammonium acetate. 10 μL of HeLa cell suspension (1.5 × 106 cells/mL) in 200 mM ammonium acetate was added to the droplet and agitated to distribute cells evenly throughout (Figure 1C). Single cells near the inlet could be directed into the capillary with the low flow generated by the hydrodynamic pressure difference between the two ends of the capillary (a video clip of a single cell loading process was included in Supporting Information). For MS analysis, the single cells were sandwiched between two plugs of 75% FA manually loaded in the same manner by lowering the sprayer tip end of the capillary by ~45 cm with respect to the capillary inlet for 30 s. The outlet of the capillary was kept at the same height as the capillary inlet to avoid the possible introduction of air bubbles into the capillary when switching between injections of cell suspension and 75% FA. After the injections of the plugs of lysis buffer and selected individual cells, the capillary cartridge was immediately placed into the CESI 8000 instrument (SCIEX, Brea, CA). To avoid the introduction of air bubbles, care was taken to ensure the inlet and outlet ends of the separation capillary were at the same height during transfer to minimize flow within the capillary. The sprayer tip of the capillary was held at a higher elevation (~25–35 cm) above the inlet for ~10 s before immersing the capillary inlet into the background electrolyte to expel any bubbles possibly introduced during the brief cartridge transfer. For fluorescent imaging, cells were stained with 0.005 mg/mL acridine orange, and 75% FA was not injected onto the capillary prior to cell loading to preserve cell integrity and morphology for visualization.
Figure 1.
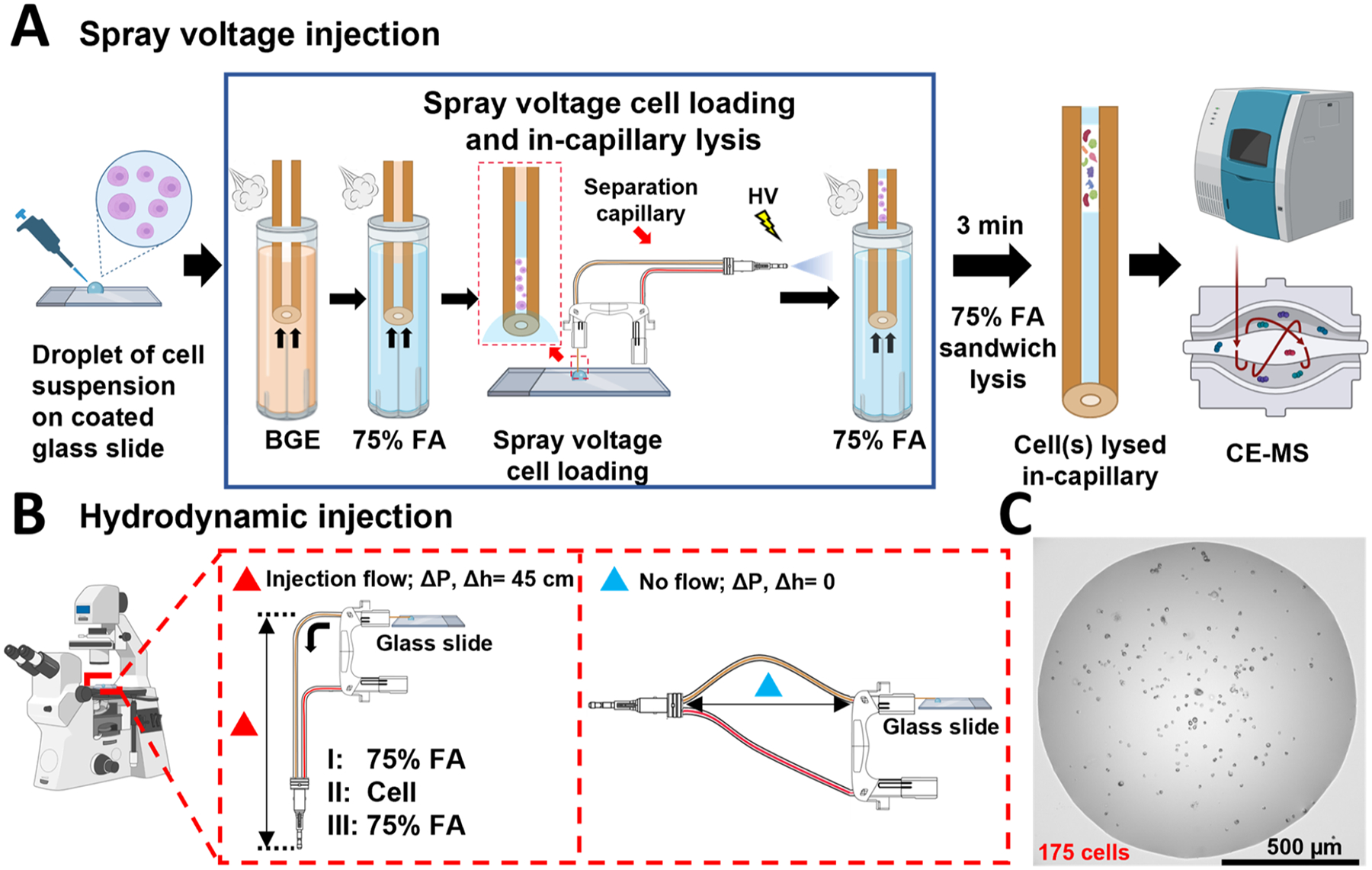
Overview of intact cell injection workflows and cell suspension droplets. (A) Schematic of spray voltage injection workflow. The separation capillary is prepared for cell lysis and CE-MS/MS analysis by filling in first with BGE and then with a plug of lysis buffer. The capillary inlet is positioned in cell suspension (500 nL) pipetted on a glass slide (the zoomed capillary inlet in the droplet is shown in the red-frame insert), and electrospray voltage is applied at the MS inlet, drawing cells into the capillary. (B) Schematic of manual injection of single cells with hydrodynamic loading using height difference between the capillary inlet and outlet to generate flow and draw in the cell. In the injection flow panel, a 75% FA plug, (I) a single cell, (II) and another 75% FA plug (III) were loaded in this order for single-cell injection and lysis. (C) Representative image cell suspension droplet used for single-cell loading.
Sheathless CE-ESI-MS/MS.
Separation was performed using a CESI 8000 (SCIEX, Brea, CA) on a bare fused silica capillary (90 cm, 30 μm ID, 150 μm OD) with a sheathless electrospray interface (SCIEX OptiMS cartridge). A 3 min ramp was applied to reach the separation voltage of +20 kV in normal polarity. MS acquisition was started after the initial 3 min CE voltage ramp up and ended at the start of a 5 min CE voltage ramp down. The BGE used was 40% acetic acid, and the conductive line was filled with 10% acetic acid. A supplemental pressure of 1 psi was applied throughout the 75 min run time. The capillary was interfaced with a Nanospray Flex ion source mounted at the front end of an Orbitrap Fusion Lumos Tribrid mass spectrometer (both Thermo Fisher Scientific).
Mass Spectrometry Settings.
The mass spectrometer was operated in positive ionization mode with a spray voltage of +1.3 kV during data collection. The total data collection time was 70 min. The mass spectrometer was operated in intact protein mode (low pressure). For MS1, full scan data were collected at 120,000 resolution (at 200 m/z) and 10 μscans were averaged. An in-source CID of 15 V was applied. Data-dependent MS2 scans were analyzed in the Orbitrap with 60,000 resolution (at 200 m/z), while 4 μscans were averaged.
Data Analysis.
All raw files were analyzed using TopPIC Suite22 and searched against the UniProtKB/Swiss-Prot human database (downloaded on 3/30/2022, containing 20,378 sequences). A decoy database with a 1% false discovery rate (FDR) was used for spectrum and proteoform level cutoff. An additional e-value cutoff of 0.01 was applied to the results. The numbers of protein and proteoform identifications for each sample with 1% FDR alone and with the additional two-level filtering are reported in Table S1. Additional search parameters are detailed in the Supporting Information (Tables S2 and S3).
The match-between-runs (MBR) approach was employed semimanually for detected and identified CE-MS features across all single-cell files. Precursor masses reported from TopPIC proteoform identifications were used to extract peaks from all replicate files using Progenesis QI for Proteomics software v 3.0 (Nonlinear Dynamics). Proteoforms were used as vectors to align the migration time of all samples. Peak picking was performed automatically by the software, using a signal intensity threshold of 1000 and a maximum charge state of 35. After isotopic features were selected, all features that matched proteoform identifications were manually inspected for further validation. If no isotopic feature envelope within the charge distribution corresponding to the TopPIC identification was observed in a given sample, the corresponding proteoform was labeled as a missing value. Proteoform raw abundances reported by Progenesis QI were used for the following analyses.
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE23 partner repository with the data set identifier PXD035339. Additional experimental details about materials, reagents, methods, mass spectrometry parameters, and data analysis are provided in the Supporting Information.
RESULTS AND DISCUSSION
Assessment of Spray Injection of Intact HeLa Cells.
First, we assessed the capability of injecting intact cells from a small volume of cell suspension using electrospray voltage to generate a low flow through the separation capillary toward the MS inlet. Previously, an electrospray capillary device has been described for reproducible injection of ultra-low volumes21 that has enabled single-cell level metabolite analysis by CE-MS in plant (onion) cells.24 Here, we employ a similar approach to a sheathless CE-MS capillary interface by applying spray voltage for different time periods to inject cultured mammalian cancer cells onto the capillary from a 500 nanoliter droplet of cell suspension (Figure 1A). Briefly, an electrospray voltage of +2 kV was applied for 1 or 2 min with the inlet of the separation capillary positioned at the bottom of the cell suspension droplet and the sheathless sprayer tip positioned ~2 mm from the MS inlet. Following the injection, the capillary was removed from the instrument and examined under the microscope at 10× magnification to count cells (Figure 2A,B). For 1 min injection, cell suspension plugs appeared to load ~1–2 mm of the capillary, and all injected cells could be identified close to the capillary inlet (Figure 2A). In the process of removing the capillary cartridge from the instrument to the microscope, efforts were made to prevent any additional gravity-driven flow within the capillary that would move cells further into the capillary. However, logistically, this is a phenomenon that could not be entirely avoided with the current setup for cell visualization, leading to some variability in the lengths of injections imaged. The observed 2 min injection cell suspension plugs were longer, and cells traveled approximately twice further into the capillary. Images shown in Figure 2B include instances where cells clumped together and could all be visualized in a 1 mm section of the capillary. Cells were stained with acridine orange, which fluoresces when bound to nucleic acids, in order to obtain fluorescent images and match them with bright-field images. These images were overlaid to validate that cells were injected and could be effectively and unequivocally detected, counted, and imaged using both fluorescence and bright-field microscopy inside the capillary (Figure 2C). Since the fluorescent dye would interfere with MS analyses, cell counts were verified for non-labeled cells using the same injection conditions and only bright-field microscopy for CE-MS analyses. Replicate injections (N = 5) at 1 and 2 min injections showed 2 ± 1 and 7 ± 2 cells injected, respectively (Figure 2D), demonstrating the level of injection reproducibility.
Figure 2.
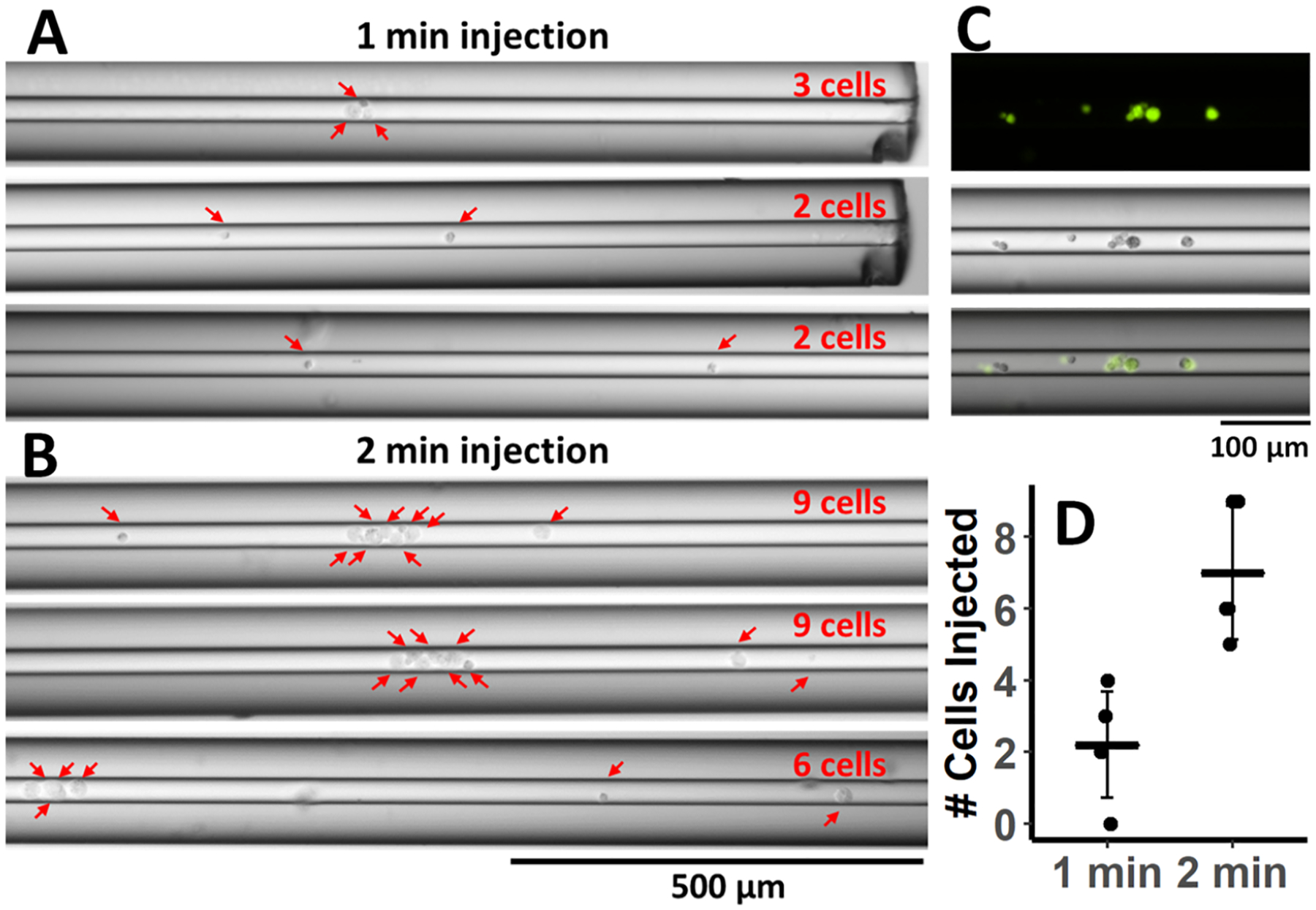
Assessment of spray voltage injection of intact cells. Microscope images at 10× magnification of cells inside the capillary from (A) 1 min (N = 3) and (B) 2 min injection (N = 3). Red arrows indicate individual cells loaded inside the capillary. (C) Fluorescent (top) and bright-field images (middle) of cells stained with acridine orange inside the capillary. Overlaid images are shown at the bottom. (D) Cells counted in the capillary with 1 and 2 min spray injections. Mean counts ± STDEV are displayed.
Assessment of Hydrodynamic Injection of Single HeLa Cells and On-capillary Lysis.
For the analysis of single HeLa cells, intact single cells were manually injected from a droplet of cell suspension (Figure 1C) by lowering the spray tip end of the capillary by ~45 cm with respect to the inlet to generate a flow in the capillary (estimated to be ~140 pL/s) (Figure 1B), as described in the experimental section. The process of the single-cell injection was monitored using the microscope to ensure only a single cell was loaded onto the separation capillary. To further demonstrate this process and validate that a single intact cell was loaded onto the capillary, cells were stained with a fluorescent dye prior to manual injection (Figure 3A). On most capillaries, patches of fluorescence were observed at the inlet of the capillary before cell injections were performed and could not be eliminated even after extensive washing of the capillary inner wall and outer surfaces. These patches at the inlet are likely due to imperfections in the glass surface and do not correspond to cells or cell debris.
Figure 3.

Demonstrating single-cell injection and cell lysis inside the capillary. (A) Fluorescent (top) and bright-field (middle) images of a representative single HeLa cell injected into the capillary by hydrodynamic pressure. Overlaid images are shown at the bottom. (B) Time course of lysis a single cell inside the capillary by a single plug of 75% FA. (C) Zoomed in images of the cell lysing by 75% FA at different time points.
Following injection, 75% FA was loaded onto the capillary by the same mechanism, and images were taken at 0, 20, 30, and 60 s to observe the cell lysing in the capillary (Figure 3B,C). A single plug of FA was used to make the visualization at a manageable time scale possible. With a cell or cells sandwiched between two plugs of 75% FA, the cells lost their integrity much faster, which made the visualization of the process challenging without using a specialized high-speed camera. Similarly, on-capillary lysis efficiency was observed for ESI-voltage driven injections of several cells, which was challenging to capture in high-resolution images with the available equipment. To visualize cells with fluorescence, a section of polyimide coating ~0.8 cm from the end of the capillary inlet was burned and removed to expose just the bare fused silica. This also had the benefit of reducing cell adsorption to the capillary inlet.
TDP CE-MS/MS Analysis with Spray Injection of Intact HeLa Cells.
Next, we attempted to lyse the injected non-labeled cultured cancer cells on the capillary to perform TDP analysis by CE-MS/MS. To achieve efficient cell lysis, it was determined that a plug of 75% FA before and after the injected cells was needed. Additionally, a waiting period of 3 min preceding CE-MS/MS analysis was used to ensure that the FA could diffuse through the sample plug to conduct cell lysis. The current instability in CE was noted when cell lysis was insufficient. Replicate analyses of 1 or 2 min spray injections with +2 kV (N = 4) demonstrated averages of 29 ± 10 proteins and 43 ± 16 proteoforms identified from 1 min injection and 51 ± 13 proteins and 74 ± 20 proteoforms identified from the 2 min injection. The ammonium acetate cell suspension buffer was also collected from the same cell suspensions and analyzed to assess the extracellular protein content that may be present from cells ruptured in the suspension and other contaminants. Briefly, this control was collected by gently centrifuging the cells at 500g for 5 min to form a pellet and removing the supernatant. The results show that a very low contribution of the protein signal could be attributed to the supernatant, indicating that cell lysis and protein secretion are minimal during the time required to perform the experiments, and increasing confidence that the observed proteins were originated from the injected intact cells (Figures 4A, and S1). Blank injections of 200 mM ammonium acetate were also analyzed to determine a minimal level of carryover that occurred between runs, which emphasized the benefits of the open tubular CE separation modality and the efficiency of capillary washes and regeneration between CE-MS runs (Figure 4A).
Figure 4.
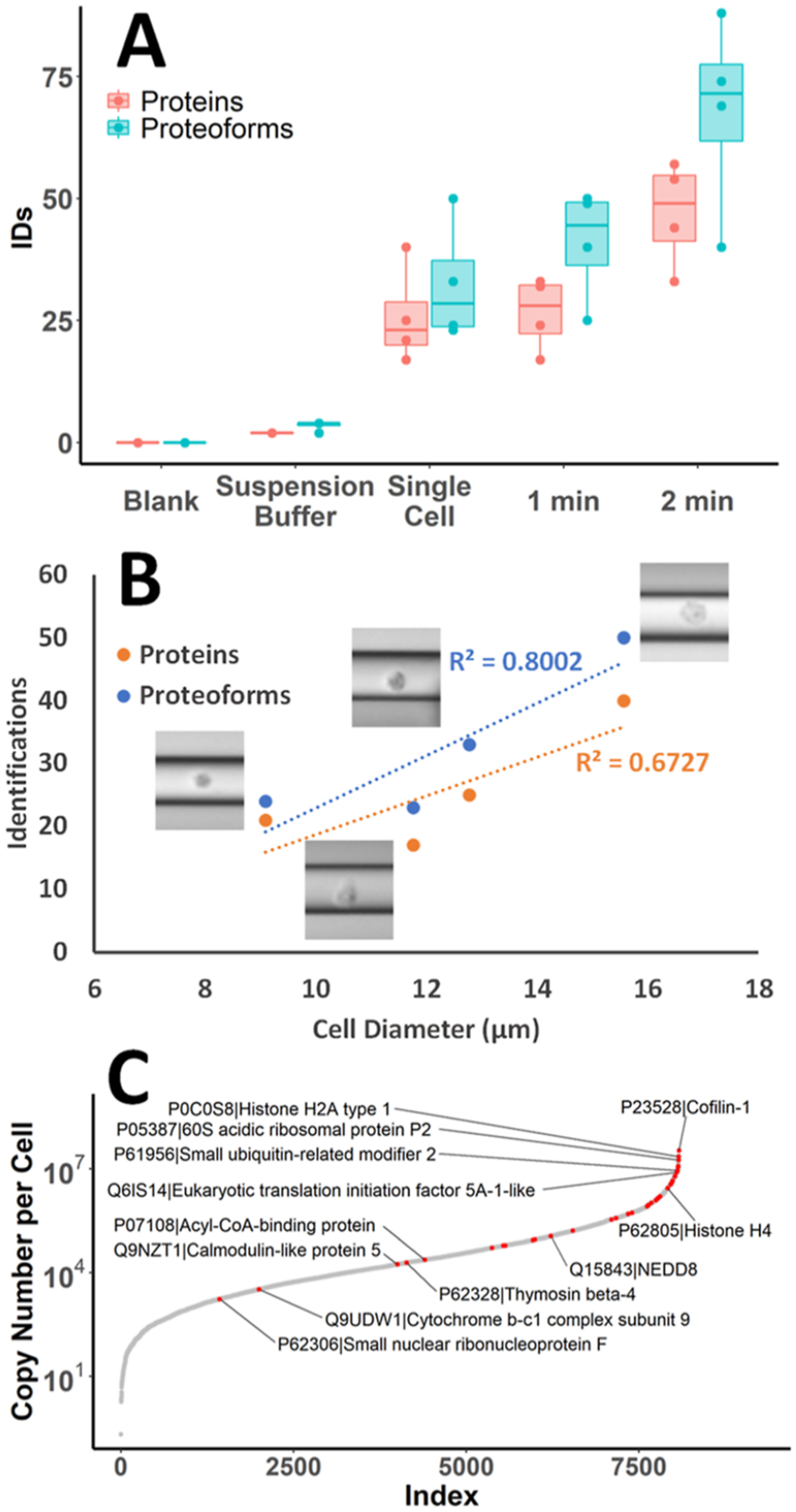
Protein and proteoform identifications from single-cell and spray voltage injections analyzed by CE-MS/MS. (A) Boxplots show the range and median of protein and proteoform identifications for each injection mode tested, as well as blank sample buffer and suspension buffer alone (N = 4). Dots indicate values from individual samples. (B) Single HeLa cell protein and proteoform identifications plotted against measured cell diameters. Linear trendlines demonstrate the correlation between protein and proteoform identifications with cell size. (C) S-curve of protein copy numbers per cell from a previously reported deep proteomic study of HeLa,29 with proteins identified from a single HeLa cell TDP in this study mapped in red. Selected proteins of interest are labeled.
TDP CE-MS/MS Analysis of Single HeLa Cells.
Single HeLa cells were analyzed by CE-MS/MS after manual hydrodynamic loading as described in previous sections. Protein and proteoform identifications from single HeLa cells (N = 4) ranged from 17 to 40 and 24 to 50 and averaged 26 ± 10 and 33 ± 12, respectively (Figure 4A). The data were searched with 1% FDR; however, to increase confidence in reported identifications, proteoforms with e-values ≥0.01 were also filtered out for all downstream analyses (Table S1). The distribution of the remaining e-values had a median value of 3.75 × 10−11. Approximate cell diameter values could be determined from the microscopy images of cells inside the capillary (Figure S2). HeLa cells have been reported to have average diameters of ~17 μm;25 however, we observed smaller diameters (average ~12 μm) and a more spherical shape for HeLa cells cultured in suspension in our experiments.26 Images were taken immediately following successful cell loading to ensure an accurate representation of cell size since the cells were observed to swell shortly after entering the capillary due to the start of lysis in the 75% FA loaded into the capillary. Based on our initial experiments, the number of protein and proteoform identifications could be correlated with a cell diameter, demonstrating R-squared values of 0.80 and 0.67, respectively (Figure 4B). This result could be expected due to a correlation between cell size and protein content.
The HeLa proteome has been extensively studied with MS-based proteomic techniques. Deep proteome coverage has enabled comprehensive documentation of HeLa cell protein abundances.27,28 Nagaraj et al. reported previously that the most abundant 600 proteins comprise ~75% of the HeLa cell proteome mass.29 Here, we mapped our protein identifications from TDP analysis of a single HeLa cell to protein copy numbers per cell for 8078 proteins characterized and quantified in that study (Figure 4C). Out of 56 proteins identified from single HeLa cell experiments (N = 4), 40 were matched to copy numbers from this list; however, 16 proteins identified in this work were not found to be quantitatively assessed in the earlier report. Of the proteins where copy numbers per cell could be matched, ~60% were in the top 600 most abundant proteins, including 9 histone proteins and 3 ribosomal proteins. Other proteins of interest that were identified include ubiquitin-like proteins (Ubls), NEDD8, and SUMO2, which play an important role in the regulation of multiple biological processes, such as proteasome-mediated degradation of proteins.30 Even a few proteins with lower copy numbers per cell could be identified from a single cell. Among these lower abundance proteins identified were small nuclear ribonucleoprotein F (snRNP), a core component of spliceosomal assembly, and subunit 9 of the cytochrome b-c1 complex, a component of the mitochondrial respiratory chain. Several modifications, such as oxidation, phosphorylation, dimethylation, and butyrylation, could also be observed in the proteoforms identified from single HeLa cells. Many proteoforms were observed with the common modification of N-terminal methionine truncation and acetylation. Unique proteoforms of histone 4 with modification mass shifts on the fragment containing lysine 17 consistent with butyrylation (+70 Da) or dimethylation (+28 Da) were confidently identified (Figure 5A–C). These modifications on lysine 17 are listed on UniProt as having been confirmed by other studies.
Figure 5.
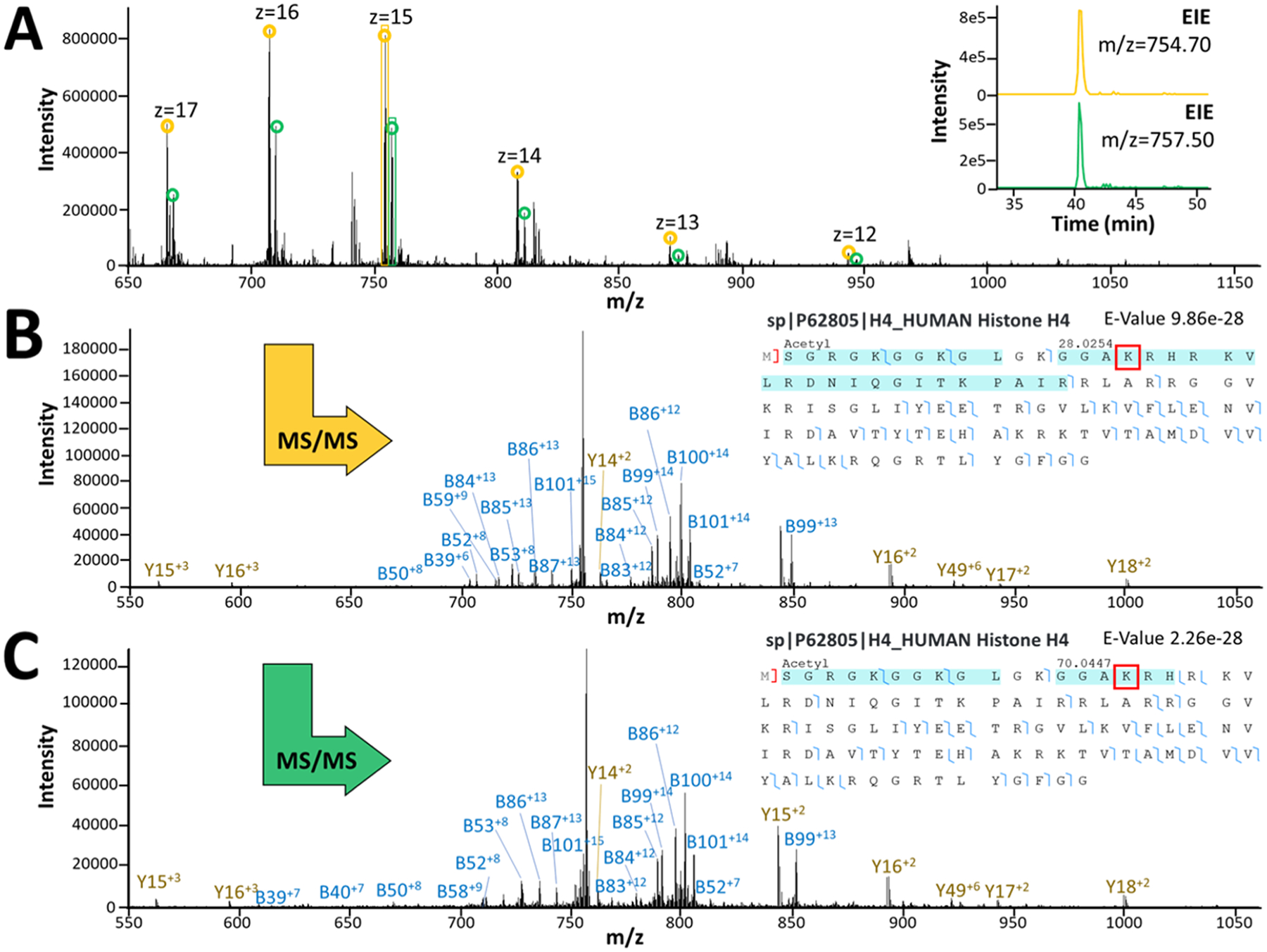
Representative MS1 and MS2 spectra from confidently identified histone proteoforms with different PTMs. (A) MS1 spectrum for co-migrating histone 4 proteoforms with dimethyl (yellow) and butyryl (green) PTM assignments. The top insert shows the extracted peaks for each proteoform. (B) MS2 spectrum with mapped fragments and fragmentation coverage indicating dimethylated (+28 Da) histone 4, identified in TDP analysis of a single HeLa cell. Putative modified lysine residue indicated with a red box. (C) MS2 spectrum with mapped fragments and fragmentation coverage indicating butyrylated (+70 Da) histone 4 identified in TDP analysis of a single HeLa cell. Putative modified lysine residue indicated with a red box.
TDP CE-MS/MS Analysis of Single Monocytes.
To demonstrate the utility of our TDP approach for assessing heterogeneity between single cells, we investigated an additional cell line isolated from human monocytic leukemia, THP-1. SCP analysis was performed on THP-1 cells as described previously for HeLa cells. An average of 16 ± 9 proteins and 18 ± 9 proteoforms were identified in single THP-1 cells (N = 3, see single THP-1 cell protein and proteoform identification results in Supporting Information), which is comparatively fewer than the identifications in HeLa cells (N = 4, see single HeLa cell protein and proteoform identification results in Supporting Information). Monocytes are smaller than HeLa cells and have been reported to have an average diameter of 8.13 μm,25 which may explain the lower number of identifications from THP-1 cells.
MBRs for HeLa and THP-1 Single-cell Analyses.
Next, we investigated an MBR approach, which is commonly used in bottom-up proteomics approaches, to improve identification rates, enhance quantification, and fill in missing values for multiple comparative analyses. Since our current top-down method requires relatively long ion fill times and scan averaging in the mass spectrometer, particularly for low sample amounts, stochastic sampling presents a challenge for the reproducibility of proteoform identifications across replicates. Our data indicate poor overlap (10%) of proteoform identifications between single HeLa cell analyses (Figure 6B). MBR has become a commonly used strategy31,32 for addressing the problem of missing data points in bottom-up proteomics that first aligns chromatograms by retention time and then matches MS1 scans within a narrow mass and retention time window to assign identifications from one run to others with missing or insufficient MS/MS information. A similar algorithm for TDPs has not yet been made widely available, according to our knowledge. As such, we used Progenesis QI for Proteomics software to pick peaks and semimanually perform MBR for proteoforms with at least one confident proteoform-spectrum match from all HeLa and THP-1 single-cell CE-MS analysis files. For the single-cell data collected in this study, migration times between runs varied greatly (up to 8 min) due to the manual nature of the injection and the time differences related to aspirating individual cells in different experiments. Volumes of sample buffer injected along with the cell, as well as the distance that the cell traveled into the capillary during the injection, were difficult to control for and consequently led to expected differences in the observed migration times from run to run. Progenesis QI for Proteomics software was used to align migration times across all samples before peak picking (Figure S3). Migration time alignment largely alleviated the issues related to the variability in injection volume in the analysis of individual cells. The list of precursors with confident proteoform identifications in TopPIC was matched with the peaks picked by the software and manually validated to confirm the correct proteoform assignment based on the examination of isotope and charge distributions. Figure S3 demonstrates the validation of MS1 features detected in all single-cell analyses and confidently matched by MS2 in one analysis for a variant of prothymosin alpha with N-terminal methionine excision and acetylation containing a mass shift of −129 Da. This mass shift is consistent with a loss of a glutamic acid residue indicated by the fragmentation data to be at position 40, which corresponds to the previously reported isoform 2 of the protein, which arises from alternative splicing.33 In some cases, after migration time alignment, precursor masses for several proteoforms were indistinguishable using MS1 alone. In these cases, proteoforms were grouped into clusters of similar features. Using the described MBR approach, the efficiency of protein and proteoform identifications was improved for all HeLa and THP-1 single-cell analyses (Figures 6A and S4), and the overlap in proteoform identifications between cell types was increased from 18 to 48 proteoforms (Figure 6C). The overlap for HeLa cell analyses was also improved, demonstrating an increase from 10 to 49% after applying MBR (Figure 6B).
Figure 6.
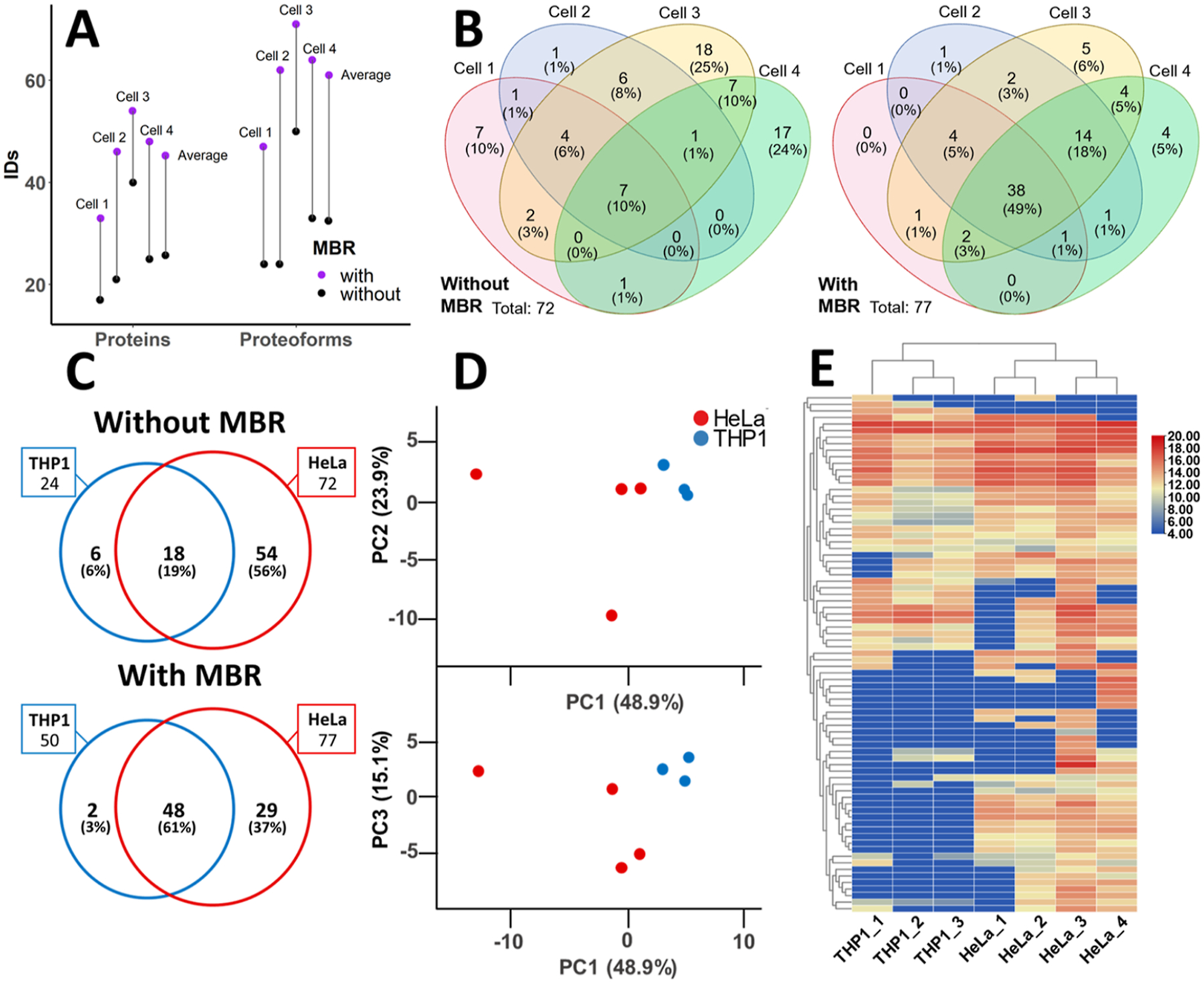
Assessment of MBR and comparison of HeLa and THP-1 single cell proteoform identifications. (A) Protein and proteoform identifications with and without MBR are shown for each single HeLa cell analyzed. The average number of identification with and without MBR is also shown. (B) Proteoform overlap between HeLa single cells shown for identifications without and with MBR. (C) Proteoform overlap between HeLa (N = 4) and THP-1 (N = 3) cells without and with MBR. (D) Principal components PC1 vs PC2 and PC1 vs PC3 are shown for PCA analysis of 79 proteoform abundances determined by MBR between HeLa and THP-1 cells. (N = 7 data points). (E) Heat map of proteoforms identified in single THP-1 (N = 3) and HeLa (N = 4) cells. Unsupervised hierarchical clustering was performed based on Euclidean distance using average linkage. Proteoform peak abundances are log2 transformed. Missing values for PCA and heat map clustering were imputed as 10% of the minimum observed value.
Gene ontology (GO) enrichment analysis of proteins identified in HeLa and THP-1 single cells after MBR showed a high representation of nuclear proteins identified in both cell types, with 63 and 61% of all proteins identified in HeLa and THP-1 cells, respectively, annotated to the nucleus cellular component. Cytoplasm- and cytosol-related proteins comprised the following identified cell component GO term enriched in the conducted SC-TDP analyses. At this level of proteome coverage from single cells, the similarity in protein identifications across these two cell types necessitates the investigation of proteoform identifications, including their corresponding quantitative values, to determine differences in molecular phenotypes of the examined cell types.
Comparing Single-cell TDP Results from Two Different Cell Lines.
Principal component analysis (PCA) was performed on the proteoform abundances for all 79 proteoform identifications confirmed with manual MBR across the HeLa and THP-1 single-cell data sets (Figure 6D). Principal components 1, 2, and 3 explain 88% of the variance between samples. PC2 (23.9%) and PC3 (15.1%) were plotted against PC1 (48.9%) to demonstrate clustering between different cell types. The HeLa cells showed a larger variation than the THP-1 cells, which clustered closely. It appears that the variation in PC1 is primarily driven by HeLa cell number3, which was the largest of the cells measured and had the highest number of identified proteoforms. Some clustering by cell type can be observed when examining PC3, indicating that a certain variation in proteoform abundances specific to cell type is present in the data. Unsupervised hierarchical clustering was also conducted for proteoform abundances to demonstrate the clustering of THP-1 cells and HeLa cells (Figure 6E). Tighter clustering was observed for the two smaller HeLa cells and the two larger HeLa cells analyzed, indicating a significant contribution from total protein content. However, overall clustering based on cell type was also observed.
CONCLUSIONS
Here we have demonstrated two techniques for injection of individual and small groups of <10 intact mammalian cancer cells, subsequent on-capillary cell lysis, and TDP analysis by ultrasensitive CE-MS/MS. Spray injection of 1–9 cells from a nL-scale droplet on a glass microslide presents a promising, relatively low labor-intensive method for the introduction of limited cell or tissue samples for CE-MS/MS analysis. Alternatively, for greater control of cell selection, hydrodynamic cell loading may be performed manually under the microscope. Automation of injection will be an important future advancement to enable higher-throughput sampling and collection of larger data sets. The developed method allowed the highest identification rates for single-cell analysis and analysis of small populations of injected cells that have been reported, enabling up to 40 unique proteins and 50 unique proteoform identifications, including several PTMs, from a single HeLa cell. The number of identifications could be improved by employing an MBR strategy to reach 48 protein and 64 proteoform identifications from a single HeLa cell. The number of protein and proteoform identifications from single cells was variable and showed a positive correlation with cell size. We suggest that the field of single-cell TDPs (SC-TDP) could alternatively be referred to as TD-SCP, depending on the emphasis and the preference of the study.
Simple, clean lysis with FA enables direct TDP analysis without introducing high concentrations of salts or detergents that can interfere with CE separation and MS analysis. However, lysis conditions with alternative buffers, including MS-friendly detergents or lower concentrations of chaotropes not studied in this work, may enable higher efficiency cell lysis without causing a detrimental effect on the analysis.
We have also shown that MBR can increase the proteoform identification rate in TDP analysis. Manual confirmation of CE-MS peaks as well as charge and isotope distribution envelopes matched between runs is time-consuming and prohibitive of large-scale implementation. Further development of TDP data processing tools to include MBR algorithms that incorporate verification of correct isotopic and charge distributions for the precursor ions and an assessment of FDR would dramatically increase the throughput and reliability of MBR in TDP of higher complexity data sets. This is especially significant for SCP because the ability to perform meaningful analysis of heterogeneity at the single-cell level relies on the ability to obtain many high-confidence measurements.
The generated SC-TDP CE-MS data acquired for a few selected cultured cells of two different cancer cell lines allowed us to distinguish the cells using PCA and unsupervised hierarchical clustering based on their quantitative proteoform profiles. These pilot results pose a promise for the developed CE-MS-based SC-TDP approach to detect differences in molecular phenotypes at the intact protein and proteoform levels for individual cells of various types as well as of various activation, differentiation, cell cycle, or health/pathology states. Although the proteome coverage presented here is rather modest, we believe that the shown initial results of our proof-of-concept experiments demonstrate the first steps toward enabling informative and deep proteome-scale CE-MS-based TDP characterization of individual cells, organelles, and other μm- and nm-scale biological structures. Further advances in sample preparation, cell procurement and injection, separation, interfacing of the separation to MS (e.g., using FAIMS34 or other ion mobility spectrometry approaches), MS data acquisition, and data analysis will be needed to make SC-TDP more thorough and informative, which, we believe, will enable new breakthroughs in biological and clinical studies.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health under the award numbers R01CA218500 (A.R.I.) and R35GM136421 (A.R.I.). We acknowledge the team of Thermo Fisher Scientific for its support through a technology alliance partnership program. The authors thank SCIEX for providing CESI capillaries used in this study and insightful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.2c03045.
Experimental methods, data analysis parameters, acquired data, and cell images (PDF)
Single HeLa cell protein and proteoform identification results (XLSX)
Single THP-1 cell protein and proteoform identification results (XLSX)
Single-cell loading process (MP4)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.2c03045
The authors declare no competing financial interest.
Contributor Information
Kendall R. Johnson, Barnett Institute of Chemical and Biological Analysis, Department of Chemistry and Chemical Biology, Northeastern University, Boston, Massachusetts 02115, United States.
Yunfan Gao, Barnett Institute of Chemical and Biological Analysis, Department of Chemistry and Chemical Biology, Northeastern University, Boston, Massachusetts 02115, United States.
Michal Greguš, Barnett Institute of Chemical and Biological Analysis, Department of Chemistry and Chemical Biology, Northeastern University, Boston, Massachusetts 02115, United States.
Alexander R. Ivanov, Barnett Institute of Chemical and Biological Analysis, Department of Chemistry and Chemical Biology, Northeastern University, Boston, Massachusetts 02115, United States
REFERENCES
- (1).Kelly RT Mol. Cell. Proteomics 2020, 19, 1739–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Budnik B; Levy E; Harmange G; Slavov N Genome Biol. 2018, 19, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhu Y; Clair G; Chrisler WB; Shen Y; Zhao R; Shukla AK; Moore RJ; Misra RS; Pryhuber GS; Smith RD; Ansong C; Kelly RT Angew. Chem., Int. Ed. Engl 2018, 57, 12370–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ctortecka C; Stejskal K; Krššáková G; Mendjan S; Mechtler K Anal. Chem 2022, 94, 2434–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Li S; Plouffe BD; Belov AM; Ray S; Wang X; Murthy SK; Karger BL; Ivanov AR Mol. Cell. Proteomics 2015, 14, 1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cong Y; Motamedchaboki K; Misal SA; Liang Y; Guise AJ; Truong T; Huguet R; Plowey ED; Zhu Y; Lopez-Ferrer D; Kelly RT Chem. Sci 2021, 12, 1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Specht H; Emmott E; Petelski AA; Huffman RG; Perlman DH; Serra M; Kharchenko P; Koller A; Slavov N Genome Biol. 2021, 22, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Johnson KR; Greguš M; Kostas JC; Ivanov AR Anal. Chem 2022, 94, 704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Smith LM; Kelleher NL; Kelleher NL Nat. Methods 2013, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Edwards RL; Griffiths P; Bunch J; Cooper HJ J. Am. Soc. Mass Spectrom 2012, 23, 1921–1930. [DOI] [PubMed] [Google Scholar]
- (11).Zhang J; Guy MJ; Norman HS; Chen YC; Xu Q; Dong X; Guner H; Wang S; Kohmoto T; Young KH; Moss RL; Ge YJ Proteome Res. 2011, 10, 4054–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gregorich ZR; Ge Y Proteomics 2014, 14, 1195–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dang X; Scotcher J; Wu S; Chu RK; Tolić N; Ntai I; Thomas PM; Fellers RT; Early BP; Zheng Y; Durbin KR; LeDuc RD; Wolff JJ; Thompson CJ; Pan J; Han J; Shaw JB; Salisbury JP; Easterling M; Borchers CH; Brodbelt JS; Agar JN; Paša-Tolić L; Kelleher NL; Young NL Proteomics 2014, 14, 1130–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).McCool EN; Lubeckyj RA; Shen X; Chen D; Kou Q; Liu X; Sun L Anal. Chem 2018, 90, 5529–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yu D; Wang Z; Cupp-Sutton KA; Liu X; Wu SJ Am. Soc. Mass Spectrom 2019, 30, 2502–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Drown BS; Jooß K; Melani RD; Lloyd-Jones C; Camarillo JM; Kelleher NL J. Proteome Res 2022, 21, 1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhou M; Uwugiaren N; Williams SM; Moore RJ; Zhao R; Goodlett D; Dapic I; Paša-Tolić L; Zhu Y Anal. Chem 2020, 92, 7087–7095. [DOI] [PubMed] [Google Scholar]
- (18).Václavek T; Foret F Electrophoresis 2022, DOI: 10.1002/elps.202100379. [DOI] [PubMed] [Google Scholar]
- (19).Valaskovic GA; Kelleher NL; McLafferty FW Science 1996, 273, 1199–1202. [DOI] [PubMed] [Google Scholar]
- (20).Hofstadler SA; Swanek FD; Gale DC; Ewing AG; Smith RD Anal. Chem 1995, 67, 1477–1480. [DOI] [PubMed] [Google Scholar]
- (21).Huang L; Wang Z; Cupp-Sutton KA; Smith K; Wu S Anal. Chem 2020, 92, 640–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kou Q; Xun L; Liu X Bioinformatics 2016, 32, 3495–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; Pérez E; Uszkoreit J; Pfeuffer J; Sachsenberg T; Yılmaz S; Tiwary S; Cox J; Audain E; Walzer M; Jarnuczak AF; Ternent T; Brazma A; Vizcaíno JA Nucleic Acids Res. 2019, 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Huang L; Fang M; Cupp-Sutton KA; Wang Z; Smith K; Wu S Anal. Chem 2021, 93, 4479–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Milo R; Jorgensen P; Moran U; Weber G; Springer M Nucleic Acids Res. 2010, 38, D750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Abdeen SH; Abdeen AM; El-Enshasy HA; Shereef AA J. Biol. Sci 2011, 11, 124–134. [Google Scholar]
- (27).Zeiler M; Straube WL; Lundberg E; Uhlen M; Mann M Mol. Cell. Proteomics 2012, 11, O111009613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bekker-Jensen DB; Kelstrup CD; Batth TS; Larsen SC; Haldrup C; Bramsen JB; Sørensen KD; Høyer S; Ørntoft TF; Andersen CL; Nielsen ML; Olsen JV Cell Syst. 2017, 4, 587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Nagaraj N; Wisniewski JR; Geiger T; Cox J; Kircher M; Kelso J; Pääbo S; Mann M Mol. Syst. Biol 2011, 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hochstrasser M Nature 2009, 458, 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tyanova S; Temu T; Cox J Nat. Protoc 2016, 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- (32).Yu F; Haynes SE; Nesvizhskii AI Mol. Cell. Proteomics 2021, 20, 100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Eschenfeldt WH; Berger SL Proc. Natl. Acad. Sci. U.S.A 1986, 83, 9403–9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Greguš M; Kostas JC; Ray S; Abbatiello SE; Ivanov AR Anal. Chem 2020, 92, 14702–14712. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.