

Abstract
Acinetobacter is present in the livestock environment, but little is known about their antibiotic resistance and pathogenic species in the farm groundwater. Here we investigated antibiotic resistance of Acinetobacter in the swine farm groundwater (JZPG) and residential groundwater (JZG) of a swine farming village, in comparison to a nearby (3.5 km) non-farming village (WTG) using metagenomic and culture-based approaches. Results showed that the abundance of antibiotic resistome in some JZG and all JZPG (~3.4 copies/16S rRNA gene) was higher than that in WTG (~0.7 copies/16S rRNA gene), indicating the influence of farming activities on both groundwater types. Acinetobacter accounted for ~95.7% of the bacteria in JZG and JZPG, but only ~8.0% in WTG. They were potential hosts of ~95.6% of the resistome in farm affected groundwater, which includes 99 ARG subtypes against 23 antibiotic classes. These ARGs were associated with diverse intrinsic and acquired resistance mechanisms, and the predominant ARGs were tetracyclines and fluoroquinolones resistance genes. Metagenomic binning analysis elucidated that non-baumannii Acinetobacter including A. oleivorans, A. beijerinckii, A. seifertii, A. bereziniae and A. modestus might pose environmental risks because of multidrug resistance, pathogenicity and massive existence in the groundwater. Antibiotic susceptibility tests showed that the isolated strains were resistant to multiple antibiotics including sulfamethoxazole (resistance ratio: 96.2%), levofloxacin (42.5%), gatifloxacin (39.0%), ciprofloxacin (32.6%), tetracycline (32.0%), doxycycline (29.0%) and ampicillin (12.0%) as well as last-resort polymyxin B (31.7%), colistin (24.1%) and tigecycline (4.1%). The findings highlight potential prevalence of groundwater-borne antibiotic-resistant pathogenic Acinetobacter in the livestock environment.
Subject terms: Environmental sciences, Water microbiology
Introduction
The massive use of antibiotics has exacerbated antimicrobial resistance (AMR) prevalence in the environment [1]. It is estimated that antibiotic-resistant bacteria (ARB) caused 1.27 million deaths in 2019, and the mortality is predicted to reach 10 million by 2050 [1, 2]. Given this great threat, AMR has been designated as an urgent public health threat by World Health Organization (WHO) [3], and is highlighted as one of the top six emerging environmental issues by United Nations Environment Programme [4, 5]. Livestock environments are hotspots for AMR development, transmission and prevalence [6]. ARB and antibiotic resistance genes (ARGs) have been detected prevalent in animal wastes, waste management systems [7–9] and the farm affected environments including water [10], agricultural soil [7], atmosphere [11] and food chains [12]. Under the “One-Health” concept, they will pose unpredictable risks to the public health [13].
AMR has become a growing concern in groundwater systems [14]. In rural China, groundwater is generally used for animal breeding processes. However, seepage of animal wastes will contaminate the surrounding groundwater, leading to the development of ARB and ARGs [15]. In northern China, carbapenem-resistant Enterobacteriaceae can be isolated from 5.3% animal farm groundwater [16]. High levels of ARGs are detected in aquaculture farm groundwater of central China [17]. Meanwhile, ARGs are prevalent in the swine farm groundwater of southern China [10]. These researches underline potential public health threats of groundwater-borne AMR, since groundwater acts as a transmission route of AMR from the environment to human body through drinking and daily use [14].
Our previous study indicates that Acinetobacter can be predominant in the swine farm groundwater with a positive correlation to the abundance of ARGs [10]. In clinical environments, these Gram-negative, non-fermented, rod-shaped bacteria are well-known opportunistic pathogens for their capabilities of escaping antibiotic biocidal actions and mutually representing new paradigms in pathogenesis, transmission, and resistance [18–21]. The isolates in clinical settings are reported resistant to most commonly used antibiotics, thus have become formidable obstacles in treating infectious diseases [22]. To minimize the public health threats, WHO recommends a routine detection of Acinetobacter in drinking water sources [23], for which antibiotic susceptibility tests have been standardized for this genus by Clinical and Laboratory Standards Institute (CLSI) [24]. Animal guts are habitats and environmental sources of antibiotic-resistant Acinetobacter [7, 9, 25]. Carbapenem-resistant A. baumannii and A. calcoaceticus as well as tigecycline-resistant A. towneri can be isolated from swine feces [26, 27]. Imipenem- and meropenem-resistant A. junii have been detected in the farm soil [28]. The Acinetobacter strains resistant to multiple first-line and newly approved antibiotics have been isolated from poultry manure, wastewater and soil in the poultry farming regions of China [29]. These studies highlight AMR risks of Acinetobacter in the livestock environment. However, groundwater is currently a blind spot of Acinetobacter-associated researches. Few studies have yet involved their abundance in non-farm groundwater [30, 31], and none analyzed their composition, resistance and pathogenicity. This limit further assessing environmental risks in the groundwater transmission route.
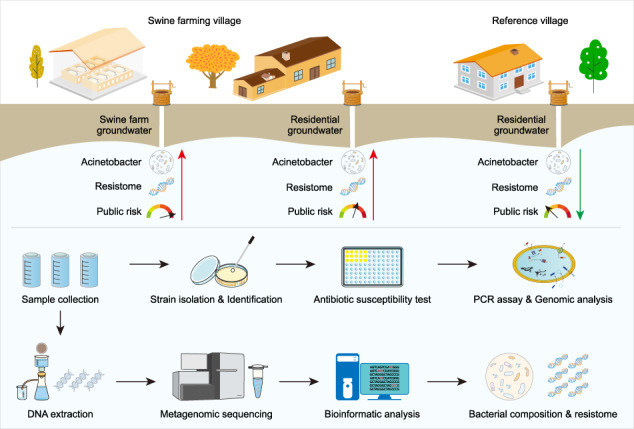
This study aims to elucidate the antibiotic resistance of Acinetobacter in the groundwater of the swine farming village using metagenomic and culture-dependent approaches, in comparison to the nearby non-farming village. Metagenomic assembling methods were used to profile the taxonomic composition and (acquired) antibiotic resistome of Acinetobacter in the groundwater. Metagenomic binning methods were used to analyze the antibiotic resistance and virulence potentials of Acinetobacter community, coupled with their genomic abundance in the groundwater. “High-risk” species were highlighted based on their antibiotic resistance, virulence and abundance. Meanwhile, Acinetobacter strains were isolated from the groundwater of the swine farming village. Their resistance profiles to 13 antibiotics were subsequently tested following the CLSI guideline. For polymyxins resistance, PCR assays were implemented to detect the associated genes, and whole genomes of highly resistant strains were sequenced to analyze potential mechanisms. The findings will expand knowledges of antibiotic-resistant Acinetobacter in the livestock environment.
Materials and methods
Study area and sample collection
In this study, a swine farming village (JZ) and a nearby (3.5 km) non-farming village (WT) in Guangxi of southern China were selected as the study area. In this distance, WT is considered having similar geographic and sanitary conditions with JZ, but limited influence by swine farming activities. JZ had 200 conventional semi-confined swine farms with a production of 50,000 heads/year, while WT only had a few animals for self-sufficient use. The swine farms lacked advanced waste treatment systems. Swine wastes were cleaned by farmers manually, and deposited in storage tanks. Then, the wastes were used for agricultural fertilization or transported to treatment plants for centralized treatment. To this end, the water systems, soil and air had been contaminated by farming activities [7, 10, 32, 33].
In December of 2018, 22 groundwater samples (average 6-m depth, the only water source in both villages) were collected from 3 well water groups: 13 wells in JZ residential houses (JZG), 5 wells in JZ swine farms (JZPG) and 4 wells in WT residential houses (WTG, Supplementary Fig. S1 of Supporting Information 1). It is noted that there were 200 farms in the village, therefore JZG were also very close to the farms despite located in residential houses. The groundwater in both JZG and JZPG were influenced by farming activities with antibiotic and ammonia contaminations [10]. Meanwhile, WT is much smaller than JZ, and 4 wells can represent the groundwater in the village. The water samples were collected in 10L sterilized plastic bottles, and then transported to the laboratory on ice. Basic water quality and antibiotic concentrations are listed in Supplementary Table S1 of Supporting Information 2.
DNA extraction and metagenomic sequencing
Groundwater samples were filtered with sterilized membranes (0.22-μm pore size, 50-mm diameter). Total DNA on each membrane was extracted using DNeasy® PowerSoil® kit (QIAGEN, Germany) according to the manufacturer’s instruments. DNA yields were measured using a Qubit 2.0 fluorometer (Thermo Fisher Scientific, USA). Water volume and DNA yield of each sample are listed in Supplementary Table S2 of Supporting Information 2. Before sequencing, DNA was fragmented into 300 bp using an ultrasound machine (Covaris M220, USA), and then pair-end DNA library was conducted using TruSeq™ DNA Sample Prep Kit (Illumina, USA). Collectively, twenty-one groundwater samples (one WTG sample was not sequenced because of low DNA yields) were metagenomically sequenced on a Novaseq 6000 platform (Illumina, USA) by Majorbio Bio-Pharm Technology (Shanghai, China) with the sequence production of over 12 Gb/sample.
Reads-based profiling of bacterial community and antibiotic resistome in the groundwater
Raw sequencing reads were quality-controlled and trimmed using KneadData v0.7.4 (https://huttenhower.sph.harvard.edu/kneaddata/) with default parameters. About 37.7–52.5 million clean reads were obtained for each sample (Table S2 of Supporting Information 2). The antibiotic resistome in the groundwater was analyzed using SARGs-OAP v2.0 (set: -l 25 -d 80 -e 1e-10) with the Structured Antibiotic Resistance Genes database [34]. Bacterial community composition was obtained using kraken2 classifier with the Genome Taxonomy Database (GTDB v202) [35, 36]. For each taxon, the maximum relative abundance lower than 0.001 were regarded as not detected.
Assembly-based analysis of ARGs’ hosts
Metagenomic reads in all samples were merged and assembled to contiguous sequences (contigs) using Megahit v1.2.9 (default parameters) [37]. Open reading frames (ORFs) on the contigs were predicted using Prodigal v2.6.3 (set: -c -p meta) [38]. The ORFs were then clustered and dereplicated using CD-HIT v4.8.1 (set: -aS 0.9 -c 0.95) [39]. Salmon v0.13.1 was used to estimate and normalize ORFs’ abundance into the transcripts per million reads (TPM) unit [40]. Potential ARGs were searched against the SARGFam database using hmmscan v3.3.2 (set: --cut_ga -noali) [34]. The taxonomy of ARG-ORFs was determined using kraken2 with the GTDB database.
Assembly-based analysis of the antibiotic resistome in Acinetobacter
The gene set of Acinetobacter was obtained by annotating ORFs’ taxonomy against the GTDB database using DIAMOND blastp v0.9.22 (set: --very-sensitive --id 90 --query-cover 90) [41]. Potential ARGs in the gene set were searched using RGI v5.1.1 (perfect and strict hits) with the Comprehensive Antibiotic Resistance Database (homolog model, v3.1.3) [42]. Potential acquired ARGs were searched using ResFinder v4.0 (set: -acq -t 0.8 -l 0.8) [43]. The taxonomy of ARG-contigs (a contig carrying at least one acquired ARG) were determined using kraken2 with the GTDB database. Functional genes and mobile gene elements (MGEs) in the contigs were searched against the NCBI nr database (downloaded on 2021.11.24) using DIAMOND blastp (set: --very-sensitive --id 80 --query-cover 80).
Metagenomic binning analysis
Metagenomic assembled genomes (MAGs) were generated from each sample by Metabat2, Maxbin2 and CONCOCT assemblers in MetaWRAP pipelines v1.3 [44]. The output MAGs were filtered by bin_refinement module (set: -c 70 -x 5). The bacterial or archaeal genomic taxonomy was classified by GTDB-Tk v1.4.1 with universal marker genes [36]. Their genomic information including assembly quality, completion and contamination are provided in Table S3 of Supporting Information 2. Potential ARGs in each MAG were searched using DeepARG, a deep learning-based approach for ARG annotation (set: --model LS --model-version v2 --arg-alignment-overlap 0.8 --type prot --arg-alignment-identity 80 --arg-alignment-evalue 1e-10 --arg-num-alignments-per-entry 1000) [45]. Potential VFGs and MGEs were annotated against the Virulence Factor Database (VFDB, downloaded on 2021.02.05) [46] and the MGEs90 database (https://bench.cs.vt.edu/ftp/data/databases/) using DIAMOND blastp (set: --very-sensitive --id 80 --query-cover 80). It is considered that one VF exists when at least one VFG is detected in the genomes. The genomic coverage (as relative abundance) of each MAG was estimated using Bowtie2 and SAMtools [47, 48], and normalized by the genomic size (Mbp) and sequencing file size (Gb).
Strain isolation and identification
Metagenomic analysis indicated that Acinetobacter was much more prevalent in the groundwater of JZ than that in WT (see result section), therefore culture-based methods were subsequently conducted for testing the antibiotic resistance of Acinetobacter in JZ groundwater. The Acinetobacter strains were isolated from 15 JZ groundwater samples (no colonies grew in the other three samples) using the Leeds Acinetobacter Medium (LAM) agar plate [49]. The purified isolates were then confirmed by Gram’s stain (negative), catalase (positive), nitrate reductase (negative) and oxidase tests (negative, Supplementary Fig. S2 of Supporting Information 1). Their 16S rRNA gene sequences were amplified and sequenced using universal primers (27F/1492R) by the Beijing Genomics Institute (BGI, China). The taxonomy was classified by aligning sequences to the reported species using NCBI Blastn online tools (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Antibiotic susceptibility tests
The broth microdilution method was used to test antibiotic susceptibility following the CLSI guidelines v2018. Briefly, the strains were inoculated (5 × 105 cells/mL in triplicates) in the cation-adjusted Mueller-Hinton broth (a final volume of 200 μL in 96-well plates) with each of 12 antibiotics at the breakpoint concentrations (ampicillin: 32 μg/mL; meropenem: 8 μg/mL; colistin: 4 μg/mL; polymyxin B: 4 μg/mL; gentamicin: 16 μg/mL; tobramycin: 16 μg/mL; doxycycline: 16 μg/mL; tetracycline: 16 μg/mL; ciprofloxacin: 4 μg/mL; levofloxacin: 8 μg/mL; gatifloxacin: 8 μg/mL; and sulfamethoxazole: 76 μg/mL). Since over 24.0% of the strains were resistant to polymyxins (colistin and polymyxin B, see result section), the minimum inhibitory concentration (MIC) for the resistant strains were determined by the two-fold serial dilution method. In addition, MICs of tigecycline for all strains were tested at a dilution range of 0.25–4 μg/mL. The resistance breakpoint was delineated as 1.0 μg/mL according to the European Committee on Antimicrobial Susceptibility Testing [50], since CLSI lacks the criteria for this antibiotic. Escherichia coli ATCC 25922 was used as the quality-control strain.
Detection of intrinsic polymyxins resistance genes using PCR and whole-genomic sequencing methods
Polymyxins resistance can be mediated by plasmid-borne mcr genes and chromosomally encoded by two-component systems pmrABC and lipopolysaccharide (LPS) biosynthesis genes (Supplementary Fig. S3 of Supporting Information 1) [51]. In this study, seven intrinsic resistance genes including pmrABC, lpxACD and lpsB were tested for the strains using PCR methods [52]. The primers and PCR programs are summarized in Supplementary Table S4 of Supporting Information 2. The mcr genes were not tested since they were not detected by metagenomic analysis. Meanwhile, the draft genomes of ten highly resistant strains were sequenced on the HiSeq X Ten platform (Illumina, USA) by Majorbio Bio-Pharm Technology (details shown in Supplementary Text S1 of Supporting Information 1). The LPS biosynthesis genes including lpxACD and lpsB were searched in the genomes using DIAMOND blastp (set: --very-sensitive --id 80 --query-cover 80) with the VFDB database.
Data visualization
R v4.0.3 was used for data statistics and visualization. Bar, stack and box plots were presented by ggplot2 v3.3.6. Venn plots were generated using venn v2.5–6. Sankey plots were drawn using networkD3 v0.4. Phylogenetic trees for MAGs were generated by gtdbtk infer module in GTDB-Tk v1.4.1. Phylogenetic relationships (16S rRNA gene) of the isolated strains were analyzed using MegaX with muscle alignment kit, maximum likelihood method, 100 bootstrap and kimura 2-parameter model. The phylogenetic trees were drawn by iTOL (https://itol.embl.de/). Genetic environments were drawn using gggenes v0.4.1. Schematic plots and figure layout were conducted by Adobe Illustrator CC v2019.
Results
Antibiotic resistome and bacterial community in the groundwater
In this study, 485 ARGs against 16 antibiotic classes were detected in the groundwater using the metagenomic reads-based methods (Fig. 1a, Supplementary Table S5 of Supporting Information 2). The ARGs encoding multidrug resistance (average 64.6% of the resistome, hereafter), tetracyclines resistance (10.5%) and aminoglycosides resistance (9.9%) were prevalent in all samples. The abundance of the resistome in all JZPG (1.4–3.4 copies/16S rRNA gene) and some JZG (0.3–0.7 copies/16S rRNA gene) was much higher than that in WTG (0.3–0.7 copies/16S rRNA gene). Over 180 ARGs were shared by three groups, while 212 ARGs including 140 beta-lactam resistance genes were only detected in JZ groundwater (Fig. 1b). This suggests an increased abundance and diversity of the resistome in the farm affected groundwater.
Fig. 1. Antibiotic resistome and bacterial community composition in the groundwater.

a Relative abundance of ARG types in the groundwater. b Unique and shared ARG subtypes in the groups. Values in the venn plot represent ARG numbers, and the percentages in pie plot are proportion of the ARGs only detected in JZG and JZPG. c Relative abundance of top ten genera with the Acinetobacter contribution to the resistome. ARG antibiotic resistance gene, MLS macrolide-lincosamide-streptogramin, JZG residential groundwater in the swine farming village, JZPG swine farm groundwater in the swine farming village, WTG residential groundwater in the reference village.
In terms of microbial community, 63.6%-95.8% of metagenomic reads were classified to bacteria in the groundwater. Of the 158 detected genera (Supplementary Table S6 of Supporting Information 2), LX47W (maximum 71.1% of the bacteria in all samples, hereafter), Zoogloea (65.3%), Pseudogulbenkiania (55.6%), Paraburkholderia (22.4%), Azohydromonas (20.5%), Azonexus (15.8%), Burkholderia (16.8%) and Limnohabitans_A (6.5%) were detected in higher abundance (Fig. 1c). Comparatively, Acinetobacter was abundant in all JZPG (54.4–95.7%) and 8 JZG (~91.1%), while the abundance was much lower in WTG (0.2%–8.0%).
Assembly-retrieved antibiotic resistome in Acinetobacter
A total of 99 ARGs against 23 antibiotic classes (multidrugs were separated into corresponding classes) were detected in Acinetobacter using the metagenomic assembling methods (Supplementary Table S7 of Supporting Information 2). They accounted for ~95.6% of total resistome in Acinetobacter dominant groundwater (Fig. 1c). The major hosts were A. baumannii (18.2% of Acinetobacter resistome, hereafter), A. pittii (9.9%), A. johnsonii (6.7%), A. seifertii (6.5%) and A. junii (3.8%) (Fig. 2). The predominant resistance mechanisms were antibiotic efflux (66.49%), antibiotic inactivation (22.34%) and antibiotic target alteration (6.85%). Tetracyclines resistance genes (50.18%) and fluoroquinolones resistance genes (53.61%) were the major ARG types (Fig. 2). Efflux pump genes associated with resistance-nodulation-cell division (RND, 49.82%), major facilitator superfamily pump (MFS, 9.39%) and multidrug and toxic compound extrusion transporter (MATE, 4.51%) were dominant gene families (Fig. 2). Meanwhile, OXA beta-lactamase (11.55%) and fluoroquinolone resistant parC (6.86%) families were also prevalent in Acinetobacter.
Fig. 2. The antibiotic resistome in Acinetobacter community.

The predominant six species are presented in the left. The percentages below species names are their proportions in Acinetobacter community. The right columns show the ARGs detected in these species, while the percentages are proportions of the ARGs in all Acinetobacter species. The complete results are summarized in Supplementary Table S7. Asterisk-marked antibiotic category includes macrolide, fluoroquinolone, lincosamide, carbapenem, cephalosporin, tetracycline, rifamycin, diaminopyrimidine, phenicol and penem.
A total of 70 contigs carrying acquired ARGs were detected in the groundwater (Fig. 3a, Supplementary Table S8 of Supporting Information 2). Therein, 30 contigs were classified to 18 Acinetobacter species. Two monooxygenase gene variants tet(X3) and tet(X6) conferring resistance to tigecycline were detected with higher abundance in Acinetobacter dominant groundwater (e.g., tet(X3) in JZPG5 and tet(X6) in JZPG4, Supplementary Table S8 of Supporting Information 2). In the NCBI nt database, the conservative genetic environment of tet(X3) has been reported in the plasmids of seven Acinetobacter species (Fig. 3b). Some ARGs conferring resistance to other antibiotic classes, such as sul2, aph(3’) and mph(E)/(G) were colocalized with this structure. Meanwhile, tet(X6) has been detected in the chromosomes and plasmids of six species including A. baumannii, A. towneri, A. indicus, A. piscatorial, A. schindleri and A. pseudolwoffii. Besides tet(X), trimethoprim resistance gene dfrA were also observed colocalized with multiple ARGs and MGEs in Acinetobacter and other bacteria (Supplementary Fig. S4 of Supporting Information 1). Meanwhile, several other ARGs including mph(E), msr(E), blaADC, cmlB, ant(2”), aac(3), aph(3), aph(6) and tet(Y) were also colocalized with MGEs.
Fig. 3. Profiles of acquired ARGs in the groundwater.

a Potential hosts of the contigs in the groundwater. Values in brackets are contig numbers. Asterisk-marked ARGs mean that there were MGEs colocalized. b Genetic environments of tet(X3)-contig in the published Acinetobacter species. The accession number of each species is given in the bracket. Genes in a shadow have the same genetic environment.
MAG-retrieved antibiotic resistance and pathogenicity in Acinetobacter
A total of 172 bacterial MAGs and 8 archaeal MAGs were obtained in the groundwater (Supplementary Table S3 of Supporting Information 2). The bacterial MAGs were classified to 15 phyla, and therein 81.4% were classified to Proteobacteria (Fig. 4a). There were 26 MAGs classified to 15 Acinetobacter species. They were abundant (~68.4 copies/Mbp/Gb) in JZ groundwater, and carried more ARGs (a sum of 30 subtypes), MGEs and VFGs than other bacteria (Fig. 4a, Supplementary Table S9 of Supporting Information 2). Specifically, A. oleivorans carried 20 ARGs, which was more than the other Acinetobacter species (Fig. 4b). A. modestus carried 14 ARGs, and was dominant in JZPG2 sample (93.1% of metagenomic reads). A. bereziniae was detected in four groundwater samples, and carried eight multidrug efflux pump genes (adeABJKR, mexT, ompR and tet39) and four beta-lactamase resistance genes (blaOXA, blaOXA-355, blaOXA-228 and blaOXA-356). Over 30 MGE classes involving recombinase, integrase and transposase were detected in Acinetobacter (Supplementary Table S9 of Supporting Information 2). These MGEs accounted for 0.1–1.9% of the genes in each MAG. The IS gene family was the most prevalent, and therein IS3 was detected in all species. Several ARGs including aac(6’)-I (found in A. wuhouensis), ompR (found in A. brisouii), and adeIJK (found in multiple species) were colocalized with MGEs.
Fig. 4. The genomic antibiotic resistance and pathogenicity of Acinetobacter in the groundwater.

a The phylogenetic tree of the MAGs. Values in brackets are genome numbers of bacteria and Acinetobacter. Acinetobacter genomes are highlighted in the shadow. Rings represent phylum, group, genomic coverage as well as numbers of VFGs, MGEs and ARGs from the innermost to the outermost. b ARG compositions in Acinetobacter genomes. ARG antibiotic resistance gene, VFG virulence factor gene, MGE mobile gene element, JZG residential groundwater in the swine farming village, JZPG swine farm groundwater in the swine farming village, and WTG residential groundwater in the reference village.
In this study, 99 VFGs referring to 14 VF classes were detected in Acinetobacter genomes (Fig. 5, Supplementary Table S9 of Supporting Information 2). The VFGs associated with capsule (ten species), LPS (ten species), two-component system BfmRS (nine species), EF-Tu (nine species) and outer membrane protein (nine species) were commonly detected, while PNAG, phospholipase C/D and quorum sensing were only found in A. oleivorans. In addition, ten VF classes including pbpG, adeFGH efflux pump, type IV pili, heme utilization, LPS, outer membrane protein, capsule, BfmRS, catalase and EF-Tu were detected in A. beijerinckii. The latter six were also detected in A. bereziniae. Collectively, A. oleivorans, A. bereziniae, A. modestus and A. beijerinckii might pose potential health risks since they were abundant (>1copy/Mbp/Gb) in the groundwater, and carried multiple (≥10 subtypes) ARGs and VFGs.
Fig. 5.

Potential virulence factors in Acinetobacter community.
Antibiotic susceptibility of the isolated strains
In this study, 341 strains were isolated from the groundwater. They were classified to 11 species including A. ursingii, A. modestus/junii (strains might be either of them since their 16S rRNA gene sequences were very similar (> 99.0%), hereafter), A. beijerinckii, A. baumannii, A. seifertii, A. pittii/oleivorans, A. rudis/xiamenensis, and A. bereziniae. Their resistance profiles to 13 antibiotics were presented in Fig. 6. Detailly, over 80.0% of the strains were resistant to at least two antibiotics, and nearly 70.0% were multidrug-resistant (≥three antibiotics). These multidrug-resistant strains were mainly classified to A. beijerinckii, A. pittii/oleivorans, A. baumannii, A. seifertii and A. bereziniae. Among all strains, the resistance ratios were higher to sulfamethoxazole (resistance ratio: 96.2%, covering all species), fluoroquinolones including levofloxacin (42.5%, none of A. ursingii, A. modestus/junii and A. beijerinckii), gatifloxacin (39.0%, none of A. modestus/junii and only one A. baumannii) and ciprofloxacin (32.6%, none of A. modestus/junii), as well as tetracyclines including tetracycline (32.0%, none of A. ursingii and A. beijerinckii) and doxycycline (29.0%, none of A. modestus/junii and only one A. baumannii). A few strains were resistant to ampicillin (12.0%, all classified to A. bereziniae) and tobramycin (1.8%, A. seifertii and A. bereziniae). However, none strains were resistant to gentamicin and meropenem. The MICs of tigecycline for most strains (87.1%) were lower than 0.25 μg/mL, while 14 strains including two A. ursingii, one A. baumannii, seven A. pittii/oleivorans and four A. bereziniae were tigecycline-resistant (MIC = 1 μg/mL).
Fig. 6. The phylogenetic relationship of Acinetobacter strains with the antibiotic resistance phenotype.

The strains are clustered into eight groups using 16S rRNA gene sequences with 99% similarity and 95% coverage. Rings represent samples as well as resistance profiles to sulfamethoxazole, ciprofloxacin, gatifloxacin, levofloxacin, doxycycline, tetracycline, polymyxin B, colistin, gentamicin, tobramycin, ampicillin, meropenem and tigecycline from the innermost to the outermost. Percentages in brackets are resistance ratios. Blue represents resistance, while yellow means MIC = 0.5 μg/mL of tigecycline, and red means MIC = 1.0 μg/mL of tigecycline.
Over 24.0% of the strains were resistant to colistin or polymyxin B (Fig. 6). Notably, thirty-one strains were resistant to both antibiotics (MICs ≥16 μg/mL, Supplementary Fig. S5 of Supporting Information 1). PCR results showed that the detection ratios of seven intrinsic resistance genes were inconsistent across the strains (Supplementary Fig. S6 of Supporting Information 2). Meanwhile, genomic analysis indicated that the amino acid (aa) sequences of lpsB and lpxACD in ten resistant strains significantly varied to the reference genome (Supplementary Table S10 of Supporting Information 2). Detailly, lpsB with 13 aa mismatches was only detected in three genomes by PCR and whole-genome sequencing. LpxA with 50 aa mismatches was detected in all genomes, but not detected by the PCR method. Similarly, lpxC was detected in all genomes with 28 aa mismatches, but detected in only four strains using the PCR method. However, lpxD was detected in two genomes by the PCR method, but not detected in the draft genomes. In addition, the sequences of lpxD in three genomes were greatly distinct (59 aa mismatches) to the reference genome.
Discussion
This study detected multiple ARGs and Acinetobacter species in the swine farm groundwater and the affected residential groundwater. Their abundance was much higher in the groundwater of the farming village than that of the nearby non-farming village, implying the influence of farming activities to both bacterial community and antibiotic resistome. The composition of Acinetobacter species was spatially heterogenous in the groundwater, and several non-A. baumannii species including A. oleivorans, A. bereziniae, A. modestus and A. beijerinckii were prevalent in farm-affected groundwater. Generally, Acinetobacter is ubiquitous existence in natural environments including soil, fresh water, ocean and sediment, as well as animal, plant and human body [53, 54]. However, its abundance may be higher in human- (urban sewage and hospitals) or animal-contaminated (livestock wastewater) environments [9, 25, 31], in where Acinetobacter is commonly associated with AMR prevalence, and as an important vector of VFs causes an increased risk to circulatory and respiratory systems [55]. Besides environmental factors, several cellular traits contribute to its prevalence in the environment, including 1) bacterial capsules and extracellular substances against environmental stress [55, 56]; 2) quorum sensing and biofilm formation for population density maintenance [57]; 3) micronutrient acquisition systems for acquiring resources in oligotrophic environments [58]; and 4) bacterial toxins directly killing other bacteria [59]. These traits not only endow Acinetobacter with robust competitive advantages for resources, but are also associated with pathogenicity to human and intrinsic resistance to antimicrobial agents [60]. Collectively, Acinetobacter might threaten the local public health via the transmission route of drinking the groundwater.
In this study, the antibiotic resistance varied across the Acinetobacter species. For example, all ampicillin-resistant strains were classified to A. bereziniae; and all A. modestus/junii strains were susceptible to fluoroquinolones. The isolated strains were widely resistant to several antibiotic classes including sulfonamides, quinolones, tetracyclines and polymyxins, but was more susceptible to meropenem, ampicillin, gentamicin and tobramycin. Comparatively, the China Antimicrobial Resistance Surveillance System (http://www.carss.cn/, 2021) reported that over half of the clinic-relevant Acinetobacter strains were resistant to imipenem (resistance ratio: 65.6%), meropenem (66.5%), levofloxacin (56.0%), ciprofloxacin (66.5%), gentamicin (62.3%) and ampicillin-sulbactam (59.1%), but they were more susceptible to colistin (1.6%), polymyxin B (0.7%) and tigecycline (2.5%). These adverse results indicated a different AMR development process between the livestock and clinical environment.
The antibiotic resistome in Acinetobacter are currently limited studied as a whole. A recent study summarized ARG contents in 21 non-A. baumannii species, and elucidated that beta-lactams and aminoglycosides resistance genes as well as the efflux pump resistance mechanisms are commonly detected in Acinetobacter [61]. The beta-lactams resistance, especially last-resort carbapenems, has obtained growing concerns in livestock-associated Acinetobacter researches [62]. Imipenem-resistant A. baumannii, carbapenem-resistant A. calcoaceticus, imipenem- and meropenem-resistant A. junii have been isolated from animal feces and farm-affected soil [26–28], suggesting that livestock environments are potential hotspots for carbapenem-resistant Acinetobacter prevalence. In terms of aminoglycosides resistance, aph(6), ant(3”), aph(3”) and strA are the most prevalent ARGs in Acinetobacter [61]. The former three are detected in A. pittii, A. indicus and A. haemolyticus, while the latter one is carried by A. pittii, A. nosocomialis, A. radioresistens, A. seifertii, A. haemolyticus, A. towneri, A. johnsonii and A. ursingii. However, despite beta-lactams and aminoglycosides resistance genes were detected using the metagenomic methods, the isolated strains in this study were rarely resistant to meropenem and aminoglycosides, implying weak activities of the relevant ARGs. Four efflux pump families were detected in multiple Acinetobacter species. The impressive genetic plasticity of the genomes can enhance the intrinsic resistance attributable to these efflux pumps or introduce new resistance by rapid gene mutation, recombination and integration [22, 63]. Meanwhile, Acinetobacter can acquire antibiotic resistance through obtaining plasmid-borne ARGs colocalized with MGEs [64]. These might contribute to the multidrug resistance in Acinetobacter of this study.
A growing amount of polymyxins-resistant Acinetobacter strains have been isolated from different regions of the world [65]. Although polymyxins have yet been used in animal breeding since 2016, the resistance ratio in this study was at a relatively higher level in comparison to the reported environments (resistant ratios: 0.2–53.1%) [65], implying that other environmental or cellular factors might influence the resistance. The polymyxins resistance in Acinetobacter can be mediated by both acquired and intrinsic resistance mechanisms [51]. Since the acquired resistance genes (the mcr family) were not detected using the metagenomic methods, the intrinsic mechanisms might contribute to the resistance. The processes involve 1) adding phosphoethanolamine (PetN) to lipid A; 2) mutations of lipid A biosynthesis genes leading to its complete loss; 3) low expression of proteins for outer membrane stability; and 4) deficient expression of LPS biosynthesis cofactors [51]. Diverse detection of the key genes in these processes suggested that the polymyxin resistance might be increased by gene mutation, recombination and insertion through influencing LPS structures and biosynthesis [66].
With the global spread of carbapenems and polymyxins resistance genes, tigecycline has been raised to be another last-line regimen for treating a vast of clinical infections caused by multidrug-resistant bacteria [67]. However, tigecycline resistance introduced by tet(X) inhibits clinical effectiveness of tigecycline [67]. In the GeneBank database, three tet(X) variants including tet(X3), tet(X5) and tet(X6) are observed in several Acinetobacter species, which are all associated with livestock environments [27]. They are generally colocalized with several MGEs referring to insertion, transposon and integron, and thereby can be transferred across species [27]. In the livestock environment, tigecycline-resistant Acinetobacter strains carrying tet(X) have been isolated from animal feces, wastewater and farm-affected soil [27], but have yet to be reported in farm affected groundwater. This study highlights the needs of future larger-scale investigation on tigecycline-resistant Acinetobacter in the livestock groundwater. However, the breakpoint value of tigecycline for Acinetobacter has not been standardized by the international communities: CLSI lacks the criteria for this antibiotic, and the breakpoint value is delineated as 1.0 μg/mL [50], 2.0 μg/mL [68] and 8.0 μg/mL [29] in different standards. Therefore, standardizing the global criteria is urgent for surveilling and controlling tigecycline-resistant Acinetobacter in the environment.
One of the most arresting features of Acinetobacter is their powerful capability to cause infectious diseases [69]. Consensus supports that multi-factorial and combinatorial strategies with at least 16 gene islands are associated with virulence [70]. Among these VFs, quorum sensing, biofilm formation and efflux pumps are associated with self-protection and population density regulation when facing environmental stress [70]. LPS, capsule, outer membrane proteins and phospholipase are associated with cellular virulence [70]. Complex pathogenicity traits also confer them competitive advantages and anti-stress abilities [71]. In the clinical environment, A. baumannii is one of the most notorious pathogens for their high rates of pathogenicity and antibiotic resistance [2]. However, this study detected multiple VFGs relating to 11 virulence factors in non-A. baumannii species, such as A. oleivorans, A. beijerinckii, A. seifertii, A. bereziniae and A. modestus. This implies that these species might also pose public health risks in the livestock environment.
Conclusively, this study reported notable prevalence of Acinetobacter with severe antibiotic resistance in the swine farm and nearby residential groundwater. Compared with A. baumannii in the clinical environment, more species in the groundwater deserve our concerns because of their prevalence, antibiotic resistance and pathogenicity. Complex intrinsic and acquired mechanisms conferred Acinetobacter resistant to multiple first-line and last-resort antibiotics. Additionally, diverse VFs might endow them with invasive abilities to human body and competitive advantages in groundwater ecosystems. Future studies are suggested to investigate the antibiotic resistance of groundwater-borne Acinetobacter at a larger geographical scale, and to assess the public health risks arising from multidrug-resistant pathogenic Acinetobacter using the “One Health” methods.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC 42030703, U1701242 and 42177226), and National Key Research and Development Program of China (2020YFC1806901), as well as Chinese Postdoctoral Science Foundation (2022M711215) and Guangdong Basic and Applied Basic Research Foundation (2019A1515110131). We would like to thanks team members involved in field campaigns.
Author contributions
F-ZG: Conceptualization, Investigation, Formal analysis, Data curation, Writing - original draft. L-YH: Validation. XC: Investigation, Resources. J-LC: Investigation. XY: Review. L-XH: Investigation, Resources. X-YH: Methodology. Z-YC: Investigation. HB: Investigation. MZ: Investigation. Y-SL: Project administration. G-GY: Conceptualization, Funding acquisition, Supervision, Review & editing.
Data availability
The metagenomic and draft genomic data in this study are available in the National Microbiology Data Center (http://nmdc.cn/) with the project accession number of NMDC10017956.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Liang-Ying He, Email: liangying.he@m.scnu.edu.cn.
Guang-Guo Ying, Email: guangguo.ying@m.scnu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s43705-023-00240-w.
References
- 1.O’Neill LJ. Antimicrobial resistance: tackling a crisis for the health and wealth of nations. Rev Antimicrob Resist. 2014;1:1–16. [Google Scholar]
- 2.Murray CJL, et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet. 2022;399:629–55. doi: 10.1016/S0140-6736(21)02724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization (WHO). Antimicrobial resistance global report on surveillance: 2014 summary. Geneva, Switzerland: WHO; 2014.
- 4.Larsson DGJ, Flach CF. Antibiotic resistance in the environment. Nat Rev Microbiol. 2021;20:257–69. doi: 10.1038/s41579-021-00649-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.UNEP. Frontiers 2017 emerging issues of environmental concern. Nairobi: United Nations Environment Programme; 2017.
- 6.Zhao Y, et al. Antibiotic resistome in the livestock and aquaculture industries: status and solutions. Crit Rev Environ Sci Technol. 2021;51:2159–96. doi: 10.1080/10643389.2020.1777815. [DOI] [Google Scholar]
- 7.Gao FZ, et al. Untreated swine wastes changed antibiotic resistance and microbial community in the soils and impacted abundances of antibiotic resistance genes in the vegetables. Sci Total Environ. 2020;741:140482. doi: 10.1016/j.scitotenv.2020.140482. [DOI] [PubMed] [Google Scholar]
- 8.Zhang M, et al. Variation of antibiotic resistome during commercial livestock manure composting. Environ Int. 2020;136:105458. doi: 10.1016/j.envint.2020.105458. [DOI] [PubMed] [Google Scholar]
- 9.Zhang M, et al. Variations of antibiotic resistome in swine wastewater during full-scale anaerobic digestion treatment. Environ Int. 2021;155:106694. doi: 10.1016/j.envint.2021.106694. [DOI] [PubMed] [Google Scholar]
- 10.Gao FZ, et al. Swine farming elevated the proliferation of Acinetobacter with the prevalence of antibiotic resistance genes in the groundwater. Environ Int. 2020;136:105484. doi: 10.1016/j.envint.2020.105484. [DOI] [PubMed] [Google Scholar]
- 11.Bai H, et al. Spread of airborne antibiotic resistance from animal farms to the environment: dispersal pattern and exposure risk. Environ Int. 2022;158:106927. doi: 10.1016/j.envint.2021.106927. [DOI] [PubMed] [Google Scholar]
- 12.Davis GS, et al. Antibiotic-resistant Escherichia coli from retail poultry meat with different antibiotic use claims. BMC Microbiol. 2018;18:174. doi: 10.1186/s12866-018-1322-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McEwen SA, Collignon PJ. Antimicrobial resistance: a one health perspective. Microbiol Spectr. 2018;6:6.2.10. doi: 10.1128/microbiolspec.ARBA-0009-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zainab SM, Junaid M, Xu N, Malik RN. Antibiotics and antibiotic resistant genes (ARGs) in groundwater: a global review on dissemination, sources, interactions, environmental and human health risks. Water Res. 2020;187:116455. doi: 10.1016/j.watres.2020.116455. [DOI] [PubMed] [Google Scholar]
- 15.Chee-Sanford JC, Aminov RI, Krapac IJ, Garrigues-Jeanjean N, Mackie RI. Occurrence and diversity of tetracycline resistance genes in lagoons and groundwater underlying two swine production facilities. Appl Environ Microbiol. 2001;67:1494–502. doi: 10.1128/AEM.67.4.1494-1502.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu C, et al. Clonal and plasmid-mediated dissemination of environmental carbapenem-resistant Enterobacteriaceae in large animal breeding areas in northern China. Environ Pollut. 2022;297:118800. doi: 10.1016/j.envpol.2022.118800. [DOI] [PubMed] [Google Scholar]
- 17.Tong L, et al. Antibiotic resistance gene profiling in response to antibiotic usage and environmental factors in the surface water and groundwater of Honghu Lake. China. Environ Sci Pollut R. 2020;27:31995–2005. doi: 10.1007/s11356-020-09487-5. [DOI] [PubMed] [Google Scholar]
- 18.Nhu NTK, et al. The induction and identification of novel Colistin resistance mutations in Acinetobacter baumannii and their implications. Sci Rep. 2016;6:28291. doi: 10.1038/srep28291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pendleton JN, Gorman SP, Gilmore BF. Clinical relevance of the ESKAPE pathogens. Expert Rev Anti Infect Ther. 2013;11:297–308. doi: 10.1586/eri.13.12. [DOI] [PubMed] [Google Scholar]
- 20.Rice LB. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J Infect Dis. 2008;197:1079–81. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 21.Visca P, Seifert H, Towner KJ. Acinetobacter infection-an emerging threat to human health. IUBMB Life. 2011;63:1048–54. doi: 10.1002/iub.534. [DOI] [PubMed] [Google Scholar]
- 22.Vrancianu CO, Gheorghe I, Czobor IB, Chifiriuc MC. Antibiotic resistance profiles, molecular mechanisms and innovative treatment strategies of Acinetobacter baumannii. Microorganisms. 2020;8:935. doi: 10.3390/microorganisms8060935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.WHO. Guidelines for drinking-water quality, 4th ed. Geneva: WHO Press; 2011.
- 24.CLSI. Performance standards for antimicrobial susceptibility testing, 28th ed. CLSI supplement M100. Wayne, PA: Clinical and Laboratory Standards Institute; 2018.
- 25.Al Atrouni A, Joly-Guillou ML, Hamze M, Kempf M. Reservoirs of Non-baumannii Acinetobacter Species. Front Microbiol. 2016;7:49. doi: 10.3389/fmicb.2016.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al Bayssari C, Dabboussi F, Hamze M, Rolain JM. Emergence of carbapenemase-producing Pseudomonas aeruginosa and Acinetobacter baumannii in livestock animals in Lebanon. J Antimicrob Chemother. 2015;70:950–1. doi: 10.1093/jac/dku469. [DOI] [PubMed] [Google Scholar]
- 27.Cheng YY, et al. Sporadic dissemination of tet (X3) and tet (X6) mediated by highly diverse plasmidomes among livestock-associated Acinetobacter. Microbiol Spectr. 2021;9:e0114121. doi: 10.1128/Spectrum.01141-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang B, Sun D. Detection of NDM-1 carbapenemase-producing Acinetobacter calcoaceticus and Acinetobacter junii in environmental samples from livestock farms. J Antimicrob Chemother. 2015;70:611–3. doi: 10.1093/jac/dku405. [DOI] [PubMed] [Google Scholar]
- 29.Cui CY, et al. Co-occurrence of plasmid-mediated tigecycline and carbapenem resistance in Acinetobacter spp. from waterfowls and their neighboring environment. Antimicrob Agents Chemother. 2020;64:e02502–19.. doi: 10.1128/AAC.02502-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong PY, Yannarell AC, Dai Q, Ekizoglu M, Mackie RI. Monitoring the perturbation of soil and groundwater microbial communities due to pig production activities. Appl Environ Microbiol. 2013;79:2620–9. doi: 10.1128/AEM.03760-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Zhang Y, Ding Y, Song H, Liu T. Analysis of microbial community resistance mechanisms in groundwater contaminated with SAs and high NH4(+)-Fe-Mn. Sci Total Environ. 2022;817:153036. doi: 10.1016/j.scitotenv.2022.153036. [DOI] [PubMed] [Google Scholar]
- 32.Gao FZ, et al. Airborne bacterial community and antibiotic resistome in the swine farming environment: metagenomic insights into livestock relevance, pathogen hosts and public risks. Environ Int. 2023;172:107751. doi: 10.1016/j.envint.2023.107751. [DOI] [PubMed] [Google Scholar]
- 33.Gao FZ, et al. The variations of antibiotics and antibiotic resistance genes in two subtropical large river basins of south China: anthropogenic impacts and environmental risks. Environ Pollut. 2022;312:119978. doi: 10.1016/j.envpol.2022.119978. [DOI] [PubMed] [Google Scholar]
- 34.Yin X, et al. ARGs-OAP v2.0 with an expanded SARG database and hidden markov models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics. 2018;34:2263–70. doi: 10.1093/bioinformatics/bty053. [DOI] [PubMed] [Google Scholar]
- 35.Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019;36:1925–7. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 38.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 40.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–9. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 42.Alcock BP, et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020;48:D517–D525.. doi: 10.1093/nar/gkz935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bortolaia V, et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother. 2020;75:3491–500. doi: 10.1093/jac/dkaa345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6:158. doi: 10.1186/s40168-018-0541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arango-Argoty G, Garner E, Pruden A, Heath LS, Vikesland P, Zhang L. DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome. 2018;6:23. doi: 10.1186/s40168-018-0401-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu B, Zheng D, Jin Q, Chen L, Yang J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019;47:D687–D692.. doi: 10.1093/nar/gky1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jawad A, Hawkey PM, Heritage J, Snelling AM. Description of Leeds Acinetobacter Medium, a new selective and differential medium for isolation of clinically important Acinetobacter spp., and comparison with Herellea agar and Holton’s agar. J Clin Microbiol. 1994;32:2353–8. doi: 10.1128/jcm.32.10.2353-2358.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.EUCAST. The European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters, version 10.0. 2020. http://www.eucast.org/clinical_breakpoints/.
- 51.Lima WG, Alves MC, Cruz WS, Paiva MC. Chromosomally encoded and plasmid-mediated polymyxins resistance in Acinetobacter baumannii: a huge public health threat. Eur J Clin Microbiol Infect Dis. 2018;37:1009–19. doi: 10.1007/s10096-018-3223-9. [DOI] [PubMed] [Google Scholar]
- 52.Lean SS, et al. Prevalence and genetic characterization of carbapenem-and polymyxin-resistant Acinetobacter baumannii isolated from a tertiary hospital in Terengganu, Malaysia. ISRN Microbiol. 2014;2014:953417. doi: 10.1155/2014/953417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jung J, Park W. Acinetobacter species as model microorganisms in environmental microbiology: current state and perspectives. Appl Microbiol Biot. 2015;99:2533–48. doi: 10.1007/s00253-015-6439-y. [DOI] [PubMed] [Google Scholar]
- 54.Adewoyin MA, Okoh AI. The natural environment as a reservoir of pathogenic and non-pathogenic Acinetobacter species. Rev Environ Health. 2018;33:265–272. doi: 10.1515/reveh-2017-0034. [DOI] [PubMed] [Google Scholar]
- 55.Doi Y, Murray GL, Peleg AY. Acinetobacter baumannii: evolution of antimicrobial resistance-treatment options. Semin Respir Crit Care Med. 2015;36:85–98. doi: 10.1055/s-0034-1398388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh JK, Adams FG, Brown MH. Diversity and function of capsular polysaccharide in Acinetobacter baumannii. Front Microbiol. 2018;9:3301. doi: 10.3389/fmicb.2018.03301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Whiteley M, Diggle SP, Greenberg EP. Progress in and promise of bacterial quorum sensing research. Nature. 2017;551:313–20. doi: 10.1038/nature24624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sheldon JR, Skaar EP. Acinetobacter baumannii can use multiple siderophores for iron acquisition, but only acinetobactin is required for virulence. PLoS Pathog. 2020;16:e1008995. doi: 10.1371/journal.ppat.1008995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weber BS, Kinsella RL, Harding CM, Feldman MF. The secrets of Acinetobacter secretion. Trends Microbiol. 2017;25:532–45. doi: 10.1016/j.tim.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Asif M, Alvi IA, Rehman SU. Insight into Acinetobacter baumannii: pathogenesis, global resistance, mechanisms of resistance, treatment options, and alternative modalities. Infect Drug Resist. 2018;11:1249–60. doi: 10.2147/IDR.S166750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baraka A, Traglia GM, Montana S, Tolmasky ME, Ramirez MS. An Acinetobacter non-baumannii population study: antimicrobial resistance genes (ARGs) Antibiotics. 2020;10:16. doi: 10.3390/antibiotics10010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Isler B, Doi Y, Bonomo RA, Paterson DL. New treatment options against carbapenem-resistant Acinetobacter baumannii infections. Antimicrob Agents Chemother. 2019;63:e01110–18. doi: 10.1128/AAC.01110-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kyriakidis I, Vasileiou E, Pana ZD, Tragiannidis A. Acinetobacter baumannii antibiotic resistance mechanisms. Pathogens. 2021;10:373. doi: 10.3390/pathogens10030373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnson T, et al. Clusters of antibiotic resistance genes enriched together stay together in swine agriculture. mBio. 2016;7:e02214–15. doi: 10.1128/mBio.02214-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oikonomou O, Sarrou S, Papagiannitsis CC, Georgiadou S, Mantzarlis K, Zakynthinos E, et al. Rapid dissemination of colistin and carbapenem resistant Acinetobacter baumannii in Central Greece: mechanisms of resistance, molecular identification and epidemiological data. BMC Infect Dis. 2015;15:559. doi: 10.1186/s12879-015-1297-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kamoshida G, Akaji T, Takemoto N, Suzuki Y, Sato Y, Kai D, et al. Lipopolysaccharide-deficient Acinetobacter baumannii due to colistin resistance is killed by neutrophil-produced lysozyme. Front Microbiol. 2020;11:573. doi: 10.3389/fmicb.2020.00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He T, et al. Emergence of plasmid-mediated high-level tigecycline resistance genes in animals and humans. Nat Microbiol. 2019;4:1450–6. doi: 10.1038/s41564-019-0445-2. [DOI] [PubMed] [Google Scholar]
- 68.Chang YY, et al. Impact of reduced tigecycline susceptibility on clinical outcomes of Acinetobacter bacteremia. J Microbiol Immunol. 2018;51:148–52. doi: 10.1016/j.jmii.2017.08.024. [DOI] [PubMed] [Google Scholar]
- 69.Moubareck CA, Halat DH. Insights into Acinetobacter baumannii: A review of microbiological, virulence, and resistance traits in a threatening nosocomial pathogen. Antibiotics. 2020;9:119. doi: 10.3390/antibiotics9030119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smith MG, et al. New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev. 2007;21:601–14. doi: 10.1101/gad.1510307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Greene C, Wu J, Rickard AH, Xi C. Evaluation of the ability of Acinetobacter baumannii to form biofilms on six different biomedical relevant surfaces. Lett Appl Microbiol. 2016;63:233–9. doi: 10.1111/lam.12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The metagenomic and draft genomic data in this study are available in the National Microbiology Data Center (http://nmdc.cn/) with the project accession number of NMDC10017956.