

Abstract
Restless legs syndrome (RLS) is a sensorimotor disorder that severely affects sleep. It is characterized by an urge to move the legs, which is often accompanied by periodic limb movements during sleep. RLS has a high prevalence in the population and is usually a life-long condition. While its origins remain unclear, RLS is initially highly responsive to treatment with dopaminergic agonists that target D2-like receptors, in particular D2 and D3, but the long-term response is often unsatisfactory. Over the years, several putative animal models for RLS have been developed, mainly based on the epidemiological and neurochemical link with iron deficiency, treatment efficacy of D2-like dopaminergic agonists, or genome-wide association studies that identified risk factors in the patient population. Here, we present the first systematic review of putative animal models of RLS, provide information about their face and construct validity, and report their role in deciphering the underlying pathophysiological mechanisms that may cause or contribute to RLS. We propose that identifying the causal links between genetic risk factors, altered organ functions, and changes to molecular pathways in neural circuitry will eventually lead to more effective new treatment options that bypass the side effects of the currently used therapeutics in RLS, especially for long-term therapy.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13311-022-01334-4.
Keywords: Dopamine, Iron, Striatum, Spinal cord, Genetics
Introduction
Restless legs syndrome (RLS) is a prevalent sensorimotor disorder characterized by an urge to move the legs that is often accompanied by periodic limb movements during sleep (PLMS). First characterized in 1672 and 1685 [1, 2] and redefined in 1960 [3], RLS is now clinically characterized by the following five essential criteria, which were originally developed by the International Restless Legs Syndrome Study group (IRLSSG) in 2003 [4] and updated in 2014 [5]: (1) an urge to move the legs usually but not always accompanied by, or felt to be caused by, uncomfortable and unpleasant sensations in the legs; (2) the urge to move the legs and any accompanying unpleasant sensations begin or worsen during periods of rest or inactivity such as lying down or sitting; (3) the urge to move the legs and any accompanying unpleasant sensations are partially or totally relieved by movement, such as walking or stretching, at least as long as the activity continues; (4) the urge to move the legs and any accompanying unpleasant sensations during rest or inactivity only occur or are worse in the evening or night than during the day; (5) the occurrence of the above features is not solely accounted for as symptoms primary to another medical or a behavioral condition (e.g., myalgia, venous stasis, leg edema, arthritis, leg cramps, positional discomfort, habitual foot tapping). The sensations generally establish themselves in the lower limbs first and are felt mostly during quiet wakefulness, when attempting to sleep, or both [6]. Consequently, RLS may have a negative impact on sleep and result in a wide range of significantly associated comorbidities.
Despite a strong and growing research interest in understanding the mechanisms that cause or contribute to RLS, the underlying causes of the disorder remain opaque, highlighting the importance of fostering RLS research. RLS is familial in about 50% of patients [7] but may be related to acquired conditions, especially iron deficiency, pregnancy, and chronic renal failure. Several predisposing candidate genes have been identified through genome-wide association studies (GWAS) [8]. Evidence suggests that RLS is associated with low intracerebral iron stores due to as yet unclear defects in iron homeostatic mechanisms [9]. Increased dopaminergic and glutamatergic and decreased adenosinergic transmission may also play a role in the pathophysiologic mechanism of the disorder [10].
RLS can manifest itself in either idiopathic/sporadic or secondary forms. Both forms express the same symptomatology, but in its idiopathic form, the causes for RLS are unknown or non-obvious and the symptoms usually do not resolve. In secondary RLS, underlying causes usually can be identified, and symptoms may abate or disappear after the resolution of the underlying health conditions or over time. Secondary RLS may also develop as a function of other neurological disorders (e.g., axonal neuropathy, Parkinson’s disease, spinocerebellar ataxia), other medical disorders (including uremia, end-stage renal failure, fibromyalgia, varicose veins, and/or iron deficiency), during the 3rd trimester of pregnancy, or as a result of drug use and abuse [11, 12].
Animal models have the potential to accelerate research on causal pathophysiological mechanisms and to foster the development of new therapeutic options for human diseases. With these aims, a range of different putative animal models have also been proposed for RLS and the resulting information has been the subject of several narrative reviews [13–18]. Still, a formal systematic scoping review of these models is missing. Recent consensus guidelines on face validity [19] and construct validity [20] criteria for developing animal models of RLS have been laid out. These guidelines focused on rodent models and have not included a systematic overview of the published previous literature.
Here, we aimed to fill this gap by performing the first systematic scoping review of putative animal models of RLS, highlighting the critical contributions (or lack thereof) of each model to our current understanding of RLS pathophysiology. To avoid reporting bias and to include research on non-rodent models, we did not restrict our search to studies satisfying the face [19] and construct [20] validity consensus criteria for rodent models. Rather, we elected to perform a broad and comprehensive search of the available knowledge base on putative animal models of RLS and then discuss the compliance of putative rodent models with the published validity guidelines [19, 20].
Methods
For the literature search aspect of this work, we followed the recently published guidelines for systematic scoping reviews [21–23].
A search of the scientific literature was conducted on PubMed.gov and Scopus.com (last access: Feb 01, 2022). Searches were limited to primary research papers published in English, and we excluded abstract-only presentations, editorials, and studies with human subjects. Review papers were also retrieved for the purpose of furthering the search for primary research papers in their reference lists. The following search terms were used: “restless legs” AND “animal model”; RLS AND “animal model”; “restless legs” AND “translational”; “restless legs” AND “rodent”; “restless legs” AND “invertebrate”; “restless legs” AND “fly”; “restless legs” AND “worm”; “restless legs” AND “elegans”; “restless legs” AND “mouse”; “restless legs” AND “rat.”
We obtained 259 hits from https://Pubmed.gov and 728 hits from https://Scopus.com. Figure 1 provides an overview of the flowchart of the selection process. After the removal of duplicates (step 1), we identified 335 unique papers that were then screened by two independent authors. To this aim, the 335 papers were split into five groups, with two reviewers independently carrying out the evaluations of each selected group. The first-pass screening evaluated if any keywords were present in the abstract and if so, the full paper was evaluated for inclusion or exclusion in this review (step 2). Of these 335 papers, 90 were identified by the authors to have met the inclusion criteria (congruency for inclusion), while the authors identified 209 papers not to have met these criteria (congruency for exclusion). The remaining 36 papers that did not meet congruency for inclusion or exclusion between the two reviewers of each group were then presented to all authors for evaluation in a consensus meeting. A simple majority vote for these 36 papers was carried out after two rounds of discussion, leading to 14 of these papers being accepted by the majority and 22 papers being rejected (step 3). In addition, 2 more recent publications were brought forward by individuals and, following additional expert evaluation, accepted to meet the parameters for inclusion in this review (step 4). The interim tallies for the acceptance or rejection were 106 papers accepted for inclusion and 231 papers rejected (step 5).
Fig. 1.

Summary of workflow to retrieve and select the existing literature on RLS animal models
Of the 106 accepted papers, 69 presented original research studies and 37 were review papers. In the subsequent steps (steps 6–9), we probed the 37 review papers for additional references of primary research literature not included in the previous searches (step 7). We identified 14 such papers (step 7), 12 of which were accepted by the authors for inclusion, and two were rejected (step 8). The final sub tallies for the number of primary research literature thus increased to 81 (step 9).
This final list of 81 primary research papers was used as the baseline for the ensuing systematic review of putative animal models of RLS, and the identified manuscripts are listed in Table 1. These papers were then complemented in the main text by additional studies that allowed the authors to provide the contextual background for these primary animal model studies concerning the pathophysiological mechanisms relevant to RLS.
Table 1.
Summary of primary research papers identified
| First author | Y | Publication | Species | GENET | Iron | DA | OT | STR | SC | PRE | FV | CV |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allen | 2020 | Sleep Med 71:141–8 | Mouse | + | + | + | ||||||
| Ashkenazi | 1982 | Pharmacol Biochem Behav 17(S1):43–7 | Rat | + | + | + | ||||||
| Baier | 2002 | J Neurol Sci 98:71–7 | Rat | + | ||||||||
| Barraud | 2010 | Plos One 5:e13306 | Monkey | + | + | NA | NA | |||||
| Beard | 1994 | Pharmacol Biochem Behav 48:621–4 | Rat | + | + | + | + | |||||
| Ben-Shachar | 1985 | J Neurochem 45:999–1005 | Rat | + | + | + | + | |||||
| Berteotti | 2021 | J Sleep Res 30:e13255 | Mouse | H | ||||||||
| Bianco | 2008 | J Neurochem 106:205–15 | Rat | + | + | + | + | |||||
| Bianco | 2009 | Chronobiol Int 26:447–63 | Mouse | + | + | + | + | |||||
| Carkaci-Salli | 2012 | J Neurosci Res 90:1583–8 | Mouse | + | + | |||||||
| Cathiard | 2021 | J Sleep Res 30:e13311 | Mouse | Meis1 | + | + | + | + | ||||
| Catoire | 2011 | Ann Neurol 70:170–5 | Worm | Meis1 | + | NA | NA | |||||
| Chen | 1995 | J Nutr 125:1529–35 | Rat | + | + | |||||||
| Clemens | 2004 | J Neurosci 24:11337–45 | Mouse | + | + | |||||||
| Clemens | 2005 | Neuroscience 133:353–7 | Mouse | + | + | |||||||
| Connor | 2008 | J Neurosci Res 86:3194–202 | Mouse | + | + | + | + | |||||
| Dean | 2006 | Sleep Med 7:634–40 | Mouse | + | ||||||||
| DeAndrade | 2012 | Hum Mol Genetics 21:3984–92 | Mouse | Btbd9 | + | + | + | + | + | |||
| DeAndrade | 2012 | Plos One 7:e35518 | Mouse | Btbd9 | + | |||||||
| Dinkins | 2017 | Sleep Med 40:47–52 | Mouse | + | + | |||||||
| Dowling | 2009 | J Nutr 139:2087–92 | Mouse | + | + | + | ||||||
| Dowling | 2011 | J Neurosci 31:70–7 | Mouse | + | + | + | ||||||
| Drgonova | 2015 | Mol Med 21:717–25 | Mouse | Ptprd | + | |||||||
| Earley | 2020 | Sleep Med 71:135–40 | Mouse | + | + | + | ||||||
| Erikson | 2000 | J Nutr 130:2831–7 | Rat | + | + | + | ||||||
| Erikson | 2001 | Physiol Pharmacol Behav 69:409–18 | Rat | + | + | + | + | |||||
| Esteves | 2013 | J Motor Behav 45:487–93 | Rat | + | ||||||||
| Esteves | 2016 | Brain Res 1639:47–57 | Rat | + | + | |||||||
| Franco | 2021 | J Neurosci Res 99:3325–38 | Rat | + | + | + | ||||||
| Frank | 2016 | Physiol Behav 154:161–8 | Rat | + | + | |||||||
| Freeman | 2012 | Curr Biol 22:1142–8 | Fly | Btbd9 | + | + | NA | NA | ||||
| Gao | 2022 | ACS Omega 7:11839–52 | Mouse | Btbd9 | + | |||||||
| Glover | 1972 | Br J Med 2:627–8 | Rat | + | + | |||||||
| Gulyani | 2009 | Exp Neurol 215:236–42 | Mouse | + | A | + | + | |||||
| Guo | 2017 | Sci Rep 7:9905 | Rat | + | + | + | ||||||
| Huang | 2021 | Front Aging Neurosci 13:651638 | Mouse | Map2k5 | + | + | + | |||||
| Hunt | 1994 | Am J Clin Nutr 59:413–8 | Rat | + | + | |||||||
| Hyacinthe | 2015 | Neuroscience 290:621–35 | Monkey | + | + | + | NA | NA | ||||
| Jellen | 2013 | Neuroscience 252:13–23 | Mouse | + | + | |||||||
| Keeler | 2012 | Exp Neurol 238:273–83 | Mouse | + | + | + | ||||||
| Koo | 2008 | Mov Disord 23:1234–42 | Rat | + | ||||||||
| Lai | 2008 | Neuroscience 154:431–43 | Cat | + | NA | NA | ||||||
| Lai | 2017 | Mov Disord 32:1687–93 | Rat | + | + | + | + | |||||
| Lai | 2020 | Sleep 43:zsz223 | Rat | + | H | + | + | |||||
| Lai | 2022 | Sleep 45:zsac110 | Rat | + | + | H | + | + | ||||
| Lo Martire | 2018 | Front Physiol 9:1818 | Mouse | + | + | |||||||
| Lopes | 2012 | Mov Disord 27:413–20 | Rat | + | + | |||||||
| Luo | 2011 | Sleep Med 12:41–6 | Mouse | + | + | + | ||||||
| Lyu | 2019 | Behav Brain Res 374:112123 | Mouse | + | + | O | + | + | ||||
| Lyu | 2019 | eNeuro 6:ENEURO.0277–19.2019 | Mouse | Btbd9 | + | + | + | |||||
| Lyu | 2020 | Brain Struct Funct 225:1743–60 |
Worm Mouse |
Btbd9 Btbd9 |
+ + |
+ | NA |
NA + |
||||
| Lyu | 2020 | Exp Neurol 323:113111 | Mouse | Btbd9 | + | + | + | + | ||||
| Lyu | 2020 | J Neurochem 155:522–37 |
Worm Mouse |
Meis1 Meis1 |
+ |
+ + |
+ |
NA + |
NA + |
|||
| Lyu | 2020 | J Neurosci Res 98:1532–48 | Mouse | + | + | O | + | + | ||||
| Lyu | 2020 | Neuroscience 440:85–96 | Mouse | Btbd9 | + | + | ||||||
| Mariano | 2014 | Rev Bras Gin Obst 36:436–41 | Rat | + | + | |||||||
| Meneely | 2018 | Front Behav Neurosci 12:199 | Mouse | Meis1 | + | + | O | + | + | |||
| Morais | 2021 | J Sleep Res 30:e13216 | Rat | Ptprd | + | + | + | |||||
| Muramatsu | 2019 | Sci Rep 9:16344 | Mouse | Btbd9 | + | + | ||||||
| Nelson | 1997 | J Nutr 127:2282–8 | Rat | + | + | + | + | |||||
| Ondo | 2000 | Mov Disord 15:154–8 | Rat | + | + | |||||||
| Pappas | 2008 | Brain Res 1214:1–10 | Mouse | + | + | + | ||||||
| Qu | 2006 | Exp Brain Res 168:152–6 | Mouse | + | + | |||||||
| Quiroz | 2010 | Exp Neurol 224:292–8 | Rat | + | A | + | + | |||||
| Quiroz | 2016 | Neuropharmacol 111:160–8 |
Mouse Rat |
+ + |
+ + |
A A |
+ + |
+ + |
||||
| Reis | 2011 | Spinal Cord 49:361–4 | Rat | + | + | |||||||
| Rivera-Oliver | 2019 | Mol Neurobiol 56:797–811 | Mouse | + | H | + | ||||||
| Salminen | 2017 | Dis Mod Mech 10:981–91 | Mouse | Meis1 | + | + | + | |||||
| Salminen | 2018 | J Sleep Res 27:e12557 | Mouse | Meis1 | + | + | ||||||
| Schulte | 2014 | Am J Hum Gen 95:85–95 | Fish | Meis1 | NA | NA | ||||||
| Silvani | 2015 | J Sleep Res 24:695–701 |
Mouse Rat |
|||||||||
| Spieler | 2014 | Genome Res 24:592:603 | Mouse | Meis1 | + | |||||||
| Sun | 2011 | J Neurol Sci 304:93–101 | Rat | + | + | O | + | |||||
| Unger | 2008 | J Nutr 138:2487–94 | Rat | + | + | + | + | |||||
| Unger | 2009 | J Appl Physiol 106:187–93 | Mouse | + | + | |||||||
| Unger | 2013 | Neuroscience 246:179–85 | Mouse | + | + | + | ||||||
| Unger | 2014 | Exp Neurol 261:462–8 | Rat | + | + | + | + | |||||
| Wang | 2004 | J Neurol Sci 220:59–66 | Rat | + | ||||||||
| Yepes | 2017 | Ann Neurol 82:951–60 | Rat | + | + | G | + | + | ||||
| Zhao | 2007 | J Neurosci Res 85:1065–76 | Mouse | + | + | + | + | |||||
| Zhu | 2008 | J Comp Physiol A 194:957–62 | Mouse | + | + |
Y year of publication, GENET genetic factors, DA dopamine, OT other transmitters (A adenosine, H histamine, Glu glutamate, O opioids), STR striatum, SC spinal cord, P pregnancy, FV face validity, CV construct validity, NA not applicable
The compliance of each of the primary research papers that dealt with putative rodent models of RLS with recently published consensus criteria for face [19] and construct [20] validity of rodent models of RLS was evaluated by two scorers, with face or construct validity scored in case the paper satisfied at least one of the respective criteria. Discrepancies between scorers were resolved by consensus. No attempt was made to assess other types of validity, such as predictive validity, and the validity of non-rodent RLS models, for which consensus validity criteria are presently lacking.
Overview of Research Papers on Putative Animal Models of RLS
Species and Publication Years
The publication year of the retrieved papers ranged from 1972 to 2022. Of the 81 primary research papers retrieved on putative animal models of RLS, most (77 papers) studied rodent models, particularly mice (Mus musculus, 44 papers) and rats (Rattus norvegicus, 33 papers). Research on vertebrate models also included two papers on non-human primates (Macaca mulatta in 2010 and Macaca fascicularis in 2015), one on cats (Felis silvestris, published in 2008) and one on Zebrafish (Danio rerio, published in 2014). Four papers studied invertebrate models, particularly worm (Caenorhabditis elegans, three papers published in 2011, 2020, and 2020) and fly (Drosophila melanogaster, one paper published in 2012) models. Four papers studied more than one species (mice and rats: two papers; mice and worms: two papers). Taken together, these data do not show a clearcut time trend in the animal model species of choice for studies on RLS, with most research still focusing on rodent models. Compared with research on mice, research on rats started earlier (first published paper in 1972 vs. 2004; median publication year: 2011 vs. 2015) but remains actual to date (the last paper on either species was published in 2022).
Targeted Pathophysiological Mechanisms
The primary research papers retrieved on putative animal models of RLS could be broadly categorized as targeting seven main pathophysiological mechanisms: alterations in dopaminergic signaling (55 papers), iron deficiency (46 papers), genetic risk variants (20 papers), striatal circuits (35 papers), spinal cord circuits (16 papers), other transmitter signaling pathways (12 papers), and pregnancy (one paper). The targeted genetic risk variants were BTBD9 (nine papers), MEIS1 (eight papers), PTPRD (two papers), and MAP2K5 (one paper). The targeted transmitters other than dopamine were opioids (four papers), histamine (four papers), adenosine (three papers), and glutamate (one paper). Most papers targeted multiple mechanisms (Table 1), with a substantial overlap in particular among the three most targeted mechanisms concerning dopaminergic signaling, iron deficiency, and striatal circuits (Fig. 2). This overlap was largely due to studies targeting the effects of iron deficiency on dopaminergic transmission in the striatum. There was also substantial overlap between studies targeting dopaminergic signaling and those targeting genetic risk variants (12 papers), spinal cord circuits (14 papers), and other transmitters (eight papers).
Fig. 2.
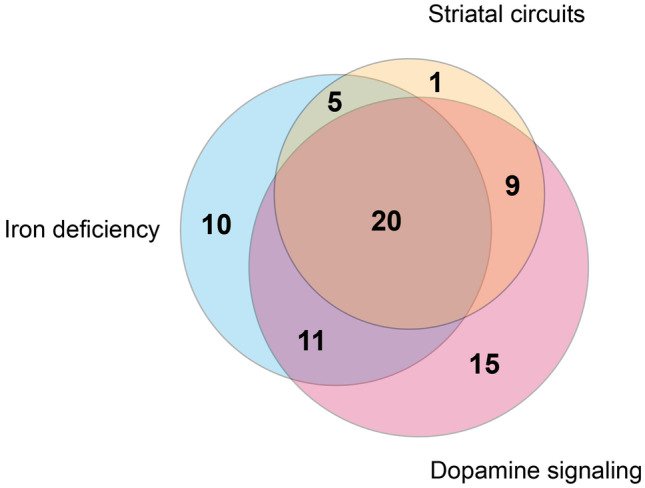
Repartition of selected manuscripts and their reported focus on iron deficiency, striatal circuits, and dopamine-related signaling in the RLS animal models
Face and Construct Validity
The vast majority of the previously published work on putative rodent models of RLS lacked either face or construct validity as defined in recently published consensus guidelines. In particular, out of the 75 papers on putative models of RLS that included rodent models, only 11 (14%) and 47 (63%) satisfied at least one face validity criterion [19] or at least one construct validity criterion [20], respectively (Table 1). Only six papers (8%) satisfied at least one face validity criterion as well as at least one construct validity criterion.
Putative Animal Models of RLS Targeting Dopaminergic Mechanisms
Overview of Dopaminergic Mechanisms in RLS Pathophysiology
Strong evidence for an important role of dopamine transmission in RLS stems from the excellent short-term pharmacological response to D2-like dopaminergic agonist medications [24, 25]. The efficacy of D2-like receptor agonists, such as pramipexole, ropinirole, and rotigotine, has been further confirmed in evidence-based reports [25, 26].
The 4 major brain dopamine pathways are the nigrostriatal, mesolimbic, mesocortical, and tuberoinfundibular pathways [27]. Dopamine receptors can be found both presynaptically [28–31] and postsynaptically [32–36]. The presence of these receptors on both sides of the synapse provides an opportunity for the nervous system to finetune dopamine-mediated responses, but it also makes it difficult to ascribe specific modulatory actions to any given receptor subtype or location. Dopamine receptors are strongly expressed in the striatum, basal nuclei [37–39], and spinal cord [40]. Dopamine receptors are divided into two classes, the D1-like (D1R and D5R) and D2-like (D2R, D3R, and D4R) receptors [27], and differentially regulate second messenger pathways by either increasing (D1-like: through the coupled Gs/olf G protein) or decreasing (D2-like: through the coupled Gi/o G protein) the activity of adenylate cyclase [27]. The affinity of the different receptor subtypes for dopamine is in the following order: D3R > D4R ~ D5R > D2R > D1R (data compiled from https://pdsp.unc.edu/databases/kidb.php, accessed Jul 29, 2022). Dopamine receptors can act independently or form heteromers (both heterodimers and heterotetramers). It is conceivable that at low levels of dopamine release, inhibitory actions promoted by D2-like receptors, and in particular by D3R, dominate overall outcome effects, while excitatory effects mediated by D1-like receptors occur only at higher local dopamine concentrations [41, 42].
In principle, a failure of diencephalo-spinal modulation induced by a compromised A11 hypothalamic dopamine neuron function could also be involved in RLS pathophysiology by causing a hypodopaminergic state at the spinal cord level (“Insights from Putative Animal Models of RLS on the Effects of Dopaminergic A11 Cell Dysfunction”). However, human autopsy data failed to reveal a neuropathological difference in the A11 region between RLS and age-matched control cases regarding the volume of tyrosine hydroxylase positive cells, general histological examination, or any signs of significant inflammation [43]. In addition, there is evidence for a hyperdopaminergic state in RLS, and the success of dopaminergic agents could then be ascribed to a preferential pre-synaptic effect [20]. Accordingly, in patients, brain imaging with positron emission tomography showed evidence of decreased dopamine transporter (DAT) and D2R binding potential in the striatum of subjects with RLS, consistent with an increase in nigrostriatal synaptic dopaminergic activity [44, 45]. One study reported abnormally increased levels of 3-ortho-methyldopa (3-OMD), an L-DOPA metabolite, and homovanillic acid (HVA), a dopamine catabolite, in the cerebrospinal fluid (CSF) of untreated RLS patients, indicating abnormally increased dopamine synthesis [46]. Interestingly, patients with increased 3-OMD CSF levels also had lower CSF ferritin compared to controls [46]. These results should be taken with caution, however, as one previous study did not find any changes in 3-OMD and HVA CSF levels in patients with RLS [47]. Nevertheless, autopsy data showed a significant decrease in D2R in the putamen of subjects with RLS, which correlated with disease severity, as well as increased levels of tyrosine hydroxylase and its phosphorylated (active) form in both the substantia nigra and putamen [48]. Increased levels of tetrahydrobiopterin (BH4, an essential cofactor of tyrosine hydroxylase) were also reported in the CSF of patients with RLS [49].
Rodent Model Validity Criteria Concerning Dopaminergic Mechanisms in RLS
Of the 55 selected studies dealing with putative animal models of dopaminergic mechanisms of RLS, 49 concerned rodent models. Of these, 28 studies were found to satisfy at least one criterion of construct validity [20], while 7 studies satisfied at least one criterion of face validity [19] (Table 1).
Published consensus criteria of the construct validity of rodent models of RLS included increased levels of BH4 and 3-OMD in the brain, increased activity of tyrosine hydroxylase in the substantia nigra and striatum, and decreased binding potential of striatal D2R, as well as exposure to L-DOPA to study the augmentation phenomenon [20]. Interventions on dopamine signaling were also considered relevant to the face validity of rodent models of RLS. The face validity criteria included RLS-like phenotype rescue at least initially with dopaminergic treatment as well as RLS-like phenotype aggravation with D2-like dopamine receptor antagonists or after short-term dopaminergic treatment withdrawal, with the possible exception of sleep-related RLS-like phenotype [19].
Rodent models of RLS addressing tyrosine hydroxylase and/or striatal D2R studied in combination with iron or genetic mechanisms will be discussed in the respective sections to avoid repetition. Here, we will focus on insights obtained from models of lesions of the dopaminergic A11 region and of D3R signaling impairment. Neither models were included among those with sufficient construct validity in consensus guidelines [20]. This was mainly due to the lack of evidence of A11 cell loss at autopsy in patients with RLS [43]. Moreover, there is no clinical or genetic evidence for alterations in D3R expression or function in RLS.
Insights from Putative Animal Models of RLS on the Effects of Dopaminergic A11 Cell Dysfunction
The A11 diencephalic area was shown to be the major source of L-DOPA in the non-human primate spinal cord, where it may play a role in the modulation of sensorimotor integration through D2R and D3R, either directly or indirectly, via dopamine synthesis in spinal dopa-decarboxylase-positive cells [40]. A targeted lesion of the A11 dopaminergic neurons led to an increase in locomotor activities in a rodent model that was reversed by treatment with a dopamine D2-like receptor agonist [50]. In addition, there is evidence of spinal projections from the dopaminergic A10 neurons of the ventral tegmental area [51]. Together, these data suggest that descending dopamine pathways into the spinal cord exist and that they can alter locomotor behavior. A failure of spinal cord modulation by the A11 hypothalamic dopamine neuron cluster was found to lead to hyperactive behavior in rats [52]. These results were later replicated and expanded with polysomnographic characterization, showing increased wakefulness and limb motor activity during sleep, particularly at the end of the dark (active) period in rats with 6-hydroxydopamine lesion of the A11 area [53], with a protective effect of physical exercise [54]. Descending A11 projections were reported to contribute to the suppression of motor activity during sleep and sleep–wake transitions in rats, with their disruption eliciting RLS-like movements, although similar conclusions were also reached for cerebello-rubro-spinal projections [55]. Further discussion of putative RLS models with A11 dysfunction is provided in “Putative Animal Models of RLS with Diet-induced Brain Iron Deficiency” and “Insights from Putative Animal Models of RLS on the Effects of Iron Deficiency on Alterations in Dopamine Transmission” in combination with iron deficiency.
Insights from Putative Animal Models of RLS on the Effects of D3R Signaling Impairment
Drugs such as pramipexole and ropinirole that show clinical effectiveness for RLS have more affinity for D3R than for other dopamine receptor subtypes. However, at clinically relevant doses, these drugs most probably occupy and activate a significant proportion of D4R and D2R, particularly the short isoform of D2R (D2SR). In addition, D3R, D2SR, and D4R are preferentially pre-synaptic, acting as autoreceptors and heteroreceptors that modulate dopamine and glutamate release. Nevertheless, the clinical efficacy of D2-like dopaminergic treatments is at least compatible with the role of D3R in mediating RLS symptoms [56–58].
The D3R knockout (KO) mouse (D3R-KO), originally developed by the introduction of a premature chain-termination mutation in the gene coding for D3R, showed hyperactivity in an exploration test as well as increased locomotor activity and rearing behavior, suggesting that the D3R mediates inhibition of the overall behavioral phenotype [59]. The disruption of the D3R was also associated with renin-dependent hypertension [60]. The gradual decline in responsiveness to long-term treatment with D2-like receptor agonists in mice could be reversed or rescued by the adjuvant block of D1R in animals that were no longer responsive to the D2-like receptor agonist alone, supporting the hypothesis that upregulation of D1R may play a role in the augmentation phenotype of RLS [61]. If replicable in the clinic, the mechanisms leading to augmentation in RLS patients after long-term use of D2-like receptor agonists might then be based on interactions between D2-like and D1-like receptors, as previously suggested to occur through receptor heteromerization [61]. This hypothesis would point to a synergistic role of D1R and D3R in RLS, and it could provide an alternative to the D2-like receptor-based treatment options currently employed [62].
Closing Remarks
The clinical studies suggest an important role of dopamine signaling in RLS. Most putative animal models of RLS addressed dopaminergic dysfunction in combination with other mechanisms, mainly iron deficiency and genetic alterations. While it has been suggested that D3R are the main targets for the therapeutic effects of currently used dopaminergic agents in RLS, animal models with D3R alterations have only incompletely been able to capture and replicate the full phenotype of RLS. It remains unclear whether these models may help understand the pathophysiology of augmentation. On the other hand, while animal models have provided proof or principle of functional behavioral effects of lesions of the dopaminergic A11 cell group, their relevance to RLS pathophysiology is uncertain due to the lack of consistent evidence of A11 region pathology at autopsy in patients with RLS. Nevertheless, it is conceivable, but still unproven, that a dysfunction of the descending A11 system may affect its projections in the spinal cord while sparing the A11 cell bodies.
Putative Animal Models of RLS Targeting Iron-related Mechanisms
Overview of Iron-related Mechanisms in RLS Pathophysiology
Investigating the complex relationship between iron and RLS has generated the most important body of literature on RLS. Based on the evidence of deficient iron storage in RLS patients and improvement in some patients with iron supplementation [63, 64], it is likely that brain iron status may play a primary role in the pathogenesis of RLS [65]. In patients with RLS, low CSF iron and ferritin concentrations and increased CSF transferrin suggesting brain iron insufficiency were documented [66, 67]. Direct magnetic resonance imaging (MRI) assessment of brain regional iron concentration further supported these findings, albeit with some inconsistencies [68–75]. Pathological autopsy studies also showed a decreased iron staining in the neuropil consistent with MRI data and with impaired brain iron acquisition in RLS [48]. Brain imaging and post-mortem brain studies of patients with RLS evidenced a reduction in myelin, loss of myelin integrity, and decreased ferritin and transferrin in the myelin fractions [76]. The relatively high prevalence (24%, 9 times higher than in the general population) of clinically significant RLS among subjects with iron-deficient anemia [77] also suggests that peripheral iron deficiency represents a risk or triggering factor for RLS, enabling the development of brain iron deficiency in susceptible individuals [20].
Rodent Model Validity Criteria Concerning Iron-related Mechanisms in RLS
Of the 46 selected studies dealing with putative animal models of iron-related mechanisms of RLS, 43 concerned rodent models. Of these, 31 studies were found to satisfy at least one criterion of construct validity [20], while 6 studies satisfied at least one criterion of face validity [19] (Table 1). Consensus criteria of the construct validity of rodent models of RLS included peripheral iron deficiency, as estimated by hemoglobin or serum ferritin levels; low levels of CSF ferritin, high levels of CSF transferrin and a reduced CSF ferritin-to-plasma ferritin ratio; decrease in brain iron content, particularly in the substantia nigra, striatum, pallidal complex, and thalamus; decreased myelination, and reduced iron and H-ferritin content in oligodendrocytes, which are the myelin-producing cells in the central nervous system (CNS); and increased levels of serum hepcidin [20]. A few studies on rodent models of iron deficiency did not fully meet RLS construct validity criteria (Table 1) due to limited or absent documentation of peripheral and/or central iron deficiency, which complicated discrimination between these two conditions in animal models. Consensus criteria of face validity for RLS rodent models also recommended checking for the effectiveness of an iron-deficient diet or iron treatment on the iron content of brain structures of interest [19]. The face validity criteria also included RLS-like phenotype manifestation or aggravation with iron-deficiency as well as RLS-like phenotype rescue with iron treatment for conditions of iron deficiency. In this context, the RLS-like phenotype referred particularly to increased activity and decreased resting in the last part of the active period in the home cage, possibly excepting PLMS-related phenotype [19].
Putative Animal Models of RLS with Diet-induced Brain Iron Deficiency
Research on iron-deficient animal models of RLS has been performed mostly on wild-type (WT) rats or mice fed iron-deficient diets from weaning. Rats are more susceptible to developing iron-deficiency anemia due to dietary iron deprivation than are at least some commonly used laboratory mouse strains such as C57BL/6. Distinct differences in the susceptibility to dietary iron deficiency also occur between rat strains, such as Fischer 344, Wistar, and Sprague Dawley [78]. In Sprague–Dawley rats, brain iron deficiency was demonstrated after as few as 2 weeks of the iron-deficient diet with 2 ppm iron during the postweaning period [79] or after 4 weeks of the diet with 3 ppm iron [80]. After 40 days of a diet with 4 ppm iron, Sprague–Dawley rats developed decreased hemoglobin and brain iron content and markers of oligodendrocyte dysfunction [81]. In C57BL/6 mice, spinal cord iron was decreased by as few as 6 weeks of the diet with 3.5 ppm iron [82, 83]. On the other hand, a much longer 24-week period of the diet with a less severe iron deficiency (< 9 ppm iron) decreased liver and serum iron concentration but did not affect either brainstem iron content or blood hemoglobin concentration in C57BL/6 mice [84]. Normative studies clarifying the effects on brain iron of different levels of dietary iron intake as a function of the duration of the dietary treatment are, to our knowledge, not available in rats or mice. Therefore, as a cautionary measure, the administration of a diet with minimal (2–3 ppm) iron content from weaning appears advisable to elicit CNS iron deficiency in rats and mice. These dietary regimens were also shown to elicit what appear as compensatory changes in iron-management proteins in the brain. A diet with 2 ppm iron increased brain cytosolic transferrin concentration in rats [79], whereas, in mice, an iron-deficient diet with 4 ppm iron increased striatal transferrin receptors [85]. Studies on WT mice fed with intermediate levels of dietary iron from weaning [86–88] have also reported distinct behavioral and molecular effects of the diets, including increased transferrin receptors in the cortex and striatum, albeit without direct demonstration of brain iron deficiency.
A set of studies on recombinant inbred BXD mouse strains, generated by crossing C57BL/6 and DBA/2 J WT mice and then inbreeding the F2 progeny for 20 consecutive generations, shed light on differences in the regulation of iron content among different structures of the CNS as well as between these structures and peripheral organs. These studies demonstrated that the regulation of central and peripheral iron content shows sex-related differences and distinct biological rhythms. This is a very pertinent finding relevant to RLS, as the RLS risk is twice as high in women as in men in the general population [89] (cf. also “Rodent Model Validity Criteria Concerning Genetic Mechanisms in RLS” below). Interestingly, studies on BXD mice failed to detect significant genetic correlations between the iron content of the ventral midbrain, which includes the substantial nigra pars reticulata, which has a much higher content of iron than the pars compacta [90], and the iron content of either the liver or the prefrontal cortex, in the face of significant correlations between ventral midbrain iron content and iron content in the caudate-putamen and nucleus accumbens [91]. These results suggested that peripheral and central iron regulatory systems are largely independent. One study using six strains of BXD mice to test the effects of iron deficiency on gene expression in the ventral midbrain revealed several iron deficiency-induced expression changes [92]. In line with different strain susceptibility to iron deficiency in the brain, large strain-dependent differences were found in three genes with direct ties to nigrostriatal dopamine functioning, two of which are implicated in an iron-dopamine-pathway: stromal cell-derived factor 1 (Cxcl12), a ferritin regulator and potent dopamine neuromodulator, and hemoglobin beta adult chain 1 (Hbb-b1), a gene recently shown to play a functional role in dopamine neurons [92].
Differences also emerged between brain structures in response to iron supplementation: In female BXD40 mice, iron content in the ventral midbrain and nucleus accumbens decreased with an iron-deficient diet and quickly recovered after iron supplementation, whereas iron content in the prefrontal cortex, caudate-putamen, cerebellum, and pons did not significantly change [92, 93]. In addition, variations in ventral midbrain iron concentrations strongly and positively correlated with Btbd9 expression [94]. Unfortunately, the studies on BXD mice published to date did not address the relationships between brain and spinal cord iron content. The latter was shown to be decreased by dietary iron deficiency in mice, with the effect enhanced by unilateral chemical lesions of the A11 region, which provides descending projections to the spinal cord [82]. The link between spinal cord function and iron metabolism may be bidirectional, as chronic spinal cord injury was found to decrease plasma iron and transferrin levels in rats [95].
Regarding biological rhythms, in the BXD40 mouse strain, which has midrange iron content in the ventral midbrain among the BXD recombinant inbred strains, liver iron was increased and plasma iron was decreased in the dark period compared with the light period [96]. Administration of an iron-deficient diet eliminated this variation in liver iron in male and female mice and in plasma iron in male mice. Dietary iron deficiency decreased ventral midbrain and nucleus accumbens iron during the dark and the light period, whereas it decreased whole-brain iron only during the light period [96]. Accordingly, an iron-deficient diet decreased iron content in the dorsal striatum in BXD40 male and female mice only during the light phase [97]. Day-night rhythms in serum and CSF iron have also been reported in Macaca fascicularis, a non-human primate, and found to be in counterphase [98]. It remains to be addressed whether these day-night rhythms in iron regulation represent true circadian rhythms, i.e., endogenous rhythms with a near-24-h free-running period that can be entrained to external time signals such as light.
Insights from Putative Animal Models of RLS on the Effects of Iron Deficiency on Alterations in Dopamine Transmission
In rats, dietary iron deficiency was consistently found to increase striatal extracellular levels of dopamine and its metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and HVA [79, 99–101]. This was not explained either by intracellular dopamine levels, which were found to decrease in rats with iron deficiency [101], or by anemia per se [102]. Rather, the increase in extracellular dopamine levels in the striatum of iron-deficient rats was associated with altered striatal dopamine uptake, which was decreased and showed impaired feedback enhancement by agonists of D2R autoreceptors [101]. The changes in dopamine levels in response to dopamine reuptake blockade with cocaine were also significantly attenuated in iron-deficient rats [102]. These alterations could be traced down to decreased levels of DAT at the mRNA and protein level, particularly in the membrane fraction of the striatum [80, 100, 103]. In agreement with this picture, repeated blood withdrawal to induce iron deficiency increased striatal dopamine and DOPAC levels in Macaca fascicularis [98]. Moreover, an iron-deficient diet decreased DAT levels in the striatum of BXD40 recombinant inbred mice [97] and in the nucleus accumbens but not the striatum of rats with neonatal iron deficiency [103]. However, increased striatal DAT in rats [104] and mice [105] with dietary iron deficiency was also reported. Sedentary offspring of dams with dietary iron deficiency since weaning also had increased striatal DAT protein compared with exercised offspring of dams with a standard diet or iron restriction during pregnancy [106]. On the other hand, research on rats suggested that the release of dopamine, and not just its reuptake, may be affected by iron deficiency. In rats with dietary iron deficiency, iron infusion at physiological concentrations into the ventral midbrain reversed the increase in extracellular dopamine level and the decrease in intracellular dopamine level in the striatum but had no effect on alterations in dopamine uptake [101]. Significant increases in tyrosine hydroxylase, the rate-limiting enzyme of dopamine biosynthesis, and in its phosphorylated active form were also seen both in rat pups that were iron-deficient during development and in iron-chelated PC12 cells, a cell model of iron insufficiency [107]. This is in line with RLS autopsy data [48] and with increased pre-synaptic dopaminergic activity in patients with RLS, as discussed in “Overview of Dopaminergic Mechanisms in RLS Pathophysiology.”
In WT rats, early studies found that dietary iron deficiency decreased D2R binding in the caudate nucleus [108, 109]. Although the effect was reproduced by incubation of caudate nucleus membrane homogenates with iron chelators, it could not be rescued by incubation of caudate nucleus membrane homogenates from iron-deficient rats with iron salts [109]. Nevertheless, dietary iron deficiency was also found to decrease D2R protein expression in the striatum and nucleus accumbens in rats [110]. These findings were later confirmed [103] and linked to preferential loss of membrane D2R protein, with less of a decrease in cytosolic D2R protein and no significant changes in D2R mRNA [96, 111, 112]. Results concerning D2R have been less consistent in iron-deficient mice, with downregulation in the striatum [88], no significant change in the striatum [105], or no significant change in the lumbar spinal cord [82]. Similarly, results concerning D1R have been inconsistent in iron-deficient rats and mice, with a downregulation in the striatum in rats in the absence of significant effects on D1R mRNA expression [111], no significant effects in the nucleus accumbens, striatum and prefrontal cortex in another experiment on rats [103], and upregulation in the striatum [105] or lumbar spinal cord [82] in mice. Interestingly, the impact of iron deficiency on dopamine was more pronounced in male than female animals, suggesting sex-related differences in the sensitivity to nutrient deprivation [111].
Further evidence supporting interactions between iron deficiency and dopamine signaling concerned behavioral phenotype. After an iron-deficient dietary treatment, the peak increase in activity was anticipated from Zeitgeber Time (time in hours from lights on, ZT) 23–24 to ZT19-22 in D3R KO mice with respect to WT mice [113]. In a different study on BXD40 recombinant inbred mice, the increase in activity late in the dark period with iron-deficient dietary treatment was blunted by L-DOPA or by quinpirole, a D2-like receptor agonist [114]. Moreover, mice with dietary iron deficiency and A11 lesions showed an increase in movements, which was attenuated by acute and chronic treatment with pramipexole, a D2-like receptor agonist with a preference for D3R [83]. Pramipexole also decreased tibialis anterior electromyogram (EMG) bursts during sleep, akin to PLMS, in iron-deficient rats [104, 115].
Finally, mice KO for Thy-1, a CNS glycosyl-phosphatidylinositol-linked integral membrane protein coded by a gene responsive to iron deficiency [116], showed increased striatal levels of dopamine, D1R and D2R [105] but no change in the expression or activity of tyrosine hydroxylase [117]. Six-month-old rats born to dams maintained on an iron-deficient diet from gestational day 5 to weaning also showed a decreased Thy-1 expression in brain homogenates [107].
Insights from Putative Animal Models of RLS on Behavioral Effects of Iron Deficiency
Despite the wealth of published data on animal models of iron deficiency, there is still a lack of firm connections between iron-dependent neurochemical events and behavioral changes. In early work and contrary to what is expected with regard to RLS, rats with dietary iron deficiency exhibited decreased motor activity, reduced levels of spontaneous movement and exploration of a novel environment, and markedly diminished behavioral responses to centrally acting drugs [103, 108, 118, 119]. The reduced levels of activity in these iron-deficient animals were in line with the reduced spontaneous activity observed in humans with iron-deficiency anemia [120]. The decrease in activity in these rats occurred mainly during the dark period [119], which is physiologically the more active, and could be so marked as to cause the reversal of the day-night activity rhythm [118]. Alterations in behavioral tests were also noted [121], particularly with increased anxiety-like behavior. More recent data on young adult rats (approximately 3 months of age) with dietary iron deficiency did not report a generalized decrease in activity but, rather, a reduction in REM sleep time, increased sleep fragmentation, and the occurrence of tibialis anterior EMG bursts akin to human PLMS [104, 115, 122].
It is unclear whether differences in the reported activity phenotype between earlier and more recent studies on rats with iron deficiency resulted from differences in the research protocol or the timing of the behavioral tests. One possible explanation is that iron deficiency does not affect all brain regions equally, since the caudate-putamen shows a 30% loss of iron concentration in iron-deficient rats, while other regions, including the substantia nigra, may be unaffected [123]. Another explanation could be related to the fact that brain iron deficiency has a differential effect on dopaminergic signaling at different biological times of the day and night [97]. The PLMS-like activity was reported in 40% of a small sample of old (16.2–20.5 months of age) WT rats fed an iron-sufficient diet, but not in younger (1.4–1.6 months of age) rats [124]. The PLMS-like activity was also reported in young adult spontaneously hypertensive rats together with a significant reduction in sleep efficiency during the dark period, with both alterations being rescued by pramipexole, a preferential D2-like receptor agonist [125]. On the other hand, young adult WT mice and rats showed a pattern of short-interval hindlimb EMG activity during sleep, which is akin to short-interval leg movements during sleep in human subjects [126, 127].
A generalized decrease in activity was also not reported in studies on iron-deficient mice. Rather, in WT mice, dietary iron deficiency was found to increase wakefulness at ZT 20–24 [86, 113] due to reductions in non-REM sleep and REM sleep time [86]. A study on female mice of the recombinant inbred BXD40 strain found an earlier increase in activity in the active period (ZT 20) with an iron-deficient diet [114]. The activity increase in the late active period was also correlated with decreases in ventral midbrain iron content during iron-deficient dietary treatment in different recombinant inbred mouse strains [128]. A more overt effect of iron deficiency on locomotion was shown in iron-deprived C57BL/6 mice with bilateral 6-hydroxydopamine lesioning in the A11 nucleus of the diencephalospinal pathway [50]. In this model, locomotor activity was significantly increased both in the mice that were iron-deprived and in the A11-lesioned mice compared with controls. The combination of iron deprivation and A11 lesions further significantly augmented activity, which was normalized after treatment with a D2-like receptor agonist, supporting the role of the spinal dopamine neurotransmission in the pathophysiology of RLS [50]. Finally, research on iron-deficient WT mice highlighted significant increases in pain sensitivity [87, 113], which may also be relevant to the pathophysiology of RLS.
Closing Remarks
Evidence suggests that the decrease of iron in the patient brain tissue could be a major contributing factor to RLS pathophysiology [63, 68, 129]. Figure 3 provides an overview of the behavioral and physiological changes reported in the different brain iron-deficient models. No consistent relationship could be found between peripheral and brain iron regulation and content in RLS [130], and the factors mediating the link between systemic and CNS iron deficiency are far from being established. Research on iron-deficient putative animal models of RLS has been built upon research on peripheral iron deficiency and shifted the focus toward the consequences of CNS iron deficiency. These models were generally far from explaining the pathophysiology responsible for brain iron deficiency, as mostly based on feeding iron-deficient diets from weaning to induce brain iron deficiency. Nevertheless, these models have provided proof of the principle of causal links between iron deficiency, alterations in dopamine transmission generally consistent with the hyperdopaminergic hypothesis of RLS, and RLS-like behaviors.
Fig. 3.

Overview of the impact of the brain iron deficiency models on behavior and physiological functions. Blue arrows indicate an upregulation of the phenotype; red arrows indicate a downregulation
Putative Animal Models of RLS Targeting Genetic Mechanisms
Overview of Genetic Mechanisms in RLS Pathophysiology
The GWAS for RLS implicated up to 23 risk loci, including BTBD9, MEIS1, PTPRD, and MAP2K5, as genetic risk factors [131–135]. BTBD9 gene variants have been identified among those imparting the highest risk for RLS [133, 136, 137]. The odds of having RLS with the BTBD9 variant are 126 to 100 [127]. Recently, BTBD9 has also been associated with sleep duration, timing, and daytime sleepiness [138, 139]. BTBD9 codes for a protein belonging to the BTB (POZ) protein family that modulates transcription, cytoskeletal arrangement, ion channels, and protein ubiquitination [140–142]. A seminal GWAS paper published on RLS also showed an association of the BTBD9 variant with serum ferritin concentration [136], with serum ferritin levels decreased by 13% per variant allele. This finding was partially replicated by others [143–145]. Interestingly, lower CSF ferritin levels have been reported in patients with RLS [66, 67]. Although these studies did not detect changes in the serum ferritin level, low ferritin levels in the CSF and serum may occur together in RLS patients, particularly in those with iron deficiency anemia [130].
In addition to BTBD9, GWAS implicated MEIS1 as the top hit among the 23 risk loci of RLS [131–135]. The odds of having RLS with the MEIS1 variant are 192 to 100 [131]. Interestingly, two independent GWAS nominated MEIS1 with an increased risk of having insomnia [146, 147]. Furthermore, MEIS1 was found to be associated with sleep duration, efficiency, and timing [138]. Taken together, these studies thus supported the role of MEIS1 in sleep regulation. MEIS1 is widely expressed in the CNS and periphery during embryogenesis and after birth [148, 149], including in the basal nuclei and the midbrain. The MEIS1 protein acts as a transcription factor and belongs to the TALE [Three Amino-acid Loop Extension]-class homeobox protein [150].
PTPRD, coding for protein tyrosine phosphatase receptor type delta, is another RLS gene nominated by GWAS [151]. The odds of having RLS with the PTPRD variant are 144 to 100 [151]. PTPRD variants make pleiotropic contributions of differing strength to several brain phenotypes and disorders [152], and there is a moderate expression of PTPRD ligands in the striatum, many thalamic nuclei, and the hypothalamus [153]. Ptprd, the mouse ortholog of human PTPRD, is implicated in motoneuron axon targeting during development [154].
The MAP2K5/SKOR1 locus has also been associated with RLS by GWAS [137, 155–157]. MAP2K5 codes for mitogen-activated protein kinase 5, which phosphorylates ERK5 and regulates the expression NF-κB and the resulting inflammatory response associated with RLS [158]. SKOR1 codes for SKI family transcriptional corepressor 1 and regulates the development of spinal cord interneurons [159]. The odds of having RLS with the MAP2K5/SKOR1 variants are 152 to 100 [137].
Rodent Model Validity Criteria Concerning Genetic Mechanisms in RLS
Of the 20 selected studies dealing with putative animal models of genetic mechanisms of RLS, 17 concerned rodent models. Of these, 16 studies were found to satisfy at least one criterion of construct validity [20], while 6 studies satisfied at least one criterion of face validity [19] (Table 1). Genetic and sex-related factors were included in the consensus criteria of the construct validity of rodent models of RLS, with specific reference to common and rare genetic variants identified with GWAS [20]. However, the guidelines remarked that the conventional KO strategy may not faithfully recapitulate the human genetic alterations [20], even for variants associated with decreased gene expression. Both construct [20] and face [19] validity guidelines recommended that experiments be performed on rodent models of both sexes. On the other hand, face validity guidelines refrained from recommending fixed age ranges for the study of mouse and rat RLS models [19].
Insights from Putative Animal Models of RLS on the Effects of BTBD9 Deficiency
The mouse ortholog of BTBD9 was the first among the RLS risk genes nominated by GWAS to be genetically inactivated in animal models. In Btbd9 complete KO mice, hippocampal synaptic transmission and plasticity were altered, with enhanced memory and long-term potentiation [160]. Moreover, loss of the BTBD9 ortholog increased motor activity, decreased dopamine levels, and disrupted sleep [161]. Similarly, Btbd9 KO mice showed motor restlessness, thermal hypersensitivity, and a disruption in sleep structure [162–164]. Treatment with a D2-like receptor agonist restored the sensory deficit of the Btbd9 KO mice [162] and sleep fragmentation in the KO fly [161]. In addition, the conditional KO of BTBD9 ortholog in dopamine neurons recapitulated the sleep deficits seen in the complete KO model [161]. However, similar experiments in mice only produced a mild sleep deficit, suggesting differential vulnerability to dopamine signaling disruption in these two genetic models [165]. Finally, the D2R protein and mRNA levels were altered in Btbd9 KO mice in a circadian-dependent manner, possibly through the action of dynamin-1 [165].
Conditional knockout of Btbd9 in various brain regions or cell types has been conducted to determine brain regions involved in the pathogenesis of RLS. This approach was complemented with manganese-enhanced magnetic resonance imaging to determine neural activity changes in specific brain regions [166–169]. Mn2+ is a Ca2+ analog, enters active synapses through voltage-dependent calcium channels [170, 171], and is sequestered and trans-synaptically transported antero- and retrogradely across active neural circuits [172, 173]. The presence of the paramagnetic Mn2+ ion in the brain increases longitudinal relaxation rates, enhances signal intensity in T1-weighted scans, and is utilized for functional mapping of synaptic activity [174]. With this technique, the Btbd9 KO mice were found to show increased neural activity in the cerebral cortex and striatum, indicating an increased cortical input to the striatum [169]. In these experiments, Btbd9 was also selectively knocked out in the medium spiny neurons, which account for over 95% of striatal neurons, to obtain Btbd9 sKO mice [169]. In a 30-min open-field activity assay, Btbd9 sKO mice were found hyperactive, which fits with the characteristic urge to move of RLS patients. Btbd9 sKO mice also showed a significantly increased probability of waking and voluntary activity in the long-term open-field and wheel running tests. These changes were restricted to the light phase, when mice usually sleep or rest, and did not occur in the dark phase, when mice are typically active [169]. Even with opposite day-night rhythms to humans, therefore, Btbd9 sKO mice show motor restlessness and a similar circadian predominance to patients. Finally, uncomfortable sensations in the lower limbs are another common phenotype of RLS [175]. The Btbd9 sKO mice had a higher sensitivity to the heat stimuli in the tail-flick test, indicating altered thermal sensation similar to complete Btbd9 KO mice [175]. Cholinergic neuron-specific Btbd9 KO (ChKO) and cerebral cortex-specific Btbd9 KO (cKO) mice have also been generated and characterized. The ChKO mice did not show any sleep and sensory deficits [169]. In contrast, the cKO mice showed a rest-phase specific motor restlessness, a thinner primary somatosensory and primary motor cortex, and decreased thermal sensation, which is opposite to the findings in complete Btbd9 KO mice [169, 176]. Furthermore, dopamine neuron- and Purkinje cell-specific conditional Btbd9 KO mice have been generated. Both lines of conditional KO mice failed to show sensory deficits [165, 177]. These conditional KO experiments suggest that the loss of BTBD9 function in the medium spiny neurons is sufficient to cause RLS-like phenotypes and highlight the importance of striatum or caudate/putamen in the pathogenesis of RLS. This conclusion is supported by an analysis of 376 post-stroke patients, which identified the body of the caudate nucleus as playing a role in the development of post-stroke RLS [178].
Finally, evidence from animal models also suggests that BTBD9 functions are related to iron metabolism. Genetic mapping of BXD recombinant inbred mouse strains revealed that Btbd9 gene expression correlates with ventral midbrain iron level [94]. Btbd9 KO mice also showed increased serum iron levels with no difference in striatal iron content [163]. BTBD9 overexpression in HEK cells revealed that BTBD9 acts together with Cullin-3 to regulate levels of iron-responsive element-binding protein 2 levels in the cell, which control ferritin expression and iron metabolism [162].
Insights from Putative Animal Models of RLS on the Effects of MEIS1 Deficiency
A MEIS1 intronic haplotype linked to RLS risk was associated with decreased protein expression [179], suggesting the variant causes haploinsufficiency. Accordingly, GWAS-driven experiments on zebrafish embryos revealed a significant excess of functionally null variants of MEIS1 in individuals with RLS compared with control subjects [180]. The lack of both alleles of Meis1, the mouse ortholog of human MEIS1, is embryonic lethal [149]. However, heterozygous Meis1 KO (Meis1+/−) mice are viable and have been used to study human RLS and other sleep disorders. So far, three lines of Meis1+/− mice have been characterized, and all three lines show some RLS-like phenotypes. The first Meis1+/− mouse model was created by knocking-in the modified estrogen receptor hormone-binding (ERT2) domain in-frame with the MEIS1a isoform coding region, while disrupting the MEIS1b coding region and the C-terminal of MEIS1 protein [149]. The model was tested for sleep disturbance and other RLS-like phenotypes. It showed hyperactivity, specifically at the beginning of the rest period, impaired pre-pulse inhibition of startle response, and a trend of reduced delta power in the electroencephalogram [181–183]. However, as pointed out later [184], the MEIS1 protein with C-terminal deletion lacks transcription-active function but retains the ability of DNA binding and complex formation [185]. Therefore, this line of Meis1+/− mice has a novel Meis1aERT2 fusion protein, which may have a toxic gain-of-function effect. With ERT2 fusion, the function of proteins normally interacting with MEIS1 protein may also have changed. It is challenging, therefore, to interpret the results obtained with this line of Meis1+/− mice and link the reported deficits unambiguously to the reduction of MEIS1 function.
The second line of Meis1+/− mice was derived from Meis1 loxP mice [loxP sites flanking exon 8] [186, 187]. This line does not have complications as the earlier mouse model and is predicted to decrease both MEIS1a and MEIS1b isoforms. These Meis1+/− mice were also found to be hyperactive [184] and to have an increased probability of waking during the light (rest) phase but not during the dark (activity) phase. In addition, these Meis1+/− mice did not have changes in serum iron and transferrin levels or in the iron content of the substantia nigra, but exhibited increased ferritin levels [188]. Nevertheless, a link between MEIS1 and iron metabolism was suggested by experiments with RNA interference for the Meis-1 ortholog, which increased ferritin expression (FTL-1 gene) in the presence of iron in the growth medium [189]. This second line of Meis1+/− mice also had increased dopamine turnover and decreased tyrosine hydroxylase levels in the striatum [188].
The third line of Meis1+/− mice was developed with the insertion of a gene coding for an enhanced cyan fluorescent protein into the first exon of the Meis1 gene [190]. This mouse line also showed spontaneous motor hyperactivity, including at the beginning of the rest period [191]. The Meis1 gene has also been conditionally knocked out in D2R-positive neurons. However, the resulting conditional KO mice did not exhibit RLS-like symptoms as assayed by open-field test and actimetry [191], suggesting MEIS1 in these D2R-positive neurons plays a less critical role in the pathogenesis of RLS. Other neuronal types and brain regions should be targeted in the future to determine the key neural circuits in which MEIS1 is involved in RLS pathogenesis.
Insights from Putative Animal Models of RLS on the Effects of PTPRD Deficiency
Analysis of 119 post-mortem brain samples revealed that the two RLS variants of PTPRD show a nominally significant association with lower PTPRD mRNA levels, supporting the use of Ptprd KO mice as a model of RLS [192]. The Ptprd KO mice showed growth retardation and semi-lethality, but with special care, most of them survived for a year or longer [192]. Like Btbd9 KO mice [160], the Ptprd KO mice showed enhanced hippocampal long-term potentiation and impaired learning and memory [192]. Finally, the Ptprd KO mice showed enhanced locomotion and reduced sleep, consistent with other target mouse models of RLS mentioned above [193]. On the other hand, no significant differences in PTPRD expression at the mRNA and protein levels in the striatum and spinal cord were detected in spontaneously hypertensive rats [194], which have been proposed as a possible model of sleep-related movement disorders [125, 195].
Insights from Putative Animal Models of RLS on the Effects of MAP2K5 Deficiency
Experiments attempting KO of Map2k5, the mouse ortholog of human MAP2K5, led to embryonic lethality [196, 197]. However, the heterozygous Map2k5+/− mice are viable, show decreased levels of Map2k5 mRNA [197], and can be studied to test RLS-related phenotypes. Female Map2k5+/− mice showed motor deficits at the Rotarod test and decreased locomotion during the rest period, which was opposite to changes observed in most genetic RLS models analyzed. Male Map2k5+/− mice displayed impairments in open-field exploration and pre-pulse inhibition of acoustic startle response. Interestingly, Map2k5 KO mice also showed decreased dopamine cell survival and tyrosine hydroxylase levels, highlighting the role of MAP2K5 in dopamine neuron function [197].
Closing Remarks
The most important contribution of the research on putative models of RLS with genetic alterations is the validation of the RLS risk genes based on GWAS nomination. Figure 4 provides an overview of the behavioral and physiological changes reported in the different genetic models. Mutation of risk gene orthologs in mice showed phenotypes relevant to RLS, such as alterations in iron metabolism, dopamine function, sensory perception, and sleep, affirming their involvement in RLS pathogenesis. Furthermore, it contributed to the identification and dissection of molecular pathways, cellular mechanisms, cell types, and brain circuits potentially involved in the RLS pathophysiology. Some animal models showed amelioration of RLS-like phenotypes in response to D2-like dopaminergic treatment. These models will likely be suitable for future preclinical RLS drug testing.
Fig. 4.

Overview of the impact of the different transgenic RLS models on behavior and physiological functions. Blue arrows indicate an upregulation of the phenotype; red arrows indicate a downregulation
Putative Animal Models of RLS Targeting Alterations in Striatal and Spinal Neural Circuits
Overview of Striatal and Spinal Circuit Alterations in RLS
The clinically defined symptoms of RLS include an urge to move the legs, usually accompanied or caused by uncomfortable and unpleasant sensations in the legs. The urge to move the legs indicates the involvement of brain circuits that integrate sensations and control locomotor output, while the sensory aspects of the symptoms and the activation of leg movements point to an involvement of spinal cord circuits.
One key hypothesis on the mechanisms that may underlie RLS is focused on a dysfunction of the circuits in the basal ganglia (cerebral nuclei) associated with changes to the central dopamine signaling, which in turn might lead to the urge to move and to the PLMS observed in RLS. The basal ganglia may be considered to include the substantia nigra pars compacta and reticulata, the globus pallidus with its internal and external segments, and the striatum with its caudate and putamen components (Fig. 5A). Within the basal ganglia, the striatum is the primary recipient of inputs from the cerebral – but not visual or auditory – cortex, intralaminar thalamic nuclei, and substantia nigra. Direct GABAergic projections from the striatum to the internal segment of the globus pallidus and substantia nigra pars reticulata mainly express D1R. These projections are likely involved in the facilitation of voluntary movement. In contrast, the indirect GABAergic projections to the internal segment of the globus pallidus and substantia nigra pars reticulata mainly express D2R and are thought to suppress voluntary movements. Alterations in striatal circuits may be responsible, at least in part, for the finding of increased excitability of the motor cortex of patients with RLS with transcranial magnetic stimulation [198, 199]. Brain imaging also provided evidence of thalamic and cerebellar relative hyperactivity in patients with RLS [74].
Fig. 5.

Model of key CNS circuits involved in RLS. A Supraspinal components. Ascending fibers project to the thalamus and cortex, where the sensation of the “urge to move” the limbs is established. Descending motor pathways from the basal nuclei initiate the motor commands that underlie the leg movements and PLMS and are under modulatory control from the substantia nigra but not the dopaminergic A11 cluster. This model does not predict the origin of RLS; rather, it models how an increase in sensory or motor drive might explain the increased excitability of the neural circuitry in the brain. B Spinal cord components. Sensory afferents, with their cell bodies located in the dorsal root ganglia (DRG), project to sensory neurons (SNs) in the dorsal horn, where the incoming signals are integrated and forwarded to the brain. In parallel, the sensory signals can either directly activate spinal motoneurons (MNs) in the ventral horn or activate the spinal central pattern generators (CPGs) in the ventral intermedial areas of the spinal cord. The CPGs on both sides of the spinal cord mutually inhibit each other, thus coordinating left–right leg movements. Descending pathways from the brain, including from the dopaminergic A11 cluster, have access to and can modulate each of the spinal cord circuits independently to adjust sensory excitability, CPGs, and motor outputs
The spinal cord is also thought to play an important role in both RLS and PLMS, which are associated with RLS in the vast majority of subjects. PLMS resemble the spinal defense reflex mechanism that results from enhanced excitability in the flexor-reflex arc [200]. PLMS have been regarded as the result of an activated “central pattern generator” (CPG) for locomotion, characterized by stereotyped motor patterns produced by subcortical networks and modulated by phylogenetically recent neocortical structure [201]. Supraspinal pathways might act in a complex and coordinated synergy, producing an excitatory effect onto the spinal CPG that controls locomotion [202] (Fig. 5B). PLMS are also observed in humans with complete transverse spinal cord lesions, responding well to D2-like receptor agonists [203]. This further strengthens the idea that the spinal cord per se contains the structures sufficient to generate PLMS and is the target of dopamine innervation that modulates these motor behaviors. Moreover, several spinal cord diseases, such as multiple sclerosis [204] and spinocerebellar atrophy, have been associated with secondary forms of RLS or PLMS [205]. In addition, human reflex studies indicate the involvement of spinal structures and, specifically, of alterations in spinal motoneurons but not in sensory neurons in RLS [206]. This is in line with the recent promising effects of spinal cord electric stimulation in refractory RLS [207].
Rodent Model Validity Criteria Concerning Striatal and Spinal Circuit Alterations in RLS
Of the 48 selected studies dealing with putative animal models of supraspinal and spinal circuit alterations in RLS, 46 concerned rodent models. Of these, 28 studies were found to satisfy at least one criterion of construct validity [20], while 8 studies satisfied at least one criterion of face validity (Table 1) [19]. The consensus criteria of the construct validity of rodent models of RLS included electrophysiological mechanisms such as hyperexcitability of the motor cortex, increased neuronal activity in the thalamus and cerebellum, and lower threshold of spinal reflexes [20]. Striatal or spinal lesion models were not considered as sufficiently supported by the currently available evidence [20].
Insights from Putative Animal Models of RLS on the Effects of Striatal and Other Supraspinal Circuit Lesions
Evidence obtained on rats indicates the role of the pallidocortical projections from the external part of the globus pallidus to the motor cortex in regulating RLS-like movements [55]. Rats on a control diet and with unilateral NMDA striatal lesions and intact rats with an iron-deficient diet showed movements akin to PLMS, decreased rapid-eye movement (REM) sleep time, and fragmentation of non-REM sleep. These sleep–wake and motor alterations did not further increase in lesioned iron-deficient rats [122]. RLS-like motor activity during sleep could be elicited by a targeted lesion of the striatum in rats [122]. An experimental study on cats found that changes in wake-sleep state and PLMS-like motor activity reminiscent of RLS could also be induced by lesions of the rostrolateral ventral mesopontine junction, including the caudal portion of the dopaminergic retrorubral nucleus, substantia nigra, and ventral tegmental area, with no correlation with the number of dopaminergic neurons lost [208].
Insights from Putative Animal Models of RLS on the Effects of Spinal Circuit Alterations
Putative animal models of RLS shed light on the dopaminergic modulation of spinal circuits, particularly concerning dopamine receptor expression, sex- and age-related differences, and potential functional implications.
All dopamine receptors have been detected in the rodent spinal cord [209–211]. Conversely, the spinal cord of the non-human primate is void of the D1R subtype [40], suggesting that the functionally similar D5R subtype may have taken on the role of the D1R subtype. In rodents, there is a substantial overlap between D1R and D3R distribution [209], which would allow for D1R-D3R interactions, possibly with functional heterodimer formation [62]. The finding of increased D1R protein expression in the spinal cord of D3R KO mice [212], similar to what occurs in MEIS1+/− mice [184], also supports the interplay between D1R and D3R. In mice, moreover, D1R expression increased with age in both the striatum and spinal cord, while D3R expression remains stable in the striatum or becomes slightly decreased in the spinal cord. The resulting D1R-to-D3R ratio indicates a strong relative upregulation of D1R-mediated signaling in old animals, which is particularly pronounced in the lumbar spinal cord [213]. These data suggest that aging may be associated with a shift in D1R- and D3R-mediated signaling in the striatum and spinal cord, which in turn could be an underlying factor in the emergence of RLS and its increased prevalence in the elderly [213]. Animal models also pointed to sex-related differences in the dopaminergic modulation of spinal circuits. In particular, dopamine concentrations in the lumbar spinal cord were higher in male than in female mice [214].
Extracellular electrophysiological studies in the isolated lumbar spinal cord show that dopamine modulates sensorimotor reflexes in a dose-dependent manner, with its effects being mediated primarily by D3R [56]. The D3R plays an important role in the spinal cord in pain modulation [42, 212, 215], reducing overall sensory [215] and motor excitability in the isolated spinal cord [216]. Functional in vitro studies in the spinal cord have shown that the disruption of D3R signaling resulted in increased spinal reflex amplitudes and converted dopamine inhibitory effects to excitatory effects [56]. A study on D3R-KO mice revealed reversed circadian rhythms of tyrosine hydroxylase in the intermediolateral nucleus of the thoracic spinal cord [217]. This study suggested that the activity of spinal preganglionic sympathetic neurons may change with altered activity of descending dopamine fibers through D3R. The interplay of D1-like and D2-like dopamine receptor subtypes can yield opposing outcomes, with activation of D2R and D3R reducing locomotor-like behavior and with activation of D1-like receptors promoting locomotion [216, 218, 219]. Experiments on motoneurons from mouse lumbar spinal cord also revealed the existence of functional heteromers including D1R and adenosine A1 receptors (A1R), by which adenosine may tonically inhibit D1R-mediated signaling and exert significant control of the motoneuron excitability [220].
Closing Remarks
The putative animal models of RLS discussed in this section contributed to widen our knowledge based on the behavioral consequences of supraspinal circuit lesions and on the dopaminergic modulation of spinal cord neurophysiology. While both of these mechanisms may conceivably contribute to RLS pathophysiology, neither mechanism is clearly supported by the evidence on patients with RLS available to date.
Putative Animal Models of RLS Targeting Alterations in Opioid and Other Transmitter Signaling
Overview of Alterations in Opioid and Other Transmitter Signaling in RLS
The use of opioids as a treatment for RLS is approved in Europe and other countries. Post-mortem analysis of the brains of RLS patients showed a deficiency of cells expressing β-endorphin and met-enkephalin, which are endogenous opioids, in the thalamus [221]. Treatment with deferoxamine, an iron-chelating agent, was shown to decrease the survival of cultured dopamine neurons, and cell death was counteracted by applying an endogenous opioid analog before iron chelation [222]. This suggests interactions among opioid signaling, iron deficiency, and dopaminergic dysfunction. However, the contribution of opioids to the pathophysiology of RLS is still a matter of debate.
Concerning transmitters other than opioids, the role of histamine in RLS is also a matter of investigation (cf. [223]). On the other hand, clinical evidence supported the effectiveness on sensory and motor symptoms of RLS of perampanel, a glutamate AMPA receptor antagonist [224], and of dipyridamole, which leads to increased extracellular adenosine due to non-selective inhibition of the equilibrative nucleoside transporters (ENT1 and ENT2) that promote the uptake of adenosine from the extracellular space [225]. While the contribution of still other transmitters to RLS pathophysiology cannot be discounted, evidence in that respect is lacking.
Rodent Model Validity Criteria Concerning Opioid and Other Transmitter Signaling in RLS
All of the 12 selected studies dealing with putative animal models of alterations in opioid and other transmitter signaling in RLS concerned rodent models. Of these, 7 studies were found to satisfy at least one criterion of construct validity [20], while 2 studies satisfied at least one criterion of face validity [19] (Table 1). Models based on alterations in opioid transmission were not considered as sufficiently supported by the currently available evidence for inclusion among consensus criteria for RLS construct validity in rodents [20]. Exposure to antihistamines was considered as a construct validity criterion [20], and aggravation of RLS-like phenotype with H1 histamine receptor antagonists that cross the blood–brain barrier was included among rodent face validity criteria for RLS, with the possible exception of PLMS-like behavior [19]. Other transmitters were not considered either in terms of construct or of face validity of rodent models of RLS.
Insights from Putative Animal Models of RLS on the Effects of Alterations in Opioid and Other Transmitter Signaling
Opioid receptor KO models have been used to study the role of opioids in the pathogenesis of RLS [226, 227]. The KO of all three opioid receptor genes (mu opioid receptor, MOR; delta opioid receptor, DOR; kappa opioid receptor, KOR) led to hyperactivity, a trend of increased probability of waking, and decreased thermal sensation during the rest period [226]. MOR KO mice were also hyperactive and had increased thermal sensitivity in wakefulness [227]. Therefore, studies on both the triple-OR KO and MOR KO mouse models have shown motor and sensory deficits similar to the human RLS patients, suggesting that the opioid receptors may play a role in mediating the core features of RLS. The triple-OR KO mice also had decreased hemoglobin and hematocrit levels, indicative of anemia, together with decreased serum iron and ferritin, but no change in striatal iron [226]. This picture would be generally suggestive of iron deficiency anemia, except there is no increase in transferrin [226]. MOR KO mice showed a similar decrease in red blood cells, hemoglobin and hematocrit, indicative of anemia. However, the serum iron and transferrin levels were decreased, while ferritin was increased [227], which is different from the findings in triple-OR KO mice [226]. In addition, MOR KO mice showed microcytic and hypochromic red blood cells and increased iron levels in the spleen and liver, but again with no significant change in striatal iron [227]. Overall, the abnormal iron homeostasis in the MOR KO mice was considered to suggest a state of inflammation [227]. The phenotype of the triple-OR KO mouse model thus appears more similar to that of human RLS, whereas the MOR KO mouse model may be useful for investigating the association of RLS with inflammatory disorders [158]. These results suggest that different opioid receptor subtypes may contribute individually or in combination to produce various aspects of the RLS spectrum (cf. Fig. 4).
Concerning other transmitters, RLS-like motor activity during sleep could be elicited by the intracerebroventricular injection of alpha-melanocyte stimulation hormone (alpha-MSH) or of adrenocorticotropic hormone (ACTH) in rats [228]. On the other hand, a study on histidine decarboxylase KO mice, which lack a key enzyme for histamine signaling, did not find an increase in PLMS-like activity. Rather, the study uncovered a significant decrease in tibialis anterior EMG activity during sleep, mainly consisting of EMG events separated by short-time intervals < 10 s [223]. These results do not support the hypothesis that preventing brain histamine signaling may promote RLS and rather suggest that short-interval limb motor activity during sleep may represent a manifestation of subcortical arousal requiring the integrity of histamine signaling [223].
The occurrence of interactions between iron deficiency and adenosine, histamine, or glutamate signaling is supported by experimental evidence on putative animal models. In WT mice, the density of striatal adenosine 2a receptors (A2AR) increased with an iron-deficient diet [85]. Rats with dietary iron deficiency also showed increased density of A2AR in the striatum but not in the cortex or basal mesencephalon, with pharmacological data supporting increased pre-synaptic and post-synaptic effects of adenosine [229]. In mice, mild (7 ppm iron) and severe (4 ppm iron) dietary iron deficiency downregulated A1R in the cortex and striatum, whereas only severe dietary iron deficiency upregulated A2AR in the striatum [88]. Locomotor activity was increased by KW-6002, an A2AR antagonist, in mice with severe dietary iron deficiency and was decreased by CPT, an A1R antagonist, but not by caffeine, in rats with severe dietary iron deficiency [88].
Dietary iron deficiency was also found to increase striatal H3R in rats [115]. Alpha-methylhistamine, an H3R agonist, increased the occurrence of tibialis anterior EMG bursts akin to PLMS in rats fed a control diet, whereas thioperamide, an H3R antagonist, decreased these bursts in control rats as well as in rats fed an iron-deficient diet [115]. Thioperamide also decreased these PLMS-like EMG bursts in rats with dietary iron deficiency and unilateral striatal lesions [122].
Hypersensitivity of corticostriatal glutamatergic terminals, revealed by a significantly lower frequency of the optogenetic stimulation needed to evoke glutamate release, was observed in male Sprague–Dawley rats with dietary iron deficiency [230]. Interestingly, hypersensitivity of these terminals was reversed by the two classes of first-line drugs used to treat RLS: D2-like receptor agonists and ligands of the α2δ subunit of voltage-gated calcium channels [230].
Closing Remarks
The putative animal models of RLS discussed in this section provided proof of principle for an effect of opioid and histamine signaling on iron metabolism and/or motor behavior, as well as for causal links between iron deficiency and alterations in histamine, adenosine, and glutamate transmission. These data provide pathophysiological background to explain the therapeutic effects of opioids and of drugs modulating glutamate and adenosine transmission. However, the precise role played by these mechanisms in RLS pathophysiology is still a matter of investigation.
The Role of Pregnancy in the Pathophysiology of RLS
Pregnancy is a clear risk factor for RLS [231], as noted originally by Ekbom himself [3]. Several studies observed that around 25% of women experience RLS symptoms during the pregnancy; these data have been confirmed worldwide, except for a lower prevalence rate in Asian countries [231]. The incidence rate of RLS increases across gestation, reaching a peak at the end of the second trimester and in the third one, then falls around delivery, and remains low during puerperium [232]. Around 40% of the women who suffer from RLS during pregnancy have already experienced RLS before in their life; however, more than half of them describe their symptoms as new [232]. Women with RLS during pregnancy report having a positive family member with RLS more often than those who do not experience RLS. These observations, together with the fact that women with RLS during pregnancy have a four-fold higher risk of developing an idiopathic form of RLS later in their life than women with no pregnancy-related RLS, suggest that a genetic predisposition might play an important role in RLS during pregnancy [233]. However, genetic mapping of the allelic variants associated with RLS during pregnancy still needs to be performed. In addition to genetic factors, vitamin or iron deficiency, changes in hormone levels and motor habits have been postulated as contributing to RLS during pregnancy [234]. Accordingly, estrogens and progesterone are well-known neuromodulators, with estrogens acting as inhibitors of DAT in nucleus accumbens and downregulators of D2R in the caudate nucleus [235], and with progesterone modulating dopaminergic pathways and enhancing spontaneous dopamine release in the striatum [236]. However, a difference in hormone levels or in iron deficiency between pregnant women with or without RLS has never been shown, and the role of estrogen and progesterone in RLS is still a matter of investigation.
Despite being interesting and well delimited, the topic of pregnancy-related RLS has been little investigated in animal models. Studying rodent dams in the second half of their pregnancy was included among the rodent construct validity criteria for RLS [20]. In the single selected study concerning pregnancy, results from the open-field activity paradigm showed that pregnant rats exhibit fewer rearing events, increased grooming time, and reduced immobilization time with respect to non-pregnant animals, while the total ambulation events and the distance traveled were similar [237]. These results warrant replication to improve their face validity for RLS, for instance including experiments also in the home cage [19].
General Conclusions
Despite a substantial number of scientific studies, our understanding of the pathophysiology of RLS is incomplete. Alterations in brain iron homeostasis, with a decrease in iron levels, particularly in the substantia nigra, and consequent dopamine neurotransmission abnormalities mainly at the level of the striatal circuits are thought to play a central role in the pathogenesis of RLS. These changes may occur along with alterations in other areas (i.e., spinal cord) and in other transmitters, mainly glutamate, adenosine, and histamine, and were targeted by extensive basic research literature on the topic. However, while the remarkable efficacy of D2-like receptor agonists has been attested in numerous clinical trials, the benefit of iron supplements is limited [238]. Opioid pathway dysfunction and reduced thalamocortical connectivity have also been put forward to explain the sensory symptoms of RLS, although clear support is still lacking, and genetic disposition to the RLS is believed to play a role in all these underlying mechanisms.
On the one hand, these multiple pathophysiological theories create challenges for researchers seeking to model RLS in animals with a high-level construct validity [20]. On the other hand, the subjective nature of the disease symptoms brings additional difficulties that prevent promoting of a unique disease animal model that recapitulates salient clinical symptoms [19]. Nevertheless, iron-deficient rodent models supported the occurrence of dopaminergic and glutamatergic transmission changes and predicted adenosinergic and histaminergic involvement, thus paving the way for novel treatment options.
Face [19] and construct [20] validity guidelines for RLS were developed to serve the research community by fostering consistency in future research. However, these guidelines should not be misinterpreted as a dogmatic attempt to cancel or silence scientific evidence they do not fit with. This is particularly relevant concerning construct validity, which is inherently a work in progress depending on our current understanding of the pathophysiology of RLS. It is this very knowledge that a virtuous cycle of animal model experimentation and clinical work is expected to increase, ultimately leading to better and more effective treatments to improve the lives of patients with RLS and their families.
Supplementary Information
Below is the link to the electronic supplementary material.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Willis T. De Anima Brutorum. London: Davis, R.; 1672.
- 2.Willis T. Of the diseases of the brain and the genus nervosum, in The London Practice of Physick. Bassett and Crooke: London; 1685.
- 3.Ekbom KA. Restless legs syndrome. Neurology. 1960;10:868–873. doi: 10.1212/WNL.10.9.868. [DOI] [PubMed] [Google Scholar]
- 4.Allen RP, et al. Restless legs syndrome: diagnostic criteria, special considerations, and epidemiology. A report from the restless legs syndrome diagnosis and epidemiology workshop at the National Institutes of Health. Sleep Med. 2003;4(2):101–119. doi: 10.1016/S1389-9457(03)00010-8. [DOI] [PubMed] [Google Scholar]
- 5.Allen RP, et al. Restless legs syndrome/Willis-Ekbom disease diagnostic criteria: updated International Restless Legs Syndrome Study Group (IRLSSG) consensus criteria–history, rationale, description, and significance. Sleep Med. 2014;15(8):860–873. doi: 10.1016/j.sleep.2014.03.025. [DOI] [PubMed] [Google Scholar]
- 6.Odin P. Diagnostic criteria. In: Chaudhuri KR, Odin P, Olanow CW, editors. Restless Legs Syndrome. London / New York: Taylor & Francis; 2004. pp. 9–20. [Google Scholar]
- 7.Winkelmann J, et al. Clinical characteristics and frequency of the hereditary restless legs syndrome in a population of 300 patients. Sleep. 2000;23(5):597–602. doi: 10.1093/sleep/23.5.1b. [DOI] [PubMed] [Google Scholar]
- 8.Winkelmann J, et al. Genetics of restless legs syndrome. Sleep Med. 2017;31:18–22. doi: 10.1016/j.sleep.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 9.Earley CJ, et al. Connectome and molecular pharmacological differences in the dopaminergic system in restless legs syndrome (RLS): plastic changes and neuroadaptations that may contribute to augmentation. Sleep Med. 2017;31:71–77. doi: 10.1016/j.sleep.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferre S, et al. Pivotal role of adenosine neurotransmission in restless legs syndrome. Front Neurosci. 2017;11:722. doi: 10.3389/fnins.2017.00722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Byrne R, Sinha S, Chaudhuri KR. Restless legs syndrome: diagnosis and review of management options. Neuropsychiatr Dis Treat. 2006;2(2):155–164. doi: 10.2147/nedt.2006.2.2.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ondo W. Secondary restless legs syndrome. In: Chaudhuri KR, Odin P, Olanow CW, editors. Restless Legs Syndrome. London / New York: Taylor & Francis; 2004. pp. 57–84. [Google Scholar]
- 13.Allen RP, et al. Animal models of RLS phenotypes. Sleep Med. 2017;31:23–28. doi: 10.1016/j.sleep.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen P, et al. Caenorhabditis elegans and its applicability to studies on restless legs syndrome. Adv Pharmacol. 2019;84:147–174. doi: 10.1016/bs.apha.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ondo WG, Zhao HR, Le WD. Animal models of restless legs syndrome. Sleep Med. 2007;8(4):344–348. doi: 10.1016/j.sleep.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 16.Baier PC, Ondo WG, Winkelmann J. Animal studies in restless legs syndrome. Mov Disord. 2007;22(Suppl 18):S459–S465. doi: 10.1002/mds.21605. [DOI] [PubMed] [Google Scholar]
- 17.Earley CJ, Jones BC, Ferre S. Brain-iron deficiency models of restless legs syndrome. Exp Neurol. 2022;356:114158. doi: 10.1016/j.expneurol.2022.114158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manconi M, et al. On the pathway of an animal model for restless legs syndrome. Neurol Sci. 2007;28(Suppl 1):S53–60. doi: 10.1007/s10072-007-0738-8. [DOI] [PubMed] [Google Scholar]
- 19.Salminen AV, et al. Consensus guidelines on rodent models of restless legs syndrome. Mov Disord. 2021. [DOI] [PMC free article] [PubMed]
- 20.Salminen AV, et al. Consensus guidelines on the construct validity of rodent models of restless legs syndrome. Dis Model Mech. 2022;15(8). [DOI] [PMC free article] [PubMed]
- 21.Peters MD, et al. Guidance for conducting systematic scoping reviews. Int J Evid Based Healthc. 2015;13(3):141–146. doi: 10.1097/XEB.0000000000000050. [DOI] [PubMed] [Google Scholar]
- 22.Munn Z, et al. Systematic review or scoping review? Guidance for authors when choosing between a systematic or scoping review approach. BMC Med Res Methodol. 2018;18(1):143. doi: 10.1186/s12874-018-0611-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tricco AC, et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): checklist and explanation. Ann Intern Med. 2018;169(7):467–473. doi: 10.7326/M18-0850. [DOI] [PubMed] [Google Scholar]
- 24.Winkelman JW, et al. Practice guideline summary: treatment of restless legs syndrome in adults: report of the guideline development, dissemination, and implementation subcommittee of the American Academy of Neurology. Neurology. 2016;87(24):2585–2593. doi: 10.1212/WNL.0000000000003388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winkelmann J, et al. Treatment of restless legs syndrome: evidence-based review and implications for clinical practice (Revised 2017)(section sign) Mov Disord. 2018;33(7):1077–1091. doi: 10.1002/mds.27260. [DOI] [PubMed] [Google Scholar]
- 26.Gossard TR, et al. Restless legs syndrome: contemporary diagnosis and treatment. Neurotherapeutics. 2021;18(1):140–155. doi: 10.1007/s13311-021-01019-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63(1):182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 28.Bonaventura J, et al. Key role of the dopamine D4 receptor in the modulation of corticostriatal glutamatergic neurotransmission. Sci Adv. 2017;3(1):e1601631. doi: 10.1126/sciadv.1601631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kondo M, Fujiwara H, Tanaka C. Dopamine release and presynaptic dopaminergic regulation in guinea pig spinal cord. Jpn J Pharmacol. 1986;41(1):39–46. doi: 10.1254/jjp.41.39. [DOI] [PubMed] [Google Scholar]
- 30.Jackisch R, et al. Presynaptic opioid receptors on dopaminergic nerves in the rabbit caudate nucleus: coupling to pertussis toxin-sensitive G-proteins and interaction with D2 autoreceptors? Naunyn Schmiedebergs Arch Pharmacol. 1994;349(3):250–258. doi: 10.1007/BF00169291. [DOI] [PubMed] [Google Scholar]
- 31.Wu J, Dougherty JJ, Nichols RA. Dopamine receptor regulation of Ca2+ levels in individual isolated nerve terminals from rat striatum: comparison of presynaptic D1-like and D2-like receptors. J Neurochem. 2006;98(2):481–494. doi: 10.1111/j.1471-4159.2006.03901.x. [DOI] [PubMed] [Google Scholar]
- 32.Huang Q, et al. Immunohistochemical localization of the D1 dopamine receptor in rat brain reveals its axonal transport, pre- and postsynaptic localization, and prevalence in the basal ganglia, limbic system, and thalamic reticular nucleus. Proc Natl Acad Sci U S A. 1992;89(24):11988–11992. doi: 10.1073/pnas.89.24.11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Navarro M, et al. Modifications of striatal D2 dopaminergic postsynaptic sensitivity during development of morphine tolerance-dependence in mice. Pharmacol Biochem Behav. 1992;43(2):603–608. doi: 10.1016/0091-3057(92)90197-N. [DOI] [PubMed] [Google Scholar]
- 34.Olianas MC, Dedoni S, Onali P. Potentiation of dopamine D1-like receptor signaling by concomitant activation of delta- and mu-opioid receptors in mouse medial prefrontal cortex. Neurochem Int. 2012;61(8):1404–1416. doi: 10.1016/j.neuint.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 35.Centonze D, et al. Receptor subtypes involved in the presynaptic and postsynaptic actions of dopamine on striatal interneurons. J Neurosci. 2003;23(15):6245–6254. doi: 10.1523/JNEUROSCI.23-15-06245.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swant J, Stramiello M, Wagner JJ. Postsynaptic dopamine D3 receptor modulation of evoked IPSCs via GABA(A) receptor endocytosis in rat hippocampus. Hippocampus. 2008;18(5):492–502. doi: 10.1002/hipo.20408. [DOI] [PubMed] [Google Scholar]
- 37.Gerfen CR, et al. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250(4986):1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- 38.Aizman O, et al. Anatomical and physiological evidence for D1 and D2 dopamine receptor colocalization in neostriatal neurons. Nat Neurosci. 2000;3(3):226–230. doi: 10.1038/72929. [DOI] [PubMed] [Google Scholar]
- 39.Bertran-Gonzalez J, et al. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28(22):5671–5685. doi: 10.1523/JNEUROSCI.1039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barraud Q, et al. Neuroanatomical study of the A11 diencephalospinal pathway in the non-human primate. PLoS ONE. 2010;5(10):e13306. doi: 10.1371/journal.pone.0013306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barriere G, et al. The restless legs syndrome. Prog Neurobiol. 2005;77(3):139–165. doi: 10.1016/j.pneurobio.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 42.Trenkwalder C, et al. Comorbidities, treatment, and pathophysiology in restless legs syndrome. Lancet Neurol. 2018;17(11):994–1005. doi: 10.1016/S1474-4422(18)30311-9. [DOI] [PubMed] [Google Scholar]
- 43.Earley CJ, et al. The dopaminergic neurons of the A11 system in RLS autopsy brains appear normal. Sleep Med. 2009;10(10):1155–1157. doi: 10.1016/j.sleep.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Earley CJ, et al. Increased synaptic dopamine in the putamen in restless legs syndrome. Sleep. 2013;36(1):51–57. doi: 10.5665/sleep.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Earley CJ, et al. The dopamine transporter is decreased in the striatum of subjects with restless legs syndrome. Sleep. 2011;34(3):341–347. doi: 10.1093/sleep/34.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allen RP, et al. Abnormally increased CSF 3-ortho-methyldopa (3-OMD) in untreated restless legs syndrome (RLS) patients indicates more severe disease and possibly abnormally increased dopamine synthesis. Sleep Med. 2009;10(1):123–128. doi: 10.1016/j.sleep.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stiasny-Kolster K, et al. Normal dopaminergic and serotonergic metabolites in cerebrospinal fluid and blood of restless legs syndrome patients. Mov Disord. 2004;19(2):192–196. doi: 10.1002/mds.10631. [DOI] [PubMed] [Google Scholar]
- 48.Connor JR, et al. Altered dopaminergic profile in the putamen and substantia nigra in restless leg syndrome. Brain. 2009;132(Pt 9):2403–2412. doi: 10.1093/brain/awp125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Earley CJ, Hyland K, Allen RP. CSF dopamine, serotonin, and biopterin metabolites in patients with restless legs syndrome. Mov Disord. 2001;16(1):144–149. doi: 10.1002/1531-8257(200101)16:1<144::AID-MDS1009>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 50.Qu S, et al. Locomotion is increased in a11-lesioned mice with iron deprivation: a possible animal model for restless legs syndrome. J Neuropathol Exp Neurol. 2007;66(5):383–388. doi: 10.1097/nen.0b013e3180517b5f. [DOI] [PubMed] [Google Scholar]
- 51.Qu S, et al. Projections of diencephalic dopamine neurons into the spinal cord in mice. Exp Brain Res. 2006;168(1–2):152–156. doi: 10.1007/s00221-005-0075-1. [DOI] [PubMed] [Google Scholar]
- 52.Ondo WG, et al. Clinical correlates of 6-hydroxydopamine injections into A11 dopaminergic neurons in rats: a possible model for restless legs syndrome. Mov Disord. 2000;15(1):154–158. doi: 10.1002/1531-8257(200001)15:1<154::AID-MDS1025>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 53.Lopes C, et al. Evaluation of periodic limb movements in a putative animal model of restless leg syndrome. Mov Disord. 2012;27(3):413–420. doi: 10.1002/mds.24058. [DOI] [PubMed] [Google Scholar]
- 54.Esteves AM, et al. Can physical exercise have a protective effect in an animal model of sleep-related movement disorder? Brain Res. 2016;1639:47–57. doi: 10.1016/j.brainres.2016.02.036. [DOI] [PubMed] [Google Scholar]
- 55.Guo CN, et al. Targeted disruption of supraspinal motor circuitry reveals a distributed network underlying restless legs syndrome (RLS)-like movements in the rat. Sci Rep. 2017;7(1):9905. doi: 10.1038/s41598-017-10284-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clemens S, Hochman S. Conversion of the modulatory actions of dopamine on spinal reflexes from depression to facilitation in D3 receptor knock-out mice. J Neurosci. 2004;24(50):11337–11345. doi: 10.1523/JNEUROSCI.3698-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clemens S, Rye D, Hochman S. Restless legs syndrome: revisiting the dopamine hypothesis from the spinal cord perspective. Neurology. 2006;67(1):125–130. doi: 10.1212/01.wnl.0000223316.53428.c9. [DOI] [PubMed] [Google Scholar]
- 58.Clemens S, Ghorayeb I. D3 and D1 receptors: the Yin and Yang in the treatment of restless legs syndrome with dopaminergics. Adv Pharmacol. 2019;84:79–100. doi: 10.1016/bs.apha.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 59.Accili D, et al. A targeted mutation of the D3 dopamine receptor gene is associated with hyperactivity in mice. Proc Natl Acad Sci U S A. 1996;93(5):1945–1949. doi: 10.1073/pnas.93.5.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Asico LD, et al. Disruption of the dopamine D3 receptor gene produces renin-dependent hypertension. J Clin Invest. 1998;102(3):493–498. doi: 10.1172/JCI3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dinkins M-L, Lallemand P, Clemens S. Long-term treatment with dopamine D3 receptor agonists induces a behavioral switch that can be rescued by blocking the dopamine D1 receptor. Sleep Med. 2017;40:47–52. doi: 10.1016/j.sleep.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 62.Clemens S. D3 receptors and restless legs syndrome. Curr Top Behav Neurosci. 2022. [DOI] [PubMed]
- 63.Allen RP, et al. Evidence-based and consensus clinical practice guidelines for the iron treatment of restless legs syndrome/Willis-Ekbom disease in adults and children: an IRLSSG task force report. Sleep Med. 2018;41:27–44. doi: 10.1016/j.sleep.2017.11.1126. [DOI] [PubMed] [Google Scholar]
- 64.Hogl B, et al. Restless legs syndrome: a community-based study of prevalence, severity, and risk factors. Neurology. 2005;64(11):1920–1924. doi: 10.1212/01.WNL.0000163996.64461.A3. [DOI] [PubMed] [Google Scholar]
- 65.O'Keeffe ST, Gavin K, Lavan JN. Iron status and restless legs syndrome in the elderly. Age Ageing. 1994;23(3):200–203. doi: 10.1093/ageing/23.3.200. [DOI] [PubMed] [Google Scholar]
- 66.Earley CJ, et al. Abnormalities in CSF concentrations of ferritin and transferrin in restless legs syndrome. Neurology. 2000;54(8):1698–1700. doi: 10.1212/WNL.54.8.1698. [DOI] [PubMed] [Google Scholar]
- 67.Mizuno S, et al. CSF iron, ferritin and transferrin levels in restless legs syndrome. J Sleep Res. 2005;14(1):43–47. doi: 10.1111/j.1365-2869.2004.00403.x. [DOI] [PubMed] [Google Scholar]
- 68.Allen RP, et al. MRI measurement of brain iron in patients with restless legs syndrome. Neurology. 2001;56(2):263–265. doi: 10.1212/WNL.56.2.263. [DOI] [PubMed] [Google Scholar]
- 69.Astrakas LG, et al. T2 relaxometry and fMRI of the brain in late-onset restless legs syndrome. Neurology. 2008;71(12):911–916. doi: 10.1212/01.wnl.0000325914.50764.a2. [DOI] [PubMed] [Google Scholar]
- 70.Moon HJ, et al. T2 relaxometry using 3.0-tesla magnetic resonance imaging of the brain in early- and late-onset restless legs syndrome. J Clin Neurol. 2014;10(3):197–202. doi: 10.3988/jcn.2014.10.3.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rizzo G, et al. Imaging brain functional and metabolic changes in restless legs syndrome. Curr Neurol Neurosci Rep. 2013;13(9):372. doi: 10.1007/s11910-013-0372-1. [DOI] [PubMed] [Google Scholar]
- 72.Godau J, et al. Multiregional brain iron deficiency in restless legs syndrome. Mov Disord. 2008;23(8):1184–1187. doi: 10.1002/mds.22070. [DOI] [PubMed] [Google Scholar]
- 73.Knake S, et al. Normal regional brain iron concentration in restless legs syndrome measured by MRI. Nat Sci Sleep. 2010;2:19–22. [PMC free article] [PubMed] [Google Scholar]
- 74.Margariti PN, et al. Investigation of unmedicated early onset restless legs syndrome by voxel-based morphometry, T2 relaxometry, and functional MR imaging during the night-time hours. AJNR Am J Neuroradiol. 2012;33(4):667–672. doi: 10.3174/ajnr.A2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li X, et al. Brain iron deficiency in idiopathic restless legs syndrome measured by quantitative magnetic susceptibility at 7 tesla. Sleep Med. 2016;22:75–82. doi: 10.1016/j.sleep.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Connor JR, et al. Postmortem and imaging based analyses reveal CNS decreased myelination in restless legs syndrome. Sleep Med. 2011;12(6):614–619. doi: 10.1016/j.sleep.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Allen RP, et al. The prevalence and impact of restless legs syndrome on patients with iron deficiency anemia. Am J Hematol. 2013;88(4):261–264. doi: 10.1002/ajh.23397. [DOI] [PubMed] [Google Scholar]
- 78.Wang X, Garrick MD, Collins JF. Animal models of normal and disturbed iron and copper metabolism. J Nutr. 2019;149(12):2085–2100. doi: 10.1093/jn/nxz172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen Q, Connor JR, Beard JL. Brain iron, transferrin and ferritin concentrations are altered in developing iron-deficient rats. J Nutr. 1995;125(6):1529–1535. doi: 10.1093/jn/125.6.1529. [DOI] [PubMed] [Google Scholar]
- 80.Erikson KM, Jones BC, Beard JL. Iron deficiency alters dopamine transporter functioning in rat striatum. J Nutr. 2000;130(11):2831–2837. doi: 10.1093/jn/130.11.2831. [DOI] [PubMed] [Google Scholar]
- 81.Beard JL, Wiesinger JA, Connor JR. Pre- and postweaning iron deficiency alters myelination in Sprague-Dawley rats. Dev Neurosci. 2003;25(5):308–315. doi: 10.1159/000073507. [DOI] [PubMed] [Google Scholar]
- 82.Zhao H, et al. Spinal cord dopamine receptor expression and function in mice with 6-OHDA lesion of the A11 nucleus and dietary iron deprivation. J Neurosci Res. 2007;85(5):1065–1076. doi: 10.1002/jnr.21207. [DOI] [PubMed] [Google Scholar]
- 83.Luo F, et al. The long-term effects of the dopamine agonist pramipexole in a proposed restless legs syndrome animal model. Sleep Med. 2010. [DOI] [PubMed]
- 84.Lo Martire V, et al. Sleep and tibialis anterior muscle activity in mice with mild hypoxia and iron deficiency: implications for the restless legs syndrome. Front Physiol. 2018;9:1818. doi: 10.3389/fphys.2018.01818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gulyani S, et al. Diminished iron concentrations increase adenosine A(2A) receptor levels in mouse striatum and cultured human neuroblastoma cells. Exp Neurol. 2009;215(2):236–242. doi: 10.1016/j.expneurol.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dean T, Jr, et al. The effects of dietary iron deprivation on murine circadian sleep architecture. Sleep Med. 2006;7(8):634–640. doi: 10.1016/j.sleep.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 87.Dowling P, et al. Iron-deficiency sensitizes mice to acute pain stimuli and formalin-induced nociception. J Nutr. 2009;139(11):2087–2092. doi: 10.3945/jn.109.112557. [DOI] [PubMed] [Google Scholar]
- 88.Quiroz C, et al. Adenosine receptors as markers of brain iron deficiency: implications for restless legs syndrome. Neuropharmacology. 2016;111:160–168. doi: 10.1016/j.neuropharm.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Berger K, et al. Sex and the risk of restless legs syndrome in the general population. Arch Intern Med. 2004;164(2):196–202. doi: 10.1001/archinte.164.2.196. [DOI] [PubMed] [Google Scholar]
- 90.Snyder AM, Connor JR. Iron, the substantia nigra and related neurological disorders. Biochim Biophys Acta. 2009;1790(7):606–614. doi: 10.1016/j.bbagen.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 91.Jones BC, et al. Quantitative genetic analysis of ventral midbrain and liver iron in BXD recombinant inbred mice. Nutr Neurosci. 2003;6(6):369–377. doi: 10.1080/10284150310001624192. [DOI] [PubMed] [Google Scholar]
- 92.Jellen LC, et al. Iron deficiency alters expression of dopamine-related genes in the ventral midbrain in mice. Neuroscience. 2013;252:13–23. doi: 10.1016/j.neuroscience.2013.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Unger EL, et al. Effects of IV iron isomaltoside-1000 treatment on regional brain iron status in an iron-deficient animal. Neuroscience. 2013;246:179–185. doi: 10.1016/j.neuroscience.2013.04.049. [DOI] [PubMed] [Google Scholar]
- 94.Jellen LC, Beard JL, Jones BC. Systems genetics analysis of iron regulation in the brain. Biochimie. 2009;91(10):1255–1259. doi: 10.1016/j.biochi.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reis FM, et al. Plasma iron levels appraised 15 days after spinal cord injury in a limb movement animal model. Spinal Cord. 2011;49(3):361–364. doi: 10.1038/sc.2010.121. [DOI] [PubMed] [Google Scholar]
- 96.Unger EL, Earley CJ, Beard JL. Diurnal cycle influences peripheral and brain iron levels in mice. J Appl Physiol. 2009;106(1):187–193. doi: 10.1152/japplphysiol.91076.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bianco LE, et al. Iron deficiency alters the day-night variation in monoamine levels in mice. Chronobiol Int. 2009;26(3):447–463. doi: 10.1080/07420520902820905. [DOI] [PubMed] [Google Scholar]
- 98.Hyacinthe C, et al. Blood withdrawal affects iron store dynamics in primates with consequences on monoaminergic system function. Neuroscience. 2015;290:621–635. doi: 10.1016/j.neuroscience.2015.01.057. [DOI] [PubMed] [Google Scholar]
- 99.Beard JL, et al. Altered monamine metabolism in caudate-putamen of iron-deficient rats. Pharmacol Biochem Behav. 1994;48(3):621–624. doi: 10.1016/0091-3057(94)90323-9. [DOI] [PubMed] [Google Scholar]
- 100.Bianco LE, et al. Iron deficiency alters dopamine uptake and response to L-DOPA injection in Sprague-Dawley rats. J Neurochem. 2008;106(1):205–215. doi: 10.1111/j.1471-4159.2008.05358.x. [DOI] [PubMed] [Google Scholar]
- 101.Unger EL, et al. Low brain iron effects and reversibility on striatal dopamine dynamics. Exp Neurol. 2014;261:462–468. doi: 10.1016/j.expneurol.2014.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nelson C, et al. In vivo dopamine metabolism is altered in iron-deficient anemic rats. J Nutr. 1997;127(12):2282–2288. doi: 10.1093/jn/127.12.2282. [DOI] [PubMed] [Google Scholar]
- 103.Beard JL, Erikson KM, Jones BC. Neurobehavioral analysis of developmental iron deficiency in rats. Behav Brain Res. 2002;134(1–2):517–524. doi: 10.1016/S0166-4328(02)00092-X. [DOI] [PubMed] [Google Scholar]
- 104.Lai YY, et al. Motor hyperactivity of the iron-deficient rat – an animal model of restless legs syndrome. Mov Disord. 2017;32(12):1687–1693. doi: 10.1002/mds.27133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Connor JR, et al. Comparative study of the influence of Thy1 deficiency and dietary iron deficiency on dopaminergic profiles in the mouse striatum. J Neurosci Res. 2008;86(14):3194–3202. doi: 10.1002/jnr.21758. [DOI] [PubMed] [Google Scholar]
- 106.Franco B, et al. Iron deficiency in pregnancy: influence on sleep, behavior, and molecular markers of adult male offspring. J Neurosci Res. 2021;99(12):3325–3338. doi: 10.1002/jnr.24968. [DOI] [PubMed] [Google Scholar]
- 107.Wang X, et al. Thy1 expression in the brain is affected by iron and is decreased in restless legs syndrome. J Neurol Sci. 2004;220(1–2):59–66. doi: 10.1016/j.jns.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 108.Ashkenazi R, Ben-Shachar D, Youdim MB. Nutritional iron and dopamine binding sites in the rat brain. Pharmacol Biochem Behav. 1982;17(Suppl 1):43–47. doi: 10.1016/0091-3057(82)90509-3. [DOI] [PubMed] [Google Scholar]
- 109.Ben-Shachar D, Finberg JP, Youdim MB. Effect of iron chelators on dopamine D2 receptors. J Neurochem. 1985;45(4):999–1005. doi: 10.1111/j.1471-4159.1985.tb05514.x. [DOI] [PubMed] [Google Scholar]
- 110.Youdim MB, Yehuda S, Ben-Uriah Y. Iron deficiency-induced circadian rhythm reversal of dopaminergic-mediated behaviours and thermoregulation in rats. Eur J Pharmacol. 1981;74(4):295–301. doi: 10.1016/0014-2999(81)90048-0. [DOI] [PubMed] [Google Scholar]
- 111.Erikson KM, et al. Iron deficiency decreases dopamine D1 and D2 receptors in rat brain. Pharmacol Biochem Behav. 2001;69(3–4):409–418. doi: 10.1016/S0091-3057(01)00563-9. [DOI] [PubMed] [Google Scholar]
- 112.Unger EL, et al. Dopamine D2 receptor expression is altered by changes in cellular iron levels in PC12 cells and rat brain tissue. J Nutr. 2008;138(12):2487–2494. doi: 10.3945/jn.108.095224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dowling P, et al. Dopamine D3 receptor specifically modulates motor and sensory symptoms in iron-deficient mice. J Neurosci. 2011;31(1):70–77. doi: 10.1523/JNEUROSCI.0959-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Allen RP, et al. Iron-deficiency and dopaminergic treatment effects on RLS-like behaviors of an animal model with the brain iron deficiency pattern of the restless legs syndrome. Sleep Med. 2020;71:141–148. doi: 10.1016/j.sleep.2020.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lai YY, et al. Striatal histamine mechanism in the pathogenesis of restless legs syndrome. Sleep. 2020;43(2). [DOI] [PMC free article] [PubMed]
- 116.Ye Z, Connor JR. Identification of iron responsive genes by screening cDNA libraries from suppression subtractive hybridization with antisense probes from three iron conditions. Nucleic Acids Res. 2000;28(8):1802–1807. doi: 10.1093/nar/28.8.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Carkaci-Salli N, et al. Gender-specific regulation of tyrosine hydroxylase in thymocyte differentiation antigen-1 knockout mice. J Neurosci Res. 2012;90(8):1583–1588. doi: 10.1002/jnr.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Glover J, Jacobs A. Activity pattern of iron-deficient rats. Br Med J. 1972;2(5814):627–628. doi: 10.1136/bmj.2.5814.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hunt JR, et al. Severe or marginal iron deficiency affects spontaneous physical activity in rats. Am J Clin Nutr. 1994;59(2):413–418. doi: 10.1093/ajcn/59.2.413. [DOI] [PubMed] [Google Scholar]
- 120.Angulo-Kinzler RM, et al. Spontaneous motor activity in human infants with iron-deficiency anemia. Early Hum Dev. 2002;66(2):67–79. doi: 10.1016/S0378-3782(01)00238-9. [DOI] [PubMed] [Google Scholar]
- 121.Weinberg J, Dallman PR, Levine S. Iron deficiency during early development in the rat: behavioral and physiological consequences. Pharmacol Biochem Behav. 1980;12(4):493–502. doi: 10.1016/0091-3057(80)90179-3. [DOI] [PubMed] [Google Scholar]
- 122.Lai YY, et al. Striatal mechanism of the restless legs syndrome. Sleep. 2022. [DOI] [PMC free article] [PubMed]
- 123.Erikson KM, et al. Regional brain iron, ferritin and transferrin concentrations during iron deficiency and iron repletion in developing rats. J Nutr. 1997;127(10):2030–2038. doi: 10.1093/jn/127.10.2030. [DOI] [PubMed] [Google Scholar]
- 124.Baier PC, et al. Assessment of spontaneously occurring periodic limb movements in sleep in the rat. J Neurol Sci. 2002;198(1–2):71–77. doi: 10.1016/S0022-510X(02)00078-3. [DOI] [PubMed] [Google Scholar]
- 125.Esteves AM, et al. Spontaneously hypertensive rats: possible animal model of sleep-related movement disorders. J Mot Behav. 2013;45(6):487–493. doi: 10.1080/00222895.2013.833079. [DOI] [PubMed] [Google Scholar]
- 126.Silvani A, et al. Physiological time structure of the tibialis anterior motor activity during sleep in mice, rats and humans. J Sleep Res. 2015;24(6):695–701. doi: 10.1111/jsr.12319. [DOI] [PubMed] [Google Scholar]
- 127.Ferri R, et al. Sequence analysis of leg movements during sleep with different intervals (<10, 10–90 and >90 s) in restless legs syndrome. J Sleep Res. 2017;26(4):436–443. doi: 10.1111/jsr.12500. [DOI] [PubMed] [Google Scholar]
- 128.Earley CJ, et al. Developing a behavioral model of restless legs syndrome utilizing mice with natural variances in ventral midbrain iron. Sleep Med. 2020;71:135–140. doi: 10.1016/j.sleep.2019.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Connor JR, et al. Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology. 2003;61(3):304–309. doi: 10.1212/01.WNL.0000078887.16593.12. [DOI] [PubMed] [Google Scholar]
- 130.Connor JR, et al. Iron and restless legs syndrome: treatment, genetics and pathophysiology. Sleep Med. 2017;31:61–70. doi: 10.1016/j.sleep.2016.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schormair B, et al. Identification of novel risk loci for restless legs syndrome in genome-wide association studies in individuals of European ancestry: a meta-analysis. Lancet Neurol. 2017;16(11):898–907. doi: 10.1016/S1474-4422(17)30327-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jimenez-Jimenez FJ, et al. Genetics of restless legs syndrome: an update. Sleep Med Rev. 2018;39:108–121. doi: 10.1016/j.smrv.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 133.Akcimen F, et al. Genetic and epidemiological characterization of restless legs syndrome in Quebec. Sleep. 2020;43(4). [DOI] [PubMed]
- 134.Didriksen M, et al. Large genome-wide association study identifies three novel risk variants for restless legs syndrome. Commun Biol. 2020;3(1):703. doi: 10.1038/s42003-020-01430-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ergun U, et al. Genome-wide association and whole exome sequencing studies reveal a novel candidate locus for restless legs syndrome. Eur J Med Genet. 2021;64(4):104186. doi: 10.1016/j.ejmg.2021.104186. [DOI] [PubMed] [Google Scholar]
- 136.Stefansson H, et al. A genetic risk factor for periodic limb movements in sleep. N Engl J Med. 2007;357(7):639–647. doi: 10.1056/NEJMoa072743. [DOI] [PubMed] [Google Scholar]
- 137.Winkelmann J, et al. Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat Genet. 2007;39(8):1000–1006. doi: 10.1038/ng2099. [DOI] [PubMed] [Google Scholar]
- 138.Jones SE, et al. Genetic studies of accelerometer-based sleep measures yield new insights into human sleep behaviour. Nat Commun. 2019;10(1):1585. doi: 10.1038/s41467-019-09576-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang H, et al. Genome-wide association analysis of self-reported daytime sleepiness identifies 42 loci that suggest biological subtypes. Nat Commun. 2019;10(1):3503. doi: 10.1038/s41467-019-11456-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Stogios PJ, Prive GG. The BACK domain in BTB-kelch proteins. Trends Biochem Sci. 2004;29(12):634–637. doi: 10.1016/j.tibs.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 141.Stogios PJ, et al. Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005;6(10):R82. doi: 10.1186/gb-2005-6-10-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chaharbakhshi E, Jemc JC. Broad-complex, tramtrack, and bric-a-brac (BTB) proteins: critical regulators of development. Genesis. 2016;54(10):505–518. doi: 10.1002/dvg.22964. [DOI] [PubMed] [Google Scholar]
- 143.Sorensen E, et al. A genetic risk factor for low serum ferritin levels in Danish blood donors. Transfusion. 2012;52(12):2585–2589. doi: 10.1111/j.1537-2995.2012.03629.x. [DOI] [PubMed] [Google Scholar]
- 144.Ji Y, et al. Genetic factors associated with iron storage in Australian blood donors. Blood Transfus. 2018;16(2):123–129. doi: 10.2450/2016.0138-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fawzy MS, et al. Molecular analysis of homeostatic iron regulator, transmembrane protease serine-6, and BTB domain-containing protein-9 variants and iron parameters in blood donors. Biosci Rep. 2021;41(1). [DOI] [PMC free article] [PubMed]
- 146.Hammerschlag AR, et al. Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits. Nat Genet. 2017;49(11):1584–1592. doi: 10.1038/ng.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lane JM, et al. Genome-wide association analyses of sleep disturbance traits identify new loci and highlight shared genetics with neuropsychiatric and metabolic traits. Nat Genet. 2017;49(2):274–281. doi: 10.1038/ng.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hisa T, et al. Hematopoietic, angiogenic and eye defects in Meis1 mutant animals. EMBO J. 2004;23(2):450–459. doi: 10.1038/sj.emboj.7600038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Azcoitia V, et al. The homeodomain protein Meis1 is essential for definitive hematopoiesis and vascular patterning in the mouse embryo. Dev Biol. 2005;280(2):307–320. doi: 10.1016/j.ydbio.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 150.Huret JL, et al. Atlas of genetics and cytogenetics in oncology and haematology in 2013. Nucleic Acids Res. 2013;41(Database issue):D920–D924. doi: 10.1093/nar/gks1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schormair B, et al. PTPRD (protein tyrosine phosphatase receptor type delta) is associated with restless legs syndrome. Nat Genet. 2008;40(8):946–948. doi: 10.1038/ng.190. [DOI] [PubMed] [Google Scholar]
- 152.Uhl GR, Martinez MJ. PTPRD: neurobiology, genetics, and initial pharmacology of a pleiotropic contributor to brain phenotypes. Ann N Y Acad Sci. 2019;1451(1):112–129. doi: 10.1111/nyas.14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shishikura M, et al. Expression of receptor protein tyrosine phosphatase delta, PTPdelta, in mouse central nervous system. Brain Res. 2016;1642:244–254. doi: 10.1016/j.brainres.2016.03.030. [DOI] [PubMed] [Google Scholar]
- 154.Uetani N, et al. Mammalian motoneuron axon targeting requires receptor protein tyrosine phosphatases sigma and delta. J Neurosci. 2006;26(22):5872–5880. doi: 10.1523/JNEUROSCI.0386-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kemlink D, et al. Replication of restless legs syndrome loci in three European populations. J Med Genet. 2009;46(5):315–318. doi: 10.1136/jmg.2008.062992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yang Q, et al. Association studies of variants in MEIS1, BTBD9, and MAP2K5/SKOR1 with restless legs syndrome in a US population. Sleep Med. 2011;12(8):800–804. doi: 10.1016/j.sleep.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li G, et al. Association of BTBD9 and MAP2K5/SKOR1 with restless legs syndrome in Chinese population. Sleep. 2017;40(4). [DOI] [PubMed]
- 158.Weinstock LB, Walters AS, Paueksakon P. Restless legs syndrome – theoretical roles of inflammatory and immune mechanisms. Sleep Med Rev. 2012;16(4):341–354. doi: 10.1016/j.smrv.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 159.Mizuhara E, et al. Corl1, a novel neuronal lineage-specific transcriptional corepressor for the homeodomain transcription factor Lbx1. J Biol Chem. 2005;280(5):3645–3655. doi: 10.1074/jbc.M411652200. [DOI] [PubMed] [Google Scholar]
- 160.DeAndrade MP, et al. Enhanced hippocampal long-term potentiation and fear memory in Btbd9 mutant mice. PLoS ONE. 2012;7(4):e35518. doi: 10.1371/journal.pone.0035518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Freeman A, et al. Sleep fragmentation and motor restlessness in a Drosophila model of restless legs syndrome. Curr Biol. 2012;22(12):1142–1148. doi: 10.1016/j.cub.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.DeAndrade MP, et al. Motor restlessness, sleep disturbances, thermal sensory alterations and elevated serum iron levels in Btbd9 mutant mice. Hum Mol Genet. 2012;21(18):3984–3992. doi: 10.1093/hmg/dds221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Muramatsu K, et al. Rotigotine suppresses sleep-related muscle activity augmented by injection of dialysis patients’ sera in a mouse model of restless legs syndrome. Sci Rep. 2019;9(1):16344. doi: 10.1038/s41598-019-52735-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gao Z, et al. Integrative proteome and ubiquitinome analyses reveal the substrates of BTBD9 and its underlying mechanism in sleep regulation. ACS Omega. 2022;7(14):11839–11852. doi: 10.1021/acsomega.1c07262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lyu S, et al. BTBD9 and dopaminergic dysfunction in the pathogenesis of restless legs syndrome. Brain Struct Funct. 2020;225(6):1743–1760. doi: 10.1007/s00429-020-02090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chiu CH, et al. Effect of MDMA-induced axotomy on the dorsal raphe forebrain tract in rats: an in vivo manganese-enhanced magnetic resonance imaging study. PLoS ONE. 2015;10(9):e0138431. doi: 10.1371/journal.pone.0138431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dudek M, et al. Brain activation induced by voluntary alcohol and saccharin drinking in rats assessed with manganese-enhanced magnetic resonance imaging. Addict Biol. 2015;20(6):1012–1021. doi: 10.1111/adb.12179. [DOI] [PubMed] [Google Scholar]
- 168.Perrine SA, et al. Cocaine-induced locomotor sensitization in rats correlates with nucleus accumbens activity on manganese-enhanced MRI. NMR Biomed. 2015;28(11):1480–1488. doi: 10.1002/nbm.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lyu S, et al. The role of BTBD9 in striatum and restless legs syndrome. eNeuro. 2019;6(5). [DOI] [PMC free article] [PubMed]
- 170.Fukuda J, Kawa K. Permeation of manganese, cadmium, zinc, and beryllium through calcium channels of an insect muscle membrane. Science. 1977;196(4287):309–311. doi: 10.1126/science.847472. [DOI] [PubMed] [Google Scholar]
- 171.Narita K, Kawasaki F, Kita H. Mn and Mg influxes through Ca channels of motor nerve terminals are prevented by verapamil in frogs. Brain Res. 1990;510(2):289–295. doi: 10.1016/0006-8993(90)91379-U. [DOI] [PubMed] [Google Scholar]
- 172.Saleem KS, et al. Magnetic resonance imaging of neuronal connections in the macaque monkey. Neuron. 2002;34(5):685–700. doi: 10.1016/S0896-6273(02)00718-3. [DOI] [PubMed] [Google Scholar]
- 173.Murayama Y, et al. Tracing neural circuits in vivo with Mn-enhanced MRI. Magn Reson Imaging. 2006;24(4):349–358. doi: 10.1016/j.mri.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 174.Duong TQ, et al. Functional MRI of calcium-dependent synaptic activity: cross correlation with CBF and BOLD measurements. Magn Reson Med. 2000;43(3):383–392. doi: 10.1002/(SICI)1522-2594(200003)43:3<383::AID-MRM10>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 175.Garcia Borreguero D, Winkelmann J, Allen RP. Introduction: towards a better understanding of the science of RLS/WED. Sleep Med. 2017;31:1–2. doi: 10.1016/j.sleep.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 176.Lyu S, et al. The role of BTBD9 in the cerebral cortex and the pathogenesis of restless legs syndrome. Exp Neurol. 2020;323:113111. doi: 10.1016/j.expneurol.2019.113111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lyu S, et al. The role of BTBD9 in the cerebellum, sleep-like behaviors and the restless legs syndrome. Neuroscience. 2020;440:85–96. doi: 10.1016/j.neuroscience.2020.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wu X, Xu J, Lu B. Acute post-stroke restless legs syndrome: the body of caudate nucleus considerations. Sleep Med. 2020;70:66–70. doi: 10.1016/j.sleep.2019.11.1253. [DOI] [PubMed] [Google Scholar]
- 179.Xiong L, et al. MEIS1 intronic risk haplotype associated with restless legs syndrome affects its mRNA and protein expression levels. Hum Mol Genet. 2009;18(6):1065–1074. doi: 10.1093/hmg/ddn443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Schulte EC, et al. Targeted resequencing and systematic in vivo functional testing identifies rare variants in MEIS1 as significant contributors to restless legs syndrome. Am J Hum Genet. 2014;95(1):85–95. doi: 10.1016/j.ajhg.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Spieler D, et al. Restless legs syndrome-associated intronic common variant in Meis1 alters enhancer function in the developing telencephalon. Genome Res. 2014;24(4):592–603. doi: 10.1101/gr.166751.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Salminen AV, et al. Meis1: effects on motor phenotypes and the sensorimotor system in mice. Dis Model Mech. 2017;10(8):981–991. doi: 10.1242/dmm.030080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Salminen AV, et al. Sleep disturbance by pramipexole is modified by Meis1 in mice. J Sleep Res. 2017. [DOI] [PubMed]
- 184.Meneely S, et al. Differential dopamine D1 and D3 receptor modulation and expression in the spinal cord of two mouse models of restless legs syndrome. Front Behav Neurosci. 2018;12:199. doi: 10.3389/fnbeh.2018.00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Huang H, et al. MEIS C termini harbor transcriptional activation domains that respond to cell signaling. J Biol Chem. 2005;280(11):10119–10127. doi: 10.1074/jbc.M413963200. [DOI] [PubMed] [Google Scholar]
- 186.Kocabas F, et al. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood. 2012;120(25):4963–4972. doi: 10.1182/blood-2012-05-432260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Unnisa Z, et al. Meis1 preserves hematopoietic stem cells in mice by limiting oxidative stress. Blood. 2012;120(25):4973–4981. doi: 10.1182/blood-2012-06-435800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lyu S, et al. Deficiency of Meis1, a transcriptional regulator, in mice and worms: neurochemical and behavioral characterizations with implications in the restless legs syndrome. J Neurochem. 2020;155(5):522–537. doi: 10.1111/jnc.15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Catoire H, et al. Restless legs syndrome-associated MEIS1 risk variant influences iron homeostasis. Ann Neurol. 2011;70(1):170–175. doi: 10.1002/ana.22435. [DOI] [PubMed] [Google Scholar]
- 190.Gonzalez-Lazaro M, et al. Two new targeted alleles for the comprehensive analysis of Meis1 functions in the mouse. Genesis. 2014;52(12):967–975. doi: 10.1002/dvg.22833. [DOI] [PubMed] [Google Scholar]
- 191.Cathiard L, et al. Investigation of dopaminergic signalling in Meis homeobox 1 (Meis1) deficient mice as an animal model of restless legs syndrome. J Sleep Res. 2021;30(5):e13311. doi: 10.1111/jsr.13311. [DOI] [PubMed] [Google Scholar]
- 192.Uetani N, et al. Impaired learning with enhanced hippocampal long-term potentiation in PTPdelta-deficient mice. EMBO J. 2000;19(12):2775–2785. doi: 10.1093/emboj/19.12.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Drgonova J, et al. Mouse model for protein tyrosine phosphatase D (PTPRD) Associations with restless leg syndrome or Willis-Ekbom disease and addiction: reduced expression alters locomotion, sleep behaviors and cocaine-conditioned place preference. Mol Med. 2015;21(1):717–725. doi: 10.2119/molmed.2015.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Morais MA, et al. PTPRD as a candidate druggable target for therapies for restless legs syndrome? J Sleep Res. 2020: e13216. [DOI] [PubMed]
- 195.Frank MK, et al. Sleep-related movement disorder symptoms in SHR are attenuated by physical exercise and an angiotensin-converting enzyme inhibitor. Physiol Behav. 2016;154:161–168. doi: 10.1016/j.physbeh.2015.11.026. [DOI] [PubMed] [Google Scholar]
- 196.Wang X, et al. Targeted deletion of mek5 causes early embryonic death and defects in the extracellular signal-regulated kinase 5/myocyte enhancer factor 2 cell survival pathway. Mol Cell Biol. 2005;25(1):336–345. doi: 10.1128/MCB.25.1.336-345.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Huang Y, et al. Map2k5-deficient mice manifest phenotypes and pathological changes of dopamine deficiency in the central nervous system. Front Aging Neurosci. 2021;13:651638. doi: 10.3389/fnagi.2021.651638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Salas RME, et al. Connecting clinical aspects to corticomotor excitability in restless legs syndrome: a TMS study. Sleep Med. 2018;49:105–112. doi: 10.1016/j.sleep.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 199.Lanza G, et al. Distinctive patterns of cortical excitability to transcranial magnetic stimulation in obstructive sleep apnea syndrome, restless legs syndrome, insomnia, and sleep deprivation. Sleep Med Rev. 2015;19:39–50. doi: 10.1016/j.smrv.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 200.Rye DB, Trotti LM. Restless legs syndrome and periodic leg movements of sleep. Neurol Clin. 2012;30(4):1137–1166. doi: 10.1016/j.ncl.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 201.Tassinari CA, et al. Neuroethological approach to frontolimbic epileptic seizures and parasomnias: the same central pattern generators for the same behaviours. Rev Neurol (Paris) 2009;165(10):762–768. doi: 10.1016/j.neurol.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 202.Ferri R. Two legs, one heart, one sleeping brain. Sleep Med. 2006;7(3):299–300. doi: 10.1016/j.sleep.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 203.Salminen AV, et al. Disconnection between periodic leg movements and cortical arousals in spinal cord injury. J Clin Sleep Med. 2013;9(11):1207–1209. doi: 10.5664/jcsm.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Manconi M, et al. Restless legs syndrome is a common finding in multiple sclerosis and correlates with cervical cord damage. Mult Scler. 2008;14(1):86–93. doi: 10.1177/1352458507080734. [DOI] [PubMed] [Google Scholar]
- 205.Abele M, et al. Restless legs syndrome in spinocerebellar ataxia types 1, 2, and 3. J Neurol. 2001;248(4):311–314. doi: 10.1007/s004150170206. [DOI] [PubMed] [Google Scholar]
- 206.Czesnik D, et al. Ih contributes to increased motoneuron excitability in restless legs syndrome. J Physiol. 2019;597(2):599–609. doi: 10.1113/JP275341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Holland MT, et al. Epidural spinal cord stimulation: a novel therapy in the treatment of restless legs syndrome. World Neurosurg. 2016;92:582.e15–582.e18. doi: 10.1016/j.wneu.2016.05.077. [DOI] [PubMed] [Google Scholar]
- 208.Lai YY, et al. Neurotoxic lesions at the ventral mesopontine junction change sleep time and muscle activity during sleep: an animal model of motor disorders in sleep. Neuroscience. 2008;154(2):431–443. doi: 10.1016/j.neuroscience.2008.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zhu H, et al. Expression and distribution of all dopamine receptor subtypes (D1–D5) in the mouse lumbar spinal cord: a real-time polymerase chain reaction and non-autoradiographic in situ hybridization study. Neuroscience. 2007;149:885–897. doi: 10.1016/j.neuroscience.2007.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zhu H, et al. Unaltered D1, D2, D4, and D5 dopamine receptor mRNA expression and distribution in the spinal cord of the D3 receptor knockout mouse. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 2008;194(11):957–962. doi: 10.1007/s00359-008-0368-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Levant B, McCarson KE. D(3) dopamine receptors in rat spinal cord: implications for sensory and motor function. Neurosci Lett. 2001;303(1):9–12. doi: 10.1016/S0304-3940(01)01692-5. [DOI] [PubMed] [Google Scholar]
- 212.Brewer KL, et al. Dopamine D3 receptor dysfunction prevents anti-nociceptive effects of morphine in the spinal cord. Front Neural Circuits. 2014;8:62-01–62-10. doi: 10.3389/fncir.2014.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Keeler BE, et al. Opposing aging-related shift of excitatory dopamine D1 and inhibitory D3 receptor protein expression in striatum and spinal cord. J Neurophysiol. 2016;115(1):363–369. doi: 10.1152/jn.00390.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Pappas SS, et al. Lack of D2 receptor mediated regulation of dopamine synthesis in A11 diencephalospinal neurons in male and female mice. Brain Res. 2008;1214:1–10. doi: 10.1016/j.brainres.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 215.Keeler BE, et al. Increased excitability of spinal pain reflexes and altered frequency-dependent modulation in the dopamine D3-receptor knockout mouse. Exp Neurol. 2012;238(2):273–283. doi: 10.1016/j.expneurol.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 216.Sharples SA, et al. Dopaminergic modulation of locomotor network activity in the neonatal mouse spinal cord. J Neurophysiol. 2015;113(7):2500–2510. doi: 10.1152/jn.00849.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Clemens S, Sawchuk MA, Hochman S. Reversal of the circadian expression of tyrosine-hydroxylase but not nitric oxide synthase levels in the spinal cord of dopamine D3 receptor knockout mice. Neuroscience. 2005;133(2):353–357. doi: 10.1016/j.neuroscience.2005.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Clemens S, et al. Opposing modulatory effects of D1- and D2-like receptor activation on a spinal central pattern generator. J Neurophysiol. 2012;107:2250–2259. doi: 10.1152/jn.00366.2011. [DOI] [PubMed] [Google Scholar]
- 219.Sharples SA, et al. Dopamine: a parallel pathway for the modulation of spinal locomotor networks. Front Neural Circuits. 2014;8:55. doi: 10.3389/fncir.2014.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Rivera-Oliver M, et al. Adenosine A1-dopamine D1 receptor heteromers control the excitability of the spinal motoneuron. Mol Neurobiol. 2019;56(2):797–811. doi: 10.1007/s12035-018-1120-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Walters AS, et al. Does the endogenous opiate system play a role in the restless legs syndrome? A pilot post-mortem study. J Neurol Sci. 2009;279(1–2):62–65. doi: 10.1016/j.jns.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 222.Sun YM, et al. Opioids protect against substantia nigra cell degeneration under conditions of iron deprivation: a mechanism of possible relevance to the restless legs syndrome (RLS) and Parkinson’s disease. J Neurol Sci. 2011;304(1–2):93–101. doi: 10.1016/j.jns.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 223.Berteotti C, et al. Tibialis anterior electromyographic bursts during sleep in histamine-deficient mice. J Sleep Res. 2021;30(4):e13255. doi: 10.1111/jsr.13255. [DOI] [PubMed] [Google Scholar]
- 224.Garcia-Borreguero D, Cano I, Granizo JJ. Treatment of restless legs syndrome with the selective AMPA receptor antagonist perampanel. Sleep Med. 2017;34:105–108. doi: 10.1016/j.sleep.2017.03.012. [DOI] [PubMed] [Google Scholar]
- 225.Garcia-Borreguero D, et al. A randomized, placebo-controlled crossover study with dipyridamole for restless legs syndrome. Mov Disord. 2021;36(10):2387–2392. doi: 10.1002/mds.28668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lyu S, et al. Hyperactivity, dopaminergic abnormalities, iron deficiency and anemia in an in vivo opioid receptors knockout mouse: implications for the restless legs syndrome. Behav Brain Res. 2019;374:112123. doi: 10.1016/j.bbr.2019.112123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Lyu S, et al. Mu opioid receptor knockout mouse: phenotypes with implications on restless legs syndrome. J Neurosci Res. 2020;98(8):1532–1548. doi: 10.1002/jnr.24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Koo BB, et al. Alpha-melanocyte stimulating hormone and adrenocorticotropic hormone: an alternative approach when thinking about restless legs syndrome? Mov Disord. 2008;23(9):1234–1242. doi: 10.1002/mds.22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Quiroz C, et al. Up-regulation of striatal adenosine A(2A) receptors with iron deficiency in rats: effects on locomotion and cortico-striatal neurotransmission. Exp Neurol. 2010;224(1):292–298. doi: 10.1016/j.expneurol.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Yepes G, et al. Targeting hypersensitive corticostriatal terminals in restless legs syndrome. Ann Neurol. 2017;82(6):951–960. doi: 10.1002/ana.25104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Chen SJ, et al. Prevalence of restless legs syndrome during pregnancy: a systematic review and meta-analysis. Sleep Med Rev. 2018;40:43–54. doi: 10.1016/j.smrv.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 232.Manconi M, et al. Restless legs syndrome and pregnancy. Neurology. 2004;63(6):1065–1069. doi: 10.1212/01.WNL.0000138427.83574.A6. [DOI] [PubMed] [Google Scholar]
- 233.Cesnik E, et al. Transient RLS during pregnancy is a risk factor for the chronic idiopathic form. Neurology. 2010;75(23):2117–2120. doi: 10.1212/WNL.0b013e318200d779. [DOI] [PubMed] [Google Scholar]
- 234.Prosperetti C, Manconi M. Restless legs syndrome/Willis-Ekbom disease and pregnancy. Sleep Med Clin. 2015;10(3):323–329. doi: 10.1016/j.jsmc.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 235.Chavez C, et al. The effect of estrogen on dopamine and serotonin receptor and transporter levels in the brain: an autoradiography study. Brain Res. 2010;1321:51–59. doi: 10.1016/j.brainres.2009.12.093. [DOI] [PubMed] [Google Scholar]
- 236.Sun J, et al. Progesterone: the neglected hormone in schizophrenia? A focus on progesterone-dopamine interactions. Psychoneuroendocrinology. 2016;74:126–140. doi: 10.1016/j.psyneuen.2016.08.019. [DOI] [PubMed] [Google Scholar]
- 237.Mariano MO, et al. Changes in motor behavior during pregnancy in rats: the basis for a possible animal model of restless legs syndrome. Rev Bras Ginecol Obstet. 2014;36(10):436–441. doi: 10.1590/SO100-720320140005105. [DOI] [PubMed] [Google Scholar]
- 238.Zhou X, et al. The efficacy and safety of pharmacological treatments for restless legs syndrome: systemic review and network meta-analysis. Front Neurosci. 2021;15:751643. doi: 10.3389/fnins.2021.751643. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.