

Abstract
Diffusion MRI is the dominant non-invasive imaging method used to characterize white matter organization in health and disease. Increasingly, fiber-specific properties within a voxel are analyzed using fixels. While tools for conducting statistical analyses of fixel-wise data exist, currently available tools support only a limited number of statistical models. Here we introduce ModelArray, an R package for mass-univariate statistical analysis of fixel-wise data. At present, ModelArray supports linear models as well as generalized additive models (GAMs), which are particularly useful for studying nonlinear effects in lifespan data. In addition, ModelArray also aims for scalable analysis. With only several lines of code, even large fixel-wise datasets can be analyzed using a standard personal computer. Detailed memory profiling revealed that ModelArray required only limited memory even for large datasets. As an example, we applied ModelArray to fixel-wise data derived from diffusion images acquired as part of the Philadelphia Neurodevelopmental Cohort (n = 938). ModelArray revealed anticipated nonlinear developmental effects in white matter. Moving forward, ModelArray is supported by an open-source software development model that can incorporate additional statistical models and other imaging data types. Taken together, ModelArray provides a flexible and efficient platform for statistical analysis of fixel-wise data.
Keywords: Fixel-based analysis, Statistical analysis, Software, Development, Big data, MRI
1. Introduction
Diffusion MRI (dMRI) is the dominant method used to non-invasively study white matter organization in the human brain. The most commonly used method for modeling the diffusion signal is diffusion tensor imaging (DTI; Basser and Pierpaoli, 1996). However, DTI cannot effectively model two or more crossing fibers within a given voxel; crossing fibers are thought to comprise up to ~90% of white matter (WM) voxels (Jeurissen et al., 2013; Schilling et al., 2018; Yeh et al., 2013). One method for addressing crossing fibers that is increasingly ascendant is fixel-based analysis (FBA; Raffelt et al., 2017; Dhollander et al., 2021). A fixel refers to a specific fiber population in a voxel; with FBA, multiple distinct fiber populations can be estimated within a voxel and multiple fiber-specific properties can be quantified (Raffelt et al., 2015, 2017; Dhollander et al., 2021). An example fixel-wise image is shown in Fig. 1. Similar to voxel-wise images, each fixel can have associated measure(s), just like a voxel-wise image represents a measure at each voxel. However, there are also distinct differences between fixels and voxels: unlike the regular 3D voxel grid, there can be zero, one, or more fixels within a single voxel. In other words, the specific information regarding a varying number of fixels at each spatial location is not simply another image dimension (i.e., four dimensions instead of three); it creates unique challenges in the analysis of fixel-wise data (Dhollander et al., 2021).
Fig. 1.
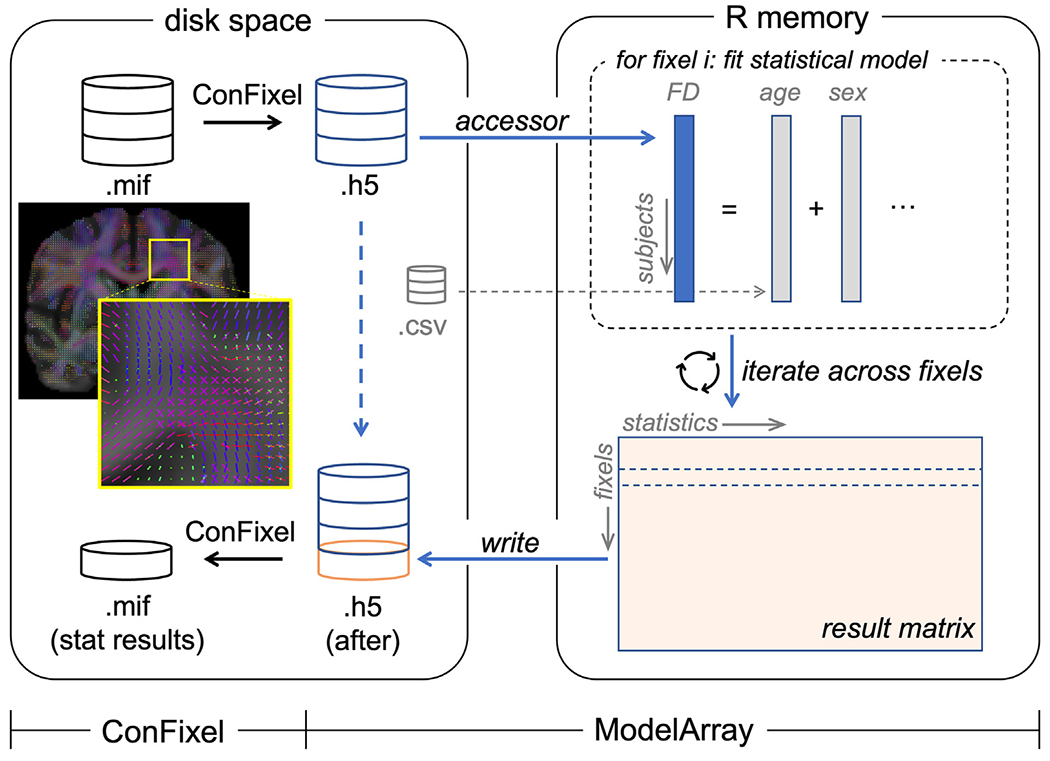
Schematic of ModelArray and its companion converter ConFixel. The original fixel-wise data (.mif files; see the example fixel-wise image) are first converted into an HDF5 file (.h5) using ConFixel (top of the left box). ModelArray provides easy access to fixel-wise data in the HDF5 file (“accessor”). When performing statistical analysis of each fixel (top of the right box), to reduce memory usage, only a limited block of fixel-wise data is read into the memory. Using the phenotypes of interest (e.g.,: age, sex; provided by a CSV file), ModelArray fits a statistical model and calculates statistical output for each fixel. After iterating across fixels, the result matrix is generated (bottom of the right box) and saved to the original HDF5 file on disk by ModelArray (“write”). Finally, ConFixel converts the result matrix in this HDF5 file into a list of .mif files ready to be viewed (bottom of the left box). The fixels in the fixel-wise image are colored by fixel orientation (red: left–right, green: anterior–posterior, blue: inferior–superior), and background image is the fiber orientation distribution (FOD) template.
The FBA pipeline typically includes two parts. First, fixel-wise data is generated for each participant in a sample and quantified according to standard measures like fiber density (FD), fiber-bundle cross-section (FC), or their combination – fiber density and cross-section (FDC). Second, the high-dimensional fixel-wise data from a sample is often analyzed in template space using mass-univariate hypothesis testing; this often relies upon connectivity-based fixel enhancement (CFE; Raffelt et al., 2015) as implemented in MRtrix (https://www.mrtrix.org/; Tournier et al., 2019). Like threshold-free cluster enhancement (TFCE) for voxel-wise data (Smith and Nichols, 2009), CFE is a statistics enhancement method that can be applied to fixel-wise data. Instead of using simple 3D voxel neighborhoods in TFCE, CFE incorporates the fixel-to-fixel connectivity information to define the cluster extent in fixel-wise data.
However, the statistical models supported by MRtrix for FBA are currently limited to the general linear model (GLM). This may not be optimal for modeling nonlinear effects which are often of interest in lifespan studies (e.g., Bethlehem et al., 2022; Lebel et al., 2012). Ideally, a statistical analysis toolset should be extensible to incorporate diverse statistical models. (https://www.R-project.org; R Core Team, 2021) is a popular open-source statistical programming software, and it supports a myriad of statistical functionality. R is also widely used by statisticians, with a constantly expanding repertoire of functionality. Generalized additive models (GAMs; Wood, 2001, 2004) are among the most widely used approaches to model nonlinear effects of interest in R. GAMs can rigorously model both linear and nonlinear effects by applying a penalty that helps avoid over-fitting; this approach is particularly valuable in high-dimensional data settings – cases when hundreds of thousands of fixels are present – where it is difficult to conduct detailed model diagnostics. Providing extensibility to diverse statistical models in R for the analysis of fixel-wise data is the primary motivation for developing ModelArray.
In addition, ModelArray also aims for memory efficiency. The memory required for statistical analysis of neuroimaging data usually scales by image resolution and sample size (e.g., Raffelt et al., 2015). These memory requirements impede the statistical analysis of large-scale data resources that include thousands of participants; e.g., the Philadelphia Neurodevelopmental Cohort (PNC; Satterthwaite et al., 2014), the Human Connectome Project (HCP; Van Essen et al., 2013), or the Healthy Brain Network (HBN; Alexander et al., 2017). When faced with such large data resources, investigators often opt to reduce the dimensionality of the data and use regional summary measures, even if it is not scientifically optimal. ModelArray aims to facilitate running large-scale fixel-wise statistical analysis on a typical personal computer (e.g., a laptop).
To address these limitations, we introduce ModelArray (https://pennlinc.github.io/ModelArray/), a memory-efficient R package for statistical analysis of fixel-wise data. To maximize memory efficiency, ModelArray does not load the entire fixel-wise data into the memory. Instead, it only reads a limited block of data when needed by leveraging the Hierarchical Data Format 5 (HDF5) file format and DelayedArray package in R (Pagès et al., 2021), At present, ModelArray supports linear models and GAMs, but it is by design extensible and can incorporate many statistical models implemented in R. To demonstrate ModelArray’s scalability, functionality, and extensibility, we profiled its memory usage and applied it to examine nonlinear patterns of brain development using fixel-wise data from the PNC (n = 938). As described below, ModelArray allows for efficient and flexible analysis of fixel-wise data in large scale data resources.
2. Materials and methods
2.1. Overview
ModelArray is an R package for mass-univariate hypothesis testing of fixel-wise data that is designed to be scalable for large datasets. We chose R as the platform as it is among the most widely used platforms for statistical computing. This feature facilitates the potential to easily incorporate diverse statistical models. ModelArray takes the fixel-wise data as input, after it has been converted to the HDF5 format by its companion software ConFixel (https://github.com/PennLINC/ConFixel). Fixel-wise data with metrics such as FD, FC, and FDC can be calculated in existing software such as MRtrix (Tournier et al., 2019). ModelArray performs statistical analysis for each fixel based on the statistical formula a user provides, and finally saves statistical output as images via ConFixel. These output images can then be viewed in widely-used visualization tools such as MRView from MRtrix (https://www.mrtrix.org/; Tournier et al., 2019).
2.2. Software design and memory efficiency
We capitalized upon the R package DelayedArray (Pagès et al., 2021) to maximize memory efficiency. Of note, the term “memory” is used in this paper to refer to the computer’s memory (RAM) used by software (including data loaded into the memory), and “disk” or “disk space” refers to the hard disk space where the files (e.g., an HDF5 file) are stored. ModelArray wraps fixel-wise data on disk into a DelayedArray object, allowing common array operations such as indexing (e.g., extracting values of a specific fixel from a matrix) or transposing to be performed without loading the on-disk object into memory. DelayedArray objects store their component data in an HDF5 file, and operations on a DelayedArray object are executed in a memory-efficient, “delayed” way (where most R operations are processed on-demand and en masse). The result is a memory-efficient and easy-to-use R interface for a large and hierarchical on-disk dataset. After being generated by ConFixel (see below), an HDF5 file of fixel-wise data contains a scalar matrix (fixels by participants), basic information of fixels and voxels (e.g., lookup tables of the directions of fixels and the coordinates of voxels that contain fixels), and, once calculated by ModelArray, one or more result matrices (fixels by statistical metrics). Leveraging DelayedArray, HDF5 format, and the supporting R package HDF5Array (Pagès, 2021), the on-disk fixel-wise data can be accessed and manipulated while minimizing memory requirements.
2.3. ModelArray workflow
ModelArray is packaged with the companion software ConFixel for converting fixel-wise data to the expected file format (see Fig. 1). Specifically, ConFixel is Python-based command-line interface software, and it converts between the original MRtrix image format (.mif) and the HDF5 file format (.h5) used for ModelArray. After the file format conversion, ModelArray generates a ModelArray-class object for representing the on-disk HDF5 file. ModelArray uses the S4 Object Oriented Programming (OOP) model which gives users easy access to the scalar matrix, the source .mif file list, one or more results matrices (if any), and the file path to the HDF5 file. When fitting models, ModelArray iterates across all fixels in the scalar matrix but only reads a limited block of data for each current fixel in order to reduce memory usage. For each fixel, the software fits a model for the participant-level phenotypes of interest – such as age, sex, or diagnosis, which are loaded from a separate CSV file provided by the user – and generates the statistical outputs for each fixel, such as p-values, coefficient estimations, and t-statistics. After generating the result matrix of fixel-wise statistics, ModelArray will calculate corrected p-values using the False Discovery Rate (FDR) and export the final result matrix back into the input HDF5 file. Finally, ConFixel converts the HDF5 file’s results matrix into a list of .mif files that are readable by widely-used visualization tools such as MRView from MRtrix (https://www.mrtrix.org/; Tournier et al., 2019).
2.4. ModelArray functions
ModelArray provides functions for model fitting and writing statistical results. At present, ModelArray supports linear models (ModelArray.lm()) as well as GAMs with and without penalized splines (ModelArray.gam()). Model fitting can be accelerated by requesting more CPU cores for parallel computing. ModelArray writes the rich statistical output of R into an HDF5 file using the writeResults() function. This HDF5 file is then converted to a list of .mif files with ConFixel for viewing, as described above. Default statistical output from ModelArray includes several maps for each model term (e.g., coefficient, t-statistic, raw and FDR-corrected p-values), as well as maps regarding the overall model fit (e.g., adjusted R-squared, raw and FDR-corrected p-values from the model F-test in linear models). New statistical models can be easily added by any GitHub contributor following the same workflow as existing ones (ModelArray.lm() and ModelArray.gam()); see developer documentation at: https://pennlinc.github.io/ModelArray/articles/doc_for_developer.html. Thus, ModelArray is extensible to many diverse statistical methods used in R.
2.5. Evaluation data
To evaluate ModelArray, we used the fixel-wise data generated from the Philadelphia Neurodevelopmental Cohort (PNC; Satterthwaite et al., 2014). Here we provide a brief summary of the data and methods including participant inclusion, image acquisition, image quality assurance, diffusion MRI preprocessing, and fixel-based analysis. In total, we included n = 938 participants (521 female, 417 male) aged 8–23 years old. Participants were excluded due to lack of diffusion imaging data, abnormalities in brain structure, major health conditions, missing B0 field map, poor image quality, etc. All the dMRI data underwent a rigorous manual and automated quality assessment as previously described (Roalf et al., 2016).
MRI scans were acquired on a Siemens TIM Trio 3T scanner. Diffusion MRI scans were acquired with a twice-refocused spin-echo (TRSE) single-shot echo-planar imaging (EPI) sequence. The sequence included 64 diffusion-weighted images of b = 1000s/mm2 as well as 7 interspersed b = 0 images; these images were acquired over two scan runs. The in-plane resolution was 1.875×1.875 mm2 , slice thickness was 2 mm without gap. In addition, a B0 field map was also acquired for distortion correction of dMRI data. In-scanner motion during the dMRI scan was quantified as the root mean squared displacement (mean relative RMS); this was calculated from 7 b = 0 volumes interspersed over the course of the dMRI scan (Roalf et al., 2016). Motion was included as a covariate when modeling age effects using GAMs (described below). Diffusion images were processed with QSIPrep (https://github.com/PennBBL/qsiprep; Cieslak et al., 2021). This process included MP-PCA denoising (using dwidenoise from MRtrix; Veraart et al., 2016), Gibbs unringing (using mrdegibbs from MRtrix; Kellner et al., 2016), B1 field inhomogeneity correction (using dwibiascorrect from MRtrix with the N4 algorithm; Tustison et al., 2010), signal drift correction (Vos et al., 2017), susceptibility distortion correction (using prelude from FSL; https://fsl.fmrib.ox.ac.uk/fsl/fslwiki/FUGUE), eddy current-induced correction and head motion correction (using Eddy from FSL, with outlier replacement; Andersson and Sotiropoulos, 2016; Andersson et al., 2016). Finally, the images were resampled to AC-PC alignment with 1.25 mm isotropic voxels.
Following preprocessing, fixel-based analysis was performed using MRtrix (https://www.mrtrix.org/, version v3.0RC3) (Dhollander et al., 2021; Raffelt et al., 2017; Tournier et al., 2019). Briefly, study-specific response functions for single-fiber white matter, gray matter and cerebrospinal fluid (CSF) were calculated via a robust and fully automated unsupervised method (Dhollander et al., 2016, 2019) using data from 30 representative participants across ages (15 M/15F). Fiber orientation distributions (FODs) for all participants were then estimated using Single-Shell 3-Tissue Constrained Spherical Deconvolution (SS3T-CSD) (Dhollander and Connelly, 2016) from MRtrix3Tissue (https://3Tissue.github.io), a fork of MRtrix3 (Tournier et al., 2019). A study-specific FOD template was generated, and participants’ FOD images were registered to this study template. After defining fixels, FDC was quantified and chosen as the metric of interest as it combines both FD and FC and may be more sensitive than FD or FC alone (Dhollander et al., 2021). Finally, the FDC values were smoothed with “connected” nearby fixels to increase the signal-to-noise ratio (Raffelt et al., 2015). To smooth the data, a whole-brain probabilistic tractogram with 2 million streamlines was generated from the FOD template, and a fixel-fixel connectivity matrix based on this tractogram was computed. Lastly, FDC values were smoothed based on this matrix. This procedure yielded fixel-wise data in template space for each participant, which included 602,229 fixels. This fixel-wise data was used by ModelArray for memory profiling and application of GAM.
2.6. Memory profiling
We evaluated the memory efficiency of ModelArray. Memory profiling was completed using a Linux system by Working Set Size (WSS) Tools for Linux (https://www.brendangregg.com/wss.html). We used a virtual machine on a standalone computer to avoid interference from other users, with memory allocated to the virtual machine = 55 Gigabytes (GB) and total RAM on the computer = 64 GB. Specifically, the resident set size (RSS) – real memory pages currently mapped – was captured by WSS and recorded. We sampled the RSS once every second for both parent and any child processes (if more than one CPU core was used). The total RSS from all processes was calculated by summing the interpolated RSS values at each second, and the maximum RSS used over time was calculated.
We used a simple linear model for memory profiling: FDC = intercept + age. To evaluate how memory usage scaled with data size, we examined the full sample (n = 938) as well as subsamples of different sizes (n = 30, n = 100, n = 300, n = 500, and n = 750). Furthermore, memory profiling over different parallelization factors was also performed. During the memory profiling for ModelArray, up to four CPU cores were made available. In all cases, memory profiling was run three times for each use case, and the median value was reported. Note that the memory profiling and the application of GAM (next section) were done in local R, without using the Docker image of ModelArray.
2.7. Application using generalized additive models
The memory benchmarking studies were conducted using linear models. However, in addition, we also demonstrated the use of GAMs in ModelArray for modeling nonlinear developmental effects. Notably, existing tools such as MRtrix only support GLMs and do not easily allow users to model nonlinear developmental effects using GAMs. This application illustrates the extensibility of ModelArray to incorporate diverse statistical models.
For this application, data from all participants (n = 938) was used. Age was modeled as a smooth term s(age) with four basis functions (k = 4); sex and in-scanner motion (mean relative RMS displacement) were included as covariates. As in prior work (Pines et al., 2022), the effect size of the age term was quantified as , where was the adjusted R-squared in the full model, and was that in a reduced model that did not include the age term.
2.8. Open-source software development and release
ModelArray has been developed on GitHub with version controls and all code is openly available on GitHub (see Data and code availability statements). Continuous Integration (CI) testing is used to ensure stability and quality assurance. Specifically, we use CircleCI to perform unit tests for all major features of ModelArray. These tests ensure the consistency between the statistical results calculated in ModelArray fitting loop and those calculated in standard R. Once updated code is committed to GitHub, CircleCI automatically builds the software and runs unit tests. If there are any errors, CircleCI will alert the developers to this failure immediately, assuring that updates do not alter software performance.
To enhance the portability of ModelArray and its companion converter ConFixel, we also provide a Docker image of ModelArray and ConFixel (publicly available at https://hub.docker.com/r/pennlinc/modelarray_confixel). With this Docker image, users do not need to install ModelArray, ConFixel or their dependent R or Python packages. Documentation of how to use this Docker image is available at https://pennlinc.github.io/ModelArray/articles/container.html. This Docker image is automatically built by CircleCI and pushed to Docker Hub.
2.9. Data and code availability statements
ModelArray documentation can be found at https://pennlinc.github.io/ModelArray. All code used to perform memory profiling and application of GAMs is available at https://github.com/PennLINC/ModelArray_paper. The source code for ModelArray is available at https://github.com/PennLINC/ModelArray, and the source code for ConFixel is available at https://github.com/PennLINC/ConFixel. The version of ModelArray used for benchmarking and demonstration was commit SHA-1 0911c4f. We also provide a Docker image of ModelArray and ConFixel (available at https://hub.docker.com/r/pennlinc/modelarray_confixel). The PNC dataset used in this paper is available on dbGAP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000607.v3.p2). As part of the software tutorial, example fixel-wise data from 100 PNC participants is openly shared on OSF (https://doi.org/10.17605/OSF.IO/JVEHY).
2.10. Ethics statement
No new data were collected specifically for this paper. The Philadelphia Neurodevelopmental Cohort (PNC; Satterthwaite et al., 2014) was approved by IRBs of the University of Pennsylvania and Children’s Hospital of Philadelphia. All adult participants in the PNC provided informed consent to participate; minors provided assent alongside the informed consent of their parents or guardian.
3. Results
3.1. Software walkthrough
Before using ModelArray, two files need to be prepared by the user: an HDF5 (.h5) file of fixel-wise data (example filename here: example.h5), and a CSV file of participant’s phenotypes of interest (e.g., age, sex, etc.; example filename here: example.csv). The HDF5 file can be obtained by applying ConFixel to convert the original fixel-wise data (.mif files) into required HDF5 file format. Although ConFixel is implemented in Python, it is used via a command-line interface. After installation, users can directly run data conversion in a terminal console, and there is no need to open a Python console. An example of the usage of ModelArray is displayed in Fig. 2. After loading the package ModelArray in R (code line #3 in Fig. 2), a ModelArray-class object modelarray was created with the function ModelArray(); it represents the fixel-wise data in the HDF5 (.h5) file on disk, including the scalar matrix (fixels by participants) (code line #5). As the entire data was not loaded into memory, this object only required less than 1 Megabytes (MB) for complete n = 938 evaluation data, much less than the HDF5 file size on the disk (2.1 GB). After the data frame of phenotypes was loaded into R (code line #6), mass-univariate analyses using linear models and GAMs were performed with ModelArray.lm() and ModelArray.gam(), respectively (code line #9–10). The statistical outputs lm.outputs and gam.outputs were saved back to the original HDF5 file with the function writeResults() (code line #13–14). These outputs saved in the HDF5 file can be converted back to .mif files by ConFixel for viewing.
Fig. 2.
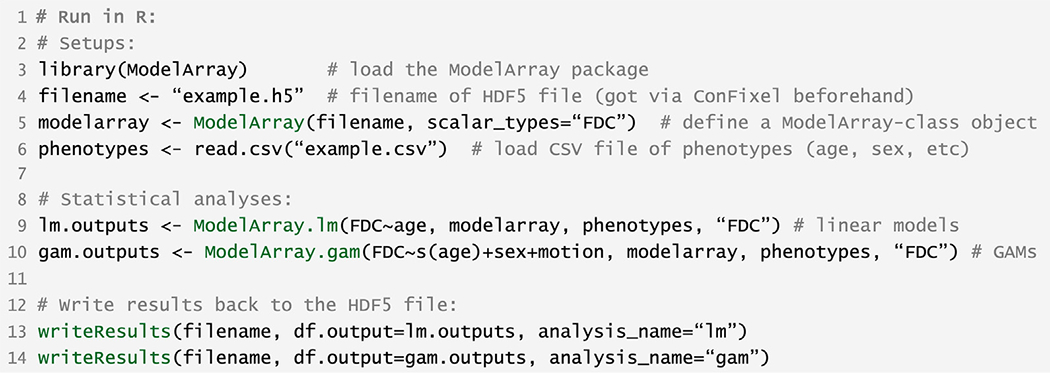
Example R code for executing analysis using ModelArray. ModelArray functions are highlighted in green.
For further details, as part of the comprehensive online documentation, please see the “Walkthrough” of ModelArray and ConFixel (https://pennlinc.github.io/ModelArray/articles/walkthrough.html). This walkthrough can be used in conjunction with openly-shared fixel-wise data from 100 PNC participants, which is available on OSF (https://doi.org/10.17605/OSF.IO/JVEHY).
3.2. ModelArray is memory-efficient and robust to dataset size
We profiled the memory usage of ModelArray over a range of input data sizes (e.g., number of participants) and parallelization settings. As a first step, we evaluated both the full dataset (n = 938) as well as five smaller sub-samples. This initial evaluation was completed using four CPU cores. As the number of participants analyzed increased, ModelArray memory usage only changed minimally (Fig. 3).
Fig. 3.
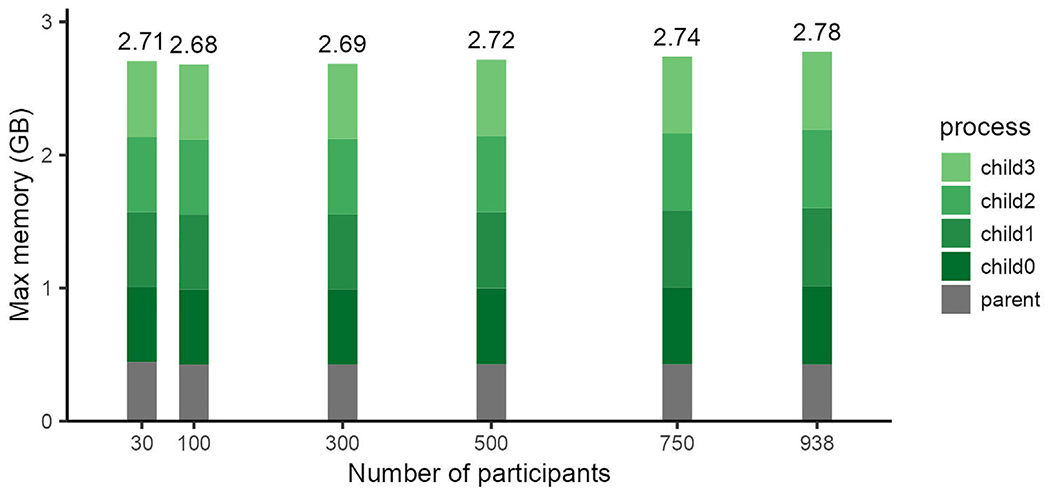
Memory required by ModelArray does not vary by sample size. The maximal memory required by a linear model executed using ModelArray.lm() was evaluated when analyzing a range of sample sizes. All models were performed with a parallelization factor of 4.
Next, we examined how parallelization options impacted memory use. As expected, when ModelArray requested more CPUs for analysis of samples of either small (n = 30, Fig. 4A) or large number of participants (n = 938, Fig. 4B), the memory required scaled by the parallelization factor. However, even when 4 CPU cores were requested, ModelArray consumed less than 3GB of memory.
Fig. 4.
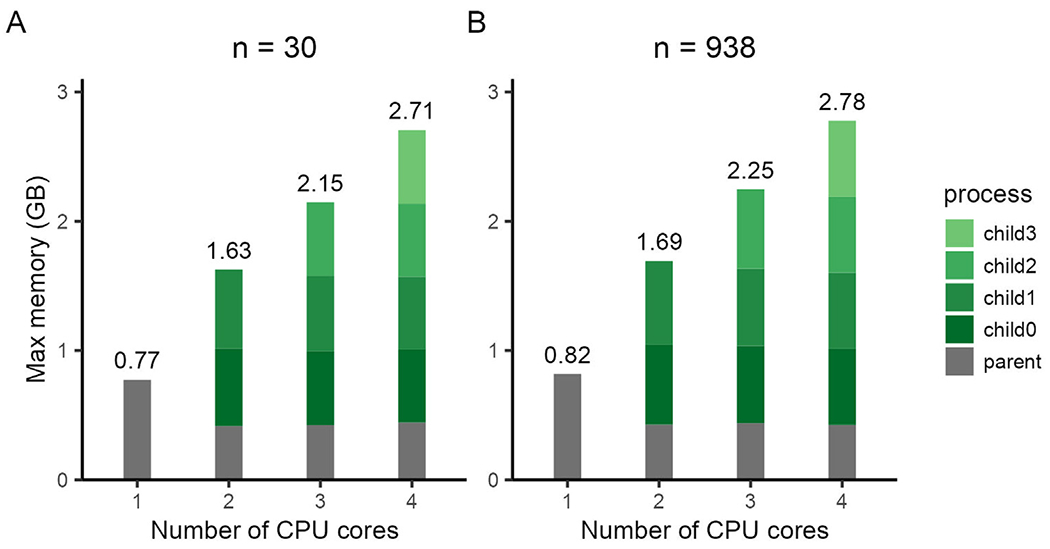
ModelArray is memory-efficient even under different parallelization configurations. Maximal memory usage for a linear model run using ModelArray.lm() was evaluated across a sample of n = 30 (A) and n = 938 (B) with varying numbers of CPU cores requested.
3.3. ModelArray captures nonlinear developmental effects
As a final illustration of ModelArray’s functionality and extensibility to diverse statistical models, we also examined nonlinear developmental effects in the PNC using GAMs. Robust nonlinear age effects can be observed in white matter tracts including the corpus callosum (CC) and tracts in the brainstem even at very high statistical thresholds (p-value of s(age) < 1 ×10−15 , Fig. 5). To visualize the nonlinear age effects, a cluster in CC was defined with above statistical threshold, and a GAM was fit for FDC averaged in an example 2D slice of this cluster (highlighted in Fig. 5A by a white arrow). The averaged FDC of these fixels increased throughout childhood and adolescence but then plateaued in young adulthood (Fig. 5B). The effect size (change in adjusted R2) of age in this fitted GAM was 0.204.
Fig. 5.
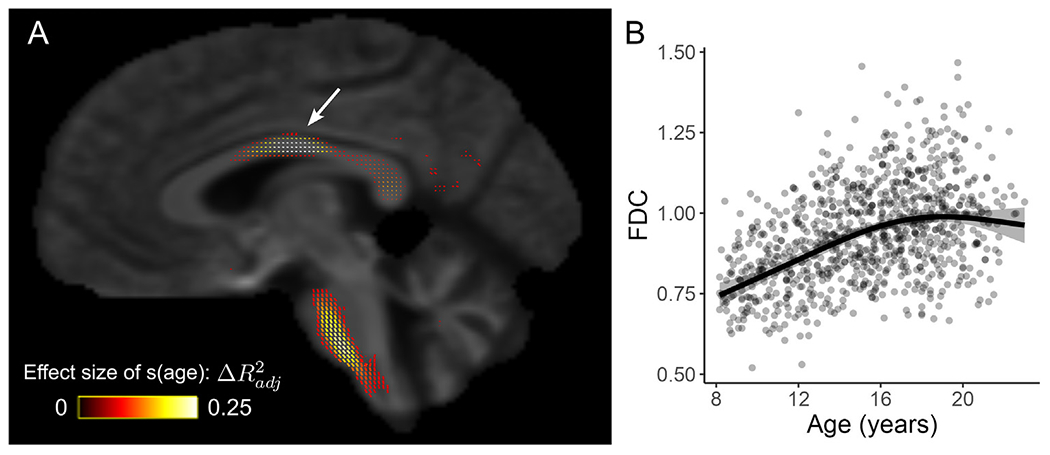
ModelArray allows the estimation of nonlinear effects. Fixel-wise GAM fitted with ModelArray.gam() revealed nonlinear FDC changes with age in childhood and adolescence (n = 938). The GAM also included sex and motion quantification as covariates. (A) Fixels whose FDC was significantly associated with age (p-value of s(age) < 1 × 10−15); fixels are colored by effect size of s(age). Background image is the FOD template. (B) GAM fit for FDC averaged in the 2D slice of the cluster in CC highlighted in panel A by a white arrow.
4. Discussion
Despite the advantages of representing diffusion imaging data as fixels, FBA is a relatively new framework compared to voxel-based analysis, and relatively few analytic tools are currently available for statistical analysis of fixel-wise data. ModelArray is an R package for mass-univariate statistical analysis of fixel-wise data. As discussed below, ModelArray allows for both linear and nonlinear modeling of fixel-wise data in large datasets while only requiring modest amounts of memory.
4.1. Extensibility to diverse statistical models
Brain changes across the lifespan are often nonlinear. One of the most-widely used statistical models to capture both linear and nonlinear effects is the GAM. GAMs use smooth functions to flexibly model linear and nonlinear effects; these smooth functions can be penalized to avoid over-fitting (Wood, 2004, 2011). The incorporation of GAMs in ModelArray and the extensibility to other models available in R represent an advance over existing tools, which at present only support the GLM. It should be noted that GLM in existing tools can also fit nonlinear models using a polynomial function; however, such an approach may not support the penalization of nonlinearity to avoid over-fitting (as in GAMs). As ModelArray is built within R, using GAMs is easy with standard model syntax. More importantly, ModelArray has the potential to leverage the myriad of statistical models that R provides. Indeed, additional statistical models can be added to ModelArray using the same workflow described in the developer documentation (https://pennlinc.github.io/ModelArray/articles/doc_for_developer.html). This extensibility will allow for ongoing enhancements – by both the original developers and the broader community – to extend ModelArray’s functionality to a wide variety of statistical models.
Some previous FBA studies focused on specific macroscopic WM pathways, calculated the average of fixel-wise metrics (e.g., FDC) in specific regions of interest, and built statistical models upon the average values in software such as R to facilitate the use of diverse statistical models (e.g., Singh et al., 2022; Genc et al., 2020; Chahal et al., 2021). ModelArray offers opportunities to directly apply statistical models that R provides, without the need of this data reduction step. ModelArray also allows users retrieve statistical metrics for each fixel without data dimensionality reduction.
4.2. Scalability to large-scale data resources
Large-scale neuroimaging datasets enhance statistical power and the reliability of findings in studies of individual differences (Marek et al., 2022). However, as data size grows, memory requirements often become quite large when performing group-level statistical analysis. To address this challenge, we designed ModelArray to minimize memory requirements by only reading data into memory as needed. Our benchmarking studies illustrated that ModelArray memory requirements were low even when analyzing hundreds of participants, and only had minimal change when the number of participants increased. This scalability facilitates fixel-wise statistical analyses of large-scale data resources even on a laptop or a workstation that only has a limited amount of memory. It also makes the exploration of statistical models easy and cheap. This memory efficiency is limited to the statistical analysis step provided by ModelArray; steps such as preprocessing dMRI data and preparing fixel-wise data may require more memory than personal laptops or workstations can provide. However, these processing steps are out of the scope of ModelArray and can be performed on High Performance Computing (HPC) clusters.
4.3. Limitations and future directions
Several limitations of ModelArray should be noted. First, ModelArray is configured to only analyze fixel-wise data. Moving forward, it may be generalized to allow for analyses of other imaging data types such as voxel (NIfTI) and surface (CIFTI) data, akin to other group-level analysis resources that are compatible with different data types, e.g., Permutation Analysis of Linear Models (PALM; Winkler et al., 2014) from FSL (https://fsl.fmrib.ox.ac.uk/fsl/fslwiki). Such extensions could leverage ModelArray’s modular I/O interface, which would only require additional companion converters (i.e., ConVoxel instead of ConFixel).
Second, it is important to note the differences between ModelArray and existing tools for statistical analysis of fixel-wise data, e.g., fixelcfestats for FBA from MRtrix (Raffelt et al., 2015; Tournier et al., 2019). In contrast to CFE implemented in fixelcfestats from MRtrix, ModelArray does not incorporate information of fixel-fixel connectivity, which limits the ability of ModelArray to conduct statistical inference exploiting connectivity information. However, the control of multiple comparisons using methods such as FDR is commonly used in large-scale studies and is currently implemented in ModelArray. Future releases of ModelArray may incorporate CFE. It should be noted that, users should be careful and aware of the differences when interpreting the statistical results from fixelcfestats from MRtrix and those from ModelArray, as the former one seeks to control family-wise error, whereas ModelArray’s default behavior aims to control the FDR.
5. Conclusion
ModelArray is a memory-efficient R package for fixel-wise statistical analysis. It offers both linear and nonlinear modeling with substantial extensibility. Taken together, ModelArray facilitates the statistical analysis of fixel-wise data in large-scale dMRI datasets.
Acknowledgements
This study was supported by grants from the National Institutes of Health: R01MH112847 (R.T.S., T.D.S.), R01MH120482 (T.D.S.), R37MH125829 (T.D.S.), R01EB022573 (T.D.S.), R01MH113550 (T.D.S.), R01MH123550 (R.T.S.), RF1MH116920 (T.D.S.), 5T32DC000038 (S.L.M.), K99MH127293 (B.L.), R01MH119185 (D.R.R), R01MH120174 (D.R.R.). V.J.S. was supported by a National Science Foundation Graduate Research Fellowship (DGE-1845298). Additional support was provided by the AE Foundation and the Penn/CHOP Lifespan Brain Institute.
Declaration of competing interest
R.T.S. has consulting income from Octave Bioscience. A.M.V. did not receive funding or consulting fees as it pertains to this work but is currently an employee of Genentech, Inc.. The remaining authors declare no competing interests.
Footnotes
Credit authorship contribution statement
Chenying Zhao: Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Writing – original draft, Writing – review & editing, Visualization. Tinashe M. Tapera: Conceptualization, Methodology, Software, Validation, Formal analysis, Writing – review & editing, Visualization. Joëlle Bagautdinova: Validation, Writing – review & editing. Josiane Bourque: Formal analysis, Writing – review & editing. Sydney Covitz: Validation, Writing – review & editing. Raquel E. Gur: Investigation, Writing – review & editing, Funding acquisition. Ruben C. Gur: Investigation, Writing – review & editing, Funding acquisition. Bart Larsen: Methodology, Writing – review & editing, Funding acquisition. Kahini Mehta: Validation, Writing – review & editing. Steven L. Meisler: Software, Validation, Writing – review & editing, Funding acquisition. Kristin Murtha: Validation, Writing – review & editing. John Muschelli: Methodology, Writing – review & editing. David R. Roalf: Formal analysis, Investigation, Data curation, Writing – review & editing, Funding acquisition. Valerie J. Sydnor: Validation, Writing – review & editing, Funding acquisition. Alessandra M. Valcarcel: Methodology, Writing – review & editing. Russell T. Shinohara: Methodology, Writing – review & editing, Funding acquisition. Matthew Cieslak: Conceptualization, Methodology, Software, Formal analysis, Writing – review & editing, Visualization, Supervision. Theodore D. Satterthwaite: Conceptualization, Methodology, Formal analysis, Investigation, Writing – original draft, Writing -review & editing, Visualization, Supervision, Funding acquisition.
Data and code availability statements
ModelArray documentation can be found at https://pennlinc.github.io/ModelArray. All code used to perform memory profiling and application of GAMs is available at https://github.com/PennLINC/ModelArray_paper. The source code for ModelArray is available at https://github.com/PennLINC/ModelArray, and the source code for ConFixel is available at https://github.com/PennLINC/ConFixel. The version of ModelArray used for benchmarking and demonstration was commit SHA-1 0911c4f. We also provide a Docker image of ModelArray and ConFixel (available at https://hub.docker.com/r/pennlinc/modelarray_confixel). The PNC dataset used in this paper is available on dbGAP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000607.v3.p2). As part of the software tutorial, example fixel-wise data from 100 PNC participants is openly shared on OSF (https://doi.org/10.17605/OSF.IO/JVEHY).
References
- Alexander LM, Escalera J, Ai L, Andreotti C, Febre K, Mangone A, Vega-Potler N, Langer N, Alexander A, Kovacs M, Litke S, O’Hagan B, Andersen J, Bronstein B, Bui A, Bushey M, Butler H, Castagna V, Camacho N, …, Milham MP, 2017. An open resource for transdiagnostic research in pediatric mental health and learning disorders. Sci. Data, 4 doi: 10.1038/sdata.2017.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson JLR, Graham MS, Zsoldos E, Sotiropoulos SN, 2016. Incorporating outlier detection and replacement into a non-parametric framework for movement and distortion correction of diffusion MR images. Neuroimage 141, 556–572. doi: 10.1016/j.neuroimage.2016.06.058. [DOI] [PubMed] [Google Scholar]
- Andersson JLR, Sotiropoulos SN, 2016. An integrated approach to correction for off-resonance effects and subject movement in diffusion MR imaging. Neuroimage 125, 1063–1078. doi: 10.1016/j.neuroimage.2015.10.019 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basser PJ, Pierpaoli C, 1996. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J. Magnet. Resonance, Ser. B 111 (3), 209–219. doi: 10.1006/jmrb.1996.0086. [DOI] [PubMed] [Google Scholar]
- Bethlehem RAI, Seidlitz J, White SR, Vogel JW, Anderson KM, Adamson C, Adler S, Alexopoulos GS, Anagnostou E, Areces-Gonzalez A, Astle DE, Auyeung B, Ayub M, Bae J, Ball G, Baron-Cohen S, Beare R, Bedford SA, Benegal V, …, Alexander-Bloch AF, 2022. Brain charts for the human lifespan. Nature 1–11. doi: 10.1038/s41586-022-04554-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahal R, Delevich K, Kirshenbaum JS, Borchers LR, Ho TC, Gotlib IH, 2021. Sex differences in pubertal associations with fronto-accumbal white matter morphometry: implications for understanding sensitivity to reward and punishment. Neuroimage 226, 117598. doi: 10.1016/j.neuroimage.2020.117598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieslak M, Cook PA, He X, Yeh F-C, Dhollander T, Adebimpe A, Aguirre GK, Bassett DS, Betzel RF, Bourque J, Cabral LM, Davatzikos C, Detre JA, Earl E, Elliott MA, Fadnavis S, Fair DA, Foran W, Fotiadis P, …., Satterthwaite TD, 2021. QSIPrep: an integrative platform for preprocessing and reconstructing diffusion MRI data. Nat. Methods 18 (7), 775–778. doi: 10.1038/s41592-021-01185-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhollander T, Clemente A, Singh M, Boonstra F, Civier O, Duque JD, Egorova N, Enticott P, Fuelscher I, Gajamange S, Genc S, Gottlieb E, Hyde C, Imms P, Kelly C, Kirkovski M, Kolbe S, Liang X, Malhotra A, …., Caeyenberghs K, 2021. Fixel-based analysis of diffusion MRI: methods, applications, challenges and opportunities. Neuroimage 241, 118417. doi: 10.1016/j.neuroimage.2021.118417. [DOI] [PubMed] [Google Scholar]
- Dhollander T, & Connelly A (2016). A novel iterative approach to reap the benefits of multi-tissue CSD from just single-shell (+b=0) diffusion MRI data. Proceedings of the 24th annual meeting of the International Society of Magnetic Resonance in Medicine. pp. 3010. [Google Scholar]
- Dhollander T, Mito R, Raffelt D, & Connelly A (2019). Improved white matter response function estimation for 3-tissue constrained spherical deconvolution. Proceedings of the 27th annual meeting of the International Society of Magnetic Resonance in Medicine, pp. 555. [Google Scholar]
- Dhollander T, Raffelt D, Connelly A, 2016. Unsupervised 3-tissue response function estimation from single-shell or multi-shell diffusion MR data without a co-registered T1 image. In: ISMRM Workshop on Breaking the Barriers of Diffusion MRI, p. 5. [Google Scholar]
- Genc S, Malpas CB, Gulenc A, Sciberras E, Efron D, Silk TJ, Seal ML, 2020. Longitudinal patterns of white matter fibre density and morphology in children are associated with age and pubertal stage. Dev. Cogn. Neurosci 45, 100853. doi: 10.1016/j.dcn.2020.100853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeurissen B, Leemans A, Tournier JD, Jones DK, Sijbers J, 2013. Investigating the prevalence of complex fiber configurations in white matter tissue with diffusion magnetic resonance imaging. Hum. Brain Mapp 34 (11), 2747–2766. doi: 10.1002/hbm.22099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner E, Dhital B, Kiselev VG, Reisert M, 2016. Gibbs-ringing artifact removal based on local subvoxel-shifts. Magn. Reson. Med 76 (5), 1574–1581. doi: 10.1002/mrm.26054. [DOI] [PubMed] [Google Scholar]
- Lebel C, Gee M, Camicioli R, Wieler M, Martin W, Beaulieu C, 2012. Diffusion tensor imaging of white matter tract evolution over the lifespan. Neuroimage 60 (1), 340–352. doi: 10.1016/j.neuroimage.2011.11.094. [DOI] [PubMed] [Google Scholar]
- Marek S, Tervo-Clemmens B, Calabro FJ, Montez DF, Kay BP, Hatoum AS, Donohue MR, Foran W, Miller RL, Hendrickson TJ, Malone SM, Kandala S, Feczko E, Miranda-Dominguez O, Graham AM, Earl EA, Perrone AJ, Cordova M, Doyle O, …., Dosenbach NUF, 2022. Reproducible brain-wide association studies require thousands of individuals. Nature 1–7. doi: 10.1038/s41586-022-04492-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès H (2021). HDF5Array: HDF5 backend for DelayedArray objects (R package version 1.20.0) [Computer software]. https://bioconductor.org/packages/HDF5Array.
- Pagès H, Hickey P, & Lun A (2021). DelayedArray: a unified framework for working transparently with on-disk and in-memory array-like datasets. (R package version 0.18.0) [Computer software]. https://bioconductor.org/packages/DelayedArray.
- Pines AR, Larsen B, Cui Z, Sydnor VJ, Bertolero MA, Adebimpe A, Alexander-Bloch AF, Davatzikos C, Fair DA, Gur RC, Gur RE, Li H, Milham MP, Moore TM, Murtha K, Parkes L, Thompson-Schill SL, Shanmugan S, Shinohara RT, …, Satterthwaite TD, 2022. Dissociable multi-scale patterns of development in personalized brain networks. Nat. Commun 13 (1), 2647. doi: 10.1038/s41467-022-30244-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team, 2021. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing (R version 4.1.2) [Computer software]. [Google Scholar]
- Raffelt DA, Smith RE, Ridgway GR, Tournier JD, Vaughan DN, Rose S, Henderson R, Connelly A, 2015. Connectivity-based fixel enhancement: whole-brain statistical analysis of diffusion MRI measures in the presence of crossing fibres. Neuroimage 117, 40–55. doi: 10.1016/j.neuroimage.2015.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffelt DA, Tournier JD, Smith RE, Vaughan DN, Jackson G, Ridgway GR, Connelly A, 2017. Investigating white matter fibre density and morphology using fixel-based analysis. Neuroimage 144 (Pt A), 58–73. doi: 10.1016/j.neuroimage.2016.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roalf DR, Quarmley M, Elliott MA, Satterthwaite TD, Vandekar SN, Ruparel K, Gennatas ED, Calkins ME, Moore TM, Hopson R, Prabhakaran K, Jackson CT, Verma R, Hakonarson H, Gur RC, Gur RE, 2016. The impact of quality assurance assessment on diffusion tensor imaging outcomes in a large-scale population-based cohort. Neuroimage 125, 903–919. doi: 10.1016/j.neuroimage.2015.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterthwaite TD, Elliott MA, Ruparel K, Loughead J, Prabhakaran K, Calkins ME, Hopson R, Jackson C, Keefe J, Riley M, Mentch FD, Sleiman P, Verma R, Davatzikos C, Hakonarson H, Gur RC, Gur RE, 2014. Neuroimaging of the Philadelphia neurodevelopmental cohort. Neuroimage 86, 544–553. doi: 10.1016/j.neuroimage.2013.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling KG, Daducci A, Maier-Hein K, Poupon C, Houde J-C, Nath V, Anderson AW, Landman BA, Descoteaux M, 2018. Challenges in diffusion MRI tractography – Lessons learned from international benchmark competitions. Magn. Reson. Imag 57, 194–209. doi: 10.1016/j.mri.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Skippen P, He J, Thomson P, Fuelscher I, Caeyenberghs K, Anderson V, Nicholson JM, Hyde C, Silk TJ, 2022. Longitudinal developmental trajectories of inhibition and white-matter maturation of the fronto-basal-ganglia circuits. Dev. Cogn. Neurosci 58, 101171. doi: 10.1016/j.dcn.2022.101171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Nichols TE, 2009. Threshold-free cluster enhancement: addressing problems of smoothing, threshold dependence and localisation in cluster inference. Neuroimage 44 (1), 83–98. doi: 10.1016/j.neuroimage.2008.03.061. [DOI] [PubMed] [Google Scholar]
- Tournier J-D, Smith R, Raffelt D, Tabbara R, Dhollander T, Pietsch M, Christiaens D, Jeurissen B, Yeh C-H, Connelly A, 2019. MRtrix3: a fast, flexible and open software framework for medical image processing and visualisation. Neuroimage 202, 116137. doi: 10.1016/j.neuroimage.2019.116137. [DOI] [PubMed] [Google Scholar]
- Tustison NJ, Avants BB, Cook PA, Zheng Y, Egan A, Yushkevich PA, Gee JC, 2010. N4ITK: improved N3 bias correction. IEEE Trans. Med. Imag 29 (6), 1310–1320. doi: 10.1109/tmi.2010.2046908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Essen DC, Smith SM, Barch DM, Behrens TEJ, Yacoub E, Ugurbil K, Consortium, for the W.-M.H, 2013. The WU-Minn human connectome project: an overview. Neuroimage 80, 62–79. doi: 10.1016/j.neuroimage.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veraart J, Novikov DS, Christiaens D, Ades-aron B, Sijbers J, Fieremans E, 2016. Denoising of diffusion MRI using random matrix theory. Neuroimage 142, 394–406. doi: 10.1016/j.neuroimage.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SB, Tax CMW, Luijten PR, Ourselin S, Leemans A, Froeling M, 2017. The importance of correcting for signal drift in diffusion MRI. Magn. Reson. Med 77 (1), 285–299. doi: 10.1002/mrm.26124. [DOI] [PubMed] [Google Scholar]
- Winkler AM, Ridgway GR, Webster MA, Smith SM, Nichols TE, 2014. Permutation inference for the general linear model. Neuroimage 92 (100), 381–397. doi: 10.1016/j.neuroimage.2014.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SN, 2001. In: mgcv: GAMs and Generalized Ridge Regression for R. R News, 1/2, pp. 20–25. [Google Scholar]
- Wood SN, 2004. Stable and efficient multiple smoothing parameter estimation for generalized additive models. J. Am. Stat. Assoc 99 (467), 673–686. doi: 10.1198/016214504000000980. [DOI] [Google Scholar]
- Wood SN, 2011. Fast stable restricted maximum likelihood and marginal likelihood estimation of semiparametric generalized linear models. J. R. Stat. Soc.: Ser. B (Stat. Methodol.) 73 (1), 3–36. doi: 10.1111/j.1467-9868.2010.00749.x. [DOI] [Google Scholar]
- Yeh F-C, Verstynen TD, Wang Y, Fernández-Miranda JC, Tseng W-YI, 2013. Deterministic diffusion fiber tracking improved by quantitative anisotropy. PLoS ONE 8 (11), e80713. doi: 10.1371/journal.pone.0080713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
ModelArray documentation can be found at https://pennlinc.github.io/ModelArray. All code used to perform memory profiling and application of GAMs is available at https://github.com/PennLINC/ModelArray_paper. The source code for ModelArray is available at https://github.com/PennLINC/ModelArray, and the source code for ConFixel is available at https://github.com/PennLINC/ConFixel. The version of ModelArray used for benchmarking and demonstration was commit SHA-1 0911c4f. We also provide a Docker image of ModelArray and ConFixel (available at https://hub.docker.com/r/pennlinc/modelarray_confixel). The PNC dataset used in this paper is available on dbGAP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000607.v3.p2). As part of the software tutorial, example fixel-wise data from 100 PNC participants is openly shared on OSF (https://doi.org/10.17605/OSF.IO/JVEHY).
ModelArray documentation can be found at https://pennlinc.github.io/ModelArray. All code used to perform memory profiling and application of GAMs is available at https://github.com/PennLINC/ModelArray_paper. The source code for ModelArray is available at https://github.com/PennLINC/ModelArray, and the source code for ConFixel is available at https://github.com/PennLINC/ConFixel. The version of ModelArray used for benchmarking and demonstration was commit SHA-1 0911c4f. We also provide a Docker image of ModelArray and ConFixel (available at https://hub.docker.com/r/pennlinc/modelarray_confixel). The PNC dataset used in this paper is available on dbGAP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000607.v3.p2). As part of the software tutorial, example fixel-wise data from 100 PNC participants is openly shared on OSF (https://doi.org/10.17605/OSF.IO/JVEHY).