

Abstract
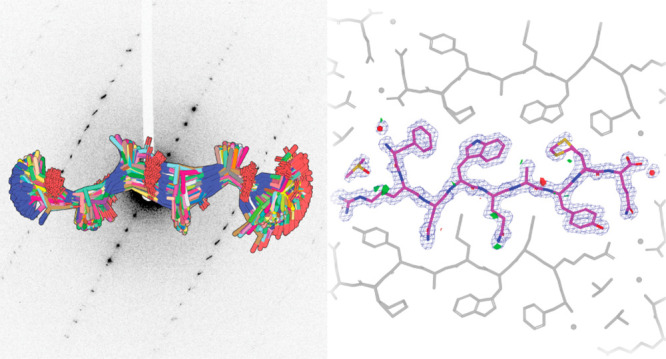
Electron diffraction (MicroED/3DED) can render the three-dimensional atomic structures of molecules from previously unamenable samples. The approach has been particularly transformative for peptidic structures, where MicroED has revealed novel structures of naturally occurring peptides, synthetic protein fragments, and peptide-based natural products. Despite its transformative potential, MicroED is beholden to the crystallographic phase problem, which challenges its de novo determination of structures. ARCIMBOLDO, an automated, fragment-based approach to structure determination, eliminates the need for atomic resolution, instead enforcing stereochemical constraints through libraries of small model fragments, and discerning congruent motifs in solution space to ensure validation. This approach expands the reach of MicroED to presently inaccessible peptide structures including fragments of human amyloids, and yeast and mammalian prions. For electron diffraction, fragment-based phasing portends a more general phasing solution with limited model bias for a wider set of chemical structures.
Keywords: fragment-based phasing, ab initio, MicroED, nanocrystal, peptide, cryo-EM, ARCIMBOLDO
Introduction
Crystallography has played a momentous role in our understanding of peptidic structures.1 Microcrystal electron diffraction (MicroED) is expanding its scope by delivering atomic structures from peptide crystals less than a micrometer in thickness.2−4 Electron diffraction leverages the strong interaction of electrons with matter, capturing diffraction signal that would be missed by conventional X-ray crystallography.5 Some molecules of high biological or chemical importance are only known to grow nanocrystals, demanding structural methods of extreme sensitivity, as seen in the amyloid peptide structures of the toxic core of the Parkinson’s-associated protein α-synuclein3 or the ultrahigh-resolution structure of a prion protofibril.4 Likewise, the technique has determined structures of complex bioderived or post-translationally modified peptides such as an amyloid-β core with a racemized residue,6 the cyclic peptide antibiotic thiostrepton7 and a synthetic tetrapeptide natural product analogue.8
Determination of MicroED structures presently follows one of two routes: ab initio phasing through direct methods9 if data resolution is atomic, or molecular replacement (MR)10 when a highly similar structure is known.3,11−13 MR is challenged by unknown peptide structures that contain uncharacterized backbone geometries or a substantial fraction of unnatural amino acids. Without atomic resolution data, novel phasing solutions are still needed for MicroED targets of uncertain geometry, identity, or chemical connectivity.
Fragment-based phasing (FBP)14 yields accurate solutions relying on the computational search for defined subsets of a target structure to obviate the need for atomic resolution data. Fragments are located by likelihood-based molecular replacement15 and expanded through density modification and map interpretation.16 The ARCIMBOLDO programs substitute the atomicity constraint underlying direct methods with stereochemical constraints.17 For a structure containing defined fragments of constant geometry a single model fragment is appropriate, and model alpha-helices have been particularly successful.18 General cases require joint evaluation of libraries of fragments, representing variations of a structural hypothesis. Relying on secondary and tertiary structure fragments extracted from the Protein Data Bank (PDB)19 or from distant homologues,20 ARCIMBOLDO_BORGES has been broadly used in phasing protein structures determined by X-ray crystallography. ARCIMBOLDO has also been used on MicroED data, to phase a 1.6 Å structure of Proteinase K from distant homologues.21 Fragments placed accurately contribute to solutions despite accounting for a few percent of the scattering atoms in a structure.22 Since any experimental or calculated fragment may be used as input, fragment-based phasing could prove powerful for the general determination of peptidic or other chemical structures by electron diffraction.
Here, we demonstrate fragment-based phasing for ab initio structure determination of novel peptide structures from MicroED data in the absence of atomic resolution. Our approach is based on the development of new fragment library methods tailored to sample structural variability, while profiting from the reduced size of active peptide structures to preclude model bias. We validate its success on known and novel structures obtained from nanocrystallites formed by diverse amyloid peptides.
Results
High-Resolution MicroED Data from Peptide Nanocrystals
The limited crystal size and directional growth exhibited by some peptides of high biological or chemical interest renders electron diffraction a necessary choice for structure determination. However, faced with a growing number of MicroED data sets from peptide crystals, for which direct methods and molecular replacement solutions were unavailable, we set out to develop dedicated fragment phasing approaches for this set of substrates.
Nanocrystals from each of five peptide segments summarized in Table 1 were preserved on grids in a frozen-hydrated state; crystals of each were visually identified and diffracted as previously described.3 Ideal candidates for MicroED yielded better than 2 Å diffraction. Diffraction data from several crystals were merged to improve completeness. Structural determination via direct methods with SHELXD9 succeeded for peptides whose crystals diffracted to atomic resolution: a synthetic mammalian prion segment (QYNNENNFV) [1] and a sequence variant of a repeat segment of the yeast prion New1p (NYNNYQ) [2] as well as a plate polymorph of the functional mammalian prion, CPEB3 (QIGLAQTQ) [3]. Direct methods solutions were unattainable for the needle polymorph of [3], a segment of the human amyloid protein LECT2 (GSTVYAPFT) [4], and a segment from the human zinc finger protein (ZFP) 292 (FRNWQAYMQ) [5].
Table 1.
| GSTVYAPFT (7N2I) | QIGLAQTQ plate polymorph (7N2F) | QIGLAQTQ needle polymorph (7N2G) | NYNNYQ (7N2K) | QYNNENNFV (7N2J) | FRNWQAYMQ (7N2D) | |
|---|---|---|---|---|---|---|
| Data Collection and Processing | ||||||
| resolution (A) | 7.67–1.40(1.40) | 7.09–1.2(1.2) | 7.61–1.20(1.20) | 8.91–1.30(1.30) | 7.42–1.5(1.5) | 19.39–1.50(1.50) |
| no. crystals | 3 | 3 | 1 | 4 | 6 | 4 |
| electron dose (e–/Å2) | <5 | <5 | <5 | <5 | <5 | <5 |
| space group | C2 | P21 | P212121 | P31 | P1 | C2 |
| a, b, c (Å) | 58.4, 4.73, 19.63 | 4.83, 16.29, 29.02 | 4.82, 20.48, 45.61 | 27.2, 27.2, 4.83 | 4.87, 10.06, 30.66 | 43.12, 4.84, 34.9 |
| α, β, γ (degrees) | 90.00, 105.01, 90.00 | 90.00, 94.61, 90.00 | 90.00, 90.00, 90.00 | 90.00, 90.00, 120.00 | 94.85, 90.26, 99.98 | 90, 115.934, 90 |
| no. total reflections | 4159 | 6437 | 3963 | 6870 | 4236 | 4017 |
| no. unique reflections | 985(106) | 1129 (105) | 1357 (123) | 969(84) | 730(74) | 961(70) |
| Rmerge | 15.9 | 20.0 | 12.8 | 21.2 | 18.5 | 20.2 |
| CC1/2 | 98.7 (89.5) | 98.1 (97.6) | 98.0 (53.3) | 98.5 (60.4) | 98.8 (81.3) | 97.7 (78.0) |
| ⟨I/σI⟩ | 5.16 | 8.31 | 4.79 | 6.16 | 7.01 | 4.32 |
| completeness | 80.3 (85.5) | 78.2 (78.52) | 81.6 (83.1) | 97.68 (87.9) | 97.8 (82.7) | 75.8 (51.2) |
| multiplicity | 4.22 | 5.70 | 2.92 | 7.09 | 5.71 | 4.18 |
| phasing success using alternative librariesa | RL: 45% correct solutions | CL: 16% correct solutions | CL: 10% correct solutions | CL:0.4% correct solutions | CL: 3% correct solutions | RL: 1% correct solutions |
| GL: 2% correct solutions | GL: 2% correct solutions | GL: 5% correct solutions | GL: 2% correct solutions | GL: 4% correct solutions | GL: 3% correct solutions | |
| residues placed | 6 | 7 | 6 | 6 | 7 | 8 |
| fragments placed | 1 | 1 | 1 | 1 | 1 | 1 |
| LLG | 47.10 | 175.90 | 62.80 | 27.43 | 60.30 | 46.9 |
| TFZ | 4.60 | 4.80 | 6.90 | 5.30 | 7.90 | 5.3 |
| final CC (%) | 21.85 | 57.14 | 30.54 | 20.21 | 34.12 | 30.29 |
| Refinement | ||||||
| Rwork (%) | 19.36 (26.52) | 19.8 (21.2) | 19.24 (32.32) | 16.14 (22.74) | 17.44 (22.90) | 21.20(38.46) |
| Rfree (%) | 19.23 (45.49) | 22.9 (18.1) | 23.65 (31.41) | 18.54 (30.74) | 22.65 (18.79) | 23.87(25.69 |
| RSCC | ||||||
| no. waters ligand atoms | 1 | 0 | 1 | 3 | 1 | 2 |
| average B-factor: | ||||||
| protein | 8.78 | 3.22 | 3.85 | 7.05 | 4.68 | 8.07 |
| water | 13.05 | 10.66 | 20.01 | 6.15 | 12.7 | |
| ligand | 18.35 | |||||
| r.m.s.d. bonds (A) | 0.033 | 0.027 | 0.008 | 0.099 | 0.012 | 0.015 |
| r.m.s.d. angles (degrees) | 2.13 | 2.69 | 0.91 | 0.55 | 1.18 | 1.41 |
| Ramachandran (ouliers,favored) (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Clashscore | 0.00 | 0.00 | 0.00 | 0.00 | 6.76 | 0 |
GL: general library CL: custom library RL: modeled library. Correct solutions are defined as those with an initial wMPE below 60°.
Phasing with ARCIMBOLDO Using General Local Fold Libraries
For ab initio macromolecular phasing, ARCIMBOLDO_BORGES exploits fragment libraries representing a common local fold as found in a vast number of PDB structures. Such libraries can be derived from millions of fragments clustered to describe the geometrical variation within the radius of convergence of the method. Fragment superposition allows joint statistical analysis of all phasing attempts as a single experiment. Since typical fragments in macromolecular libraries contain more residues than our peptide structures,23 we devised dedicated libraries that handled the high abundance of motifs exhibited by short peptides; for example, two short antiparallel beta strands. Weighing overall and local properties when superposing such small models also presented a challenge that was solved experimentally, simulating data from a template and refining the location of all library models against the calculated data.
Single-strand libraries yielded partial solutions for all peptides in Table 1. However, the best solutions identified by this approach did not benefit from the statistical or phase combination algorithms enabled by prior superposition.24 Instead, optimal solutions were obtained by correct placement of the single most accurate fragments. As peptidic MicroED data sets tend to be small and thus amenable to a large number of calculations from different starting fragments, we decided to exploit competing hypotheses to address the more pressing concern of model bias. With that in mind, we devised libraries encompassing dissimilar, nonsuperimposable models.
Broad, Knowledge-Based Libraries to Phase Nanocrystals
To develop diverse, knowledge-based libraries, we began with ideal cases: atomic resolution data from crystals of two peptide segments that yielded structures by direct methods. The first data set represented five crystals of mammalian prion segment [1]. The full 0.9 Å data were included in our set as a gold standard (Figure 1A). When intentionally truncated to a resolution of 1.5 Å, both direct methods and molecular replacement using models of closely related prion sequences4 were unsuccessful. A library of 249 polyglycine hexapeptides derived from previously determined amyloid peptide structures were used as inputs for ARCIMBOLDO_BORGES (Figure 1B). This library contained models that could be clearly discriminated as potential solutions, scoring above 25% in their initial correlation coefficient22 (Figure 1C). These same solutions would later be found to exhibit low errors relative to the phases calculated from a final structure (Table S2). The best solution placed six residues and was sufficient to build the remainder of the peptide based on difference density (Table 1, Figure 1D,E).
Figure 1.

Workflow for using peptide fragments for phasing MicroED data. A) Electron diffraction pattern reaching atomic resolution for QYNNENNFV. Rings designate resolution ranges while arrowhead designates highest resolution spot. B) All fragments comprising the polyglycine hexapeptide library aligned in pymol. C) LLG vs InitCC plot for the fragments screened in ARCIMBOLDO-BORGES. Color indicates the wMPE of the fragment relative to the phases calculated from the final structure. Inset shows the fragment chosen for SHELXE expansion overlaid on the final structure. D) Output solution from ARCIMBOLDO-BORGES following SHELXE expansion is shown overlaid on the final structure. Maps are shown after one round of refinement in Phenix. E) Final structure and potential map for the QYNNENNFV peptide with symmetry related chains shown in gray.
Data collected from crystals of the New1p segment [2] presented an increased challenge. Although microcrystals of [2] yielded an X-ray crystallographic structure (Figures S2 and S3B), the same condition also produced nanocrystals requiring MicroED. The 1.1 Å data set obtained combining four crystals of the latter polymorph rendered a direct methods structure different from that originally determined by X-ray diffraction (Figure S3A,B). The data set truncated to 1.3 Å served as a second test for ARCIMBOLDO_BORGES. A library holding polyglycine pentapeptides yielded a single promising solution that could be fully extended and matched the direct methods solution (Table 1, Figure S4).
Structure from a Segment of the Prion Domain of CPEB3 [3]
We next sought to determine novel peptide structures from data that were not suitable for direct methods. Crystallization of a segment from the prion domain of CPEB3 [3] produced crystal slurries ideal for MicroED (Figure 2). Screening them in overfocused diffraction mode revealed two distinct morphologies, suggesting the presence of multiple structures (Figure 2A,E). Crystals of plate morphology belonged to P21, while those of the relatively rare needle morphology presented space group P212121 (Table 1, Figure 2E).
Figure 2.
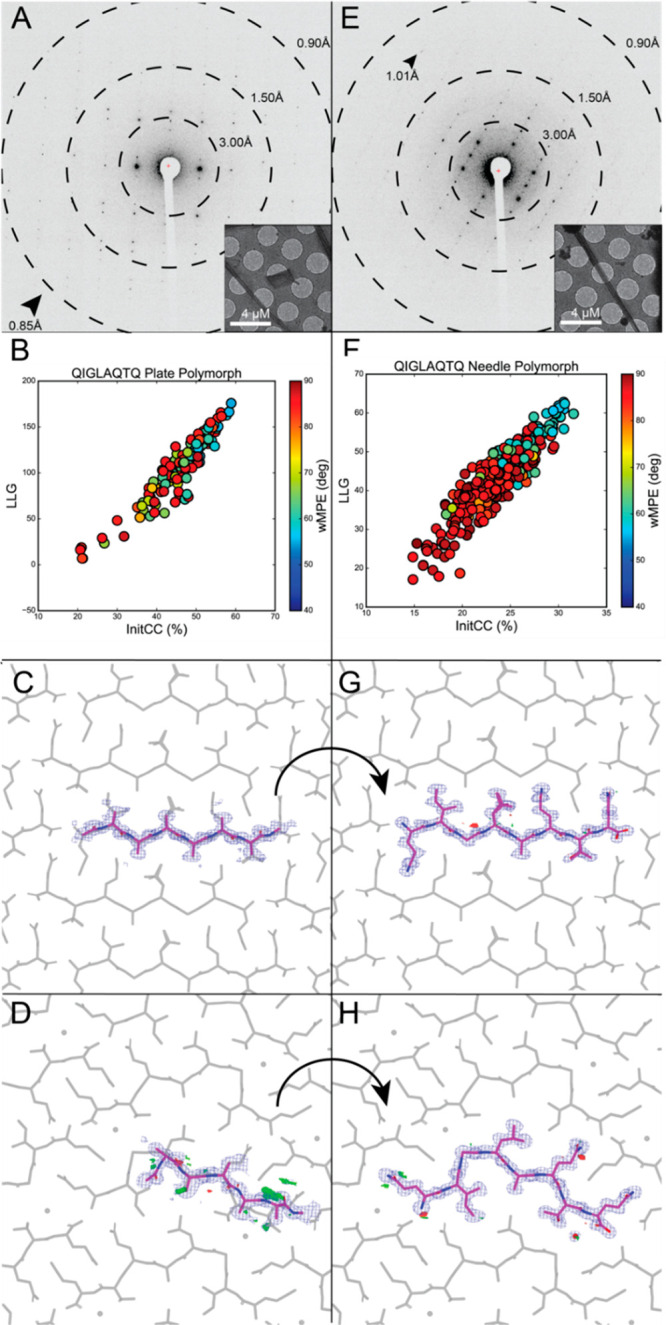
FBP of MicroED data from CPEB3 peptide QIGLAQTQ reveals two polymorphs. A) Electron diffraction pattern reaching atomic resolution for QIGLAQTQ plate morphology. Rings designate resolution ranges while arrowheads designate the highest resolution spot. Overfocused diffraction image of plate morphology crystal. B) Postmortem analysis from ARCIMBOLDO-BORGES plotting LLG vs InitCC (P21). C) Initial output potential maps following SHELXE expansion by ARCIMBOLDO-BORGES for plate polymorph overlaid on final solution (gray). Buildable density visible on several residues. D) Final density maps of QIGLAQTQ plate polymorph asymmetric unit with symmetry mates shown in gray. E) Diffraction pattern for QIGLAQTQ needle morphology. Rings designate resolution ranges while arrowhead designates highest resolution spot. F) Postmortem analysis from ARCIMBOLDO-BORGES plotting LLG vs IniCC (P212121). G) Initial output potential maps following SHELXE expansion by ARCIMBOLDO-BORGES for needle polymorph overlaid on final solution (gray). H) Final density maps of QIGLAQTQ needle polymorph asymmetric unit with symmetry mates shown in gray and water molecule displayed as red sphere.
Merged data from 3 crystals of the plate polymorph were phased by direct methods. Its structure, an unkinked beta strand, was refined at 1.0 Å (Figure S3C). In contrast, a single crystal of the rare needle polymorph diffracted to ∼1.1 Å resolution, generated a data set that was 81.5% complete at 1.2 Å (Figure 2). Despite its high resolution, neither SHELXD nor molecular replacement with the structure derived from the plate polymorph truncated to poly alanine yielded a solution. Instead, the needle polymorph data set was successfully phased using a 270-fragment polyglycine library of tetrapeptides in ARCIMBOLDO-BORGES (Table 1, Figure S1A,B). An initial solution containing four alanine residues led to a fully refined model (Figure 2G,H). Alternatively, applying the same procedure with an 89-fragment polyglycine library of pentapeptides (Figure S1C,D) to data from the plate polymorph also resulted in a number of possible solutions. In both cases, nonrandom solutions with the highest LLG and Initial CC scores were identified by ARCIMBOLDO-BORGES (Table S2, Figure 2B,F).
Structures Determined by Modeled Fragment Libraries
To overcome the limitation of requiring prior structural knowledge for fragment libraries, we generated atomic models computationally. Such libraries would be ideally suited for determining structures with unanticipated local geometries and could be broadly applicable to a variety of small molecules. We computed fragments using PyRosetta25,26 starting from a known peptide backbone as a template onto which sequences of interest were threaded and modeled27 (Figure 3). This scheme was parallelized to generate libraries containing hundreds of fragments, which successfully facilitated FBP of several unknown structures.
Figure 3.

Rosetta library generation and wMPE analysis for GSTVYAPFT and FRNWQAYMQ peptide structures. 1) NNQQNY peptide structure used as a template steric zipper for Rosetta modeling. 2) NNQQNY peptide stripped to glycine residues in preparation for threading. 3) One example of a Rosetta-generated steric zipper structure after threading, repacking, and relaxing in iterative cycles to reach a calculated energy minimum. 4) Rosetta-generated structures are stripped to alanine residues and have hydrogen atoms removed. 5) Individual chains are isolated and are used as the fragment library for phasing with ARCIMBOLDO-BORGES. The LLG vs InitCC plots for A) GSTVYAPFT and B) FRNWQAYMQ are shown below. Inset shows the fragment that was chosen for SHELXE expansion, leading to the correct solution, overlaid with the final structures. C) Final potential maps of GSTVYAPFT asymmetric unit with symmetry mates shown in gray. D) Final potential maps of FRNWQAYMQ asymmetric unit with symmetry mates shown in gray.
Crystals grown from a segment of the LECT2 protein [4] diffracted to only 1.4 Å by MicroED, and the data set combined from three such crystals failed to yield solutions from direct methods or molecular replacement with prior fragment libraries or a closely related peptide structure (Table 1). We generated a new hexapeptide poly alanine library using Rosetta in an attempt to approximate the structure of [4] in close packing while including the internal proline residue, an uncommon feature in amyloid peptides (Figure S1G,H). The full sequence of the LECT2 protein was used to generate hexapeptide models, that were subsequently threaded pairwise onto the two backbones of the known structure of the peptide NNQQNY.27 Models were allowed to repack and relax to energy minima in Rosetta. Then, 111 models containing the GSTVYAPFT sequence were truncated to alanine and used in ARCIMBOLDO_BORGES (Figure 3), yielding a discriminated solution (Table S2).
A segment from the human protein ZFP-292 [5] presented the most severe challenge to conventional phasing. Due to a high degree of orientation bias of crystals on EM grids, merging data from four crystals achieved an overall completeness of 75.8% at 1.5 Å resolution (Table 1). As in the case of [4], this segment could not be phased by direct methods or standard molecular replacement, and no solutions were found when attempting FBP using our fragment libraries from known structures. We again turned to Rosetta in this case to populate a library of fragments that approximated the structure of [5], relying only on the 9-residue sequence of the peptide to generate paired hexapeptides for threading. These threaded segments were then evaluated in Rosetta to generate 20 models per pair (Figure 3). This library of 640 models produced in ARCIMBOLDO_BORGES (Figure S1) yielded low wMPE solutions, one of which facilitated a refined solution (Figure 3B,D, Table S2).
Outlook and Conclusions
To satisfy the need for new ab initio phasing solutions for MicroED, we have developed and deployed new fragment-based phasing strategies using ARCIMBOLDO_BORGES and determined five novel structures. The variation in diffraction quality we observed is representative of the spectrum typically encountered in chemical structure analysis, including examples of relatively low completeness, crystals with low solvent content, and lack of atomic resolution. While the six structures determined here represent a small sampling of the greater universe of peptidic molecules, each of these structures revealed challenges that could be generalized. All analyzed peptides had a high aggregation propensity, contained little to no disordered solvent, and naturally produced nanocrystallites instead of larger crystals. Polymorphism was encountered and in one case revealed differences in atomic structure.
In these cases, and particularly where atomic resolution is not available, information from pre-existing solutions is a lifeline for the phasing process. However, preventing the propagation of errors derived from model bias becomes even more pressing in such cases. Hence, ARCIMBOLDO jointly evaluates large libraries, where competing hypotheses are compared to provide a safeguard against erroneous solutions. The fragments used in our approach were successfully selected by Phaser, based on their LLG, identified with SHELXE CC scores and subsequently expanded by SHELXE into accurate initial solutions. While we observe examples of high phase error models scoring well in preliminary steps, the discrimination of competing potential solutions revealed an unambiguous solution in all cases.
Verification through competition28 is particularly promising in chemical crystallography when exhaustive searches of solution space are manageable. The libraries we exploited for macromolecular phasing hold variations around a common local fold and yielded correct solutions in all instances. In all cases, single fragment solutions were found to outperform combined solutions. Broadening the base of hypotheses through the use of heterogeneous libraries helped address model bias. All solutions were verified through clear discrimination between conflicting models and similarly high scores for structurally compatible hypotheses. Knowledge-based and modeled libraries rendered higher Z-scores than general libraries, discriminating as illustrated in Figure 2B,F. This makes the present strategy amenable to exploring distinct secondary structure motifs, including primarily α-helical peptides or structures with more than one type of secondary structure.29,30
Our trials demonstrate that computed libraries were beneficial when applied to our most challenging cases [4–5] and could further benefit from new advancements in machine learning. Recently, AlphaFold harnessed the vast structural diversity available in the PDB using deep neural networks to achieve correct prediction of protein folds with unexpectedly high accuracy.31 However, small, chemically and geometrically diverse structures still require dedicated development. Exploration of the rich structural expanse of chemical space will require methods that accurately select structural fragments while excluding bias artifacts to achieve structural solutions.
Summary
We expand the ab initio phasing toolkit for electron diffraction, overcoming the need of atomic resolution diffraction to produce de novo solutions. Using ARCIMBOLDO-BORGES and libraries of both known and computed structures, we determine six novel atomic structures of peptide segments. The structures determined using this method are accurate and represent varied geometries and sequences. Model bias is precluded by parallel assessment of a large collection of structural hypotheses providing a baseline. These methods successfully establish a three-dimensional structure from samples that were previously intractable and open a road to structural solutions for small molecules from near atomic-resolution MicroED/3DED data.
Experimental Considerations
Collection of Microfocus X-ray Data and Structure Determination
Crystal clusters of NYNNYQ were grown at room temperature in a 96-well Wizard screen, using a nominally 24.5 mM aqueous solution of the peptide. The crystallization condition chosen for further optimization in 24-well, hanging drop trays consists of 20% 2-methyl-2,4-pentanediol (MPD), buffered by 0.1 M sodium acetate pH 4.5). The peptide crystals were harvested from hanging drops using CryoLoops from Hampton Research with no additional cryoprotectant other than the MPD already present and flash-frozen in liquid nitrogen. Diffraction data sets were collected under cryogenic conditions (100 K) on beamline 24-ID-E at the Advanced Photon Source (APS) equipped with an ADSC Q315 CCD detector, using a 5 μm beam with a wavelength of 0.979 Å. The data were collected via manual vector scanning. 56 diffraction images were collected over three scans from one crystal and one scan from a different crystal. All images have an oscillation range of 5° and were indexed and integrated by XDS.32 The reflection list outputted by XDS was sorted and merged in XSCALE. SHELXD33 was able to reach an ab initio solution. The atomic coordinates from SHELXD were used to generate a Fcalc map with SHELXL.34 An atomic model commensurate with the generated electron density was built in Coot and refined in PHENIX against measured data. The refinement statistics of the final structure are listed in Table S1.
Preparation of Peptide Nanocrystals
Lyophilized, synthetic peptides were purchased from Genscript. Crystals of each peptide were grown as follows: the QYNNENNFV peptide was dissolved in water at 0.88 mM. Crystals were grown using the hanging drop method where 1.5 μL of peptide was added to 1.5 μL of well solution (0.1 M Li2SO4, 2.5 M NaCl, 0.1 M NaOAc adjusted to pH 4.5 with acetic acid) in 24-well trays over 500 μL of well solution. QIGLAQTQ was prepared at 64.5 mM in water. Crystals were grown via 24-well hanging-drop vapor diffusion at 27 mM in 14% polyethylene glycol (PEG) 20,000, 100 mM MES, and 3% DMSO. The crystal slurry was briefly sonicated and 0.5 μL was used to seed a new 3 μL drop and repeated three times. FRNWQAYMQ peptide from ZFP-292 was prepared by dissolving at 1.61 mM in water and 3% DMSO. Crystals were grown in batch containing 35% MPD, 100 mM MES, and 200 mM Li2SO4, in 1:1 ratio of peptide to buffer. GSTVYAPFT peptide was prepared at 21.3 mM concentration dissolved in water. Crystals were grown in a 10 μL batch in 0.7 M sodium formate with 100 mM sodium acetate pH 4.6. All crystals appeared within 24 h and were identified by light microscopy, and subsequently, EM. Batch crystals of NYNNYQ were prepared by dissolving the peptide in water, resulting in a 24.5 mM solution. An equal volume of the crystallization reagent (20% MPD, buffered by 0.1 M of sodium acetate to pH 4.5) was added to the peptide solution. The solution was then seeded with crushed crystals grown in hanging drop experiments described in the microfocus X-ray data collection section above.
Collection of MicroED Data
For GSTVYAPFT and QYNNENNFV, 2 μL of crystal slurry was applied to each side of a glow-discharged holey carbon grid (Quantifoil, R 1/4 300 mesh Cu, Electron Microscopy Sciences) followed by plunging into liquid ethane using an FEI Vitrobot Mark IV set to a blot time of 22 and a blot force of 22 for QYNNENNFV and 24 for GSTVYAPFT. For QIGLAQTQ, 1.8 μL of crystal slurry was applied to each side of a glow-discharged holey carbon grid (Quantifoil, R 2/1 200 mesh Cu, Electron Microscopy Sciences) and plunge frozen into liquid ethane using a FEI Vitrobot Mark IV using a blot time of 25–30 s and a blot force of 22. For NYNNYQ, 2 μL of crystal slurry was applied to each side of a glow-discharged holey carbon grid (Quantifoil, R 2/1 200 mesh Cu, Electron Microscopy Sciences) and plunge frozen into liquid ethane using a FEI Vitrobot Mark IV, blotting with the force set at 22 for 20–30 s.
For GSTVYAPFT, QYNNENNFV, and FRNWQAYMQ, diffraction patterns and crystal images were collected under cryogenic conditions using a FEI Tecnai F20 operated at 200 keV in diffraction mode. Diffraction patterns were recorded while continuously rotating at 0.3 deg/s (GSTVYAPFT) or 0.25 deg/s (QYNNENNFV, FRNWQAYMQ) using a bottom mount TemCam-F416 CMOS camera (TVIPS). Individual image frames were acquired with 2 s exposures per image for all peptides and 5 s exposures for some FRNWQAYMQ data sets to increase signal. A selected area aperture corresponding to approximately 4 or 6 μm at the sample plane was selected depending on the crystal. For QIGLAQTQ, diffraction patterns were collected under cryogenic conditions using a Thermo-Fisher Talos Arctica electron microscope operating at 200 keV and a Thermo-Fisher CetaD CMOS detector in rolling shutter mode. Individual frames were acquired with 3 s exposures rotating at 0.3 deg/s using selected area apertures of 100, 150, or 200 μm, as needed to match the size of the crystal. A total of 28 movies were collected from QIGLAQTQ crystals of two distinct morphologies, 15 from crystals of needle morphology and 13 from crystals of plate morphology.
MicroED Data Processing
The collected TVIPS movies were converted to individual images in Super Marty View (SMV) format, which are compatible with X-ray data processing software. The diffraction images were indexed and integrated with XDS. The indexing raster size and scan pattern as well as integration in XDS were optimized to minimize contributions by background and intensities from secondary crystal lattices. The reflection outputs from XDS were sorted and merged in XSCALE. For the linear P21 QIGLAQTQ structure, three partial data sets, containing 252 diffraction images, were merged to produce a final data set with acceptable completeness (∼80%) up to 1.00 Å, which was truncated to 1.2 Å for phasing and refinement with ARCIMBOLDO. For the kinked P212121 QIGLAQTQ structure, one data set, consisting of 82 diffraction images, was sufficient to produce a final merged data set with high completeness up to 1.2 Å for phasing and refinement by ARCIMBOLDO. For the NYNNYQ structure, four partial data sets, composed of 297 diffraction images, were merged to produce a final data set with high completeness up to 1.10 Å, which was truncated to 1.3 Å for phasing and refinement with ARCIMBOLDO. For the GSTVYAPFT structure, three partial data sets, comprised of 327 diffraction images, were merged to produce a final data set with high completeness up to 1.3 Å. For the QYNNENNFV structure, six partial data sets, containing 931 diffraction images, were merged to produce a final data set with high completeness up to 0.9 Å, which was truncated to 1.5 Å for phasing and refinement with ARCIMBOLDO. For FRNWQAYMQ structure, four partial data sets composed of 224 diffraction images were merged to produce a final data set with acceptable completeness out to 1.5 Å. The statistics for each merge are presented in Table 1.
Structure Determination by Direct Methods and Refinement
Electron diffraction data for NYNNYQ, QIGLAQTQ (plate), and QYNNENNFV were of high enough resolution to yield direct methods solutions. SHELXD was able to reach ab initio solutions with all three data sets. The atomic coordinates from SHELXD and corresponding reflection files were used as inputs for SHELXL34 to generate calculated density maps for each solution. Atomic models consistent with the generated density maps were built in Coot and refined in PHENIX against measured data, using electron scattering form factors. The refinement statistics of the final structures are listed in Table S1.
Generation and Use of Peptide Fragment Libraries for Phasing MicroED Peptide Structures Using ARCIMBOLDO-BORGES
In ARCIMBOLDO, fragments are identified by likelihood-based molecular replacement15 and expanded through density modification and map interpretation.16 A library of amyloid peptide structures determined by both X-ray and electron diffraction was assembled to provide a diverse collection of backbone conformations with potential in phasing novel structures. To take advantage of these probes, a high-throughput, fragment-based phasing methodology in the form of the ARCIMBOLDO software was used. The ARCIMBOLDO suite of programs uses secondary structure fragments as initial probes for molecular replacement carried out by Phaser.35 These fragments undergo rotation and translation analysis and are scored based on log-likelihood gain (LLG) and an initial correlation coefficient (InitCC) to identify potentially accurate starting models (Figure 1C). Rather than making an arbitrary choice on how to direct the superposition, determining the best average for the whole model or of a core, or tolerating outlier atoms to be excluded, an empirical answer was drawn simulating data from a template and refining the location of all other models against the calculated data.
Following this, initial maps are calculated and improved by density modification using the sphere-of-influence algorithm in SHELXE.16 Finally, main chain autotracing is performed and solutions are scored by correlation coefficient.36 Generally, a final CC greater than 25% is indicative of a correct solution. The ability to expand partial solutions in SHELXE permits the use of smaller, potentially more accurate, molecular replacement probes that are identified by Phaser. All runs were carried out using ARCIMBOLDO_BORGES version 2020 Phaser version 2.8.3, and the distributed SHELXE version 2019. Electron scattering factors were used for Phaser analysis, and information including molecular weight and predicted solvent content are available upon request to the authors.
Generation of Custom and Rosetta Fragment Libraries
The fragment libraries used as probes were generated in multiple ways. Polyglycine libraries of varying sizes, four to six residues in length, were generated by extracting fragments from a collection of previously solved amyloid peptide structures (supplementary citations). These fragments were separated into custom libraries only containing fragments of the same length, aligned in PyMOL, and used as probes in ARCIMBOLDO_BORGES. The peptides QIGLAQTQ (plate) and NYNNYQ were phased using a polyglycine library of amino acid pentapeptides containing 89 fragments. The peptide QIGLAQTQ (needle) was phased using a polyglycine library of amino acid tetrapeptides containing 270 fragments. The peptide QYNNENNFV was phased using a polyglycine library of amino acid hexapeptides containing 249 fragments.
Additionally, two libraries of poly alanine fragments were generated using the program Rosetta37 by modeling the packing of the peptide sequences threaded over the steric zipper structure of a previously determined amyloid peptide, NNQQNY.27 After simple threading over the backbone, the side chains were packed, and the chains were allowed to relax to a calculated energy minimum over iterative cycles. These models were isolated as individual fragments six amino acids long, stripped to polyalanine side chains, and aligned in PyMOL to complete the Rosetta libraries. The Rosetta library for GSTVYAPFT was generated by threading all possible six amino acid segments of the LECT2 sequence against each other and then extracting only those which modeled the peptide sequence of interest. This library contained 111 fragments. The Rosetta library for FRNWQAYMQ was generated by threading all six amino acid long permutations of the peptide sequence against each other pairwise over the NNQQNY backbone. The threading, packing, and relaxation steps were done 20 separate times for each pair of sequences and the resulting library contained 640 single chain fragments.
General Strand Libraries Generated with ALEPH
The general libraries with strands generated by ALEPH23 for use in ARCIMBOLDO_BORGES and distributed in CCP4 are all larger in size than any of the peptides investigated, as they contained a minimum of three strands forming a sheet in parallel or antiparallel arrangement. To obtain libraries of one or two strands, we started from the distributed three antiparallel strand (udu) library. Using a template of only two strands, we extracted all compatible models (around 24000) using ALEPH. Using the template model, we then generated an artificial data set in space group P1 to a resolution of 2.0 Å. As the models come from a standard library already, they were superposed originally based on their three strands and also clustered geometrically. To achieve that, we performed a Phaser rigid body refinement against the simulated data, using an rmsd error tolerance of 1.0 Å. We then selected the top LLG-scoring fragments for each of the geometric clusters and took them as our representatives for the two-strand library (246 models). Of those, outliers that did not superimpose well were removed. Only 108 models remained, each a pair of antiparallel beta strands. Then we generated two libraries of a single strand by extracting into separate PDB files from the parent library. This procedure yielded two general libraries to use in our experiments.
Structure Refinement
All structures were refined using Phenix version 1.16–3549–00038,39 and Coot version 0.8.9.40 All refinements used standard settings and the built-in electron scattering tables in Phenix. For each peptide, the final model output, called the best.pdb, from the ARCIMBOLDO-BORGES run was used directly as the model for the first round of refinement in Phenix. One exception was the QIGLAQTQ plate polymorph structure. In this case, the model from ARCIMBOLDO-BORGES was run through an additional round of Phaser molecular replacement to correct for what appeared to be a translational shift in the chain. This could be due to the extremely tight packing of the chains and the lack of any solvent, ordered or disordered, making initial chain placement difficult.
Acknowledgments
We thank Duilio Cascio (UCLA) for discussions and helpful analysis and the David Eisenberg laboratory (UCLA) for generous access to all of their amyloid peptide structures. This work was performed as part of STROBE, an NSF Science and Technology Center through Grant DMR-1548924. This work is also supported by DOE Grant DE-FC02-02ER63421 and NIH-NIGMS Grant R35 GM128867 and P41GM136508. L.S.R. is supported by the USPHS National Research Service Award 5T32GM008496. M.D.F. was funded by Eugene V. Cota-Robles Fellowship and Ruth L. Kirschstein NRSA GM007185 and is currently funded by a Whitcome Pre-Doctoral Fellowship and a National Science Foundation Graduate Research Fellowship. C.G. was funded by Ruth L. Kirschstein NRSA GM007185 and is currently funded by Ruth L. Kirschstein Predoctoral Individual NRSA, 1F31 AI143368. R.J.B. received fellowship from FAPESP (16/24191-8 and 17/13485-3). C.M. is grateful to MICINN for her BES-2015-071397 scholarship associated with the Structural Biology Maria de Maeztu Unit of Excellence. This work was supported by grants PGC2018-101370-B-I00 and PID2021-128751NB-I00 (MICINN/AEI/FEDER/UE) and Generalitat de Catalunya (2017SGR-1192) to I.U. J.A.R. is supported as a Pew Scholar, a Beckman Young Investigator, and a Packard Fellow. The Northeastern Collaborative Access Team beamline is funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Data Availability Statement
All structures determined in this work have been deposited to the PDB. The deposited ARCIMBOLDO structures of peptides and their associated PDB IDs are QYNNENNFV (7N2J), NYNNYQ (7N2K), QIGLAQTQ_plate (7N2F), QIGLAQTQ_needle (7N2G), GSTVYAPFT (7N2I), and FRNWQAYMQ (7N2D). The deposited direct methods structures of peptides are QYNNENNFV (7N2L), NYNNYQ (7N2H), and QIGLAQTQ_plate (7N2E). The truncated .mtz file for the MicroED structure of NYNNYQ is available from the authors upon request.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomedchemau.2c00082.
Data showing statistics and structures of peptides phased ab initio, summaries of fragment-based phasing statistics, and details for peptide fragment libraires utilized (PDF)
Author Contributions
# L.S.R., M.D.F., and C.M. contributed equally to this work. J.A.R. and I.U. directed the research. L.S.R., M.D.F., and C.M. generated fragment libraries and performed phasing with ARCIMBOLDO. L.S.R, M.D.F, C.G., and J.A.R. crystallized peptides and prepared samples. C.G., M.G.J., and J.A.R. collected diffraction data. M.D.F., L.S.R., C.Z., C.G., M.R.S., and M.G.J processed diffraction data. C.M., I.U., and R.J.B. developed and evaluated MicroED specific features in the ARCIMBOLDO framework. All authors helped to write and provided critical feedback on the article. CRediT: Logan S Richards data curation (lead), formal analysis (lead), investigation (lead), methodology (lead), validation (lead), visualization (lead), writing-original draft (lead), writing-review & editing (lead); Maria D Flores data curation (lead), formal analysis (lead), investigation (lead), methodology (lead), validation (lead), visualization (lead), writing-original draft (lead), writing-review & editing (lead); Claudia Millan data curation (lead), formal analysis (lead), investigation (lead), methodology (lead), resources (equal), software (lead), validation (lead), visualization (lead), writing-original draft (lead), writing-review & editing (lead); Calina Glynn data curation (equal), formal analysis (equal), investigation (equal), validation (equal), writing-review & editing (equal); Marcus Gallagher-Jones data curation (equal), formal analysis (equal), investigation (equal), validation (equal), writing-review & editing (equal); Rafael J. Borges data curation (equal), formal analysis (equal), investigation (equal), methodology (equal), software (equal), writing-review & editing (equal); Isabel Usón conceptualization (lead), data curation (lead), formal analysis (lead), funding acquisition (lead), investigation (lead), methodology (lead), project administration (lead), resources (lead), software (lead), supervision (lead), validation (lead), visualization (lead), writing-original draft (lead), writing-review & editing (lead).
The authors declare the following competing financial interest(s): J.A.R. is an equity stake holder of Medstruc, Inc.
Supplementary Material
References
- Brink C.; Hodgkin D. C.; Lindsey J.; Pickworth J.; Robertson J. H.; White J. G. Structure of Vitamin B12: X-Ray Crystallographic Evidence on the Structure of Vitamin B12. Nature 1954, 174 (4443), 1169–1171. 10.1038/1741169a0. [DOI] [PubMed] [Google Scholar]
- Sawaya M. R.; Rodriguez J.; Cascio D.; Collazo M. J.; Shi D.; Reyes F. E.; Hattne J.; Gonen T.; Eisenberg D. S. Ab Initio Structure Determination from Prion Nanocrystals at Atomic Resolution by MicroED. Proc. Natl. Acad. Sci. 2016, 113 (40), 11232–11236. 10.1073/pnas.1606287113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J. A.; Ivanova M. I.; Sawaya M. R.; Cascio D.; Reyes F. E.; Shi D.; Sangwan S.; Guenther E. L.; Johnson L. M.; Zhang M.; Jiang L.; Arbing M. A.; Nannenga B. L.; Hattne J.; Whitelegge J.; Brewster A. S.; Messerschmidt M.; Boutet S.; Sauter N. K.; Gonen T.; Eisenberg D. S. Structure of the Toxic Core of α-Synuclein from Invisible Crystals. Nature 2015, 525 (7570), 486–490. 10.1038/nature15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher-Jones M.; Glynn C.; Boyer D. R.; Martynowycz M. W.; Hernandez E.; Miao J.; Zee C.-T.; Novikova I. V.; Goldschmidt L.; McFarlane H. T.; Helguera G. F.; Evans J. E.; Sawaya M. R.; Cascio D.; Eisenberg D. S.; Gonen T.; Rodriguez J. A. Sub-Ångström Cryo-EM Structure of a Prion Protofibril Reveals a Polar Clasp. Nat. Struct. Mol. Biol. 2018, 25 (2), 131–134. 10.1038/s41594-017-0018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R. The Potential and Limitations of Neutrons, Electrons and X-Rays for Atomic Resolution Microscopy of Unstained Biological Molecules. Q. Rev. Biophys. 1995, 28 (2), 171–193. 10.1017/S003358350000305X. [DOI] [PubMed] [Google Scholar]
- Warmack R. A.; Boyer D. R.; Zee C.-T.; Richards L. S.; Sawaya M. R.; Cascio D.; Gonen T.; Eisenberg D. S.; Clarke S. G. Structure of Amyloid-β (20–34) with Alzheimer’s-Associated Isomerization at Asp23 Reveals a Distinct Protofilament Interface. Nat. Commun. 2019, 10 (1), 1–12. 10.1038/s41467-019-11183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. G.; Martynowycz M. W.; Hattne J.; Fulton T. J.; Stoltz B. M.; Rodriguez J. A.; Nelson H. M.; Gonen T. The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination. ACS Cent. Sci. 2018, 4 (11), 1587–1592. 10.1021/acscentsci.8b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting C. P.; Funk M. A.; Halaby S. L.; Zhang Z.; Gonen T.; van der Donk W. A. Use of a Scaffold Peptide in the Biosynthesis of Amino Acid Derived Natural Products. Science 2019, 365 (6450), 280–284. 10.1126/science.aau6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M.; Gilmore C. J.; Hauptman H. A.; Weeks C. M.; Miller R.; Usón I.. Ab Initio Phasing. In International Tables for Crystallography; Brock C. P., Hahn T., Wondratschek U., Müller U., Shmueli U., Prince E., Auhier A., Kopský V., Litvin D. B., Arnold E.. et al. , Eds.; John Wiley & Sons, Ltd., 2012; pp 413–432. [Google Scholar]
- Rossmann M. G. The Molecular Replacement Method. Acta Crystallogr. A 1990, 46, 73–82. 10.1107/S0108767389009815. [DOI] [PubMed] [Google Scholar]
- Shi D.; Nannenga B. L.; Iadanza M. G.; Gonen T. Three-Dimensional Electron Crystallography of Protein Microcrystals. eLife 2013, 2, e01345 10.7554/eLife.01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krotee P.; Rodriguez J. A.; Sawaya M. R.; Cascio D.; Reyes F. E.; Shi D.; Hattne J.; Nannenga B. L.; Oskarsson M. E.; Philipp S.; Griner S.; Jiang L.; Glabe C. G.; Westermark G. T.; Gonen T.; Eisenberg D. S. Atomic Structures of Fibrillar Segments of HIAPP Suggest Tightly Mated β-Sheets Are Important for Cytotoxicity. eLife 2017, 6, e19273 10.7554/eLife.19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz M. J.; Hattne J.; Shi D.; Seidler P.; Rodriguez J.; Reyes F. E.; Sawaya M. R.; Cascio D.; Weiss S. C.; Kim S. K.; Hinck C. S.; Hinck A. P.; Calero G.; Eisenberg D.; Gonen T. Atomic Resolution Structures from Fragmented Protein Crystals by the CryoEM Method MicroED. Nat. Methods 2017, 14 (4), 399–402. 10.1038/nmeth.4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez D. D.; Grosse C.; Himmel S.; González C.; de Ilarduya I. M.; Becker S.; Sheldrick G. M.; Usón I. Crystallographic Ab Initio Protein Structure Solution below Atomic Resolution. Nat. Methods 2009, 6 (9), 651–653. 10.1038/nmeth.1365. [DOI] [PubMed] [Google Scholar]
- Read R. J.; McCoy A. J. A Log-Likelihood-Gain Intensity Target for Crystallographic Phasing That Accounts for Experimental Error. Acta Crystallogr., Sect. D: Struct. Biol. 2016, 72, 375–387. 10.1107/S2059798315013236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usón I.; Sheldrick G. M. An Introduction to Experimental Phasing of Macromolecules Illustrated by SHELX; New Autotracing Features. Acta Crystallogr. Sect. Struct. Biol. 2018, 74 (2), 106–116. 10.1107/S2059798317015121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millán C.; Sammito M.; Usón I. Macromolecular Ab Initio Phasing Enforcing Secondary and Tertiary Structure. IUCrJ 2015, 2, 95–105. 10.1107/S2052252514024117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammito M.; Millán C.; Frieske D.; Rodríguez-Freire E.; Borges R. J.; Usón I. ARCIMBOLDO_LITE: Single-Workstation Implementation and Use. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015, 71, 1921–1930. 10.1107/S1399004715010846. [DOI] [PubMed] [Google Scholar]
- Sammito M.; Millán C.; Rodríguez D. D.; de Ilarduya I. M.; Meindl K.; De Marino I.; Petrillo G.; Buey R. M.; de Pereda J. M.; Zeth K.; Sheldrick G. M.; Usón I. Exploiting Tertiary Structure through Local Folds for Crystallographic Phasing. Nat. Methods 2013, 10 (11), 1099–1101. 10.1038/nmeth.2644. [DOI] [PubMed] [Google Scholar]
- Millán C.; Sammito M. D.; McCoy A. J.; Nascimento A. F. Z.; Petrillo G.; Oeffner R. D.; Domínguez-Gil T.; Hermoso J. A.; Read R. J.; Usón I. Exploiting Distant Homologues for Phasing through the Generation of Compact Fragments, Local Fold Refinement and Partial Solution Combination. Acta Crystallogr. Sect. Struct. Biol. 2018, 74 (4), 290–304. 10.1107/S2059798318001365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards L. S.; Millán C.; Miao J.; Martynowycz M. W.; Sawaya M. R.; Gonen T.; Borges R. J.; Usón I.; Rodriguez J. A. Fragment-Based Determination of a Proteinase K Structure from MicroED Data Using ARCIMBOLDO_SHREDDER. Acta Crystallogr. Sect. Struct. Biol. 2020, 76 (8), 703–712. 10.1107/S2059798320008049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn A.; Sheldrick G. M. Extending Molecular-Replacement Solutions with SHELXE. Acta Crystallogr. D Biol. Crystallogr. 2013, 69 (11), 2251–2256. 10.1107/S0907444913027534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina A.; Triviño J.; Borges R. J.; Millán C.; Usón I.; Sammito M. D. ALEPH: A Network-Oriented Approach for the Generation of Fragment-Based Libraries and for Structure Interpretation. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2020, 76, 193–208. 10.1107/S2059798320001679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millán C.; Jiménez E.; Schuster A.; Diederichs K.; Usón I. ALIXE: A Phase-Combination Tool for Fragment-Based Molecular Replacement. Acta Crystallogr. Sect. Struct. Biol. 2020, 76 (3), 209–220. 10.1107/S205979832000056X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alford R. F.; Leaver-Fay A.; Jeliazkov J. R.; O’Meara M. J.; DiMaio F. P.; Park H.; Shapovalov M. V.; Renfrew P. D.; Mulligan V. K.; Kappel K.; Labonte J. W.; Pacella M. S.; Bonneau R.; Bradley P.; Dunbrack R. L.; Das R.; Baker D.; Kuhlman B.; Kortemme T.; Gray J. J. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13 (6), 3031–3048. 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S.; Lyskov S.; Gray J. J. PyRosetta: A Script-Based Interface for Implementing Molecular Modeling Algorithms Using Rosetta. Bioinforma. Oxf. Engl. 2010, 26 (5), 689–691. 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt L.; Teng P. K.; Riek R.; Eisenberg D. Identifying the Amylome, Proteins Capable of Forming Amyloid-like Fibrils. Proc. Natl. Acad. Sci. 2010, 107 (8), 3487–3492. 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero I.; Sammito M.; Millán C.; Lebedev A.; Soler N.; Usón I. ARCIMBOLDO on Coiled Coils. Acta Crystallogr. Sect. Struct. Biol. 2018, 74 (3), 194–204. 10.1107/S2059798317017582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayeb-Fligelman E.; Tabachnikov O.; Moshe A.; Goldshmidt-Tran O.; Sawaya M. R.; Coquelle N.; Colletier J.-P.; Landau M. The Cytotoxic Staphylococcus Aureus PSMα3 Reveals a Cross-α Amyloid-like Fibril. Science 2017, 355 (6327), 831–833. 10.1126/science.aaf4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas N.; Tayeb-Fligelman E.; Sammito M. D.; Bloch D.; Jelinek R.; Noy D.; Usón I.; Landau M. The Amphibian Antimicrobial Peptide Uperin 3.5 Is a Cross-α/Cross-β Chameleon Functional Amyloid. Proc. Natl. Acad. Sci. 2021, 118 (3), e2014442118 10.1073/pnas.2014442118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior A. W.; Evans R.; Jumper J.; Kirkpatrick J.; Sifre L.; Green T.; Qin C.; Žídek A.; Nelson A. W. R.; Bridgland A.; Penedones H.; Petersen S.; Simonyan K.; Crossan S.; Kohli P.; Jones D. T.; Silver D.; Kavukcuoglu K.; Hassabis D. Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 2020, 577 (7792), 706–710. 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Integration, Scaling, Space-Group Assignment and Post-Refinement. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 133–144. 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usón I.; Sheldrick G. M. Advances in Direct Methods for Protein Crystallography. Curr. Opin. Struct. Biol. 1999, 9 (5), 643–648. 10.1016/S0959-440X(99)00020-2. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71 (1), 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga M.; Read R. J. Experiences with a New Translation-Function Program. J. Appl. Crystallogr. 1987, 20 (6), 517–521. 10.1107/S0021889887086102. [DOI] [Google Scholar]
- Leaver-Fay A.; Tyka M.; Lewis S. M.; Lange O. F.; Thompson J.; Jacak R.; Kaufman K.; Renfrew P. D.; Smith C. A.; Sheffler W.; Davis I. W.; Cooper S.; Treuille A.; Mandell D. J.; Richter F.; Ban Y.-E. A.; Fleishman S. J.; Corn J. E.; Kim D. E.; Lyskov S.; Berrondo M.; Mentzer S.; Popović Z.; Havranek J. J.; Karanicolas J.; Das R.; Meiler J.; Kortemme T.; Gray J. J.; Kuhlman B.; Baker D.; Bradley P. Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods Enzymol. 2011, 487, 545–574. 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D.; Afonine P. V.; Baker M. L.; Bunkóczi G.; Chen V. B.; Croll T. I.; Hintze B.; Hung L. W.; Jain S.; McCoy A. J.; et al. Macromolecular Structure Determination Using X-Rays, Neutrons and Electrons: Recent Developments in Phenix. Acta Crystallogr. Sect. Struct. Biol. 2019, 75, 861–877. 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Echols N.; Headd J. J.; Moriarty N. W.; Mustyakimov M.; Terwilliger T. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. Towards Automated Crystallographic Structure Refinement with Phenix.Refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68 (4), 352–367. 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All structures determined in this work have been deposited to the PDB. The deposited ARCIMBOLDO structures of peptides and their associated PDB IDs are QYNNENNFV (7N2J), NYNNYQ (7N2K), QIGLAQTQ_plate (7N2F), QIGLAQTQ_needle (7N2G), GSTVYAPFT (7N2I), and FRNWQAYMQ (7N2D). The deposited direct methods structures of peptides are QYNNENNFV (7N2L), NYNNYQ (7N2H), and QIGLAQTQ_plate (7N2E). The truncated .mtz file for the MicroED structure of NYNNYQ is available from the authors upon request.