

Abstract

Methyllycaconitine (MLA), 1, is a naturally occurring norditerpenoid alkaloid that is a highly potent (IC50 = 2 nM) selective antagonist of α7 nicotinic acetylcholine receptors (nAChRs). Several structural factors affect its activity such as the neopentyl ester side-chain and the piperidine ring N-side-chain. The synthesis of simplified AE-bicyclic analogues 14–21 possessing different ester and nitrogen side-chains was achieved in three steps. The antagonist effects of synthetic analogues were examined on human α7 nAChRs and compared to that of MLA 1. The most efficacious analogue (16) reduced α7 nAChR agonist responses [1 nM acetylcholine (ACh)] to 53.2 ± 1.9% compared to 3.4 ± 0.2% for MLA 1. This demonstrates that simpler analogues of MLA 1 possess antagonist effects on human α7 nAChRs but also indicates that further optimization may be possible to achieve antagonist activity comparable to that of MLA 1.
Keywords: antagonist, human α7 nAChR, methyllycaconitine (MLA), 2-methylsuccinimido benzoate ester, nicotinic acetylcholine receptors (nAChR), nicotinic competitive antagonist, norditerpenoid alkaloid
Introduction
Nicotinic acetylcholine receptors (nAChRs) are members of a superfamily of ligand-gated ion channels and are receptors for the neurotransmitter acetylcholine (ACh). They are oligomeric proteins in which five transmembrane subunits coassemble to form a central cation-selective pore. Agonists, such as ACh, bind to a site on the extracellular region of nAChRs and, in doing so, cause a conformational change in the receptor that results in the opening of the transmembrane ion channel and the influx of cations.1,2 Nicotinic receptors are located at postsynaptic sites (for example, on nerve and muscle cells), where they can mediate rapid neuronal or neuromuscular signaling, but they are also located at presynaptic sites (for example, in the brain), where they can play a more modulatory role. Sixteen nAChR subunits are expressed in humans (α1−α7, α9, α10, β1−β4, γ, δ, and ε) and these can coassemble into a diverse array of both homomeric and heteromeric nAChR subtypes with distinct physiological and pharmacological properties.3 One nAChR subtype that has attracted particular attention is the α7 nAChR, a homomeric receptor containing five copies of the α7 subunit. It is expressed in several regions of the brain and has been implicated in a range of neurological disorders.4
Signaling through nAChRs can be blocked by the binding of antagonists acting either at the orthosteric agonist binding site (competitive antagonists) or at distinct allosteric sites (noncompetitive antagonists).5 Methyllycaconitine (MLA), 1, is one example of a nAChR competitive antagonist that is highly potent and highly selective for α7 nAChRs.6 It forms part of a broader family of norditerpenoid alkaloids (NDAs) from Delphinium and Aconitum, which are highly oxygenated hexacyclic systems and can exert a variety of pharmacological effects by modulating transmembrane proteins such as nAChRs and voltage gated sodium channels (VGSCs).7,8 In addition, the potential therapeutic use of MLA 1 has been examined in connection with disorders such as cerebral palsy and Parkinson’s disease.8 MLA 1 and other Delphinium alkaloids are also responsible for livestock intoxication9 due to their action on α1 nAChRs expressed at neuromuscular junctions.10 However, it has been reported previously that MLA 1 has higher affinity for α7 nAChRs compared to other nAChR subtypes.6,11,12
Several structural features of MLA 1 have been studied in our ongoing structure–activity relationship (SAR) studies. For example, it was found that the nitrogen atom plays a key role in the pharmacological action of NDAs.13 Also, the ester side-chain is an important moiety as MLA 1 lost 1000-fold of its activity when converted to neopentyl alcohol lycoctonine.14−16 Furthermore, the side-chain on the nitrogen atom affects the interaction with the target nAChR. Several piperidine (ring E) analogues of MLA have been synthesized (Figure 1) with different N-side-chains (methyl, ethyl, n-butyl, 2-phenylethyl, 3-phenylpropyl, diethyl ether, and 2-phenylethyl ether) and tested on bovine adrenal α3β4 nAChRs, where the best analogue had a 3-phenylpropyl N-side-chain.17 This analogue system was tested on the α7 nAChR in a competition binding experiment on rat brain preparations using [125I]αBGT, where the best analogue (3-phenylpropyl N-side-chain) showed little inhibition with an IC50 = 177 μM, while other analogues showed no inhibition with IC50 > 300 μM.18
Figure 1.

E-ring analogue system of MLA 1.
The aim of this study was to synthesize AE-bicyclic analogues of MLA 1 with different nitrogen and ester side-chains (Scheme 1) and to examine their ability to modulate the activity of human α7 nAChRs, with the aim of obtaining a better SAR understanding of these compounds.
Scheme 1. Relationship of MLA 1 to the Target AE-Bicyclic System.

Results and Discussion
Synthesis of the AE-Bicyclic Core
The synthesis of MLA analogues starts with the core synthesis using the classical double Mannich reaction18,19 where different amines were used to obtain different N-side-chains. The side-chains (methyl, ethyl, benzyl, 2-phenylethyl, 3-phenylpropyl, and 4-phenylbutyl) (Scheme 2) were selected to investigate the importance of the hydrophobic interactions. The reaction was accomplished by heating the reactants under reflux in ethanol for 4 h. As the boiling point of the methylamine solution (40 wt % in water) is 48 °C, the synthesis of compound 3 was also achieved at 20 °C for 2 d with no significant drop in yield.
Scheme 2. Synthesis of AE-Bicyclic Analogues 14–21.

Reduction of the AE-Bicyclic Core Using LiAlH4 (LAH)
The reduction of the AE-bicyclic compounds 3–8 was performed using LAH in anhydrous THF under N2 gas (Scheme 2), and the reaction was monitored by TLC and quenched after 7 h using the Fieser method, where X mL (X = grams of LAH) of water was added slowly followed by X mL of 15% aq. sodium hydroxide solution and then 3X mL of water. The resulting mixture was stirred with magnesium sulfate for 10 min and then filtered over Celite and evaporated to dryness. The reduction results in epimeric secondary alcohol at position 9. As an example, cyclohexanone 4 was reduced to get both epimers at position 9. The 1H NMR methyl triplet signals of the NCH2CH3 showed that the ratio of the isomers is 3:1 (Figure 2). The 1H NMR spectrum also shows the difference in the intensity of 9-H signals in both epimers (3.60 ppm vs 3.68 ppm) (Figure 2). The full 1H and 13C NMR spectra also showed different intensities of signals for the 3:1 isomeric ratio (SI Figure S1).
Figure 2.
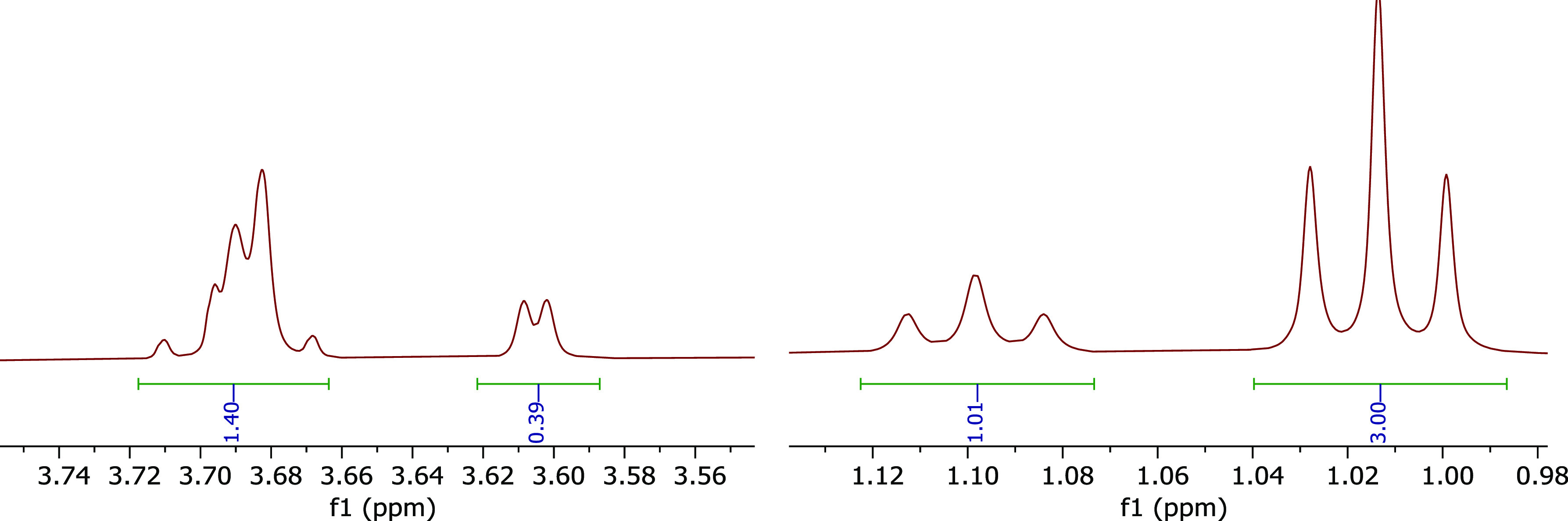
1H NMR (500 MHz) of 9-H proton (left) and the methyl of NCH2CH3 (right) of the epimeric mixture after reduction of cyclohexanone 4.
The mixture was purified using column chromatography to obtain the major isomer, diol 9. The β-alcohol was established by NOESY as the proton at position 9 showed a correlation with protons 2ax and 4ax (Figure 3). The methylene protons (CH2OH) resonate as two adjacent doublets (3.35 and 3.39 ppm) while they showed as a multiplet in the epimeric mixture (Figure 2). The compounds 3 and 5–8 were reduced and used in the esterification step without purification.
Figure 3.

NOESY correlation between 9-Heq and 2-Hax and 4-Hax in diol 9.
Synthesis of the Carboxylic Acid Side-Chains
MLA 1 is a potent nAChR antagonist. Lappaconitine 10 is the most clinically successful NDA where its hydrobromide salt (Allapinin) is used as an antiarrhythmic drug.8 Therefore, their side-chains (Figure 4) were chosen to be attached to the analogues.
Figure 4.

Side-chains (red) of methyllycaconitine 1 and lappaconitine 10.
Synthesis of Lappaconitine Side-Chain
The synthesis of 2-acetamidobenzoic acid 11 was accomplished through refluxing anthranilic acid and acetic anhydride in anhydrous tetrahydrofuran under nitrogen gas for 4 h. The reaction was quenched using 1 M aq. HCl, and the product was recrystallized from water and ethanol (1:1).
Synthesis of Methyllycaconitine Side-Chain
The first step of MLA 1 side-chain synthesis was performed by neat fusion of anthranilic acid and citraconic anhydride at 140 °C under nitrogen gas for 24 h (Scheme 3).19−21 Chiral hydrogenation of 12 to get the S-enantiomer was tried with (S)-ruthenium diacetate (2,2′-bis(diphenylphosphino)-1,1′-binaphthyl) (S-Ru(OAc)2BINAP) without success. Then it was performed using (2S,4S)-1-Boc-4-diphenylphosphino-2-(diphenylphosphinomethyl)pyrrolidine (BPPM) coupled with rhodium cyclooctadiene chloride dimer (Rh(COD)Cl)2.22−24 The optical rotation was −12.0°, consistent with the literature value.25
Scheme 3. Synthesis of MLA Side-Chain.

The 13C NMR of 13 showed doubling phenomena at 25 °C when measured in CDCl3, which could be due to an intramolecular interaction that hindered the free rotation of the methyl succinimide group. Variable temperature (VT) NMR experiments were performed, and the doubling phenomena disappeared on increasing the temperature to 55 °C where the molecule has more energy to rotate freely, and then reappeared upon cooling down to 25 and 15 °C (Figure 5). To explain the hindrance that results in the NMR doubling, the 3D models in Figure 5 show that the clash happens between the carboxylic acid and the methylsuccinimide moiety.
Figure 5.

13C NMR (125 MHz) in CDCl3 of 13 at various temperatures.
NMR of 13 was also measured in CD3OD to check if the doubling happens due to intramolecular H-bonding or steric clash. 13C NMR spectra in CD3OD again showed doubling of the signals (SI Figure S2) consistent with steric hindrance in methylsuccinimido anthranilate 13.
Synthesis of the Analogues by Esterification
The reduced AE-bicycles were esterified with the naturally occurring NDA side-chains (11 and 13) using N,N′-dicyclohexyl-carbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) in anhydrous acetonitrile at 40 °C under anhydrous nitrogen gas (Scheme 2). The reaction was monitored and stopped after 24 h, and the crude material was purified to homogeneity to yield analogues 14–21.
The stereochemistry of the hydroxy group at position 9 was determined to be axial by NOESY spectrum as the 9-Heq showed correlation with 2-Hax and 4-Hax. Analogues 14 and 21 were taken as examples, and SI Figure S3 shows the NOE correlations in both of them.
The NMR spectra of the analogues were similar, the only major difference observed was for the protons at carbon 1′, which showed the roofing effect in analogues 20 and 21 with the 2-acetamido-benzoic acid side-chain where this could be caused by a steric hindrance effect from the side-chain. They merge into one multiplet signal in analogues 14–19 with the 2-methylsuccinimidobenzoic acid side-chain. SI Figure S4 shows the 1H NMR signal at position 1′ in analogues 14–15 and 20–21.
As we have reported with some naturally occurring NDAs,26 these simple analogues show the effect of steric compression on the axial proton of position 7. The equatorial protons resonate usually at a higher frequency due to the anisotropic effect of the C–C bond.27 In these bicyclic compounds, the axial proton at position 7 is further downfield due to the interaction with the nitrogen lone pair of electrons. The chemical shift difference that was observed in the bicyclic compounds 3–9 and 14–21 is ∼1–1.5 ppm. The axial protons at position 6 and 8 also resonate at a higher chemical shift, which is probably due to 1–3 interactions through space with substituents at positions 1 and 9.28
Antagonist Activity of MLA Analogues on Human α7 nAChRs
The antagonistic activity of MLA 1 and the analogues 14–21 has been tested on human α7 nAChRs heterologously expressed in Xenopus oocytes. The level of antagonism was measured by the coapplication of the analogues [1 nM] with an EC50 concentration of ACh [100 μM], after a preapplication of the analogues for 2 min. Responses to ACh in the presence of MLA analogues were normalized to responses to an EC50 concentration of ACh [100 μM] applied in the absence of analogues (Figure 6). MLA 1 inhibited the receptor response to 3.4 ± 0.2% (n = 4) of normalized responses. In addition, all of the analogues exhibited antagonist effects at human α7 nAChRs, resulting in significantly reduced agonist responses (P < 0.0001; Figure 6). Compounds 16, 19, and 17 showed the highest levels of antagonism, agonist responses to 53.2 ± 1.9% (n = 3), 56.7 ± 2.5% (n = 3), and 64.3 ± 2.2% (n = 3) of normalized responses, respectively (Figure 6).
Figure 6.

Normalized agonist (ACh) response of α7 nAChRs in the presence of MLA or analogues 14–21. Data were generated from cloned human α7 nAChRs expressed in Xenopus oocytes. Data are mean ± SEM of at least three independent experiments.
The antagonist activity of these analogues showed little advantage of the (S)-2-methylsuccinimido benzoate ester side-chain especially when comparing analogue 14 with 20. The data for analogues 14–19 highlights the effect of the N-side-chain on the antagonist activity at human α7 nAChR where the activity is in the following order: benzyl > 4-phenylbutyl > 2-phenylethyl > 3-phenylpropyl > methyl > ethyl (Figure 7). These data indicate that a bulkier N-side-chain (with phenyl moiety) enhances the activity compared to alkane side-chains. The E-ring analogue system developed by Bergmeier, McKay and co-workers17,18,29−31 was tested on bovine adrenal α3β4 nAChRs and showed that the best analogue, 3-phenylpropyl N-side-chain, inhibits the nicotine-stimulated catecholamine secretion [50 μM] by around 86%17 with IC50 = 11.4 μM18 compared to 95% inhibition of the nicotine-stimulated catecholamine secretion [50 μM]17 with IC50 = 2.6 μM18 for MLA 1. In addition, this analogue system was tested on α7 nAChRs in a competition binding experiment on rat brain preparations using [125I]αBGT where the best analogue (3-phenylpropyl N-side-chain) showed only a little inhibition with IC50 = 177 μM compared to 0.01 μM for MLA 1.18 The AE-bicyclic analogues showed better activity compared to the reported one (E) ring system where the best analogue 16, benzyl N-side-chain, inhibits the agonist response at human α7 nAChR to around 53% [1 nM].
Figure 7.

The three most active analogues, 16, 17, and 19.
Conclusions
Several MLA 1 AE-bicyclic analogues were synthesized with different N-side-chains and ester side-chains. Antagonist effects of synthetic analogues were examined on human α7 nAChRs and compared to that of MLA 1. The antagonist activity of these analogues showed little advantage of the (S)-2-methylsuccinimido benzoate ester side-chain especially when comparing analogue 14 with 20. The data from analogues 14–19 highlight the effect of the N-side-chain on the antagonist activity at human α7 nAChRs, where a bulkier N-side-chain (with a phenyl moiety) enhanced the antagonist activity compared to alkane side-chains. The pharmacological results achieved with these AE-bicyclic analogues, synthesized in three steps, showed better activity compared to the reported one ring system. The best analogue 16, containing a benzyl N-side-chain, inhibited the agonist response at human α7 nAChRs to around 53% [1 nM]. However, these are significantly less efficacious than MLA 1 so further optimization will be required to achieve comparable antagonist activity. In addition, it may be of interest to undertake further studies to examine the selectivity of these novel compounds for α7 nAChRs by examining their influence on a broader range of nAChR subtypes.
Experimental Section
General Methods
Analytical thin layer chromatography (TLC) was performed using aluminum backed sheet precoated silica gel plates (Merck Kieselgel 60 F254). Compounds were visualized by UV light or by staining with iodine, ninhydrin, and p-anisaldehyde. Column chromatography was performed over silica gel 200–400 mesh (purchased from Sigma-Aldrich).1H NMR spectra were recorded with a Bruker Avance III (500 MHz) spectrometer at 25 °C. Chemical shifts are given in parts per million (ppm), referenced to the residual solvent peak, and reported as position (δ), multiplicity (s = singlet, br = broad, d = doublet, dd = doublet of doublets, t = triplet, dt = doublet of triplets, tt = triplet of triplets, q = quartet, qd = quartet of doublets, qt = quartet of triplets, quin = quintet, m = multiplet), relative integral, assignment, and coupling constant (J in Hz). 13C NMR spectra were recorded with a Bruker Avance III (125 MHz) spectrometer at 25 °C with complete proton decoupling. Chemical shifts are expressed in parts per million (ppm) referenced to the used solvent, and reported as position (δ). In addition, 1H–1H COSY, 1H–13C HMBC, and 1H–13C HSQC correlation spectra were used for the complete assignment of the proton and carbon resonances. 1H–1H NOESY NMR spectra were recorded in special cases to determine the stereochemistry of diastereoisomers. High Resolution Time-of Flight (HR TOF) mass spectra (MS) were obtained on a Bruker Daltonics “micrOTOF” mass spectrometer using electrospray ionization (ESI) (loop injection +ve and −ve mode). A PerkinElmer 65 spectrum FT-IR spectrometer was used to obtain the IR spectra. Optical rotations were recorded on an Optical Activity LTD high performance polarimeter using halogen spectral line 589 nm. The final compounds tested for biological activity were all >98% pure; indeed analytical HPLC showed that the purity of 18 was 98%; all seven other analogues were >99% pure (HPLC traces for compounds 14–21 are provided in the SI). These compounds were also all homogeneous by TLC and NMR.
Ethyl 3-Methyl-9-oxo-3-azabicyclo[3.3.1]nonane-1-carboxylate (3)
A solution of ethyl cyclohexanone-2-carboxylate (4.44 mmol, 0.748 mL, 95%), 2.2 equiv of formaldehyde (9.768 mmol, 0.713 mL, 38% aq v/v) and 1.1 equiv of methylamine (4.88 mmol, 0.608 mL, 33% in EtOH) in ethanol (25 mL) was stirred at 40 °C for 2 d under N2. Then the solution was concentrated under vacuum and purified by column chromatography using 12.5% EtOAc in petroleum ether to yield the title compound 3 (280 mg, 28%) as a yellow oil. Rf = 0.36 (12.5% EtOAc in petroleum ether). HRMS (ESI): m/z calcd. for C12H20NO3: 226.1443, found: 226.1443 [M + H]+ and m/z calcd. for C12H19NO3Na: 248.1263, found: 248.1262 [M + Na]+. νmax (NaCl)/cm–1 1733 (ester, C=O), 1717 (ketone, C=O). 1H NMR (500 MHz, CDCl3): δ (ppm) = 1.26 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.49–1.57 (m, 1H, H7eq), 2.00–2.09 (m, 1H, H6eq), 2.10–2.17 (m, 1H, H6ax), 2.15–2.29 (m, 1H, H8eq), 2.25 (s, 3H, NCH3), 2.40–2.45 (m, 1H, H5eq), 2.50 (dddd, J = 14.2, 12.3, 6.3, 2.0 Hz, 1H, H8ax), 2.59 (dd, J = 11.2, 3.8 Hz, 1H, H4ax), 2.76–2.89 (m, 1H, H7ax), 2.96 (dd, J = 11.3, 1.9 Hz, 1H, H2ax), 3.04 (dt, J = 11.2, 2.3 Hz, 1H, H4eq), 3.11 (dd, J = 11.5, 2.2 Hz, 1H, H2eq), 4.19 (q, J = 7.1 Hz, 2H, OCH2CH3). 13C NMR (125 MHz, CDCl3): δ (ppm) = 13.8 (OCH2CH3), 20.2 (C7), 34.0 (C6), 36.8 (C8), 44.8 (N CH3), 47.1 (C5), 58.5 (C1), 60.9 (OCH2CH3), 62.3 (C4), 64.0 (C2), 170.9 (ester), 212.3 (C9).
Ethyl 3-Ethyl-9-oxo-3-azabicyclo[3.3.1]nonane-1-carboxylate (4)
A solution of ethyl cyclohexanone-2-carboxylate (25.07 mmol, 4.09 mL, 98%), 2.2 equiv of formaldehyde (55.154 mmol, 2.19 mL, 38% aq v/v), and 1.1 equiv of ethylamine (27.577 mmol, 3.99 mL, 70% aq v/v) in ethanol (170 mL) was heated under reflux for 3 h under N2. Then the solution was cooled and concentrated under vacuum, and purified by column chromatography using 10% EtOAc in petroleum ether to yield the title compound 4 (3.75 g, 63%) as a yellow oil. Rf = 0.25 (10% EtOAc in petroleum ether). HRMS (ESI): m/z calcd. for C13H22NO3: 240.1600, found: 240.1600 [M + H]+ and m/z calcd. for C13H21NO3Na: 262.1419, found: 262.1418 [M + Na]+. νmax (NaCl)/cm–1 1733 (ester, C=O), 1716 (ketone, C=O). 1H NMR (500 MHz, CDCl3): δ (ppm) = 1.10 (t, J = 7.2 Hz, 3H, NCH2CH3), 1.28 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.46–1.57 (m, 1H, H7eq), 2.00–2.18 (m, 2H, H6ax and H6eq), 2.19–2.28 (m, 1H, H8eq), 2.37–2.60 (m, 5H, NCH2CH3, H5, H8ax and H4ax), 2.78–2.90 (m, 1H, H7ax), 2.94 (d, J = 12.0 Hz, 1H, H2ax), 3.15 (d, J = 11.1 Hz, 1H, H4eq), 3.22 (d, J = 11.4 Hz, 1H, H2eq), 4.21 (q, J = 7.1 Hz, 2H, OCH2CH3). 13C NMR (125 MHz, CDCl3): δ (ppm) = 12.7 (NCH2CH3), 14.1 (OCH2CH3), 20.5 (C7), 34.1 (C6), 36.8 (C8), 47.2 (C5), 51.1 (NCH2CH3), 58.8 (C1), 59.9 (C4), 61.0 (OCH2CH3), 61.6 (C2), 171.1 (ester), 212.6 (C9).
Ethyl 3-Benzyl-9-oxo-3-azabicyclo[3.3.1]nonane-1-carboxylate (5)
A solution of ethyl cyclohexanone-2-carboxylate (9.95 mmol, 1.62 mL, 98%), 2.2 equiv of formaldehyde (21.89 mmol, 1.6 mL, 38% aq v/v), and 1.1 equiv of benzylamine (10.9 mmol, 1.2 mL, 99%) in ethanol (70 mL) was heated under reflux for 3 h under N2. Then the solution was cooled and concentrated under vacuum, and purified by column chromatography using 10% EtOAc in petroleum ether to yield the title compound 5 (750 mg, 25%) as a yellow oil. Rf = 0.29 (10% EtOAc in petroleum ether). HRMS (ESI): m/z calcd. for C18H24NO3: 302.1756, found: 302.1755 [M + H]+ and m/z calcd. for C18H23NO3Na: 324.1576, found: 324.1573 [M + Na]+. νmax (NaCl)/cm–1 1732 (ester, C=O), 1717 (ketone, C=O). 1H NMR (500 MHz, CDCl3): δ (ppm) = 1.27 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.55- 1.65 (m, 1H, H7eq), 2.02–2.19 (m, 2H, H6ax and H6eq), 2.20–2.27 (m, 1H, H8eq), 2.44–2.48 (m, 5H), 2.54 (dddd, J = 14.1, 12.2, 6.4, 1.9 Hz, 1H, H8ax), 2.63 (dd, J = 10.9, 2.5 Hz, 1H, H4ax), 2.92–3.06 (m, 2H, H2ax and H7ax), 3.13 (d, J = 11.2, 2.4 Hz, 1H, H4eq), 3.20 (dd, J = 11.5, 2.4 Hz, 1 H, H2eq), 3.52 (s, 2H, NCH2Ph), 4.19 (qd, J = 7.2, 3.0 Hz, 2H, OCH2CH3), 7.27–7.36 (m, 5H, NCH2Ph). 13C NMR (125 MHz, CDCl3): δ (ppm) = 14.1 (OCH2CH3), 20.7 (C7), 34.1 (C6), 36.7 (C8), 47.2 (C5), 58.9 (C1), 60.3 (C4), 61.1 (OCH2CH3), 61.8 (C2), 62.1 (NCH2Ph), 127.2 (C4 arom), 128.4, 128.7 (C2 arom, C3 arom, C5 arom and C6 arom), 138.3 (C1 arom), 170.9 (ester), 212.4 (C9).
Ethyl 3-(2-Phenylethyl)-9-oxo-3-azabicyclo[3.3.1]nonane-1-carboxylate (6)
A solution of ethyl cyclohexanone-2-carboxylate (3.17 mmol, 0.517 mL, 98%), 2.2 equiv of formaldehyde (7 mmol, 0.525 mL, 38% aq. v/v), and 1.1 equiv of 2-phenylethyl amine (3.49 mmol, 0.446 mL, 99%) in ethanol (20 mL) was heated under reflux for 3 h under N2. Then the solution was cooled and concentrated under vacuum and purified by column chromatography using 10% EtOAc in petroleum ether to yield the title compound 6 (640 mg, 64%) as a yellow oil. Rf = 0.30 (10% EtOAc in petroleum ether). HRMS (ESI): m/z calcd. for C19H26NO3: 316.1913, found: 316.1912 [M + H]+ and m/z calcd. for C19H25NO3Na: 338.1732, found: 338.1731 [M + Na]+. νmax (NaCl)/cm–1 1732 (ester, C=O), 1716 (ketone, C=O). 1H NMR (500 MHz, CDCl3): δ (ppm) = 1.28 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.37–1.45 (m, 1H, H7eq), 1.97–2.11 (m, 2H, H6ax and H6eq), 2.12–2.20 (m, 1H, H8eq), 2.42–2.53 (m, H5 and H8ax), 2.55–2.68 (m, 4H, H7ax, H4ax and NCH2CH2Ph), 2.82 (t, J = 7.5 Hz, 2H, NCH2CH2Ph), 3.02 (d, J = 11.3 Hz, 1H, H2ax), 3.18 (d, J = 11.0 Hz, 1H, H4eq), 3.27 (d, J = 11.4 Hz, 1 H, H2eq), 4.21 (q, J = 7.1 Hz, 2H, OCH2CH3), 7.18–7.32 (m, 5H, NCH2CH2Ph). 13C NMR (125 MHz, CDCl3): δ (ppm) = 14.1 (OCH2CH3), 20.2 (C7), 33.8 (NCH2CH2Ph), 34.1 (C6), 36.8 (C8), 47.2 (C5), 58.5 (NCH2CH2Ph), 58.8 (C1), 60.2 (C4), 61.1 (OCH2CH3), 61.8 (C2), 126.0 (C4 arom), 128.3 (C2 arom and C6 arom), 128.6 (C3 arom and C5 arom), 140.1 (C1 arom), 171.1 (ester), 212.5 (C9).
Ethyl 3-(3-Phenylpropyl)-9-oxo-3-azabicyclo[3.3.1]nonane-1-carboxylate (7)
A solution of ethyl cyclohexanone-2-carboxylate (3 mmol, 0.49 mL, 99%), 2.2 equiv of formaldehyde (6.6 mmol, 0.48 mL, 38% aq. v/v), and 1.1 equiv of 3-phenylpropyl amine (3.3 mmol, 0.48 mL, 99%) in ethanol (20 mL) was heated under reflux for 3 h under N2. Then the solution was concentrated under vacuum and purified by column chromatography using 10% EtOAc in petroleum ether to yield the title compound 7 (540 mg, 54%) as a yellow oil. Rf = 0.34 (10% EtOAc in petroleum ether). HRMS (ESI): m/z calcd. for C20H28NO3: 330.2069, found: 330.2068 [M + H]+ and m/z calcd. for C20H27NO3Na: 352.1889, found: 352.1887 [M + Na]+. νmax (NaCl)/cm–1 1732 (ester, C=O), 1716 (ketone, C=O). 1H NMR (500 MHz, CDCl3): δ (ppm) = 1.28 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.52- 1.62 (m, 1H, H7eq), 1.83 (quin, J = 7.2 Hz, 1H, NCH2CH2CH2Ph), 2.05–2.16 (m, 2H, H6ax and H6eq), 2.21–2.29 (m, 1H, H8eq), 2.36 (t, J = 7.0 Hz, 2H, NCH2CH2CH2Ph), 2.45–2.49 (m, 1H, H5), 2.51–2.61 (m, 2H, H4ax and H8ax), 2.7 (t, J = 7.7 Hz, 2H, NCH2CH2CH2Ph), 2.84–2.97 (m, 2H, H2ax and H7ax), 3.15 (dt, J = 11.1, 2.4 Hz, 1H, H4eq), 3.21 (dd, J = 11.4, 2.3 Hz, 1 H, H2eq), 4.21 (q, J = 7.1 Hz, 2H, OCH2CH3), 7.16–7.22, 7.26–7.32 (m, 5H, NCH2CH2CH2Ph). 13C NMR (125 MHz, CDCl3): δ (ppm) = 14.1 (OCH2CH3), 20.6 (C7), 29.1 (N CH2CH2CH2Ph), 33.5 (NCH2CH2CH2Ph) 34.2 (C6), 36.8 (C8), 47.2 (C5), 56.4 (NCH2CH2CH2Ph), 58.8 (C1), 60.4 (C4), 61.1 (OCH2CH3), 62.0 (C2), 125.8 (C4 arom), 128.37, 128.40 (C2 arom, C6 arom, C3 arom and C5 arom), 142.0 (C1 arom), 171.1 (ester), 212.5 (C9).
Ethyl 3-(4-Phenylbutyl)-9-oxo-3-azabicyclo[3.3.1]nonane-1-carboxylate (8)
A solution of ethyl cyclohexanone-2-carboxylate (2.91 mmol, 0.476 mL, 95%), 2.2 equiv of formaldehyde (5.83 mmol, 0.425 mL, 38% aq v/v), and 1.1 equiv of 4-phenylbutylamine (3.205 mmol, 0.52 mL, 98%) in ethanol (20 mL) was heated under reflux for 3 h under N2. Then the solution was cooled and concentrated under vacuum and purified by column chromatography using 10% EtOAc in petroleum ether to yield the title compound 8 (600 mg, 60%) as a yellow oil. Rf = 0.35 (10% EtOAc in petroleum ether). HRMS (ESI): m/z calcd. for C21H30NO3: 344.2226, found: 344.2227 [M + H]+ and m/z calcd. for C21H29NO3Na: 366.2045, found: 366.2044 [M + Na]+. νmax (NaCl)/cm–1 1733 (ester, C=O), 1717 (ketone, C=O). 1H NMR (500 MHz, CDCl3): δ (ppm) = 1.28 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.50- 1.68 (m, 3H, H7eq and NCH2CH2CH2CH2Ph), 1.66–1.74 (m, 2H, NCH2CH2CH2CH2Ph), 2.02–2.16 (m, 2H, H6ax and H6eq), 2.19–2.25 (m, 1H, H8eq), 2.35 (td, J = 7.0, 1.4 Hz, 2H, NCH2CH2CH2CH2Ph), 2.42–2.46 (m, 1H, H5), 2.49–2.57 (m, 2H, H4ax and H8ax), 2.65 (t, J = 7.6 Hz, 2H, NCH2CH2CH2CH2Ph), 2.80–2.88 (m, 1H, H7ax), 2.91 (dd, J = 11.5, 2.0 Hz, 1H, H2ax) 3.10 (dt, J = 11.2, 2.4 Hz, 1 H, H4eq), 3.17 (dd, J = 11.4, 2.3 Hz, 1H, H2eq), 4.20 (q, J = 7.1 Hz, 2H, OCH2CH3), 7.16–7.21, 7.26–7.31 (m, 5H, NCH2CH2CH2CH2Ph). 13C NMR (125 MHz, CDCl3): δ (ppm) = 14.1 (OCH2CH3), 20.5 (C7), 26.7 (NCH2CH2CH2CH2Ph), 29.0 (NCH2CH2CH2CH2Ph), 34.1 (C6), 35.6 (NCH2CH2CH2CH2Ph), 36.8 (C8), 47.2 (C5), 56.8 (NCH2CH2CH2CH2Ph), 58.8 (C1), 60.4 (C4), 61.0 (OCH2CH3), 62.0 (C2), 125.7 (C4 arom), 128.26 (C2 arom and C6 arom), 128.31 (C3 arom and C5 arom), 142.4 (C1 arom), 171.1 (ester), 212.6 (C9).
(9R)-3-Ethyl-1-hydroxymethyl-3-azabicyclo[3.3.1]nonan-9-ol (9)
LiAlH4 (1.756 mmol, 66.6 mg) was added to a solution of cyclohexanone 4 (0.878 mmol, 210 mg) (which was dried under high vacuum for 24 h) in anhydrous THF (5 mL), and the reaction stirred for 7 h at 19 °C under N2. Then the mixture was quenched with 66 μL of water, followed by 66 μL of sodium hydroxide solution (15%w/v) and then 200 μL water. The resulting mixture was stirred with anhydrous magnesium sulfate for 15 min and filtered over Celite. The filtrate was concentrated under vacuum and purified over column chromatography with 5–20% MeOH in DCM to yield the title compound 9 (60 mg, 34%) as a yellow oil. Rf = 0.27 (20% MeOH in DCM). HRMS (ESI): m/z calcd. for C11H22NO2: 200.1651, found: 200.1648 [M + H]+ and m/z calcd. for C11H21NO2Na: 222.1470, found: 222.1488 [M + Na]+. νmax (NaCl)/cm–1 3413 (OH). 1H NMR (500 MHz, CDCl3): δ (ppm) = 1.03 (t, J = 7.2 Hz, 3H, NCH2CH3), 1.26 (dd, J = 13.0, 5.6 Hz, 1H, H8eq), 1.43–1.54 (m, 2H, H7eq and H6eq), 1.80–2.02 (m, 4H, H8ax, H6ax, H5 and H2ax), 2.18 (dt, J = 11.1, 2.4 Hz, 1H, H4ax), 2.20–2.28 (m, 2H, NCH2CH3), 2.52–2.61 (m, 1H, H7ax), 2.63 (dd, J = 11.0, 1.5 Hz, 1H, H2eq), 2.66–2.93 (br, 2H, 9-OH and 1 × OH), 2.96 (dt, J = 11.1, 2.1 Hz, 1H, H4eq), 3.35 (d, J = 10.8, 1H, CHaHbOH), 3.39 (d, J = 10.8, 1H, CHaHbOH), 3.70 (d, J = 3.6 Hz, 1H, 9-H). 13C NMR (125 MHz, CDCl3): δ (ppm) = 12.7 (NCH2CH3), 20.5 (C7), 23.9 (C6), 26.5 (C8), 36.1 (C5), 38.1 (C1), 52.3 (NCH2CH3), 58.3 (C4), 60.4 (C2), 70.9 (C1), 75.1 (C9).
2-Acetamidobenzoic Acid (11)
Anthranilic acid (28 mmol, 3.92 g, 98%) was heated under reflux with 5 equiv of acetic anhydride (140 mmol, 13.23 mL) and 1 equiv of anhydrous triethylamine (28 mmol, 3.94 mL, 99%) in THF (20 mL) under nitrogen for 4 h. The reaction mixture was cooled to 19 °C and then in an ice bath, then 20 mL of 1 M aq. HCl was added gradually while the reaction mixture was on ice. The precipitate was filtered and washed with ice-cold water. The product was recrystallized from water and ethanol to yield the title compound 11 (4.2 g, 84%) as pale brown crystals. Rf = 0.42 (10% MeOH in DCM). HRMS (ESI): m/z calcd. for C9H8NO3: 178.0504, found: 178.0505 [M – H]− and m/z calcd. for C10H10NO5: 224.0559, found: 224.0606 [M + HCOO]−. 1H NMR (500 MHz, CD3OD): δ (ppm) = 2.18 (s, 3H, COCH3), 7.11 (td, J = 7.7, 1.2 Hz, 1H, H5 arom), 7.52 (ddd, J = 8.7, 7.7, 1.7 Hz, 1H, H4 arom), 8.05 (dd, J = 7.7, 1.7 Hz, 1H, H6 arom), 8.52 (d, J = 8.4 Hz, 1H, H3 arom). 13C NMR (125 MHz, CD3OD): δ (ppm) = 25.1 (COCH3), 117.4 (C1 arom), 121.4 (C3 arom), 123.9 (C5 arom), 132.5 (C6 arom), 135.1 (C4 arom), 142.3 (C2 arom), 171.24 (COOH), 171.39 (NHCOCH3).
2-(3-Methyl-2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)benzoic Acid (12)
Neat anthranilic acid (21.6 mmol, 3.02 g, 98%) was stirred with 1 equiv of citraconic anhydride (21.6 mmol, 1.98 mL, 98%) at 140 °C for 24 h under nitrogen then cooled to 19 °C. After that, the crude mixture was dissolved in EtOAc (30 mL). The organic layer was washed sequentially with 1 M HCl (2 × 20 mL), water (1 × 20 mL), and brine (1 × 20 mL). The organic layer was dried (MgSO4) and filtered, and the filtrate was concentrated under vacuum and purified over column chromatography with 10% MeOH in DCM to yield the title compound 12 (4.0 g, 80%) as a brownish yellow powder. Rf = 0.32 (10% MeOH in DCM). HRMS (ESI): m/z calcd. for C12H8NO4: 230.0453, found: 230.0458 [M – H]− and m/z calcd. for C13H10NO6: 276.0508, found: 276.0528 [M + HCOO]−. 1H NMR (500 MHz, CDCl3): δ (ppm) = 2.18 (d, J = 1.7 Hz, 3H, 5′ CH3), 6.51 (d, J = 1.9 Hz, 1H, H3′), 7.32 (dd, J = 7.9, 1.2 Hz, 1H, H3 arom), 7.52 (td, J = 7.7, 1.2 Hz, 1H, H5 arom), 7.69 (td, J = 7.7, 1.6 Hz, 1H, H4 arom), 8.16 (dd, J = 7.9, 1.5 Hz, 1H, H6 arom). 13C NMR (125 MHz, CDCl3): δ (ppm) = 11.3 (5′, CH3), 127.20 (C1 arom), 128.05 (C3′), 129.15 (C5 arom), 130.54 (C3 arom), 132.16 (C2 arom), 132.44 (C6 arom), 134.25 (C4 arom), 146.4 (C2′), 169.84 (COOH), 170.20 (C4′), 170.87 (C1′).
(S)-2-(3-Methyl-2,5-dioxopyrrolidin-1-yl)benzoic Acid (13)
(2S,4S)-1-Boc-4-diphenylphosphino-2-(diphenylphosphinomethyl)pyrrolidine (BPPM) (0.649 mmol (5 mol %), 359 mg) and rhodium cyclooctadiene chloride dimer (Rh(COD)Cl)2 (0.649 mmol (5 mol %), 326 mg, 98%) were stirred together in anhydrous toluene (10 mL) under nitrogen gas for 30 min. Then the flask was vacuumed, and hydrogen was introduced. Compound 12 (0.01298 mol, 3 g) was dissolved in anhydrous methanol (10 mL) and added to the mixture. The reaction was monitored by TLC and stopped after 24 h. The mixture was concentrated under vacuum and purified over column chromatography with 10% MeOH in DCM to yield the title compound 13 (2.9 g, 95%) as a brownish yellow powder. Rf = 0.31 (10% MeOH in DCM). [α]D −12.0° (c 1.0, CHCl3). HRMS (ESI): m/z calcd. for C12H10NO4: 232.0610, found: 232.0613 [M – H]−. 1H NMR (500 MHz, CDCl3): δ (ppm) = 1.44 (d, J = 6.8 Hz, 3H, 5′ CH3), 2.53 (d, J = 17.9 Hz, 1H, H3′A), 3.01–3.16 (m, 2H, H2′ and H3′ B), 7.27 (d, J = 7.7 Hz, 1H, H3 arom), 7.54 (t, J = 7.7 Hz,1H, H5 arom), 7.70 (t, J = 7.7 Hz, 1H, H4 arom), 8.19 (d, J = 7.7 Hz, 1H, H6 arom). 13C NMR (125 MHz, CDCl3): δ (ppm) = 16.5 (5′), 35.13 and 35.5 (3′), 37.0 (2′), 125.5 (C1 arom), 129.5 (C5 arom), 129.9 (C3 arom), 132.5 (C6 arom), 132.9 (C2 arom), 134.4 (C4 arom), 146.4 (C2′), 169.2 and 169.4 (COOH), 176.0 and 176.1 (C4′), 179.9 and 180.0 (C1′).
((9R)-9-Hydroxy-3-methyl-3-azabicyclo[3.3.1]nonan-1-yl)methyl 2-((S)-3-Methyl-2,5-dioxopyrrolidin-1-yl)benzoate (14)
Compound 3 (0.89 mmol, 200 mg) was reduced using LAH (1.78 mmol, 84.2 mg) as described for compound 9. The crude product was used for the esterification step without purification. Compound 13 (0.189 mmol, 44 mg) was stirred with DCC (0.189 mmol, 39.4 mg, 99%) and DMAP (0.0189 mmol, 2.3 mg, 99%) in anhydrous acetonitrile under nitrogen gas at 40 °C for 20 min, and then the crude amino alcohol (35 mg) was added. The reaction was monitored by TLC and stopped after 24 h. The mixture was concentrated under vacuum and purified over column chromatography with 5% MeOH in DCM to yield the title compound 14 (18 mg, 24%) as a yellow oil. Rf = 0.4 (5% MeOH in DCM). HRMS (ESI): m/z calcd. for C22H29N2O5: 401.2077, found: 401.2073 [M + H]+. 1H NMR (500 MHz, CD3OD): δ (ppm) = 1.41 (d, J = 6.7 Hz, 3H, 5‴), 1.44–1.56 (m, 3H, H6eq, H7eq, H8eq), 1.72–1.81 (m, 1H, H8ax), 1.86 (br s, 1H, H5), 1.99–2.07 (m, 1H, H6ax), 2.14–2.21 (m, 4H, N–CH3, H2ax), 2.34 (d, J = 11.4,1H, H4ax), 2.45–2.63 (m, 2H, H7ax, H3‴A), 2.85–2.91 (m, 1H, H2eq), 2.96 (d, J = 11.4 Hz, 1H, H4eq), 3.04–3.15 (m, 2H, H2‴ and H3‴B), 3.59–3.66 (m, 1H, H9), 3.96–4.11 (m, 2H, H1′ A and B), 7.35 (d, J = 7.4 Hz, 1H, H3″), 7.61 (d, J = 7.4 Hz, 1H, H5″), 7.73 (d, J = 7.4 Hz, 1H, H4″), 8.12 (d, J = 7.4 Hz, 1H, H6″). 13C NMR (125 MHz, CDCl3): δ (ppm) = 16.2 (5‴), 21.5 (C7), 25.0 (C6), 28.0 (C8), 36.18 (C2‴), 37.33 (C5), 38.00 (C3‴), 39.51 (C1), 46.4 (NCH3), 62.30 (C4), 64.44 (C2), 71.78 (C9), 71.80 (C1′), 128.99 (C1″), 130.42 (C5″), 131.10 (C3″), 132.07 (C6″), 133.72 (C2″), 134.50 (C4″), 165.6 (ester), 173.1 (C4‴), 181.8 (C1‴).
((9R)-3-Ethyl-9-hydroxy-3-azabicyclo[3.3.1]nonan-1-yl)methyl 2-((S)-3-methyl-2,5-dioxopyrrolidin-1-yl)benzoate (15)
Compound 13 (0.778 mmol, 181 mg) was stirred with DCC (0.778 mmol, 162 mg, 99%) and DMAP (0.0778 mmol, 9.6 mg, 99%) in anhydrous acetonitrile under nitrogen gas at 40 °C for 20 min, and then compound 9 (155 mg) was added. The reaction was monitored by TLC and stopped after 24 h. The mixture was concentrated under vacuum and purified over column chromatography with 5% MeOH in DCM to yield the title compound 15 (130 mg, 40%) as a yellow oil. Rf = 0.44 (5% MeOH in DCM). HRMS (ESI): m/z calcd. for C23H31N2O5: 415.2233, found: 415.2233 [M + H]+. 1H NMR (500 MHz, CD3OD): δ (ppm) = 1.19 (m, 3H, NCH2CH3), 1.41 (d, J = 7.0 Hz, 3H, 5‴), 1.44–1.55 (m, 3H, H6eq, H7eq, H8eq), 1.66–1.74 (m, 1H, H8ax), 1.80–1.87 (m, 1H, H5), 1.97–2.05 (m, 1H, H6ax), 2.07–2.12 (m, 1H, H2ax), 2.14–2.29 (m, 3H, H4ax and NCH2CH3), 2.46–2.57 (m, 1H, H3‴A), 2.58–2.69 (m, 1H, H7ax), 2.89–2.96 (m, 1H, H2eq), 3.00–3.06 (m,1H, H4eq), 3.05–3.14 (m, 2H, H2‴ and H3‴B), 3.76 (br s, 1H, H9), 4.00–4.16 (m, 2H, H1′ A and B), 7.36 (d, J = 7.8 Hz, 1H, H3″), 7.61 (d, J = 7.8 Hz, 1H, H5″), 7.74 (d, J = 7.8 Hz, 1H, H4″), 8.13 (d, J = 7.8 Hz, 1H, H6″). 13C NMR (125 MHz, CDCl3): δ (ppm) = 11.3 (NCH2CH3), 15.8 (5‴), 21.3 (C7), 24.8 (C6), 27.9 (C8), 35.7 (C2‴), 37.18 (C5), 37.67 (C3‴), 39.5 (C1), 53.1 (NCH2CH3), 59.4 (C4), 61.7 (C2), 70.7 (C1′), 72.0 (C9), 128.79 (C1″), 130.50 (C5″), 131.21 (C3″), 132.10 (C6″), 133.82 (C2″), 134.62 (C4″), 165.4 (ester), 175.5 (C4‴), 182.1 (C1‴).
((9R)-3-Benzyl-9-hydroxy-3-azabicyclo[3.3.1]nonan-1-yl)methyl 2-((S)-3-Methyl-2,5-dioxopyrrolidin-1-yl)benzoate (16)
Compound 5 (1.66 mmol, 500 mg) was reduced using LAH (4.15 mmol, 157.5 mg) as described for compound 9. The crude product was used for the esterification step without purification. Compound 13 (0.593 mmol, 138 mg) was stirred with DCC (0.593 mmol, 123.6 mg, 99%) and DMAP (0.0593 mmol, 7.3 mg, 99%) in anhydrous acetonitrile under nitrogen gas at 40 °C for 20 min, and then the crude amino alcohol (155 mg) was added. The reaction was monitored by TLC and stopped after 24 h. The mixture was concentrated under vacuum and purified over column chromatography with 5% MeOH in DCM to yield the title compound 16 (140 mg, 50%) as a yellow oil. Rf = 0.48 (5% MeOH in DCM). HRMS (ESI): m/z calcd. for C28H33N2O5: 477.2390, found: 477.2385 [M + H]+. 1H NMR (500 MHz, CD3OD): δ (ppm) = 1.37–1.43 (m, 3H, 5‴), 1.42–1.53 (m, 3H, H6eq, H7eq, H8eq), 1.67–1.81 (m, 1H, H8ax), 1.82–1.86 (m, 1H, H5), 1.95–2.06 (m, 1H, H6ax), 2.08–2.15 (m, 1H, H2ax), 2.21–2.30 (m,1H, H4ax), 2.43–2.63 (m, 1H, H3‴A), 2.72–2.83 (m, 1H, H7ax), 2.83–2.91 (m, 1H, H2eq), 2.90–2.96 (m,1H, H4eq), 3.01–3.14 (m, 2H, H2‴ and H3‴B), 3.34–3.41 (m, 2H, NCH2Ph), 3.60–3.68 (m, 1H, H9), 3.95–4.13 (m, 2H, H1′ A and B), 7.18–7.30 (m, 5H, NCH2Ph), 7.31–7.35 (m, 1H, H3″), 7.56 (td, J = 7.7, 1.3 Hz, 1H, H5″), 7.72 (td, J = 7.7, 1.5 Hz, 1H, H4″), 7.99 (dd, J = 7.7, 1.6 Hz, 1H, H6″). 13C NMR (125 MHz, CDCl3): δ (ppm) = 16.4 (5‴), 22.1 (C7), 25.1 (C6), 28.2 (C8), 36.31 (C2‴), 37.73 (C5), 38.16 (C3‴), 39.54 (C1), 60.1 (C4), 62.40 (C2), 64.38 (NCH2Ph), 71.20 (C1′), 72.63 (C9), 127.92 (C4 Ph), 128.87 (C1″), 129.26, 129.75 (C2 Ph, C3 Ph, C5 Ph, C6 Ph), 130.48 (C5″), 131.12 (C3″), 132.00 (C6″), 133.97 (C2″), 134.48 (C4″), 140.5 (C1 Ph), 165.7 (ester), 174.9 (C4‴), 181.6 (C1‴).
((9R)-9-Hydroxy-3-(2-phenethyl)-3-azabicyclo[3.3.1]nonan-1-yl)methyl 2-((S)-3-Methyl-2,5-dioxopyrrolidin-1-yl)benzoate (17)
Compound 6 (0.634 mmol, 200 mg) was reduced using LAH (1.585 mmol, 60.2 mg) as described for compound 9. The crude product was used for the esterification step without purification. Compound 13 (0.2 mmol, 46.6 mg) was stirred with DCC (0.2 mmol, 41.6 mg, 99%) and DMAP (0.02 mmol, 2.5 mg, 99%) in anhydrous acetonitrile under nitrogen gas at 40 °C for 20 min, and then the crude amino alcohol (55 mg) was added. The reaction monitored by TLC and stopped after 24 h. The mixture was concentrated under vacuum and purified over column chromatography with 5% MeOH in DCM to yield the title compound 17 (25 mg, 26%) as a yellow oil. Rf = 0.54 (5% MeOH in DCM). HRMS (ESI): m/z calcd. for C29H35N2O5: 491.2546, found: 491.2549 [M + H]+. 1H NMR (500 MHz, CD3OD): δ (ppm) = 1.37–1.52 (m, 6H, H5‴, H6eq, H7eq, H8eq), 1.67–1.80 (m, 1H, H8ax), 1.91 (br s, 1H, H5), 1.97–2.08 (m, 1H, H6ax), 2.09–2.18 (m, 1H, H7ax), 2.34–2.42 (m, 1H, H2ax), 2.45–2.60 (m, 2H, H4ax and H3‴A), 2.63–2.73 (m, 2H, NCH2CH2Ph), 2.80–2.89 (m, 2H, NCH2CH2Ph), 3.02–3.16 (m, 2H, H2‴ and H3‴B), 3.18–3.28 (m, 2H, H2eq and H4eq), 3.63–3.72 (m, 1H, H9), 4.00–4.12 (m, 2H, H1′ A and B), 7.12–7.30 (m, 5H, NCH2CH2Ph), 7.35 (d, J = 7.5 Hz, 1H, H3″), 7.61 (td, J = 7.5, 1.3 Hz, 1H, H5″), 7.74 (td, J = 7.5, 1.5 Hz, 1H, H4″), 8.13 (dd, J = 7.5, 1.6 Hz, 1H, H6″). 13C NMR (125 MHz, CDCl3): δ (ppm) = 16.1 (5‴), 20.6 (C7), 24.3 (C6), 27.2 (C8), 33.45 (NCH2CH2Ph), 35.76 (C2‴), 36.62 (C5), 37.48 (C3‴), 39.58 (C1), 59.46 (C4), 60.81 (NCH2CH2Ph), 61.30 (C2), 70.00 (C1′), 70.85 (C9), 127.07 (C4 Ph), 128.96 (C1″), 129.29, 129.73 (C2 Ph, C3 Ph, C5 Ph, C6 Ph), 130.50 (C5″), 131.04 (C3″), 132.15 (C6″), 133.26 (C2″), 134.47 (C4″), 139.67 (C1 Ph), 164.5 (ester), 177.9 (C4‴), 180.8 (C1‴).
((9R)-9-Hydroxy-3-(3-phenylpropyl)-3-azabicyclo[3.3.1]nonan-1-yl)methyl 2-((S)-3-Methyl-2,5-dioxopyrrolidin-1-yl)benzoate (18)
Compound 7 (0.91 mmol, 300 mg) was reduced using LAH (2.28 mmol, 86.3 mg) as described for compound 9. The crude product was used for the esterification step without purification. Compound 13 (0.17 mmol, 40.3 mg) was stirred with DCC (0.17 mmol, 36 mg, 99%) and DMAP (0.017 mmol, 2.1 mg, 99%) in anhydrous acetonitrile under nitrogen gas at 40 °C for 20 min, and then the crude amino alcohol (50 mg) was added. The reaction was monitored by TLC and stopped after 24 h. The mixture was concentrated under vacuum and purified over column chromatography with 5% MeOH in DCM to yield the title compound 18 (26 mg, 30%) as a yellow oil. Rf = 0.57 (5% MeOH in DCM). HRMS (ESI): m/z calcd. for C30H37N2O5: 505.2703, found: 505.2701 [M + H]+. 1H NMR (500 MHz, CD3OD): δ (ppm) = 1.41 (d, J = 7.5 Hz, H5‴), 1.49–1.74 (m, 3H, H6eq, H7eq, H8eq), 1.66–1.88 (m, 1H, H8ax), 1.89–1.96 (m, 1H, H5), 1.96–2.02 (m, 2H, NCH2CH2CH2Ph), 2.04–2.12 (m, 1H, H6ax), 2.13–2.26 (m, 1H, H7ax), 2.29–2.39 (m, 1H, H2ax), 2.42–2.55 (m, 2H, H4ax and H3‴A), 2.57–2.63 (m, 2H, NCH2CH2CH2Ph), 2.64–2.74 (m, 2H, NCH2CH2CH2Ph), 3.01–3.06 (m, 1H, H2eq), 3.05–3.16 (m, 3H, H4eq, H2‴ and H3‴B), 3.63–3.68 (m, 1H, H9), 4.04–4.16 (m, 2H, H1′ A and B), 7.12–7.30 (m, 5H, NCH2CH2CH2Ph), 7.34 (dd, J = 7.7, 1.3 Hz, 1H, H3″), 7.58 (td, J = 7.7, 1.3 Hz, 1H, H5″), 7.72 (td, J = 7.7, 1.6 Hz, 1H, H4″), 8.10 (dd, J = 7.7, 1.6 Hz, 1H, H6″). 13C NMR (125 MHz, CDCl3): δ (ppm) = 16.5 (5‴), 21.0 (C7), 24.3 (C6), 27.6 (C8), 32.0 (NCH2CH2CH2Ph), 34.0 (NCH2CH2CH2Ph), 36.64 (C2‴), 36.72 (C5), 38.10 (C3‴), 39.61 (C1), 59.88 (C4), 60.53 (NCH2CH2CH2Ph), 61.42 (C2), 70.57 (C1′), 71.67 (C9), 126.98 (C4 Ph), 128.87 (C1″), 129.33, 129.56 (C2 Ph, C3 Ph, C5 Ph, C6 Ph), 130.43 (C5″), 131.06 (C3″), 132.36 (C6″), 133.57 (C2″), 134.43 (C4″), 143.0 (C1 Ph), 166.4 (ester), 177.4 (C4‴), 182.0 (C1‴).
((9R)-9-Hydroxy-3-(4-phenylbutyl)-3-azabicyclo[3.3.1]nonan-1-yl)methyl 2-((S)-3-Methyl-2,5-dioxopyrrolidin-1-yl)benzoate (19)
Compound 8 (0.58 mmol, 200 mg) was reduced using LAH (1.54 mmol, 55 mg) as described for compound 9. The crude product was used for the esterification step without purification. Compound 13 (0.158 mmol, 36.8 mg) was stirred with DCC (0.158 mmol, 32.9 mg, 99%) and DMAP (0.0158 mmol, 1.9 mg, 99%) in anhydrous acetonitrile under nitrogen gas at 40 °C for 20 min, and then the crude amino alcohol (48 mg) was added. The reaction was monitored by TLC and stopped after 24 h. The mixture was concentrated under vacuum and purified over column chromatography with 5% MeOH in DCM to yield the title compound 19 (20 mg, 25%) as a yellow oil. Rf = 0.61 (5% MeOH in DCM). HRMS (ESI): m/z calcd. for C31H39N2O5: 519.2859, found: 519.2851 [M + H]+. 1H NMR (500 MHz, CD3OD): δ (ppm) = 1.41 (d, J = 7.1 Hz, H5‴), 1.46–1.74 (m, 3H, H6eq, H7eq, H8eq), 1.79–1.97 (m, 4H, H5, H8ax and NCH2CH2CH2CH2Ph), 1.98–2.10 (m, 3H, H6ax and NCH2CH2CH2CH2Ph), 2.17–2.29 (m, 1H, H7ax), 2.33–2.42 (m, 1H, H2ax), 2.43–2.52 (m, 2H, H4ax and H3‴A), 2.54–2.61 (m, 2H, NCH2CH2CH2CH2Ph), 2.62–2.77 (m, 2H, NCH2CH2CH2CH2Ph), 2.99–3.05 (m, 1H, H2eq), 3.06–3.16 (m, 3H, H4eq, H2‴ and H3‴B), 3.62–3.66 (m, 1H, H9), 4.01–4.18 (m, 2H, H1′ A and B), 7.10–7.30 (m, 5H, NCH2CH2CH2Ph), 7.34 (dd, J = 7.9, 1.3 Hz, 1H, H3″), 7.58 (td, J = 7.9, 1.3 Hz, 1H, H5″), 7.72 (td, J = 7.9, 1.6 Hz, 1H, H4″), 8.10 (dd, J = 7.9, 1.6 Hz, 1H, H6″). 13C NMR (125 MHz, CDCl3): δ (ppm) = 16.5 (5‴), 21.1 (C7), 24.4 (C6), 28.8 (C8), 32.0 (NCH2CH2CH2CH2Ph), 34.71 (NCH2CH2CH2CH2Ph), 36.33 (C2‴), 36.59 (NCH2CH2CH2CH2Ph), 37.19 (C5), 38.05 (C3‴), 39.54 (C1), 60.00 (C4), 60.48 (NCH2CH2CH2CH2Ph), 61.26 (C2), 70.35 (C1′), 71.70 (C9), 127.07 (C4 Ph), 128.50 (C1″), 129.35, 129.50 (C2 Ph, C3 Ph, C5 Ph, C6 Ph), 130.45 (C5″), 131.05 (C3″), 132.35 (C6″), 133.35 (C2″), 134.47 (C4″), 143.0 (C1 Ph), 166.5 (ester), 177.4 (C4‴), 181.9 (C1‴).
((9R)-9-Hydroxy-3-methyl-3-azabicyclo[3.3.1]nonan-1-yl)methyl 2-Acetamidobenzoate (20)
Compound 3 (0.89 mmol, 200 mg) was reduced using LAH (1.78 mmol, 84.2 mg) as described for compound 9. The crude product was used for the esterification step without purification. Compound 11 (0.189 mmol, 33.8 mg) was stirred with DCC (0.189 mmol, 39.4 mg, 99%) and DMAP (0.0189 mmol, 2.3 mg, 99%) in anhydrous acetonitrile under nitrogen gas at 40 °C for 20 min, and then the crude amino alcohol (35 mg) was added. The reaction was monitored by TLC and stopped after 24 h. The mixture was concentrated under vacuum and purified over column chromatography with 5% MeOH in DCM to yield the title compound 20 (29 mg, 45%) as a yellow oil. Rf = 0.6 (5% MeOH in DCM). HRMS (ESI): m/z calcd. for C19H27N2O4: 347.1971, found: 347.1969 [M + H]+. 1H NMR (500 MHz, CD3OD): δ (ppm) = 1.43–1.59 (m, 3H, H6eq, H7eq, H8eq), 1.78–1.83 (m, 1H, H8ax), 1.86 (br s, 1H, H5), 1.99–2.08 (m, 1H, H6ax), 2.18 (s, 3H, N–CH3), 2.19–2.22 (m, 4H, NHCOCH3, H2ax), 2.32 (d, J = 11.4 Hz,1H, H4ax), 2.53–2.61 (m, 1H, H7ax), 2.92 (d, J = 11.3 Hz, 1H, H2eq), 2.95 (d, J = 11.4 Hz, 1H, H4eq), 3.67 (d, J = 3.8 Hz, 1H, H9), 4.08 (d, J = 11.0 Hz, 1H, H1′A), 4.14 (d, J = 11.0 Hz, 1H, H1′B), 7.19 (td, J = 7.7, 1.3 Hz, 1H, H5″), 7.58 (ddd, J = 8.6, 7.7, 1.3 Hz, 1H, H4″), 8.05 (dd, J = 7.7, 1.3 Hz, 1H, H6″), 8.46 (d, J = 8.6 Hz, 1H, H3″). 13C NMR (125 MHz, CDCl3): δ (ppm) = 21.3 (C7), 24.48 (NHCOCH3), 24.77 (C6), 28.0 (C8), 37.35 (C5), 39.44 (C1), 46.1 (NCH3), 61.9 (C4), 64.2 (C2), 71.09 (C1′), 72.00 (C9), 116.6 (C1″), 121.5 (C3″), 124.1 (C5″), 131.3 (C6″), 134.8 (C4″), 140.4 (C2″), 167.5 (ester), 169.9 (amide).
((9R)-3-Ethyl-9-hydroxy-3-azabicyclo[3.3.1]nonan-1-yl)methyl 2-Acetamidobenzoate (21)
Compound 11 (0.8 mmol, 143.9 mg) was stirred with DCC (0.8 mmol, 167 mg, 99%) and DMAP (0.08 mmol, 9.9 mg, 99%) in anhydrous acetonitrile under nitrogen gas at 40 °C for 20 min, and then compound 9 (160 mg) was added. The reaction was monitored by TLC and stopped after 24 h. The mixture was concentrated under vacuum and purified over column chromatography with 5% MeOH in DCM to yield the title compound 21 (130 mg, 45%) as a yellow oil. Rf = 0.64 (5% MeOH in DCM). HRMS (ESI): m/z calcd. for C20H29N2O4: 361.2127, found: 361.2122 [M + H]+. 1H NMR (500 MHz, CD3OD): δ (ppm) = 1.20 (t, J = 7.2 Hz, 3H, NCH2CH3), 1.52–1.66 (m, 3H, H6eq, H7eq, H8eq), 1.81–1.92 (m, 1H, H8ax), 1.99–2.16 (m, 2H, H5 and H6ax), 2.20 (s, 3H, NHCOCH3), 2.24–2.34 (m, 1H, H7ax), 2.58–2.80 (m, 4H, H2ax, H4ax and NCH2CH3), 3.27–3.36 (m, 2H, H2eq and H4eq), 3.82 (d, J = 3.5 Hz, 1H, H9), 4.09 (d, J = 11.2 Hz, 1H, H1′A), 4.21 (d, J = 11.2 Hz, 1H, H1′B), 7.20 (t, J = 7.7 Hz, 1H, H5″), 7.58 (ddd, J = 8.7, 7.7, 1.7 Hz, 1H, H4″), 8.07 (dd, J = 7.7, 1.3 Hz, 1H, H6″), 8.42 (d, J = 8.7 Hz, 1H, H3″). 13C NMR (125 MHz, CDCl3): δ (ppm) = 11.2 (NCH2CH3), 20.5 (C7), 23.76 (C6), 24.53 (NHCOCH3), 26.9 (C8), 36.4 (C5), 39.4 (C1), 54.3 (NCH2CH3), 58.90 (C4), 60.63 (C2), 69.88 (C9), 70.16 (C1′), 118.2 (C1″), 121.8 (C3″), 123.9 (C5″), 131.6 (C6″), 135.0 (C4″), 141.5 (C2″), 169.0 (ester), 171.5 (amide).
Electrophysiological Methods
Oocyte expression studies employed the human α7 nAChR subunit in plasmid pSP64GL.32 Oocytes were isolated from adult female Xenopus laevis and defolliculated by treatment with collagenase (2.5 mg/mL; Gibco, ThermoFisher Scientific) in calcium-free Barth’s solution containing 88 mM NaCl, 2.4 mM NaHCO3, 1 mM KCl, 0.82 mM MgSO4, and 15 mM Tris, pH 7.5, as described previously.33 Heterologous expression was achieved by cytoplasmic injection of in vitro transcribed cRNA. Prior to in vitro synthesis of cRNA plasmid, cDNA was linearized by restriction enzyme digestion and purified with QIAQuik PCR purification kit (Qiagen). In vitro synthesis of cRNA was performed using mMessage mMachine SP6 transcription kit (ThermoFisher Scientific).
Oocytes were injected with approximately 9 ng of cRNA using a Drummond variable volume microinjector. After injection, oocytes were incubated at 14 °C in calcium-containing Barth’s solution (composition as above but with 0.77 mM CaCl2) supplemented with antibiotics (100 units/mL penicillin, 100 μg/mL streptomycin, 4 μg/mL kanamycin, and 50 μg/mL tetracycline). Experiments were performed on oocytes after 3 to 5 d of incubation. Oocytes were placed in a recording chamber and continuously perfused with a modified Ringer’s solution (115 mM NaCl, 2.5 mM KCl, 1.8 mM BaCl2, and 10 mM HEPES, pH 7.3) with a flow rate of approximately 15 mL/min. Two-electrode voltage-clamp recordings were performed using a Warner Instruments OC-725C amplifier (Harvard Apparatus) with the oocyte membrane potential held at −60 mV, as described previously.34,35 Application of compounds was controlled by LabChart software (AD Instruments) using a BPS-8 solenoid valve solution exchange system (ALA Scientific Inc.). Compounds were preapplied for 2 min before coapplication with EC50 concentration of agonist (100 μM ACh) and normalized to responses to the EC50 concentration of agonist in the absence of the compound on the same oocyte. Data are presented as mean ± SEM of at least three independent experiments, that were conducted on separate oocytes. For multiple comparisons, statistical significance was determined with an unpaired one-way analysis of variance (ANOVA).
Acknowledgments
We acknowledge Zarqa University, Jordan, for funding the studentship of Ashraf M. A. Qasem, and a BBSRC Industrial CASE PhD studentship associated with the London Interdisciplinary Doctoral Program (LIDo) and in collaboration with Syngenta [Grant BB/M009513/1] awarded to Victoria R. Sanders.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomedchemau.2c00057.
1H and 13C NMR spectra of a mixture of epimers from reduction of cyclohexanone 4, 13C NMR spectra of chiral 13, NOESY of analogues 14 and 21, expanded 1H NMR spectra at 1′ on 14, 15, 20, and 21, and HPLC traces for target compounds 14–21 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Galzi J. L.; Revah F.; Bessis A.; Changeux J.-P. Functional architecture of the nicotinic acetylcholine receptor: from electric organ to brain. Annu. Rev. Pharmacol. Toxicol. 1991, 31, 37–72. 10.1146/annurev.pa.31.040191.000345. [DOI] [PubMed] [Google Scholar]
- Changeux J.-P. The nicotinic acetylcholine receptor: A typical ‘Allosteric Machine’. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170174. 10.1098/rstb.2017.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar N. S.; Gotti C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacol. 2009, 56 (1), 237–246. 10.1016/j.neuropharm.2008.07.041. [DOI] [PubMed] [Google Scholar]
- Borroni V.; Barrantes F. J. Homomeric and heteromeric α7 nicotinic acetylcholine receptors in health and some Central Nervous System diseases. Membranes 2021, 11 (9), 664. 10.3390/membranes11090664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzidaki A.; Millar N. S. Allosteric modulation of nicotinic acetylcholine receptors. Biochem. Pharmacol. 2015, 97, 408–417. 10.1016/j.bcp.2015.07.028. [DOI] [PubMed] [Google Scholar]
- Wonnacott S.; Albuquerque E. X.; Bertrand D. MLA discriminates between nicotinic receptor subclasses. Methods Neurosciences 1993, 263–275. [Google Scholar]
- Shen Y.; Liang W. J.; Shi Y. N.; Kennelly E. J.; Zhao D. K. Structural diversity, bioactivities, and biosynthesis of natural diterpenoid alkaloids. Nat. Prod. Rep. 2020, 37 (6), 763–796. 10.1039/D0NP00002G. [DOI] [PubMed] [Google Scholar]
- Qasem A. M. A.; Zeng Z.; Rowan M. G.; Blagbrough I. S. Norditerpenoid alkaloids from Aconitum and Delphinium: Structural relevance in medicine, toxicology, and metabolism. Nat. Prod. Rep. 2022, 39, 460–473. 10.1039/D1NP00029B. [DOI] [PubMed] [Google Scholar]
- Panter K. E.; Manners G. D.; Stegelmeier B. L.; Lee S.; Gardner D. R.; Ralphs M. H.; Pfister J. A.; James L. F. Larkspur poisoning: Toxicology and alkaloid structure-activity relationships. Biochem. Systematics and Ecology 2002, 30 (2), 113–128. 10.1016/S0305-1978(01)00123-5. [DOI] [Google Scholar]
- Dobelis P.; Madl J. E.; Pfister J. A.; Manners G. D.; Walrond J. P. Effects of Delphinium alkaloids on neuromuscular transmission. J. Pharm. Exp. Therap. 1999, 291 (2), 538–546. [PubMed] [Google Scholar]
- Jennings K. R.; Brown D. G.; Wright D. P. Methyllycaconitine, a naturally occurring insecticide with a high affinity for the insect cholinergic receptor. Experientia 1986, 42 (6), 611–613. 10.1007/BF01955557. [DOI] [Google Scholar]
- Trigg W. J.; Hardick D. J.; Grangier G.; Wonnacott S.; Lewis T.; Rowan M. G.; Potter B. V. L.; Blagbrough I. S.. Selective probes for nicotinic acetylcholine receptors from substituted AE-bicyclic analogs of methyllycaconitine, Synthesis and Chemistry of Agrochemicals V Baker D. R., Fenyes J. G., Basarab G. S., Hunt D. A., Eds.; ACS Symposium Series; American Chemical Society: 1998; Vol. 686, pp 194–205.
- Manners G. D.; Panter K. E.; Pelletier S. W. Structure-activity relationships of norditerpenoid alkaloids occurring in toxic larkspur (delphinium) species. J. Nat. Prod. 1995, 58 (6), 863–869. 10.1021/np50120a007. [DOI] [PubMed] [Google Scholar]
- Hardick D. J.; Blagbrough I. S.; Cooper G.; Potter B. V. L.; Critchley T.; Wonnacott S. Nudicauline and elatine as potent norditerpenoid ligands at rat neuronal α-bungarotoxin binding sites: Importance of the 2-(methylsuccinimido)benzoyl moiety for neuronal nicotinic acetylcholine receptor binding. J. Med. Chem. 1996, 39 (24), 4860–4866. 10.1021/jm9604991. [DOI] [PubMed] [Google Scholar]
- Blagbrough I. S.; Coates P. A.; Hardick D. J.; Lewis T.; Rowan M. G.; Wonnacott S.; Potter B. V. L. Acylation of lycoctonine: semi-synthesis of inuline, delsemine analogues and methyllycaconitine. Tetrahedron Lett. 1994, 35, 8705–8708. 10.1016/S0040-4039(00)78477-2. [DOI] [Google Scholar]
- Hardick D. J.; Cooper G.; Scott-Ward T.; Blagbrough I. S.; Potter B. V. L.; Wonnacott S. Conversion of the sodium channel activator aconitine into a potent α7-selective nicotinic ligand. FEBS Lett. 1995, 365, 79–82. 10.1016/0014-5793(95)00426-A. [DOI] [PubMed] [Google Scholar]
- Bergmeier S. C.; Lapinsky D. J.; Free R. B.; McKay D. B. Ring E analogs of methyllycaconitine (MLA) as novel nicotinic antagonists. Bioorg. Med. Chem. Lett. 1999, 9 (15), 2263–2266. 10.1016/S0960-894X(99)00378-9. [DOI] [PubMed] [Google Scholar]
- Bryant D. L.; Free R. B.; Thomasy S. M.; Lapinsky D. J.; Ismail K. A.; McKay S. B.; Bergmeier S. C.; McKay D. B. Structure-activity studies with ring E analogues of methyllycaconitine on bovine adrenal α3β4* nicotinic receptors. Neuroscience Res. 2002, 42 (1), 57–63. 10.1016/S0168-0102(01)00304-2. [DOI] [PubMed] [Google Scholar]
- Coates P. A.; Blagbrough I. S.; Rowan M. G.; Potter B. V. L.; Pearson D. P. J.; Lewis T. Rapid and efficient entry to substituted 2-succinimidobenzoate-3-azabicyclo[3.3.1]nonanes: AE-bicyclic analogues of methyllycaconitine. Tetrahedron Lett. 1994, 35, 8709–8712. 10.1016/S0040-4039(00)78478-4. [DOI] [Google Scholar]
- Grangier G.; Trigg W. J.; Lewis T.; Rowan M. G.; Potter B. V. L.; Blagbrough I. S. Synthesis of C5-substituted AE-bicyclic analogues of lycoctonine, inuline and methyllycaconitine. Tetrahedron Lett. 1998, 39, 889–892. 10.1016/S0040-4039(97)10648-7. [DOI] [Google Scholar]
- Barker D.; Brimble M. A.; McLeod M. D. Synthesis of simple analogues of methyllycaconitine — an efficient method for the preparation of the N-substituted anthranilate pharmacophore. Tetrahedron 2004, 60 (28), 5953–5963. 10.1016/j.tet.2004.05.024. [DOI] [Google Scholar]
- Coates P. A.; Blagbrough I. S.; Hardick D. J.; Rowan M. G.; Wonnacott S.; Potter B. V. L. Rapid and efficient isolation of the nicotinic receptor antagonist methyllycaconitine from Delphinium: assignment of the methylsuccinimide absolute stereochemistry as S. Tetrahedron Lett. 1994, 35, 8701–8704. 10.1016/S0040-4039(00)78476-0. [DOI] [Google Scholar]
- Hardick D. J.; Blagbrough I. S.; Potter B. V. L. Isotopic enrichment by asymmetric deuteriation. An investigation of the synthesis of deuteriated (S)-(−)-methylsuccinic acids from itaconic acid. J. Am. Chem. Soc. 1996, 118 (25), 5897–5903. 10.1021/ja950597w. [DOI] [Google Scholar]
- Barker D.; Brimble M. A.; McLeod M. D. A convenient synthesis of 2-(3-methyl-2,5-dioxopyrrolidin-1-yl)benzoic acid. Synthesis 2003, 5, 656–658. 10.1055/s-2003-38065. [DOI] [Google Scholar]
- Ismail K. A.; Bergmeier S. C. Structure–activity studies with ring E analogues of methyllycaconitine. Synthesis and evaluation of enantiopure isomers of selective antagonist at the α3 nicotinic receptor. Eur. J. Med. Chem. 2002, 37, 469–474. 10.1016/S0223-5234(02)01353-3. [DOI] [PubMed] [Google Scholar]
- Zeng Z.; Qasem A. M. A.; Kociok-Köhn G.; Rowan M. G.; Blagbrough I. S. The 1α-hydroxy-A-rings of norditerpenoid alkaloids are twisted-boat conformers. RSC Adv. 2020, 10 (32), 18797–18805. 10.1039/D0RA03811C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood L. M.; Claridge T. D. W.. Introduction to Organic Spectroscopy; Oxford University Press: 1997; p 39. [Google Scholar]
- Fleming I.; Williams D. H.. Spectroscopic Methods in Organic Chemistry, 7th ed.; Springer: Basel, 2019; p 231, 10.1007/978-3-030-18252-6. [DOI] [Google Scholar]
- Bergmeier S. C.; Ismail K. A.; Arason K. M.; McKay S.; Bryant D. L.; McKay D. B. Structure activity studies of ring E analogues of methyllycaconitine. Part 2: Synthesis of antagonists to the α3β4* nicotinic acetylcholine receptors through modifications to the ester. Bioorg. Med. Chem. Lett. 2004, 14 (14), 3739–3742. 10.1016/j.bmcl.2004.05.001. [DOI] [PubMed] [Google Scholar]
- McKay D. B.; Chang C.; González-Cestari T. F.; McKay S. B.; El-Hajj R. A.; Bryant D. L.; Zhu M. X.; Swaan P. W.; Arason K. M.; Pulipaka A. B.; Orac C. M.; Bergmeier S. C. Analogs of methyllycaconitine as novel noncompetitive inhibitors of nicotinic receptors: Pharmacological characterization, computational modeling, and pharmacophore development. Mol. Pharmacol. 2007, 71 (5), 1288–1297. 10.1124/mol.106.033233. [DOI] [PubMed] [Google Scholar]
- Huang J.; Orac C. M.; McKay S.; McKay D. B.; Bergmeier S. C. The synthesis of 5-substituted ring E analogs of methyllycaconitine via the Suzuki-Miyaura cross-coupling reaction. Bioorg. Med. Chem. 2008, 16 (7), 3816–3824. 10.1016/j.bmc.2008.01.050. [DOI] [PubMed] [Google Scholar]
- Broadbent S.; Groot-Kormelink P. J.; Krashia P. A.; Harkness P. C.; Millar N. S.; Beato M.; Sivilotti L. G. Incorporation of the β3 subunit has a dominant-negative effect on the function of recombinant central-type neuronal nicotinic receptors. Mol. Pharmacol. 2006, 70, 1350–1356. 10.1124/mol.106.026682. [DOI] [PubMed] [Google Scholar]
- Young G. T.; Zwart R.; Walker A. S.; Sher E.; Millar N. S. Potentiation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 14686–14691. 10.1073/pnas.0804372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young G. T.; Broad L. M.; Zwart R.; Astles P. C.; Bodkin M.; Sher E.; Millar N. S. Species selectivity of a nicotinic acetylcholine receptor agonist is conferred by two adjacent extracellular β4 amino acids that are implicated in the coupling of binding to channel gating. Mol. Pharmacol. 2007, 71, 389–397. 10.1124/mol.106.030809. [DOI] [PubMed] [Google Scholar]
- Gill J. K.; Dhankher P.; Sheppard T. D.; Sher E.; Millar N. S. A series of α7 nicotinic acetylcholine receptor allosteric modulators with close chemical similarity but diverse pharmacological properties. Mol. Pharmacol. 2012, 81, 710–718. 10.1124/mol.111.076026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.