

Significance
Therapeutic failure is a major problem for cancer patients. Chemotherapy often induces DNA damage or arrest of DNA synthesis, leading to the abnormal presence of DNA in the cytoplasm, which activates the cGAS–STING pathway. STAT2 binds to STING, modulating its function by inhibiting the expression of IRF3-dependent antitumor genes, but not of NF-κB–dependent protumor genes, thus helping cancer cells to resist DNA damage. This function of STAT2 is dependent on its phosphorylation on T404, a high level of which is seen in about 25% of human lung cancer specimens. Our data reveal a mechanism, quite distinct from its regulation of transcription, through which STAT2 reshapes STING activation and promotes malignancy.
Keywords: STAT2, STING, DNA damage
Abstract
In cancer cells, endogenous or therapy-induced DNA damage leads to the abnormal presence of DNA in the cytoplasm, which triggers the activation of cGAS (cyclic GMP–AMP synthase) and STING (stimulator of interferon genes). STAT2 suppresses the cGAMP-induced expression of IRF3-dependent genes by binding to STING, blocking its intracellular trafficking, which is essential for the full response to STING activation. STAT2 reshapes STING signaling by inhibiting the induction of IRF3-dependent, but not NF-κB–dependent genes. This noncanonical activity of STAT2 is regulated independently of its tyrosine phosphorylation but does depend on the phosphorylation of threonine 404, which promotes the formation of a STAT2:STING complex that keeps STING bound to the endoplasmic reticulum (ER) and increases resistance to DNA damage. We conclude that STAT2 is a key negative intracellular regulator of STING, a function that is quite distinct from its function as a transcription factor.
Stimulator of interferon genes (STING) plays a crucial role in regulating the activation of innate and adaptive immunity. By directly binding to cyclic dinucleotides (CDNs), STING induces the expression of host defense genes whose products mediate essential antimicrobial and antitumor immunity (1–3). CDNs induce conformational changes of STING, which lead to its departure from the endoplasmic reticulum (ER) (4). This intracellular trafficking is essential for the full response to STING activation (4). STING migrates from the ER to ERGIC (ER-Golgi intermediate compartment) and the Golgi body, where it recruits and activates TBK1, which phosphorylates S366 of STING (5, 6). Phosphorylated S366 is a docking site for IRF3 and is essential for the subsequent induction of Interferons (IFNs) (7, 8). Our previous work demonstrated that IRF3 binds to phosphorylated STING in the late endosomal compartment, where TBK1 phosphorylates and activates IRF3 (9).
Additional proteins regulate the trafficking of STING. STIM1 (stromal interaction molecule 1), a transmembrane protein that mediates Ca2+ influx, inhibits STING activation by causing its retention in the ER (10, 11). In contrast, iRhom2 (inactive rhomboid protein 2) and STEEP interact with STING and facilitate its trafficking from the ER and its functions (12, 13). In the present study, we show that STAT2 is a negative regulator of STING intracellular trafficking, which is essential for the full response to its activation. Furthermore, we show that E316 of STING and the phosphorylation of T404 in the DNA-binding domain (DBD) of STAT2 are both required to form the complex with STAT2 that blocks STING trafficking.
It is well known that STAT2 helps to transduce type I IFN-dependent signals when its tyrosine residue Y690 is phosphorylated (14). STAT2 also contributes to tumorigenesis in various types of cancer (15). We have shown that elevated levels of tyrosine-unphosphorylated STAT2 and IRF9 enhance the expression of a subset of NF-κB–dependent genes, including IL-6 (16, 17). Importantly, a high level of STAT2 drives a gene signature that is associated with therapeutic resistance and poor prognosis (18, 19). Here, we report a protumor function of STAT2 that inhibits the activation of STING in cancer cells. STING activation leads in turn to the activation of IRF3 and NF-κB (20). The antitumor activities of STING depend primarily on IRF3 activation (21, 22). STING senses therapy-induced or endogenous DNA damage and consequently stimulates antitumor immune responses, including the recruitment and activation of cytotoxic T-cells (23, 24). STING also activates NF-κB, which promotes progression and resistance to therapy by inducing the production of inflammatory cytokines (25), including IL-6, which is required for the survival of cancer cells following genotoxic treatments (26). Moreover, we found that STAT2 reshapes STING signaling by suppressing the induction of IRF3-dependent but not NF-κB–dependent genes. The phosphorylation of STAT2 on T404 inhibits the DNA damage response that is induced by exposure of lung cancer cells to cisplatin and may correlate with the infiltration of fewer CD8+ T cells in lung cancers. Our data indicate that tumor cells reshape and evade STING-mediated antitumor effects by manipulating the expression level and T404 phosphorylation of STAT2.
Results
STAT2 Inhibits IFN-β Induction in Response to STING Ligands.
Mammalian cells detect DNA viruses through the cGAS–STING pathway (3). cGAS recognizes cytoplasmic DNA and generates 2′, 3′-Cyclic guanosine monophosphate–adenosine monophosphate (cGAMP), an intracellular second messenger that directly binds to and activates STING to induce the transcription of IRF3- and NF-κB–dependent genes, whose products elicit antiviral and proinflammatory responses (20, 27, 28). We find that high expression of STAT2 compromises the antiviral response to Herpes Simplex Virus 1 (HSV-1) (Fig. 1A), a DNA virus, due to insufficient production of IFN-β (Fig. 1B). We propose that high expression of STAT2 compromises the antiviral defense against HSV by inhibiting STING activation. When cells expressing a high level of STAT2 were treated with 2′, 3′-cGAMP, the induction of IFN-β expression was substantially inhibited (Fig. 1C). Interestingly, STAT2 preferentially inhibits the expression of IRF3-dependent genes but not NF-κB–dependent genes (Fig. 1C). Similar results were observed with Calu-1 (nonsmall cell lung cancer cells, SI Appendix, Fig. S1A), and THP1 (immortalized human monocytes, SI Appendix, Fig. S1B). By directly binding to microbial (CDNs), STING also connects microbial sensing with the induction of multiple host defense genes (29). Consistently, the STING ligands cdi-cyclic dimeric guanosine monophosphate (GMP) and 2′, 3′-cGAMP gave similar results (SI Appendix, Fig. S1C). In summary, a function of STAT2 inhibits STING activation in response to various ligands.
Fig. 1.

STAT2 interacts with STING and inhibits induction of IFN-β. (A) HMEs (human mammary epithelial cells) stably expressing STAT2 were infected with HSV-1 (MOI = 1) for 20 h. Whole-cell lysates were analyzed by the Western blot method. (B) HMEs stably expressing STAT2 were infected with HSV-1 (MOI = 1) for 6 h. Total RNAs were analyzed by qPCR. (C) HMEs overexpressing parental or STAT2 were stimulated with 2′3′cGAMP (8 μg/mL) for 4 h, followed by RT-PCR analysis. (D) IP analysis of endogenous STAT2–STING interaction in HT1080 cells treated with 8 µg/mL cGAMP for 3 h.
STAT2 Interacts with STING and Inhibits STING-Dependent Signaling.
STAT2 binds to STING upon stimulation with 2′, 3′-cGAMP (Fig. 1D), stimulating an investigation of how STAT2 interacts with STING and inhibits its activation. Using a set of mGST-fusion proteins with different STING truncations, we detected direct interaction between residues 265 to 379 of STING and STAT2 (SI Appendix, Fig. S2A). Analysis of several deletions within the 265 to 379 region revealed that residues 309 to 316 are necessary for binding to STAT2 (SI Appendix, Fig. S2 B and C).
Furthermore, the single STING mutation E316A disrupts the interaction (SI Appendix, Fig. S2D). Critically, E316 is highly conserved among species (SI Appendix, Fig. S2E). The negative charge of E316 is critical for the intramolecular interaction between the CDN-binding domain and the C-terminal tail, which keeps STING in a dormant state (30). Loss of this negative charge might change its conformation and switch STING to a hyper-active state (31). We made HT1080 cells deficient in STING using CRISPR-Cas9 and then restored STING expression with either wild-type (WT) or the E316A mutant. Upon HSV-1 infection, E316A-STING drove a more robust induction of the expression of IFNB, an IRF3-dependent gene, but not of CCL20, an NF-κB–dependent gene (SI Appendix, Fig. S2F). Consequently, HSV replication is lower in cells expressing E316A-STING than WT-STING (SI Appendix, Fig. S2G). This altered gene expression pattern repeats the finding that STAT2 specifically inhibits IRF3-dependent gene induction. Our discovery agrees with previous findings indicating that E316 is essential to enable STING to interact with STAT2, thus strengthening the dormant state of STING. Therefore, STAT2 is a vital negative regulator of STING-dependent signaling, especially with regard to IFN-β production and antiviral activity.
STAT2 Impedes STING Trafficking from the ER.
The ligand-induced formation of STING dimers and oligomers enables full activation of downstream signaling. STING translocates from the ER through the ER–Golgi and Golgi-to-post-Golgi compartments (4, 5). During its translocation, STING mediates the activation of several kinases, including TANK-binding kinase 1 (TBK1) and IκB Kinases (IKKs), which mediate downstream transcription. Since intracellular trafficking is essential for complete STING activation (4), we investigated whether STAT2 colocalizes with STING, using Bimolecular Fluorescence Complementation. STAT2 interacts with STING primarily in the cytosol, especially in the ER (SI Appendix, Fig. S3A), where STING resides in the absence of stimulation (3). Interestingly, confocal microscopy revealed that cGAMP enhances the translocation of STAT2 to the ER in Calu-1 cells (Fig. 2A and SI Appendix, Fig. S3B) and primary human peripheral blood mononuclear cells (SI Appendix, Fig. S3C).
Fig. 2.
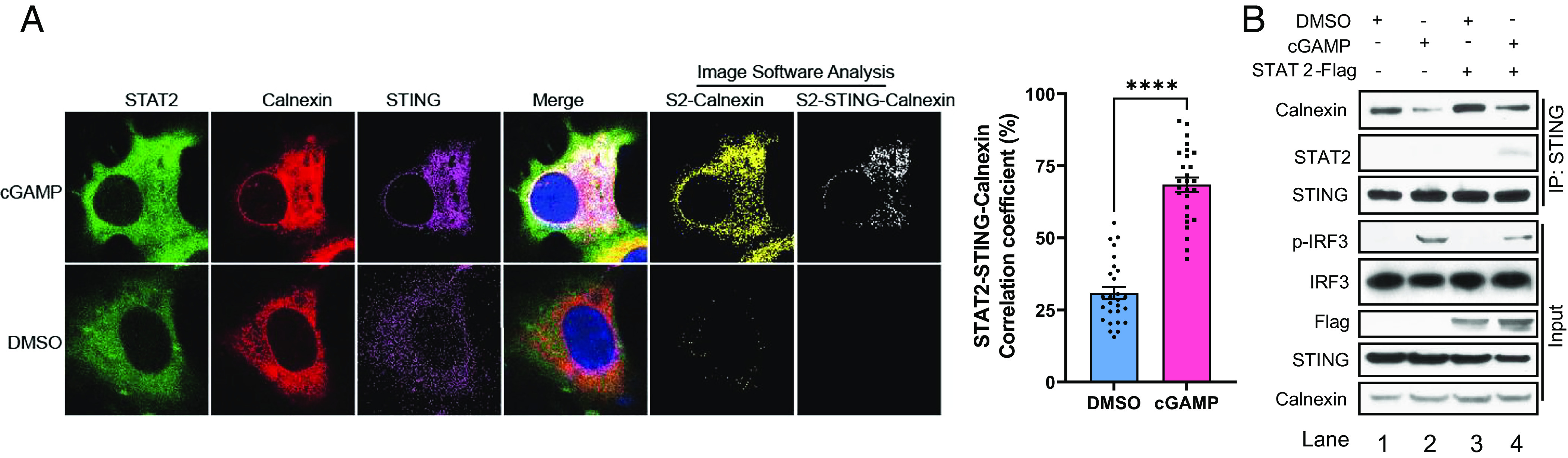
STAT2 impedes STING trafficking from ER. (A) Calu-1 cells were transfected with STAT2–GFP (green) and STING–HA for 24 h and treated with cGAMP for 1 h. The cells were fixed and stained with calnexin antibody (red) and HA antibody (purple). Imaging data from confocal fluorescence microscopy were analyzed by Fuji software to reveal colocalization, shown as white dots. (B) THP1 cells over expressing vector or STAT2-flag (WT) were stimulated with 2′3′cGAMP (8 μg/mL) for 3 h, followed IP analysis of STAT2–STING–calnexin interactions.
To confirm that STAT2 translocates to the ER upon treatment with a STING ligand, Co-IP experiments were performed, showing that both STING and STAT2 bind to calnexin, a chaperone protein located on the ER membrane (SI Appendix, Fig. S3D). We speculate that STAT2 does not enter the ER lumen and may interact with the C-terminus of calnexin, which faces the cytoplasm, while its N-terminus faces the ER lumen (32). Furthermore, the CCD domain of STAT2 is needed for this interaction (SI Appendix, Fig. S3E). In THP1 cells, treatment with cGAMP decreased the association of STING with calnexin, revealing the departure of STING from the ER (Fig. 2B, comparing lanes 1 and 2). Exogenously increased expression of STAT2 prevents the relocation of STING from the ER, as shown by a strengthened interaction between STING and calnexin (Fig. 2B, comparing lanes 2 and 4). These data reveal that STAT2 inhibits STING activation by suppressing its trafficking.
Phosphorylation of T404 Drives the Inhibition of STING by STAT2.
To determine whether the inhibition of STING activation depends on the phosphorylation of STAT2 on Y690, we expressed Y690F or WT STAT2 in STAT2-deficient Calu-1 cells, finding that the ability of STAT2 to blunt gene expression in response to 2′, 3′-cGAMP is independent of tyrosine phosphorylation (SI Appendix, Fig. S4). By studying deletions of STAT2, we found that the DBD of STAT2 is required for the STAT2:STING complex to form (Fig. 3A). IKKi phosphorylates STAT2 on T404, which is in the DBD (33), and IKKi is activated in response to STING activation (34). Stimulation with 2′, 3′-cGAMP induced T404 phosphorylation (SI Appendix, Fig. S5A). To determine whether T404 phosphorylation of STAT2 is critical for its interaction with STING, WT, T404A, or T404E, WT STAT2 or the two mutants were expressed in STAT2-deficient Calu-1 cells. Upon treatment with cGAMP, the T404A mutation, which prevents phosphorylation, blocked the ability to bind to STING. In contrast, the phospho-mimetic T404E mutation led to stronger binding (Fig. 3B). In addition, STING recruited less IRF3 in cells expressing T404E-STAT2 (Fig. 3B). The induction of phosphorylation of STING on S366 of IRF3 on S396 was inhibited in T404E compared to WT or T404A cells (Fig. 3C). We also found that IRF3 nuclear translocation (Fig. 3D and SI Appendix, Fig. S5B) and the induction of IRF3-dependent genes (Fig. 3E) were substantially inhibited in cells expressing T404E-STAT2. These results are consistent with preferential inhibition of these genes, mediated by STAT2 T404 phosphorylation in response to cGAMP and cdi-GMP in THP1 cells (SI Appendix, Fig. S5C). In summary, the phosphorylation of T404 of STAT2 increases its affinity for STING, which inhibits STING-dependent IRF3 activation, resulting in less IFN production.
Fig. 3.
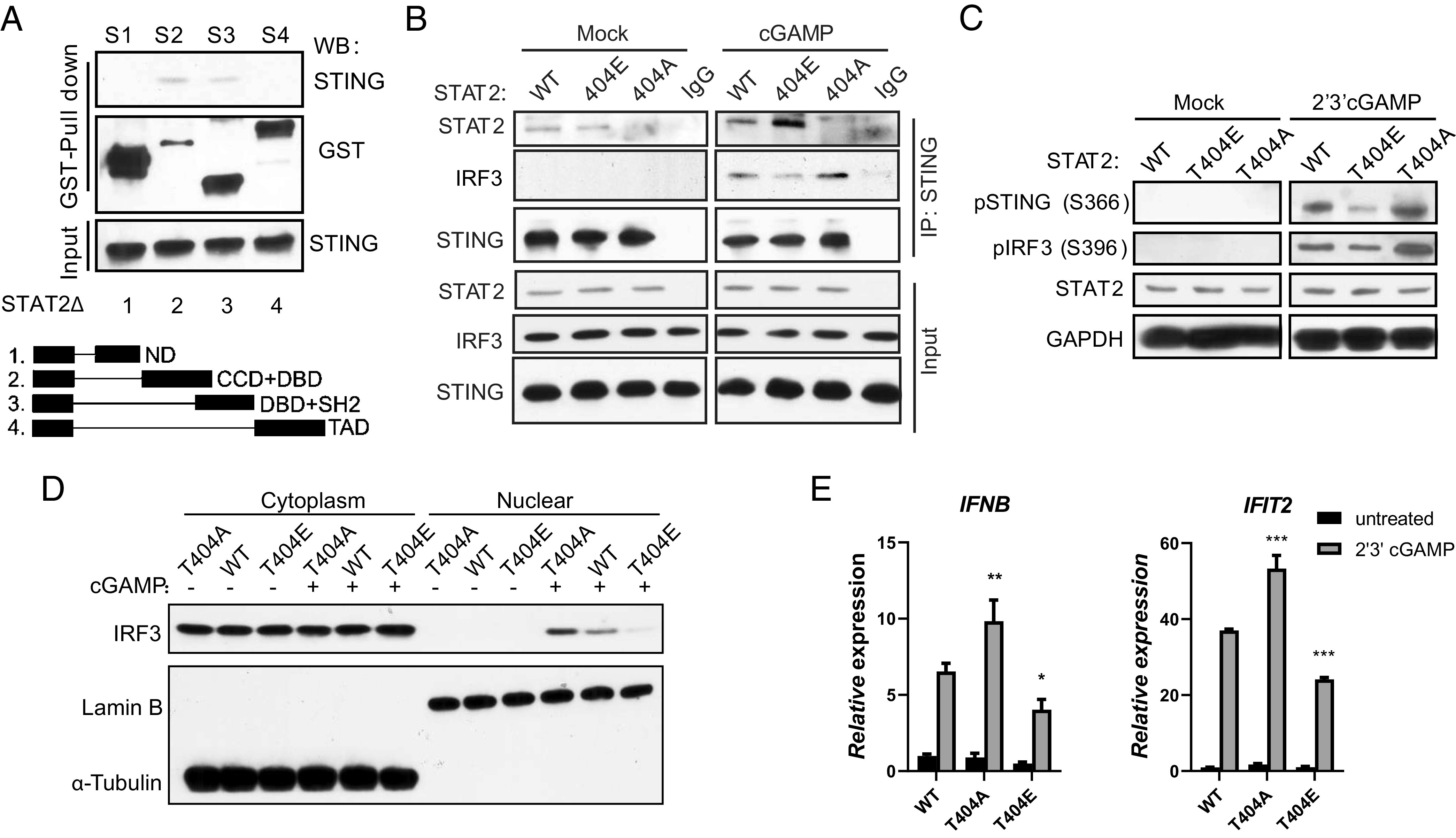
Phosphorylation of T404 drives STAT2 inhibition of STING. (A) GST-pull-down assay using HEK293T cells transfected with GST-tagged truncations of STAT2. N-terminal domain, CCD: coil-coiled domain, DBD, trans-activation domain; (B) STAT2-deficient Calu-1 cells restored with WT, T404A, or T404E STAT2 were stimulated with 2′3′cGAMP (8 μg/mL) for 3 h, followed by IP analysis of the STAT2–STING–IRF3 interaction. (C) STAT2-deficient Calu-1 cells restored with WT, T404A, or T404E STAT2 were stimulated with 2′3′cGAMP (8 μg/mL) for 3 h, followed by Western analysis. (D) STAT2-deficient Calu-1 cells restored with WT, T404A, or T404E STAT2 were stimulated with 2′3′cGAMP (8 μg/mL) for 3 h, followed by nuclear fractionation and analysis by the Western method. (E) STAT2-deficient Calu-1 cells restored with WT, T404A, or T404E STAT2 were stimulated with 2′3′cGAMP (8 μg/mL) for 4 h, followed by RT-PCR analysis.
How does the phosphorylation of STAT2 on T404 mediate its ability to inhibit the STING–IRF3 pathway? Our previous study (9) revealed that the Y245 phosphorylation of STING is a marker for late endosome trafficking and allows STING to form a tripartite complex with TBK1 and IRF3. We now find that cGAMP induces a higher level of Y245 phosphorylation in cells expressing WT or T404A-STAT2 than in cells expressing the T404E mutant (SI Appendix, Fig. S5D). These data reveal that STAT2 phosphorylated on T404 controls the trafficking of STING to late endosomes, limiting IRF3 activation and IFN production in response to cGAMP. Upon cGAMP treatment, IRF3 forms foci and colocalizes with STING and CD63, a marker of late endosomes (SI Appendix, Fig. S5E). Most importantly, we observed more STING–IRF3–CD63 foci, with a distinct pattern, in cells expressing T404A-STAT2 compared to cells expressing WT- or T404E-STAT2 (SI Appendix, Fig. S5E). Consequently, IRF3 nuclear translocation was inhibited in cells expressing T404E-STAT2 (SI Appendix, Fig. S5F). These data demonstrate that the phosphorylation of T404 drives the ability of STAT2 to inhibit STING by impeding STING trafficking from the ER.
STAT2-Dependent Inhibition of STING Promotes Resistance to Chemotherapy and Tumor Progression.
Cancer cells frequently downregulate the expression and function of cGAS and STING, thus promoting malignancy (21, 35). Many studies have revealed the tumor-suppressive functions of these proteins in different types of cancer, including lung adenocarcinomas (SI Appendix, Fig. S6 A and B). Consistent with our new findings on the ability of STAT2 to inhibit STING-dependent signaling, there is a significant association between high levels of STAT2 in tumors and shorter overall survival in patients with lung adenocarcinomas (SI Appendix, Fig. S6C). Interestingly, in the same patient population, elevated expression of the IFN-I receptor subunit IFNAR1 correlated with better prognosis (SI Appendix, Fig. S6D), revealing an additional role of STAT2 that is independent of its tyrosine phosphorylation.
STING activation increases the kinase activity of TBK1 and IKKi (5, 6, 34). Interestingly, we found an inverse correlation between the expression of IKKi and TBK1 with patient prognosis in lung adenocarcinomas (SI Appendix, Fig. S6 E and F). IKKi is the dominant kinase for T404 phosphorylation (33), while TBK1, but not IKKi, is essential for IRF3 activation, which is critical for antitumor activity (21, 34, 35). Based on our finding that STAT2 preferentially inhibits the induction of IRF3-dependent genes, we propose that the IKKi-induced T404 phosphorylation of STAT2 mediates tumor progression in lung adenocarcinoma by inhibiting the tumor-suppressive function of the cGAS–STING pathway.
Sufficient activation of the cGAS–STING pathway is essential for the success of DNA-damage mediated therapy in cancer (21, 25). STING activation increases tumor cell death in response to DNA damaging agents, including cisplatin, a standard-of-care treatment for lung cancer (1, 36, 37). To investigate the role of STAT2 in resistance to cisplatin, we employed three lung cancer cell lines with different expression levels of STAT2: low in A549 cells and high in H196 and Calu-1 cells (SI Appendix, Fig. S7A). Increased expression of STAT2 attenuated cisplatin-induced cell death in A549 cells (SI Appendix, Fig. S7B), and downregulation of STAT2 expression increased cell death in Calu-1 and H196 cells (SI Appendix, Fig. S7 C and D). We found that cisplatin induces T404 phosphorylation (Fig. 4A), and the prevention of this phosphorylation by the T404A mutation promotes IRF3 nuclear translocation, which is opposite in the cells expressing the phospho-mimetic T404E mutation (Fig. 4B). Consequently, expression of T404A-STAT2 enhances the production of IRF3-dependent cytokines, such as IFN-β and CXCL10, which mediate potent immunogenic cell death (Fig. 4C) (38–40). To further evaluate whether the ability of STAT2 to mediate resistance to cisplatin depends on T404 phosphorylation, WT, T404A, T404E, or Y690F STAT2 were expressed in STAT2-deficient Calu-1 and H196 cells. The phosphorylation-deficient mutation T404A inhibited cisplatin resistance in Calu-1 cells, which have a high basal level of T404 phosphorylation when the WT protein is present (SI Appendix, Fig. S7E). In contrast, the GOF T404E mutation increased resistance to cisplatin in phosphorylation H196 cells, which have a low level of T404 phosphorylation when the WT protein is present (SI Appendix, Fig. S7F).
Fig. 4.
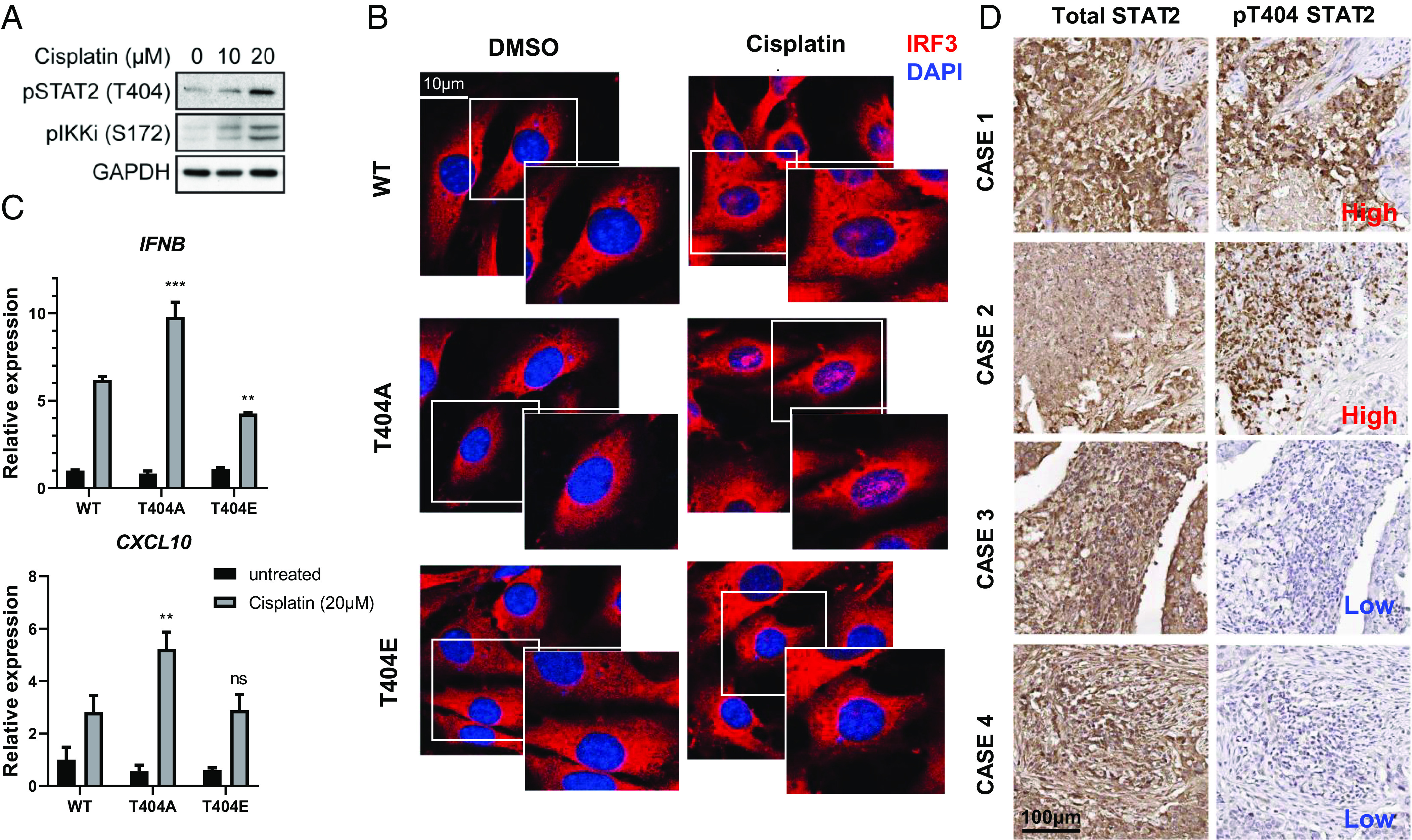
T404 phosphorylation of STAT2 promotes resistance to cisplatin in lung cancer cell lines and inhibits IRF3 nuclear translocation in response to cisplatin. (A) Calu-1 cells were treated with cisplatin for 24 h, and whole-cell lysates were analyzed by the Western method. (B) STAT2-deficient Calu-1 cells restored with WT, T404A, or T404E STAT2 were treated with cisplatin for 24 h. The cells were fixed then stained with IRF3 antibody (red) and DAPI for confocal analysis. (C) STAT2-deficient Calu-1 cells restored with WT, T404A, T404E, or Y690F STAT2 were treated with cisplatin (20 μM) for 24 h, followed by RT-PCR analysis. (D) Representative images of immunohistochemical staining for P-T404 STAT2 and STAT2 in specimens from human lung adenocarcinoma patients.
Using an antibody that specifically recognizes phosphorylated T404 (33), we analyzed T404 phosphorylation in a human tissue array that includes various types of lung cancer (Fig. 4D and SI Appendix, Fig. S8 A and B). T404 phosphorylation is readily detected in about half of these specimens, with about 20% of the samples showing a high level (SI Appendix, Fig. S8C). STING also connects genotoxic with immunogenic responses in tumor progression (41, 42). Activation of the STING pathway promotes the infiltration of T cells into tumors (43–45). Interestingly, about 20% of the human lung cancer specimens analyzed in SI Appendix, Fig. S8D showed noticeable infiltration of CD8+ cells. However, in tumors with a high level of T404 phosphorylation, only 7% of those had cytotoxic T cells (SI Appendix, Fig. S8D), revealing a trend toward a negative correlation between T404 phosphorylation and antitumor immunity (P = 0.12 by the chi-square test).
Discussion
Insight into STING-Dependent Signaling.
STING mediates three important innate immune functions: Inducing antiviral genes by activating IRF3, inducing inflammatory cytokines by activating NF-κB, and inducing autophagy to degrade intracellular pathogens or heterologous dsDNA (4, 46, 47). The binding of ligands permits STING to depart from the ER to different subcellular organelles and to generate the different signals noted above. Intracellular trafficking plays a crucial role in the complete activation of STING, including induction of IFNs, chemokines, and cytokines, and in initiating autophagy. Current studies have revealed two trafficking routes for STING: The first triggers the formation of autophagosomes by the translocation of STING to the ERGIC, allowing autophagy to clear cytosolic DNA and pathogens. In the second, STING departs from the ERGIC to the Golgi network and then to late endosomes, where it activates transcription factors, including IRF3, to induce the expression of genes encoding proteins important for defense against pathogens. These two routes will eventually introduce STING into autolysosomes, leading to its eventual degradation (4, 9).
The translocation of STING is regulated by posttranslational modifications and the interaction of STING with other proteins. For example, palmitoylation of C88 and C91 results in the translocation of STING to the Golgi apparatus, which is required for IRF3 activation (6). Furthermore, the K224R mutant of STING stays in the ER, thus failing to induce IFN production (48). Several proteins, including STIM1, NPC1, and SREBP2, influence trafficking by binding to STING, providing potential targets for future clinical research (11, 49). Our study demonstrates that STAT2 binds to STING and delays its trafficking, thus limiting the induction of IRF3-dependent genes, including IFNB and CXCL10. We show that STAT2 inhibits the phosphorylation of STING S366, which is required for the binding and subsequent activation of IRF3, but not NF-kB. Since STAT2 inhibits the transcription of IRF3-dependent, but not NF-kB–dependent genes, STAT2 may impair the ability of TBK1 to phosphorylate IRF3 by disrupting the IRF3–STING interaction. On the other hand, STAT2 has a strong nuclear export signal when Y690 is not phosphorylated (50). Therefore, in addition to its effect on IRF3 phosphorylation, if it binds to a STING–IRF3 complex, STAT2 may also inhibit the nuclear translocation of IRF3 and thus its transcriptional function.
Moreover, we reveal a unique negative feedback mechanism mediated by the phosphorylation of T404 of STAT2, induced by TBK1/IKKi following the binding of specific ligands to STING, resulting in less STING translocation to late endosomes. The STAT2–STING interaction reveals a crosstalk between cGAS–STING and STATs signaling that needs further investigation to reveal additional mechanistic details and therapeutic opportunities.
Insight into Disease.
When cGAS binds to cytoplasmic DNA, it produces 2′, 3′-cGAMP which, in turn, activates STING. This pathway is a prominent topic in immuno-oncology research (51–53). In combination with immune checkpoint blockade, injection of irradiated tumor cells into mice promoted tumor clearance in a STING-dependent manner (54). In addition to enabling immune surveillance in the tumor microenvironment, STING agonists promote tumor regression by activating NLRP3 inflammasomes (55, 56). To avoid being recognized by STING-mediated innate immune responses, cancer cells frequently evolve to impede the cGAS–STING signaling pathway, including, but not limited to, downregulating the expression of key signaling proteins (57), limiting STING translocation to the Golgi (6, 46), and mutating the IFN-I locus (48). A recent study demonstrates that cGAS–STING drives IL-6–STAT3 signaling, which promotes the survival of chromosomally unstable cancers (26). Genotoxic stress induces IL-6 via STING-mediated activation of NF-κB, rather than IRF3. We find that high expression and T404 phosphorylation of STAT2 inhibit STING subcellular translocation and reduce the production of IRF3-dependent cytokines, including IFNs and T cell chemotaxis, without inhibiting IL-6 production, thus promoting tumor cell survival.
Since the function of STING in sensing pathogens and inducing antimicrobial innate immune responses is crucial, pathogens have evolved to evade host immunity by attenuating STING activation. Some proteins from pathogens specifically hinder STING trafficking, including M152 from Murine Cytomegalovirus (MCMV) (46) and IpaJ from Shigella flexneri (5, 58), Therefore, these proteins share a similar effect with STAT2 inhibition on STING activation, which affects only IRF3-dependent gene expression but not the expression of genes that depend on NF-κB activation. In addition to infectious diseases, gain-of-function (GOF) mutations of STING have been identified in STING-associated vasculopathy with onset in infancy (SAVI), as well as in patients with lupus (59–61). These mutations constitutively activate STING regardless of ligand binding (5). Evidence from genetic experiments in mice demonstrates that STING-associated lung diseases are independent of IRF3 but rely on T cells (62, 63). Wu et al. have shown that T cell cytopenia is due to disrupted calcium homeostasis caused by the constitutive ER departure of a GOF STING mutant (64). Even though IFN-I signaling is not thought to contribute to the onset of SAVI (62), Janus Kinase (JAK) 1/2 inhibitors inhibit STING signaling in vitro and in several patients carrying GOF STING mutations (65, 66) by an unknown mechanism. We find that JAK-induced tyrosine phosphorylation of STAT2 is not required for its ability to inhibit STING. Thus, we hypothesize that in addition to ameliorating interferonopathy, inhibiting JAKs could skew STAT2 away from ISGF3 to instead complex with STING and prevent its departure from the ER.
In summary, our study reveals that STAT2 is an ER retention factor that arrests STING trafficking. Further dissection of the mechanism underlying the maintenance of STING–STAT2 complexes, such as additional structural information, may inform the design of specific therapeutic approaches for balancing STING-mediated innate immune responses.
Materials and Methods
Cell Culture and Transfection.
Calu (HTB-54), A549 (CCL-185), H196 (NCI-H196), THP1 (TIB-202), L929 (CCL-1), HT1080 (CCL-121), HEK293T (CRL-11268), HeLa (CRM-CCL-2), and BJ cells (CRL-2522) were purchased from American Type Culture Collection (ATCC) (Vanassas, VA, USA), HT1080 STING−/− cells were made by using Crispr/Cas9 sgRNA. U6A STAT2−/− cells were previously described (67). The Barber lab graciously provided us with Primary STING−/− Mouse Embryonic Fibroblasts (MEFs) (9, 48), while SI Appendix, Table S1 showcases a comprehensive list of the utilized cell lines. Plasmid transfections were conducted with Lipofectamine 2,000, following the manufacturer's protocol. In contrast, polyethyleneimine (PEI) was used for 2′, 3′-cGAMP transfections. To achieve this, 1 μg of 2′, 3′-cGAMP (final concentration: 8 μg/mL) and 2.25 μL of PEI (1 mg/mL, pH 7.0) were separately incubated in 0.25 mL of serum-free medium for 5 min. The two were then combined for 15 min before being added dropwise to the cells as previously described (9). HSV-1 strain KOS propagation and infection were performed as previously described (68, 69), with virus titers being determined via plaque assay.
Western Analyses.
Following lysis in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1 mM DTT, 1 mM Phenylmethylsulfonyl Fluoride) and cocktail (Roche) at 4 °C for 15 min, the cells were centrifuged at 12,000× g at 4 °C for 10 min. The resulting cell lysates were either analyzed via SDS-PAGE or used for IP. After electrophoresis of the proteins in SDS-PAGE gels, they were transferred to Polyvinylidene Fluoride (PVDF) membranes (Bio-Rad). Subsequently, the membranes were immersed in 5% skim milk in Tris-buffered saline with Tween (TBST) buffer (150 mM NaCl; Tris, pH 7.4; and 0.1% Tween 20) for about an hour at room temperature and then left to incubate with the primary antibody in a cold room overnight. Western experiments were conducted with the designated antibodies and visualized through the use of Super-Signal West Pico Chemiluminescent substrate (Pierce Chemical), with the Western images being crafted with Adobe Illustrator.
Immunoprecipitation.
For immunoprecipitations, cells were lysed in a buffer consisting of 20 mM HEPES, pH 7.5, 150 mM NaCl, 10 mM NaF, 1.5 mM MgCl2, 10 mM β-glycerophosphate, 2 mM EGTA, 1 mM Na3VO4, 1% (v/v) Triton X-100, 0.2% NP-40, and protease inhibitors (Roche Applied Science, Indianapolis, IN, USA). After centrifuging whole-cell lysates at 12,000× g for 10 min at 4 °C, the supernatant suspensions were used for immunoprecipitation, involving Protein A/G PLUS Agarose (Santa Cruz) and overnight incubation with 3 μg of mouse or rabbit monoclonal antibody at 4 °C. The related beads were washed six times with IP buffer before being heated in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) buffer for 10 min at 95 °C as previously described (9). The samples were then separated in SDS-PAGE gels and transferred onto PVDF membranes (Bio-Rad) for Western experiments performed with the designated antibodies and visualized through the use of Super-Signal West Pico Chemiluminescent substrate (Pierce Chemical).
RT-PCR.
To extract cDNA, the RNeasy Mini Kit (Qiagen) was employed, and 0.5 ng of cDNA was utilized for Realtime PCR on a 384-well plate, using Applied Biosystem’s Power SYBR Green PCR mix in Roche LightCycler 480 II. The levels of induced mRNAs were standardized to 18S rRNA. The specificity of the primers was verified by analyzing the melting curves of the PCR products. The primer sequences used are provided in SI Appendix, Table S1.
Immunofluorescence Staining and Confocal Microscopy.
Cells were cultured on 4-well slides and transfected with specified plasmids using lipo2000 for 24 h. The fixed cells were treated with 4% paraformaldehyde overnight at a low temperature and then permeabilized in Phosphate-Buffered Saline (PBS) containing 0.1% Triton X-100 for 10 min at room temperature. After being washed four times with PBS, the cells were blocked with 10% Bovine Serum Albumin (BSA) in PBS for 1.5 h at room temperature. The antibody was incubated with the cells overnight in a cold room, followed by three washes with PBS for 10 min. The cells were then treated with Goat anti-rabbit Alexa Fluor 594 (Invitrogen), anti-mouse Alexa Fluor 594 (Invitrogen), anti-rabbit Alexa Fluor 647 (Invitrogen), or Donkey Anti-Goat IgG H&L (Alexa Fluor® 488) (Thermo Fisher). Images were captured by confocal laser scanning microscopy (Leica True Confocal Scanning SP8) and processed with Leica LCS software as previously described (9). Fiji (ImageJ) software Co-localization plugin was utilized to identify protein colocalization, and Pearson’s colocalization coefficient was determined using Fiji software (ImageJ). Random images were collected for each condition, and higher parameters were set to generate clear results, resulting in fewer white spots for control and negative groups.
Quantification and Statistical Analysis.
For qPCR analysis, the SD were calculated from three or more independent biological experiments and represented by error bars. Results from Western analyses, co-immunoprecipitations, and confocal experiments were considered representative of at least two independent experiments unless otherwise noted. The two-tailed t test using Prism 5 software (GraphPad) was used to determine statistical significance between two groups. All values represent means ± SD of the indicated independent experiments. NS, P > 0.05; *P < 0.05; **P < 0.01, ***P < 0.001.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was funded by NIH Grants HL160639 (to Y.W.), AI153085 and CA062220 (to G.R.S.), CA062220 and CA068782 (to G.C.S.), and Fundamental Research Funds for the Central Universities Grant lzujbky-2021-44 (to J.N.).
Author contributions
C.W., X.L., G.C.S., G.R.S., and Y.W. designed research; C.W., J.N., E.H.-B., S.W., and Y.W. performed research; C.W., X.C., M.-B.L., J.Z., and Y.W. contributed new reagents/analytic tools; C.W., X.C., J.Z., X.L., G.C.S., G.R.S., and Y.W. analyzed data; and C.W., G.R.S., and Y.W. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: G.B., University of Miami; and D.E.L., New York University School of Medicine.
Contributor Information
Ganes C. Sen, Email: seng@ccf.org.
George R. Stark, Email: starkg@ccf.org.
Yuxin Wang, Email: wangy7@ccf.org.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Hayman T. J., et al. , STING enhances cell death through regulation of reactive oxygen species and DNA damage. Nat. Commun. 12, 2327 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lohard S., et al. , STING-dependent paracriny shapes apoptotic priming of breast tumors in response to anti-mitotic treatment. Nat. Commun. 11, 259 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng Z., et al. , The interactions between cGAS-STING pathway and pathogens. Signal Transduct. Target. Ther. 5, 91 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gui X., et al. , Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobbs N., et al. , STING activation by translocation from the ER Is associated with infection and autoinflammatory disease. Cell Host Microbe 18, 157–168 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mukai K., et al. , Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 7, 11932 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu S., et al. , Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Tanaka Y., Chen Z. J., STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal 5, ra20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang C., et al. , EGFR-mediated tyrosine phosphorylation of STING determines its trafficking route and cellular innate immunity functions. EMBO J. 39, e104106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu J., Yan N., STIM1 moonlights as an anchor for STING. Nat. Immunol. 20, 112–114 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Srikanth S., et al. , The Ca(2+) sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat. Immunol. 20, 152–162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo W. W., et al. , iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat. Immunol. 17, 1057–1066 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Zhang B. C., et al. , STEEP mediates STING ER exit and activation of signaling. Nat. Immunol. 21, 868–879 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stark G. R., Cheon H., Wang Y., Responses to cytokines and Interferons that depend upon JAKs and STATs. Cold Spring Harb. Perspect. Biol. 10, a028555 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gamero A. M., et al. , STAT2 contributes to promotion of colorectal and skin carcinogenesis. Cancer Prev. Res. (Phila.) 3, 495–504 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nan J., Wang Y., Yang J., Stark G. R., IRF9 and unphosphorylated STAT2 cooperate with NF-κB to drive IL6 expression. Proc. Natl. Acad. Sci. U.S.A. 115, 3906–3911 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W., et al. , Unphosphorylated ISGF3 drives constitutive expression of interferon-stimulated genes to protect against viral infections. Sci. Signal. 10, eaah4248 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Weichselbaum R. R., et al. , An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. U.S.A. 105, 18490–18495 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheon H., et al. , IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 32, 2751–2763 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hopfner K. P., Hornung V., Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 21, 501–521 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Barber G. N., STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 15, 760–770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arwert E. N., et al. , STING and IRF3 in stromal fibroblasts enable sensing of genomic stress in cancer cells to undermine oncolytic viral therapy. Nat. Cell Biol. 22, 758–766 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y., et al. , Cancer cell-intrinsic STING is associated with CD8 + T-cell infiltration and might serve as a potential immunotherapeutic target in hepatocellular carcinoma. Clin. Transl. Oncol. 23, 1314–1324 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Xu N., et al. , STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J. Exp. Med. 218, e20200844 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yum S., Li M., Fang Y., Chen Z. J., TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. U.S.A. 118, e2100225118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong C., et al. , cGAS-STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature 607, 366–373 (2022). [DOI] [PubMed] [Google Scholar]
- 27.Ishikawa H., Barber G. N., STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishikawa H., Ma Z., Barber G. N., STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pandey S., Kawai T., Akira S., Microbial sensing by toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol. 7, a016246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin Q., et al. , Cyclic di-GMP sensing via the innate immune signaling protein STING. Mol. Cell 46, 735–745 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burdette D. L., et al. , STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ho S. C., et al. , Membrane anchoring of calnexin facilitates its interaction with its targets. Mol. Immunol. 36, 1–12 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Wang Y., et al. , A virus-induced conformational switch of STAT1-STAT2 dimers boosts antiviral defenses. Cell Res. 31, 206–218 (2021), 10.1038/s41422-020-0386-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balka K. R., et al. , TBK1 and IKKepsilon act redundantly to mediate STING-induced NF-kappaB responses in myeloid Cells. Cell Rep. 31, 107492 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Kwon J., Bakhoum S. F., The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 10, 26–39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maekawa H., et al. , Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. 29, 1261–1273.e1266 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Grabosch S., et al. , Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 38, 2380–2393 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gulen M. F., et al. , Signalling strength determines proapoptotic functions of STING. Nat. Commun. 8, 427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murthy A. M. V., Robinson N., Kumar S., Crosstalk between cGAS-STING signaling and cell death. Cell Death Differ. 27, 2989–3003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang-Bishop L., et al. , Potent STING activation stimulates immunogenic cell death to enhance antitumor immunity in neuroblastoma. J. Immunother. Cancer 8, e000282 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu Y., et al. , STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 18, 152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lv M., et al. , Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. 30, 966–979 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Demaria O., et al. , STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. U.S.A. 112, 15408–15413 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pantelidou C., et al. , PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. 9, 722–737 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sivick K. E., et al. , Magnitude of therapeutic STING activation determines CD8(+) T cell-mediated anti-tumor immunity. Cell Rep. 25, 3074–3085.e3075 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Stempel M., et al. , The herpesviral antagonist m152 reveals differential activation of STING-dependent IRF and NF-kappaB signaling and STING’s dual role during MCMV infection. EMBO J. 38, e100983 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Decout A., Katz J. D., Venkatraman S., Ablasser A., The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 21, 548–569 (2021), 10.1038/s41577-021-00524-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ni G., Konno H., Barber G. N., Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci. Immunol. 2, eaah7119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chu T. T., et al. , Tonic prime-boost of STING signalling mediates niemann-pick disease type C. Nature 596, 570–575 (2021), 10.1038/s41586-021-03762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Banninger G., Reich N. C., STAT2 nuclear trafficking. J. Biol. Chem. 279, 39199–39206 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Li T., Chen Z. J., The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 215, 1287–1299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reislander T., Groelly F. J., Tarsounas M., DNA damage and cancer immunotherapy: A STING in the tale. Mol. Cell 80, 21–28 (2020), 10.1016/j.molcel.2020.07.026. [DOI] [PubMed] [Google Scholar]
- 53.Sen T., et al. , Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 9, 646–661 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harding S. M., et al. , Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaidt M. M., et al. , The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell 171, 1110–1124.e1118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swanson K. V., et al. , A noncanonical function of cGAMP in inflammasome priming and activation. J. Exp. Med. 214, 3611–3626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xia T., Konno H., Ahn J., Barber G. N., Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 14, 282–297 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burnaevskiy N., et al. , Proteolytic elimination of N-myristoyl modifications by the shigella virulence factor IpaJ. Nature 496, 106–109 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan N., Immune diseases associated with TREX1 and STING dysfunction. J. Interferon Cytokine Res. 37, 198–206 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Melki I., et al. , Disease-associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J. Allergy Clin. Immunol. 140, 543–552.e545 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Jeremiah N., et al. , Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J. Clin. Invest. 124, 5516–5520 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luksch H., et al. , STING-associated lung disease in mice relies on T cells but not type I interferon. J. Allergy Clin. Immunol. 144, 254–266.e258 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Warner J. D., et al. , STING-associated vasculopathy develops independently of IRF3 in mice. J. Exp. Med. 214, 3279–3292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu J., et al. , STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J. Exp. Med. 216, 867–883 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu Y., et al. , Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 371, 507–518 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rodero M. P., Fremond M. L., Rice G. I., Neven B., Crow Y. J., JAK inhibition in STING-associated interferonopathy. Ann. Rheum. Dis. 75, e75 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Leung S., Qureshi S. A., Kerr I. M., Darnell J. E. Jr., Stark G. R., Role of STAT2 in the alpha interferon signaling pathway. Mol. Cell Biol. 15, 1312–1317 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang X., et al. , STING requires the adaptor TRIF to trigger innate immune responses to microbial infection. Cell Host Microbe. 20, 329–341 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blaho J. A., Morton E. R., Yedowitz J. C., Herpes simplex virus: Propagation, quantification, and storage. Curr. Protoc. Microbiol. Chapter 14(Unit) 14E 11 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.