

Abstract
Immunotherapy has significantly improved treatment outcomes in various cancer entities. To enhance immunogenicity and efficacy, and to further broaden its applicability, co-administration of anti-tumor vaccines is considered as a promising strategy. Here, we introduce adeno-associated virus (AAV) vectors, widely used for in vivo gene therapy, as a potent cancer vaccine platform. Our AAV vector-based vaccine combines antigen display on the capsid surface with a vector-mediated antigen overexpression targeting different components of the immune system in a unique chronological order by a single intramuscular application. Thereby, both profound and long-lasting antigen-specific T and B cell immune responses were induced. Moreover, mice receiving the vaccine were protected against tumor growth, demonstrating its efficacy in two tumor models, including the low immunogenic and aggressive B16/F10-Ova melanoma model. Remarkably, this approach was even effective in conditions of a late tumor challenge, i.e., 80 days post-vaccination, between 88% (B16/F10-Ova melanoma) and 100% (EG7 thymoma) of mice remained tumor free. Thus, decorating AAV vector particles with antigens by capsid engineering represents a potent vaccine concept for applications in cancer immunotherapy. Its modular and versatile "plug-and-play" framework enables the use of tumor antigens of choice and the easy implementation of additional modifications to enhance immunogenicity further.
Keywords: AAV capsid engineering, cancer immunotherapy, prime-boost, single administration, vaccine
Graphical abstract
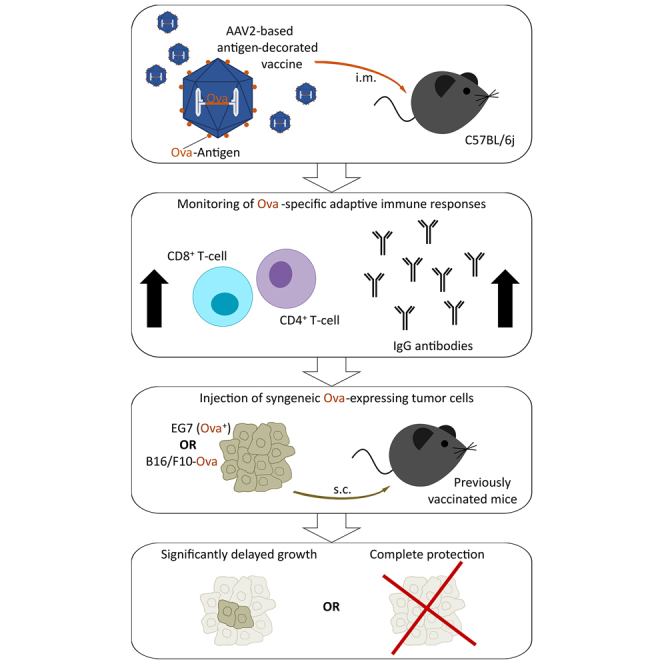
Büning and colleagues report on antigen-decorated adeno-associated virus vectors as new class of vaccine for cancer immunotherapy that induces potent and long-lasting antigen-specific adaptive immune responses after a single intramuscular injection. Vaccination protected mice from tumor growth after subcutaneous challenge with syngeneic EG7 (Ova+) thymoma or B16/F10-Ova melanoma cells.
Introduction
Immunotherapy has become an important strategy in cancer therapy. Clinical success is mainly based on antibodies targeting immune checkpoint molecules1 and on chimeric antigen receptor (CAR) T cells.2,3 To improve tumor immunogenicity through triggering efficient humoral and T cell-mediated immune responses,4 thus complementing or enhancing the activity of immune checkpoint inhibitors,5 or to support prolonged CAR T cell efficacy,6 cancer vaccines are being developed.
Current approaches encompass DNA-, RNA-, and vector-based vaccines using different tumor antigens and adjuvants.7 To achieve efficient and long-lasting immune responses, repeated vaccine administrations, i.e., prime-boost vaccination schemes, are generally required. This strategy poses a challenge to vector-based vaccines due to the induction of neutralizing antibodies that interfere with vector re-administration.4 To circumvent this challenge, heterologous prime-boost approaches are in use,8 which have the clear disadvantage of being logistically (e.g., manufacturing, application) more complex than a "single-shot" approach.
In response to these challenges, we developed a novel vaccine concept for cancer immunotherapy that is based on the adeno-associated virus (AAV). AAVs are members of the parvovirus family and have become prevalent as vectors in gene therapy due to their favorable safety profile and efficiency.9 They deliver a single-stranded DNA genome within an icosahedral non-enveloped protein capsid that mediates cell binding and transduction.10 The capsid is composed of 60 subunits, assembled in a 1:1:10 ratio from three capsid proteins, VP1, VP2, and VP3.10 All capsid proteins share the VP3 amino acid sequence (common VP3 region), with an additional 65 amino acids at the N terminus shared by VP1 and VP2 (VP1/VP2 common region) and a further 137 amino acids unique to VP1 (VP1u).10 The capsids convey AAV vector particles with high stability, a favorable feature concerning manufacturing, storage, and shipment.9
Since the viral capsid accepts even extensive genetic modifications, we and others hypothesized that the capsid could be decorated with antigens by genetic engineering for a protein-based immunization.11,12 If these particles are not used as "virus-like-particles" but deliver a vector genome encoding the same antigen, the capsid-displayed antigen serves the immune priming phase, followed by a “booster” immunization when the vector genome-encoded antigen is produced and secreted from AAV vector-transduced cells.12 Since the vector genome is a single-stranded DNA that needs to be converted into a transcriptionally active DNA double-strand prior to the onset of transcription,13 newly produced antigens are released with a significant temporal delay, enabling a "prime-boost scenario" by a single vector administration.12
We initially tested our vaccine concept in the context of Mycobacteria tuberculosis, aiming for an antigen-specific humoral immune response.12 Indeed, with our new vaccine concept, we observed a faster onset in antigen-specific antibody production compared with a conventional vector-based approach.12 The antigen-specific humoral immune response was long-lasting, and antibodies showed a higher avidity, arguing that a single administration of our antigen-decorated AAV vector-based vaccine is sufficient to induce affinity maturation, a feature conventionally seen in prime-boost scenarios.12 However, the efficacy of vaccines in cancer immunotherapy relies particularly on efficient and long-lasting antigen-specific T cell responses.14,15
Here, we report on the impressive potential of our capsid-engineered AAV vector-based vaccine for cancer immunotherapy exploring ovalbumin (Ova) as model antigen. The antigen was encoded as a single-stranded vector genome of AAV serotype 2 (AAV2) vectors, which were, in addition, decorated on the capsid by genetic insertion of major histocompatibility complex class I (MHC class I)- or MHC class II-restricted Ova epitopes. A single intramuscular administration of our vaccine in mice induced robust and long-lasting antigen-specific humoral and CD4+ and CD8+ T cell immune responses. Moreover, both short- and long-term protective effects were demonstrated in the immunogenic EG7 (Ova+) and the far more aggressive and poorly immunogenic B16/F10-Ova tumor models.
Results
Ova-antigen-displaying capsids can be efficiently produced
We designed four Ova-antigen-decorated capsids, which differed in the location chosen for antigen display, the amount of antigen per capsid, and the size of the displayed antigen. Precisely, in analogy to our previous work,12 we fused the cytoplasmic version of Ova (cOva) as a full-length protein to the N terminus of VP2 (AAV-Vac_cOvaVP2). As an alternative design, the MHC class I-restricted epitope Ova257-264 (SIINFEKL) and the MHC class II-restricted epitope Ova323-339 (ISQAVHAAHAEINEAGR) were fused to VP2 (AAV-Vac_Ova4+8VP2). In unmodified capsids, the N termini of VP2 are located inside the capsid.16 However, when fusing foreign sequences to the N terminus of VP2, the modified N termini are protruded through pores of the capsid and thus become accessible for recognition on the capsid surface.16,17 VP2, however, is one of the two minor capsid proteins contributing with a maximum of five subunits to the capsid.18 To increase the number of antigens displayed per capsid, we turned to I-587 as an alternative insertion position. I-587 is located in the VP3 common region, corresponding to the tip of the second-highest protrusion at the 3-fold axis of symmetry.18 At this position, we inserted either the MHC class I-restricted Ova epitope SIINFEKL (AAV-Vac_Ova8587) or the MHC class II-restricted Ova epitope ISQAVHAAHAEINEAGR (AAV-Vac_Ova4587).
To test whether the four different engineered capsids can be produced as genome-containing vector particles, we packaged the capsids with vector genomes encoding for enhanced green fluorescent protein (eGFP). As a reference, AAV2 vectors with unmodified, i.e., wild-type capsids delivering the same vector genome, were produced. Western blot analysis revealed a shift in bands for genetically modified VPs (Figure S1) compared with wild-type VPs of AAV2, as reported previously by us and others.19,20 Furthermore, AAV2, AAV-Vac_Ova8587, and AAV-Vac_Ova4587 capsids showed the expected ratio of VP1:VP2:VP3. In contrast, both VP2-fusion-containing capsids showed a weaker signal for the VP2 fusion proteins and a slightly stronger band for VP1 compared with AAV2 (Figure S1).
None of the capsid modifications impacted capsid assembly or packaging efficiency (Table S1). However, concerning the infectivity, we observed a significant reduction in the transduction efficiency of vector particles with AAV-Vac_Ova8587 and AAV-Vac_Ova4587 capsids (Table S1). This reduction can be explained by the loss of function of the heparan sulfate proteoglycan binding motif, which is required for cell attachment of AAV2,21,22 because the two key residues of this binding motif, arginine 585 and arginine 588, become separated by the insertion of peptides at I-587.23
To restore the infectivity of the I-587-based vaccine candidates, we decided to produce these capsids as hybrid capsids, i.e., approximately half of the capsid subunits contain the epitope (either SIINFEKL in the case of AAV-Vac_Ova8587 or ISQAVHAAHAEINEAGR in the case of AAV-Vac_Ova4587), while the other half are wild-type capsid protein subunits. Capsids were packaged with a single-stranded DNA vector genome encoding the secreted form of Ova (sOva). Similarly, the full-length-cOva-VP2-fusion (AAV-Vac_cOvaVP2) and the Ova-epitopes-VP2-fusion (AAV-Vac_Ova4+8VP2) capsids, as well as the AAV2 wild-type capsids (AAV2::sOva), were packaged with the same vector genome. Vaccine candidates and AAV2::sOva were produced with comparable titers (Table 1). Western blot analysis confirmed the expected capsid composition (Figure 1). Specifically, for AAV-Vac_Ova8587 and AAV-Vac_Ova4587, wild-type and modified VPs were detectable as double bands for each of the three VPs, and for the VP2 fusion constructs, again a weaker signal compared with AAV2 was detected for the fusion proteins (Figure 1).
Table 1.
Characterization of sOva encoding AAV preparations
| Name | Capsid | Vector genome | Vector genomes/μL | Capsids/μL | Cap/vg |
|---|---|---|---|---|---|
| AAV2::sOva | unmodified | sOva, ss | 6.15 ×108 | 6.60 × 108 | 1.1 |
| AAV-Vac_Ova8587a | Ova CD8 epitope, position I-587 | sOva, ss | 4.68 × 108 | 8.60 × 108 | 1.8 |
| AAV-Vac_Ova4587a | Ova CD4 epitope, position I-587 | sOva, ss | 6.34 × 108 | 1.17 × 109 | 1.8 |
| AAV-Vac_Ova4+8VP2 | Ova CD4 + CD8 epitope, VP2 fusion | sOva, ss | 1.58 × 108 | 4.49 × 107 | 0.3 |
| AAV-Vac_cOvaVP2 | full-length cOva, VP2 fusion | sOva, ss | 2.02 × 108 | 3.16 × 108 | 1.6 |
sOva, secreted full-length ovalbumin; cOva, cytoplasmic ovalbumin; ss, single-stranded vector-genome conformation; a, produced as hybrids with a mixture of modified and wild-type AAV2 capsid proteins.
Following iodixanol density gradient ultracentrifugation and AVB-column chromatography purification, including a dialysis step, the genomic and capsid titers of the indicated sOva-encoding AAV preparations were determined by qPCR and ELISA, respectively. Based on these values, the packaging efficiency as capsids (Cap)/vector genome (vg) was calculated.
Figure 1.

Western blot analysis of sOva encoding AAV preparations
Vector genome-containing particles (1 × 1010) of the indicated AAV preparations, encoding for sOva, were separated on an 8% SDS-PAGE. The capsid protein-specific antibody B1 was used for visualizing the three different capsid proteins VP1, VP2, and VP3 in their wild-type as well as modified form (VP1, 87 kDa; VP2, 72 kDa; VP3, 62 kDa; Ova8+4-eGFP-VP2, 98 kDa; cOva-VP2, 96 kDa). AAV-Vac_Ova4587 or AAV-Vac_Ova8587 encoding for sOva were produced as hybrid capsids of wild-type and epitope-containing capsid proteins (MHC class I Ova epitope in the case of AAV-Vac_Ova8587 and MHC class II Ova epitope in the case of AAV-Vac_Ova4587). Therefore, double bands are visible in lanes 3 and 4.
Ideally, both MHC class I- and MHC class II-restricted epitopes should be present during the priming phase to induce humoral and T cell responses
All our vaccine candidates encode for sOva but differ in the Ova antigen displayed on the capsid (Table 1). To investigate whether it is sufficient that either MHC class I- or MHC class II-restricted epitopes are presented during the priming phase or whether both epitopes are required to support antigen-specific adaptive immune responses, we vaccinated C57BL/6j mice either with AAV-Vac_Ova4587 or AAV-Vac_Ova8587, or a 1:1 mixture of both vaccine candidates (Table 1; Figure 2A). Immune responses were compared with AAV2::sOva, our reference (Table 1). All cohorts were closely monitored for the induction of Ova257-264-specific CD8+ T cells and antigen-specific IgG antibodies through dextramer staining and ELISA, respectively.
Figure 2.

Presence of both capsid-displayed MHC class I- and MHC class II-restricted epitopes is more effective in supporting the induction of adaptive immune responses than the presence of either epitope
Experimental design as depicted (A). C57BL/6j mice (7 to 8 weeks old; n = 7 per group) received via intramuscular injection AAV2::sOva, AAV-Vac_Ova4587, or AAV-Vac_Ova8587 at a dose of 3 × 109 vector genome-containing particles (vg) per animal, or a 1:1 mixture of AAV-Vac_Ova4587 and AAV-Vac_Ova8587 (termed AAV-Vac_Ova4+8587) at a total dose of 6 × 109 vg per animal. Particles encoding for sOva (see Table 1). Blood and serum samples were collected on days (D) 14, 21, 43, and 63. Blood samples were evaluated by flow cytometry after staining with H-2Kb/Ova257-264 dextramer for the presence of Ova-specific CD8+ T lymphocytes (B): D14 + 21 + 63, AAV2::sOva, AAV-Vac_Ova4587, and AAV-Vac_Ova8587 n = 6; D43, AAV2::sOva and AAV-Vac_Ova4587 n = 5 and Vac_Ova8587 n = 6, and serum samples by ELISA for the presence of Ova-specific IgG antibodies (C). Statistical analysis: Kruskal-Wallis test with Dunns post test; ∗p ≤ 0.05, ∗∗p ≤ 0.01. Data are represented as mean with SEM.
As indicated in Figure 2B, vaccination with either the AAV-Vac_Ova4587 or the AAV-Vac_Ova8587 vaccine or the 1:1 mixture of both vaccine candidates (AAV-Vac_Ova4+8587) induced an antigen-specific CD8+ T cell response that peaked already at day (D) 21. In the AAV-Vac_Ova4+8587 cohort, the highest level was detected: approximately 16% of the total CD8+ population were antigen-specific CD8+ T cells (Figure 2B, right panel). The AAV-Vac_Ova4+8587 cohort thereby reached a significantly higher level than the AAV2::sOva control cohort (Figure 2B, right panel). The latter showed a peak in CD8+ T cell response on D43 (Figure 2B, left panel). The level of antigen-specific CD8+ T cells progressively declined after D21 in all cohorts vaccinated with the antigen-decorated AAV vector-based vaccine candidates, as expected for cellular immune responses but remained apparent until the experimental endpoint at D63, suggesting a persistent and robust immune response (Figure 2B, left panel). A decline was also observed for AAV2::sOva after D43, reaching nearly background level at D63 (Figure 2B, left panel).
Also, concerning the humoral immune response, differences between the cohorts receiving our antigen-decorated AAV vector-based vaccine candidates and the AAV2:sOva group were prominent (Figure 2C). The anti-Ova IgG response, measured in the AAV-Vac_Ova4+8587 cohort, was, for example, significantly higher compared with the reference AAV2::sOva and increased until D63 (Figure 2C).
Based on these results, we decided to use the 1:1 mixture of AAV-Vac_Ova4587 and AAV-Vac_Ova8587 vaccine candidates (AAV-Vac_Ova4+8587) in all subsequent experiments.
Insertion at position I-587 of the AAV2 capsid is superior to the N-terminal fusion to VP2 for induction of antigen-specific humoral as well as T cell responses
Next, we compared AAV-Vac_Ova4+8587 with the two VP2 fusion vaccine candidates, AAV-Vac_Ova4+8VP2 and AAV-Vac_cOvaVP2 (Table 1; Figure 3A). Ova257-264-specific CD8+ T cell response and antigen-specific IgG antibodies were again monitored by dextramer staining and ELISA, respectively. In addition, splenocytes were isolated at the experimental endpoint (D60) and analyzed via enzyme-linked immunospot (ELISpot) assays.
Figure 3.

Display of MHC class I- and MHC class II-restricted Ova epitopes at capsid position I-587 is superior in supporting the induction of adaptive immune responses compared with antigen insertions at the VP2 N terminus
Experimental design as depicted (A). C57BL/6j mice (7 to 8 weeks old; n = 7 per group) received via intramuscular injection AAV2::sOva, AAV-Vac_Ova4+8VP2, or AAV-Vac_cOvaVP2 at a dose of 3 × 109 vector genome-containing particles (vg) per animal, or AAV-Vac_Ova4+8587 at a total dose of 6 × 109 vg per animal. Blood and serum samples were collected on days (D) 14, 21, 28, 40, and 60. Blood samples were evaluated by flow cytometry after staining with H-2Kb/Ova257-264 dextramer for the presence of Ova-specific CD8+ T lymphocytes (B) (D14, Vac_Ova4+8587 n = 6). Splenocytes were isolated at D60 post-vaccine administration and evaluated by ELISpot assays. Quantification of specific T cells was performed by detection of INF-γ secretion upon in vitro re-stimulation with Ova immune-dominant peptides. Cells were re-stimulated with the MHC class I-restricted peptide Ova257-264 to determine the level of anti-Ova CD8+ T cell response (C) (left graph) or with the MHC class II-restricted peptide Ova323-339 to determine the level of anti-Ova CD4+ T cell response (C) (right graph). The number of spot-forming cells (SFCs) per 1 × 106 splenocytes is depicted. Serum samples were evaluated by ELISA for the presence of Ova-specific IgG antibodies (D). D60, Vac_Ova4+8587 and AAV-Vac_Ova4+8VP2 n = 6. Statistical analysis: Kruskal-Wallis test with Dunns post test; ∗p ≤ 0.05, ∗∗p ≤ 0.01. Data are represented as mean with SEM.
Mice vaccinated with our reference AAV2::sOva or with AAV-Vac_Ova4+8VP2 failed to induce antigen-specific T cells (Figure 3B). In contrast, mice receiving AAV-Vac_Ova4+8587 developed a significant Ova-specific CD8+ T cell response, which, as before, peaked at D21, representing this time approximately 6% of the total CD8+ T cell population. ELISpot assays confirmed these data on a functional level, as interferon-gamma (IFN-γ) secretion from Ova-specific CD8+ T cells was only detected in mice vaccinated with AAV-Vac_Ova4+8587 (Figure 3C, left panel). ELISpot assays also confirmed the presence and function of anti-Ova CD4+ T cells in this cohort (Figure 3C, right panel).
The mouse cohort receiving AAV-Vac_cOvaVP2 also showed an Ova-specific CD8+ T cell response, which peaked on D21 (Figure 3B). However, this response was weaker compared with the AAV-Vac_Ova4+8587-induced response, as confirmed by the near absence of CD8+ and CD4+ T cells in the AAV-Vac_cOvaVP2 group in the ELISpot assay at D60 (Figure 3C).
A humoral immune response above background was only detectable in the group vaccinated with AAV-Vac_Ova4+8587, starting from D29 post-vaccination onward, with a significantly increased level of anti-Ova IgG antibodies compared with the other vaccine candidates or AAV2::sOva (Figure 3D).
Thus, following a single intramuscular administration, AAV-Vac_Ova4+8587 induced a robust cellular and humoral immune response, out-competing our reference (AAV2::sOva) and the VP2 fusion containing vaccine candidates.
Single vaccine administration is sufficient for inducing anti-tumor protection against the immunogenic EG7 thymoma
To determine whether our antigen-decorated AAV vector-based vaccine can induce a protective, antigen-specific immune response against tumor engraftment by a single intramuscular administration, we challenged vaccinated C57/BL6j mice with syngeneic Ova-expressing EG7 thymoma cells (Figure 4). In detail, mice were vaccinated with AAV-Vac_Ova4+8587 at a total dose of 6 × 109 vaccine particles per mouse (AAV-Vac_Ova4+8587-high cohort) or with half the dose (AAV-Vac_Ova4+8587-low cohort). Again, we vaccinated a control cohort with our reference AAV2::sOva. A second control cohort received an unrelated vector, an AAV2 vector encoding for eGFP (AAV2::eGFP). On D15 post-vaccination, 1 × 106 EG7 tumor cells were injected subcutaneously into the flank, and tumor development was monitored for another 15 days (Figure 4A).
Figure 4.

Antigen-decorated AAV vector-based vaccine-induced Ova-specific immune responses potently inhibit the growth of EG7 tumor cells inoculated 15 days post-vaccination
Experimental design as depicted (A). C57BL/6j mice (7 to 8 weeks old; n = 8 per group) received via intramuscular injection AAV2::sOva or the negative control AAV2::eGFP at a dose of 3 × 109 vector genome-containing particles (vg) per animal, or AAV-Vac_Ova4+8587 at two different doses—either a total dose of 3 × 109 vg per animal (AAV-Vac_Ova4+8587-low) or a total dose of 6 × 109 vg per animal (AAV-Vac_Ova4+8587-high). Fifteen days after vaccination, 1 × 106 Ova-expressing EG7 syngeneic thymoma cells were injected subcutaneously into the flank of each mouse. Blood samples were collected on days (D) 14, 21, and 30, and serum samples on D30. Blood samples were evaluated by flow cytometry after staining with H-2Kb/Ova257-264 dextramer for the presence of Ova-specific CD8+ T lymphocytes (B). Splenocytes were isolated at D30 post-vaccination and evaluated by ELISpot assays for the IFN-γ secretion after in vitro re-stimulation. Cells were re-stimulated with the MHC class I-restricted peptide Ova257-264 to determine the level of anti-Ova CD8+ T cell response (C) (left graph) or with the MHC class II-restricted peptide Ova323-339 to determine the level of anti-Ova CD4+ T cell response (C) (right graph). Serum samples were evaluated by ELISA for the presence of Ova-specific IgG antibodies (D). Tumor growth was measured over time using a digital caliper. The mean tumor volumes in each indicated group as well as the percentage and number of tumor-bearing mice are depicted (E). Statistical analysis: Kruskal-Wallis test with Dunns post test; ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001. Data are represented as mean with SEM.
A robust and persistent Ova-specific CD8+ T cell response was only induced in the AAV-Vac_Ova4+8587-high and AAV-Vac_Ova4+8587-low cohorts, peaking at D14 and D21, respectively (Figure 4B). ELISpot assays confirmed the functionality of the Ova-specific CD8+ T cells (Figure 4C, left panel). In addition, both AAV-Vac_Ova4+8587-vaccinated groups showed higher levels of functional Ova-specific CD4+ T cells compared with the control groups (Figure 4C, right panel). These cohorts, in contrast to the AAV2::sOva cohort, also showed a significantly stronger IgG response at D30 (Figure 4D).
In line, we observed remarkable differences in tumor growth (Figure 4E). The most prominent tumor growth was observed—as expected—in the AAV2::eGFP cohort, where all eight mice (100%) developed tumors, closely followed by the AAV2::sOva cohort with tumors in seven of eight mice (88%). In both control cohorts, tumors were detectable from D5 post-tumor cell injection onward. In contrast, cohorts receiving AAV-Vac_Ova4+8587 were completely protected against tumor development. Indeed, even a thorough post-mortem examination did not reveal any signs of tumor engraftment in these two groups.
Single vaccine administration is sufficient for inducing anti-tumor protection against the aggressive B16/F10-Ova melanoma
Next, we tested our new vaccine platform in the poorly immunogenic and more aggressive B16/F10-Ova melanoma model. We inoculated mice with 5 × 105 B16/F10-Ova cells 15 days post-vaccination and monitored tumor growth for 15 more days (Figure 5A). As before, the cohorts receiving our antigen-decorated AAV vector-based vaccine differed significantly from the control cohorts regarding the level of Ova-specific CD8+ T cell responses (Figure 5B). ELISpot assays confirmed the presence of IFN-γ-secreting Ova-specific CD8+ T cells with significant differences between the AAV-Vac_Ova4+8587 cohorts and the AAV2::sOva control cohort (Figure 5C). The anti-Ova IgG responses in the AAV-Vac_Ova4+8587 cohorts were also more pronounced than in the AAV2::sOva-treated group (Figure 5D).
Figure 5.

Antigen-decorated AAV vector-based vaccine-induced Ova-specific immune responses potently inhibit the growth of B16/F10-Ova tumor cells inoculated 15 days post-vaccination
Experimental design as depicted (A). C57BL/6j mice (7 to 8 weeks old; n = 8 per group) received via intramuscular injection AAV2::sOva or the negative control AAV2::eGFP at a dose of 3 × 109 vector genome-containing particles (vg) per animal, or AAV-Vac_Ova4+8587 at two different doses—either a total dose of 3 × 109 vg per animal (AAV-Vac_Ova4+8587-low) or a total dose of 6 × 109 vg per animal (AAV-Vac_Ova4+8587-high). Fifteen days after immunization, 5 × 105 B16/F10-Ova syngeneic melanoma cells were injected subcutaneously into the flank of each mouse. Blood and serum samples were collected on days (D) 14, 21, and 30. Blood samples were evaluated by flow cytometry after staining with H-2Kb/Ova257-264 dextramer for the presence of Ova-specific CD8+ T lymphocytes (B). Splenocytes were isolated at D30 post-vaccination and evaluated by ELISpot assays to determine the number of anti-Ova CD8+ T cells secreting IFN-γ upon in vitro re-stimulation (C). Serum samples were evaluated by ELISA for the presence of Ova-specific IgG antibodies (D). Tumor growth was measured over time using a digital caliper. The mean tumor volumes in each indicated group as well as the percentage and number of tumor-bearing mice are depicted (E). Statistical analysis: Kruskal-Wallis test with Dunns post test; ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001. Data are represented as mean with SEM.
In line, all eight mice receiving AAV2::eGFP developed fast-growing tumors, which were detectable 7 days after tumor inoculation (Figure 5E). In the AAV2::sOva cohort, six out of eight mice (75%) developed tumors, although of lower volume compared with the AAV2::eGFP group. In contrast, in the AAV-Vac_Ova4+8587-low cohort injected with a total dose of 3 × 109 vaccine particles per animal, a tumor became detectable in two of eight mice (25%) on D13 post-tumor cell inoculation. In the AAV-Vac_Ova4+8587-high cohort receiving 6 × 109 vaccine particles per animal, only a single animal (13%) developed a detectable tumor, albeit already from D9 post-tumor cell inoculation onward.
In contrast to the EG7 model, post-mortem examinations of the B16/F10-Ova-treated animals at the endpoint (D30 post-vaccination) revealed additional small tumors, which were not visible prior to dissection (Figure S2). Thus, post-mortem tumors were detectable in all mice receiving AAV2::sOva (100%). In the cohorts vaccinated with the antigen-decorated AAV vector-based vaccine, 75% of the mice in the AAV-Vac_Ova4+8587-low cohort (six of eight mice) and 50% in the AAV-Vac_Ova4+8587-high cohort (four of eight mice) showed tumors. The growth of these tumors was, however, significantly suppressed (Figure S2).
In summary, a single intramuscular administration of the antigen-decorated AAV vector-based vaccine protected mice against tumor development in two different models, including the poorly immunogenic and aggressive B16/F10-Ova melanoma.
Majority of mice are protected against tumor development when challenged 80 days post-vaccination
We next examined the long-term protective potential of our antigen-decorated AAV vector-based vaccine in both tumor models by injecting the Ova-expressing tumor cells at D80 instead of D15 post-vaccination (Figures 6A [EG7] and 7A [B16/F10-Ova]). We opted for this time point as, in previous experiments, the level of Ova-specific CD8+ T cells had already substantially declined until D60 post-vaccination, representing the endpoint in these experiments (Figures 2 and 3).
Figure 6.

Antigen-decorated AAV vector-based vaccine-induced Ova-specific immune responses potently inhibit the growth of EG7 tumor cells injected 80 days post-vaccination
Experimental design as depicted (A). C57BL/6j mice (7 to 8 weeks old; n = 8 per group) received via intramuscular injection AAV2::sOva or the negative control AAV2::eGFP at a dose of 3 × 109 vector genome-containing particles (vg) per animal, or AAV-Vac_Ova4+8587 at two different doses—either a total dose of 3 × 109 vg per animal (AAV-Vac_Ova4+8587-low) or a total dose of 6 × 109 vg per animal (AAV-Vac_Ova4+8587-high). Eighty days after immunization, 1 × 106 Ova-expressing EG7 syngeneic thymoma cells were injected subcutaneously into the flank of each mouse. Blood samples were collected on days (D) 14, 21, 28, 73, 82, and 88, and serum samples on D17, 24, 31, and 95. Blood samples were evaluated by flow cytometry after staining with H-2Kb/Ova257-264 dextramer for the presence of Ova-specific CD8+ T lymphocytes (B). Splenocytes were isolated at D95 post-vaccination and evaluated by ELISpot assays to determine the number of anti-Ova CD8+- (C), (left graph) and anti-Ova CD4+- (C) (right graph) specific T cells secreting IFN-γ upon in vitro re-stimulation. Serum samples were evaluated by ELISA for the presence of Ova-specific IgG antibodies (D). Percentage and number of mice that remained tumor-free until day 10 post-tumor challenge are depicted (E). Statistical analysis: Kruskal-Wallis test with Dunns post test; ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001. Data are represented as mean with SEM.
Figure 7.

Antigen-decorated AAV vector-based vaccine-induced Ova-specific immune responses potently inhibit the growth of B16/F10-Ova tumor cells inoculated 80 days post-vaccination
Experimental design as depicted (A). C57BL/6j mice (7 to 8 weeks old; n = 8 per group) received via intramuscular injection AAV2::sOva or the negative control AAV2::eGFP at a dose of 3 × 109 vector genome-containing particles (vg) per animal, or AAV-Vac_Ova4+8587 at two different doses—either a total dose of 3 × 109 vg per animal (AAV-Vac_Ova4+8587-low) or a total dose of 6 × 109 vg per animal (AAV-Vac_Ova4+8587-high). Eighty days after vaccination, 5 × 105 B16/F10-Ova syngeneic melanoma cells were injected subcutaneously into the flank of each mouse. Blood and serum samples were collected on days (D) 14, 21, 28, 60, and 95. Blood samples were evaluated by flow cytometry after staining with H-2Kb/Ova257-264 dextramer for the presence of Ova-specific CD8+ T lymphocytes (B) (D60, AAV2::sOva n = 7; D95, AAV2::eGFP n = 7 and AAV2::sOva n = 6). Splenocytes were isolated at D95 post-vaccination and evaluated by ELISpot assays as in previous figures to determine the number of anti-Ova CD8+- (C) (left graph) and anti-Ova CD4+- (C) (right graph) specific T cells secreting IFN-γ upon in vitro re-stimulation Due to technical issues, not all samples could be analyzed (AAV2::eGFP n = 5, AAV2::sOva n = 2, AAV-Vac_Ova4+8587 low n = 5, AAV-Vac_Ova4+8587 n = 6). Serum samples were evaluated by ELISA for the presence of Ova-specific IgG antibodies (D). Percentage and number of mice that remained tumor-free until day 11 post-tumor challenge are depicted (E). Statistical analysis: Kruskal-Wallis test with Dunns post test; ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001. Data are represented as mean with SEM.
As in the previous experiments, the level of anti-Ova CD8+ T cells detected in the blood of AAV-Vac_Ova4+8587-treated mice peaked at D21, while mice vaccinated with the AAV2::sOva reference vector developed a weaker immune response that was detectable only after D28 and remained at a lower level over time (Figure 6B). ELISpot assays at study termination confirmed these results, as higher levels of immune responses were observed in the AAV-Vac_Ova4+8587-vaccinated cohort (Figure 6C). Similarly, AAV-Vac_Ova4+8587-vaccinated animals also showed a faster onset of the humoral immune response with higher levels of anti-Ova IgG titers than mice that received AAV2::sOva (Figure 6D).
On D10 post-EG7 inoculation (=D90 post-vaccination), two mice (25%) in the AAV2::eGFP cohort and five of eight mice (63%) in the AAV2::sOva cohort remained tumor free. In contrast, six of eight mice (75%) in the AAV-Vac_Ova4+8587-low cohort, and all the animals of the AAV-Vac_Ova4+8587-high cohort remained tumor free (100%) (Figure 6E). These results are in line with the levels of cellular and humoral immune responses detected over time and at the study’s endpoint (D95 post-vaccination) (Figures 6B–6E).
The same experimental scheme was used to evaluate a possible long-term protective effect in the more aggressive B16/F10-Ova melanoma tumor model (Figure 7A). Mice injected with the antigen-decorated AAV vector-based vaccine developed a robust anti-Ova CD8+ T cell response that peaked at D21 (Figure 7B). In contrast, mice vaccinated with the AAV2::sOva control vector developed a weaker immune response that became detectable from D28 onward and persisted at a lower level over time (Figure 7B). Also, ELISpot assays at study termination confirmed higher immune responses in the two AAV-Vac_Ova4+8587 cohorts (Figure 7C). Similar to the CD8+ T cell response, anti-Ova-specific IgG levels were detected earlier and reached higher levels in the cohorts treated with the AAV-Vac_Ova4+8587 vaccine compared with the AAV2::sOva control cohort (Figure 7D).
On D11 post-tumor cell inoculation (=D91 post-vaccination), all mice in the AAV2::eGFP cohort had visible tumors, while five of eight mice (63%) in the AAV2::sOva cohort remained tumor free (Figure 7E). In contrast, seven out of eight mice (88%) in both AAV-Vac_Ova4+8587 cohorts remained tumor free (Figure 7E).
Thus, a single intramuscular injection of our antigen-decorated AAV vector-based vaccine is sufficient to induce a long-lasting and functional antigen-specific adaptive immune response that confers tumor protection, even nearly 3 months post-vaccination.
Discussion
The use of AAV vectors per se is so far rather uncommon in cancer immunotherapy-related strategies.24,25,26,27,28 Similarly, if explored as a vaccine, AAV vectors are mainly used to overexpress antigens from vector genomes,29,30 like our reference AAV2::sOva. Here, we report on a novel concept, the use of antigen-decorated AAV vector particles to induce an efficient anti-tumor immune response by a single administration aiming to empower cancer immunotherapy approaches. Specifically, we demonstrated the induction of potent antigen-specific adaptive T cell and humoral immune responses. These specific immune responses protected mice from tumor development, even when mice were inoculated with tumor cells as late as 80 days post-vaccination.
AAV vectors are commonly considered poorly immunogenic, particularly when applied intramuscularly.31 Nevertheless, the viral capsid and the viral vector genome can be recognized through Toll-like receptor 2 (TLR2) and TLR9, respectively.32,33 The latter was proven to result in anti-capsid T cell immune responses when TLR9 signaling occurs in the presence of dendritic cells.34 TLR2 might serve a similar function as TLR2 engagement on dendritic cells was shown to play an important role in the induction of T cell responses.35 In the context of our antigen-decorated AAV vector-based vaccine, which displays the antigen within the symmetric capsid scaffold, TLR2-mediated sensing of the capsid by antigen-presenting cells (APCs) might contribute to the immune response priming in an adjuvant-like manner. Moreover, local inflammation and immune cell infiltration induced upon intramuscular injection36 are involved in the initial immune priming. Besides conventional APCs, muscle cells might contribute as they can function as non-professional APCs presenting incoming antigens via MHC class II to T cells in vivo.37
As we used a single-stranded AAV vector genome, transgene production occurs with a delay since second-strand synthesis must be accomplished prior to transgene expression. In muscle-directed gene therapy approaches, this fact represents a clear disadvantage since it takes up to 4 weeks to reach steady-state transgene expression levels.38 In contrast, this delay is beneficial in our setting, as it enables a time-based delimitation of the priming phase induced by the antigen-decorated capsid from the immune boost mediated by the antigen produced in and subsequently secreted from vaccine-transduced muscle cells.
The vaccine dose is an important parameter to consider in vaccine development. AAV vectors used in the clinics have proven an excellent safety profile even at high doses (e.g., 1 × 1012 vg/kg for AAV1 vector based, intramuscularly applied gene therapy against lipoprotein lipase deficiency).39 For the present study, significant lower AAV doses (i.e., 3–6 × 109 vg/animal) were sufficient to achieve significant protection, and doses are even lower than in other studies that have explored AAV as a vaccine platform.28,40,41
We introduced the overall concept of an antigen-decorated AAV vector-based vaccine in the context of infectious diseases, demonstrating that insertion of the Ag85A antigen as N-terminal fusion to VP2 induced in BALB/c mice a fast and long-lasting antigen-specific humoral immune response.12 Unexpectedly, neither full-length cOva nor the combination of MHC class I- and MHC class II-restricted Ova epitopes as an N-terminal fusion to VP2 induced an efficient antigen-specific humoral immune response in our present study (Figure 3). Differences between antigens (i.e., Ova vs. Ag85A) and mouse strains (i.e., C57BL/6 vs. BALB/c) may have played a role. As an additional factor, differences in the amount of antigen displayed on the capsid should be considered, as VP2 is not essential for capsid assembly.20 Consequently, our AAV VP2-fusion-derived preparations contained antigen-proficient and antigen-deficient particles.42 Albeit no direct comparison was possible due to the lack of samples, western blot analyses of the VP2-fusion-derived vaccines from our previous12 and current study (Figure 1) suggested that the Ag85A-VP2-fusion vaccine preparation contained more antigen-proficient particles than the full-length-cOva- or Ova-epitopes-VP2-fusion vaccine preparation.
Even with the possibility of enriching antigen-proficient VP2-based vaccine particles by applying affinity chromatography, the amount of antigen displayed per capsid would remain low compared with the display of epitopes at position I-587. Nieto et al. were the first to explore positions I-587 and I-453 for antigen display in the context of vaccination, focusing on virus-like particles (i.e., without delivering a vector genome) and reporting on antigen-specific humoral immune responses.11 We opted for I-587 based on the better performance reported by Nieto et al. and our experience with this position as an acceptor site in cell surface targeting.11,43 However, I-587 is neighboring the two main residues of the HSPG binding epitope of AAV2.21,22 Therefore, insertion of foreign sequences at I-587 is prone to destroy the HSPG binding motif, thereby impacting the transduction efficiency of AAV vectors23 or, as in our case, of an AAV vector-based vaccine. Thus, to restore the transduction efficiency while using I-587 to decorate our capsid with antigens, we had to opt for hybrid capsids in which we combined capsid subunits with wild-type sequence and capsid subunits containing the Ova epitope (MHC class I epitope in the case of AAV-Vac_Ova8587 and MHC class II epitope in the case of AAV-Vac_Ova4587).
Concerning its potency, our antigen-decorated AAV vector-based vaccine might be best compared with a recently reported study by Krotova et al.40 Like in our study, they aimed to protect mice from tumor growth by inducing an antigen-specific adaptive immune response.40 They used the same model antigen (Ova), administration route, and tumor model (B16/F10-Ova). However, the two studies differed as Krotova et al. focused on transducing dendritic cells by an AAV6 vector encoding for Ova and being optimized for transducing dendritic cells.40,44 In contrast, our study employed an Ova epitope-decorated AAV2 vector encoding for sOva in addition and focusing on muscle transduction. Krotova et al. obtained a higher level of antigen-specific CD8+ T cells in the blood on D14 post-vaccination (13%40 vs. 3%–10% in our study). Despite this higher level, they observed tumor growth in all their mice when administering tumor cells on D14 post-vaccination.40 Our results for the tumor challenge experiment on D15 post-vaccination revealed that up to 88% (Figure 5)—after dissection of the animals, still up to 50% (Figure S2) —of mice vaccinated with a single administration of AAV-Vac_Ova4+8587 remained tumor free. Even more impressive, when challenging our AAV-Vac_Ova4+8587-vaccinated mice with tumor cells at D80 post-vaccination, 88% of the mice remained tumor free, suggesting the development of a long-lasting T cell response (Figure 7).
We observed that the AAV2::sOva cohort showed better protection in the long-term (i.e., D80; Figures 6 and 7) than in the short-term (i.e., D15; Figures 4 and 5) experiments, which correlates very well with the slower onset of the T cell response, which peaked in these groups close to D80. Thus, the immune system of AAV2::sOva-treated animals in the long-term setting was at least partially sensitized to respond to the "incoming" tumor cells. This finding may indicate that a prolonged antigen expression, even without a delineated priming phase by antigen-decorated capsids, may also confer some protection by inducing a low-level T cell response re-stimulated over time by antigen re-encounter. However, also in the long-term setting, the cellular and humoral immune responses, as well as the functionality of T cells at study termination (i.e., as assessed by IFN-γ ELISpot assays but also by their efficiency to reject tumor cells), were impressively higher in the cohorts vaccinated with the antigen-decorated AAV vector-based vaccine than in the AAV2::sOva-treated cohort (Figures 6 and 7). A detailed characterization of the immunological response during the initial priming phase induced by our antigen-decorated AAV vector-based vaccine, which caused this difference, cannot be provided since we did not include cohorts receiving antigen-decorated AAV vectors encoding for an unrelated protein or antigen-decorated empty capsids. Nevertheless, our results highlight the advantage of our novel vaccine platform for cancer immunotherapy compared with the conventional design.
Taking another viewpoint, we estimated—based on the outcome of the here reported results—the probability of developing tumors (Figure S3) when vaccinated by a single administration of our antigen-decorated AAV vector-based vaccine compared with AAV2::sOva, representing the conventional vaccine design, and compared with the negative control (AAV2::eGFP). This estimation was performed for all four conditions, i.e., two tumor models in a short- and long-term setting, and again highlights the advantage of using an antigen-decorated AAV vector-based vaccine (AAV-Vac_Ova4+8587).
Taken together, in this study, we introduce the proof-of-concept of a novel AAV vector-based vaccine strategy facilitating the induction of anti-tumor immune responses. We demonstrated that a single administration of an antigen-decorated AAV vector-based vaccine induces protective antigen-specific adaptive immune responses in two distinct tumor models. Given the general weak anti-capsid immune responses seen in mouse models34,45 compared with human clinical trials,46 it can be anticipated that results with our new vaccine design might be even more impressive in humans. Furthermore, our strategy of decorating the capsids of antigen-encoding AAV vectors with epitopes of the same antigen that are MHC class I and class II specific can be easily translated to other antigens as algorithms have been developed that predict respective epitopes in conjunction with HLA haplotypes.47,48 From this experimental background, the switch from model antigens to tumor-associated antigens (i.e., epitopes), such as gp100 and tyrosinase,40 as well as combinations with other immunotherapy approaches, such as immune checkpoint inhibition, appear to be promising next steps.
Methods
Animals
Animal care and experimental procedures were approved by the Lower Saxony State Office for Consumer Protection and Food Safety (LAVES, Germany; reference number 33.12-42502-04-18/3060) or by the local institutional ethic committee (Cenomexa, project no. 32592) and were in compliance with the European directive 2010/63/EU. All possible efforts were taken to minimize the suffering of the animals.
C57BL/6j mice (7- to 8-week-old females) were purchased from Charles River (Sulzfeld, Germany) and housed at Hannover Medical School’s Animal Facility, or were purchased from Janvier Labs (Le Genest-Saint-Isle, France) and housed in the dedicated animal facility of UMRs1234 Labs (University of Rouen Normandie, Rouen, France).
After an acclimatization period of around 2 weeks, mice were immunized by intramuscular injection of the indicated AAV preparations in a total volume of 50 or 100 μL PBS into hind leg muscles. Animals received a total dose of either 3 × 109 or 6 × 109 vg per animal, as indicated. Blood and serum samples were taken at the indicated time points via retrobulbar punction. Due to technical issues, for some animals it was not possible to get sufficient blood for the analyses. The reduction in the number of animals in some experimental groups is indicated in the figure legends. For other reasons, reduced numbers of animals have always been indicated along with the reason in the figure legend. No other than the stated criteria were applied for including or excluding data points during the analysis.
At the endpoint, animals were euthanized by gradual CO2 inhalation followed by cervical dislocation. For endpoint analysis, blood, serum, and splenocytes were isolated. Tumors were inoculated on D15 or D80 by injecting 5 × 105 B16/F10-Ova or 1 × 106 EG7 tumor cells49,50 subcutaneously in a total volume of 200 μL PBS. The tumor growth was monitored for 15 days and measured with a digital caliper, with the endpoint analysis on D30 or D95, respectively.
Cloning of antigen-decorated AAV capsids
All capsid variants were based on AAV serotype 2 (AAV2). The MHC class I- (OVA257–264 SIINFEKL) or MHC class II-restricted (OVA323–339 ISQAVHAAHAEINEAGR) epitopes of Ova were cloned as oligonucleotides into pRC'99 (resulting in linker sequences: AS-epitope-PA) to produce the helper plasmids pRC-Ova_CD8 and pRC-Ova_CD4.51,52 The same Ova epitopes, separated by five glycine residues, were fused via an alanine linker to the coding sequence of eGFP of the peGFP-VP2 plasmid53 to produce the helper plasmid pOva_CD4_CD8-EGFP-VP2. We decided to use eGFP as a stuffer sequence to ensure that the two relatively small Ova epitopes are well displayed on the capsid surface when fused to the N terminus of VP2. Moreover, usage of the glycine and alanine linkers is based on Masuko et al.51 The coding sequence of a cytoplasmic form of Ova (where the sequence coding for amino acids 20 to 145 was removed to delete the internal signal sequence54) was fused to the VP2 N terminus instead of the eGFP coding sequence in the peGFP-VP2 plasmid53 to produce the helper plasmid pcOva-VP2.
AAV vector production
For the experiments described in Figure S1 and Table S1, the indicated capsids were packaged with a self-complementary (sc) AAV vector genome encoding for eGFP under transcriptional control of the cytomegalovirus (CMV) promoter. In contrast, a single-stranded (ss) AAV vector genome conformation was used for all other experiments. The vector plasmids were pCMV-sOva, a plasmid encoding for a secreted form of Ova (sOva) under transcriptional control of the CMV promoter,49 or pCMV-GFP, which was based on the pCMV-sOva plasmid but with the sOva coding sequence replaced by the eGFP coding sequence.
AAV vectors were produced and purified as described.55,56 For AAV2::sOva and AAV2::eGFP (scGFP or ssGFP), the helper plasmid pRC alone was used57 and combined with pCMV-sOva, pscGFP,58 or pCMV-GFP, respectively, and pXX6.55 AAV-Vac_Ova4587 or AAV-Vac_Ova8587 encoding for sc eGFP (Figure S1; Table S1) were produced with unique capsids by using pRC-Ova_CD4 or pRC-Ova_CD8, respectively, combined with pscGFP and pXX6. In all vaccination experiments, AAV-Vac_Ova4587 or AAV-Vac_Ova8587 were produced as hybrid capsids to restore transduction efficiency (see above and Table 1). Therefore, AAV-Vac_Ova4587 or AAV-Vac_Ova8587, encoding for sOva, were produced using a 1:1 M ratio of pRC (providing wild-type AAV2 capsid subunits) and one of the two helper plasmids (pRC-Ova_CD4 or pRC-Ova_CD8) combined with pCMV-sOva and pXX6. For the production of the VP2 fusion AAV vaccine candidates encoding for sc eGFP (Figure S1; Table S1), pRC-VP2ko53 and pcOva-VP2 or pOva_CD4_CD8-EGFP-VP2, pscGFP, and pXX6 were transfected into HEK293 in a 1:1:1:1 M ratio. For the production of the VP2 fusion AAV vaccine candidates encoding for sOva (Figure 3; Table 1) pRC-VP2ko53 and pcOva-VP2 or pOva_CD4_CD8-EGFP-VP2, pCMV-sOva, and pXX6 were transfected.
After purification by discontinuous iodixanol gradient ultracentrifugation, further purification and concentration steps were employed. Specifically, we used either affinity chromatography purification using an AVB Sepharose HP column (GE Healthcare, Solingen, Germany) with subsequent dialysis (Slide-A-Lyzer Dialysis Cassette G2; Thermo Fisher Scientific, Waltham, MA) (Figures 2 and 3) or ultrafiltration using Amicon Ultra-15 centrifugal filter units (Merck Millipore, Darmstadt, Germany) (Figures 4, 5, 6, and 7), both with 1× PBS/1 mM MgCl2/2.5 mM KCl2 as the final buffer.
Characterization of AAV preparations
Genomic titers were determined by absolute qPCR quantification using the Roche LightCycler 96 system (Roche Diagnostics, Mannheim, Germany) with transgene-specific primers (b-OVA-pA_fw GCTGAAAAACTCTGTCCCTTCC; b-OVA-pA_rev GCCACCCGTAGATCTCTCGAG; Ova_fw AAGCAGGCAGAGAGGTGGTA; Ova_rev GAATGGATGGTCAGCCCTAA) and a corresponding AAV vector plasmid standard curve. Capsid titers were quantified using the AAV2 Titration ELISA Kit (Progen, Heidelberg, Germany). Transducing titers were determined for the eGFP encoding vectors (Table S1) by incubating HEK293 cells with a serial dilution of the indicated AAV preparations in dilution steps of 1:3 (1–0.0003 μL in a total volume of 1 mL medium per 12-well plate). After 48 h, cells were analyzed by flow cytometry. Transducing titers were determined from the dilution that resulted in approximately 10% of transgene-expressing cells.19 For western blot analyses, the capsid protein-specific antibody B1 (Progen) and the goat anti-mouse IgG HRP secondary antibody (polyclonal; Cayman Chemical, Ann Arbor, MI) were used as described previously.19
Monitoring of immune responses
Flow cytometric analysis
The number of antigen-specific T cells was determined by dextramer staining. Specifically, 50–100 μL of whole blood or 1 × 106 isolated splenocytes were stained according to the manufacturer’s instructions with 2 μL/sample MHC I dextramer reagent (SIINFEKL dye: PE; Immudex, Virum, Denmark) in combination with 2 μL/sample TruStain FcX (anti-mouse CD16/32; clone 93; BioLegend, San Diego, CA). In addition, staining with PE/Cy7-CD44 (clone IM7), APC/Cy7-CD4 (clone RM4-5), APC-CD8a (clone 53–6.7), and FITC-CD45 (clone 30-F11) (BioLegend) was performed at a concentration of 0.25 μg/sample for each antibody. Flow cytometry measurement was conducted with the BD LSR II (BD Biosciences, Franklin Lakes, NJ) (Figures 5 and 7) or the BD LSRFortessa (BD Biosciences) (Figures 2, 3, 4, and 6), and analysis was performed with FlowJo 10 software.
ELISA
Antigen-specific humoral immune response was determined by ELISA. Therefore, Nunc MaxiSorp Flat-Bottom Plates (Thermo Fisher Scientific) were coated o/n at 4°C with 10 μg/mL ovalbumin in carbonate/bicarbonate coating buffer. All further incubation steps are done for 1 h at RT. As blocking buffer, 5% milk powder in 1× DPBS and, as washing buffer, 0.1% Tween 20 in 1× DPBS were used. Serum samples were pre-diluted at 1:800 with a subsequent 1:3 dilution row (8 dilutions per sample). Purified anti-chicken ovalbumin antibody (clone TOSG1C6; BioLegend) was equally diluted and served as a positive and inter-plate control. Goat anti-mouse IgG (H + L) Poly-HRP Secondary Antibody (polyclonal; Thermo Fisher Scientific) was used at a concentration of 0.25 μg/mL. The ready-to-use TMB substrate (Sigma/Merck, Darmstadt, Germany) was incubated on the plate for 15 min and stopped by the addition of 1 M H2SO4. Absorbance was measured at 450 nm with the Photometer SPECTRAmax 340PC 96-well plate reader (Molecular Devices, San Jose, CA) (Figures 5 and 7) or with the Multiskan FC photometer (Thermo Fisher Scientific) (Figures 2, 3, 4, and 6). The presented anti-Ova IgG titers correspond to the dilution yielding the half-maximal optical density obtained using the positive control.
In vitro T cell assay
T cell function was measured by ELISpot. Specifically, MultiScreen-IP ELISpot plates (Millipore, Darmstadt, Germany) were coated o/n at 4°C with purified anti-mouse IFN-γ antibody (clone AN-18; BioLegend) at a concentration of 4 μg/mL in 1× DPBS. Blocking of the plates was done with complete RPMI-1640 for at least 2 h in a humidified incubator at 37°C. The different antigens (ovalbumin CD8-epitope SIINFEKL (SL-8), ovalbumin CD4-epitope ISQAVHAAHAEINEAGR (IR-17); GeneCust, Boynes, France) were used at a final concentration of 10 μg/mL. Freshly isolated splenocytes were seeded at the indicated densities and incubated for 16 h o/n in a humidified incubator at 37°C and 5% CO2. For washing, 0.05% Tween 20 in 1× DPBS was used. The biotinylated IFN-γ monoclonal detection antibody (clone R4-6A2; Thermo Fisher Scientific) was diluted with 1× ELISA/ELISpot Diluent (Thermo Fisher Scientific) to reach a concentration of 1 μg/mL and incubated on the plates for 2 h at RT. Streptavidin-ALP (Mabtech, Nacka Strand, Sweden) was diluted 1:1,000 in 1× ELISA/ELISpot Diluent and incubated for 1 h at RT on the plate. For detection of spots, ready-to-use BCIP/NBT-plus substrate for ELISpot (Mabtech) was incubated for 10–15 min on the plates. Spots were counted with the ImmunoSpot ELISpot reader (CTL, Shaker Heights, OH) (Figures 5 and 7) or the S6 Macro M2 ELISpot reader (CTL) (Figures 2, 3, 4, and 6).
Statistical analysis
Statistical analysis was performed using Kruskal-Wallis tests with Dunn’s multiple comparison post tests. Data are presented as SEM. Significance values are defined as ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
All statistical analyses were performed in GraphPad Prism 5 (GraphPad Software, Northside, San Diego, CA).
Probability values for Figure S3 were generated with the function "dbinom()" in R:Base 4.3, with the experimentally observed frequencies as probabilities.
Data availability
This study includes no data deposited in external repositories.
Acknowledgments
The authors thank Prof. Jude Samulski (University of North Carolina at Chapel Hill) for providing pXX6. This work was funded by the Wilhelm Sander Foundation (2018.046.1 and 2018.046.2) to H.B., U.T.H., and S.A., and the BMBF and MWK Lower Saxony-funded Professorinnenprogramm Niedersachsen to H.B.
Author contributions
A.-C.F., R.H., L.P., M.B., M.D., N.M.J., and T.C.H. performed the experiments. A.-C.F. and R.H., together with S.A., U.T.H., and H.B., analyzed the data. A.-C.F. created the figures. A.-C.F. and P.J.-N. calculated the statistics. H.B., U.T.H., and S.A. designed the study, acquired funding, and supervised the experiments. H.B. wrote the initial manuscript.
Declaration of interests
H.B. is an inventor on patent applications focusing on AAV capsid engineering. The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.03.010.
Supplemental information
References
- 1.Wei S.C., Duffy C.R., Allison J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 2.Fesnak A.D., June C.H., Levine B.L. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer. 2016;16:566–581. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu S., Yi M., Qin S., Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol. Cancer. 2019;18:125. doi: 10.1186/s12943-019-1057-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Igarashi Y., Sasada T. Cancer vaccines: toward the next breakthrough in cancer immunotherapy. J. Immunol. Res. 2020;2020 doi: 10.1155/2020/5825401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sahin U., Oehm P., Derhovanessian E., Jabulowsky R.A., Vormehr M., Gold M., Maurus D., Schwarck-Kokarakis D., Kuhn A.N., Omokoko T., et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. 2020;585:107–112. doi: 10.1038/s41586-020-2537-9. [DOI] [PubMed] [Google Scholar]
- 6.Reinhard K., Rengstl B., Oehm P., Michel K., Billmeier A., Hayduk N., Klein O., Kuna K., Ouchan Y., Woll S., et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science. 2020;367:446–453. doi: 10.1126/science.aay5967. [DOI] [PubMed] [Google Scholar]
- 7.Saxena M., van der Burg S.H., Melief C.J.M., Bhardwaj N. Therapeutic cancer vaccines. Nat. Rev. Cancer. 2021;21:360–378. doi: 10.1038/s41568-021-00346-0. [DOI] [PubMed] [Google Scholar]
- 8.He Q., Mao Q., An C., Zhang J., Gao F., Bian L., Li C., Liang Z., Xu M., Wang J. Heterologous prime-boost: breaking the protective immune response bottleneck of COVID-19 vaccine candidates. Emerg. Microbes Infect. 2021;10:629–637. doi: 10.1080/22221751.2021.1902245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li C., Samulski R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020;21:255–272. doi: 10.1038/s41576-019-0205-4. [DOI] [PubMed] [Google Scholar]
- 10.Venkatakrishnan B., Yarbrough J., Domsic J., Bennett A., Bothner B., Kozyreva O.G., Samulski R.J., Muzyczka N., McKenna R., Agbandje-McKenna M. Structure and dynamics of adeno-associated virus serotype 1 VP1-unique N-terminal domain and its role in capsid trafficking. J. Virol. 2013;87:4974–4984. doi: 10.1128/JVI.02524-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nieto K., Weghofer M., Sehr P., Ritter M., Sedlmeier S., Karanam B., Seitz H., Müller M., Kellner M., Hörer M., et al. Development of AAVLP(HPV16/31L2) particles as broadly protective HPV vaccine candidate. PLoS One. 2012;7 doi: 10.1371/journal.pone.0039741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rybniker J., Nowag A., Janicki H., Demant K., Hartmann P., Büning H. Incorporation of antigens into viral capsids augments immunogenicity of adeno-associated virus vector-based vaccines. J. Virol. 2012;86:13800–13804. doi: 10.1128/JVI.01708-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrari F.K., Samulski T., Shenk T., Samulski R.J. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J. Virol. 1996;70:3227–3234. doi: 10.1128/Jvi.70.5.3227-3234.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borst J., Ahrends T., Bąbała N., Melief C.J.M., Kastenmüller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018;18:635–647. doi: 10.1038/s41577-018-0044-0. [DOI] [PubMed] [Google Scholar]
- 15.Ostroumov D., Fekete-Drimusz N., Saborowski M., Kühnel F., Woller N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. Life Sci. 2018;75:689–713. doi: 10.1007/s00018-017-2686-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sonntag F., Bleker S., Leuchs B., Fischer R., Kleinschmidt J.A. Adeno-associated virus type 2 capsids with externalized VP1/VP2 trafficking domains are generated prior to passage through the cytoplasm and are maintained until uncoating occurs in the nucleus. J. Virol. 2006;80:11040–11054. doi: 10.1128/JVI.01056-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Münch R.C., Janicki H., Völker I., Rasbach A., Hallek M., Büning H., Buchholz C.J. Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol. Ther. 2013;21:109–118. doi: 10.1038/mt.2012.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie Q., Bu W., Bhatia S., Hare J., Somasundaram T., Azzi A., Chapman M.S. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. USA. 2002;99:10405–10410. doi: 10.1073/pnas.162250899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boucas J., Lux K., Huber A., Schievenbusch S., von Freyend M.J., Perabo L., Quadt-Humme S., Odenthal M., Hallek M., Büning H. Engineering adeno-associated virus serotype 2-based targeting vectors using a new insertion site-position 453-and single point mutations. J. Gene Med. 2009;11:1103–1113. doi: 10.1002/jgm.1392. [DOI] [PubMed] [Google Scholar]
- 20.Warrington K.H., Jr., Gorbatyuk O.S., Harrison J.K., Opie S.R., Zolotukhin S., Muzyczka N. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J. Virol. 2004;78:6595–6609. doi: 10.1128/JVI.78.12.6595-6609.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kern A., Schmidt K., Leder C., Müller O.J., Wobus C.E., Bettinger K., Von der Lieth C.W., King J.A., Kleinschmidt J.A. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J. Virol. 2003;77:11072–11081. doi: 10.1128/JVI.77.20.11072-11081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Opie S.R., Warrington K.H., Jr., Agbandje-McKenna M., Zolotukhin S., Muzyczka N. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J. Virol. 2003;77:6995–7006. doi: 10.1128/JVI.77.12.6995-7006.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perabo L., Goldnau D., White K., Endell J., Boucas J., Humme S., Work L.M., Janicki H., Hallek M., Baker A.H., Büning H. Heparan sulfate proteoglycan binding properties of adeno-associated virus retargeting mutants and consequences for their in vivo tropism. J. Virol. 2006;80:7265–7269. doi: 10.1128/JVI.00076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hacker U.T., Bentler M., Kaniowska D., Morgan M., Büning H. Towards clinical implementation of adeno-associated virus (AAV) vectors for cancer gene therapy: current status and future perspectives. Cancers. 2020;12 doi: 10.3390/cancers12071889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kratky W., Reis e Sousa C., Oxenius A., Spörri R. Direct activation of antigen-presenting cells is required for CD8+ T-cell priming and tumor vaccination. Proc. Natl. Acad. Sci. USA. 2011;108:17414–17419. doi: 10.1073/pnas.1108945108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandya M., Britt K., Hoffman B., Ling C., Aslanidi G.V. Reprogramming immune response with capsid-optimized AAV6 vectors for immunotherapy of cancer. J. Immunother. 2015;38:292–298. doi: 10.1097/CJI.0000000000000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steel J.C., Di Pasquale G., Ramlogan C.A., Patel V., Chiorini J.A., Morris J.C. Oral vaccination with adeno-associated virus vectors expressing the Neu oncogene inhibits the growth of murine breast cancer. Mol. Ther. 2013;21:680–687. doi: 10.1038/mt.2012.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hensel J.A., Khattar V., Ashton R., Ponnazhagan S. Recombinant AAV-CEA tumor vaccine in combination with an immune adjuvant breaks tolerance and provides protective immunity. Mol. Ther. Oncolytics. 2019;12:41–48. doi: 10.1016/j.omto.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nieto K., Salvetti A. AAV vectors vaccines against infectious diseases. Front. Immunol. 2014;5:5. doi: 10.3389/fimmu.2014.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zabaleta N., Dai W., Bhatt U., Hérate C., Maisonnasse P., Chichester J.A., Sanmiguel J., Estelien R., Michalson K.T., Diop C., et al. An AAV-based, room-temperature-stable, single-dose COVID-19 vaccine provides durable immunogenicity and protection in non-human primates. Cell Host Microbe. 2021;29:1437–1453.e8. doi: 10.1016/j.chom.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brockstedt D.G., Podsakoff G.M., Fong L., Kurtzman G., Mueller-Ruchholtz W., Engleman E.G. Induction of immunity to antigens expressed by recombinant adeno-associated virus depends on the route of administration. Clin. Immunol. 1999;92:67–75. doi: 10.1006/clim.1999.4724. [DOI] [PubMed] [Google Scholar]
- 32.Hösel M., Broxtermann M., Janicki H., Esser K., Arzberger S., Hartmann P., Gillen S., Kleeff J., Stabenow D., Odenthal M., et al. Toll-like receptor 2-mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology. 2012;55:287–297. doi: 10.1002/hep.24625. [DOI] [PubMed] [Google Scholar]
- 33.Zhu J., Huang X., Yang Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J. Clin. Invest. 2009;119:2388–2398. doi: 10.1172/JCI37607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogers G.L., Shirley J.L., Zolotukhin I., Kumar S.R.P., Sherman A., Perrin G.Q., Hoffman B.E., Srivastava A., Basner-Tschakarjan E., Wallet M.A., et al. Plasmacytoid and conventional dendritic cells cooperate in crosspriming AAV capsid-specific CD8(+) T cells. Blood. 2017;129:3184–3195. doi: 10.1182/blood-2016-11-751040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chandran S.S., Verhoeven D., Teijaro J.R., Fenton M.J., Farber D.L. TLR2 engagement on dendritic cells promotes high frequency effector and memory CD4 T cell responses. J. Immunol. 2009;183:7832–7841. doi: 10.4049/jimmunol.0901683. [DOI] [PubMed] [Google Scholar]
- 36.Liang F., Loré K. Local innate immune responses in the vaccine adjuvant-injected muscle. Clin. Transl. Immunology. 2016;5:e74. doi: 10.1038/cti.2016.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin Y.Y., Belle I., Blasi M., Huang M.N., Buckley A.F., Rountree W., Klotman M.E., Cara A., Negri D. Skeletal muscle is an antigen reservoir in integrase-defective lentiviral vector-induced long-term immunity. Mol. Ther. Methods Clin. Dev. 2020;17:532–544. doi: 10.1016/j.omtm.2020.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCarty D.M., Fu H., Monahan P.E., Toulson C.E., Naik P., Samulski R.J. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther. 2003;10:2112–2118. doi: 10.1038/sj.gt.3302134. [DOI] [PubMed] [Google Scholar]
- 39.Gaudet D., Méthot J., Déry S., Brisson D., Essiembre C., Tremblay G., Tremblay K., de Wal J., Twisk J., van den Bulk N., et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther. 2013;20:361–369. doi: 10.1038/gt.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krotova K., Day A., Aslanidi G. An engineered AAV6-based vaccine induces high cytolytic anti-tumor activity by directly targeting DCs and improves Ag presentation. Mol. Ther. Oncolytics. 2019;15:166–177. doi: 10.1016/j.omto.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin J., Calcedo R., Vandenberghe L.H., Bell P., Somanathan S., Wilson J.M. A new genetic vaccine platform based on an adeno-associated virus isolated from a rhesus macaque. J. Virol. 2009;83:12738–12750. doi: 10.1128/JVI.01441-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Münch R.C., Muth A., Muik A., Friedel T., Schmatz J., Dreier B., Trkola A., Plückthun A., Büning H., Buchholz C.J. Off-target-free gene delivery by affinity-purified receptor-targeted viral vectors. Nat. Commun. 2015;6:6246. doi: 10.1038/ncomms7246. [DOI] [PubMed] [Google Scholar]
- 43.Büning H., Srivastava A. Capsid modifications for targeting and improving the efficacy of AAV vectors. Mol. Ther. Methods Clin. Dev. 2019;12:248–265. doi: 10.1016/j.omtm.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pandya J., Ortiz L., Ling C., Rivers A.E., Aslanidi G. Rationally designed capsid and transgene cassette of AAV6 vectors for dendritic cell-based cancer immunotherapy. Immunol. Cell Biol. 2014;92:116–123. doi: 10.1038/icb.2013.74. [DOI] [PubMed] [Google Scholar]
- 45.Martino A.T., Basner-Tschakarjan E., Markusic D.M., Finn J.D., Hinderer C., Zhou S., Ostrov D.A., Srivastava A., Ertl H.C.J., Terhorst C., et al. Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. Blood. 2013;121:2224–2233. doi: 10.1182/blood-2012-10-460733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verdera H.C., Kuranda K., Mingozzi F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol. Ther. 2020;28:723–746. doi: 10.1016/j.ymthe.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mei S., Li F., Leier A., Marquez-Lago T.T., Giam K., Croft N.P., Akutsu T., Smith A.I., Li J., Rossjohn J., et al. A comprehensive review and performance evaluation of bioinformatics tools for HLA class I peptide-binding prediction. Brief. Bioinform. 2020;21:1119–1135. doi: 10.1093/bib/bbz051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Racle J., Michaux J., Rockinger G.A., Arnaud M., Bobisse S., Chong C., Guillaume P., Coukos G., Harari A., Jandus C., et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat. Biotechnol. 2019;37:1283–1286. doi: 10.1038/s41587-019-0289-6. [DOI] [PubMed] [Google Scholar]
- 49.Hardet R., Chevalier B., Dupaty L., Naïmi Y., Riou G., Drouot L., Jean L., Salvetti A., Boyer O., Adriouch S. Oral-tolerization prevents immune responses and improves transgene persistence following gene transfer mediated by adeno-associated viral vector. Mol. Ther. 2016;24:87–95. doi: 10.1038/mt.2015.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hubert S., Rissiek B., Klages K., Huehn J., Sparwasser T., Haag F., Koch-Nolte F., Boyer O., Seman M., Adriouch S. Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2-P2X7 pathway. J. Exp. Med. 2010;207:2561–2568. doi: 10.1084/jem.20091154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masuko K., Wakita D., Togashi Y., Kita T., Kitamura H., Nishimura T. Artificially synthesized helper/killer-hybrid epitope long peptide (H/K-HELP): preparation and immunological analysis of vaccine efficacy. Immunol. Lett. 2015;163:102–112. doi: 10.1016/j.imlet.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 52.Nicklin S.A., Buening H., Dishart K.L., de Alwis M., Girod A., Hacker U., Thrasher A.J., Ali R.R., Hallek M., Baker A.H. Efficient and selective AAV2-mediated gene transfer directed to human vascular endothelial cells. Mol. Ther. 2001;4:174–181. doi: 10.1006/mthe.2001.0424. [DOI] [PubMed] [Google Scholar]
- 53.Lux K., Goerlitz N., Schlemminger S., Perabo L., Goldnau D., Endell J., Leike K., Kofler D.M., Finke S., Hallek M., Büning H. Green fluorescent protein-tagged adeno-associated virus particles allow the study of cytosolic and nuclear trafficking. J. Virol. 2005;79:11776–11787. doi: 10.1128/JVI.79.18.11776-11787.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boyle J.S., Koniaras C., Lew A.M. Influence of cellular location of expressed antigen on the efficacy of DNA vaccination: cytotoxic T lymphocyte and antibody responses are suboptimal when antigen is cytoplasmic after intramuscular DNA immunization. Int. Immunol. 1997;9:1897–1906. doi: 10.1093/intimm/9.12.1897. [DOI] [PubMed] [Google Scholar]
- 55.Xiao X., Li J., Samulski R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zolotukhin S., Byrne B.J., Mason E., Zolotukhin I., Potter M., Chesnut K., Summerford C., Samulski R.J., Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 57.Rossi A., Dupaty L., Aillot L., Zhang L., Gallien C., Hallek M., Odenthal M., Adriouch S., Salvetti A., Büning H. Vector uncoating limits adeno-associated viral vector-mediated transduction of human dendritic cells and vector immunogenicity. Sci. Rep. 2019;9:3631. doi: 10.1038/s41598-019-40071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hacker U.T., Wingenfeld L., Kofler D.M., Schuhmann N.K., Lutz S., Herold T., King S.B.S., Gerner F.M., Perabo L., Rabinowitz J., et al. Adeno-associated virus serotypes 1 to 5 mediated tumor cell directed gene transfer and improvement of transduction efficiency. J. Gene Med. 2005;7:1429–1438. doi: 10.1002/jgm.782. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study includes no data deposited in external repositories.