

Abstract
After herpes simplex virus type 1 (HSV-1) infection, the cytosolic sensor cyclic GMP-AMP synthase (cGAS) recognizes DNA and catalyzes synthesis of the second messenger 2′3′-cGAMP. cGAMP binds to the ER-localized adaptor protein MITA (also known as STING) to activate downstream antiviral responses. Conversely, HSV-1-encoded proteins evade antiviral immune responses via a wide variety of delicate mechanisms, promoting viral replication and pathogenesis. Here, we identified HSV-1 envelop protein UL56 as a negative regulator of cGAS-mediated innate immune responses. Overexpression of UL56 inhibited double-stranded DNA-triggered antiviral responses, whereas UL56-deficiency increased HSV-1-triggered induction of downstream antiviral genes. UL56-deficiency inhibited HSV-1 replication in wild-type but not MITA-deficient cells. UL56-deficient HSV-1 showed reduced replication in the brain of infected mice and was less lethal to infected mice. Mechanistically, UL56 interacted with cGAS and inhibited its DNA binding and enzymatic activity. Furthermore, we found that UL56 homologous proteins from different herpesviruses had similar roles in antagonizing cGAS-mediated innate immune responses. Our findings suggest that UL56 is a component of HSV-1 evasion of host innate immune responses by antagonizing the DNA sensor cGAS, which contributes to our understanding of the comprehensive mechanisms of immune evasion by herpesviruses.
Keywords: HSV-1, UL56, cGAS, Antiviral innate immunity, Immune evasion
Graphical abstract
One important feature of HSV-1 is to establish long-term persistent infection. Zheng et al. reveal that HSV-1 protein UL56 interacts with cGAS and inhibits its enzymatic activity, leading to suppression of cGAS-mediated innate antiviral response.
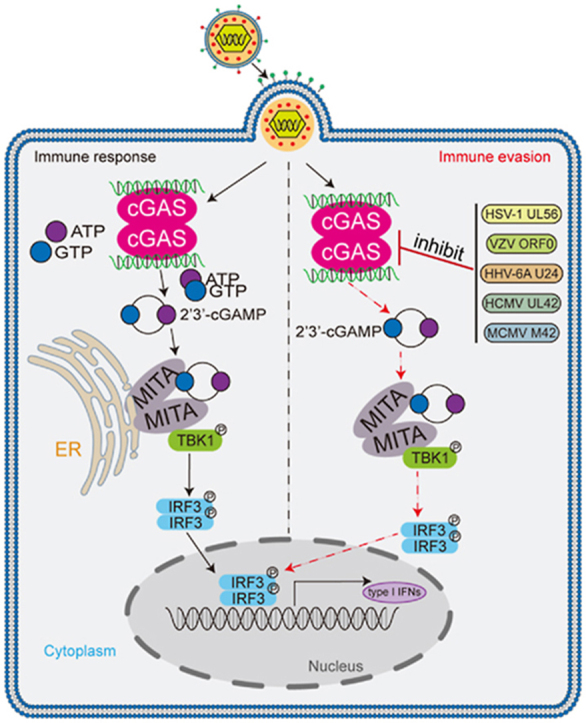
Highlights
-
•
HSV-1 UL56 negatively regulates cGAS-mediated innate immune response.
-
•
UL56-deficiency inhibits HSV-1 replication in cells and mice.
-
•
UL56 interacts with cGAS and inhibits its DNA binding and enzymatic activity.
-
•
Herpesvirus UL56 homologs are conserved in evading cGAS-mediated innate immunity.
1. Introduction
Upon infection of microbial pathogens, their conserved structural components called pathogen-associated molecular patterns (PAMPs) are recognized by host cellular pattern recognition receptors (PRRs) (Akira et al., 2006). This results in activation of downstream signaling cascades that lead to induction of downstream antiviral effector genes including type I interferons (IFNs) and proinflammatory cytokines, which promote innate and adaptive immune responses to clear pathogens (Akira et al., 2006; Hu and Shu, 2018; Medzhitov and Janeway, 2000).
Viral genomic DNA is a major PAMP of DNA virus. Upon infection, viral DNA is sensed by several cellular proteins (Atianand and Fitzgerald, 2013; Sun et al., 2013; Takaoka et al., 2007; Unterholzner et al., 2010; Zhang et al., 2011). Among them, cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) synthase (cGAS) has been shown to be a general cytosolic sensor of viral double-strand (ds) DNA (Li et al., 2013; Sun et al., 2013). After binding to dsDNA, cGAS utilizes ATP and GTP as substrates to catalyze synthesis of the second messenger 2′3′-cGAMP (Li et al., 2013; Wu et al., 2013). Subsequently, cGAMP binds to the endoplasmic reticulum (ER)-located adaptor protein MITA (also known as STING/MPYS/ERIS) (Ishikawa and Barber, 2008; Ishikawa et al., 2009; Jin et al., 2008; Sun et al., 2009; Zhong et al., 2008), which then traffics from the ER via Golgi apparatus to nuclear punctate structures. In these processes, MITA recruits TANK-binding kinase 1 (TBK1) and IκB kinase (IKK), leading to activation of the transcription factors interferon regulatory factor 3 (IRF3) and nuclear factor-kB (NF-κB), and ultimate induction of downstream antiviral genes (Barber, 2014; Hu and Shu, 2020; Zhang and Zhong, 2022, in press).
Herpes simplex virus type 1 (HSV-1), a member of the alpha herpesvirus family, infects about two-third of the world populations, which causes various mild and life-threatening diseases, including oral sores, herpetic keratitis, and sometimes meningitis and encephalitis (Looker et al., 2015; Wagner and Bloom, 1997). HSV-1 is an enveloped virus with a linear, large dsDNA genome encoding at least 84 proteins (Whitley and Roizman, 2001). Upon infection, HSV-1 triggers innate immune responses with distinct mechanisms. It has been shown that HSV-1 genomes can be sensed by cGAS in the cytoplasm, which signals MITA/STING-dependent innate antiviral response (Li et al., 2013; Sun et al., 2013). HSV-1 DNA can be sensed by the nuclear sensor IFI16, which then signals through MITA/STING by unknown mechanisms (Unterholzner et al., 2010). Several studies have also suggested that TLR3 plays a critical role in innate immune responses to HSV-1 (Lafaille et al., 2012; Zhang et al., 2007). In addition, HSV-1 infection can result in release of mitochondrial DNA into the cytoplasm, which is sensed by cGAS (He et al., 2021; West et al., 2015). No matter how HSV-1 is sensed by the innate immune system, various studies have demonstrated that the cGAS-MITA/STING pathways are critically involved in innate immune responses to HSV-1 in various cell types and mice (Hu et al., 2016; Ishikawa and Barber, 2008; Ishikawa et al., 2009; Li et al., 2013; Sun et al., 2013; Wu et al., 2013). In most individuals, HSV-1 causes latent infection and is activated under certain circumstances, suggesting that HSV-1 evolves strategies to counteract the host immune responses (Lin and Zheng, 2019; Su et al., 2016). It has been demonstrated that HSV-1 nuclear protein ICP0 impairs the nuclear relocalization of IFI16 and its stabilization (Orzalli et al., 2012); HSV-1 ICP27 interacts with activated TBK1 via its RGG motif to prevent phosphorylation of IRF3 (Christensen et al., 2016); HSV-1 ICP6 suppresses necroptosis of infected cells to facilitate viral replication by targeting RIP1/RIP3 (Guo et al., 2015; Huang et al., 2015; Wang et al., 2014).
Previously, it has been shown that HSV-1 HFEM strain, which has a deletion in the promoter region of its UL56 gene, was apathogenic for tree shrews and mice (Kehm et al., 1996). Replacement of the UL56 gene in the genome of the avirulent HSV-1 HFEM strain with that of virulent HSV-1 strain 17 or F, restores the virulent phenotype of the recombinant viruses (Kehm et al., 1996). These studies suggest that HSV-1 UL56 is a critical determinant of its virulence.
HSV-1 UL56 protein consists of 234 amino acids, with three PPxY motifs in its N-terminus and a hydrophobic domain between amino acid residues 217 and 234 (Kehm et al., 1996; Koshizuka et al., 2002). The UL56 protein is shown as a tail-anchored type II transmembrane protein and the C-terminal hydrophobic domain is required for anchoring it to cytoplasmic membranes (Koshizuka et al., 2002). It has also been shown that the N-terminus of UL56 is crucial for its localization to the Golgi apparatus and cytoplasmic vesicles (Koshizuka et al., 2002). As a component of HSV-1 virions, UL56 anchors to the inside of the viral envelope (Kehm et al., 1994, 1998).
In this study, we identified HSV-1 UL56 as a negative regulator of cGAS-mediated innate immune signaling. UL56 interacted with cGAS, and inhibited its binding to DNA and its enzymatic activity. We also found that UL56 homologs in other herpesviruses had conserved roles in evading cGAS-mediated innate antiviral responses. Our findings reveal a general mechanism on how HSV-1 and other herpesviruses evade innate immunity.
2. Results
2.1. HSV-1 UL56 inhibits viral DNA-triggered antiviral response
It has also been shown that HSV-1 UL56 is a critical determinant of its virulence (Berkowitz C. et al., 1993; Kehm et al., 1996). Therefore, we attempted to investigate whether HSV-1 UL56 is involved in antagonizing innate immune response to HSV-1. Since cGAS is an important innate sensor for HSV-1 infection (Li et al., 2013; Sun et al., 2013), we firstly determined whether HSV-1 UL56 regulates cGAS-mediated signaling. As shown in Fig. 1A, overexpression of UL56 markedly inhibited activation of the IFNβ promoter, ISRE and NF-κB mediated by overexpression of cGAS and MITA in 293 cells.
Fig. 1.

HSV-1 UL56 is an inhibitor of viral DNA-triggered innate antiviral response. (A) UL56 inhibits cGAS-MITA-mediated activation of the IFNβ promoter, ISRE and NF-κB. HEK293-MITA cells (1×105) were transfected with the IFNβ promoter (50 ng), ISRE (50 ng) or NF-κB (2 ng) reporter plasmids, and expression plasmids for cGAS (100 ng), or an empty vector as well as the UL56 plasmids for 24 h before luciferase assays were performed. The immunoblot analysis was performed with the indicated antibodies. (B) UL56 inhibits HSV-1- and HCMV-, but not SeV-triggered transcription of antiviral genes in HFFs. HFFs stably-expressing UL56 or control cells were left uninfected or infected with HSV-1 (MOI=1), HCMV (MOI=1) or SeV (MOI=1) for the indicated times before qPCR analysis. (C) UL56 inhibits HSV-1-triggered transcription of antiviral genes. MLFs stably-expressing UL56 or control cells were left uninfected or infected with HSV-1 (MOI=1) for the indicated times before qPCR analysis. (D) Both the N-terminal PPxY motifs and C-terminal transmembrane domain of UL56 are essential for its inhibition of HSV-1-triggered transcription of antiviral genes. MLFs stably-expressing UL56, transmembrane domain-deletion (ΔTM) or PY motif-mutated (P/A) mutant were left uninfected or infected with HSV-1 (MOI=1) for 6 h before qPCR analysis. (E) UL56 inhibits UV-inactivated HSV-1-triggered transcription of antiviral genes. HFFs stably-expressing UL56 were left uninfected or infected with untreated or UV-inactivated HSV-1 (MOI=1) for the indicated times before qPCR analysis. (F) UL56 inhibits dsDNA-triggered transcription of antiviral genes. HFFs and MLFs stably-expressing UL56 were mock-transfected or transfected with HSV120 for the indicated times, or with HT-DNA or DNA90 for 4 h before qPCR analysis. (G) UL56 inhibits dsDNA-triggered phosphorylation of MITA, TBK1, and IRF3. MLFs stably-expressing UL56 were mock-transfected or transfected with HSV120 for the indicated times before immunoblot analysis was performed with the indicated antibodies.
The data shown are means ± SD (A–F) from one representative experiment performed in triplicates (3 technical repeats). Similar data were obtained from at least two independent experiments. ∗, p < 0.05; ∗∗, p < 0.01 (Student's unpaired t-test).
Previously, it has been shown that HSV-1 infection activates the cGAS-MITA pathways in human primary foreskin fibroblasts (HFFs) and SV40-immortalized murine lung fibroblasts (MLFs) (Li and Shu, 2020; Li et al., 2013). We established stable UL56-expressing HFFs and MLFs by lentiviral-mediated transduction. qPCR analysis indicated that ectopic expression of UL56 inhibited transcription of downstream antiviral genes including IFNB1, ISG56, CXCL10 and IL6 induced by infection of HSV-1 and HCMV in HFFs (Fig. 1B). In these experiments, UL56 did not affect the RNA virus Sendai virus (SeV)-induced transcription of downstream genes (Fig. 1B). Similarly, UL56 also inhibited HSV-1-triggered transcription of downstream antiviral genes in MLFs (Fig. 1C). Deletion of the C-terminal transmembrane domain or mutation of the N-terminal PPxY motifs of UL56 impaired its inhibitory effects on HSV-1-induced transcription of downstream genes (Fig. 1D), suggesting that the proper activity and cellular localization of UL56 is important for its inhibition of innate antiviral responses. Furthermore, we investigated the effects of UL56 on transcription of antiviral genes induced by UV-inactivated HSV-1, which can infect host cells but does not undergo viral transcription and translation after infection. We found that UV-inactivated HSV-1 induced significantly higher mRNA levels of IFNB1, ISG56, CXCL10 and IL6 genes than un-treated HSV-1, and ectopic expression of UL56 inhibited transcription of these antiviral genes induced by both UV-treated and un-treated HSV-1 (Fig. 1E). These results suggest that UL56 inhibits viral DNA-triggered innate immune responses. Consistently, UL56 markedly inhibited transcription of downstream antiviral genes induced by transfection of synthetic dsDNAs, including 120-mer dsDNA representing the genome of HSV-1 (HSV120), dsDNA of approximately 90 bp (dsDNA90), and herring testis DNA (HT-DNA) (Fig. 1F). In addition, UL56 also inhibited phosphorylation of MITA, TBK1, and IRF3 induced by transfected HSV120 (Fig. 1G). These results suggest that UL56 negatively regulates DNA-induced innate immune responses.
2.2. UL56-deficiency potentiates HSV-1-triggered antiviral response
To further evaluate the effects of UL56 on HSV-1 immune evasion, we generated a UL56-deficient HSV-1 strain (HSV-1ΔUL56) by introducing a point mutation (C→A) at the 8th nucleotide of the UL56 coding sequence by bacterial artificial chromosome (BAC) genetic operating system, which resulted in a premature stop codon at this position. This mutation was confirmed by sequencing of the parental HSV-1 and HSV-1ΔUL56 genomes (Fig. S1). Sequence alignment indicated that no other sites in the HSV-1ΔUL56 genome were mutated (Fig. S1).
We then examined the effects of UL56-deficiency on HSV-1-triggered transcription of downstream antiviral genes in various cells including human monocytic THP-1 cells, HFFs, MLFs, and mouse bone marrow-derived macrophages (BMDMs). The results indicated that mRNA levels of antiviral genes induced by HSV-1ΔUL56 were markedly higher than those induced by wild-type HSV-1 in these cells (Fig. 2A). Consistently, UL56-deficiency also increased HSV-1-induced phosphorylation of MITA, TBK1, and IRF3 in THP-1 and MLF cells, (Fig. 2B). However, UL56-deficiency did not affect the course of viral gene expression in this stage (Fig. 2C). These results suggest that UL56 plays a pivotal role in suppression of innate immune responses against HSV-1 infection.
Fig. 2.

UL56-deficiency enhances HSV-1-induced antiviral response. (A) UL56-deficiency enhances HSV-1-induced transcription of antiviral genes. The indicated cells were infected with HSV-1 (MOI=1) or HSV-1ΔUL56 (MOI=1) for the indicated times before qPCR analysis. (B-C) UL56-deficiency enhances HSV-1-induced phosphorylation of MITA, TBK1, and IRF3. THP-1 and MLF cells were infected with HSV-1 (MOI=1) or HSV-1ΔUL56 (MOI=1) for the indicated times before immunoblot analysis was performed with the indicated antibodies (B). The lower panels were results of RT-PCR analysis for viral and cellular mRNA (C).
The data shown are means ± SD (A) from one representative experiment performed in triplicates (3 technical repeats). Similar data were obtained from at least two independent experiments. ∗, p < 0.05; ∗∗, p < 0.01 (Student's unpaired t-test).
We next investigated the effects of UL56 on HSV-1 immune evasion. Plaque assays indicated that overexpression of UL56 markedly enhanced HSV-1 replication (Fig. 3A & S2A). In contrast, HSV-1ΔUL56 produced less progeny virions than wild-type HSV-1 after infection in MLFs and HFFs (Fig. 3B & S2B). To determine whether UL56 promotes HSV-1 replication by impairing cGAS-MITA-mediated innate immune responses, we infected MITA-deficient MLFs and HFFs with wild-type or UL56-deficient HSV-1. As shown in Fig. 3C & S2C, comparable amounts of progeny virions of wild-type and UL56-deficient HSV-1 were detected in MITA-deficient cells, suggesting that UL56 enhances HSV-1 replication by antagonizing cGAS-MITA-mediated signaling. Consistently, similar kinetics of production of wild-type and UL56-deficient HSV-1 virions were observed in Vero cells that are interferon defective (Fig. 3D). Additionally, qPCR experiments indicated that UL56-deficiency had no effects on transcription of HSV-1 genome in Vero cells (Fig. 3E). These results suggest that UL56 does not affect HSV-1 replication in IFN-deficient cells, which is consistent with previous reports (Soh et al., 2020; Ushijima et al., 2008). We also investigated whether UL56 affects HSV-1 infection during the early phase. qPCR analysis of viral genomes as indicated by UL1, UL19, UL36, and UL49 genes showed similar levels in cells infected with wild-type and UL56-deficient HSV-1 (Fig. 3F & S2D), suggesting that UL56 has no marked effects on HSV-1 infection at the early phase. Taken together, these results suggest that UL56 promotes HSV-1 replication by antagonizing cGAS-mediated innate immune responses.
Fig. 3.

UL56 promotes HSV-1 replication. (A) UL56 promotes replication of HSV-1. MLFs stably-expressing UL56 were infected with HSV-1 (MOI=1) for 48 h. The infective supernatant was collected for viral plaque assays. (B) UL56-deficiency impairs the replication of HSV-1. MLFs were infected with HSV-1 (MOI=1) or HSV-1ΔUL56 (MOI=1) for 48 h, then the infective supernatant was collected for viral plaque assays. (C) UL56-deficiency impairs the replication of HSV-1 via the cGAS-MITA axis. MITA-deficient (Mita-KO) MLFs were infected with HSV-1 (MOI=1) or HSV-1ΔUL56 (MOI=1) for 48 h, then the infective supernatant was collected for viral plaque assays. (D) Effects of UL56-deficiency on HSV-1 replication in Vero cells. Vero cells were infected with HSV-1 (MOI=1) or HSV-1ΔUL56 (MOI=1) for the indicated times. The infective supernatant was collected for viral plaque assays. (E) Effects of UL56-deficiency on HSV-1 gene expression. Vero cells were infected with HSV-1 (MOI=1) or HSV-1ΔUL56 (MOI=1) for the indicated times before qPCR analysis. (F) Effects of UL56-deficiency on HSV-1 infection. MLF cells were infected with HSV-1 (MOI=1) or HSV-1ΔUL56 (MOI=1) for the indicated times. DNA was extracted for examination of HSV-1 genes by qPCR analysis. (G) Effects of UL56-deficiency on HSV-1-induced death of mice. Age- and gender-matched C57BL/6 mice (n=10 per strain, 7–9 weeks old) were intra-nasally infected with HSV-1 or HSV-1ΔUL56 (2.5×107 PFU per mouse), and the survival of infected mice were monitored daily. (H) Measurements of viral genomic copy numbers. Age- and gender-matched C57BL/6 mice (n=3 per strain, 7–9 weeks old) were intra-nasally infected with HSV-1 or HSV-1ΔUL56 (2.5×107 PFU per mouse) for 3 days. The brains were collected, HSV-1 copy numbers were quantified by qPCR with primers targeting viral gene UL27. Each symbol represents an individual mouse, and the copy number of HSV-1 in the brains of mice infected wild-type HSV-1 was normalized to 1. (I) Effects of UL56-deficiency on HSV-1-induced transcription of antiviral genes in the brains of infected mice. Age- and gender-matched C57BL/6 mice (n=8 per strain, 7–9 weeks old) were intra-nasally infected with HSV-1 or HSV-1ΔUL56 (2.5×107 PFU per mouse) for 2 days. The brains were collected for qPCR analysis. Each symbol represents an individual mouse, and the transcription of antiviral genes in the brains of mice infected wild-type HSV-1 was normalized to 1. (J) Effects of UL56-deficiency on serum levels of CXCL10, IL-6 and TNF-α induced by HSV-1. Age- and gender-matched C57BL/6 mice (n=10 per strain, 7–9 weeks old) were intra-nasally infected with HSV-1 or HSV-1ΔUL56 (2.5×107 PFU per mouse) or uninfected (n=5) for 10 h before measurement of the indicated serum cytokines by ELISA. Each symbol represents an individual mouse.
The data shown are means ± SD (A–F) from one representative experiment performed in triplicates (3 technical repeats). Similar data were obtained from at least two independent experiments. ∗, p < 0.05; ∗∗, p < 0.01 (Student's unpaired t-test for A-F or log-rank test for G-J).
To further demonstrate the importance of UL56 in HSV-1 infection and pathogenicity in vivo, we intra-nasally infected C57BL/6 mice with wild-type HSV-1 and HSV-1ΔUL56. We found that mice infected with wild-type HSV-1 developed ataxia and paralysis, and all died by 6 days after infection. In contrast, mice infected with HSV-1ΔUL56 virus had less pathogenic symptoms and half of the infected mice survived at 10 days after infection (Fig. 3G). In addition, the amounts of HSV-1 genomic copy numbers of HSV-1ΔUL56 were ∼2% of that of wild-type HSV-1 in the brains of infected mice (Fig. 3H). We also examined the mRNA levels of antiviral genes including Ifnb1, Isg56 and Tnf in the brains of infected mice. As shown in Fig. 3I, the mRNA level of Ifnb1, Isg56 and Tnf was markedly increased in the brains of mice infected with HSV-1ΔUL56. Moreover, ELISA results showed that, for mice infected with HSV-1ΔUL56, the level of serum cytokines such as TNF, IL-6 and CXCL10 was higher than those infected with wild-type HSV-1 (Fig. 3J). These results suggest that UL56 acts as a virulence factor for HSV-1 by antagonizing host innate immune responses.
2.3. UL56 impairs cGAS activation
To further investigate the mechanisms on UL56-mediated inhibition of innate immune responses, we performed co-immunoprecipitation experiments to identify its potential cellular targets. The results indicated that UL56 interacted with cGAS but not MITA, TBK1, IRF3, IKKβ, or P65 (Fig. 4A). Confocal microscopy showed that ectopically-expressed UL56 was associated with cGAS in the cytoplasm (Fig. 4B). Endogenous co-immunoprecipitation experiments indicated that UL56 interacted with cGAS following HSV-1 infection in MLFs (Fig. 4C). GST pull-down assays with recombinant proteins indicated that UL56 directly bound to cGAS in vitro (Fig. 4D).
Fig. 4.

UL56 interacts with cGAS. (A) Association of UL56 with cGAS. HEK293T cells were transfected with the indicated plasmids for 24 h. Co-immunoprecipitation and immunoblot analysis were performed with the indicated antibodies. (B) Co-localization of UL56 with cGAS. HeLa cells stable expressing Flag-cGAS (green) were transfected with HA-UL56 (red) for 20 h before confocal microscopy. Nuclei were stained with DAPI (blue). The cells were observed with a Nikon confocal microscope under a 60 × oil objective. (C) Endogenous association of UL56 with cGAS following HSV-1 infection. MLFs were infected with HSV-1 (MOI=0.5) for the indicated times. Co-immunoprecipitation and immunoblot analysis were performed with the indicated antibodies. (D) UL56 directly interacts with cGAS. GST or GST-UL56 was bound to glutathione agarose beads and incubated with purified His-cGAS for 3 h. Immunoblot analysis was performed with the indicated antibodies.
Previous studies have demonstrated that DNA binding of cGAS is necessary for its enzymatic activity and the downstream signaling (Civril et al., 2013; Li et al., 2013). Pull-down assays indicated that UL56 inhibited the binding of cGAS to the synthetic dsDNA HSV120 in vitro (Fig. 5A). It is noted that UL56 could also bound to HSV120 in these experiments (Fig. 5A). Microscale thermophoresis technology (MST) experiments indicated that cGAS could bind to the Cy5-dyed synthetic dsDNA HSV60 with an affinity of Kd = 372 ± 32 nM, whereas the presence of UL56 caused an approximately 4.5-fold decrease of the affinity (Kd =1660 ± 30 nM) between cGAS and dsDNA (Fig. 5B). We also examined whether UL56 inhibits binding of cGAS to viral DNA in HSV-1 infected cells by “foot-print” experiments. After HSV-1 infection, cGAS was immunoprecipitated, and its-bound viral DNA was extracted and analyzed by qPCR with primers targeting various regions of HSV-1 genome. The results indicated that overexpression of UL56 inhibited binding of cGAS to HSV-1 DNA (Fig. 5C). Consistently, the levels of cGAS-bound viral DNA of HSV-1 ΔUL56 were marked higher than those of wild-type HSV-1 following infection of MLFs (Fig. 5D). These results suggest that UL56 impairs the recognition of viral DNA by cGAS.
Fig. 5.

UL56 impairs cGAS binding to viral DNA. (A) UL56 impairs the binding of cGAS to dsDNA. HEK293T cells were transfected with the indicated plasmids. Twenty-four hours later, the cell extracts were incubated with biotinylated-HSV120 and streptavidin agarose for 3 h. The bound proteins were analyzed by immunoblots with the indicated antibodies. (B) MST measurement of binding affinities. Binding affinities between GST-cGAS and the synthetic dsDNA HSV60 in the presence of UL56 or BSA were measured by MST. (C) UL56 impairs the binding of cGAS to HSV-1 DNA. HEK293T cells were transfected with HA-cGAS and Flag-UL56 or an empty vector for 20 h. After transfection, cells were infected with HSV-1 (MOI=1) for 3 h. The cell lysate was then immunoprecipitated with control mouse IgG or anti-HA. The protein-bound DNA was extracted and analyzed by qPCR analysis. Specific cGAS-bound DNA amount in each sample was calculated by subtraction of anti-HA-precipitated DNA minus control mouse IgG-precipitated DNA. The specific cGAS-bound DNA amount from the empty vector-transfected sample was treated as 1.0, and the relative specific cGAS-bound DNA amount from UL56-transfected sample was calculated by dividing the specific raw DNA amount with the specific cGAS-bound DNA amount from the empty vector-transfected sample. (D) Deficiency of UL56 increases binding of endogenous cGAS to HSV-1 DNA. MLFs were infected with HSV-1 (MOI=1) or HSV-1ΔUL56 (MOI=1) for 12 h. The cell lysate was then immunoprecipitated with control mouse IgG or anti-cGAS (mouse). The protein-bound DNA was extracted and analyzed by qPCR analysis. Specific cGAS-bound DNA amount in each sample was calculated by subtraction of anti-cGAS-precipitated DNA minus control IgG-precipitated DNA. The specific cGAS-bound DNA amount from HSV-1-infected sample was treated as 1.0, and the relative specific cGAS-bound DNA amount from HSV1ΔUL56-infected sample was calculated by dividing the specific raw DNA amount with the specific cGAS-bound DNA amount from the HSV-1-infected sample. (E) UL56 inhibits HT-DNA- or HSV-1-induced synthesis of cGAMP. THP-1 cells stably-expressing UL56 or control cells were transfected with HT-DNA for 4 h or infected with HSV-1 (MOI=1) for the indicated times. cGAMP in the cell extracts was measured by ELISA. (F) UL56 inhibits cGAS enzymatic activity in vitro. Purified His-cGAS and His-UL56 or BSA were mixed with ATP, GTP, and HT-DNA in the reaction buffer. After incubation, samples were collected for Mono Q analysis of cGAMP production. (G) Effects of UL56 on cGAMP-induced transcription of antiviral genes. Digitonin-permeabilized MLFs stably-expressing UL56 or control cells (1×106) were transfected with cGAMP (0.1 μg) for 3 h before qPCR analysis.
The data shown are means ± SD (C-E, G) from one representative experiment performed in triplicates (3 technical repeats). Similar data were obtained from at least two independent experiments. ∗, p < 0.05; ∗∗, p < 0.01 (Student's unpaired t-test).
Previously, it has been shown that cGAS binding to DNA induces its enzymatic activation (Li et al., 2013). We found that synthesis of cGAMP induced by transfected HT-DNA or HSV-1 infection was markedly reduced by ectopic expression of UL56 in THP-1 cells (Fig. 5E). In vitro enzymatic assays also indicated that UL56 inhibited the synthesis of cGAMP by cGAS (Fig. 5F). However, qPCR assays showed that UL56 had no marked effects on cGAMP-induced transcription of Ifnb1, Il6, and Cxcl10 genes (Fig. 5G). Taken together, these results suggest that UL56 inhibits cGAS enzymatic activity but not downstream signaling events.
2.4. Conserved functions of herpesvirus UL56 homologs in antagonizing cGAS
The N-terminal PPxY motifs and C-terminal transmembrane domain of HSV-1 UL56 are conserved with proteins of other herpesviruses, such as ORF0 of Varicella-zoster virus (VZV), U24 of Human herpesvirus 6A (HHV-6A), UL42 of Human cytomegalovirus (HCMV), and M42 of Murine cytomegalovirus (MCMV) (Koshizuka et al., 2016, 2018) (Fig. 6A). To determine whether UL56 homologs in herpesviruses have conserved functions, we examined their abilities to inhibit cGAS-mediated innate immune responses. We found that HSV-1 UL56 homologs, including VZV ORF0, HHV-6A U24, HCMV UL42 and MCMV M42, inhibited cGAS-mediated activation of the IFNβ promoter, ISRE and NF-κB in reporter assays (Fig. 6B). qPCR analysis indicated that ectopic expression of UL56 homologs markedly inhibited transcription of IFNB1 and CXCL10 genes induced by HSV-1 infection (Fig. 6C) or transfection of HT-DNA (Fig. 6D). Co-immunoprecipitation experiments indicated that cGAS interacted with these UL56 homologs (Fig. 6E). Pull-down assays indicated that the UL56 homologs also impaired cGAS binding to dsDNA (Fig. 6F). These results suggest that herpesvirus UL56 homologs have an evolutionarily conserved function in antagonizing cGAS-mediated innate immune responses.
Fig. 6.

Herpesvirus UL56 homologs antagonize cGAS-mediated antiviral responses. (A) Herpesvirus UL56 homologs. PPxY motifs and the transmembrane domain are indicated. (B) UL56 homologs inhibit cGAS-MITA-mediated signaling. HEK293T cells (1×105) were transfected with the IFNβ promoter (50 ng), ISRE (50 ng) or NF-κB (2 ng) reporter plasmids, and expression plasmids for cGAS (10 ng), MITA (15 ng) and the herpesvirus UL56 homolog for 24 h before luciferase assays were performed. (C) UL56 homologs inhibit HSV-1-triggered transcription of antiviral genes. THP-1 cells stably-expressing UL56, ORF0, U24 or UL42 and MLFs stably-expressing M42 were left uninfected or infected with HSV-1 (MOI=1) for the indicated times before qPCR analysis. (D) UL56 homologs inhibit HT-DNA-triggered transcription of antiviral genes. THP-1 cells stably-expressing UL56, ORF0, U24, UL42 and MLFs stably-expressing M42 were transfected with HT-DNA for 4 h before qPCR analysis. (E) Association of UL56 homologs with cGAS. HEK293T cells were transfected with the indicated plasmids for 24 h. Coimmunoprecipitation and immunoblot analysis was performed with the indicated antibodies. (F) UL56 homologs impair the binding of cGAS to dsDNA. HEK293T cells were transfected with the indicated plasmids. Twenty-four hours later, the cell extracts were incubated with biotinylated-HSV120 and streptavidin agarose for 3 h. The bound proteins were analyzed by immunoblots with the indicated antibodies.
The data shown are means ± SD (B–D) from one representative experiment performed in triplicates (3 technical repeats). Similar data were obtained from at least two independent experiments. ∗, p < 0.05; ∗∗, p < 0.01 (Student's unpaired t-test).
3. Discussion
Previous studies suggest that UL56 is a critical determinant of virulence of HSV-1 (Berkowitz C. et al., 1993; Kehm et al., 1996). In this study, we found that UL56 directly targeted cGAS and inhibited cGAS-mediated innate immune responses. UL56-deficient HSV-1 virus had much reduced replication in infected mice and was significantly less pathogenic and lethal to mice. Our findings suggest that UL56 plays a critical role in evading innate immune responses by HSV-1, and provide an explanation on the mechanisms of UL56-mediated virulence of HSV-1.
Several experiments suggest that UL56 directly impairs cGAS binding to viral DNA and its enzymatic activity. Firstly, ectopic expression of UL56 inhibited transcription of downstream antiviral genes induced by DNA viruses including HSV-1 and HCMV but not by the RNA virus SeV. Second, UL56 interacted with cGAS in vivo and in cells. This interaction impaired cGAS binding to viral DNA as well as its enzymatic activity. Since UL56 can also bind to viral DNA, it is possible that competition for viral DNA binding contributes to UL56-mediated inhibition of cGAS activation. Our experiments also indicated that UL56 inhibited cGAMP synthesis induced by viral DNA or HSV-1 infection. Consistently, ectopic expression of UL56 promoted HSV-1 replication, whereas UL56-deficient HSV-1 showed reduced replication than its wild-type counterpart. These findings suggest that UL56 plays an important role in antagonizing cGAS-mediated innate immune responses and contributing to the virulence of HSV-1. Interestingly, we also found that herpesvirus UL56 homologous proteins also impaired cGAS-mediated antiviral immune responses, suggesting an evolutionarily conserved mechanism for the inhibition of cGAS activity within herpesvirus.
Previously, it has been reported that HSV-1 deamidase UL37 inhibits cGAS enzyme activity by deamidation (Zhang et al., 2018). HSV-1 UL41 gene encodes the virion host shutoff (vhs) protein, and destabilizes cellular RNAs including cGAS mRNA (Esclatine et al., 2004; Su and Zheng, 2017). It has also been shown that other herpesviruses employ a range of strategies to antagonize cGAS-mediated innate immune responses, such as HCMV UL31 (Huang et al., 2018) and KSHV ORF52 (Wu et al., 2015). The simplest explanation is that these viral proteins antagonize cGAS-mediated innate immune responses at different phases of infection and/or targets distinct steps of cGAS-mediated signaling events. Nevertheless, our identification of UL56 as a critical inhibitor of cGAS provides insight into the mechanisms of innate immune evasion and virulence of HSV-1 as well as other herpesviruses.
4. Materials and methods
4.1. Reagents, antibodies, cells, and viruses
The following reagents were purchased from the indicated companies. Lipofectamine 2000 (Invitrogen); FuGene (Promega); puromycin (Thermo Fisher); SYBR Green Supermix (BIO-RAD); polybrene (Millipore); digitonin and HT-DNA (Sigma-Aldrich); 2′3′-cGAMP (Invivogen); ATP and GTP (TaKaRa); EZ-LINK PSORALEN-PEG3-biotin (Thermo Fisher); protein G sepharose (GE Healthcare); streptavidin agarose resin (Thermo Fisher); BSA (Sigma-Aldrich); dual-specific luciferase assay kit (Promega); 2′3′-cGAMP ELISA kit (Cayman Chemical); ELISA kits for murine IL-6 and TNF-α (BioLegend), and CXCL10 (BOSTER); TIANamp Genomic DNA kit (TIANGEN).
Mouse antibodies against Flag, and β-actin (Sigma-Aldrich), His and HA (OriGene), Myc (9B11) (Cell Signaling Technology), HSV-1 ICP27 and ICP0 (Abcam); rabbit monoclonal antibodies against HA, phosphor-MITA (Ser 366), mouse cGAS, phosphor-TBK1/NAK (Ser 172) and phosphor-IRF3 (Ser 396) (Cell Signaling Technology), cGAS (MB21D1) (Sigma-Aldrich), TBK1 (Abcam), IRF3 (Proteintech) were purchased from the indicated companies. Antisera against MITA and UL56 were generated by immunizing rabbit or mice with purified recombinant MITA (151–379) and UL56 proteins.
HFFs were provided by Dr. Min-Hua Luo (Wuhan Institute of Virology). Human embryonic kidney HEK293T (Cat # CRL-11268) and human peripheral blood monocyte THP-1 (Cat # TIB-202) cells were purchased from ATCC. MLFs and MITA-deficient MLFs were previously described (Luo et al., 2016). These cells were cultured in DMEM (Hyclone) or RAPI supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Thermo Fisher Scientific) at 37 °C with 5% CO2.
HCMV (AD169) was provided by Dr. Min-Hua Luo (Wuhan Institute of Virology, CAS). HCMV stock was prepared on HFFs and the viral titer was determined by standard TCID50 assays. HSV-1 stock was prepared on Vero cells and the viral titer was determined by standard plaque assays.
4.2. Synthetic dsDNA
HSV120, DNA90 and VACV70 were synthesized based on sequences previously described (Lian et al., 2018; Zou et al., 2020).
4.3. Plasmids
The pGL3-IFNβ promotor, pGL3-ISRE and NF-κB luciferase reporter plasmids, mammalian expression plasmids for Flag-cGAS or HA-tagged cGAS, MITA, TBK1, IRF3, IKKβ, and p65 were previously described (Fu et al., 2019; Huang et al., 2018). Mammalian expression plasmids for Flag- or HA-tagged UL56, UL56 mutants including transmembrane domain-deletion mutant (ΔTM) and PPxY motif mutants (all Pro in the PPxY motifs are mutated to Ala), and UL56 homologs were constructed by standard molecular biology techniques.
4.4. Transfection and reporter assays
HFFs, MLFs, THP-1s were transfected by lipofectamine 2000. HEK293T cells were transfected by standard calcium phosphate precipitation method. Control plasmids were added to ensure that each transfection receives the same amount of total DNA. In reporter assays, pRL-TK (Renilla luciferase, 0.01 μg) reporter plasmid was transfected as an internal control. Luciferase assays were performed using a dual-specific luciferase assay kit (Promega). Firefly luciferase activities were normalized on the basis of Renilla luciferase activities.
4.5. Stable cell lines
HEK293T cells were transfected with two packaging plasmids (pSPAX2 and pMD2. G) together with empty vector, or the indicated plasmids by calcium phosphate precipitation. Twelve hours later, the medium was replaced with DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Thirty-six hours later, the recombinant virus-containing medium was filtered (0.45 μm) and added to THP-1, HFF, or MLF cells in the presence of polybrene (4–8 μg/mL). Twenty-four hours post-infection, cells were selected with puromycin (1–4 μg/mL) for 7 days before experiments.
4.6. Recombinant protein preparation
The cDNAs encoding for cGAS or UL56 was cloned into pET30c-His or pGEX-6p-1-GST plasmid respectively. The plasmid was transformed into BL21 or Rosetta E. coli strain. Expression of His-cGAS and UL56 were induced with 0.5 mM IPTG at 16 °C for 4–6 h. The recombinant proteins were purified using ÄKTA pure protein purification system (GE Healthcare) with lysis buffer (20 mM Tris-HCl [pH7.4], 0.5 M NaCl, 20% glycerin, 0.1% NP-40 and 10 mM imidazole), wash buffer (20 mM Tris-HCl [pH7.4], 0.5 M NaCl, 20% glycerin and 60 mM imidazole) and elution buffer (20 mM Tris-HCl [pH7.4], 0.5 M NaCl, 20% glycerin and 500 mM imidazole). Expression of GST-cGAS and UL56 were induced with 0.5 mM IPTG at 16 °C for 16 h, and the recombinant proteins were purified with GST resins in PBS and eluted with elution buffer (PBS, 10 mM reduced glutathione). The purified proteins were analyzed by Coomassie staining and/or immunoblotting analysis.
4.7. In vitro cGAS activity assay
In vitro cGAS activity assay was performed as previously described (Cui et al., 2017; Huang et al., 2018; Wang et al., 2017). Purified cGAS (60 μg), UL56 (6 μg) or BSA (6 μg) were mixed in 500 μL reaction buffer (20 mM HEPES [pH7.4], 5 mM MgCl2, 2 mM ATP, 2 mM GTP, and 0.1 mg/mL HT-DNA) and incubated at 37 °C for 1 h. The samples were then boiled at 95 °C for 5 min to terminate the reaction and centrifuged at 16,000 g for 10 min. The supernatant was filtrated with Amicon Ultra-0.5 mL/10 kD ultrafiltration filter (Millipore) and diluted by 20-folds. The sample was subjected to chromatography on a Mono Q 5/50 GL column (GE Healthcare), equilibrated with buffer A (20 mM Tris-HCl [pH 8.5]), and eluted with a gradient of buffer B (20 mM Tris-HCl [pH 8.5], 1 M NaCl) in buffer A from 0 to 50%. The first peak of absorbance of the elutions indicates the amount of synthetic cGAMP.
4.8. qPCR
Total RNAs were isolated from cells and reverse-transcribed to cDNA for qPCR analysis to measure mRNA levels of the indicated genes. Data shown are the relative abundance of the indicated mRNAs normalized to that of GAPDH. Sequence of primer pairs for qPCR assays are listed in Supplementary Table S1.
4.9. Co-immunoprecipitation and immunoblot analysis
HEK293T cells (5×106) or MLFs (1×107) were lysed with l mL NP-40 lysis buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 10 μg/mL aprotinin, 10 μg/mL leupeptin and 1 mM phenylmethylsulfonyl fluoride) for 30 min on ice. Cell lysates were clarified by centrifugation at 4 °C, 12,000 rpm for 15 min. For each immunoprecipitation, the cell lysate (800 μL) was incubated with the indicated antibody (0.5 μg) and protein G sepharose beads (25 μL of 1:1 slurry) at 4 °C for 3 h. The protein-bound beads were then collected and washed three times with 1 mL of lysis buffer containing 0.5 M NaCl. Immunoblotting analysis was performed by standard procedures.
4.10. Confocal microscopy
HeLa cells were transfected with the indicated plasmids by FuGene. After transfection for 20 h, the cells were fixed with 4% paraformaldehyde for 10–15 min on ice and washed with PBS for three times, then permeabilized with 0.3% Triton X-100 on ice for 10 min and blocked in 1% BSA for 20 min at room temperature. The cells were then incubated with the indicated primary antibodies overnight at 4 °C. Alexa Fluor 488- or 555-conjugated secondary antibodies were incubated with the cells for 1 h. The nuclei were stained with DAPI for 2 min before images were acquired using Nikon confocal microscope under a 60 × oil lens objective.
4.11. Generation of HSV-1 ΔUL56
HSV-1 recombinant viruses were derived from the pYEbac102 infectious clone of HSV-1 strain F, and recombination was performed in GS1783 E. coli strain. HSV-1ΔUL56 was generated using a two-step markerless recombination system as previously described (Bai et al., 2019; Tanaka et al., 2003; Tischer et al., 2010; Zou et al., 2020). The Kana cassette linear fragment was amplified with the following pair of primers: GCGCGCGGGAGTCGTGGCTTTGGGGCGCATCCATGGCTTaGGAGGCGGCGCAACCCGACGAGGATGACGACGATAAGTAGGG; and CCCCGCGCTCCATAGACCCGCGTCGGGTTGCGCCGCCTCCtAAGCCATGGATGCGCCCCACAACCAATTAACCAATTCTGATTAG).
4.12. HSV-1 genome sequencing and reads mapping
Genomic DNAs of HSV-1 and HSV-1ΔUL56 were extracted using TIANamp Genomic DNA kit (TIANGEN). The viral genomes of HSV-1 and HSV-1ΔUL56 were sequenced with the Illumina Miseq platform. The sequencing reads were mapped against the reference sequence (GU734771.1) and aligned and visualized as previously described (Huang et al., 2018).
4.13. Mice experiment
All mice were bred in specific pathogen-free (SPF) conditions and viral infection experiments in this study were performed at the biosafety level 2 facility at the Center for Animal Experiment of Wuhan Institute of Virology. Animal experiments were conducted without blinding, with 7–9-week-old age- and sex-matched female mice. All animal experiments were performed in accordance with the Guideline for Animal Care and Use of Wuhan Institute of Virology, Chinese Academy of Sciences.
C57BL/6 mice were intra-nasally (i.n.) infected with wild-type HSV-1 or HSV-1ΔUL56 virus (2.5×107 PFU for each mouse, no more than 50 μL per mice) (Lian et al., 2018; Zhang et al., 2018, 2020). Mouse survival was monitored daily for 10 days. Mice from each group were sacrificed at 2- or 3-days post-infection (dpi). Mouse brains were collected, weighed, and homogenized in TRIzol reagents. RNA was isolated, and HSV-1 copy number and the transcription of antiviral genes were determined by qPCR. Mice for each group were infected for 10 h, and then sera of mice were collected for measurement of CXCL10, IL-6 and TNF-α by ELISA.
4.14. Viral plaque assay
Infected cells were collected and freeze-thaw three times with liquid nitrogen. The suspensions were then centrifuged at 13,000 rpm for 15 min, and the supernatants were collected to measure the viral yield by plaque assays. Vero cells were seeded in 24-well plates, and the cells were infected by incubation for 2 h at 37 °C with serial dilutions of the viruses. After infection for 2 h, 0.5% methylcellulose was overlaid, and the plates were incubated for 48 h. The overlay was removed, and cells were fixed with 4% paraformaldehyde for 30 min and stained with 1% crystal violet for 30 min before plaque counting.
4.15. GST pull-down assay
GST-UL56 or GST protein was mixed with glutathione agarose beads in 1 mL PBS for 3 h at 4 °C. The beads were washed three times with 1 mL PBS, then mixed with purified His-cGAS protein (50 μg) and incubated for 3 h at 4 °C. The beads were collected and washed three times with PBS containing 0.5 M NaCl and analyzed by immunoblotting with the indicated antibodies or Coomassie blue (Fu et al., 2017).
4.16. DNA pull-down assay
HSV120 was conjugated to biotin (EZ-link Psoralen-PEG3-biotin) by UV (365 nm wavelength) irradiation for 1 h. HEK293T cells were lysed with 1 mL lysis buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% NP-40) and the cell lysate was incubated with biotinylated-HSV120 (8 μg) at 4 °C for 1 h, then followed by incubation with streptavidin agarose for 2 h at 4 °C. The agarose beads were collected and washed five times with lysis buffer containing 1 M NaCl before immunoblotting analysis.
4.17. Microscale thermophoresis technology (MST)
MST analysis was performed using a NanoTemper Monolith NT.155 instrument (NanoTemper Technologies GmbH) (Lian et al., 2018). For detecting affinity between GST-cGAS to dsDNA, 35 nM Cy5-labeled 60 bp dsDNA HSV60 (Sangon Biotech, China) was mixed with different concentrations of proteins in PBS with 100 mM Tris-HCl [pH8.8]. His-UL56 was added with dsDNA as a cofactor and BSA was used as a negative control. Samples were loaded into Premium Coated Capillaries and MST measurements were performed using 20% MST power and 40% LED power at 25 °C. Laser-on and -off times were 30 and 5 s respectively. NanoTemper Analysis 1.2.20 software was used to fit the data and to determine the Kd values.
4.18. Quantification of cGAS-bound viral DNA
HEK293T cells were transfected with HA-cGAS and Flag-UL56 or an empty vector for 20 h. The cells were then infected with HSV-1 for 1 h, washed with medium, and cultured for 2 h. Cells were lysed with NP-40 lysis buffer. The lysate was centrifuged to remove the nucleus and the supernatant was aliquoted. One aliquot was incubated with protein G sepharose beads conjugated with an anti-HA antibody or control mouse IgG at 4 °C for 4 h. The beads were washed 3 times with lysis buffer containing 1 M NaCl, treated with elution buffer (0.1 M NaHCO3, 1% SDS) containing protease K for two times at 65 °C and subjected to phenol-chloroform extraction. The aqueous phase was collected and mixed with ethanol (2.5 times volume of supernatants), 10% 3M KOAc pH 5.2, and 1.25% glycogen (5 mg/mL) and stored at −20 °C overnight. DNA was precipitated by centrifugation at 12,000 rpm for 30 min at 4 °C. The precipitated DNA was washed with 75% ethanol and air-dried before resuspension in ddH2O and used for quantification of the indicated HSV-1 genes by qPCR.
Ethics statement
All mice were housed in the specific pathogen-free facility and viral infection experiments were carried out in an ABSL-2 facility at Wuhan Institute of Virology. The experimental protocol was adhered to the International Guiding Principles for Biomedical Involving Animals. The protocol for animal experiments were approved by the Institutional Animal Care and Use Committee of Wuhan Institute of Virology (approval number WIVA31201903).
Statistics
Unpaired Student's t-test and log-rank test were used for statistical analysis with GraphPad Prism Software; ∗, P < 0.05; ∗∗, P < 0.01.
Author contributions
Z-Q.Z., Y-Z.F., and Y-Y.W. conceived, designed, and supervised the study and wrote the manuscript. Z-Q.Z., Y-Z.F., and S-Y.W. processed data analysis. H-M.Z. offered help for performing the construction of HSV-1 recombinant virus. Z-S.S. offered help for performing the animal experiment. All authors read and approved the contents of the manuscript.
Declaration of cmpeting interest
The authors have declared that no competing interests exist.
Acknowledgment
We thank Dr. Ding Gao from the Core Facility and Technical Support of Wuhan Institute of Virology for help with the confocal microscopy. We thank Fan Zhang and You-Ling Zhu from Center for Animal Experiment of Wuhan Institute of Virology for antibody preparation, and Xue-Fang An and Li-Li from Center for Animal Experiment of Wuhan Institute of Virology for help with animal experiments. We thank Drs. Ke Lan and Jun-Jie Zhang (Wuhan University) for help with the construction of HSV-1 recombinant virus. We also thank Lei Zhang from the Center for Instrumental Analysis and Metrology of Wuhan Institute of Virology for HSV-1 genome sequencing, and Zhe-Fu Huang for help with analysis of the genome sequencing data. This study was supported by the Strategic Priority Research Program (XDB29010302, awarded to Y.Y.W.), the National Natural Science Foundation of China (31800732, awarded to Y.Z.F.) and Key Research Programs of Frontier Sciences funded by the Chinese Academy of Sciences (awarded to Y.Y.W.) and Special Research Assistant Grant Program of the Chinese Academy of Sciences (, awarded to Y.Z.F.).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cellin.2022.100014.
Contributor Information
Yu-Zhi Fu, Email: yuzhi.fu@wh.iov.cn.
Yan-Yi Wang, Email: wangyy@wh.iov.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Atianand M.K., Fitzgerald K.A. Molecular basis of DNA recognition in the immune system. J. Immunol. 2013;190:1911–1918. doi: 10.4049/jimmunol.1203162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L., Dong J., Liu Z., Rao Y., Feng P., Lan K. Viperin catalyzes methionine oxidation to promote protein expression and function of helicases. Sci. Adv. 2019;5 doi: 10.1126/sciadv.aax1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber G.N. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014;35:88–93. doi: 10.1016/j.it.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Berkowitz C., Moyal M., Rosen-Wolff A., Darai G., Y., B. Herpes simplex virus type 1 (HSV-1)UL56 gene is involved in viral intraperitoneal pathogenicity to immunocompetent mice. Arch. Virol. 1993;134:73–83. doi: 10.1007/BF01379108. [DOI] [PubMed] [Google Scholar]
- Christensen M.H., Jensen S.B., Miettinen J.J., Luecke S., Prabakaran T., Reinert L.S., Mettenleiter T., Chen Z.J.J., Knipe D.M., Sandri-Goldin R.M., et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 2016;35:1385–1399. doi: 10.15252/embj.201593458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F., Deimling T., Mann C.C.D., Ablasser A., Moldt M., Witte G., Hornung V., Hopfner K.P. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498:332. doi: 10.1038/nature12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y., Yu H., Zheng X., Peng R., Wang Q., Zhou Y., Wang R., Wang J., Qu B., Shen N., et al. SENP7 potentiates cGAS activation by relieving SUMO-mediated inhibition of cytosolic DNA sensing. PLoS Pathog. 2017;13 doi: 10.1371/journal.ppat.1006156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esclatine A., Taddeo B., Evans L., Roizman B. The herpes simplex virus 1 UL41 gene-dependent destabilization of cellular RNAs is selective and may be sequence-specific. Proc. Natl. Acad. Sci. U. S. A. 2004;101:3603–3608. doi: 10.1073/pnas.0400354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.Z., Guo Y., Zou H.M., Su S., Wang S.Y., Yang Q., Luo M.H., Wang Y.Y. Human cytomegalovirus protein UL42 antagonizes cGAS/MITA-mediated innate antiviral response. PLoS Pathog. 2019;15 doi: 10.1371/journal.ppat.1007691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.Z., Su S., Gao Y.Q., Wang P.P., Huang Z.F., Hu M.M., Luo W.W., Li S., Luo M.H., Wang Y.Y., et al. Human cytomegalovirus tegument protein UL82 inhibits STING-mediated signaling to evade antiviral immunity. Cell Host Microbe. 2017;21:231–243. doi: 10.1016/j.chom.2017.01.001. [DOI] [PubMed] [Google Scholar]
- Guo H.Y., Omoto S., Harris P.A., Finger J.N., Bertin J., Gough P.J., Kaiser W.J., Mocarski E.S. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe. 2015;17:243–251. doi: 10.1016/j.chom.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W.R., Cao L.B., Yang Y.L., Hua D., Hu M.M., Shu H.B. VRK2 is involved in the innate antiviral response by promoting mitostress-induced mtDNA release. Cell. Mol. Immunol. 2021;18:1186–1196. doi: 10.1038/s41423-021-00673-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M.M., Shu H.B. Cytoplasmic mechanisms of recognition and defense of microbial nucleic acids. Annu. Rev. Cell Dev. Biol. 2018;34:357–379. doi: 10.1146/annurev-cellbio-100617-062903. [DOI] [PubMed] [Google Scholar]
- Hu M.M., Shu H.B. Innate immune response to cytoplasmic DNA: mechanisms and diseases. Annu. Rev. Immunol. 2020;38:79–98. doi: 10.1146/annurev-immunol-070119-115052. [DOI] [PubMed] [Google Scholar]
- Hu M.M., Yang Q., Xie X.Q., Liao C.Y., Lin H., Liu T.T., Yin L., Shu H.B. Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity. 2016;45:555–569. doi: 10.1016/j.immuni.2016.08.014. [DOI] [PubMed] [Google Scholar]
- Huang Z., Wu S.Q., Liang Y.J., Zhou X.J., Chen W.Z., Li L.S., Wu J.F., Zhuang Q.F., Chen C.A., Li J.X., et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe. 2015;17:229–242. doi: 10.1016/j.chom.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Huang Z.F., Zou H.M., Liao B.W., Zhang H.Y., Yang Y., Fu Y.Z., Wang S.Y., Luo M.H., Wang Y.Y. Human cytomegalovirus protein UL31 inhibits DNA sensing of cGAS to mediate immune evasion. Cell Host Microbe. 2018;24:69–80 e64. doi: 10.1016/j.chom.2018.05.007. [DOI] [PubMed] [Google Scholar]
- Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–U674. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H., Ma Z., Barber G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461 doi: 10.1038/nature08476. 788-U740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L., Waterman P.M., Jonscher K.R., Short C.M., Reisdorph N.A., Cambier J.C. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol. Cell Biol. 2008;28:5014–5026. doi: 10.1128/MCB.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehm R., Gelderblom H.R., Darai G. Identification of the UL56 protein of herpes simplex virus type 1 within the virion by immuno electron microscopy. Virus Gene. 1998;17:49–53. doi: 10.1023/a:1008053017716. [DOI] [PubMed] [Google Scholar]
- Kehm R., Lorentzen E., Rosen-Wolff A., Darai G. In vitro expression of UL56 gene of herpes simplex virus type 1; detection of UL56 gene product in infected cells and in virions. Virus Res. 1994;33:55–66. doi: 10.1016/0168-1702(94)90017-5. [DOI] [PubMed] [Google Scholar]
- Kehm R., RosenWolff A., Darai G. Restitution of the UL56 gene expression of HSV-1 HFEM led to restoration of virulent phenotype; Deletion of the amino acids 217 to 234 of the UL56 protein abrogates the virulent phenotype. Virus Res. 1996;40:17–31. doi: 10.1016/0168-1702(96)80248-6. [DOI] [PubMed] [Google Scholar]
- Koshizuka T., Goshima F., Takakuwa H., Nozawa N., Daikoku T., Koiwai O., Nishiyama Y. Identification and characterization of the UL56 gene product of herpes simplex virus type 2. J. Virol. 2002;76:6718–6728. doi: 10.1128/JVI.76.13.6718-6728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshizuka T., Kobayashi T., Ishioka K., Suzutani T. Herpesviruses possess conserved proteins for interaction with Nedd4 family ubiquitin E3 ligases. Sci Rep-Uk. 2018;8 doi: 10.1038/s41598-018-22682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshizuka T., Tanaka K., Suzutani T. Degradation of host ubiquitin E3 ligase Itch by human cytomegalovirus UL42. J. Gen. Virol. 2016;97:196–208. doi: 10.1099/jgv.0.000336. [DOI] [PubMed] [Google Scholar]
- Lafaille F.G., Pessach I.M., Zhang S.Y., Ciancanelli M.J., Herman M., Abhyankar A., Ying S.W., Keros S., Goldstein P.A., Mostoslavsky G., et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature. 2012;491:769–773. doi: 10.1038/nature11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Shu H.B. Dephosphorylation of cGAS by PPP6C impairs its substrate binding activity and innate antiviral response. Protein Cell. 2020;11:584–599. doi: 10.1007/s13238-020-00729-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Shu C., Yi G.H., Chaton C.T., Shelton C.L., Diao J.S., Zuo X.B., Kao C.C., Herr A.B., Li P.W. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity. 2013;39:1019–1031. doi: 10.1016/j.immuni.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H., Wei J., Zang R., Ye W., Yang Q., Zhang X.N., Chen Y.D., Fu Y.Z., Hu M.M., Lei C.Q., et al. ZCCHC3 is a co-sensor of cGAS for dsDNA recognition in innate immune response. Nat. Commun. 2018;9:3349. doi: 10.1038/s41467-018-05559-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Zheng C. A tug of war: DNA-sensing antiviral innate immunity and herpes simplex virus type I infection. Front. Microbiol. 2019;10:2627. doi: 10.3389/fmicb.2019.02627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looker K.J., Magaret A.S., May M.T., Turner K.M.E., Vickerman P., Gottlieb S.L., Newman L.M. Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS One. 2015;10 doi: 10.1371/journal.pone.0140765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W.W., Li S., Li C., Lian H., Yang Q., Zhong B., Shu H.B. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat. Immunol. 2016;17:1057–1066. doi: 10.1038/ni.3510. [DOI] [PubMed] [Google Scholar]
- Medzhitov R., Janeway C. Innate immune recognition: mechanisms and pathways. Immunol. Rev. 2000;173:89–97. doi: 10.1034/j.1600-065x.2000.917309.x. [DOI] [PubMed] [Google Scholar]
- Orzalli M.H., DeLuca N.A., Knipe D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. P Natl Acad Sci USA. 2012;109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh T.K., Davies C.T.R., Muenzner J., Hunter L.M., Barrow H.G., Connor V., Bouton C.R., Smith C., Emmott E., Antrobus R., et al. Temporal proteomic analysis of herpes simplex virus 1 infection reveals cell-surface remodeling via pUL56-mediated GOPC degradation. Cell Rep. 2020;33:108235. doi: 10.1016/j.celrep.2020.108235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C., Zhan G., Zheng C. Evasion of host antiviral innate immunity by HSV-1, an update. Virol. J. 2016;13:38. doi: 10.1186/s12985-016-0495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C.H., Zheng C.F. Herpes simplex virus 1 abrogates the cGAS/STING-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein. UL41. J Virol. 2017;91 doi: 10.1128/JVI.02414-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L.J., Wu J.X., Du F.H., Chen X., Chen Z.J.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W.X., Li Y., Chen L., Chen H.H., You F.P., Zhou X., Zhou Y., Zhai Z.H., Chen D.Y., Jiang Z.F. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. P Natl Acad Sci USA. 2009;106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A., Wang Z., Choi M.K., Yanai H., Negishi H., Ban T., Lu Y., Miyagishi M., Kodama T., Honda K., et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- Tanaka M., Kagawa H., Yamanashi Y., Sata T., Kawaguchi Y. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 2003;77:1382–1391. doi: 10.1128/JVI.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer B.K., Smith G.A., Osterrieder N. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 2010;634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- Unterholzner L., Keating S.E., Baran M., Horan K.A., Jensen S.B., Sharma S., Sirois C.M., Jin T., Latz E., Xiao T.S., et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushijima Y., Koshizuka T., Goshima F., Kimura H., Nishiyama Y. Herpes simplex virus type 2 UL56 interacts with the ubiquitin ligase Nedd4 and increases its ubiquitination. J. Virol. 2008;82:5220–5233. doi: 10.1128/JVI.02515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner E.K., Bloom D.C. Experimental investigation of herpes simplex virus latency. Clin. Microbiol. Rev. 1997;10:419. doi: 10.1128/cmr.10.3.419. -+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Huang L., Hong Z., Lv Z., Mao Z., Tang Y., Kong X., Li S., Cui Y., Liu H., et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog. 2017;13 doi: 10.1371/journal.ppat.1006264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Li Y., Liu S., Yu X.L., Li L., Shi C.L., He W.H., Li J., Xu L., Hu Z.L., et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. P Natl Acad Sci USA. 2014;111:15438–15443. doi: 10.1073/pnas.1412767111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A.P., Khoury-Hanold W., Staron M., Tal M.C., Pineda C.M., Lang S.M., Bestwick M., Duguay B.A., Raimundo N., MacDuff D.A., et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley R.J., Roizman B. Herpes simplex virus infections. Lancet. 2001;357:1513–1518. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- Wu J.J., Li W.W., Shao Y.M., Avey D., Fu B.S., Gillen J., Hand T., Ma S.M., Liu X., Miley W., et al. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe. 2015;18:333–344. doi: 10.1016/j.chom.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.X., Sun L.J., Chen X., Du F.H., Shi H.P., Chen C., Chen Z.J.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.Y., Liao B.W., Xu Z.S., Ran Y., Wang D.P., Yang Y., Luo W.W., Wang Y.Y. USP44 positively regulates innate immune response to DNA viruses through deubiquitinating MITA. PLoS Pathog. 2020;16 doi: 10.1371/journal.ppat.1008178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Zhao J., Xu S., Li J., He S., Zeng Y., Xie L., Xie N., Liu T., Lee K., et al. Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe. 2018;24:234–248. doi: 10.1016/j.chom.2018.07.004. e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.Y., Jouanguy E., Ugolini S., Smahi A., Elain G., Romero P., Segal D., Sancho-Shimizu V., Lorenzo L., Puel A., et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Yuan B., Bao M., Lu N., Kim T., Liu Y.J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 2011;12:959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.D., Zhong B. Regulation and function of the cGAS-MITA/STING axis in health and disease. Cell Insight. 2022 doi: 10.1016/j.cellin.2021.100001. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B., Yang Y., Li S., Wang Y.Y., Li Y., Diao F.C., Lei C.Q., He X., Zhang L., Tien P., et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor Activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Zou H.M., Huang Z.F., Yang Y., Luo W.W., Wang S.Y., Luo M.H., Fu Y.Z., Wang Y.Y. Human cytomegalovirus protein UL94 targets MITA to evade the antiviral immune response. J. Virol. 2020;94 doi: 10.1128/JVI.00022-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.