

Abstract
One obstacle to the development of gene therapies for the central nervous system is the lack of workflows for quantifying transduction efficiency in affected neural networks and ultimately predicting therapeutic potential. We integrated data from a brain cell atlas with 3D or 2D semi-automated quantification of transduced cells in segmented images to predict AAV transduction efficiency in multiple brain regions. We used this workflow to estimate the transduction efficiency of AAV2/rh.10 and AAV2.retro co-injection in the corticostriatal network affected in Huntington’s disease. We then validated our pipeline in gene editing experiments targeting both human and mouse huntingtin genes in transgenic and wild-type mice, respectively. Our analysis predicted that 54% of striatal cells and 7% of cortical cells would be edited in highly transduced areas. Remarkably, in the treated animals, huntingtin gene inactivation reached 54.5% and 9.6%, respectively. These results demonstrate the power of this workflow to predict transduction efficiency and the therapeutic potential of gene therapies in the central nervous system.
Keywords: CNS, Gene Therapy, Gene Editing, Semi-automated, Transduction Efficiency, Huntington’s Disease, Adeno-associated vector, AAV- AAV-KamiCas9
Graphical abstract
Déglon and colleagues use semi-automated quantification pipelines to show that striatal co-injection of AAV2/rh.10 and AAV2.retro efficiently transduces the corticostriatal network affected in HD. Striatal and cortical transduction efficiency predicted HTT inactivation level in both regions, demonstrating the power of these pipelines to predict the therapeutic potential of gene therapies.
Introduction
Over the last decade, the concept of permanently treating neurological diseases by a single delivery of a therapeutic gene has driven the development of central nervous system (CNS) gene therapy.1 This global effort has achieved a certain success, as attested by the recent approval of gene therapy products for spinal muscular atrophy (SMA)2 and aromatic L-amino acid decarboxylase (AADC) deficiency.3 However, notwithstanding these recent accomplishments, the development of CNS gene therapies remains challenging due to the complexity of neurodegenerative diseases. These diseases are often characterized by alterations to multiple neuronal networks in several brain regions, with a poor understanding of the underlying molecular mechanisms, associated with complex gene delivery. Adeno-associated viruses (AAVs) are the principal delivery vehicle for therapeutic gene delivery to the CNS.1 The emergence of new AAV capsids has greatly increased transduction efficiency, but there is still a lack of semi-automated quantitative workflows for assessing the transduction profile across multiple brain areas and predicting therapeutic potential. Methods based on FACS,4 or single-cell RNA sequencing5 may be useful to characterize AAV tropism and/or quantify the overall number of transduced cells in selected areas, but they are not suitable to study AAV biodistribution since the anatomical information of the transduced cells are lost. The current imaging workflows are based on the injection of AAVs expressing fluorescent reporter genes followed by the confocal imaging of immunolabeled tissue sections.6,7,8,9 These methods are time-consuming, especially when large areas of the brain are transduced, and the manual parcellation of each brain region is required to assess transduction efficiency.
Here, we took advantage of 3D and 2D semi-automated workflows for evaluating transduction efficiency in multiple brain regions, with a view to predicting therapeutic potential. This pipeline integrates data from a mouse brain cell atlas10 to predict the pattern of transduction in various anatomical brain regions. It also makes use of recently developed methods for analyzing images from mice after AAV-GFP injection. 3D images from clarified brain (CLARITY)11,12 or 2D images from coronal brain sections13,14,15 are co-registered with a mouse reference atlas (ARA)16 to evaluate brain delivery performance.
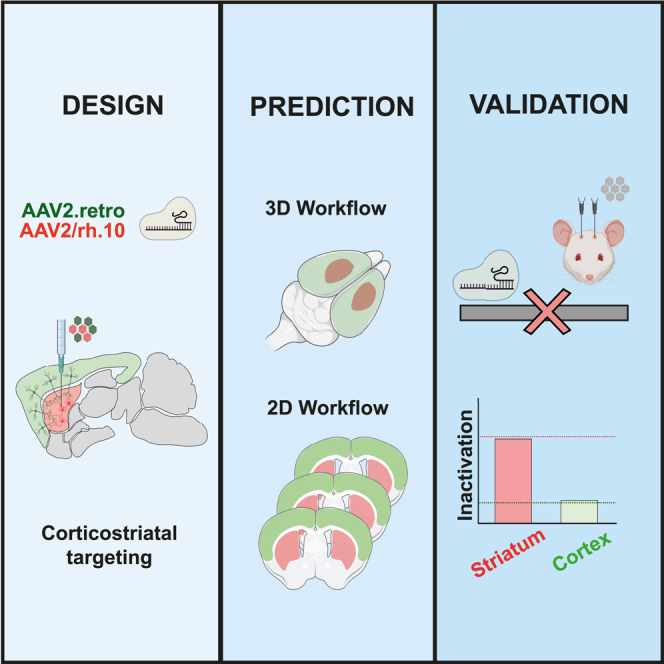
We used these workflows to optimize gene editing approaches for Huntington’s disease (HD), which is caused by a triplet repeat expansion in the huntingtin (HTT) gene. HD is characterized by a degeneration of spiny projection neurons (SPNs) in the striatum and neuronal dysfunction and cell death in the cerebral cortex and other areas of the brain.17,18 All gene editing strategies for HD to date have focused exclusively on the striatum.19,20,21,22 We developed an AAV platform based on our KamiCas9 system23 to target the corticostriatal neuronal network affected in HD. We delivered AAV2.retro24 which has strong retrograde properties, together with AAV2/rh.1025,26 which efficiently transduces the striatum, to maximize HTT gene editing. We used our 3D and 2D workflows to estimate cortical and striatal transduction efficiency and to predict the degree of HTT gene inactivation. We show that these semi-automated workflows provide accurate quantitative assessments of AAV profiles in various brain regions and demonstrate the power of these pipelines for predicting the therapeutic potential of gene editing strategies.
Results
Development of an AAV-KamiCas9 system for CNS gene editing
We previously developed a lentiviral (LV)-based self-inactivating KamiCas9 system for gene editing.23 Here, we adapted the system for AAV, which diffuse widely throughout the brain and yield high transduction efficiencies in various regions. As proof-of-principle, we first showed that the co-injection of AAV2/1-EFS-SpCas927 with an AAV2/1 expressing a sgRNA targeting the translational start site of GFP (sgGFP) led to a complete loss of GFP fluorescence in the transduced striatal area of Drd2-GFP transgenic mice28,29 (Figures 1A and 1B). We then focused on the HTT gene and developed two AAV-KamiCas9 systems (Figure S1A). The HTT-targeting sgRNA (sgHTT1)23 is under the control of the U6 promoter, whereas expression of the SpCas9 self-targeting sgRNA (sgCas9) is driven either by the U6 promoter (KamiCas9v1) or by the 7sk promoter (KamiCas9v2). High levels of HTT editing (42.5–57.9%) and SpCas9 self-editing (57.3–73.6%) were obtained with both systems in human embryonic kidney 293T (HEK293T) cells (Figure S1B). We selected the AAV2/1-CRISPR and AAV2/1-KamiCas9v1 for inactivation of the wild-type (WT) human HTT gene in the striatum of fully humanized HU18/18 transgenic mice30 (Figure 1C). The injection of AAV2/1-CRISPR and AAV2/1-KamiCas9v1 generated indels in 30.2 ± 6.3% and 21.0 ± 8.03% of HTT alleles (Figure 1D). We assessed SpCas9 self-editing, by analyzing DNA and RNA samples (Figure 1E). Löw and coworkers reported that large numbers of AAV genomes remained present as single-stranded AAV (ssAAV) several weeks after injection31 (Figure 1F). By measuring SpCas9 self-editing at the RNA level, we avoided amplifying ssAAVs, which are not a substrate for SpCas9 (Figure 1F). We showed that 50.7 ± 7.7% of the SpCas9 transcripts contained indels, whereas only 29.0 ± 8.1% of AAV genomes had been edited (ssAAV and dsAAV) (Figures 1E and 1F). It has also been suggested that the failure of the DNA repair machinery to fix double-strand breaks (DSBs) on dsAAV episomes can lead to the degradation of these structures32,33,34 (Figure 1F). We tested this hypothesis, by determining whether HTT editing was affected by cleavage of the AAV expressing the sgHTT1 (Figure S1C). HTT editing efficiency was strongly decreased by the induction of AAV cleavage (7.1 ± 1.3%), resulting in levels much lower than for the uncleavable vector (28.2 ± 3.6%) (Figure S1D). This lower rate of HTT editing is explained by the lower levels of sgHTT1 due to the cleavage-induced partial degradation of the dsAAV expressing the sgHTT1. Translating these results to the KamiCas9 system, SpCas9 self-cleavage induces a partial degradation of dsAAVs expressing SpCas9, thereby decreasing SpCas9 expression. Other AAV self-deleting systems rely exclusively on AAV degradation,32,33,34 but our AAV-KamiCas9 efficiently inactivates SpCas9 even when the DSBs are successfully repaired (Figure 1F), further decreasing the risk of off-target events. In summary, we show here that the AAV-KamiCas9 system induces high levels of target gene editing with the effective self-activation of SpCas9.
Figure 1.

Development of an AAV-KamiCas9 system for in vivo gene editing
(A) Schematic representation of the AAV-CRISPR-GFP used for in vivo gene editing. The first two AAV2/1 express mCherry or the spCas9 under the control of the short elongation factor 1α promoter (EFS). The third vector expresses a sgRNA targeting the translational start site of GFP (sgGFP) under the control of the U6 promoter. (B) Representative images of the striatum of Drd2-GFP mice with and without injection of the CRISPR-GFP system. Scale bar: 200 μm. (C) Schematic representation of AAV2/1-CRISPR and AAV2/1-KamiCas9v1 used to inactivate the human HTT gene in the striatum of HU18/18 transgenic mice. The sgRNA targeting the translational start site of the human HTT gene (sgHTT1)23 is under the control of the U6 promoter in both systems. In KamiCas9v1, there is a second sgRNA targeting the translational start site of SpCas9 itself (sgCas9). (D) Eight weeks post-injection, striatal punch specimens were collected from the GFP-positive area. The frequency of HTT indels was assessed by TIDE35 in DNA extracts (n = 6 hemispheres from 3 animals for control and CRISPR; n = 8 hemispheres from 4 animals for KamiCas9v1). (E) The frequency of indels in the SpCas9 transgene was assessed in DNA (n = 6 hemispheres from 3 animals for CRISPR; n = 8 hemispheres from 4 animals for KamiCas9v1) and RNA (n = 6 hemispheres from 3 animals for CRISPR; n = 6 hemispheres from 4 animals for KamiCas9v1) extracts. (F) Diagrams illustrating the progressive inactivation of nuclease activity in the AAV-KamiCas9 system. Most AAV genomes are not self-targeted because they are present as ssAAV. The amplification of ssAAV-SpCas9 during TIDE analysis does not reflect the functional, actively transcribed AAV-SpCas9 genome population. By contrast, TIDE analysis for the SpCas9 transgene in RNA samples detects indels only in actively transcribed dsAAV-SpCas9 genomes. Data are represented as mean ± SD.
Development of semi-automated workflows for quantifying transduction efficiency in multiple brain areas
CNS disorders are complex and often associated with alterations to the neuronal networks in multiple brain regions. New therapeutic strategies and genetic tools for the treatment of CNS disorders are continually being developed, but effective gene delivery to affected brain regions remains a major challenge. Transduction efficiency depends on the specific features of each AAV serotype (e.g., tropism, diffusion, and transport properties), the amount of AAV injected (e.g., dose, volume, and flow rate) and the surgical procedure used (e.g., cannulas, coordinates, and procedure variability). All these parameters can be optimized to maximize gene delivery, but there are currently no automated quantitative workflows for measuring transduction efficiency in various regions of the brain. As a means of optimizing CNS gene delivery and facilitating the development of new therapies, we used semi-automated 3D and 2D fluorescence-based workflows for quantifying transduction efficiency in multiple brain areas (Figure 2). The first step in this pipeline is the estimation of the maximum theoretical number of cells that can be targeted in each brain region based on the specific characteristics of the vector, such as its tropism and retrograde transport. This estimate integrates information from the Blue Brain Cell Atlas.10 For example, a vector with a neuronal tropism can potentially target 88% of the cells in the cerebellum but only 74% of the cells in the hippocampus. Once the maximum number of cells that can be targeted is known, it is possible to quantify the number of cells actually transduced in a clarified brain (3D) or in coronal sections (2D). The 3D imaging-based workflow involves brain clarification11 followed by light sheet fluorescence microscopy (LSFM) imaging (Figure 2). The multimodal image registration and connectivity analysis (MIRACL) pipeline12 were then used to register against the Allen mouse brain common coordinate framework (CCFv3) reference atlas.36 The coordinates of all transduced cells are determined with fiji37 and cataloged on segmented brain images to quantify the number of cells transduced in each brain region using the recently published map objects to atlas (MOTA) pipeline.38 In the 2D workflow, the DAPI signal from brain coronal sections is used for registration against the Allen CCFv3 reference atlas36 with the aligning big brains and atlases (ABBA) plugin in fiji13 (Figure 2). The segmented brain regions are then imported into QuPath software14 and the StarDist deep learning-based extension15 is used to quantify the number of transduced cells in the various regions.
Figure 2.

Diagram of the 2D and 3D fluorescence-based quantitative workflows for estimating transduction efficiencies in multiple brain regions
The first step of the pipeline is estimation of the theoretical maximum number of cells that can be transduced based on data from the Blue Brain Cell Atlas and the specific features of the vector (tropism, retrograde transport). The second step is quantification of the cells transduced, with either the 3D or the 2D imaging workflow. In the 3D workflow, the whole-mount brain is clarified11 and imaged by LSFM. The MIRACL pipeline12 is then used to register the 3D images against the Allen CCFv3 reference atlas36 which contains 662 annotated structures. The coordinates of every GFP-positive cell are determined in fiji and cataloged on the registered images with the MOTA pipeline.38 In the 2D workflow, 25 μm-thick brain coronal sections are subjected to whole-slide scanning. The images are aligned with the Allen CCFv3 reference atlas36 with the ABBA plugin13 in fiji. Qupath software14 and the StarDist plugin15 are then used to identify DAPI (4′,6-diamidino-2-phenylindole) signals and to quantify GFP-positive signals with a cell classifier.
Gene editing strategies for HD to date have been evaluated only in the striatum.19,20,21,22 However, a loss of striatal SPNs is not the only sign of HD; other regions of the brain are also affected in this disease, including, in particular, the corticostriatal projecting neurons (CPNs).17,18 We made use of the retrograde transport properties of AAV2.retro to achieve a broad delivery distribution and to target brain circuits affected in HD.24 We injected AAV2.retro-CBA-GFP into the striatum and then used our 3D and 2D pipelines to assess the pattern of CPN transduction (Figure 3A). We first estimated the maximum theoretical numbers of neurons that could be transduced in each isocortical region. Almost all CPNs are excitatory neurons present in cortical layers II/III, V and VI39; only these cortical cells can be transduced by AAV2.retro (Figure 3A). According to the Blue Brain Cell Atlas, the proportion of excitatory neurons in layers II/III, V and VI differs between isocortical regions (ranging from 22% to 49%) (Table S1). For example, AAV2.retro can potentially target 31.4% of the neurons in the orbital area (ORB), but only 22% of those in somatomotor areas (MO).
Figure 3.

3D and 2D workflows for quantifying transduction in various cortical regions
(A) Diagram illustrating the pipeline for determining the theoretical maximal number of excitatory neurons that could be transduced in the cortex of animals following the injection of AAV2.retro-CBA-GFP. The numbers of excitatory neurons in layers II/III, V and VI of the cortex were determined with information from the Blue Brain Cell Atlas. These neurons correspond to the cells that could be transduced with AAV2.retro-CBA-GFP. (B) Dorsoventral light sheet acquisitions of clarified brain were subjected to MIRACL pipeline for registration against the Allen CCFv3 reference atlas. Light sheet acquisitions were also processed in Fiji in parallel to determine X,Y,Z coordinates of each transduced cells using Find Maxima function. Both resulting outputs were combined using MOTA to get proper segmentation of cells within each brain region ontology. (C) Coronal brain sections were registered against the Allen CCFv3 reference atlas in fiji with the ABBA plugin, and then transferred to QuPath. The DAPI-positive nuclei were detected with Stardist, a deep learning-based 2D nucleus detection method trained with a set of fluorescent nucleus images (dsb2018_heavy_augment.pb). A cell classifier was trained to detect transduced GFP-positive cells throughout the entire section. A script was finally generated to run the analysis on all images for the project. Scale bar for low magnification: 800 μm. Scale bar for high magnification: 20 μm. (D) 3D and 2D data showing the percentage of GFP+ excitatory neurons (hence, transduction efficiency) in the 10 isocortical regions most transduced with AAV2.retro-CBA-GFP.
We investigated whether AAV2.retro transduction was optimal or whether the delivery parameters needed to be modified, by quantifying the cells transduced in a 3D clarified brain (Figures 3B and S2A, and Videos S1 and S2) and in 2D coronal sections (Figures 3C and S2B and S2C). The 3D and 2D analyses revealed that there were 417,579 and 507,408 GFP-positive neurons, respectively, in the isocortex (Table S1). Both analyses indicated that most of the transduced neurons were in more frontal cortical areas, such as the frontal pole (FRP), prelimbic area (PL), MO, ORB, agranular insular area (AI), gustatory area (GU), anterior cingulate area (ACA) and infralimbic area (ILA) (Table S1). AAV2.retro transduced 19% of the excitatory neurons in the 10 most transduced isocortical regions (Figure 3D). Remarkably, about half of the neurons that could be targeted were transduced in the FRP and 30% of those in the PL and MO areas (Figure 3D). These results are consistent with the mouse corticostriatal connectome data,40 demonstrating a high degree of connectivity between the dorsolateral striatum and the prefrontal and motor cortices. Variations in the number of GFP-positive cells in different cortical regions between 2D and 3D workflows are mainly due to small variations in the surgical procedure between animals since distinct zones of the dorsal striatum are differently connected to cortical regions.40 In summary, we demonstrated that the 2D and 3D pipelines yielded similar quantitative outcomes, and that both accurately estimated transduction efficiency.
Maximizing the targeting of the neuronal circuitry affected in HD
We have shown that AAV2.retro efficiently transduces CPNs from isocortical areas implicated in HD, such as the MO and ACA.17 However, AAV2.retro transduction at the site of injection (the striatum) was very limited, as previously reported24,41,42 (data not shown). As a means of maximizing the targeting of the neuronal circuitry affected in HD, we tested the co-injection of AAV2.retro-CBA-GFP and AAV2/rh.10-CBA-mCherry (Figure 4A). We also modified the stereotaxic coordinates, injecting the vectors bilaterally at a single striatal site per hemisphere, to reduce the variability associated with the surgical procedure and increase transduction efficacy. As expected, AAV2/rh.10-CBA-mCherry extensively transduced the striatum, whereas AAV2.retro-CBA-GFP predominantly transduced distant CPNs (Figure 4B). The minimal overlap between the fluorescent signals obtained suggests that the two AAV serotypes have complementary tropisms. We prevented confounding due to GFP-positive neuropil signals and facilitated the detection and quantification of transduced cells in the striatum, by performing a new experiment with AAV2.retro and AAV2/rh.10 expressing a nuclear green fluorescent protein (AcGFPnuc) (Figures 4C and 4D). The 2D workflow revealed that 927,725 ± 337,650 striatal cells were AcGFPnuc-positive (Figure S3), corresponding to the transduction of 32.2 ± 14.1% of the DAPI-positive nuclei (Figure 4E). An analysis of highly transduced regions in the dorsal striatum (around the injection site) indicated that 53.9 ± 5.4% of the DAPI-positive nuclei were AcGFPnuc-positive (Figure 4F). Overall, we found that the simultaneous delivery of AAV2.retro and AAV2/rh.10 into the striatum resulted in the transduction of both CPNs and SPNs, the principal neuronal cells affected in HD.
Figure 4.

Optimized gene delivery following the co-injection of AAV2.retro and AAV2/rh.10
(A) Schematic representation of the experimental setting. AAV2.retro-CBA-GFP and AAV2/rh.10-CBA-GFP were co-injected bilaterally into the dorsolateral striatum of mice to favor retrograde transport in the cortex and striatal transduction. (B) Dense cortical layers of GFP-positive CPNs are observed throughout the rostrocaudal axis, whereas mCherry-positive cells are widely distributed throughout the striatum. Scale bar: 1000 μm. (C) Schematic representation of the experimental setting. AAV2.retro-CBA-AcGFPnuc and AAV2/rh.10-CBA- AcGFPnuc were co-injected bilaterally into the dorsolateral striatum for the quantification of striatal transduction efficiency. (D) Coronal sections were co-registered against the Allen CCFv3 reference atlas to identify the striatum, and the 2D workflow described in Figure 3 was applied. Scale bar for low magnification: 800 μm. Scale bar for high magnification: 20 μm. (E) Quantitative analysis showing the percentage of DAPI-positive cells expressing AcGFPnuc in the left and right striatum of three animals. The mean transduction efficiency for the whole striatum is indicated. (F) Quantitative analysis showing the percentage of DAPI-positive cells expressing AcGFPnuc in five representative highly transduced areas of the dorsolateral striatum. The mean transduction efficiency in the highly transduced regions is indicated. Data are represented as mean ± SD.
Quantitative workflows accurately predict the outcome of a gene editing strategy targeting the corticostriatal circuitry affected in HD
We then assessed the value of the workflows in predicting HTT gene editing in fully humanized HU97/18 mice30 (Figure 5A). Three months after the co-injection of AAV2.retro- and AAV2/rh.10-KamiCas9v2, the animals were killed, and cortical and striatal punch specimens were collected from the GFP-positive areas (Figure S4A). We predicted a maximum theoretical editing level of 31.2 ± 3.6% for the cells in the mediodorsal frontal cortical regions (Figure S4A and Table S1). The 3D and 2D transduction data indicated that, on average, there were 24.6 ± 18% (3D) and 17.6 ± 13.5% (2D) GFP-positive cortical neurons, corresponding to 7.3 ± 4.9% (3D) and 5.1 ± 3.6% (2D) of all cells in a punch specimen. In the striatum, only SPNs were included in the theoretical prediction, because the EFS promoter driving SpCas9 expression displays neuronal tropism (Figure S4B). The predicted maximum theoretical number of cells that could be transduced in the striatum was 70 ± 0.4%43 and based on the 2D workflow, we expected to target 53.9 ± 5.4% of all cells in the dorsolateral striatum (Figure S4A).
Figure 5.

Validation of the workflow with a gene editing experiment targeting the HTT gene
(A). AAV2.retro- and AAV2/rh.10 KamiCas9v2 expressing the sgHTT1 and sgCas9 were injected bilaterally into fully humanized HU97/18 HD mice. Mice were killed three months post-injection and punch specimens were collected from the GFP-positive cortical and striatal areas (Figure S4A). (B-C) Cortical (n = 4 punch specimens from 2 animals for the control and n = 30 punch specimens from 5 animals for KamiCas9v2) and striatal (n = 4 punch specimens from 2 animals for the control and n = 23 punch specimens from 5 animals for KamiCas9v2) editing was assessed by TIDE on DNA extracts from the punch specimens. The green dashed line indicates the theoretical maximum for transduction, and the predicted editing efficiency based on the 2D and 3D workflows is indicated by the blue and red dashed lines, respectively. (D, E and F) Capillary-based immunoassay showing that HTT editing leads to a proportional decrease in HTT protein levels (4C8 antibody). The vinculin antibody was used as an internal standard for the quantitative analysis (n = 4 punch specimens from 2 animals for the control and n = 6 punch specimens from 3 animals for KamiCas9v2). (G) AAV2.retro- and AAV2/rh.10 KamiCas9v2 expressing sgHTT51 and sgCas9 were bilaterally injected into FVB mice. Mice were killed five months post-injection and the analysis was performed as in (A). (H and I) Cortical (n = 6 punch specimens from 2 animals for the control and n = 20 punch specimens from 4 animals for KamiCas9v2) and striatal (n = 8 punch specimens from 2 animals for the control and n = 23 punch specimens from 4 animals for KamiCas9v2) editing was assessed as in panels (B) and (C). Data are represented as mean ± SD.
The bilateral injection of the AAV-KamiCas9v2 system into HU97/18 transgenic mice with AAV2.retro and AAV2/rh.10 resulted in the inactivation of 5.2 ± 2.1% and 34.7 ± 10.6% of the HTT alleles in the cortex and striatum, respectively (Figures 5A–5C). We investigated whether the HTT indels resulted in an inactivation of HTT translation, by analyzing HTT protein levels in a capillary-based western-blot assay (Figures 5D and 5E and S5). We found that the level of HTT gene editing was correlated with the loss of striatal HTT protein (Figure 5F). The editing efficiencies actually achieved were slightly lower than the predictions from our 3D and 2D workflows, probably due to the presence of multiple, tandem copies of the human HTT (4 copies of mutant and 4 copies of WT HTT gene) in the HU97/18 transgenic mouse model (Figure S6), as recently highlighted by Shin and coworkers.44
We tested this hypothesis, by using our predictive pipeline in WT FVB mice and targeting the two copies of the mouse HTT gene (Figure 5G). There is a nucleotide mismatch between the human and mouse HTT sequences in the region targeted by sgHTT1.23 We therefore replaced sgHTT1 with sgHTT51, the sequence of which is identical to that of the mouse HTT gene. The injection of the AAV-KamiCas9v2 system expressing sgHTT51 into WT mice induced indels in 9.6 ± 6.7% and 54.4 ± 5.3% of the mouse HTT genes in the cortex and striatum, respectively (Figures 5H and 5I). These editing efficiencies are consistent with the predictions of our 3D and 2D workflows and confirm the impact of multiple copies of the HTT gene on editing efficiencies.
In summary, we demonstrate that our semi-automated quantitative workflows can be used not only to estimate transduction efficiency, but also to optimize gene delivery and to predict gene editing outcomes in brain circuits affected by neurodegenerative disorders.
Discussion
Despite intensive research to improve gene delivery to the CNS and the refinement of genetic tools to tackle neurological disorders, semi-automated analytic pipelines for evaluating gene delivery performance and gene editing outcome are still lacking. The co-registration of a reference atlas and 3D images of clarified brains have been used to study drug distribution45 neuronal activity45,46,47 and pathological disease mechanisms.12,48,49 Despite their considerable potential, these methods have been largely overlooked in the development of gene therapy products. Lopes and coworkers recently used registered LSFM-acquired images to evaluate the biodistribution of AAV2/9 transduction in the brain after intravenous (IV) administration.50 However, their analysis was limited to the measurement of GFP-positive areas in various brain regions . This measurement is a good indicator of AAV biodistribution, but provides no information about AAV transduction efficiency.
We therefore used a mouse cell atlas and the Allen Brain Reference Atlas (ARA) to develop a semi-automated workflow. The Blue Brain Cell Atlas provides information about cellular composition for 86 regions of the mouse brain.10 By considering specific characteristics of the delivery system — in this case, AAV tropism, retrograde transport, and site of injection into the brain — it is possible to estimate the maximum number of cells that can be targeted in a particular brain region. This theoretical maximum number of target cells can then be compared with experimental data, to determine the actual transduction efficiency.
In this study, we tested 3D and 2D semi-automated workflows for determining the efficiency of CPN transduction with AAV2.retro-CBA-GFP injected into the mouse striatum. We estimated that AAV2.retro would be able to transduce 19% of the excitatory CPNs in the top 10 transduced cortical regions, corresponding to approximately 5.5% of the total number of cells in these regions. Our results indicated that the number of GFP-positive cells was consistent between the 3D and 2D workflows among several cortical regions. One advantage of the 2D workflow is that it can be performed in most laboratories, whereas the 3D imaging pipeline requires LSFM and a high computational power for atlas co-registration. In addition, the examination of deeper brain structures may be more appropriate using the 2D imaging workflow due to light scattering and possible incomplete brain clearing in 3D whole-mount brains. Zhang and coworkers recently achieved constant image resolution across the whole brain with multi-scale LSFM,51 which automatically removes brain areas once they have been imaged, to ensure that the acquisition field of view is always close to the surface and to prevent light scattering through the tissue. The use of fluorescence proteins that localize to the nucleus, thereby preventing interference from neuropil signals, together with nuclear staining and StarDist-like machine learning algorithms, would also facilitate the implementation of the 3D workflow by the scientific community. Finally, although GFP fluorescence has been shown to be compatible with clearing methods11,50 and to allow successful quantification for AAV2.retro-transduced cells, brighter, more photostable reporters, such as StayGold52 could also be used to improve the detection of cells with weak fluorescent signals. Concerning the 2D workflow, the preparation of individual high-quality brain sections is time-consuming and dependent on user’s experience. Several parameters such as brain perfusion, sectioning, and mounting can affect the quality of brain sections. Although tissue deformations do not compromise quantitative analysis, they may perturb the automation of image co-registration with the Allen Brain reference atlas. In these cases, integrated tools in the ABBA pipeline (manual affine transformation and spline transformation) can be used to manually adjust brain alignment to the reference atlas.13
The current version of the mouse Blue Brain Cell Atlas is limited to cell composition in terms of oligodendrocytes, astrocytes, microglia, and excitatory and inhibitory neurons (parvalbumin (PV+), somatostatin (SST+), vasoactive intestinal peptide (VIP+) and residual (RES) GABAergic neurons). These workflows cannot, therefore, be applied to vectors yielding transgene expression restricted to a subset of cells within a brain structure. For example, the proportion of striatal Drd1/Drd2 neurons in the striatum is not yet described in the atlas. Similarly, the integration of the connectome data40 in the 3D cell atlas would certainly facilitate the evaluation of retrograde transduction efficiency. The lack of atlases providing information on cellular composition in different structures of larger brains prevents the use of these workflows in non-human primates. International initiatives such as the Brain Initiative Cell Census Network (BICCN)53 and the EBRAINS54 will provide more complex and complete atlases in the future to extend and refine the application of these workflows in multiple species.
CNS disorders are often associated with alterations to multiple neuronal networks across the brain and the targeting of various brain regions is complex. In recent years, AAV serotypes able to cross the blood-brain barrier have been characterized55 and IV administration has been considered as an option for targeting multiple brain regions. However, the IV delivery of AAVs to target the CNS requires the systemic administration of large doses, potentially leading to liver toxicity56 and the transduction in deep brain structures remains suboptimal. Intraparenchymal delivery requires lower doses of AAV, but it can be difficult to target multiple brain regions by this approach. The constant discovery of new AAV serotypes with improved and/or distinct brain biodistribution keeps expanding the ability to transduce multiple brain structures.57,58,59,60
Here, we show that combining the potent retrograde transport properties of AAV2.retro24 with the broad striatal diffusion of AAV2/rh.1025,26 makes it possible to target CPNs and striatal cells. This combinatorial approach could be particularly useful for the treatment of neurodegenerative diseases in which large areas of the brain are affected, such as Alzheimer’s disease. In addition to making it possible to transduce multiple brain regions, the use of a combination of AAV serotypes also makes it possible to target different cell types. Lin and coworkers recently produced an AAV variant capable of efficiently transducing microglia with minimal immune activation (AAV-MG).61 This work is paving the way for the combination of serotypes targeting neurons, astrocytes, and microglial cells. As these and other CNS delivery strategies are explored, the integration of semi-automated quantitative pipelines, such as those presented here, will be essential for the evaluation of their potential.
In the last part of the study, we validated our workflow for the codelivery of AAV2.retro and AAV2/rh.10-KamiCas9 vectors targeting the HTT gene. Based on the transduction efficiencies of AAV2.retro and AAV2/rh.10, we expected about 6% and 54% of the HTT alleles to be edited in the cortical and striatal regions, respectively. Rates of human HTT gene inactivation were relatively low in HU97/18 mice, but well within the predicted range for WT mice. HU97/18 mice are generated by crossing mice transgenic for the bacterial artificial chromosome (BACHD)62 and yeast artificial (YAC18)63 systems. As a result, it contains multiple copies of the human HTT gene in tandem. This artificial genomic structure of the human HTT loci in these mice probably accounts for the differences in HTT editing efficiency between the two models. These results suggest that genome editing studies should be performed preferentially in knock-in models, to prevent confounding due to genome structure-related factors.
In summary, we present here powerful semi-automated 2D and 3D quantitative workflows for the estimation of transduction efficiency throughout the mouse brain. We also demonstrate that such estimates can be used to predict editing outcomes. In addition, we propose the combination of AAV serotypes to expand gene delivery in the brain and to maximize gene editing in the corticostriatal network affected in HD.
Materials and methods
Plasmid production
Several fluorescence reporter constructs were used to the different AAV serotypes and to assess their transduction efficiency. The pAAV2ss-EFS-GFP-SynPolyA and pAAV2ss-EFS-mCherry-SynPolyA plasmids were generated by replacing the SpCas9 with the GFP and mCherry transgenes in pAAV2ss-EFS-SpCas9-synPolyA (a gift from Ryohei Yasuda; RRID: Addgene 104588)27 after AgeI and EcoRI digestion. The pAAV2ss-CBA-EGFP-WPRE-bGH plasmid was kindly provided by Prof. During, Ohio, USA. Two other reporter plasmids, pAAV2ss-CBA-AcGFPnuc-WPRE-bGH and pAAV2ss-CBA-mCherry-WPRE-bGH, were generated by transferring the AcGFPnuc and mCherry coding sequences from the entry plasmids, pENTR4-AttL1-AcGFPnuc-AttL2 and pENTR4-AttL1-mCherry-AttL2, to an AAV destination transfer vector, pAAV2ss-CBA-Gateway-WPRE-bGH.
For the development of the AAV-KamiCas9 system, we ordered, from GeneArt, a universal entry plasmid, pMK-AttL1-NotI/BamHI-U6-BsaI-tracrRNAopt-BamHI-U6-SapI-tracrRNAopt-NotI/XbaI-7sk-BsmBI-tracrRNA-XbaI-AttL2 containing three sgRNA expression cassettes, to make it possible to adopt a flexible cloning strategy for multiple sgRNAs. The U6-driven expression cassettes contain an optimized tracrRNA described by Dang and coworkers64 whereas the 7sk-driven expression cassette is associated with the original tracrRNA.23 Unique restriction sites (NotI, BamHI and XbaI) were strategically placed between the cassettes to facilitate the removal of every possible cassette combination. The spacer sequence of each sgRNA was inserted into the universal entry plasmid with overhang-compatible annealed oligos after digestion of the plasmid with type IIS restriction enzymes (BsaI, SapI or BsmBI). The sgGFP targeting the GFP transgene was cloned by SapI digestion and insertion of the annealed oligomers 5′-CACCGGGCGAGGAGCTGTTCACCG-3′ and 5′-AAACCGGTGAACAGCTCCTCGCCC-3′ to generate the pMK-AttL1-U6-sgGFP-AttL2 plasmid. Similarly, sgHTT1 and sgHTT51 were cloned by BsaI digestion and annealing of the oligos 5′-CACCGACCCTGGAAAAGCTGATGA-3′ and 5′-AAACTCATCAGCTTTTCCAGGGTC-3′ or 5′-CACCGAACCCTGGAAAAGCTGATGA-3′ and 5′-AAACTCATCAGCTTTTCCAGGGTTC-3′, respectively. A new sgCas9_2 (hereafter called sgCas9) targeting the 5′end of the open reading frame present in the pAAV2ss-EFS-SpCas9-synPolyA was inserted downstream of the 7sk promoter by BsmBI digestion and insertion of the annealed oligos 5′-CCTCGTCGCCGAAGAAAAAGCGCA-3′ and 5′-AAACTGCGCTTTTTCTTCGGCGAC-3′ (plasmids pMK-AttL1-U6-sgHTT1-7sk-sgCas9-AttL2 and pMK-AttL1-U6-sgHTT51-7sk-sgCas9-AttL2), or downstream of the U6 promoter by SapI digestion and annealing of the oligos 5′-ACCGTCGCCGAAGAAAAAGCGCA-3′ and 5′-AACTGCGCTTTTTCTTCGGCGAC-3′ (plasmid pMK-AttL1-U6-sgHTT1-U6-sgCas9-AttL2). The AAV plasmids expressing only sgRNAs, pAAV2ss-U6-sgGFP, pAAV2ss-U6-sgHTT1-7sk-sgCas9 and pAAVss-U6-sgHTT51-7sk-sgCas9, were produced by transferring the sgRNA expression cassettes from the entry plasmids into an AAV destination vector, pAAV2ss-Gateway-bGH. The AAV plasmids expressing sgRNAs together with the GFP reporter, pAAV2ss-EFS-GFP-synPolyA-U6-sgHTT1 and pAAV2ss-EFS-GFP-synPolyA-U6-sgHTT1-U6-sgCas9, were produced by classical restriction/ligation cloning with the NotI enzyme.
We also used the SIN-cPPT-PGK-SpCas9-WPRE (Addgene #87886), SIN-cPPT-U6-sgHTT1-PGK-mCherry-WPRE and SIN-cPPT-PGK-mCherry-WPRE plasmids to generate an HTT-KO clonal HEK293T cell line and an HEK293T cell line stably expressing SpCas9.
Human embryonic kidney 293T (HEK293T) cells
HEK293T cells (mycoplasma-negative, ATCC, LGC Standards GmbH, Wessel, Germany) were cultured in DMEM-Glutamax supplemented with 10% FBS and 1% penicillin/streptomycin (Gibco, Life Technologies, Zug, Switzerland) at 37°C under an atmosphere containing 5% CO2. For routine culture, cells were passaged twice weekly after trypsin treatment for dissociation (Gibco, Life Technologies, Zug, Switzerland) and plated at a density of 2 × 106 cells/cm2 in T175 flasks.
Production of lentiviral vectors
The LV vectors were produced by the calcium phosphate-mediated transfection of HEK-293T cells with a four-plasmid system.65 Human immunodeficiency virus type 1 (HIV-1) vectors were pseudotyped with the vesicular stomatitis virus glycoprotein (VSV-G) envelope, concentrated by ultracentrifugation and resuspended in phosphate-buffered saline (PBS, Gibco, Life Technologies, Zug, Switzerland) supplemented with 1% bovine serum albumin (BSA, Sigma-Aldrich, Buchs, Switzerland). The viral particle content of each batch was determined in a p24 antigen enzyme-linked immunosorbent assay (p24 ELISA, RETROtek, Kampenhout, Belgium). Viral stocks were stored at −80°C until use.
Production of adeno-associated vectors
AAVs were also produced in HEK293T cells by calcium phosphate-mediated transfection. The cells were transfected with the pAAV2ss containing the transgene of interest together with the pAd Helper-AAV (Agilent Technologies kit #240071) and pAAV-rh10_Rep_Cap (Penn Vector Core, University of Pennsylvania, School of Medicine, Philadelphia, USA) or pAAV2.retro (a gift from Alla Karpova & David Schaffer RRID: Addgene_81070). For AAV2/1, the cells were transfected with the pAAV2ss containing the transgene of interest together with the pDP1rs-RFP plasmid encoding helper functions (Plasmid Factory plasmid PF401). The cell suspension was centrifuged at 360 x g for 10 min at 4°C 72 h post-transfection. The supernatant was supplemented with 10 mM PEG (Roth, Arlesheim, Switzerland) and 0.5 M NaCl (Merck, Nottingham, UK) and was incubated at 4°C for at least 2 h. Cell pellets were pooled and incubated in lysis buffer (0.15 M NaCl, Merck, Nottingham, UK; 50 mM Tris-HCl, pH 8.5, Sigma-Aldrich, Buchs, Switzerland) for 3 consecutive freeze/thaw cycles (30 min in dry ice/ethanol followed by 30 min at 37°C). After the initial 2-h incubation period, the PEG-containing supernatant was centrifuged at 3700 x g for 20 min at 4°C and the supernatant was discarded. Cell lysate was added to the pellets and the mixture was incubated at 37°C for 1 h to ensure that the pellets were fully homogenized. The lysate was then treated with 50 U/mL Benzonase (Sigma-Aldrich, Buchs, Switzerland) and 10 mM MgCl2 (Sigma-Aldrich, Buchs, Switzerland) at 37°C for 30 min. The treated lysate was clarified by centrifugation at 3700 x g for 20 min at 4°C. AAVs were separated by iodixanol (AxonLab, Le Mont sur Lausanne, Switzerland) gradient ultracentrifugation at 255,690 g (70Ti rotor, Beckman-Coulter, Nyon, Switzerland) for 90 min at 20°C. The AAV-containing phase was harvested and loaded on an Amicon Ultra-15 PL 100 column (Millipore, Zug, Switzerland) with 0.001% Pluronic F68 D-PBS (Gibco, Thermo Fisher Scientific, Waltham, USA, Zug, Switzerland) for iodixanol cleaning and viral particle concentration. The tubes were first centrifuged at 4000 x g at 4°C until all the solution had passed through the column. Two additional washes with 0.001% Pluronic F68 D-PBS were performed and the AAV were finally suspended in 120–150 μL 0.001% Pluronic F68 D-PBS.
The viral genome content (vg/mL) for each AAV was assessed by Taqman qPCR with primers recognizing the inverted terminal repeats of the AAV2 viral genome (forward primer: 5′- GGAACCCCTAGTGATGGAGTT-3′, reverse primer: 5′-CGGCCTCAGTGAGCGA-3′, Taqman probe: 5′- FAM-CACTCCCTCTCTGCGCGCTCG-TAMRA-3′) and the KAPA probe fast qPCR universal kit (Sigma-Aldrich, Buchs, Switzerland). AAV vectors were stored at −80°C until use.
Transfection of HEK293T cells
For the transfection experiment (Figures S1A and S1B), we plated 5 × 105 cells per well in six-well plates the day before transfection. We mixed 1150 fmol plasmids (150 fmol pAAV2ss-EFS-GFP-synPolyA, 250 fmol pAAV2ss-EFS-SpCas9-synPolyA and 750 fmol sgRNA(s)-expressing plasmids) in 0.25 M CaCl2 solution and the mixture was then added dropwise to HEPES saline buffer (Sigma-Aldrich, Buchs, Switzerland, CaCl2-H2O:HEPES ratio 1:1). Cells not treated with the sgRNA-expressing plasmid were used as a negative control. The mixture was incubated at room temperature for 5 min and added dropwise to the cells (10% of the culture volume). The medium was completely replaced 6 h after transfection. We evaluated the efficiency of HTT editing by SpCas9 four days post-transfection.
Production of a clonal huntingtin knockout (HTT-KO) HEK293T cell line
For the generation of an HTT-KO clonal HEK293T cell line (Figures S4B and S4C), we plated 5 × 106 cells in a 10 cm Petri dish the day before transfection. We mixed 30 μg of plasmids (15 μg SIN-cPPT-PGK-SpCas9-WPRE and 15 μg SIN-cPPT-U6-sgHTT1-PGK-mCherry-WPRE) in 0.25 M CaCl2 solution and the mixture was then added dropwise to HEPES saline buffer (Sigma-Aldrich, Buchs, Switzerland, CaCl2-H2O:HEPES ratio 1:1). The mixture was incubated at room temperature for 5 min and added dropwise to the cells (10% of the culture volume). The medium was completely replaced 6 h after transfection. Five days post-transfection, the cells were dissociated with trypsin and serial dilutions were plated in 96-well plates. The cell clones that grew at the highest dilution conditions were sequentially expanded in 48-well plates, 24-well plates and, finally, six-well plates. We screened several clonal cell lines by Sanger sequencing and selected a single clone in which the HTT alleles had an extra adenosine nucleotide (+1, A) immediately downstream of the ATG at the sgHTT1 target site. This clonal cell line (clone F10) was then expanded and frozen in liquid nitrogen.
Transduction of HEK293T cells
For the transduction experiment (Figures S1C and S1D), we first generated a cell line stably expressing the SpCas9. We plated 5 × 105 cells per well in six-well plates the day before LV transduction. We mixed 300 ng of LV (150 ng SIN-cPPT-PGK-SpCas9-WPRE and 150 ng SIN-cPPT-PGK-mCherry-WPRE) in DMEM medium and then added the mixture dropwise to the cells (10% of the culture volume). The medium was completely replaced 24 h after transduction and the cells were passaged twice weekly. Two weeks after LV infection, we plated 1.2 × 105 cells per well in 24-well plates. Cells were infected with 1.5 × 109 vg of AAV2/1 (7.5 × 108 vg AAV2/1-EFS-GFP-SynPolyA-U6-sgHTT1 and 7.5 × 108 vg AAV2/1-U6-sgGFP5) the next day. Cells incubated without AAV2/1-U6-sgGFP5 were used as a negative control. The medium was replaced 24 h after transduction and HTT editing efficiency was evaluated seven days post-AAV transduction.
Animals
Adult male and female mice (9–15 weeks) were used for the in vivo experiments. Wild-type C57BL/6 and FVB mice were obtained from Janvier (Le Genest-Saint-Isle, France). Drd2-EGFP transgenic mice (Tg(Drd2-EGFP)S118Gsat/Mmnc, RRID:MMRRC_000230-UNC) were acquired from the Mutant Mouse Resource and Research Center (MMRRC) at the University of North Carolina at Chapel Hill. Transgenic mice expressing the full-length human wild-type HTT gene (HU18/18; YAC18++; Hdh−/−) or wild-type and mutant HTT gene (HU97/18; BACHD+; YAC18++; Hhd−/−) were kindly provided by Prof. Hayden (Vancouver, Canada)30 and BAC-GLT1-eGFP transgenic mice expressing eGFP specifically in astrocytes were provided by Prof. J Rothstein (Baltimore, MD, USA).66 Mice were housed in a specific pathogen-free (SPF) facility with IVC cages GM500 (Tecniplast) or rat R.BTM.U x/R.ICV.6 cages (Innovive, Paris, France) and Innorack rats, simple face (cat# RS.5.8.40) containing corn cob bedding, with no more than five mice per cage. The animals were maintained in a controlled-temperature room (22 ± 1°C), under a 14-h light/10-h dark cycle. The following enrichments were provided: 2 pieces of wipes, 1 cardboard tunnel, 1 cardboard or polysulfone house with 2 entrances/exits. Food (SAFE 150, Safe, Rosenberg, Germany) and water were provided ad libitum. All experimental procedures were performed in strict accordance with Swiss regulations concerning the care and use of laboratory animals (veterinary authorizations 3447 and 3682).
Measurement of HTT gene copy number
Genomic DNA was extracted from HEK293T, neural progenitor cells (NPCs) (derived from Coriell GM03621), BACHD,62 HU18/18 and HU97/18 mice30 using the 500μL TRIzol reagent (Life Technologies, Zug, Switzerland) according to the manufacturer’s protocol. Concentration and quality of DNA were evaluated using a NanoDrop ultraviolet spectrophotometer (Thermo Fisher Scientific, Reinach, Switzerland). The absolute number of human HTT copies was quantified by digital PCR (QIAcuity digital PCR, Qiagen, Basel, Switzerland) in QIAcuity 24-well Nanoplates with 8500 partitions. Separated Taqman assays targeting the SNP rs362331 (C/T) in exon 50 of the HTT gene were generated from the C_2231945_10 ThermoFisher assay (custom designed, ThermoFisher Scientific, Reinach, Switzerland) to individually analyze the number of HTT alleles containing a cytosine (VIC) or a thymine (FAM). We used the assay described by Christodoulou and colleagues targeting the poly(rC)-binding protein 2 (PCBP2) gene67 to normalize HTT amplification signals to the amount of gDNA. HTT-FAM and HTT-VIC signals were normalized to PCBP2-VIC (probe VIC-CCCTCTCCTGGCTCTAAATGTTGTGT-BHQ1) and PCBP2-FAM signals (FAM-CCCTCTCCTGGCTCTAAATGTTGTGT-BHQ1), respectively (Microsynth, Balgach, Switzerland). Each reaction was set up accordingly to the manufacturer’s instructions in a 12 μL sample volume containing 3μL of 4x QIAcuity probe PCR mastermix (Qiagen, Basel, Switzerland), 1X Taqman HTT custom assay, 0.8 μM of PCBP2 forward and reverse primers, 0.4 μM PCBP2-labeled probes (Microsynth, Balgach, Switzerland), 3 units of EcoRI-HF restriction enzyme, and 100 ng genomic DNA. Loaded plates were incubating 10 min at RT for EcoRI-HF digestion followed by DNA amplification: initial denaturation at 95°C for 2min, 40 amplification cycles at 95°C, 60°C and 72°C for 15 s each, and a final step at 40°C for 5 min. Finally, data analysis was performed with the QIAcuity Software Suite 2.0.20 (Qiagen, Basel, Switzerland).
Stereotaxic injections
Anesthesia and surgical procedures were performed as previously described.23 A total volume of 4–6 μL AAV per hemisphere (between 4.0 × 108 and 1.75 × 109 vg/hemisphere) was administered to the mouse striatum at a rate of 0.5 μL/min. AAVs administered bilaterally at a single striatal site per hemisphere were injected at the coordinates +1; ±1.8; −3.5 (+1 mm rostral to bregma; ±1.8 mm lateral to midline; and 3.5 mm ventral from the skull surface, with the tooth bar set at −3.3 mm) in mice with a C57Bl/6 background mice, and at the coordinates +0.7; ±1.9; −3.0 in mice with an FVB background. For unilateral AAV delivery to two striatal sites, we used the coordinates +1.2; ±2.0; −3.2 (site 1) and +0.26; ±2.8; −3.2 (site 2).68 The needles were left in place for 5 min after the injection and were then slowly removed. During surgery, body temperature was controlled with a warming blanket (CMA 450 Temperature Controller, Phymep, Paris, France) and the eyes were protected with 0.2% Viscotears liquid gel (Novartis, Basel, Switzerland). Post-surgery analgesic treatment (acetaminophen, Dafalgan Upsa 1000 mg/750 mL) was administered in drinking water for 72 h.
As a proof-of-concept for gene editing (Figures 1A and 1B), we injected 3.0 × 108 vg AAV2/1-EFS-SpCas9-SynPolyA, 3.0 × 108 vg AAV2/1-U6-sgGFP5 and 1.0 × 108 vg AAV2/1-EFS-mCherry-SynPolyA bilaterally, at one site per hemisphere, in Drd2-EGFP mice. Mice not receiving the SpCas9 transgene were used as a negative control. Mice were killed three weeks after the injection, for histological processing.
For comparisons of the efficiency of AAV-CRISPR and AAV-KamiCas9 to edit the human HTT gene (Figures 1C–1E), we injected 4.0 × 108 vg AAV2/1-EFS-SpCas9-SynPolyA with 4.0 × 108 vg AAV2/1-EFS-GFP-SynPolyA-U6-sgHTT1 or 4.0 × 108 vg AAV2/1-EFS-GFP-SynPolyA-U6-sgHTT1-U6-sgCas9 bilaterally, at one site per hemisphere, in HU18/18 mice. Mice not receiving such injections were used as a negative control. Mice were killed eight weeks post-injection for the extraction of DNA and RNA.
For the establishment of the pipeline (Figures 3B–3D, S2 and Videos S1 and S2), we injected 1.3 × 109vg (high dose) or 3.2 × 108vg (low dose) of AAV2.retro-CBA-EGFP-WPRE-bGH into wild-type C57BI/6 mice unilaterally, at two sites. The two mice were killed three weeks post-injection, for histological processing (low dose) or CLARITY (high dose).
We maximized the targeting of the circuitry affected in HD (Figures 4A and 4B), by injecting 2.0 × 108 vg AAV2/RH.10-CBA-mCherry-WPRE-bGH and 3.0 × 108 vg AAV2/RH.10-CBA-EGFP-WPRE-bGH bilaterally, at one site per hemisphere, in HU18/18 mice. The mice were killed three weeks post-injection for histological processing.
For the estimation of striatal transduction efficiency (Figures 4C–4E and S3), we injected 4.5 × 108 vg AAV2/RH.10-U6-sgHTT51-7sk-sgCas9, 1.5 × 107 vg AAV2/RH.10-CBA-AcGFPnuc-WPRE-bGH, 6.7 × 108 vg AAV2.retro-U6-sgHTT51-7sk-sgCas9 and 1.5 × 107 vg AAV2.retro-CBA-AcGFPnuc-WPRE-bGH bilaterally, at one site per hemisphere, in wild-type FVB mice. Mice were killed 24 weeks post-injection, for histological processing.
For the evaluation of the neuronal tropism of the AAV2/RH.10 and/or EFS promoters (Figure S4B), 4.0 × 108 vg AAV2/RH.10-EFS-mCherry-synPolyA were bilaterally injected into the striatum of BAC-GLT-eGFP mice, at one site per hemisphere. Mice not receiving such injections were used as a negative control. Mice were killed three weeks post-injection, for histological processing.
For the evaluation of mutant HTT editing efficiency in both the striatum and cortex (Figures 5A–5C and S4D and S4E), we injected 2.0 × 108 vg AAV2/RH.10-EFS-SpCas9-SynPolyA, 4.0 × 108 vg AAV2/RH.10-U6-sgHTT1-7sk-sgCas9, 1.0 × 108 vg AAV2/RH.10-CBA-AcGFPnuc-WPRE-bGH, 3.0 × 108 vg AAV2.retro-EFS-SpCas9-SynPolyA, 6.0 × 108 vg AAV2.retro-U6-sgHTT1-7sk-sgCas9 and 1.5 × 108 vg AAV2.retro-CBA-AcGFPnuc-WPRE-bGH bilaterally into HU97/18 mice, at one site per hemisphere. Mice not receiving SpCas9 transgene injections were used as a negative control. Mice were killed 14 weeks post-injection, for DNA and protein extraction.
For the evaluation of mouse HTT editing efficiency in both the striatum and cortex (Figures 5D–5H), we injected 1.5 × 108 vg AAV2/RH.10-EFS-SpCas9-SynPolyA, 4.5 × 108 vg AAV2/RH.10-U6-sgHTT51-7sk-sgCas9, 1.5 × 107 vg AAV2/RH.10-CBA-AcGFPnuc-WPRE-bGH, 2.3 × 108 vg AAV2.retro-EFS-SpCas9-SynPolyA, 6.7 × 108 vg AAV2.retro-U6-sgHTT51-7sk-sgCas9 and 1.5 × 107 vg AAV2.retro-CBA-AcGFPnuc-WPRE-bGH bilaterally into FVB mice, at one site per hemisphere. Mice not receiving SpCas9 transgene injections were used as a negative control. Mice were killed 19 weeks post-injection, for DNA and protein extraction.
CLARITY, 3D light-sheet imaging and MIRACL analysis
For estimation of the transduction efficiency of AAV2.retro with the 3D quantitative workflow (Figure 3B and Video S1), mice were killed by sodium pentobarbital overdose three weeks post-injection and transcardially perfused with PBS followed by 4% paraformaldehyde (4% PFA) (Fluka, Sigma, Buchs, Switzerland). The brain was removed, post-fixed by incubation in 4% PFA for 24 h and washed in PBS. CLARITY sample preparation and light-sheet imaging were performed as previously described.68 The clarified brain was analyzed with fiji software (Version 2.0.0-rc-69/1.52p) followed by the MIRACL registration pipeline.12 CLARITY acquisition was followed by exportation as an image sequence to generate one tif file per stack, ordered numerically and dorsoventrally. This image sequence tif file was duplicated and processed in parallel in fiji and MIRACL. In fiji, it was post-processed to determine the numbers and coordinates of GFP-positive cells after subtract background on the whole stack. GFP-positive cells can be found outside of the focal plane due to epifluorescence limitation. We therefore used only one in every six consecutive stacks (every 30 μm) to localize the cells and to ensure that the same cell was not considered twice. We obtained the coordinates of all GFP-positive cells, by using the Find Maxima/List function of fiji and merging all coordinates into a single csv file. In parallel, the MIRACL pipeline12 was run on all image sequence tif files. Briefly, the images were initially subjected to processing with the changeValue function of fiji and manual editing to enhance co-registration quality and to decrease the discrepancy between transduced and non-transduced areas. The co-registration was performed on the 25 μm Allen Regional Atlas template. ITKsnap (http://www.itksnap.org) was used to visualize MIRACL outputs. Finally, the csv coordinates of GFP-positive cells and MIRACL co-registered brain were used to map segmented cells to the Allen Regional Atlas according to the recently described MOTA pipeline.38
Histological processing and imaging acquisition for 2D analysis
For the characterization of AAV serotype transduction patterns and tropisms (Figures 1A, 1B, 4A, 4B, and S4B), the animals were killed by sodium pentobarbital injection three weeks post-injection and transcardially perfused with PBS followed by 4% paraformaldehyde (4% PFA) (Fluka, Sigma, Buchs, Switzerland). The brains of the animals were removed, post-fixed by incubation in 4% PFA for 24 h and cryoprotected by two consecutive incubations in 20% and 30% sucrose (Sigma-Aldrich, Buchs, Switzerland) for 12 h each. A cryostat (CM1850, Leica Biosystems, Muttenz, Switzerland) with a freezing stage at −20°C (SM2400; Leica Microsystems AG,Glattbrugg, Switzerland) was used to cut 25 μm-thick coronal brain sections. Sections were collected and stored in anti-freeze solution (0.2 M sodium phosphate buffer, 25% glycerol, 30% ethylene glycol) in 96-well plates at −20°C until use. If no immunohistochemical staining was to be performed, the brain sections were mounted directly on slides, in Vectashield supplemented with DAPI (Reactolab, Servion, Switzerland). Brain sections for immunohistochemical staining were rinsed in TBS (3 × 10 min) at room temperature and blocked by incubation for 1 h in TBS-T (TBS, 0.1% Triton X-100) supplemented with 5% BSA (Sigma-Aldrich, Buchs, Switzerland). For AcGFPnuc immunoblotting (Figures 4C–4E), sections were incubated overnight at 4°C with a goat polyclonal GFP primary antibody (RRID: AB_2333100; AB0020-500, Sicgen) diluted 1/250 in TBS-T supplemented with 1% BSA. They were washed three times with TBS (3 × 10 min) and were then incubated with the Alexa Fluor568-conjugated donkey anti-goat secondary antibody (A-11057, Life Technologies, Zug, Switzerland) diluted 1/1000 in TBS-T supplemented with 1% BSA for 1 h at room temperature. For the mCherry immunoblotting (Figure S4B), sections were incubated overnight at 4°C with a goat polyclonal mCherry primary antibody (RRID: AB_2333093; AB0040-500, Sicgen) diluted 1/250 in TBS-T supplemented with 1% BSA. They were washed three times with TBS (3 × 10 min) and then incubated with the Alexa Fluor568-conjugated donkey anti-goat secondary antibody (A-11057, Life Technologies, Zug, Switzerland) diluted 1/1000 in TBS-T supplemented with 1% BSA for 1 h at room temperature. Finally, for all immunoblots, the brain sections were washed three times in TBS (3 × 10 min) and mounted on slides, in Vectashield supplemented with DAPI (Reactolab, Servion, Switzerland).
Images of the full brain sections were obtained with a digital camera (3CCD Hitachi HV-F202SCL) on a slide scanner microscope (×20 objective, Zeiss axioscan Z1).
QuPath and ABBA analysis
For estimation of the cortical transduction efficiency of AAV2.retro (Figure 3C) and the striatal transduction efficiency of AAV2.retro and AAV2/rh.10 (Figures 4C–4E) with the 2D quantitative workflow, images of the brain sections were obtained with a digital camera (3CCD Hitachi HV-F202SCL) on a slide scanner microscope (×20 objective, Zeiss Axioscan Z1). We then used a fiji and QuPath workflow to estimate the number of transduced cells in various brain regions. We co-registered the coronal brain sections and the Allen CCFv3 reference atlas36 in fiji, with the Aligning Big Brains & Atlases (ABBA) plugin13 and exported the data in QuPath (https://biop.github.io/ijp-imagetoatlas/). The QuPath bioimage analysis software and StarDist extension,15 a deep learning-based method of nucleus detection with pretrained models, were then used to identify nuclei on the basis of DAPI staining.14 Finally, a cell classifier was used to quantify GFP-positive cells and to export the data.
DNA, RNA and protein extraction
For in vitro experiments (Figure S1), DNA was extracted from HEK293T cells with QuickExtract DNA extraction solution (QE09050, Lubioscience, Zürich, Switzerland), according to the manufacturer’s recommendations. Briefly, cells were dissociated with trypsin (Gibco, Life Technologies, Zug, Switzerland) and washed in PBS. Genomic DNA from approximately 3 × 105 cells was extracted by mixing the cells with 100 μL QuickExtract solution and incubating at 65°C and 98°C for 10 and 2 min, respectively. In addition, protein was extracted from wild-type and HTT-KO HEK293T cells in RIPA buffer (R0278, Sigma) supplemented with a 1/200 dilution of protease inhibitor cocktail (P8340, Sigma-Aldrich, Buchs, Switzerland) and 5 μM Z-VAD-FMK (HY-16658B, Chemie Brunschwig, Basel, Switzerland), hereafter referred to as RIPA+ buffer. Approximately 1 × 106 cells were washed in PBS and 50 μL of RIPA+ buffer was added to the cell pellet. The pellet was homogenized with a pellet mixer (VWR, Dietikon, Switzerland) and left on ice for 30 min. The protein extract was then centrifuge at 18000 x g for 15 min at 4°C and the supernatant containing solubilized proteins was collected into a new tube and stored at −80°C.
For in vivo experiments, mice were sacrificed by isoflurane overdose and brains were collected and cut into 1 mm-thick coronal slices. For the comparison of AAV-CRISPR and AAV-KamiCas9 systems (Figures 1C–1E), we collected 5–6 punches (∼1.5 mm3 each) from the GFP-positive striatum and merged them by hemisphere. The DNA and RNA from each transduced striatal hemisphere was extracted with 500μL TRIzol reagent (Life Technologies, Zug, Switzerland) according to the manufacturer’s protocol. DNA was resuspended by incubation overnight in water at RT and it was then stored at −20°C. RNA samples were first treated with RQ1 RNase-free DNase (Promega, Dübendorf, Switzerland) and then converted into cDNA using the RNA with Superscript II (Thermo Fisher, Reinach, Switzerland) according to the manufacturer’s instructions. RNA and cDNA samples were then stored at −80°C. For the evaluation of human and mouse HTT editing in both the cortex and striatum (AAV2.retro and AAV2/RH.10) (Figures 5 and S4D and S4E), we collected four to six striatal punch specimens and six to 10 cortical punch specimens from the transduced areas. The striatal punch specimens were then split into two tubes for the extraction of both DNA and protein. DNA was extracted from each individual punch specimen with the AllPrep DNA/RNA/miRNA Universal Kit (Qiagen, Hombrechtikon, Switzerland) according to the manufacturer’s recommendations. The DNA was stored at −20°C. The protein from each striatal punch specimen was extracted in 30 μL RIPA+ buffer, as described for protein extraction from HEK293T cells.
TIDE analysis
HTT and SpCas9 editing efficiencies were assessed by performing Tracking of Indels by Decomposition (TIDE) analysis35 on amplified gDNA or cDNA. The indel size range was set to 10 and the size of the decomposition window was adapted for reads of low quality or containing repetitive sequences. The significance cutoff was set to 0.05 (https://tide.nki.nl/). Nucleic acid amplification was performed on 100 ng of gDNA or 5 ng of cDNA with the KAPA HiFi Hotstart kit (KAPA Biosystems, Labgene) according to the manufacturer’s recommendations. The primers 5′-TTGCTGTGTGAGGCAGAACCTGCGG-3′ (hHTT_fwd/seq) and 5′-TGCAGCGGCTCCTCAGCCAC-3′ (hHTT_rev) were used to amplify the sgHTT1 target site of the human HTT gene from DNA samples from HEK293T cells and HD transgenic mice.23 TIDE analysis was performed on chromatograms for wild-type HTT amplicons sequenced with the primer hHTT_fwd primer, or chromatograms for mutant HTT amplicons sequenced with the 5′-GCGGGATCCATAACTTCGTA-3′ (mutHTT_seq) primer. For the mouse HTT gene, the sgHTT51 target site was amplified from WT mouse DNA samples with the primers 5′-CCTCCTCACTTCTTTTCTATCG-3′ (mHTT_fwd) and 5′-AGCATTATGTCATCCACTACC-3′ (mHTT_rev), with a melting temperature of 56°C and an extension period of 90 s. The mouse HTT amplicons were sequenced with the primer 5′-CTGTCAATTCTGCGGGTCTG-3′ (mHTT_seq) to generate the chromatograms for indel analysis. Finally, the sgCas9 target site in AAV-SpCas9 was amplified from both DNA and cDNA with the primers 5′-TTTTTCGCAACGGGTTTGCC-3′ (SpCas9_fwd) and 5′-AGAAGCTGTCGTCCACCTTG-3′ (SpCas9-rev/seq), using a melting temperature of 65°C and an extension period of 25 s. The efficiency of sgCas9 editing was evaluated by TIDE on analysis on amplicons sequenced with the SpCas9_rev/seq primer.
Virtual western blot
Protein concentration was assessed with a BCA kit (Thermo Fisher Scientific, Reinach, Switzerland) according to the recommended procedure. After dilution in 0.1x SB (ProteinSimple, Bio-Techne), 0.6 μg of total protein extract was size-separated with the Jess capillary-based immunoassay system. Samples were processed according to the manufacturer’s instructions, with the 66–440 kDa separation module (SM-W008). The primary anti-huntingtin antibody clone 1HU-4C8 (RRID: AB_2123255; MAB2166, Zug, Switzerland) (diluted 1/500) and the anti-vinculin antibody clone 3M13 (RRID: AB_2935846; ZRB1089, Merck, Nottingham, UK) (diluted 1/50) were used to target the HTT and vinculin proteins, respectively. HTT and vinculin were then detected by chemiluminescence with the anti-rabbit-HRP conjugate (043–026, ProteinSimple, Bio-Techne AG, Zug Switzerland) diluted 1/20 in the ready-to-use anti-mouse-HRP conjugate (DM-002, ProteinSimple, Bio-Techne AG, Zug, Switzerland). Compass software (version 6.1) was used for the analysis. Peaks were determined with the dropped line method and HTT and vinculin signals were used to calculate the HTT/vinculin ratios.
Statistical methods
For the statistical analyses, we tested the hypotheses of normally distributed data and equal variances to determine the most appropriate statistical test. Post-hoc analyses were performed with GraphPad Prism 9.1.0 software. ANOVA was used for comparisons of more than two groups and t-tests were used for comparisons between two groups. The results of statistical tests were considered significant if the p value obtained was below 5%. No specific method was used for sample randomization, sample-size estimation, or data inclusion/exclusion. All results are presented as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
For HTT editing in HU18/18 mice in Figure 1D, p values were calculated for Brown-Forsythe and Welch ANOVA, with F∗(DFn, DFd): 111.9(2.0, 11.9). For SpCas9 editing in HU18/18 mice in Figure 1E, p values were calculated by Brown-Forsythe and Welch ANOVA, with F∗(DFn, DFd): 100.2(3.0, 11.0).
For HTT editing in transfected HEK293T cells in Figure S1B, the p values were calculated by ordinary one-way ANOVA with F(DFn, DFd): 386.1(3, 8). For SpCas9 editing in transfected HEK293T cells in Figure S1B, the p values were calculated by ordinary one-way ANOVA with F(DFn, DFd): 3343(2, 6). For HTT editing in transduced HEK293T cells in Figure S1D, the p values were calculated by ordinary one-way ANOVA with F(DFn, DFd): 205.9(2,7).
For cortical HTT editing in HU97/18 mice in Figure 5B, p values were calculated in one-tailed unpaired t-tests with Welch’s correction (t value: 9.3; DF: 28.1). For striatal HTT editing in HU97/18 mice in Figure 5C, p values were calculated in one-tailed unpaired t tests with Welch’s correction (t value: 14.4; DF: 24.7). For cortical HTT editing in wild-type mice in Figure 5E, p values were calculated in one-tailed unpaired t tests with Welch’s correction (t value: 5.3; DF: 23.3). For striatal HTT editing in wild-type mice in Figure 5F, p values were calculated in one-tailed unpaired t tests with Welch’s correction (t value: 48.3; DF: 23.7). For the striatal HTT protein in wild-type mice in Figure 5H, p values were calculated in one-tailed unpaired t tests (t value: 11.0; DF: 8).
For the striatal HTT protein in HU97/18 mice in Figure S5D, p values were calculated in one-tailed unpaired t tests (t value: 6.1; DF: 6).
Acknowledgments
We would like to thank the Wyss Center Advanced Lightsheet Imaging Center (ALICe) and, in particular, Laura Batti, Ivana Gantar and Stéphane Pagès for their assistance in image processing with the MIRACL workflow. We also thank the University of Lausanne (UNIL) Cellular Imaging Facility (CIF), including, in particular, Luigi Bozzo and Yannick Krempp for image processing and QuPath analysis, the protein analysis facility, Manfredo Quadroni and Séverine Lorrain for Jess capillary western analyses. Finally, we would like to thank the EPFL BioImaging & Optics Core Facility, and Nicolas Chiaruttini, in particular, for assistance with image processing with the ABBA workflow. This work was supported in part by the Swiss National Science Foundation (FN 310030_184761/1), a research agreement from the CHDI Foundation (A-14069), and the European Union’s Horizon 2020 research and innovation program under grant agreement no. 31ER30_179594 (TreatPolyQ).
Author contributions
ND, MeR and FD designed the experiments. FD, SR, MaR performed the molecular biology experiments. MS and FD performed the in vivo experiments. MS, FD, performed the immunohistochemical analysis. MaR provided technical support for cell culture and viral vector production. MeR, ND, EH and FD performed the quantitative analysis. FD, ND wrote the manuscript. All the authors revised the manuscript.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.03.013.
Supplemental information
Data availability statement
The most relevant AAV plasmids have been deposited in addgene database: https://www.addgene.org/Nicole_Deglon/.
References
- 1.Hudry E., Vandenberghe L.H. Therapeutic AAV gene transfer to the nervous system: a clinical reality. Neuron. 2019;101:839–862. doi: 10.1016/j.neuron.2019.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K., et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 3.Tai C.H., Lee N.C., Chien Y.H., Byrne B.J., Muramatsu S.I., Tseng S.H., Hwu W.L. Long-term efficacy and safety of eladocagene exuparvovec in patients with AADC deficiency. Mol. Ther. 2022;30:509–518. doi: 10.1016/j.ymthe.2021.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lang J.F., Toulmin S.A., Brida K.L., Eisenlohr L.C., Davidson B.L. Standard screening methods underreport AAV-mediated transduction and gene editing. Nat. Commun. 2019;10:3415. doi: 10.1038/s41467-019-11321-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown D., Altermatt M., Dobreva T., Chen S., Wang A., Thomson M., Gradinaru V. Deep parallel characterization of AAV tropism and AAV-mediated transcriptional changes via single-cell RNA sequencing. Front. Immunol. 2021;12:730825. doi: 10.3389/fimmu.2021.730825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deverman B.E., Pravdo P.L., Simpson B.P., Kumar S.R., Chan K.Y., Banerjee A., Wu W.L., Yang B., Huber N., Pasca S.P., Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan K.Y., Jang M.J., Yoo B.B., Greenbaum A., Ravi N., Wu W.L., Sánchez-Guardado L., Lois C., Mazmanian S.K., Deverman B.E., Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanlon K.S., Meltzer J.C., Buzhdygan T., Cheng M.J., Sena-Esteves M., Bennett R.E., Sullivan T.P., Razmpour R., Gong Y., Ng C., et al. Selection of an efficient AAV vector for robust CNS transgene expression. Mol. Ther. Methods Clin. Dev. 2019;15:320–332. doi: 10.1016/j.omtm.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith E.J., Farshim P.P., Flomen R., Jones S.T., McAteer S.J., Deverman B.E., Gradinaru V., Bates G.P. Use of high-content imaging to quantify transduction of AAV-PHP viruses in the brain following systemic delivery. Brain Commun. 2021;3:105. doi: 10.1093/braincomms/fcab105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erö C., Gewaltig M.O., Keller D., Markram H. A cell atlas for the mouse brain. Front. Neuroinform. 2018;12:84. doi: 10.3389/fninf.2018.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung K., Deisseroth K. CLARITY for mapping the nervous system. Nat. Methods. 2013;10:508–513. doi: 10.1038/nmeth.2481. [DOI] [PubMed] [Google Scholar]
- 12.Goubran M., Leuze C., Hsueh B., Aswendt M., Ye L., Tian Q., Cheng M.Y., Crow A., Steinberg G.K., McNab J.A., et al. Multimodal image registration and connectivity analysis for integration of connectomic data from microscopy to MRI. Nat. Commun. 2019;10:5504–5517. doi: 10.1038/s41467-019-13374-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiaruttini N., Burri O., Haub P., Guiet R., Sordet-Dessimoz J., Seitz A. An open-source whole slide image registration workflow at cellular precision using Fiji, QuPath and elastix. Front. Comput. Sci. 2022;3:130. [Google Scholar]
- 14.Bankhead P., Loughrey M.B., Fernández J.A., Dombrowski Y., McArt D.G., Dunne P.D., McQuaid S., Gray R.T., Murray L.J., Coleman H.G., et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 2017;7:16878. doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt U., Weigert M., Broaddus C., Myers G. Medical Image Computing and Computer Assisted Intervention – MICCAI. Vol. 11071. Springer; 2018. Cell detection with star-convex polygons; pp. 265–273. [Google Scholar]
- 16.Perens J., Hecksher-Sørensen J. Digital brain maps and virtual neuroscience: an emerging role for light-sheet fluorescence microscopy in drug development. Front. Neurosci. 2022;16:866884. doi: 10.3389/fnins.2022.866884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson E.B., Ziegler G., Penny W., Rees G., Tabrizi S.J., Scahill R.I., Gregory S. Dynamics of cortical degeneration over a decade in Huntington’s disease. Biol. Psychiatry. 2021;89:807–816. doi: 10.1016/j.biopsych.2020.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tabrizi S.J., Flower M.D., Ross C.A., Wild E.J. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020;16:529–546. doi: 10.1038/s41582-020-0389-4. [DOI] [PubMed] [Google Scholar]
- 19.Ekman F.K., Ojala D.S., Adil M.M., Lopez P.A., Schaffer D.V., Gaj T. CRISPR-Cas9-Mediated genome editing increases lifespan and improves motor deficits in a Huntington’s disease mouse model. Mol. Ther. Nucleic Acids. 2019;17:829–839. doi: 10.1016/j.omtn.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monteys A.M., Ebanks S.A., Keiser M.S., Davidson B.L. CRISPR/Cas9 editing of the mutant huntingtin allele in vitro and in vivo. Mol. Ther. 2017;25:12–23. doi: 10.1016/j.ymthe.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oikemus S.R., Pfister E.L., Sapp E., Chase K.O., Kennington L.A., Hudgens E., Miller R., Zhu L.J., Chaudhary A., Mick E.O., et al. Allele-specific knockdown of mutant huntingtin protein via editing at coding region single nucleotide polymorphism heterozygosities. Hum. Gene Ther. 2022;33:25–36. doi: 10.1089/hum.2020.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang S., Chang R., Yang H., Zhao T., Hong Y., Kong H.E., Sun X., Qin Z., Jin P., Li S., Li X.J. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Invest. 2017;127:2719–2724. doi: 10.1172/JCI92087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merienne N., Vachey G., de Longprez L., Meunier C., Zimmer V., Perriard G., Canales M., Mathias A., Herrgott L., Beltraminelli T., et al. The self-inactivating KamiCas9 system for the editing of CNS disease genes. Cell Rep. 2017;20:2980–2991. doi: 10.1016/j.celrep.2017.08.075. [DOI] [PubMed] [Google Scholar]
- 24.Tervo D.G.R., Hwang B.Y., Viswanathan S., Gaj T., Lavzin M., Ritola K.D., Lindo S., Michael S., Kuleshova E., Ojala D., et al. A designer AAV variant permits efficient retrograde access to projection neurons. Neuron. 2016;92:372–382. doi: 10.1016/j.neuron.2016.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cearley C.N., Wolfe J.H. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol. Ther. 2006;13:528–537. doi: 10.1016/j.ymthe.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 26.Gao G., Vandenberghe L.H., Alvira M.R., Lu Y., Calcedo R., Zhou X., Wilson J.M. Clades of adeno-associated viruses are widely disseminated in human tissues. J. Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishiyama J., Mikuni T., Yasuda R. Virus-mediated genome editing via homology-directed repair in mitotic and postmitotic cells in mammalian brain. Neuron. 2017;96:755–768.e5. doi: 10.1016/j.neuron.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong S., Zheng C., Doughty M.L., Losos K., Didkovsky N., Schambra U.B., Nowak N.J., Joyner A., Leblanc G., Hatten M.E., Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- 29.Kramer P.F., Christensen C.H., Hazelwood L.A., Dobi A., Bock R., Sibley D.R., Mateo Y., Alvarez V.A. Dopamine D2 receptor overexpression alters behavior and physiology in drd2-EGFP mice. J. Neurosci. 2011;31:126–132. doi: 10.1523/JNEUROSCI.4287-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Southwell A.L., Warby S.C., Carroll J.B., Doty C.N., Skotte N.H., Zhang W., Villanueva E.B., Kovalik V., Xie Y., Pouladi M.A., et al. A fully humanized transgenic mouse model of Huntington disease. Hum. Mol. Genet. 2013;22:18–34. doi: 10.1093/hmg/dds397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Löw K., Aebischer P., Schneider B.L. Direct and retrograde transduction of nigral neurons with AAV6, 8, and 9 and intraneuronal persistence of viral particles. Hum. Gene Ther. 2013;24:613–629. doi: 10.1089/hum.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ibraheim R., Tai P.W.L., Mir A., Javeed N., Wang J., Rodríguez T.C., Namkung S., Nelson S., Khokhar E.S., Mintzer E., et al. Self-inactivating, all-in-one AAV vectors for precision Cas9 genome editing via homology-directed repair in vivo. Nat. Commun. 2021;12:6267. doi: 10.1038/s41467-021-26518-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krooss S.A., Dai Z., Schmidt F., Rovai A., Fakhiri J., Dhingra A., Yuan Q., Yang T., Balakrishnan A., Steinbrück L., et al. Ex vivo/in vivo gene editing in hepatocytes using “all-in-one” CRISPR-adeno-associated virus vectors with a self-linearizing repair template. iScience. 2020;23 doi: 10.1016/j.isci.2019.100764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Q., Su J., Liu Y., Jin X., Zhong X., Mo L., Wang Q., Deng H., Yang Y. In vivo PCSK9 gene editing using an all-in-one self-cleavage AAV-CRISPR system. Mol. Ther. Methods Clin. Dev. 2021;20:652–659. doi: 10.1016/j.omtm.2021.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brinkman E.K., Chen T., Amendola M., Van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Q., Ding S.L., Li Y., Royall J., Feng D., Lesnar P., Graddis N., Naeemi M., Facer B., Ho A., et al. The allen mouse brain common coordinate framework: a 3D reference atlas. Cell. 2020;181:936–953.e20. doi: 10.1016/j.cell.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abramoff M.D., Magalhaes P.J., Ram S.J. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 38.Scholler J., Pagès S., Batti L. Zenodo; 2022. MOTA: Map Objects to Atlas. [Google Scholar]
- 39.Assous M., Tepper J.M. Excitatory extrinsic afferents to striatal interneurons and interactions with striatal microcircuitry. Eur. J. Neurosci. 2019;49:593–603. doi: 10.1111/ejn.13881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hintiryan H., Foster N.N., Bowman I., Bay M., Song M.Y., Gou L., Yamashita S., Bienkowski M.S., Zingg B., Zhu M., et al. The mouse cortico-striatal projectome. Nat. Neurosci. 2016;19:1100–1114. doi: 10.1038/nn.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai A., Zheng N., Thompson G.J., Wu Y., Nie B., Lin K., Su P., Wu J., Manyande A., Zhu L., et al. Longitudinal neural connection detection using a ferritin-encoding adeno-associated virus vector and in vivo MRI method. Hum. Brain Mapp. 2021;42:5010–5022. doi: 10.1002/hbm.25596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiss A.R., Liguore W.A., Domire J.S., Button D., McBride J.L. Intra-striatal AAV2.retro administration leads to extensive retrograde transport in the rhesus macaque brain: implications for disease modeling and therapeutic development. Sci. Rep. 2020;10:6970. doi: 10.1038/s41598-020-63559-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matamales M., Bertran-Gonzalez J., Salomon L., Degos B., Deniau J.M., Valjent E., Hervé D., Girault J.A. Striatal medium-sized spiny neurons: identification by nuclear staining and study of neuronal subpopulations in BAC transgenic mice. PLoS One. 2009;4 doi: 10.1371/journal.pone.0004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shin J.W., Hong E.P., Park S.S., Choi D.E., Seong I.S., Whittaker M.N., Kleinstiver B.P., Chen R.Z., Lee J.M. Allele-specific silencing of the gain-of-function mutation in Huntington’s disease using CRISPR/Cas9. JCI Insight. 2022;7 doi: 10.1172/jci.insight.141042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salinas C.B.G., Lu T.T.H., Gabery S., Marstal K., Alanentalo T., Mercer A.J., Cornea A., Conradsen K., Hecksher-Sørensen J., Dahl A.B., et al. Integrated brain atlas for unbiased mapping of nervous system effects following liraglutide treatment. Sci. Rep. 2018;8:10310–10312. doi: 10.1038/s41598-018-28496-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansen H.H., Perens J., Roostalu U., Skytte J.L., Salinas C.G., Barkholt P., Thorbek D.D., Rigbolt K.T.G., Vrang N., Jelsing J., Hecksher-Sørensen J. Whole-brain activation signatures of weight-lowering drugs. Mol. Metab. 2021;47 doi: 10.1016/j.molmet.2021.101171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Renier N., Adams E.L., Kirst C., Wu Z., Azevedo R., Kohl J., Autry A.E., Kadiri L., Umadevi Venkataraju K., Zhou Y., et al. Mapping of brain activity by automated volume analysis of immediate early genes. Cell. 2016;165:1789–1802. doi: 10.1016/j.cell.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Detrez J.R., Maurin H., Van Kolen K., Willems R., Colombelli J., Lechat B., Roucourt B., Van Leuven F., Baatout S., Larsen P., et al. Regional vulnerability and spreading of hyperphosphorylated tau in seeded mouse brain. Neurobiol. Dis. 2019;127:398–409. doi: 10.1016/j.nbd.2019.03.010. [DOI] [PubMed] [Google Scholar]
- 49.Liebmann T., Renier N., Bettayeb K., Greengard P., Tessier-Lavigne M., Flajolet M. Three-dimensional study of alzheimer’s disease hallmarks using the iDISCO clearing method. Cell Rep. 2016;16:1138–1152. doi: 10.1016/j.celrep.2016.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lopes M.M., Paysan J., Rino J., Lopes S.M., Pereira de Almeida L., Cortes L., Nobre R.J. A new protocol for whole-brain biodistribution analysis of AAVs by tissue clearing, light-sheet microscopy and semi-automated spatial quantification. Gene Ther. 2022;29:665–679. doi: 10.1038/s41434-022-00372-z. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Z., Yao X., Yin X., Ding Z., Huang T., Huo Y., Ji R., Peng H., Guo Z.V. Multi-scale light-sheet fluorescence microscopy for fast whole brain imaging. Front. Neuroanat. 2021;15:732464. doi: 10.3389/fnana.2021.732464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirano M., Ando R., Shimozono S., Sugiyama M., Takeda N., Kurokawa H., Deguchi R., Endo K., Haga K., Takai-Todaka R., et al. A highly photostable and bright green fluorescent protein. Nat. Biotechnol. 2022;40:1132–1142. doi: 10.1038/s41587-022-01278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muñoz-Castañeda R., Zingg B., Matho K.S., Chen X., Wang Q., Foster N.N., Li A., Narasimhan A., Hirokawa K.E., Huo B., et al. Cellular anatomy of the mouse primary motor cortex. Nature. 2021;598:159–166. doi: 10.1038/s41586-021-03970-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Appukuttan S., Bologna L.L., Schürmann F., Migliore M., Davison A.P. EBRAINS live papers - interactive Resource sheets for computational studies in neuroscience. Neuroinformatics. 2023;21:101–113. doi: 10.1007/s12021-022-09598-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu D., Zhu M., Zhang Y., Diao Y. Crossing the blood-brain barrier with AAV vectors. Metab. Brain Dis. 2021;36:45–52. doi: 10.1007/s11011-020-00630-2. [DOI] [PubMed] [Google Scholar]
- 56.High-dose AAV gene therapy deaths. Nat. Biotechnol. 2020;38:910. doi: 10.1038/s41587-020-0642-9. [DOI] [PubMed] [Google Scholar]
- 57.Naidoo J., Stanek L.M., Ohno K., Trewman S., Samaranch L., Hadaczek P., O’Riordan C., Sullivan J., San Sebastian W., Bringas J.R., et al. Extensive transduction and enhanced spread of a modified AAV2 capsid in the non-human primate CNS. Mol. Ther. 2018;26:2418–2430. doi: 10.1016/j.ymthe.2018.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tordo J., O’Leary C., Antunes A.S.L., Palomar N., Aldrin-Kirk P., Basche M., Bennett A., D’Souza Z., Gleitz H., Godwin A., et al. A novel adeno-associated virus capsid with enhanced neurotropism corrects a lysosomal transmembrane enzyme deficiency. Brain. 2018;141:2014–2031. doi: 10.1093/brain/awy126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davidsson M., Wang G., Aldrin-Kirk P., Cardoso T., Nolbrant S., Hartnor M., Mudannayake J., Parmar M., Björklund T. A systematic capsid evolution approach performed in vivo for the design of AAV vectors with tailored properties and tropism. Proc. Natl. Acad. Sci. USA. 2019;116:27053–27062. doi: 10.1073/pnas.1910061116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin K., Zhong X., Li L., Ying M., Yang T., Zhang Z., He X., Xu F. AAV9-Retro mediates efficient transduction with axon terminal absorption and blood–brain barrier transportation. Mol. Brain. 2020;13:138. doi: 10.1186/s13041-020-00679-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin R., Zhou Y., Yan T., Wang R., Li H., Wu Z., Zhang X., Zhou X., Zhao F., Zhang L., et al. Directed evolution of adeno-associated virus for efficient gene delivery to microglia. Nat. Methods. 2022;19:976–985. doi: 10.1038/s41592-022-01547-7. [DOI] [PubMed] [Google Scholar]
- 62.Gray M., Shirasaki D.I., Cepeda C., André V.M., Wilburn B., Lu X.H., Tao J., Yamazaki I., Li S.H., Sun Y.E., et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hodgson J.G., Agopyan N., Gutekunst C.A., Leavitt B.R., Lepiane F., Singaraja R., Smith D.J., Bissada N., McCutcheon K., Nasir J., et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 64.Dang Y., Jia G., Choi J., Ma H., Anaya E., Ye C., Shankar P., Wu H. Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 2015;16:280. doi: 10.1186/s13059-015-0846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hottinger A.F., Azzouz M., Déglon N., Aebischer P., Zurn A.D. Complete and long-term rescue of lesioned adult motoneurons by lentiviral-mediated expression of glial cell line-derived neurotrophic factor in the facial nucleus. J. Neurosci. 2000;20:5587–5593. doi: 10.1523/JNEUROSCI.20-15-05587.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Regan M.R., Huang Y.H., Kim Y.S., Dykes-Hoberg M.I., Jin L., Watkins A.M., Bergles D.E., Rothstein J.D. Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J. Neurosci. 2007;27:6607–6619. doi: 10.1523/JNEUROSCI.0790-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Christodoulou I., Patsali P., Stephanou C., Antoniou M., Kleanthous M., Lederer C.W. Measurement of lentiviral vector titre and copy number by cross-species duplex quantitative PCR. Gene Ther. 2016;23:113–118. doi: 10.1038/gt.2015.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Humbel M., Ramosaj M., Zimmer V., Regio S., Aeby L., Moser S., Boizot A., Sipion M., Rey M., Déglon N. Maximizing lentiviral vector gene transfer in the CNS. Gene Ther. 2021;28:75–88. doi: 10.1038/s41434-020-0172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The most relevant AAV plasmids have been deposited in addgene database: https://www.addgene.org/Nicole_Deglon/.