

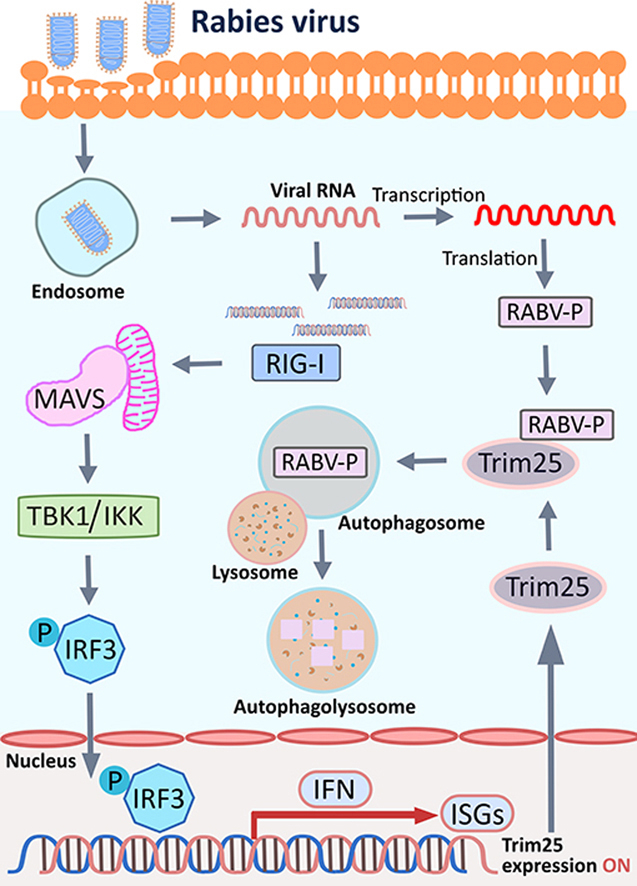
Abstract
Tripartite motif-containing protein 25 (Trim25) is an E3 ubiquitin ligase that activates retinoid acid-inducible gene I (RIG-I) and promotes the antiviral interferon response. Recent studies have shown that Trim25 can bind and degrade viral proteins, suggesting a different mechanism of Trim25 on its antiviral effects. In this study, Trim25 expression was upregulated in cells and mouse brains after rabies virus (RABV) infection. Moreover, expression of Trim25 limited RABV replication in cultured cells. Overexpression of Trim25 caused attenuated viral pathogenicity in a mouse model that was intramuscularly injected with RABV. Further experiments confirmed that Trim25 inhibited RABV replication via two different mechanisms: an E3 ubiquitin ligase-dependent mechanism and an E3 ubiquitin ligase-independent mechanism. Specifically, the CCD domain of Trim25 interacted with RABV phosphoprotein (RABV-P) at amino acid (AA) position at 72 and impaired the stability of RABV-P via complete autophagy. This study reveals a novel mechanism by which Trim25 restricts RABV replication by destabilizing RABV-P, which is independent of its E3 ubiquitin ligase activity.
Keywords: Rabies virus, Trim25, Phosphoprotein, Protein stability, Autophagy
Graphical abstract
Highlights
-
•
Trim25 is upregulated after RABV infection in vitro and in vivo.
-
•
Trim25 inhibits RABV replication in both IFN dependent and independent pathways.
-
•
The coiled-coil domain of Trim25 interacts with RABV-P.
-
•
Trim25 degrades RABV-P through complete autophagy.
1. Introduction
Rabies virus (RABV) causes rabies; an ancient disease traced back 4000 years. Rabies still poses a public health threat and causes 50,000–70,000 human deaths annually worldwide (Fisher et al., 2018; Schnell et al., 2010). RABV is a single-stranded negative-sense RNA virus belonging to the genus Lyssavirus of the family Rhabdoviridae (order Mononegavirales). The genome of RABV is approximately 12,000 nucleotides and encodes five proteins, namely, nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G), and large RNA-dependent RNA polymerase (L), in the order of 3′-N-P-M-G-L-5′ (Hidaka et al., 2018). The N, P, and L genes of RABV form a ribonucleoprotein (RNP) complex that participates in viral transcription and replication in a regulated and efficient manner (Fu et al., 1994; Mavrakis et al., 2006).
The phosphoprotein of RABV (RABV-P) consists of 297 amino acids (AA) and is highly hydrophilic. RABV-P is indispensable for the replication of RABV since it is a nonenzymatic cofactor that associates with the L protein and interacts with N to form Negri bodies (Chenik et al., 1994). The L protein alone cannot perform its polymerase function without being stabilized by RABV-P (Jacob et al., 2001). Residues 1–19 of RABV-P form the major L-binding site and residues 11–50 of P stimulate the polymerase activity of the L protein (Chenik et al., 1998; Morin et al., 2017). Nonetheless, the crystal structure of the P-L complex showed that even residues 11–50 of RABV-P could stabilize the C-terminal domains of the L protein, and a longer fragment (1–91) of RABV-P can shift the positions of these domains to a degree equivalent to full-length RABV-P, suggesting that residues 50–91 of RABV-P may also participate in L-P interactions (Horwitz et al., 2020; Inoue et al., 1993). Besides, RABV-P is identified as the primary interferon (IFN) antagonist. Moreover, residues 176–186 of RABV-P are critical for its function of antagonizing interferon regulatory factor 3 (IRF3) and IRF7 (Brzózka et al., 2005; Rieder et al., 2011). RABV-P can also interact with STATs to compromise the downstream ISG production (Brzózka et al., 2006; Lieu et al., 2013).
Tripartite motif-containing protein 25 (Trim25, also known as EFP) is a 71-kDa E3 ubiquitin ligase. It comprises a RING domain, two B-box domains, a coiled-coil (CC) dimerization domain, and a C-terminal SPRY domain (Carthagena et al., 2009). Trim25 has been shown to induce Lys63-linked ubiquitination of the N-terminal CARDs of retinoic acid-inducible gene I (RIG-I), and Trim25 in part activates the RIG-I signaling pathway to elicit host antiviral innate immunity (Gack et al., 2007). Trim25, which is a vital component that promotes IFN production, is emerging as an antagonistic target through which different types of viruses, such as influenza A virus (IAV), dengue virus, paramyxovirus, porcine reproductive and respiratory syndrome virus (PRRSV), human papillomavirus (HPV), severe fever with thrombocytopenia syndrome virus (SFTSV), JC polyomavirus (JCV), severe acute respiratory syndrome coronavirus (SARS-CoV), and SARS-CoV-2, compromise the host innate immune system (Chiang et al., 2018, 2021; Gack et al., 2009; Gori Savellini et al., 2021; Hu et al., 2017; Manokaran et al., 2015; Min et al., 2020; Sánchez-Aparicio et al., 2018; Zhao et al., 2019).
The role of Trim25 on RABV infection has not been reported. In this study, we identified that Trim25 could restrict RABV replication in vitro and in vivo. The antiviral function of Trim25 against RABV is partially independent of its ubiquitin E3 ligase activity or its ability to activate the IFN pathway. We found that Trim25 interacted with RABV-P and caused the reduced stability of RABV-P.
2. Results
2.1. Expression of Trim25 is upregulated after RABV infection in vitro and in vivo
To explore the role of Trim25 on RABV infection, groups of C57BL/6 mice were intracranially (i.c.) injected with 100 FFU lab-attenuated strain RABV CVS-B2c (CVS) or mock inoculated with the same volume of DMEM. At 3-, 6- and 9-days post-infection (d.p.i.), the mice were euthanized by CO2. The mouse brains were collected, and total RNA was extracted. Then, the mRNA levels of Trim25 in the mouse brains were quantified by qRT–PCR (qPCR). The Trim25 mRNA level was significantly upregulated at 6 and 9 d.p.i. (Fig. 1A). Furthermore, the mRNA level of Trim25 was measured in different brain regions after RABV infection. The results showed that all four regions, including the olfactory bulb, cortex, cerebellum, and brain stem, showed significantly enhanced Trim25 mRNA levels after RABV infection (Fig. 1B). As a control, the mRNA level of Trim32, which belongs to the same family as Trim25 and is also enriched in the brain, showed no noticeable change after RABV infection (Fig. 1C).
Fig. 1.

Trim25 expression is upregulated after RABV infection. C57BL/6 mice (n = 3) were intracerebrally (i.c.) inoculated with a lab-attenuated RABV strain CVS-B2c (CVS) at 100 FFU or the same volume of DMEM (mock). At 3-, 6-, and 9-days post-infection (d.p.i.), mouse brains were harvested and used for further analysis. (A) Total RNA was isolated from brain tissues, and the mRNA level of Trim25 was analyzed by qPCR. (B) Total RNA from different brain sections (harvested at 9 d.p.i.) was isolated, and the mRNA level of Trim25 was analyzed by qPCR. (C) Total RNA from different brain sections (harvested at 9 d.p.i.) was isolated, and the mRNA level of Trim32 was analyzed by qPCR. (D) The protein levels of Trim25, Trim32, and RABV-N in the cortex and cerebellum of mouse brains harvested at 9 d.p.i. were assessed by Western blotting, and β-actin was used as the control. (E) Mouse cerebellums harvested at 9 d.p.i. were fixed and sectioned. In situ Trim25 expression was analyzed by immunofluorescence assay (IFA). Scale bar, 400 μm. (F) BV2 cells were infected with CVS at an MOI of 1 for the indicated time points, and the Trim25 mRNA levels were measured using qPCR. (G) At 48 h post-RABV infection, BV2 cells were harvested, and the Trim25 protein levels were measured by Western blotting. Error bars represent standard deviation (SD), n = 3. Statistical differences between virus-infected cells and mock infected cells were determined by using Student’s t-test and are denoted as follows: ∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001. Western blotting data are representative of at least two independent experiments.
When we used Western blotting to measure the protein levels of Trim25 and Trim32 in the RABV-infected mouse cortex and cerebellum, the protein level of Trim25 but not Trim32 in the brain was upregulated after RABV infection (Fig. 1D). Similarly, immunofluorescence histochemistry analysis in situ in RABV-infected mouse cerebellum, revealed that Trim25 protein expression was upregulated in the RABV-infected brain and upregulated Trim25 aggregated at the site of RABV infection (white arrows in Fig. 1E).
To define the function of upregulated Trim25, BV2 cells (mouse microglia) were infected with CVS for an indicated time at an multiplicities of infection (MOI) of 1. Then, the total RNA was extracted from the RABV-infected BV2 harvested cells, and the mRNA of Trim25 was quantified by qPCR. Trim25 mRNA levels were significantly upregulated at 48 h post-infection (h.p.i.) (Fig. 1F). Moreover, the protein level of Trim25 in BV2 cells was measured by Western blotting. The protein level of Trim25 was consistently significantly increased after RABV infection (Fig. 1G). Therefore, Trim25 expression is upregulated by RABV infection both in vitro and in vivo.
2.2. Overexpression of Trim25 inhibits RABV replication in cultured cells
As Trim25 expression is upregulated by RABV infection, Trim25 plays a role in RABV replication. To test this hypothesis, Trim25 was cloned into the pCAGGS plasmid with a FLAG-tag in its N-terminal (named FLAG-Trim25). Then, FLAG-Trim25 was overexpressed in 293T cells by transient transfection at the indicated doses for 12 h. Then, these cells were infected with CVS for another 36 h. The supernatants and cells were harvested for further experiments. Virus titration results showed that overexpression of Trim25 reduces the virus load in the cells (Fig. 2A) and the supernatants (Fig. 2B). Consistently, immunofluorescence assay (IFA) showed the decreased protein level of RABV-N after Trim25 overexpression (Fig. 2C). We also evaluated the antiviral effects of Trim25 against two wildtype RABV strains named DRV-Mexico (DRV) and SHBRV-18 (SHBRV) (Morimoto et al., 1996; Yu et al., 2014; Zhang and Fu, 2012). Viral titration and indirect immunofluorescence assay demonstrated that Trim25 could restrict DRV and silver-haired bat rabies virus (SHBRV) replication in 293T cells (Fig. 2D and S1). Therefore, overexpression of Trim25 inhibits RABV replication in 293T cells.
Fig. 2.

Trim25 restrains RABV infection in 293T cells. (A–C) 293T cells were transfected with the expression vector pCAGGS or pCAGGS to express the FLAG-Trim25 (FLAG-Trim25) at the indicated volumes. The cells were infected with CVS at an MOI of 0.01 at 12 h post-transfection. After 36 h incubation, the cells were harvested to measure the protein levels of Trim25, RABV-N, and β-actin by Western blotting (A). The supernatants were harvested for virus titration (B). The same treated 293T cells were fixed and stained to measure RABV-N and Trim25 expression using IFA. The merged pictures are shown in the right panels, and the enlarged pictures are placed inside the white dotted box (C). Scale bar, 200 μm. (D, E) 293T cells were transfected with the expression vector pCAGGS or pCAGGS to express FLAG-Trim25 (FLAG-Trim25) at the indicated volumes. At 12 h post-transfection, the cells were infected with a wildtype RABV strain DRV (D) or SHBRV (E) at an MOI of 0.01. The supernatants were harvested for virus titration. Statistical analysis of comparisons between groups was carried out by Student’s t-test (∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001). The bar graph shows the mean ± SD, n = 3. Western blotting data are representative of at least two independent experiments. See also Fig. S1.
2.3. Trim25 expression attenuates the pathogenicity of RABV in mice
To confirm the role of the Trim25 in inhibiting the RABV replication, we constructed a recombinant RABV (rRABV) expressing Trim25 (named rRABV-Trim25), and an rRABV expressing a null Trim25 with the mutated start codon TAA instead of ATG (named rRABV-Trim25(−)) (Fig. 3A). Western blot analysis showed that Trim25 was highly expressed in BSR (Fig. 3B) and N2a (Fig. 3C) cells infected with rRABV-Trim25, but not in rRABV-Trim25(−) or rRABV-infected cells. The growth dynamics of rRABV-Trim25, rRABV-Trim25(−), and its parent virus rRABV were further characterized in cultured cells. rRABV-Trim25 replicated in a lower titer than rRABV and rRABV-Trim25(−) at the indicated time points post-infection in BSR (Fig. 3D) and N2a (Fig. 3E) cells.
Fig. 3.

Trim25 attenuates RABV pathogenicity in mice. (A) Schematic diagram of the construction of rRABV, rRABV-Trim25(−), and rRABV-Trim25. (B, C) BSR (B) and N2a (C) cells were infected with rRABV, rRABV-Trim25(−), or rRABV-Trim25 at an MOI of 0.01 for 48 h, and cells were collected for measurement of the protein levels of Trim25, RABV-P, and β-actin by Western blotting. (D, E) BSR (D) and N2a (E) cells were infected with rRABV, rRABV-Trim25(−), or rRABV-Trim25 at an MOI of 0.01 for the indicated time points, and the cell culture supernatants were harvested for virus titration. (F, G) Female C57BL/6 mice (n = 10) were intramuscularly (i.m.) infected with 8 × 105 FFU rRABV, rRABV-Trim25(−), or rRABV-Trim25. The survival ratio (F) and body weight change (G) was monitored daily for 21 continuous days (mean ± SD; ∗, P < 0.05; survival ratio was analyzed by Log-rank test). (H) Female C57BL/6 mice (n = 3) were inoculated as in (F). At 12 d.p.i., the mouse brains were harvested, prepared as paraffin sections, and analyzed by immunohistochemical staining with antibodies against RABV-P. Scale bar, 50 μm. Statistical analysis of comparisons between groups was carried out by Student’s t-test (∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001). The bar graph shows the mean ± SD, n = 3.
To assess the impact of Trim25 on viral pathogenicity in mice, rRABV, rRABV-Trim25(−), or rRABV-Trim25 (8 × 104 FFU) was intramuscularly (i.m.) inoculated into the mouse hind legs, and the changes in the body weights and survival ratios were monitored daily. The survival ratio of the rRABV-Trim25-infected mice was significantly increased compared with that of the rRABV-infected or rRABV-Trim25(−)-infected mice (Fig. 3F). The reduction in body weight of the rRABV-Trim25-infected mice was lower than that of the rRABV-infected or rRABV-Trim25(−)-infected mice (Fig. 3G). Immunohistochemistry analysis of RABV-P in various regions of the RABV-infected mouse brains at 9 d.p.i. showed that, unlike the rRABV-infected or rRABV-Trim25(−)-infected mice, almost no viral antigen could be observed in the brains of the rRABV-Trim25-infected mice, especially in the cerebrum and brain stem (Fig. 3H). Therefore, Trim25 expression causes attenuated viral pathogenicity of RABV in mice.
2.4. Trim25 restricts RABV replication independently of its E3 ligase activity
Since Trim25 can activate the RIG-I pathway by exercising its E3 ubiquitin ligase activity and then conjugating K63-polyubiquitin chains to RIG-I (Gack et al., 2007), we wondered whether the anti-RABV capacity of Trim25 was entirely dependent on its RIG-I activation ability. We used BX795, a potent and selective inhibitor of TANK-binding kinase 1 (TBK1) and IκB kinase ε (IKKε) (Clark et al., 2009) to treat Trim25-transfected cells and then infected cells with RABV. Then, the viral titer results demonstrated that Trim25 could still inhibit RABV replication with BX795 treatment, while its antiviral capacity was decreased compared with the DMSO treatment and Trim25-transfected group (Fig. S2A). We also transfected Trim25 to an IFN secretion defective Vero cell line and measured RABV replication level in Trim25-transfected Vero cells. The results showed that Trim25 expression could significantly decrease RABV replication level (Fig. S2B). These results implied that Trim25 has a mechanism to inhibit RABV replication independent of its RIG-I pathway activation ability.
To further confirm that Trim25 can inhibit RABV replication independent of the activation of the RIG-I pathway, we constructed a mutated Trim25 that was defective in activating the RIG-I pathway. The mutations in the RING domain (C13A/C16A) of Trim25 could abolish its E3 ubiquitin ligase activity, resulting in its inability to activate the RIG-I pathway, which is consistent with the previous studies (D'Cruz et al., 2013; Meyerson et al., 2017). Therefore, the amino acids (AAs) C13 and C16 of mouse Trim25 were mutated to A13 and A16, and then, the mutated Trim25 with a FLAG-tag in its N-terminus was cloned into the pCAGGS expression vector (named E3-mut-Trim25). Using RIG-I as the target protein, the ubiquitin ligase activity of E3-mut-Trim25 was evaluated by Western blotting. The results confirmed that E3-mut-Trim25 lost its ubiquitin ligase activity compared with WT Trim25 (Fig. 4A). To further testify to the inability of E3-mut-Trim25 to activate the RIG-I pathway, an IRF3 luciferase experiment was performed. The results demonstrated that E3-mut-Trim25 did not exhibit IRF3 activation ability (Fig. 4B).
Fig. 4.

Trim25 lacking its RIG-I activation ability, still inhibits RABV replication. (A) 293T cells were co-transfected with the expression vector pCAGGS or pCAGGS to express the Myc-Trim25 (Myc-Trim25) or with the expression, vector pCAGGS to express the Myc-E3-mut-Trim25 (Myc-E3-mut-Trim25). With HA-ubiquitin-K63 (HA-Ub-K63), FLAG-RIG-I for 48 h, Co-immunoprecipitation (Co-IP) was then performed using magnetic beads conjugated with antibodies against the FLAG-tag. The expression of the indicated proteins was measured by Western blotting (B) 293T cells were co-transfected with the vector plasmid or plasmids encoding Trim25 or its mutant, along with the IRF3 reporter plasmid and pRL-TK. The cells were mock infected or infected with Sendai virus (SeV) at 24 h post-transfection (h.p.t.); after infection for 16 h, the luciferase activities were measured. (C–F) 293T cells were transfected with Trim25 or their mutant and then infected with CVS at an MOI of 0.01 at 12 h.p.t. After 36 h of incubation, the cell culture supernatants were harvested for virus titration (C). The cells were either harvested for Western blotting (D) and qPCR (E) or fixed for measurement of RABV-P expression by using IFA. Scale bar, 200 μm. (F). The mean fluorescence intensity of RABV-P in the respective groups was calculated using ImageJ software (F). Data are presented as mean ± SD, n = 3. Western blotting data are representative of at least two independent experiments. See also Fig. S2.
After abolishing the RIG-I activation ability of Trim25, its anti-RABV capacity was evaluated in 293T cells. The viral load in the cells transfected with Trim25 and E3-mut-Trim25 was significantly lower than in cells transfected with the pCAGGS. Interestingly, E3-mut-Trim25 still displayed anti-RABV ability, although weaker than WT Trim25 (Fig. 4C–F), indicating that Trim25 possessed anti-RABV activities independent of its E3 ligase activity. Notably, there was a significant difference in anti-RABV ability between Trim25 and E3-mut-Trim25, suggesting that the inhibition of RABV by Trim25 is only partially dependent on the RIG-I activation, but its major anti-RABV activity would be through another mechanism. Therefore, Trim25 restricts RABV replication independently of its RIG-I pathway.
2.5. Trim25 interacts with the phosphoprotein of RABV
To further explore the mechanism by which Trim25 restricts RABV replication, we investigated the interaction of Trim25 with RABV structural proteins. After the transfection of FLAG-Trim25 into 293T cells for 12 h, cells were infected with CVS at an MOI of 2 and incubated for another 36 h. Then, the whole cell lysates were immunoprecipitated with FLAG-conjugated magnetic beads, and polyclonal antibodies against RABV were used to screen viral proteins that interacted with Trim25. The results showed that RABV-P might interact with Trim25. This result was further confirmed when an anti-P monoclonal antibody was included in determining whether RABV-P was present (Fig. 5A). Then, we used RABV-P antibodies as bait and confirmed that endogenous Trim25 interacted with RABV-P under CVS infection (Fig. 5B). Using confocal analysis, we also observed robust co-localization phenomena between endogenous Trim25 and RABV-P under CVS infection (Fig. 5C).
Fig. 5.

Trim25 directly interacts with RABV-P. (A) 293T cells were transfected with the plasmid vector pCAGGS or FLAG-Trim25. The cells were infected with CVS at an MOI of 1 at 12 h.p.t. and incubated for an additional 36 h. Then, the cells were lysed with NP-40 lysis buffer. Co-immunoprecipitation (Co-IP) was then performed using magnetic beads conjugated with antibodies against the FLAG-tag. Western blotting was applied to measure the expression of the indicated proteins. (B) 293T cells were infected with CVS at an MOI of 1 for 36 h, the cells were lysed, and IP was performed using magnetic beads conjugated with antibodies against the RABV-P protein. The expression of the indicated proteins was measured by Western blotting. (C) 293T cells were infected with CVS at an MOI of 1 for 36 h, fixed with 4% paraformaldehyde, stained with antibodies against Trim25, RABV-P, or DAPI, and observed under confocal fluorescence microscopy. The white arrow indicates the co-localization of Trim25 and RABV-P. Scale bar, 10 μm. (D) GST or GST-Trim25 was expressed in E. coli, and purified lysates containing GST or GST-Trim25 were incubated with HA-CVS-P or HA-CVS-N (expressed in 293T cells) to assess binding. (E) HA-CVS-P was expressed alone or co-expressed with FLAG-Trim25 or FLAG-E3-mut-Trim25 in 293T cells. Cell lysates were subjected to Co-IP and analyzed by Western blotting. Western blotting data are representative of at least two independent experiments. See also Fig. S3.
Transfection of FLAG-Trim25 with HA-CVS-N or HA-CVS-P was then used to detect possible interactions in cells overexpressing these proteins. The results showed that only CVS-P could precipitate Trim25, and Trim25 could precipitate both CVS-P and CVS-N (Fig. S3). Since immunoprecipitation assays detect direct and indirect interactions, GST-tagged Trim25 was then purified. GST pull-down assays were performed to determine whether there was a direct interaction between Trim25 and CVS-P or CVS-N. The results showed that GST-Trim25 directly interacts with CVS-P but not CVS-N (Fig. 5D). Additionally, we tested whether E3-mut-Trim25 could interact with CVS-P. Immunoprecipitation results showed that E3-mut-Trim25 also possessed the ability to interact with CVS-P (Fig. 5E). These results demonstrate that both Trim25 and E3-mut-Trim25 directly interact with CVS-P.
2.6. The CC dimerization domain of Trim25 interacts with RABV-P, and residue 72 of RABV-P is essential for its binding with Trim25
To pinpoint the structural domain responsible for the interaction with CVS-P, Trim25 was divided into four parts, namely, the RING, bb-boxes, CC dimerization (CCD), and SPRY domains (Fig. 6A), as described previously (D'Cruz et al., 2013; Gack et al., 2009). These domains were cloned into pCAGGS expression plasmid with a FLAG-tag in N-terminals. Then, the plasmids were transfected into 293T cells along with CVS-P. The immunoprecipitation results showed that only the CCD domain of Trim25 interacted with CVS-P (Fig. 6B).
Fig. 6.

Amino acid 72 of RABV-P is critical for its interaction with Trim25. (A) Schematic diagram of different domains of Trim25 and different HA-CVS-P truncations. (B) 293T cells were co-transfected with HA-CVS-P, and different truncations of Trim25 (RING, bb-boxes, CCD, SPRY domains) fused with a FLAG-tag. At 48 h.p.t., cell lysates were subjected to Co-IP and analyzed by Western blotting. (C–F) 293T cells were co-transfected with FLAG-Trim25, HA-CVS-P (HA-P), or pCAGGS expressing different HA-CVS-P truncations. At 48 h.p.t., Co-IP and Western blotting were performed with the indicated antibodies. (G) 293T cells were co-transfected with FLAG-Trim25 or pCAGGS and HA-CVS-P, HA-P-72m, or HA-P-73m. At 48 h.p.t., Co-IP and Western blotting were performed with the indicated antibodies. Western blotting data are representative of at least two independent experiments. See also Fig. S4.
The sequence of RABV-P required for Trim25 binding was determined using a co-immunoprecipitation (Co-IP) assay. First, we truncated RABV-P into five fragments and inserted them into the pCAGGS vector fused with an HA tag. These fragments were named HA-19-297-P, HA-52-297-P, HA-82-297-P, HA-138-297-P, and HA-172-297-P (Fig. 6A). Since different truncations of RABV-P may vary in their cellular localizations (Chenik et al., 1995; Oksayan et al., 2012), we examined their intracellular localization, and HA-CVS-P, HA-19-297-P, HA-138-297-P, and HA-172-297-P were found mostly in the cytoplasm, while HA-52-297-P and HA-82-297-P were seen in both cytoplasm and nucleus (Fig. S4A).
Then, these fragments were expressed along with FLAG-Trim25. After the deletion of residues 1–52 of RABV-P, the interaction between Trim25 and RABV-P remained, but this interaction was lost when residues 1–82 of RABV-P were deleted (Fig. 6C). Moreover, the interaction between the HA-72-297-P fragment and Trim25 was the same as that of HA-52-297-P and Trim25. In contrast, HA-82-297-P lost its ability to interact with Trim25 (Fig. 6D).
We inserted 76–297 of RABV-P into the pCAGGS vector for the Co-IP assay. The results showed that Trim25 could pull-down HA-72-297-P but not HA-76-297-P (Fig. 6E). Thus, the interaction region was narrowed to four AA residues, 72–76 of RABV-P. Therefore, we constructed HA-74-297-P to assess the interaction between Trim25 and the RABV-P truncations by Co-IP assay and found that HA-72-297-P retained its ability to interact with Trim25, while HA-74-297-P lost this ability (Fig. 6F). Therefore, AAs essential for the interaction between Trim25 and RABV-P were 72–73 of RABV-P. We also examined the intracellular localization of HA-72-297-P, HA-74-297-P, and HA-76-297-P, the results showed that these truncations of RABV-P were presented in both cytoplasm and nucleus (Fig. S4B).
Then, we constructed RABV-P carrying mutated AA at position 72 from valine to alanine (HA-P-72m) and mutated AA at position 73 from glycine to alanine (HA-P-73m), respectively. Then, the Co-IP assay showed that HA-P-73m could interact with Trim25, while HA-P-72m lost this ability (Fig. 6G). Therefore, residue 72 of RABV-P is critical for the interaction between Trim25 and RABV-P.
2.7. Trim25 disrupts RABV-P stability through its interaction with RABV-P
To elucidate the mechanism underlying the RIG-I pathway independent anti-RABV function of Trim25, we performed a series of Co-IP assays and confirmed that the Trim25-P interaction did not notably affect the P–P, P–N, or P-L interactions (Figs. S5A–S5C). Thus, we investigated whether Trim25 could reduce the RABV-P expression level. Notably, the results showed that when Trim25 was co-transfected with RABV-P or GFP, the expression level of RABV-P was significantly decreased compared with that of GFP (Fig. 7A). When we also tested whether Trim25 could function at other RABV proteins, Trim25 could not reduce the expression of RABV-N, RABV-M, or RABV-G (Figs. S5D–S5F). We then investigated whether the co-transfection of RABV-P and Trim25 could lead to a decrease in RABV-P mRNA levels. The results in Fig. 7B demonstrated that the reduction of RABV-P expression was not due to the changes in transcriptional level, suggesting that post-translational modification (PTM) may involve in the degradation of RABV-P.
Fig. 7.

Trim25 disrupts the stability of RABV-P. (A) 293T cells were co-transfected with FLAG-Trim25 (at the indicated volumes), HA-CVS-P, and HA-GFP for 48 h, and cell lysates were analyzed by Western blotting. (B) 293T cells were transfected with pCAGGS, HA-CVS-P, FLAG-Trim25 with pCAGGS, or FLAG-Trim25 with HA-CVS-P for 36 h. Then, cellular RNA was extracted, and CVS-P mRNA levels were measured. (C) 293T cells were co-transfected with FLAG-Trim25, HA-CVS-P, and HA-GFP for 48 h, and 10 μM CHX was added to the culture medium at 0, 3, 6, and 9 h before cell lysis. The cell lysates were analyzed by Western blotting. (D) 293T cells were co-transfected with FLAG-Trim25 and HA-CVS-P for 48 h and treated with 10 μM MG132 10 h before cell lysis. The cell lysates were analyzed by Western blotting. (E, F) 293T cells were co-transfected with HA-74-297-P, HA-74-297-P with FLAG-Trim25 (E) or FLAG-E3-mut-Trim25 (F), HA-CVS-P, and HA-CVS-P with FLAG-Trim25 (E) or FLAG-E3-mut-Trim25 (G) for 48 h. Then, cell lysates were analyzed by Western blotting. (G, H) 293T cells were co-transfected with HA-72m-P, HA-72m-P with FLAG-Trim25 (G) or FLAG-E3-mut-Trim25 (H), HA-CVS-P, and HA- CVS-P with FLAG-Trim25 (G) or FLAG-E3-mut-Trim25 (H) for 48 h. Then, cell lysates were analyzed by Western blotting. Western blotting data are representative of at least two independent experiments. See also Fig. S5.
Next, cycloheximide (CHX) was applied at different time points to block protein expression. However, CHX treatment did not affect the expression level of RABV-P when Trim25 was co-expressed (Fig. 7C). To further examine if the classical protease-dependent degradation pathway influenced the expression of RABV-P, a classical proteasome inhibitor MG132 (10 μM) was applied to cells co-transfected with RABV-P and Trim25. The results demonstrated that MG132 treatment also did not affect the expression of RABV-P (Fig. 7D).
Since we have pinpointed the essential AA of RABV-P that affects Trim25-P interaction (Fig. 6G), we wondered whether HA-72m-P, which could not bind with Trim25, could be degraded by Trim25. Additionally, we have included HA-74-297-P as a control, as HA-74-297-P could not interact with Trim25 (Fig. 6F). Then, Trim25 or E3-mut-Trim25 was co-transfected with the adjusted amounts of HA-72m-P, HA-74-297-P, or HA-CVS-P. The results showed that HA-72m-P or HA-74-297-P expression level was not decreased after Trim25 or E3-mut-Trim25 expression, while HA-CVS-P expression was still decreased when co-transfected with Trim25 or E3-mut-Trim25 (Fig. 7E–H). Together, these results indicate that the interaction between Trim25 and RABV-P causes the instability of RABV-P.
2.8. Trim25 degrades RABV-P through complete autophagy
We have confirmed that Trim25 degrades RABV-P through a mechanism that is independent of proteasome function, and this degradation relies on the interaction between Trim25 and RABV-P. We investigated the mechanism of P protein degradation by Trim25 from the P protein's function, most importantly, the functions that may affect RABV-P stability. It has been well studied that RABV-P binds to beclin1, an autophagy-related gene, to induce incomplete autophagy. RABV-P was co-localized with GFP-LC3 aggregates (Liu et al., 2017, 2020); we wondered whether Trim25 affected RABV-P-induced autophagy. We used chloroquine (CQ), an acidification inhibitor of lysosome degradation that could suppress autophagic flux and accumulate autophagosomes, to treat RABV-P- and Trim25-expressed cells. The results showed that CQ treatment successfully recovered the RABV-P protein level when Trim25 was co-expressed (Fig. 8A). We further analyzed the LC3-I/II and p62 levels in these groups and found that overexpression of Trim25 and CVS-P caused a significant decrease of LC3-II and p62 compared with overexpressing CVS-P alone. CQ treatment recovered LC3-II and p62 levels (Fig. 8B). Therefore, Trim25 induced complete autophagy to degrade CVS-P, and suppressing autophagic flux would prevent Trim25 from degrading CVS-P. To further clarify our hypothesis, we measured the LC3-II and p62 levels in cells expressed with CVS-P-72m and Trim25 with or without CQ treatment. CVS-P-72m was not affected by Trim25 (Fig. 7G and H). As expected, LC3-II and p62 levels remained unchanged in the CVS-P-72m- and Trim25-expressed group compared with other groups (Fig. 8C).
Fig. 8.

Trim25 mediated P degradation through autophagy. (A) 293T cells were co-transfected with FLAG-Trim25 and HA-CVS-P for 48 h and treated with 1 μM CQ 24 h before cell lysis. The cell lysates were analyzed by Western blotting. (B) 293T cells were co-transfected with FLAG-Trim25 and HA-CVS-P for 48 h and treated with 1 μM CQ 24 h before cell lysis; cell lysates were analyzed by Western blotting. (C) 293T cells were co-transfected with FLAG-Trim25 and HA-P-72m for 48 h and treated with 1 μM CQ 24 h before cell lysis; cell lysates were analyzed by Western blotting. (D) 293T cells were co-transfected with mCherry-GFP-LC3 and HA-CVS-P, or FLAG-Trim25, or HA-P-72m for 36 h and then analyzed for the co-localization of mCherry with GFP. The white arrow indicates the fluorescence reduction of GFP-LC3. Scale bar, 10 μm. Data are presented as mean ± SD, n = 3. Western blotting data are representative of at least three independent experiments.
Furthermore, we used a tandem reporter construct, mCherry-GFP-LC3; the GFP of this tandem autophagosome reporter is sensitive and attenuated in an acidic pH environment by lysosomal degradation, whereas the mCherry is not. Therefore, the fusion of the autophagosome with the lysosome will result in the loss of green fluorescence and the appearance of only red fluorescence of mCherry (Klionsky et al., 2012). Confocal microscopy showed both green and red fluorescence signals in CVS-P-expressed cells (Fig. 8D), indicating that autophagosomes did not fuse with lysosomes, whereas in CVS-P- and Trim25-expressed cells, there was less green fluorescence signal. Still, many red fluorescence signals remained detectable (Fig. 8D), indicating that autophagosomes fused with lysosomes. After CQ treatment, both green and red fluorescence signals were observed in CVS-P- and Trim25-expressed cells (Fig. 8D). Both red and green fluorescence signals were observed in CVS-P-72m- and Trim25-expressed cells (Fig. 8D). Together, above results illustrate that Trim25 degrades RABV-P protein via complete autophagy.
3. Discussion
Trim25 inhibits viral replication through two different mechanisms: RIG-I pathway-dependent and RIG-I pathway independent. The RIG-I pathway-dependent mechanism could occur via the E3 ligase activity of Trim25, which conjugates K63-polyubiquitin chains to RIG-I to help activate the RIG-I pathway, ultimately increasing IFN production, as reported previously (Gack et al., 2007). Here, we revealed a novel mechanism of Trim25 to restrict RABV replication independent of the RIG-I pathway. Especially, the CCD domain of Trim25 interacts with RABV-P, causing instability of RABV-P and reduction of its expression level. Additionally, the valine at position 72 of RABV-P is critical for its interaction with Trim25, and mutation of this AA inhibits the reduction of RABV-P by Trim25.
Recent studies have demonstrated different strategies for Trim25 to defend against different types of viruses (Atkinson et al., 2021; Jin et al., 2019; Kaikai et al., 2021; Kmiec et al., 2021; Meyerson et al., 2017; Wang et al., 2021; Zhou et al., 2020). Trim25 can target the VP3 protein of infectious bursal disease virus (IBDV) and mediate the K27-linked polyubiquitination and subsequent proteasomal degradation of the VP3 protein (Wang et al., 2021). Trim25 can also target IAV ribonucleoprotein to block the onset of RNA chain elongation, and this function is independent of its E3 ligase activity (Meyerson et al., 2017). Furthermore, recent studies have shown that Trim25 can bind to RNA through its central CC domain, and this discovery caused scientists to wonder whether Trim25 can directly bind to viral RNA to exert antiviral effects (Castello et al., 2012; Choudhury et al., 2020; Sanchez et al., 2018). Similar to the finding of this study, this anti-IAV effect occurs through a mechanism that is also independent of the E3-ligase activity of Trim25.
Many studies have verified that the unique antiviral activities of ISGs against particular viruses are generally related to the proteins encoded by each virus (Ke et al., 2019; Panayiotou et al., 2018; Tian et al., 2020; Zhang et al., 2020). In many cases, the interaction between ISG and viral proteins can provide insight into how ISG suppresses a particular virus. In this study, the CCD domain of Trim25 is responsible for its interaction with RABV-P, which is the same domain that mediates its interaction with the NS1 protein of the IAV. It has been reported that the NS1 protein blocks Trim25 from inhibiting the viral RNA synthesis of IAV (Gack et al., 2009). However, our results from the minigenome assay showed that the overexpression of Trim25 or its mutant had no significant effect on the RNP activity of RABV (Fig. S5G), which indicates that Trim25 could not inhibit RABV replication by impairing the viral RNA synthesis. We also performed Co-IP assays and investigated the potential effect of Trim25 on the interaction between truncated L protein and P protein. The results showed that Trim25 could not affect P-L interactions (Fig. S5C).
Since RABV-P performs multiple functions during the life cycle of the RABV (Zhou et al., 2019), we investigated whether Trim25 could hinder RABV replication through other mechanisms. To exclude the potential impact of Trim25 on RABV-P’s anti-IFN activity (Chelbi-Alix et al., 2006), we measured the IRF3 activation by using luciferase assay after the co-expression of E3-mut-Trim25 and RABV-P, and the results showed that E3-mut-Trim25 did not affect RABV-P’s anti-IFN activity. To evaluate the effect of Trim25 on the RABV-P dimerization (Gerard et al., 2009; Ivanov et al., 2010), which has been proven to be crucial for RABV replication, Co-IP assays were performed, and the results showed that Trim25 did not impede RABV-P dimerization (Fig. S5B). We also used bimolecular fluorescence complementation (BiFC) to measure the dimerization of RABV-P. The results demonstrated that unlike IIGP1, which has been testified to affect RABV-P’s dimerization (Tian et al., 2020), Trim25’s expression had no effect on RABV-P’s GFP signals (Fig. S5H), confirming that Trim25 did not impede RABV-P dimerization. Since RABV-P can interact with RABV-N to form a high-order phase separation structure, Negri body, we then performed Co-IP assays and investigated the potential effect of Trim25 on RABV-P’s Negri body formation (Lahaye et al., 2009; Zhou et al., 2019). However, Trim25 expression did not affect the interaction between RABV-P and RABV-N (Fig. S5A).
Moreover, Trim25 decreases the protein level of RABV-P but not the transcriptional level (Fig. 7B), indicating that Trim25 decreased RABV-P expression at the PTM level. However, CHX and MG132 treatment did not affect the RABV-P degradation caused by Trim25 (Fig. 7C–E), indicating that Trim25-mediated RABV-P degradation is unrelated to the proteasome. RABV-P could induce incomplete autophagy, and RABV-P was co-localized with some GFP-LC3 dots (Liu et al., 2020). Since many types of viruses had utilized incomplete autophagy to escape from being degraded by lysosomes, thus facilitating viral replication or egress (Ding et al., 2014; Jia et al., 2022), we successfully discovered that Trim25 degrades RABV-P through the complete autophagy. However, the detailed mechanism of how Trim25 affects RABV-P-induced autophagy is uncovered. Other researchers find that RABV protein 82-297-P (isoform P5) induces incomplete autophagy via the beclin1-mediated signaling pathway by interacting with the N-terminal residues 1–139 of beclin1 (Liu et al., 2020). While our results show that 82-297-P loses its ability to interact with Trim25 (Fig. 6C), whether Trim25 disrupts the interaction between RABV-P and beclin1 need to be clarified in the following experiments.
We have found that the AA at position 72 of RABV-P affects the interaction between Trim25 and RABV-P (Fig. 6G), we wondered whether it could affect viral pathogenicity in vivo. To confirm this hypothesis, we constructed a recombinant RABV with the valine at position 72 of RABV-P mutated to alanine, named rRABV-P-72m and confirmed that rRABV-P-72m replicated in a lower titer than rRABV in N2a and BSR cells (Figs. S6A–S6C). Then we inoculated mice with rRABV-P-72m and rRABV as in Fig. 3D. The survival ratio of the rRABV-P-72m-infected mice was significantly increased compared with that of the rRABV-infected mice (Fig. S6D). The viral loads in the cortex, olfactory bulb, cerebellum and brain stem of the rRABV-P-72m-infected mice were significantly higher than those in the rRABV-inoculated mice at 12 d.p.i. (Figs. S6F–S6I). Since RABV-P plays multiple roles in the virus replication cycle, we cannot exclude the possibility that the mutation of valine 72 to alanine 72 may impair the virus replication besides the interaction with Trim25.
In summary, our study highlighted a previously unappreciated strategy for Trim25 to suppress virus replication by interfering with viral protein stability independent of its E3 ligase activity. This study also revealed the exact region of RABV-P targeted by Trim25, which is a potential therapeutic target for treating RABV infection.
4. Materials and methods
4.1. Cells, virus, antibodies, and mice
HEK-293T (human embryonic kidney, 293T), N2a (neuroblastoma), BV2 (mouse microglioma), BSR (cloned from baby hamster kidney-21, BHK-21), and Vero (Cercopithecus aethiops kidney) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher, USA) supplemented with 10%–12% fetal bovine serum (Thermo Fisher). The lab-attenuated RABV strain CVS-B2c (CVS) (derived from CVS-24 virus by passaging in BHK-21 cells), the dog-derived RABV strain DRV-Mexico (DRV) (Yu et al., 2014; Zhang and Fu, 2012) and the silver-haired bat rabies virus (SHBRV) strain 18 (Morimoto et al., 1996) was stored in our laboratory. Sendai virus (Cantell strain, Charles River Laboratories) was used at a final concentration of 100 hemagglutinin units per ml. Monoclonal antibodies against FLAG-tag, HA tag, V5-tag, and β-actin were purchased from Medical & Biological Laboratories (MBL, Nagoya, Japan). Monoclonal antibodies (mAbs) against the RABV-N protein RABV-P were prepared in our laboratory. All experiments involving mice were performed following the recommendations in the Guide for the Care and Use of Laboratory Animals of the Ministry of Science and Technology of China and were approved by the Scientific Ethics Committee of Huazhong Agricultural University (approval number: HZAUMO-2017-053).
4.2. Virus titration
BSR, 293T, and N2a cells were infected with CVS-B2c, DRV-Mexico, SHBRV-18, rRABV, rRABV-Trim25(−), or rRABV-Trim25 at different MOIs for 1 h at 37 °C. The cells were washed with PBS three times and cultured in a cell incubator at 37 °C. The supernatants were collected 1–5 days post-infection (d.p.i.) for virus titration. RABV titers were determined by direct immunofluorescence assay as described previously (Tian et al., 2016).
4.3. Construction and rescue of the recombinant RABV
The recombinant RABV was constructed and rescued as described previously (Sui et al., 2020; Zhang et al., 2022). Briefly, the murine Trim25 gene and murine Trim25 encoding a null Trim25 ORF were inserted into the genome of the RABV CVS-B2c strain between the G and L genes by replacing the pseudogene, respectively. To rescue the viruses, a plasmid containing the full-length viral genome and the helper plasmids expressing the N, P, G, and L proteins were transfected into N2a cells using Lipofectamine 3000 (Thermo Fisher, USA), and the recombinant viruses were harvested at 5 days post-transfection. The recombinant viruses were propagated and titrated in BSR cells.
4.4. Mouse infection
Female C57BL/6 mice (6-week-old) were intramuscularly (i.m.) inoculated with 100 μL of rRABV, rRABV-Trim25(−), or rRABV-Trim25 (8 × 104 FFU) at hind legs. Changes in body weight and mortality of the mice were monitored daily. At 9 d.p.i., mice were euthanized with CO2, and their brains were harvested for immunohistochemistry. At 6, 9, and 12 d.p.i., mice were euthanized with CO2, and their brains were harvested for RNA isolation and qPCR analysis.
4.5. Quantitative real-time PCR (qPCR)
Brain tissues and cells were subjected to RNA isolation using TRIzol (Invitrogen) according to the manufacturer’s instructions, and qPCR assays were performed as described previously (Tian et al., 2016).
The primer sets used in this study are listed here: RABV-N-F: ACACCGCAACTACAAGACA; RABV-N-R: ATGGTACTCCAGTTGGCACA; β-actin-F: AGGTGACAGCATTGCTTCTG; β-actin-R: GCTGCCTCAACACCTCAAC; Trim25-F: ACAAGTTGCCCACCTTTGGA; Trim25-R: AAGCTCCGTTCTGGACTTGG. To quantify cellular RABV-N RNA levels, total RNA was transcribed using AMV Reverse Transcriptase XL (TAKARA, Kusatsu, Japan) and a primer specific for the RABV-N gene. A standard curve was generated from serially diluted plasmids carrying the N gene, and the copy number of N RNA was normalized to 1 mg of total RNA.
4.6. Western blotting
Cells were cultured in 24-well plates and lysed with NP40 lysis buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA, and 0.5% NP-40) supplemented with a protease inhibitor cocktail and PMSF (1 mM) for 30 min at 4 °C. The cell lysates were centrifuged for 10 min at 12,000 g and 4 °C. The supernatants were transferred into a new tube, resolved by sodium dodecyl sulfate gel electrophoresis (SDS–PAGE), and transferred to PVDF membranes to measure protein expression.
4.7. Confocal microscopy
293T cells seeded on 14-mm coverslips were transfected with plasmids and infected with RABV. After incubation, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and then stained with antibodies against RABV-P, Trim25, or DAPI. After being washed three times with PBS, the cells were incubated with Alexa 488-conjugated or 594-conjugated secondary antibodies for 1 h at room temperature. Staining was visualized with a ZEISS LSM 880 confocal microscope under an oil objective (Carl Zeiss AG, Oberkochen, Germany).
4.8. GST pull-down
Purified GST-tagged Trim25 (or GST) and HA-RABV-P (or HA-RABV-N) were incubated with glutathione agarose beads. After washing with PBS three times, the bound proteins were eluted with elution buffer, separated by SDS–PAGE, and subjected to Western blotting.
4.9. Co-immunoprecipitation
For the Co-immunoprecipitation assay, 293T cells were co-transfected with the mammalian expression vector pCAGGS expressing FLAG-Trim25 and truncated fragments of HA-CVS-P or mutants of HA-CVS-P. At 48 h post-transfection, the cells were washed with cold PBS and lysed with NP-40 lysis buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA, and 0.5% NP-40) supplemented with a protease inhibitor cocktail (Roche) and PMSF (1 mM) for 30 min at 4 °C. The cell lysates were centrifuged for 10 min at 12,000 g and 4 °C, and the supernatants were transferred into a new tube. After incubation with a mAb against FLAG or HA tag for 12 h, rProtein A/G MagPoly Beads (Cat#. SM015005, Smart-Lifesciences, China) were added to the cell lysates and incubated for 3 h at 4 °C while rotating. The samples were washed with NP-40 lysis buffer five times, and the bound proteins were analyzed by Western blotting with the indicated antibodies.
4.10. Immunohistochemistry (IHC) analysis
The mice were intracardially perfused with PBS and 4% paraformaldehyde. The brains were removed and incubated in 4% paraformaldehyde for over 12 h. After dehydration and wax immersion, the samples were embedded in paraffin and sectioned into 4-μm sections. For IHC analysis, the sections were processed using antigen retrieval and endogenous peroxidase quenching.
4.11. Immunofluorescence histochemistry
Mice were infected with CVS-B2c (8 × 104 FFU) for 9 days through the i.m. route. Then, their cerebellums were harvested and fixed with 4% paraformaldehyde for 24 h. The cerebellums were dehydrated in 30% (wt/vol) sucrose, embedded and frozen in OCT medium, and cut into 30-μm-thick sections. The tissue sections were transferred to microplates and stained with Trim25 antibodies and RABV-P antibodies, followed by incubation with Alexa Fluor 594- and Alexa Fluor 488-conjugated secondary antibodies for 1 h in the dark. After incubation, the tissue sections were washed three times with PBS and then placed on ultrathin glass slides for observation. Fluorescent images were captured under an EVOS FL Auto imaging system (Thermo Fisher Scientific).
4.12. Statistical analysis
The graphs of Western blotting, immunofluorescence, and immunohistochemistry were analyzed using ImageJ software (https://imagej.nih.gov/ij/). The data were expressed as the mean and standard deviation (SD). Student's t-test was performed to analyze the significant differences between the two groups. One-way ANOVA was performed to analyze the significant differences between the three groups. The survival ratio was analyzed by a Log-rank (Mantel-Cox) test. The asterisks indicate statistical significance (∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001). Graphs were plotted and analyzed using GraphPad Prism software, version 8.0 (GraphPad Software, La Jolla, CA, USA).
Acknowledgments
This research was supported by the National Natural Science Foundation of China (31872451to L.Z.).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cellin.2022.100057.
Appendix A. Supplementary data
The following is the supplementary data to this article:
References
- Atkinson S.C., Heaton S.M., Audsley M.D., Kleifeld O., Borg N.A. TRIM25 and DEAD-box RNA helicase DDX3X cooperate to regulate RIG-I-mediated antiviral immunity. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22169094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzózka K., Finke S., Conzelmann K.K. Identification of the rabies virus alpha/beta interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J. Virol. 2005;79:7673–7681. doi: 10.1128/JVI.79.12.7673-7681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzózka K., Finke S., Conzelmann K.K. Inhibition of interferon signaling by rabies virus phosphoprotein P: activation-dependent binding of STAT1 and STAT2. J. Virol. 2006;80:2675–2683. doi: 10.1128/JVI.80.6.2675-2683.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthagena L., Bergamaschi A., Luna J.M., David A., Uchil P.D., Margottin-Goguet F., Mothes W., Hazan U., Transy C., Pancino G., et al. Human TRIM gene expression in response to interferons. PLoS One. 2009;4 doi: 10.1371/journal.pone.0004894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B.M., Strein C., Davey N.E., Humphreys D.T., Preiss T., Steinmetz L.M., et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Chelbi-Alix M.K., Vidy A., El Bougrini J., Blondel D. Rabies viral mechanisms to escape the IFN system: the viral protein P interferes with IRF-3, Stat1, and PML nuclear bodies. J. Interferon Cytokine Res. 2006;26:271–280. doi: 10.1089/jir.2006.26.271. [DOI] [PubMed] [Google Scholar]
- Chenik M., Chebli K., Blondel D. Translation initiation at alternate in-frame AUG codons in the rabies virus phosphoprotein mRNA is mediated by a ribosomal leaky scanning mechanism. J. Virol. 1995;69:707–712. doi: 10.1128/jvi.69.2.707-712.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenik M., Chebli K., Gaudin Y., Blondel D. In vivo interaction of rabies virus phosphoprotein (P) and nucleoprotein (N): existence of two N-binding sites on P protein. J. Gen. Virol. 1994;75(Pt 11):2889–2896. doi: 10.1099/0022-1317-75-11-2889. [DOI] [PubMed] [Google Scholar]
- Chenik M., Schnell M., Conzelmann K.K., Blondel D. Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J. Virol. 1998;72:1925–1930. doi: 10.1128/jvi.72.3.1925-1930.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C., Dvorkin S., Chiang J.J., Potter R.B., Gack M.U. The small t antigen of JC virus antagonizes RIG-I-mediated innate immunity by inhibiting TRIM25's RNA binding ability. mBio. 2021;12 doi: 10.1128/mBio.00620-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C., Pauli E.K., Biryukov J., Feister K.F., Meng M., White E.A., Münger K., Howley P.M., Meyers C., Gack M.U. The human papillomavirus E6 oncoprotein targets USP15 and TRIM25 to suppress RIG-I-mediated innate immune signaling. J. Virol. 2018;92 doi: 10.1128/JVI.01737-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury N.R., Heikel G., Michlewski G. vol. 11. Wiley interdisciplinary reviews RNA; 2020. p. e1588. (TRIM25 and its Emerging RNA-Binding Roles in Antiviral Defense). [DOI] [PubMed] [Google Scholar]
- Clark K., Plater L., Peggie M., Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J. Biol. Chem. 2009;284:14136–14146. doi: 10.1074/jbc.M109.000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Cruz A.A., Kershaw N.J., Chiang J.J., Wang M.K., Nicola N.A., Babon J.J., Gack M.U., Nicholson S.E. Crystal structure of the TRIM25 B30.2 (PRYSPRY) domain: a key component of antiviral signalling. Biochem. J. 2013;456:231–240. doi: 10.1042/BJ20121425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B., Zhang G., Yang X., Zhang S., Chen L., Yan Q., Xu M., Banerjee A.K., Chen M. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe. 2014;15:564–577. doi: 10.1016/j.chom.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Fisher C.R., Streicker D.G., Schnell M.J. The spread and evolution of rabies virus: conquering new frontiers. Nat. Rev. Microbiol. 2018;16:241–255. doi: 10.1038/nrmicro.2018.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z.F., Zheng Y., Wunner W.H., Koprowski H., Dietzschold B. Both the N- and the C-terminal domains of the nominal phosphoprotein of rabies virus are involved in binding to the nucleoprotein. Virology. 1994;200:590–597. doi: 10.1006/viro.1994.1222. [DOI] [PubMed] [Google Scholar]
- Gack M.U., Albrecht R.A., Urano T., Inn K.S., Huang I.C., Carnero E., Farzan M., Inoue S., Jung J.U., García-Sastre A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack M.U., Shin Y.C., Joo C.H., Urano T., Liang C., Sun L., Takeuchi O., Akira S., Chen Z., Inoue S., et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- Gerard F.C., Ribeiro Ede A., Jr., Leyrat C., Ivanov I., Blondel D., Longhi S., Ruigrok R.W., Jamin M. Modular organization of rabies virus phosphoprotein. J. Mol. Biol. 2009;388:978–996. doi: 10.1016/j.jmb.2009.03.061. [DOI] [PubMed] [Google Scholar]
- Gori Savellini G., Anichini G., Gandolfo C., Cusi M.G. SARS-CoV-2 N protein targets TRIM25-mediated RIG-I activation to suppress innate immunity. Viruses. 2021;13 doi: 10.3390/v13081439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka Y., Lim C.K., Takayama-Ito M., Park C.H., Kimitsuki K., Shiwa N., Inoue K.I., Itou T. Segmentation of the rabies virus genome. Virus Res. 2018;252:68–75. doi: 10.1016/j.virusres.2018.05.017. [DOI] [PubMed] [Google Scholar]
- Horwitz J.A., Jenni S., Harrison S.C., Whelan S.P.J. Structure of a rabies virus polymerase complex from electron cryo-microscopy. Proc. Natl. Acad. Sci. U.S.A. 2020;117:2099–2107. doi: 10.1073/pnas.1918809117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Li W., Gao T., Cui Y., Jin Y., Li P., Ma Q., Liu X., Cao C. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J. Virol. 2017;91 doi: 10.1128/JVI.02143-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S., Orimo A., Hosoi T., Kondo S., Toyoshima H., Kondo T., Ikegami A., Ouchi Y., Orimo H., Muramatsu M. Genomic binding-site cloning reveals an estrogen-responsive gene that encodes a RING finger protein. Proc. Natl. Acad. Sci. U.S.A. 1993;90:11117–11121. doi: 10.1073/pnas.90.23.11117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov I., Crépin T., Jamin M., Ruigrok R.W. Structure of the dimerization domain of the rabies virus phosphoprotein. J. Virol. 2010;84:3707–3710. doi: 10.1128/JVI.02557-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob Y., Real E., Tordo N. Functional interaction map of lyssavirus phosphoprotein: identification of the minimal transcription domains. J. Virol. 2001;75:9613–9622. doi: 10.1128/JVI.75.20.9613-9622.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D., Liang Q., Liu H., Li G., Zhang X., Chen Q., Wang A., Wei T. A nonstructural protein encoded by a rice reovirus induces an incomplete autophagy to promote viral spread in insect vectors. PLoS Pathog. 2022;18 doi: 10.1371/journal.ppat.1010506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y., Jia K., Zhang W., Xiang Y., Jia P., Liu W., Yi M. Zebrafish TRIM25 promotes innate immune response to RGNNV infection by targeting 2CARD and RD regions of RIG-I for K63-linked ubiquitination. Front. Immunol. 2019;10:2805. doi: 10.3389/fimmu.2019.02805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaikai H., Zhao D., Liu Y., Liu Q., Huang X., Yang J., Zhang L., Li Y. The E3 ubiquitin ligase TRIM25 inhibits tembusu virus replication in vitro. Front. Vet. Sci. 2021;8 doi: 10.3389/fvets.2021.722113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke W., Fang L., Tao R., Li Y., Jing H., Wang D., Xiao S. Porcine reproductive and respiratory syndrome virus E protein degrades porcine cholesterol 25-hydroxylase via the ubiquitin-proteasome pathway. J. Virol. 2019;93 doi: 10.1128/JVI.00767-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J., Abdalla F.C., Abeliovich H., Abraham R.T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J.A., et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kmiec D., Lista M.J., Ficarelli M., Swanson C.M., Neil S.J.D. S-farnesylation is essential for antiviral activity of the long ZAP isoform against RNA viruses with diverse replication strategies. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahaye X., Vidy A., Pomier C., Obiang L., Harper F., Gaudin Y., Blondel D. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: evidence that NBs are sites of viral transcription and replication. J. Virol. 2009;83:7948–7958. doi: 10.1128/JVI.00554-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu K.G., Brice A., Wiltzer L., Hirst B., Jans D.A., Blondel D., Moseley G.W. The rabies virus interferon antagonist P protein interacts with activated STAT3 and inhibits Gp130 receptor signaling. J. Virol. 2013;87:8261–8265. doi: 10.1128/JVI.00989-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Liao M., Yan Y., Yang H., Wang H., Zhou J. Rabies virus phosphoprotein P5 binding to BECN1 regulates self-replication by BECN1-mediated autophagy signaling pathway. Cell Commun. Signal. : CCS. 2020;18:153. doi: 10.1186/s12964-020-00644-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Wang H., Gu J., Deng T., Yuan Z., Hu B., Xu Y., Yan Y., Zan J., Liao M., et al. BECN1-dependent CASP2 incomplete autophagy induction by binding to rabies virus phosphoprotein. Autophagy. 2017;13:739–753. doi: 10.1080/15548627.2017.1280220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manokaran G., Finol E., Wang C., Gunaratne J., Bahl J., Ong E.Z., Tan H.C., Sessions O.M., Ward A.M., Gubler D.J., et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science. 2015;350:217–221. doi: 10.1126/science.aab3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis M., Méhouas S., Réal E., Iseni F., Blondel D., Tordo N., Ruigrok R.W. Rabies virus chaperone: identification of the phosphoprotein peptide that keeps nucleoprotein soluble and free from non-specific RNA. Virology. 2006;349:422–429. doi: 10.1016/j.virol.2006.01.030. [DOI] [PubMed] [Google Scholar]
- Meyerson N.R., Zhou L., Guo Y.R., Zhao C., Tao Y.J., Krug R.M., Sawyer S.L. Nuclear TRIM25 specifically targets influenza virus ribonucleoproteins to block the onset of RNA chain elongation. Cell Host Microbe. 2017;22:627–638. doi: 10.1016/j.chom.2017.10.003. e627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min Y.Q., Ning Y.J., Wang H., Deng F. A RIG-I-like receptor directs antiviral responses to a bunyavirus and is antagonized by virus-induced blockade of TRIM25-mediated ubiquitination. J. Biol. Chem. 2020;295:9691–9711. doi: 10.1074/jbc.RA120.013973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto K., Patel M., Corisdeo S., Hooper D.C., Fu Z.F., Rupprecht C.E., Koprowski H., Dietzschold B. Characterization of a unique variant of bat rabies virus responsible for newly emerging human cases in North America. Proc. Natl. Acad. Sci. U.S.A. 1996;93:5653–5658. doi: 10.1073/pnas.93.11.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin B., Liang B., Gardner E., Ross R.A., Whelan S.P.J. An in vitro RNA synthesis assay for rabies virus defines ribonucleoprotein interactions critical for polymerase activity. J. Virol. 2017;91 doi: 10.1128/JVI.01508-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksayan S., Wiltzer L., Rowe C.L., Blondel D., Jans D.A., Moseley G.W. A novel nuclear trafficking module regulates the nucleocytoplasmic localization of the rabies virus interferon antagonist, P protein. J. Biol. Chem. 2012;287:28112–28121. doi: 10.1074/jbc.M112.374694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panayiotou C., Lindqvist R., Kurhade C., Vonderstein K., Pasto J., Edlund K., Upadhyay A.S., Överby A.K. Viperin restricts zika virus and tick-borne encephalitis virus replication by targeting NS3 for proteasomal degradation. J. Virol. 2018;92 doi: 10.1128/JVI.02054-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder M., Brzózka K., Pfaller C.K., Cox J.H., Stitz L., Conzelmann K.K. Genetic dissection of interferon-antagonistic functions of rabies virus phosphoprotein: inhibition of interferon regulatory factor 3 activation is important for pathogenicity. J. Virol. 2011;85:842–852. doi: 10.1128/JVI.01427-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Aparicio M.T., Feinman L.J., García-Sastre A., Shaw M.L. Paramyxovirus V proteins interact with the RIG-I/TRIM25 regulatory complex and inhibit RIG-I signaling. J. Virol. 2018;92 doi: 10.1128/JVI.01960-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez J.G., Sparrer K.M.J., Chiang C., Reis R.A., Chiang J.J., Zurenski M.A., Wan Y., Gack M.U., Pornillos O. TRIM25 binds RNA to modulate cellular anti-viral defense. J. Mol. Biol. 2018;430:5280–5293. doi: 10.1016/j.jmb.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell M.J., McGettigan J.P., Wirblich C., Papaneri A. The cell biology of rabies virus: using stealth to reach the brain. Nat. Rev. Microbiol. 2010;8:51–61. doi: 10.1038/nrmicro2260. [DOI] [PubMed] [Google Scholar]
- Sui B., Chen D., Liu W., Wu Q., Tian B., Li Y., Hou J., Liu S., Xie J., Jiang H., et al. A novel antiviral lncRNA, EDAL, shields a T309 O-GlcNAcylation site to promote EZH2 lysosomal degradation. Genome Biol. 2020;21:228. doi: 10.1186/s13059-020-02150-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B., Yuan Y., Yang Y., Luo Z., Sui B., Zhou M., Fu Z.F., Zhao L. Interferon-inducible GTPase 1 impedes the dimerization of rabies virus phosphoprotein and restricts viral replication. J. Virol. 2020;94 doi: 10.1128/JVI.01203-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D., Luo Z., Zhou M., Li M., Yu L., Wang C., Yuan J., Li F., Tian B., Sui B., et al. Critical role of K1685 and K1829 in the large protein of rabies virus in viral pathogenicity and immune evasion. J. Virol. 2016;90:232–244. doi: 10.1128/JVI.02050-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Yu M., Liu A., Bao Y., Qi X., Gao L., Chen Y., Liu P., Wang Y., Xing L., et al. TRIM25 inhibits infectious bursal disease virus replication by targeting VP3 for ubiquitination and degradation. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F., Zhang G., Zhong X., Han N., Song Y., Zhao L., Cui M., Rayner S., Fu Z.F. Comparison of complete genome sequences of dog rabies viruses isolated from China and Mexico reveals key amino acid changes that may be associated with virus replication and virulence. Arch. Virol. 2014;159:1593–1601. doi: 10.1007/s00705-013-1966-2. [DOI] [PubMed] [Google Scholar]
- Zhang A., Duan H., Zhao H., Liao H., Du Y., Li L., Jiang D., Wan B., Wu Y., Ji P., et al. Interferon-induced transmembrane protein 3 is a virus-associated protein which suppresses porcine reproductive and respiratory syndrome virus replication by blocking viral membrane fusion. J. Virol. 2020;94 doi: 10.1128/JVI.01350-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G., Fu Z.F. Complete genome sequence of a street rabies virus from Mexico. J. Virol. 2012;86:10892–10893. doi: 10.1128/JVI.01778-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Xing X., Long B., Cao Y., Hu S., Li X., Yu Y., Tian D., Sui B., Luo Z., et al. A spatial and cellular distribution of rabies virus infection in the mouse brain revealed by fMOST and single-cell RNA sequencing. Clin. Transl. Med. 2022;12 doi: 10.1002/ctm2.700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K., Li L.W., Jiang Y.F., Gao F., Zhang Y.J., Zhao W.Y., Li G.X., Yu L.X., Zhou Y.J., Tong G.Z. Nucleocapsid protein of porcine reproductive and respiratory syndrome virus antagonizes the antiviral activity of TRIM25 by interfering with TRIM25-mediated RIG-I ubiquitination. Vet. Microbiol. 2019;233:140–146. doi: 10.1016/j.vetmic.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.R., Liu J.H., Li H.M., Zhao Y., Cheng Z., Hou Y.M., Guo H.J. Regulatory effects of chicken TRIM25 on the replication of ALV-A and the MDA5-mediated type I interferon response. Vet. Res. 2020;51:145. doi: 10.1186/s13567-020-00870-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Su J.M., Samuel C.E., Ma D. Measles virus forms inclusion bodies with properties of liquid organelles. J. Virol. 2019;93 doi: 10.1128/JVI.00948-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.