

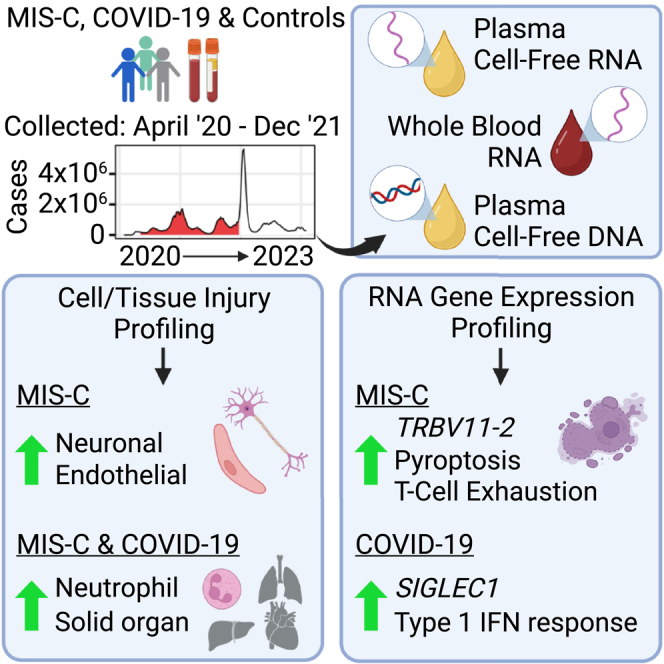
Summary
Differential host responses in coronavirus disease 2019 (COVID-19) and multisystem inflammatory syndrome in children (MIS-C) remain poorly characterized. Here, we use next-generation sequencing to longitudinally analyze blood samples from pediatric patients with COVID-19 or MIS-C across three hospitals. Profiling of plasma cell-free nucleic acids uncovers distinct signatures of cell injury and death between COVID-19 and MIS-C, with increased multiorgan involvement in MIS-C encompassing diverse cell types, including endothelial and neuronal cells, and an enrichment of pyroptosis-related genes. Whole-blood RNA profiling reveals upregulation of similar pro-inflammatory pathways in COVID-19 and MIS-C but also MIS-C-specific downregulation of T cell-associated pathways. Profiling of plasma cell-free RNA and whole-blood RNA in paired samples yields different but complementary signatures for each disease state. Our work provides a systems-level view of immune responses and tissue damage in COVID-19 and MIS-C and informs future development of new disease biomarkers.
Keywords: SARS-CoV-2, coronavirus disease 2019, cell-free DNA, cell-free RNA, whole-blood RNA, RNA sequencing, RNA-seq, clinical severity, multisystem inflammatory syndrome in children, nucleic acid sequencing, DNA damage, cell damage, tissue damage, host response, signaling pathways, disease biomarkers, immune response, systems biology, bisulfite sequencing, pediatric
Graphical abstract
Highlights
-
•
Patients with MIS-C and COVID-19 have distinct cell injury and host response profiles
-
•
Patients with MIS-C and COVID-19 have elevated solid-organ-derived cell-free DNA
-
•
Patients with MIS-C have increased endothelium- and neuron-derived cell-free RNA
-
•
Cell-free and whole-blood RNA provide complementary measurements of patient health
Loy et al. use cell-free RNA, whole-blood RNA, and cell-free DNA sequencing to characterize distinct host response and cellular injury profiles in pediatric patients with MIS-C and/or COVID-19. This study highlights the complementary information from cell-free and whole-blood RNA analyses, with broad implications for future liquid biopsy applications.
Introduction
At the onset of the coronavirus disease 2019 (COVID-19) pandemic, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was thought to only cause mild or asymptomatic infection in children. Large-scale surveillance studies have since revealed pediatric cases of severe COVID-19-associated pneumonia occurring throughout the pandemic, especially in children with underlying comorbidities.1,2 Children also are at risk for the post-infectious multisystem inflammatory syndrome in children (MIS-C) associated with COVID-19, which can manifest as severe disease with systemic hyperinflammation and multiorgan involvement.3 Diagnosis of MIS-C currently relies on clinical symptoms, and the pathogenesis of MIS-C is incompletely understood. Many MIS-C cases were initially misdiagnosed as Kawasaki disease (KD),4 another systemic inflammatory syndrome, because of overlapping clinical features, including fever, hyperinflammation, mucocutaneous involvement, and vascular endothelial dysfunction. Subsequent studies have demonstrated distinct clinical5 and immunological6 phenotypes associated with MIS-C compared with KD. Thus, a better understanding of the pathogenesis of MIS-C is critical to improve clinical diagnosis and inform targeted interventions.
Initial characterization by proteomics and RNA sequencing revealed that MIS-C has an inflammatory profile similar to KD and severe COVID-19 but with key differences, including specific increases in interferon γ (IFNγ), interleukin-6 (IL-6), IL-17, IL-10, alarmin-related proteins, and other pro-inflammatory factors.7,8,9 Autoantibody profiling revealed a unique autoantigen profile targeting organs often injured during MIS-C, but the organ and tissue damage has not yet been quantified on a system-wide level.8 Immune cell profiling showed that MIS-C is associated with an expansion of specific subsets of natural killer (NK) cells, T cells, and B cells,8,10 and gene expression profiling showed that a high fraction (∼24%) of T cells in patients with MIS-C are T cell receptor beta variable 11-2 (TRBV11-2) positive.10,11,12 T cell receptor repertoire analyses have led to the hypothesis that the SARS-CoV-2 spike protein has a superantigen effect, causing T cell dysregulation that contributes to the development of MIS-C.12 Finally, flow cytometry analyses have demonstrated that children with MIS-C have reduced virus-specific CD4+ and CD8+ T cells compared with children with COVID-19 and controls.13
Despite these prior studies, much remains unclear about the pathogenesis of MIS-C, and there is a lack of biomarkers that could be leveraged to develop diagnostic and prognostic assays. Here we performed unbiased profiling of whole-blood cellular RNA (wbRNA), plasma cell-free RNA (cfRNA), and methylated plasma cell-free DNA (cfDNA) from 205 pediatric patients diagnosed with COVID-19 or MIS-C and 23 controls from 3 pediatric hospital systems in the United States. Longitudinal sampling of these blood analytes enables a complementary and systems-level view of immune responses and cell/tissue damage associated with MIS-C and COVID-19.
Results
Clinical COVID-19 and/or MIS-C cohort
We collected 402 blood and/or plasma samples from 228 patients from the University of California, San Francisco (UCSF); Emory University/Children’s Healthcare of Atlanta (Emory), and Children’s National Hospital (CNH) (Figures 1A and 1B; Table 1; STAR Methods). All samples were stratified by diagnosis, time of collection relative to hospitalization (acute, 0–4 days after hospital admission or post acute, >4 days after hospital admission), and severity of disease (Figure 1C; Table S1). Patients were either diagnosed with COVID-19 (without MIS-C) or MIS-C or were uninfected donor control subjects. All MIS-C patients were classified as having moderate or severe disease. Patients with asymptomatic and mild COVID-19 were either outpatient cases or hospitalized for an indication other than COVID-19 (Tables S1 and S5). Control subjects were healthy outpatient children with minimal, if any, comorbidities (asthma, attention deficit hyperactivity disorder [ADHD], seasonal allergies) (Table S5). Of the 402 samples analyzed, 218 (54%) were from patients who received intravenous immunoglobulin (IVIg), of which the sample was collected after IVIg treatment in 183 (83%). Because of limited sample volume, cfRNA, cfDNA, and wbRNA profiling were performed on different numbers of samples. The demographic breakdown of the samples used for each analyte can be found in Tables S2–S4.
Figure 1.

Study design and patient characteristics
(A) Sample collection and processing overview.
(B) Distribution of samples across analytes.
(C) Distribution of disease severity for each sample group.
See also Table 1.
Table 1.
Demographics and clinical characteristics of the study cohort
| Variables | Group | Overall | COVID-19 | MIS-C | Control | p Value |
|---|---|---|---|---|---|---|
| n | 228 | 68 | 137 | 23 | ||
| Origin, n (%) | CNH | 35 (15.4) | 35 (25.5) | <0.0001 | ||
| Emory | 167 (73.2) | 47 (69.1) | 97 (70.8) | 23 (100.0) | ||
| UCSF | 26 (11.4) | 21 (30.9) | 5 (3.6) | |||
| Disease severity, n (%) | asymptomatic | 5 (2.2) | 5 (7.4) | |||
| mild | 5 (2.2) | 5 (7.4) | ||||
| moderate | 60 (26.3) | 21 (30.9) | 29 (28.5) | |||
| severe | 135 (59.2) | 37 (54.4) | 98 (71.5) | |||
| Age, mean (SD) | 11 (5) | 12 (6) | 9 (5) | 14 (2) | ||
| Sex, n (%) | female | 100 (43.9) | 37 (54.4) | 48 (35) | 15 (65.2) | 0.0028 |
| male | 128 (56.1) | 31 (45.6) | 89 (65.0) | 9 (34.8) | ||
| Race, n (%) | American Indian | 2 (0.9) | 2 (8.7) | 0.0005 | ||
| Asian | 9 (3.9) | 4 (5.9) | 1 (0.7) | 4 (17.4) | ||
| Black/African-American | 117 (51.3) | 28 (41.2) | 84 (61.3) | 5 (21.7) | ||
| White | 56 (24.6) | 16 (23.5) | 30 (21.9) | 10 (43.5) | ||
| Other/declined | 44 (19.3) | 20 (29.4) | 22 (16.1) | 2 (8.7) | ||
| Ethnicity, n (%) | Hispanic | 56 (24.6) | 22 (32.4) | 29 (21.2) | 5 (21.7) | 0.23 |
| non-Hispanic | 172 (75.4) | 46 (67.6) | 108 (78.8) | 18 (78.3) |
The p values were calculated using a Fisher’s exact test. See also Tables S1–S5.
Circulating cfRNA profiling
We performed transcriptome sequencing of plasma cfRNA on 129 samples from 121 pediatric patients. Of the 121 patients, 82 (68%) were classified as MIS-C, 27 (22%) as moderate to severe COVID-19, and 12 (10%) as negative controls (Figure 2A). We consistently observed significant increases in cfRNA from neutrophils, kidney epithelial cells, thymocytes, and solid-organ-derived cell types in moderate to severe COVID-19 compared with control individuals and further increased contributions from these cell types in MIS-C (p < 0.05 by Mann-Whitney U test) (Figures 2C and S1A). We also observed significant increases in cfRNA from endothelial cells and neuronal schwann cells specifically in children with MIS-C (p < 0.05 by Mann-Whitney U test) (Figures 2B and 2C). Platelet cfRNA was decreased in MIS-C and moderate to severe COVID-19 compared with donor controls. A comparison of cell type of origin (CTO) profiles between moderate and severe acute MIS-C did not yield any significant differences, likely because of challenges in classification of moderate versus severe MIS-C and the low number of samples (Figure S1B). We also did not observe significant differences between MIS-C and COVID-19 CTO fractions from gastrointestinally derived cell types but did observe significantly higher intestinal secretory cell--derived cfRNA in samples from patients with MIS-C versus controls (Figure S1A).
Figure 2.

Plasma cfRNA profiling
(A) Study design and analysis overview.
(B) Average cfRNA deconvolution results over various time points.
(C) cfRNA deconvolution results.
(D) Diversity of cell type contributions to the cell-free transcriptome.
(E) Dissimilarity of samples compared with controls.
(F) Scaled counts per million (CPM) values of significantly differentially abundant genes (DAGs) (DESeq2, Benjamini-Hochberg-adjusted p < 0.01, |Log2FoldChange| > 1.5). Samples and genes are clustered based on correlation.
(G) Normalized CPM values of TGM2 and the antiviral gene RSAD2 across sample groups at the acute time point.
(H) Cumulative CPM of genes in significant Gene Ontology groups (topGO, adjusted p < 0.05). Boxplots show cumulative CPM distribution of controls and acute time point MIS-C and moderate to severe COVID-19. Points represent average cumulative CPM, and bars represent standard error. Outliers are indicated with arrows and values. Asterisks indicate statistical significance by Mann-Whitney U test using Benjamini-Hochberg-adjusted p values as follows: ns, non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.001.
See also Figure S1 and Tables S2, S6, and Table S9. cfRNA COVID-19 vs. control differential abundance, related to Figure 2, Table S10. cfRNA MIS-C vs. control differential abundance, related to Figure 2, Table S11. cfRNA COVID-19 vs. MIS-C differential abundance, related to Figure 2.
Next, we sought to investigate longitudinal changes in cfRNA CTO in MIS-C. We observed significantly greater CTO diversity using the Simpson’s index14 in acute versus convalescent MIS-C (≥1 month post hospitalization) or controls (p < 0.05 by Mann-Whitney U test) (Figures 2B and 2D). In contrast, cfRNA CTO diversity was not significantly different for acute moderate to severe COVID-19 compared with controls or convalescent COVID-19 (p > 0.05 by Mann-Whitney U test) (Figures 2B and 2D). Samples from patients with acute moderate to severe COVID-19 separated into 2 groups, one group with high and one with low cfRNA CTO diversity, consistent with the previously described heterogeneity of cell types involved in COVID-19.15 We further compared the cfRNA CTO diversity of post-acute MIS-C and COVID-19. We found that samples from post-acute MIS-C patients had high cfRNA CTO diversity. while samples from post-acute moderate to severe COVID-19 patients had low cfRNA CTO diversity (Figures S1C and S1D). These trends were also observed using the Shannon diversity index.16 Next, we analyzed pairwise dissimilarity in cfRNA CTO between COVID-19 and MIS-C samples and controls. The Bray-Curtis dissimilarity of cfRNA CTO within controls was significantly lower than between controls and acute MIS-C (mean dissimilarity = 0.19 versus 0.56, p < 0.05 by Mann-Whitney U test) and between controls and acute moderate to severe COVID-19 (mean dissimilarity = 0.19 versus 0.38, p < 0.05 by Mann-Whitney U test) (Figure 2E). Among convalescent COVID-19 or MIS-C samples, only the COVID-19 1-month follow up cohort exhibited significant divergence of cfRNA CTO compared with controls (Figure 2E). Patients from 2 different hospitals (Emory and CNH) exhibited similar diversity and pairwise dissimilarity metrics (Figures 2D and 2E). These findings revealed that acute MIS-C exhibited a higher diversity of cell types than either COVID-19 or controls and that the observed cell types based on cfRNA were significantly different between MIS-C and COVID-19.
We identified differentially abundant genes (DAGs) in cfRNA associated with dead or dying cells to gain insight into disease pathogenesis and characterize potential diagnostic biomarkers.17 Patients in the discovery cohort (samples from Emory) diagnosed with acute MIS-C or moderate to severe COVID-19 were compared pairwise with controls. Using an absolute log2 fold change cutoff of 1.5 and a Benjamini-Hochberg adjusted p value of less than 0.01, 1,215 DAGs were identified between MIS-C and controls, 219 DAGs between COVID-19 and controls, and 102 DAGs between MIS-C and COVID-19 (Table S9. cfRNA COVID-19 vs. control differential abundance, related to Figure 2, Table S10. cfRNA MIS-C vs. control differential abundance, related to Figure 2, Table S11. cfRNA COVID-19 vs. MIS-C differential abundance, related to Figure 2). Unsupervised clustering revealed distinct gene expression profiles separating MIS-C and COVID-19 from controls, and incorporation of the validation cohort (samples from CNH) showed MIS-C samples clustering together regardless of hospital of origin (Figure 2F). We did not observe any impact of IVIg treatment or time relative to sample collection on sample clustering. Samples from acute MIS-C patients were assigned, using unsupervised clustering, into three groups, each with a distinct CTO profile (Figure S1E). The three groups consisted of cfRNA predominantly derived from (1) endothelial cells, NK cells, and respiratory ciliated cells; (2) monocytes, neutrophils, and myeloid progenitors; and (3) platelets. The third group clustered with samples from controls and COVID-19 patients and thus may represent a technical artifact associated with platelet lysis during sample preparation.
We analyzed significant DAGs between acute MIS-C and moderate to severe COVID-19. Acute MIS-C was associated with elevated levels of endothelial cell markers (AKAP12, CNN3, and FZD4), neuronal markers (GAS7, FEZ1, and VAT1), actin-related genes (FSCN1, AFAP1L1, and ITGA9), and an autoantigen also found in patients with celiac disease (TGM2) (Figures 2G and S1F). In contrast, acute COVID-19 was associated with elevated levels of IFN genes (IFI6, IFIT1, IFI44L, IFI27, and IFITM1), antiviral genes (RSAD2, MX1, CMPK2, and LY6E), chemokine genes (CXCL5 and CXCL3), and ciliated olfactory cell markers (OR2B6 and ENKUR) (Figures 2G and S1F). These trends in relative gene levels were observed in the discovery and validation cohorts. Next, we performed Gene Ontology analysis using the R package topGO.18 Gene Ontology terms enriched in COVID-19 patient samples included those associated with programmed cell death, response to viral infection, and regulation of the viral life cycle, while those enriched in MIS-C patients included actin cytoskeleton organization, endothelial cell migration, cytokine responses, and cell migration. To identify disease-specific pathways, we calculated counts per million (CPMs) for each Gene Ontology group by summing DAGs counts.19 Compared with controls, the cumulative CPM score for endothelial cell migration was significantly increased in acute MIS-C (p < 0.05 by Mann-Whitney U test), while scores for myeloid cell differentiation were increased in acute MIS-C and moderate to severe COVID-19 (p < 0.05 by Mann-Whitney U test) (Figure 2H). Control samples were associated with increased Gene Ontology groups related to cell division and cell communication, consistent with a baseline cfRNA signal predominantly derived from extracellular vesicles and cells undergoing apoptosis.20 Next, we performed pathway analysis using Ingenuity Pathway Analysis (IPA). Pathways enriched in MIS-C included pyroptosis, Th2 signaling, synaptogenesis, IL-6 signaling, IL-8 signaling, and interferon-associated antiviral responses, while pathways enriched in COVID-19 included macrophage production of nitric oxide, phosphatase and tensin homolog (PTEN) signaling, neutrophil extracellular trap signaling, HOX transcript antisense RNA (HOTAIR) regulation, and hypercytokinemia (Figures S1G and S1H).
wbRNA profiling by transcriptome sequencing (RNA sequencing [RNA-seq])
Whole-blood transcriptome profiling was performed on 217 samples from 187 pediatric patients, with 131 (63%) classified as MIS-C, 54 (26%) as moderate to severe COVID-19, and 23 (11%) as negative controls (Figure 3A). Samples from Emory and UCSF were used for the differential expression analysis; no batch effect based on hospital site was identified (Figure S2A). Samples from CNH were used as a validation group (Figures 3B and 3C). Using an absolute log2 fold change cutoff of 1.5 and a Benjamini-Hochberg adjusted p value of less than 0.01, 2,024 DAGs were identified between MIS-C and controls, 1,097 DAGs between COVID-19 and controls, and 84 DAGs between MIS-C and COVID-19 (Table S12. wbRNA COVID-19 vs. control differential expression, related to Figure 3, Table S13. wbRNA MIS-C vs. control differential expression, related to Figure 3, Table S14. wbRNA MIS-C vs. COVID-19 differential expression, related to Figure 3). Although we observed differences in gene expression between the discovery (Emory and UCSF) and validation (CNH) MIS-C cohorts (Figure 3D), likely because of differences in sample collection, MIS-C samples from both cohorts still clustered separate from controls, and did not group by origin when clustering with COVID-19, showing that the overall gene expression profiles were similar (Figures 3B, S2B, and S2C). Clustering was not affected by IVIg treatment or time relative to sample collection.
Figure 3.

wbRNA profiling
(A) Study design and analysis overview.
(B) Scaled CPM values of significantly differentially expressed genes (DEGs) (DESeq2, Benjamini-Hochberg-corrected p < 0.01, |Log2FoldChange| > 1.5). The total number of DEGs is indicated on the left of each heatmap. Samples and genes are clustered based on correlation.
(C) Scaled CPM values of the top 300 significantly expressed genes from each comparison (union of genes, DESeq2, Benjamini-Hochberg-adjusted p < 0.01, ranked by absolute Log2FoldChange). Samples and genes are clustered based on correlation.
(D) CPM of ADAMTS2, TRBV11-2, SIGLEC1, and KLRB1 in controls, acute MIS-C, and acute moderate to severe COVID-19. Points represent average CPM, and bars represent standard error. Asterisks indicate statistical significance by Mann-Whitney U test using Benjamini-Hochberg-adjusted p values as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.001.
(E) Top 30 differential pathways between controls and MIS-C or COVID-19 ranked by activation Z score. Lines connect matching pathways. Pathways highlighted in red are distinct to either MIS-C or COVID-19.
(F) Top 20 differential pathways between MIS-C and COVID-19 ranked by activation Z score.
See also Figures S2–S4 and Tables S3, S7, and Table S12. wbRNA COVID-19 vs. control differential expression, related to Figure 3, Table S13. wbRNA MIS-C vs. control differential expression, related to Figure 3, Table S14. wbRNA MIS-C vs. COVID-19 differential expression, related to Figure 3.
Pairwise comparisons of MIS-C and severe COVID-19 relative to controls showed a large degree of overlap in shared DEGs between MIS-C and COVID-19 (DESeq2, Benjamini-Hochberg-corrected p < 0.01, |Log2FoldChange| > 1.5) (Figures 3B, 3C, and S3A). Two of the top shared DEGs in both diseases were ADAMTS2, a metalloprotease that processes procollagen (Figures 3D and S3A), and CD177, a neutrophil activator (Figures S3A and S3B). Notably, ADAMTS2 has been implicated previously in severe COVID-19,21 whereas CD177 has been reported to be upregulated in MIS-C and COVID-19 patients.22 We also observed elevated levels of ADAMTS2 during the post-acute stage of MIS-C and COVID-19, with levels returning to baseline in children with MIS-C after 1 month but still elevated in those with COVID-19 (Figure S3C). Certain inflammatory genes, such as IFN-stimulated gene 15 (ISG15) and macrophage-associated sialic acid-binding Ig-like lectin 1 (SIGLEC1), were significantly upregulated in COVID-19 but not in MIS-C versus controls (Figures 3D and S3A). In contrast, TRBV11-2 was more highly expressed in MIS-C than in COVID-19 versus controls, which was also seen in a head-to-head comparison between MIS-C and COVID-19 (Figure 3D). This finding is consistent with two studies showing that TRBV11-2 is overexpressed by T cells in most MIS-C patients but not in patients with COVID-19, KD, or toxic shock syndrome (TSS).11,23 Differences in TRBV11-2 expression in MIS-C versus COVID-19 or controls were also observed at the post-acute time point, but not at the 1 month or 3 month or later time points (Figure S3C). Interestingly, expression of the gene paralogs KLRF1 and KLRB1, NK cell surface receptors, were found to be significantly decreased in COVID-19 and MIS-C compared with controls (Figures 3D and S3B), consistent with lower expression of these genes reported in severe COVID-19.24 Decreased KLRF1 and KLRB1 expression in COVID-19 and MIS-C was also observed at the post-acute time point but 1 month post-hospitalization returned to baseline in MIS-C but remained decreased in COVID-19 (Figure S3C). Top upregulated genes in COVID-19 compared with MIS-C were related to the antiviral type 1 IFN response pathway (e.g., IFIT2, SIGLEC1, IFI27, IFI44L, ISG15, and IFIT3) (Figure S3D; Table S14), while the top upregulated genes in MIS-C were related to multiple pathways, including those associated with cell-to-cell communication (e.g., ITGA7, CDHR1, CD177, PGF, ERFE, and MMP8) (Table S14). Rerunning the differential expression analysis using white blood cell count as a covariate yielded 3.4%, 9.6%, and 9.5% increases in DEG counts, respectively, when comparing MIS-C versus controls, COVID-19 versus controls, and MIS-C versus COVID-19 (Figures 3B and S3E). These results indicated that the differential wbRNA profiles observed were partially, but not fully, explained by differences in WBC count.
Next, we compared differentially expressed pathways in MIS-C and COVID-19 relative to controls (Figure 3E) and between each other (Figure 3F). In MIS-C and COVID-19 patients, we found increased activation of immune-related pathways, including phagosome formation, macrophage production of nitric oxide, and TREM1, IL-6, and IL-8 signaling. Pathways associated with cardiac hypertrophy and hepatic fibrosis were also upregulated in MIS-C and COVID-19. MIS-C patients showed a marked inhibition of T cell receptor, IL-2, IL-4, and focal adhesion kinase (FAK) signaling pathways, concurrent with activation of tumor environment and IL-1 signaling pathways. In contrast, COVID-19 patients showed more pronounced activation of cytokine, B cell, and adrenomedullin pathways (Figure 3F). We also observed striking differences in wbRNA pathways between MIS-C and COVID-19 compared with controls (Figure S4A). MIS-C was characterized by downregulation of inflammatory response, cell death and survival, cell-to-cell signaling, and immune cell trafficking pathways, whereas activation of these same pathways was predicted in COVID-19. These results are consistent with the downregulation of exhausted T cells that has been reported previously in children with MIS-C.25 cfRNA pathways also showed downregulation of cell-to-cell signaling, cellular movement, and immune cell trafficking pathways in MIS-C relative to COVID-19 (Figure S4B).
cfDNA methylation tissues-of-origin profiling
We performed whole-genome bisulfite sequencing of cfDNA extracted from plasma samples from 65 children with MIS-C (n = 41), COVID-19 (n = 21), or controls (n = 3) (Figure 4A) and compared the results with previously published cfDNA from an adult COVID-19 cohort.26 The highest mean levels of total cfDNA were found in children with MIS-C compared with pediatric COVID-19, adult COVID-19, or controls (4.12 ng/μL, p < 0.03 by Mann-Whitney U test) (Figure 4B). A subset of MIS-C patients (n = 3) showed a markedly elevated burden of cfDNA (>10 ng/μL), likely secondary to widespread tissue injury. There were also significantly higher mean levels of cfDNA in more severe COVID-19 pediatric and adult cases compared with adult nonsevere COVID-19 cases and controls (p < 0.05 by Mann-Whitney U test) (Figure 4B). The observations regarding cfDNA concentration in controls were consistent with previous research.26,27,28,29 Next, we compared total cfDNA abundance between samples with moderate or severe MIS-C and did not observe significant differences, likely because of limited sample sizes (Figure S5A).
Figure 4.

Plasma cfDNA TOOs by methylation profiling
(A) Study design and analysis overview.
(B) Total cfDNA concentration and solid-organ-derived cfDNA concentration.
(C) cfDNA concentration derived from innate and adaptive immune cell types.
(D) Abundance of mitochondrial derived cfDNA.
(E) Correlation of cfDNA metrics with standard clinical measurements, along with the Pearson correlation Benjamini-Hochberg-adjusted p value for each comparison.
For (B) and (C), bars display significance comparisons between the group farthest to the right on the bar and all other groups. For (B)–(D), asterisks indicate statistical significance by Mann-Whitney U test using Benjamini-Hochberg-adjusted p values as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.001. ASX, asymptomatic. See also Figure S5 and Tables S3 and S8.
We next examined the utility of cfDNA methylation tissue of origin (TOO) profiling to identify tissue damage by comparison with a reference set.26 We observed significantly elevated levels of solid-organ-derived cfDNA in pediatric acute MIS-C and pediatric moderate to severe COVID-19 compared with adult controls, pediatric acute mild or asymptomatic COVID-19, and adult mild to severe COVID-19 cases (p < 0.03, by Mann-Whitney U test) (Figure 4B). Levels of solid-organ-derived cfDNA in acute MIS-C were increased relative to acute moderate to severe COVID-19, although this difference was not significant (p = 0.12 by Mann-Whitney U test) (Figure 4B). We further observed elevated levels of cfDNA derived from innate immune cell types in acute MIS-C and moderate to severe pediatric COVID-19 compared to all other groups; however, this difference was also not significant (Figure 4C). In addition, we identified extensive heterogeneity in the TOO profiles from patients in the moderate to severe MIS-C cohort, including elevated levels of eosinophil-, neutrophil-, erythroblast-, liver-, heart-, kidney-, lung-, and spleen-derived cfDNA (Figure S5B). We did not observe differences in colon-derived cfDNA between COVID-19 and MIS-C (Figure S5C). Next, we quantified the concentration of mitochondrial cfDNA and observed a significant reduction of mitochondrial cfDNA in pediatric patients with MIS-C and COVID-19 (p < 0.04 by Mann-Whitney U test) (Figure 4D). Finally, we found that cfDNA metrics associated with affected tissues and organs mirrored organ-specific clinical laboratory parameters. Significant correlations between kidney cfDNA concentration and creatinine levels (Pearson correlation; R = 0.40, p = 0.004), total cfDNA concentration and C-reactive protein (CRP) levels in the blood (Pearson correlation; R = 0.29, p = 0.049), and liver cfDNA concentration and alanine transaminase (ALT) levels (Pearson correlation; R = 0.303, p = 0.036) were observed (Figure 4E).
Comparative analysis of plasma cfRNA and wbRNA
We compared wbRNA and cRNA profiles from paired samples (n = 86) (Figure 5A). First, we assessed the correlation of cfRNA and wbRNA for abundant genes (mean log-transformed CPM > 10) and identified significant correlations in 900 genes (Pearson coefficient, Benjamini-Hochberg-adjusted p < 0.05). Positive correlation was observed in 890 (99%) of 900 genes, predominated by genes associated with myeloid cell transcription, such as BNIP3L, HEMGN, and NFKBIA (Figures S6A and S6B). Randomized permutation testing (n = 1,000 permutations) using either randomly paired genes or samples yielded, on average, far fewer significantly correlated genes (mean = 194 and <1 with gene and sample randomization, respectively, at a Benjamini-Hochberg-adjusted p < 0.05) and decreased positive correlation (59% and 35% with gene and sample randomization, respectively), confirming the robustness of these findings.
Figure 5.

Comparison of paired wbRNA-seq and cfRNA-seq data
(A) Study design and analysis overview.
(B) Overlap of DAGs/DEGs between wbRNA-seq and cfRNA-seq using paired samples (DESeq2, Benjamini-Hochberg-adjusted p < 0.05, |Log2FoldChange| > 1). Venn diagrams represent the overlap of upregulated genes between analytes in each sample group, as indicated by fill color.
(C) Comparison of clustering topology between paired wbRNA and cfRNA samples. Samples are clustered based on correlation of DAGs/DEGs from their respective analyses. Lines between trees connect paired wbRNA and cfRNA samples. Correlation was calculated using Baker’s gamma, and p values were calculated using a Monte Carlo permutation test.
(D) Comparison of CTO diversity between cfRNA and wbRNA as calculated using Simpson’s index. Paired samples are connected with a line. Asterisks indicate statistical significance by Mann-Whitney U test using Benjamini-Hochberg-adjusted p values as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.001.
(E–G) CTO fractions, normalized to blood cell types and split by diagnosis group.
(H) CTO fractions from cfRNA and wbRNA samples split into low and high groups based on biomarker levels (CRP, 18 mg/L; WBC, 11 × 109/L; ALT, 36 U/L; ALB, 3.4 g/dL). Bars connect groups compared with a Mann-Whitney U test, and the numbers above are Benjamini-Hochberg-adjusted p values.
See also Figure S6.
Next, we examined the degree of overlap in DEGs/DAGs among patients with MIS-C (n = 31), moderate to severe COVID-19 (n = 12), or controls (n = 12) (Figure 5B). We observed a substantial overlap in DEGs/DAGs in wbRNA and cfRNA when comparing MIS-C (n = 579) or COVID-19 (n = 89) with controls but less overlap when comparing MIS-C with moderate to severe COVID-19 (n = 29). Of the 29 DEGs/DAGs that did overlap, several had been reported previously in COVID-19 (IFI6, IFI44L, RSAD2, LY6E, EPSTI1, XAF1, and MX1).30,31,32,33,34 Differential pathway analysis revealed minimal overlap in top enriched pathways between wbRNA and cfRNA (Figures S6C and S6D).
Next, we compared wbRNA and cfRNA DEGs/DAGs using unsupervised hierarchical sample clustering (Figure 5C). Samples clustered similarly in the MIS-C and COVID-19 versus control comparisons (Baker’s gamma index = 0.60 and 0.95, respectively; two-sided p < 0.01 for both; n = 43 and n = 24, respectively). The MIS-C versus COVID-19 comparison yielded clustering that was less similar but still significantly correlated (Baker’s gamma index = 0.22; two-sided p = 0.002, n = 43). The COVID-19 versus control comparison was unlikely to be an artifact from small sample size (n = 26) because differences between the originally calculated and bootstrapped Baker’s gamma index values for other comparisons were not significant (p = 0.85 for the MIS-C versus control comparison; p = 0.40 for the MIS-C versus COVID-19 comparison). Thus, despite the lack of overlap in DEGs/DAGs, cfRNA and wbRNA sample grouping by unsupervised clustering was similar for MIS-C and COVID-19 samples relative to controls, indicating that these two analytes provide complementary information.
We also compared cfDNA methylation and cfRNA gene count deconvolution results in available paired samples (n = 38; MIS-C, n = 32; COVID-19, n = 6). We compared fractions from similar tissue/cell types (n = 14 pairs; e.g., cfDNA liver with cfRNA intrahepatic cholangiocyte) and observed three pairs that were significantly correlated: (1) cfDNA and cfRNA B cells, (2) cfDNA erythroblast and cfRNA erythrocyte/erythroid progenitor cells, and (3) cfDNA liver and cfRNA intrahepatic cholangiocytes (Pearson correlation; uncorrected p < 0.02, Benjamini-Hochberg-corrected p < 0.11) (Figure S6E). Correlations between all other tissue/cell type pairs were not significant (unadjusted p values ranging from 0.28–0.88).
Next, we compared the CTOs in wbRNA and cfRNA between paired samples. We observed significantly different wbRNA and cfRNA CTO diversity in acute moderate to severe COVID-19 patients and controls (p = 0.034 and p = 0.001, respectively, by paired Wilcoxon test) (Figure 5D). However, wbRNA and cfRNA CTO diversity were not significantly different in acute MIS-C (p = 0.36 by paired Wilcoxon test) (Figure 5D). Furthermore, in unpaired analyses, we did not observe a significant difference in wbRNA CTO diversity between acute moderate to severe COVID-19 and controls or acute MIS-C and controls (Mann-Whitney U Test, Benjamini-Hochberg-adjusted p < 0.05) (Figure S6F). Conversely, we observed a significant difference in cfRNA CTO diversity in acute MIS-C and controls but not in acute moderate to severe COVID-19 and controls (Mann-Whitney U test, Benjamini-Hochberg-adjusted p < 0.01 and 0.17, respectively) (Figure S6D). Finally, we compared patterns of blood-derived CTOs in paired wbRNA and cfRNA samples from patients with MIS-C, patients with acute moderate to severe COVID-19, and controls (Figure 5E). The wbRNA and cfRNA CTO profiles corresponding to MIS-C and COVID-19 (Figure 5E), characterized by multiple blood cell types, were distinct from the control group profiles, which consisted of a single predominant baseline cell type (erythroid precursors in wbRNA and platelets in cfRNA) (Figure 5E). Within each disease group, wbRNA and cfRNA CTO profiles were also distinct. For COVID-19, wbRNA CTO profiles had higher proportions of neutrophils, NK cells, T cells, and monocytes, whereas cfRNA CTO profiles had lower proportions of these inflammatory cells (Figure 5E). In contrast, for MIS-C, wbRNA CTO profiles were predominated by neutrophils, whereas cfRNA CTO profiles had relatively higher proportions of myeloid progenitor cells, NK cells, and monocytes (Figure 5E).
Last, we compared cfRNA and wbRNA CTO profiles with common laboratory biomarkers, including CRP, white blood cell (WBC) count, ALT, and albumin (ALB) levels. For each biomarker, we separated samples into a “low” and “high” group based on established cutoffs and the distribution of values in our patient cohort (CRP, 18 mg/L; WBC, 11 × 109/L; ALT, 36 U/L; ALB, 3.4 g/dL). We observed elevated levels of endothelium-derived cfRNA in the high-CRP group, decreased platelet derived wbRNA in the high-WBC group, increased levels of intrahepatic cholangiocyte-derived cfRNA in the high-ALT group, and decreased levels of kidney epithelium-derived cfRNA in the low-ALB group (Figure 5F). These findings are consistent with the corresponding TOO for each of these laboratory markers.
Discussion
Here, we report a systems-level, longitudinal analysis of COVID-19 and MIS-C by next-generation sequencing of nucleic acids (cfRNA, wbRNA, and cfDNA) in a large multihospital study of 402 blood samples from 228 patients. Using plasma cfRNA profiling, we identify signatures associated with cellular injury and death that distinguish MIS-C and COVID-19 as well as the involvement of previously unreported cell types in MIS-C. wbRNA analysis reveals substantial overlap in pro-inflammatory pathways between MIS-C and COVID-19 but also reveals pathways that are specific to each disease state. Plasma cfDNA methylation profiling suggests increased cfDNA and solid organ involvement in MIS-C compared with COVID-19 and controls. Comparative analyses of paired cfRNA and wbRNA samples demonstrate that these analytes yield separate but complementary signatures associated with MIS-C and COVID-19 that reflect distinct CTOs. These results provide novel insights into the differential pathogenesis of MIS-C and COVID-19. They also lay the groundwork for the development of minimally invasive gene-expression-based diagnostic tests that can differentiate MIS-C from other hyperinflammatory states, including septic shock, KD, severe COVID-19 with systemic involvement, and TSS. They also underscore the potential utility of cfRNA and cfDNA biomarkers for evaluating tissue injury and monitoring recovery. Thus, diagnostic tests based on cell-free and cell-associated nucleic acids may, in the future, yield clinically actionable data that would expedite appropriate treatment and improve patient outcomes.
wbRNA sequencing (RNA-seq) of cellular RNA has traditionally been considered the gold-standard method for assaying gene expression in the blood. However, the signal from wbRNA is primarily derived from leukocytes because of sampling of circulating cells, thus directly measuring a patient’s inflammatory and immune response to an infection. In contrast, plasma cfRNA and methylated cfDNA measure levels and types of cell death from circulating cells and peripheral tissues.35,36 cfDNA enables quantification of cells that are turning over or dying, whereas cfRNA enables characterization of the gene expression pathways associated with these cells.37,38,39 cfRNA profiling leverages the extensive information available from recent large-scale human cell atlas projects, whereas reference data for cfDNA methylation profiling are more limited.36 Overall, these cell-associated (wbRNA) and cell-free (cfRNA and cfDNA) approaches complement each other. When combined, they provide a more complete picture of the dynamic, “yin-yang” interplay between host and pathogen or between cell activation or proliferation and cell death.
cfRNA is an analyte that probes cellular death and immune dynamics on a systems level. Previous analyses of MIS-C and COVID-19 from the blood have relied on single-cell or bulk RNA-seq of whole blood cells, which primarily characterizes the host immune response, or on proteomics- and cytokine-based assays, which generally use a limited number of markers or markers for which there is a lack of standardized reference data. In contrast, the signals from cfRNA are derived from any cell or vascularized tissue type, and there is a plethora of RNA-seq reference data that can be used to interpret results. Consistent with prior studies, here we observe increased levels of cfRNA from endothelial cells in MIS-C40 and from neutrophils and thymocytes in MIS-C and COVID-197,41,42 as well as increased signaling from disease-specific pathways in MIS-C (IL-6, IL-8, and Th2)8,11 and COVID-19 (nitric oxide production, PTEN, and neutrophil extracellular net signalling).43,44,45 However, the cfRNA data also uncover several previously undescribed features of MIS-C, such as enrichment of neuronal genes associated with synaptogenesis and increased cfRNA burden from Schwann cells. These findings suggest that peripheral nervous system damage may be a common feature of MIS-C. Interestingly, central and peripheral nervous system involvement in MIS-C has been described previously described,46,47 although overt clinical manifestations are infrequent. Our identification of potential gene markers of neuronal damage in MIS-C patients is also consistent with previous research that reports elevated levels of circulating spike protein in MIS-C patients11,48,49 and identifies neurotoxin-like regions in the spike protein of SARS-CoV-2 that can bind T cell receptors and mediate neurotoxicity.50 Of note, peripheral nervous system damage has also been documented in pediatric and adult COVID-19 and in post-acute sequelae such as long COVID.51,52,53 Thus, it is possible that the differences in neuronal involvement that we observe between MIS-C and COVID-19 may reflect differences in the timing of sample collection relative to SARS-CoV-2 infection rather than being a MIS-C-specific characteristic. Future studies are needed to elucidate the mechanisms and clinical spectrum of neurologic involvement in acute MIS-C and their association with long-term neurodevelopment. In addition, here we observe an enrichment of genes associated with the pyroptosis pathway in MIS-C and likely related to inflammasome activation.54 Pyroptosis is a form of rapid cellular death that occurs during highly inflammatory states.55 In KD, a systemic inflammatory syndrome similar to MIS-C, pyroptosis occurs in vascular endothelial cells.56 Taken together, these data support the likely critical role of pyroptosis and endothelial cells in MIS-C pathogenesis and may explain the overlapping clinical presentations between MIS-C and KD in acutely ill pediatric patients.
wbRNA-seq reveals a high degree of overlap in shared, largely pro-inflammatory genes and pathways between COVID-19 and MIS-C. This is expected because both diseases are caused by SARS-CoV-2 and are highly inflammatory states. However, different levels of expression are observed for certain genes, such as upregulation of ISG15 and SIGLEC1 in COVID-19 and upregulation of TRBV11-2 in MIS-C, as well as for certain pathways, such as inhibition of T cell receptor, IL-2, and FAK signaling pathways in MIS-C. The latter finding is consistent with the reported T cell exhaustion associated with downregulation of NK and CD8+ T cells in MIS-C.25 Genes showing differences in levels of gene expression (e.g., ISG15, SIGLEC1, TRBV11-2, and CREB3L) or persistence of gene expression (e.g., ADAMTS2 and KLRFB1) may be useful target biomarkers to discriminate among MIS-C, COVID-19, and other hyperinflammatory conditions.
Like cfRNA, cfDNA methylation profiling allows monitoring and quantification of tissue injury and cell death in a minimally invasive manner from the blood. Previous studies have shown that the relative concentrations of cfDNA in specific tissues vary in different disease states, including COVID-19 in adults, solid-organ transplant rejection, graft versus host disease following stem cell transplantation, urinary tract infection, and cancer.26,28,57,58,59 cfDNA methylation profiling can also be used to estimate TOOs. Here we observe an increase in cell death and high levels of heterogeneity in TOOs in MIS-C compared with COVID-19 and controls, consistent with the systemic inflammation and multi-organ involvement typically associated with MIS-C. We also report a decrease in mitochondrial cfDNA in moderate to severe MIS-C or COVID-19, in contrast to recently published studies that report increases in mitochondrial cfDNA using quantitative PCR (qPCR).60,61 We believe that this discrepancy may be explained by differences in measurement methodologies; qPCR estimates are largely based on measurement of long mitochondrial DNA molecules in plasma, whereas sequencing-based estimates are based on measurement of short mitochondrial DNA molecules. We also report a comparison (n = 38) of cfRNA and cfDNA from paired samples in MIS-C and COVID-19 and find only 3 of 14 paired cfRNA cell type/cfDNA tissue fractions to be positively correlated. There are several potential explanations for the limited correlations: (1) the cfRNA reference is derived from single-cell RNA-seq (scRNA-seq) data, whereas the cfDNA methylation reference is based on bulk whole-genome sequencing; (2) the amount of cfDNA released from a cell is fairly constant across cell types, whereas the amount of cfRNA is dependent on cell volume; and (3) the amount of cfRNA and its stability is greatly affected by the cellular excretion or cell death pathway (apoptosis, pyroptosis, necrosis, etc.) from which it is derived, resulting in potentially higher variability in cfRNA measurements.
By longitudinal sampling of cfRNA and wbRNA, we observe in patients with MIS-C and COVID-19 that most, but not all, gene measurements return to baseline levels by 1 month post hospitalization. Elevated ADAMTS2 and KLRB wbRNA levels, for instance, return to baseline in MIS-C but not in COVID-19 at 1 month post hospitalization. These findings may be related to post-acute sequelae of SARS-CoV-2 infection (“long COVID”) that have been postulated to be caused by persistent immune dysregulation.62 In contrast, despite the severity of the initial presentation, most clinical and laboratory abnormalities from MIS-C resolve quickly within a few weeks, along with normalization of inflammatory and injury biomarkers.63 In cfRNA, we found that most biomarker measurements, such as CTO values and gene module scores, persist at 1 month but return to baseline 3 months post hospitalization or later, consistent with the generally accepted time frames of recovery after MIS-C.64
Finally, we report a large-scale comparison (n = 86) of wbRNA and cfRNA profiles from paired samples in MIS-C and COVID-19. A previous study comparing paired wbRNA and plasma cfRNA from healthy individuals was limited by a small sample size (n = 3) and lack of a disease group for comparison.65 Our results reveal distinct, largely nonoverlapping sets of DAGs/DEGs associated with MIS-C and COVID-19 in wbRNA and cfRNA. Thus, both analytes provide complementary information in distinguishing MIS-C and COVID-19 from controls and from each other. These findings are consistent with the origin of wbRNA and cfRNA, with wbRNA being primarily derived from active immune cells in the blood and cfRNA from dying cells from the blood and peripheral tissues. They are also consistent with our CTO data showing predominantly platelets in cfRNA and erythrocytes in wbRNA and a greater diversity of cell types from peripheral tissues represented in cfRNA compared with wbRNA. Notably, we observed correlations between cfRNA and wbRNA levels and levels of organ-specific biomarkers, highlighting the utility of cfRNA in measuring tissue damage and wbRNA in measuring circulating cell dynamics.
Limitations of the study
This study has some limitations. First, sample sizes from asymptomatic to mild COVID-19 cases (cfDNA, n = 10), longitudinally collected samples (wbRNA, n = 61; cfRNA, n = 42), and controls (cfDNA, n = 3) were limited. Second, the accuracy of our deconvolution analyses was likely dependent on the reference set used as a comparator. The cfDNA deconvolution reference set consists of a limited number of methylomes. The cfRNA/wbRNA deconvolution reference set is based on polyadenylated mRNA transcripts, whereas here we used host ribosomal RNA depletion to enrich for transcripts. Third, we had very few samples from MIS-C “look-alike” inflammatory conditions (e.g., septic shock, KD, TSS, macrophage activation syndrome, etc.). Fourth, the demographic distribution of patients within the various cohorts was not uniformly distributed. Previous studies have shown that non-Hispanic Black children are at a higher risk of MIS-C compared with other children infected with COVID-19,66 suggesting differential immune responses, although phenotype comparisons identified no racial or ethnic differences.67 Further studies incorporating additional samples and representative sampling are needed to address these limitations.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Plasma and whole blood samples from patients diagnosed with COVID-19 or MIS-C and pediatric healthy controls | - Emory University School of Medicine and Children’s Healthcare of Atlanta - University of California, San Francisco - Children’s National Hospital |
N/A |
| Critical commercial assays | ||
| Norgen Plasma/Serum Circulating and Exosomal RNA Purification Mini Kit | Norgen | 51000 |
| DNase Turbo kit | Invitrogen | AM2238 |
| Baseline Zero DNase | Lucigen-Epicenter | DB0715K |
| Zymo RNA Clean and Concentrate | Zymo | R1015 |
| Takara SMARTer® Stranded Total RNA-Seq Kit v2 | Takara | 634418 |
| Qubit dsDNA HS Assay Kit | Ivitrogen | Q32854 |
| Agilent HS NGS Fragment Kit | Agilent | DNF-474-0500 |
| Qiagen Circulating Nucleic Acid Kit | Qiagen | 55114 |
| Zymo EZ Methylation-Gold Kit (for bisulfite conversion) | Zymo | D5005 |
| RNA/DNA Shield | Zymo | R1200 |
| Quick-RNA Whole Blood Kit | Zymo | R1201 |
| Qubit RNA HS Assay Kit | Initrogen | Q32852 |
| NEBNext rRNA Depletion Kit (Human/Mouse/Rat) | NEB | E6310 |
| NEBNext Ultra II Directional RNA Library Prep Kit | NEB | E7760 & E7765 |
| NEBNext Multiplex Oligos | NEB | E6609L |
| NEBNext Sample Purification Beads | NEB | E7103 |
| Agencourt AMPure XP | Beckman Coulter | A63880 |
| Deposited data | ||
| Gene count and Methylation data | This manuscript | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE225223 |
| GRCh38 | Gencode | https://www.gencodegenes.org/human/release_38.html |
| Tabula Sapiens scRNA-seq Atlas | Tabula Sapiens Consortium | https://tabula-sapiens-portal.ds.czbiohub.org |
| Cell-free DNA Methylation data | Cheng et al., 202126 | https://www.github.com/alexpcheng/cfDNAme |
| Oligonucleotides | ||
| cfDNA concentration control 5′- TTTAAC GCATAAACATGCGTTTTGGGTAGTGTTT TTTGGAAACACAGATCCGTGCGCACAC CTGGTGGAG-3′ |
Integrated DNA Technologies | https://www.idtdna.com/pages |
| cfDNA concentration control 2 5′- ATAAAC ATGCGTTTTGGGTAGTGTTTTTTGGAAA CACAGATCCGTGCGCACACCT-3′ |
Integrated DNA Technologies | https://www.idtdna.com/pages |
| cfDNA concentration control 3 5′- GCGTTT TGGGTAGTGTTTTTTGGAAACACAGATCC GTGCG-3′ |
Integrated DNA Technologies | https://www.idtdna.com/pages |
| cfDNA concentration control 4 5′-GGTAGT GTTTTTTGGAAACACAGAT-3′ |
Integrated DNA Technologies | https://www.idtdna.com/pages |
| Software and algorithms | ||
| Custom Scripts | This manuscript | https://github.com/conorloy/Loy-etal_CellRepMed |
| Snakemake | Molder et al., 202168 | https://snakemake.readthedocs.io/en/stable/ |
| BBDUK | Bushnell et al., 201769 | https://jgi.doe.gov/data-and-tools/software-tools/bbtools/bb-tools-user-guide/bbduk-guide/ |
| STAR | Dobin et al., 201370 | https://github.com/alexdobin/STAR |
| featureCounts | Liao et al., 201471 | https://subread.sourceforge.net |
| GATK | McKenna et al., 201072 | https://gatk.broadinstitute.org/hc/en-us/articles/360037052812-MarkDuplicates-Picard- |
| Samtools | Li et al., 200973 | https://samtools.github.io |
| Qualimap | Fernando García-Alcalde et al., 201274 | http://qualimap.conesalab.org |
| Qiagen Ingenuity Pathway Analysis (v73620684) | Qiagen | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ |
| R Statistical Computing Software | The R Foundation | https://www.r-project.org/ |
| R package BayesPrism | Chu et al., 202275 | https://github.com/Danko-Lab/BayesPrism |
| R package vegan | Oksanen et al., 200776 | https://cran.r-project.org/web/packages/vegan/index.html |
| R package Tidyverse | Wickman et al., 201977 | https://www.tidyverse.org/packages/ |
| R package DESeq2 | Love et al., 201417 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Python programming language | Python Software Foundation | https://www.python.org |
| Python Package ScanPy | ScanPy78 | https://scanpy.readthedocs.io |
| Other | ||
| Illumina NextSeq 500 | Illumina | https://illumina.com |
| Illumina NovaSeq | Illumina | https://illumina.com |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Charles Chiu (charles.chiu@ucsf.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Ethics statement
The protocols for this study were approved locally at each site by the University of California, San Francisco (UCSF) Institutional Review Board (IRB) (#21–33403), San Francisco, CA; Emory University IRB (STUDY00000723), Atlanta, GA; Children’s National Medical Center IRB (Pro00010632), Washington, DC; and Cornell University IRB for Human Participants (2012010003), New York, NY. The protocols at UCSF and Children’s National Medical Center were “no subject contact” archival biobanking protocols under which samples were processed and data extracted from the medical chart with waiver of consent. The protocol at Emory University IRB was a prospective enrollment study under which parents provided consent and children provided assent as appropriate for age. De-identified samples and patient information were shared with collaborating institutions for sample processing (UCSF and Cornell University) and analysis.
Human subjects
Pediatric patients, both male and female, who tested positive for COVID-19, were diagnosed with MIS-C, or were healthy outpatient controls were enrolled in this study. Demographic information and sample counts were reported in the main text Tables 1 and S2–S4.
Method details
Sample acquisition
As part of a “no subject contact” study with waiver of consent, IRB approval was obtained from UCSF and Children’s National Hospital for archival biobanking of residual patient samples from routine clinical testing and RNA and DNA profiling analyses. At UCSF, pediatric hospitalized patients who tested positive and negative for COVID-19 were identified from SARS-CoV-2 real-time PCR (RT-PCR) results from the UCSF Clinical Laboratories daily. Residual whole blood samples were collected in EDTA lavender top tubes and processed within 12–72 h. 250 μL of sample was aliquoted with 250 μL of 2x DNA/RNA shield (Zymo Research) for a 1:1 ratio. The remaining blood was centrifuged at 2500 rpm for 15 min and the available plasma was obtained. Samples were properly identified and added to the biobanking registry. All samples were stored at −80°C freezer until used. At Children’s National Hospital, patients with MIS-C were identified by a multidisciplinary task force according to the CDC case definition. Residual whole blood samples from this population were identified, collected, and processed 12–72 h after collection. Samples were centrifuged at 1300 xG for 5 min at room temperature. Plasma was aliquoted into a cryovial and frozen at −80°C. A DMSO-based cryopreservative (CryostorⓇ CS10) was added in a 1:1 ratio to the cell pellet and then frozen at −80°C in a controlled rate freezing container (i.e., Mr. Frosty). After freezing the pellets with Cryostor they were transferred to liquid nitrogen cryostorage within 1 week.
At Emory and Children’s Healthcare of Atlanta, pediatric patients with COVID-19, MIS-C, or controls were enrolled into a specimen collection protocol following informed consent and assent, as appropriate for age. For this study, patients were classified as having MIS-C if they met the CDC case definition, and as having COVID-19 if they had any PCR-confirmed SARS-CoV-2 infection. Controls were healthy outpatients with no known history of COVID-19 who volunteered for specimen collection. The specimen collection protocol was approved by the Emory University IRB. Residual whole blood and plasma samples were retrieved from the clinical laboratory and processed within 72 h of collection, and prospective blood samples were additionally collected in EDTA lavender top tubes and processed within 4 h of collection. Longitudinal samples were also collected at 1-month and ≥3-month timepoints for participants who returned for follow-up. From the EDTA tubes, whole blood was aliquoted, and the remaining blood was centrifuged at 2500 rpm for 15 min to obtain the available plasma. All samples were de-identified and assigned study IDs. Samples were stored at −80°C and shipped on dry ice to either UCSF or Cornell for analysis.
Characteristics of the sample cohort
Of the 402 samples analyzed, 218 (54%) were from patients who received intravenous immunoglobulin (IVIg), of which the sample was collected after IVIg treatment in 183 (83%). cfRNA and cfDNA methylation profiling by next-generation sequencing (NGS) were performed from plasma, and transcriptome RNA profiling (RNA-Seq) was performed from whole blood. Due to limited sample volume, cfRNA, cfDNA, and wbRNA profiling were each performed on different numbers of samples. Furthermore, samples from patients with asymptomatic or mild COVID-19 were only used in the cfDNA analyses to compare to a previously published adult COVID-19 dataset. The demographic breakdown of the samples used for each analyte can be found in the supplemental material (Tables S2–S4). For the cfRNA and wbRNA profiling, samples from Emory were used as a discovery cohort and samples from CNH as a validation cohort.
Clinical data
For the purposes of this study, MIS-C was defined as any patient who met the CDC case definition.3 Multidisciplinary teams which adjudicated whether a patient met the case definition of MIS-C. COVID-19 was defined as any patient with PCR-confirmed SARS-CoV-2 infection within the preceding 14 days who did not also meet the MIS-C case definition. Clinical data was abstracted from the medical record and entered into a shared REDCap79,80 database housed at UCSF.
Cell-free RNA sample processing
Plasma samples were received on dry ice and stored at −80°C until processed. Prior to extraction, plasma was thawed at room temperature and spun at 1300xg for 10 min at 4°C. The supernatant was taken and cfRNA was isolated from plasma (115–1000 μL) using the Norgen Plasma/Serum Circulating and Exosomal RNA Purification Mini Kit (51000, Norgen). Extracted RNA was DNase treated with 14 μL of 10 μL DNase Turbo Buffer (AM2238, Invitrogen), 3 μL DNase Turbo (AM2238, Invitrogen), 1μL Baseline Zero DNase (DB0715K, Lucigen-Epicenter) for 30 min at 37°C and then concentrated into 12 μL using the Zymo RNA Clean and Concentrate Kit (R1015, Zymo).
Sequencing libraries were constructed from 8 μL of concentrated RNA using the Takara SMARTer Stranded Total RNA-Seq Kit v2 – Pico Input Mammalian (634418, Takara). Briefly, extracted RNA was reverse transcribed using random priming, barcoded using the SMARTer RNA Unique Dual Index Kit (634451, Takara), rRNA depleted, and further amplified. Library concentration was quantified using a Qubit 3.0 Fluorometer (Q33216, Invitrogen) with the dsDNA HS Assay Kit (Q32854, Invitrogen). Libraries were quality-controlled using an Agilent Fragment Analyzer 5200 (M5310AA, Agilent) with the HS NGS Fragment kit (DNF-474-0500, Agilent). Libraries were pooled to equal concentrations and sent to the Cornell Genomics core for 150-base pair, paired-end sequencing on an Illumina NextSeq550 machine for an average of 10 million reads per sample.
Cell-free DNA sample processing
Plasma samples were received on dry ice and stored at −80°C until processed. Prior to extraction, plasma samples (75–650 μL) were thawed at room temperature and spun at 1300xg for 10 min at 4°C. The supernatant was taken and cfDNA was isolated from plasma using the Qiagen Circulating Nucleic Acid Kit (55114, Qiagen) and eluted to 45 μL.
Bisulfite treatment was peformed and sequencing libraries were constructed from 20 μL of extracted DNA using a single-stranded library preparation.26 Library concentration was quantified using a Qubit 3.0 Fluorometer (Q33216, Invitrogen) with the dsDNA HS Assay Kit (Q32854, Invitrogen). Libraries were quality-controlled using an Agilent Fragment Analyzer 5200 (M5310AA, Agilent) with the HS NGS Fragment kit (DNF-474-0500, Agilent). Libraries were pooled to equal concentrations and sent to the Cornell Genomics core for 150-base pair, paired-end sequencing on an Illumina NextSeq550 machine for an average of 33 million reads per sample.
Whole blood RNA sample processing
Whole blood samples were received on dry ice and stored at −80°C until processed. Before extraction, all samples were thawed and pretreated with a 1:1 ratio of 2X RNA/DNA Shield (R1200, Zymo Research) if this was not added prior to freezing. RNA was extracted from whole blood samples (400 μL) using the Quick-RNA Whole Blood kit (R1201, Zymo Research) following manufacturer’s instructions. Ribosomal depletion was not performed. RNA was eluted in 15 μL of RNase-free water and stored at −80°C until use. The concentration of eluted RNA was measured using a Qubit Flex Fluorometer (Q33326, Invitrogen) with the RNA HS Assay Kit (Q32852, Invitrogen). RNA Integrity was assessed on a subset of samples using the Agilent Bioanalyzer RNA 6000 Nano/Pico Chip (5067–4626) to determine the RNA Integrity Number (RIN). All samples analyzed were partially degraded with RIN values between 2 and 5.
Extracted RNA (7 μL or 10–200 ng) was processed using the NEBNext rRNA Depletion Kit (Human/Mouse/Rat) (E6310, NEB) and the NEBNext Ultra II Directional RNA Library Prep Kit (E7760 & E7765, NEB) following manufacturer’s specifications. Briefly, samples were first treated for ribosomal RNA depletion and DNase digestion, fragmented for 8 min, and reverse transcribed. Adapters were ligated to the purified cDNA (1:25 diluted adaptor), followed by library amplification and barcoding using NEBNext Multiplex Oligos (E6609L, NEB) sets 1 to 4. Libraries were purified using NEBNext Sample Purification Beads (E7103, NEB).
Libraries were quantified using the Qubit Flex (Q33327, Invitrogen) with the dsDNA HS Assay Kit (Q32854, Invitrogen). Libraries were pooled and sent to the UCSF Center of Advanced Technology (CAT) for sequencing on an Illumina NovaSeq 6000 Sequencing System using 150-base pair paired-end sequencing. Negative controls (nuclease-free water) were included in every run to monitor for contamination.
RNA bioinformatic processing
Sequencing data was processed using a custom bioinformatics pipeline utilizing the Snakemake workflow management system (v7.7.0) for the cfRNA samples and a bash script for the wbRNA samples. Samples were quality filtered and trimmed using BBDUK (v38.90), aligned to the Gencode GRCh38 human reference genome (v38, primary assembly) using STAR (v2.7.0f) default parameters, and features quantified using featureCount (v2.0.0). cfRNA samples were also deduplicated using Picard MarkDuplicates prior to feature quantification (v2.19.2). Mitochondrial, ribosomal, X, and Y chromosome genes were removed prior to analysis.
RNA sample quality filtering
wbRNA and cfRNA samples were filtered using different quality control metrics due to differences in RNA concentration and quality. wbRNA samples with less than 10% of reads aligning to the transcriptome were removed from all analyses. cfRNA samples were filtered on the basis of DNA contamination, rRNA contamination, number of counts, and RNA degradation. DNA contamination was estimated by calculating the ratio of reads mapping to introns and exons. Samples with an intron to exon ratio above three were removed. rRNA contamination was measured using Samtools (v1.14). Total counts were calculated using featureCounts. Degradation was estimated by calculating the 5–3′ bias as calculated by Qualimap (v2.2.1). Samples with rRNA contamination, total counts, or 5–3′ bias greater than three standard deviations from the mean were removed. Also, samples with fewer than 75,000 total counts were removed. Quality control metrics are recorded in the Tables S6 and S7.
RNA cell deconvolution and diversity
Cell type deconvolution was performed using BayesPrism (V1.1) with the Tabula Sapiens single cell RNA-seq atlas (Release 1) as a ref. 75,81. Cells from the Tabula Sapiens atlas were grouped as previously described in Vorperian et al.36 Cell types with more than 100,000 unique molecular identifiers (UMIs) were included in the reference and subsampled to 300 cells using ScanPy (v1.8.1). Providing an equal number of cells to the deconvolution ensured an unbiased prior for the Bayesian algorithm used in BayesPrism. Cell-type contribution diversity metrics were calculated using the vegan R package (v2.5.7).
RNA differential expression analysis
Comparative analysis of DEGs was performed using a negative binomial model as implemented in the DESeq2 package (wbRNA: v1.28.1, cfRNA: v1.34.0) using a Benjamini-Hochberg corrected p value cutoff <0.01, unless otherwise stated. Heatmaps were constructed using the pheatmap package in R (v1.0.12), samples and genes were clustered using correlation based hierarchical clustering. Gene ontology analysis was performed using the topGO R package (v2.46.0)19 using a Benjamini-Hochberg-corrected p value cutoff of <0.05. Cumulative CPM values for gene ontology terms were calculated by taking the sum of normalized counts from all the significant genes in each gene ontology module. Canonical pathways, diseases and functions were analyzed using QIAGEN Ingenuity Pathway Analysis (IPA) software (v90348151). Pathway analysis was run twice: (1) with all available samples and (2) with samples that have both whole blood and cell-free data available.
RNA paired sample correlation analysis
Log CPM normalized RNA counts were correlated between paired cfRNA and wbRNA samples. Genes with a low average expression (<10 CPM) in either cfRNA or wbRNA were removed. Samples with extreme cfRNA or wbRNA counts (CPM Z score >3) were removed to eliminate outlier bias. A Pearson correlation was calculated for each gene and a Benjamini-Hochberg–corrected p value cutoff <0.05 was used to determine significance.
We performed two permutation tests (n = 1000 permutations) to determine the reliability of the number of genes observed to be correlated. First, we randomly shuffled the sample labels and recalculated the total number of significant genes. Second, we randomly shuffled gene labels and recalculated the total number of significant correlations.
Entanglement analysis
Differential abundance/expression analysis was performed using only paired cfRNA and wbRNA samples as previously described, using a Benjamini-Hochberg–corrected p value cutoff <0.05. Samples were clustered using correlation based hierarchical clustering. Using the dendextend package in R (v1.15.2), dendrograms were plotted, paired samples were connected by lines, and Baker’s gamma correlation coefficient was calculated. Non-exact, two-sided p values were calculated using a Monte Carlo permutation test.
DNA concentration
Eluted DNA was quantified using a Qubit 3.0 Fluorometer (Q33216, Invitrogen) with the dsDNA HS Assay Kit (Q32854, Invitrogen). Total cfDNA concentration was estimated using the following formula:
DNA bioinformatic processing
Sequencing data was processed using a custom bioinformatics pipeline utilizing the Snakemake workflow management system (v7.7.0). Samples were quality filtered and trimmed using BBDUK (v38.46), aligned to the Gencode GRCh38 human reference genome (v38, primary assembly) and deduplicated using Bismark (v0.22.1) with default parameters, and quality filtered using samtools (v1.14). Prior to any analysis, X and Y chromosome mapped reads were removed.
DNA sample quality filtering
cfDNA samples were filtered on the basis of sequencing depth (>0.1), mapping efficiency (>0.5), and bisulfite conversion efficiency (>0.97). Sequencing depth, mapping efficiency, bisulfute conversion efficancy, and cfDNA concentration are recorded in Table S8.
DNA deconvolution
DNA tissues of origin deconvolution were performed as previously described.26 Briefly, a custom bioinformatic pipeline utilizing the Snakemake workflow management system (v7.7.0) was used to process publicly available methylation references, convert to a standard data format, and normalize across samples. Metline (v0.2-7) was used to discover differentially methylated regions and a quadratic programming algorithm estimated relative contributions of the tissues represented in the methylation references. Tissue specific cfDNA concentration was calculated for each sample by multiplying the estimated fractional contribution from each tissue by the cfDNA concentration, calculated using the formula shown above. Similarly, mitochondrial cfDNA concentration was calculated for each sample by multiplying the fraction of mitochondrial aligned reads by the cfDNA concentration.
Data visualization
Figures were created using Qiagen IPA, Adobe Illustrator, Affinity Designer, and BioRender.com software.
Quantification and statistical analysis
All statistical analyses were performed using R (cfDNA: v4.1.0, cfRNA: v4.1.0, wbRNA: v4.0.3). Data wrangling and visualization was performed using Python (3.9.1), Pandas (1.3.0) matplotlib-venn (0.11.6), R (v4.1.0), Tidyverse (v1.3.1), and ggplot2 (v3.3.5). Statistical significance was tested using Wilcoxon signed-rank tests and Mann-Whitney U tests in a two-sided manner, unless otherwise stated. All sequencing data was aligned to the GRCh38 Gencode v38 Primary Assembly and features counted using the GRCh38 Gencode v38 Primary Assembly Annotation.
Acknowledgments
We would like to acknowledge staff members at the UCSF Clinical Laboratories and the UCSF Clinical Microbiology Laboratories for help with identifying and retrieving patient whole-blood samples. We thank the Cornell Genomics Center and the UCSF Center for Advanced Technology for help with sequencing libraries. At Emory, we thank Christopher Choi, Caroline Ciric, Khalel De Castro, Theda Gibson, Hui-Mien Hsiao, Wensheng Li, Austin Lu, Lisa Macoy, Kathy Stephens, Madeline Taylor, Ashley Tippett, and the Children’s Healthcare of Atlanta Research Laboratory for contributions to specimen and data collection. We thank the patients and their families for contributing blood to further our understanding of pediatric COVID-19 and MIS-C. This work was supported by National Institutes of Health (NIH)/National Institute of Child Health and Human Development (NICHD) grant R61HD105618 (to R.D., C.A.R., I.D.V., and C.Y.C.) and CDC contract award 75D30122C14367 (to C.Y.C.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
C.J.L., R.L.D., C.A.R., I.D.V., and C.Y.C. conceived and designed the study. C.J.L., A.S.-G., V.S., J.L., A.P.C., and A.B. performed sequencing experiments. J.N., J.L., M.E.W., P.S., N.B., J.S., W.S., C.J.H., N.W., A.F., A.G. K.B., M.A.P., L.H., E.J.A., A.C., M.D., R.L.D., C.A.R., and C.Y.C. identified and collected patient samples and clinical metadata. C.J.L., A.S.-G., V.S., B.B., M.D., R.L.D., C.A.R., I.D.V., and C.Y.C. analyzed sequencing data. M.D., A.B., R.L.D., C.A.R., I.D.V., and C.Y.C. supervised the study. C.J.L., V.S., S.B., I.D.V., and C.Y.C. wrote the manuscript and prepared the figures. All authors read and edited the manuscript and agreed to its contents.
Declaration of interests
I.D.V. is a member of the Scientific Advisory Board of Karius Inc., Kanvas Biosciences, and GenDX. C.Y.C. is a founder of Delve Bio and a member of the Scientific Advisory Boards of Delve Bio, Poppy Health, BiomeSense, and Mammoth Biosciences. A.P.C. is listed as an inventor on submitted patents pertaining to cfDNA (US patent applications 63/237,367, 63/056,249, 63/015,095, and 16/500,929) and receives consulting fees from Eurofins Viracor. C.A.R.’s institution has received funding to conduct clinical research unrelated to this manuscript from BioFire Inc., GSK, MedImmune, Micron, Merck, Novavax, PaxVax, Regeneron, Pfizer, and Sanofi-Pasteur. She is a co-inventor of patented RSV vaccine technology (International PCT Application No. PCT/US2016/058976, filed 12/28/2016 by Emory University), which has been licensed to Meissa Vaccines, Inc. with royalties received. Her institution has received funding from the NIH to conduct clinical trials of Moderna and Janssen COVID-19 vaccines. E.J.A has consulted for Pfizer, Sanofi Pasteur, GSK, Janssen, and Medscape, and his institution receives funds to conduct clinical research unrelated to this manuscript from MedImmune, Regeneron, PaxVax, Pfizer, GSK, Merck, Sanofi-Pasteur, Janssen, and Micron. He also serves on a safety monitoring board for Kentucky BioProcessing, Inc. and Sanofi Pasteur. He serves on a data adjudication board for WCG and ACI Clinical. His institution has also received funding from the NIH to conduct clinical trials of Moderna and Janssen COVID-19 vaccines. A.B. is a co-founder and consultant to Personalis and NuMedii; consultant to Mango Tree Corporation, and in the recent past, Samsung, 10× Genomics, Helix, Pathway Genomics, and Verinata (Illumina); has served on paid advisory panels or boards for Geisinger Health, Regenstrief Institute, Gerson Lehman Group, AlphaSights, Covance, Novartis, Genentech, Merck, and Roche; is a shareholder in Personalis and NuMedii; is a minor shareholder in Apple, Meta (Facebook), Alphabet (Google), Microsoft, Amazon, Snap, 10× Genomics, Illumina, Regeneron, Sanofi, Pfizer, Royalty Pharma, Moderna, Sutro, Doximity, BioNtech, Invitae, Pacific Biosciences, Editas Medicine, Nuna Health, Assay Depot, and Vet24seven, and several other non-health related companies and mutual funds; and has received honoraria and travel reimbursement for invited talks from Johnson and Johnson, Roche, Genentech, Pfizer, Merck, Lilly, Takeda, Varian, Mars, Siemens, Optum, Abbott, Celgene, AstraZeneca, AbbVie, Westat, and many academic institutions, medical or disease-specific foundations and associations, and health systems. A.B. receives royalty payments through Stanford University for several patents and other disclosures licensed to NuMedii and Personalis. A.B.’s research has been funded by the NIH, Peraton (as the prime on an NIH contract), Genentech, Johnson and Johnson, the FDA, the Robert Wood Johnson Foundation, the Leon Lowenstein Foundation, the Intervalien Foundation, Priscilla Chan and Mark Zuckerberg, the Barbara and Gerson Bakar Foundation, and in the recent past the March of Dimes, the Juvenile Diabetes Research Foundation, the California Governor’s Office of Planning and Research, the California Institute for Regenerative Medicine, L’Oréal, and Progenity. The authors have declared that none of these companies or competing interests had any role in this work or manuscript.
Published: April 21, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101034.
Contributor Information
Iwijn De Vlaminck, Email: id93@cornell.edu.
Charles Y. Chiu, Email: charles.chiu@ucsf.edu.
Supplemental information
Data and code availability
-
•
The raw sequencing data reported in this study cannot be deposited in a public repository due to patient privacy concerns. De-identified RNA-seq count matrices and DNA methylation matrices have been uploaded to the NCBI (National Center for Biotechnology Information) GEO (Gene Expression Omnibus) database (GSE225223) and are publicly available. DOIs are listed in the key resources table.
-
•
Additional Supplemental Items are available from Mendeley Data at http://dx.doi.org/10.17632/5np8gym63j.1.
-
•
All original code has been depositied on GitHub: https://github.com/conorloy/Loy-etal_CellRepMed and is publicaly available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Woodruff R.C., Campbell A.P., Taylor C.A., Chai S.J., Kawasaki B., Meek J., Anderson E.J., Weigel A., Monroe M.L., Reeg L., et al. Risk factors for severe COVID-19 in children. Pediatrics. 2022;149 doi: 10.1542/peds.2021-053418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi D.S., Whitaker M., Marks K.J., Anglin O., Milucky J., Patel K., Pham H., Chai S.J., Kawasaki B., Meek J., et al. Hospitalizations of children aged 5–11 Years with laboratory-confirmed COVID-19 — COVID-NET, 14 states, March 2020–february 2022. MMWR Morb. Mortal. Wkly. Rep. 2022;71:574–581. doi: 10.15585/MMWR.MM7116E1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Information for Healthcare providers about multisystem inflammatory syndrome in children (MIS-C) | CDC. https://www.cdc.gov/mis/mis-c/hcp/index.html
- 4.Verdoni L., Mazza A., Gervasoni A., Martelli L., Ruggeri M., Ciuffreda M., Bonanomi E., D’Antiga L. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet. 2020;395:1771–1778. doi: 10.1016/S0140-6736(20)31103-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Godfred-Cato S., Abrams J.Y., Balachandran N., Jaggi P., Jones K., Rostad C.A., Lu A.T., Fan L., Jabbar A., Anderson E.J., et al. Distinguishing multisystem inflammatory syndrome in children from COVID-19, kawasaki disease and toxic shock syndrome. Pediatr. Infect. Dis. J. 2022;41:315–323. doi: 10.1097/inf.0000000000003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Consiglio C.R., Cotugno N., Sardh F., Pou C., Amodio D., Rodriguez L., Tan Z., Zicari S., Ruggiero A., Pascucci G.R., et al. The immunology of multisystem inflammatory syndrome in children with COVID-19. Cell. 2020;183:968–981.e7. doi: 10.1016/j.cell.2020.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gruber C.N., Patel R.S., Trachtman R., Lepow L., Amanat F., Krammer F., Wilson K.M., Onel K., Geanon D., Tuballes K., et al. Mapping systemic inflammation and antibody responses in multisystem inflammatory syndrome in children (MIS-C) Cell. 2020;183:982–995.e14. doi: 10.1016/j.cell.2020.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porritt R.A., Binek A., Paschold L., Rivas M.N., McArdle A., Yonker L.M., Alter G., Chandnani H.K., Lopez M., Fasano A., et al. The autoimmune signature of hyperinflammatory multisystem inflammatory syndrome in children. J. Clin. Invest. 2021;131 doi: 10.1172/JCI151520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carter M.J., Fish M., Jennings A., Doores K.J., Wellman P., Seow J., Acors S., Graham C., Timms E., Kenny J., et al. Peripheral immunophenotypes in children with multisystem inflammatory syndrome associated with SARS-CoV-2 infection. Nat. Med. 2020;26:1701–1707. doi: 10.1038/s41591-020-1054-6. [DOI] [PubMed] [Google Scholar]
- 10.Ramaswamy A., Brodsky N.N., Sumida T.S., Comi M., Asashima H., Hoehn K.B., Li N., Liu Y., Shah A., Ravindra N.G., et al. Immune dysregulation and autoreactivity correlate with disease severity in SARS-CoV-2-associated multisystem inflammatory syndrome in children. Immunity. 2021;54:1083–1095.e7. doi: 10.1016/j.immuni.2021.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sacco K., Castagnoli R., Vakkilainen S., Liu C., Delmonte O.M., Oguz C., Kaplan I.M., Alehashemi S., Burbelo P.D., Bhuyan F., et al. Immunopathological signatures in multisystem inflammatory syndrome in children and pediatric COVID-19. Nat. Med. 2022;28:1050–1062. doi: 10.1038/s41591-022-01724-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porritt R.A., Paschold L., Rivas M.N., Cheng M.H., Yonker L.M., Chandnani H., Lopez M., Simnica D., Schultheiß C., Santiskulvong C., et al. HLA class I–associated expansion of TRBV11-2 T cells in multisystem inflammatory syndrome in children. J. Clin. Invest. 2021;131 doi: 10.1172/JCI146614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh V., Obregon-Perko V., Lapp S.A., Horner A.M., Brooks A., Macoy L., Hussaini L., Lu A., Gibson T., Silvestri G., et al. Limited induction of SARS-CoV-2–specific T cell responses in children with multisystem inflammatory syndrome compared with COVID-19. JCI Insight. 2022;7 doi: 10.1172/jci.insight.155145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simpson E.H. Measurement of diversity. Nature. 1949;163:688. doi: 10.1038/163688a0. [DOI] [Google Scholar]
- 15.Suratannon N., Dik W.A., Chatchatee P., Hagen P.M.v. COVID-19 in children: heterogeneity within the disease and hypothetical pathogenesis. Asian Pac. J. Allergy Immunol. 2020;38:170–177. doi: 10.12932/ap-170720-0920. [DOI] [PubMed] [Google Scholar]
- 16.Shannon C.E. The mathematical theory of communication. MD Comput. 1963;14:306–317. [PubMed] [Google Scholar]
- 17.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/S13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexa A., Rahnenfuhrer J. 2020. topGO: Enrichment Analysis for Gene Ontology. [Google Scholar]
- 19.Rasmussen M., Reddy M., Nolan R., Camunas-Soler J., Khodursky A., Scheller N.M., Cantonwine D.E., Engelbrechtsen L., Mi J.D., Dutta A., et al. RNA profiles reveal signatures of future health and disease in pregnancy. Nature. 2022;601:422–427. doi: 10.1038/s41586-021-04249-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pös O., Biró O., Szemes T., Nagy B. Circulating cell-free nucleic acids: characteristics and applications. Eur. J. Hum. Genet. 2018;26:937–945. doi: 10.1038/s41431-018-0132-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y., Wang S., Xia H., Guo J., He K., Huang C., Luo R., Chen Y., Xu K., Gao H., et al. Identification of monocytes associated with severe COVID-19 in the PBMCs of severely infected patients through single-cell transcriptome sequencing. Engineering (Beijing) 2022;17:161–169. doi: 10.1016/J.ENG.2021.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang C.A., Huang Y.L., Chiang B.L. Innate immune response analysis in COVID-19 and kawasaki disease reveals MIS-C predictors. J. Formos. Med. Assoc. 2022;121:623–632. doi: 10.1016/J.JFMA.2021.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moreews M., le Gouge K., Khaldi-Plassart S., Pescarmona R., Mathieu A.L., Malcus C., Djebali S., Bellomo A., Dauwalder O., Perret M., et al. Polyclonal expansion of TCR Vbeta 21.3 + CD4 + and CD8 + T cells is a hallmark of multisystem inflammatory syndrome in children. Sci. Immunol. 2021;6 doi: 10.1126/SCIIMMUNOL.ABH1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalfaoglu B., Almeida-Santos J., Tye C.A., Satou Y., Ono M. T-cell hyperactivation and paralysis in severe COVID-19 infection revealed by single-cell analysis. Front. Immunol. 2020;11:589380. doi: 10.3389/fimmu.2020.589380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beckmann N.D., Comella P.H., Cheng E., Lepow L., Beckmann A.G., Tyler S.R., Mouskas K., Simons N.W., Hoffman G.E., Francoeur N.J., et al. Downregulation of exhausted cytotoxic T cells in gene expression networks of multisystem inflammatory syndrome in children. Nat. Commun. 2021;12:4854. doi: 10.1038/s41467-021-24981-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng A.P., Cheng M.P., Gu W., Sesing Lenz J., Hsu E., Schurr E., Bourque G., Bourgey M., Ritz J., Marty F.M., et al. Cell-free DNA tissues of origin by methylation profiling reveals significant cell, tissue, and organ-specific injury related to COVID-19 severity. Med. 2021;2:411–422.e5. doi: 10.1016/j.medj.2021.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng A.P., Burnham P., Lee J.R., Cheng M.P., Suthanthiran M., Dadhania D., de Vlaminck I. A cell-free DNA metagenomic sequencing assay that integrates the host injury response to infection. Proc. Natl. Acad. Sci. USA. 2019;116:18738–18744. doi: 10.1073/pnas.1906320116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng A.P., Cheng M.P., Loy C.J., Lenz J.S., Chen K., Smalling S., Burnham P., Timblin K.M., Orejas J.L., Silverman E., et al. Cell-free DNA profiling informs all major complications of hematopoietic cell transplantation. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/PNAS.2113476118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salvi S., Gurioli G., de Giorgi U., Conteduca V., Tedaldi G., Calistri D., Casadio V. Cell-free DNA as a diagnostic marker for cancer: current insights. OncoTargets Ther. 2016;9:6549–6559. doi: 10.2147/OTT.S100901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu L., Yang P., Zhao Y., Zhuang Z., Wang Z., Song R., Zhang J., Liu C., Gao Q., Xu Q., et al. Single-cell sequencing of peripheral mononuclear cells reveals distinct immune response landscapes of COVID-19 and influenza patients. Immunity. 2020;53:685–696.e3. doi: 10.1016/J.IMMUNI.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfaender S., Mar K.B., Michailidis E., Kratzel A., Boys I.N., V’kovski P., Fan W., Kelly J.N., Hirt D., Ebert N., et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat. Microbiol. 2020;5:1330–1339. doi: 10.1038/S41564-020-0769-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao X., Liu Y., Zou S., Liu P., Zhao J., Yang C., Liang M., Yang J. Genome-wide screening of SARS-CoV-2 infection-related genes based on the blood leukocytes sequencing data set of patients with COVID-19. J. Med. Virol. 2021;93:5544–5554. doi: 10.1002/JMV.27093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaath H., Vishnubalaji R., Elkord E., Alajez N.M. Single-cell transcriptome analysis highlights a role for neutrophils and inflammatory macrophages in the pathogenesis of severe COVID-19. Cells. 2020;9 doi: 10.3390/CELLS9112374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karami H., Derakhshani A., Ghasemigol M., Fereidouni M., Miri-moghaddam E., Baradaran B., Tabrizi N.J., Najafi S., Solimando A.G., Marsh L.M., et al. Weighted gene co-expression network analysis combined with machine learning validation to identify key modules and hub genes associated with sars-cov-2 infection. J. Clin. Med. 2021;10 doi: 10.3390/JCM10163567/S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehmann-Werman R., Neiman D., Zemmour H., Moss J., Magenheim J., Vaknin-Dembinsky A., Rubertsson S., Nellgård B., Blennow K., Zetterberg H., et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. USA. 2016;113:E1826–E1834. doi: 10.1073/PNAS.1519286113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vorperian S.K., Moufarrej M.N., Tabula Sapiens Consortium, Quake S.R., Krasnow M., Pisco A.O., Quake S.R., Salzman J., Yosef N., Bulthaup B., et al. Cell types of origin of the cell-free transcriptome. Nat. Biotechnol. 2022;40:855–861. doi: 10.1038/S41587-021-01188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fox-Fisher I., Piyanzin S., Ochana B.L., Klochendler A., Magenheim J., Peretz A., Loyfer N., Moss J., Cohen D., Drori Y., et al. Remote immune processes revealed by immune-derived circulating cell-free DNA. Elife. 2021;10 doi: 10.7554/ELIFE.70520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y., Li J., Zhang L., Sun H.X., Zhang Z., Xu J., Xu Y., Lin Y., Zhu A., Luo Y., et al. Plasma cell-free RNA characteristics in COVID-19 patients. Genome Res. 2022;32:228–241. doi: 10.1101/GR.276175.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan W., Ngo T.T.M., Camunas-Soler J., Song C.X., Kowarsky M., Blumenfeld Y.J., Wong R.J., Shaw G.M., Stevenson D.K., Quake S.R. Simultaneously monitoring immune response and microbial infections during pregnancy through plasma cfRNA sequencing. Clin. Chem. 2017;63:1695–1704. doi: 10.1373/CLINCHEM.2017.273888. [DOI] [PubMed] [Google Scholar]
- 40.Diorio C., Shraim R., Vella L.A., Giles J.R., Baxter A.E., Oldridge D.A., Canna S.W., Henrickson S.E., McNerney K.O., Balamuth F., et al. Proteomic profiling of MIS-C patients indicates heterogeneity relating to interferon gamma dysregulation and vascular endothelial dysfunction. Nat. Commun. 2021;12:7222. doi: 10.1038/S41467-021-27544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vella L.A., Giles J.R., Baxter A.E., Oldridge D.A., Diorio C., Kuri-Cervantes L., Alanio C., Pampena M.B., Wu J.E., Chen Z., et al. Deep immune profiling of MIS-C demonstrates marked but transient immune activation compared to adult and pediatric COVID-19. Sci. Immunol. 2021;6 doi: 10.1126/SCIIMMUNOL.ABF7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reusch N., de Domenico E., Bonaguro L., Schulte-Schrepping J., Baßler K., Schultze J.L., Aschenbrenner A.C. Neutrophils in COVID-19. Front. Immunol. 2021;12:652470. doi: 10.3389/FIMMU.2021.652470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ibrahim I.H., Ellakwa D.E.S. SUMO pathway, blood coagulation and oxidative stress in SARS-CoV-2 infection. Biochem. Biophys. Rep. 2021;26:100938. doi: 10.1016/J.BBREP.2021.100938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarma A., Christenson S.A., Byrne A., Mick E., Pisco A.O., DeVoe C., Deiss T., Ghale R., Zha B.S., Tsitsiklis A., et al. Tracheal aspirate RNA sequencing identifies distinct immunological features of COVID-19 ARDS. Nat Commun. 2021;12:5152. doi: 10.1038/s41467-021-25040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu Y., Chen X., Liu X. NETosis and Neutrophil Extracellular Traps in COVID-19: Immunothrombosis and Beyond. Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.838011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdel-Mannan O., Eyre M., Löbel U., Löbel U., Eltze C., Hameed B., Hemingway C., Hacohen Y. Neurologic and radiographic findings associated with COVID-19 infection in children. JAMA Neurol. 2020;77:1440–1445. doi: 10.1001/jamaneurol.2020.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Larovere K.L., Riggs B.J., Poussaint T.Y., Young C.C., Newhams M.M., Maamari M., Walker T.C., Singh A.R., Dapul H., Hobbs C.v., et al. Neurologic involvement in children and adolescents hospitalized in the United States for COVID-19 or multisystem inflammatory syndrome. JAMA Neurol. 2021;78:536–547. doi: 10.1001/JAMANEUROL.2021.0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zollner A., Koch R., Jukic A., Pfister A., Meyer M., Rössler A., Kimpel J., Adolph T.E., Tilg H. Postacute COVID-19 is characterized by gut viral antigen persistence in inflammatory bowel diseases. Gastroenterology. 2022;163:495–506.e8. doi: 10.1053/j.gastro.2022.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Natarajan A., Zlitni S., Brooks E.F., Vance S.E., Dahlen A., Hedlin H., Park R.M., Han A., Schmidtke D.T., Verma R., et al. Gastrointestinal symptoms and fecal shedding of SARS-CoV-2 RNA suggest prolonged gastrointestinal infection. Med (N Y) 2022;3:371–387.e9. doi: 10.1016/j.medj.2022.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng M.H., Zhang S., Porritt R.A., Noval Rivas M., Paschold L., Willscher E., Binder M., Arditi M., Bahar I. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Proc. Natl. Acad. Sci. USA. 2020;117:25254–25262. doi: 10.1073/pnas.2010722117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patti R., De Araujo Duarte C., Dalsania N., gonuguntla V.T., atuaka C., De Brito Gomes B., Somal N., Seneviratne C. Post-infectious demyelinating polyneuropathy secondary to COVID-19 infection. Chest. 2021;160:A658–A659. doi: 10.1016/J.CHEST.2021.07.626. [DOI] [Google Scholar]
- 52.Feldstein L.R., Tenforde M.W., Friedman K.G., Newhams M., Rose E.B., Dapul H., Soma V.L., Maddux A.B., Mourani P.M., Bowens C., et al. Characteristics and outcomes of US children and adolescents with multisystem inflammatory syndrome in children (MIS-C) compared with severe acute COVID-19. JAMA. 2021;325:1074–1087. doi: 10.1001/JAMA.2021.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balbi P., Saltalamacchia A., Lullo F., Fuschillo S., Ambrosino P., Moretta P., Lanzillo B., Maniscalco M. Peripheral neuropathy in patients recovering from severe COVID-19: a case series. Medicina (Kaunas) 2022;58:523. doi: 10.3390/MEDICINA58040523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vora S.M., Lieberman J., Wu H. Inflammasome activation at the crux of severe COVID-19. Nat. Rev. Immunol. 2021;21:694–703. doi: 10.1038/S41577-021-00588-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bergsbaken T., Fink S.L., Cookson B.T. Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 2009;7:99–109. doi: 10.1038/NRMICRO2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jia C., Zhang J., Chen H., Zhuge Y., Chen H., Qian F., Zhou K., Niu C., Wang F., Qiu H., et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. 2019;10:778. doi: 10.1038/S41419-019-2021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chabon J.J., Hamilton E.G., Kurtz D.M., Esfahani M.S., Moding E.J., Stehr H., Schroers-Martin J., Nabet B.Y., Chen B., Chaudhuri A.A., et al. Integrating genomic features for non-invasive early lung cancer detection. Nature. 2020;580:245–251. doi: 10.1038/S41586-020-2140-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burnham P., Dadhania D., Heyang M., Chen F., Westblade L.F., Suthanthiran M., Lee J.R., de Vlaminck I. Urinary cell-free DNA is a versatile analyte for monitoring infections of the urinary tract. Nat. Commun. 2018;9:2412. doi: 10.1038/S41467-018-04745-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Vlaminck I., Valantine H.A., Snyder T.M., Strehl C., Cohen G., Luikart H., Neff N.F., Okamoto J., Bernstein D., Weisshaar D., et al. Circulating cell-free DNA enables noninvasive diagnosis of heart transplant rejection. Sci. Transl. Med. 2014;6:241ra77. doi: 10.1126/SCITRANSLMED.3007803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andargie T.E., Tsuji N., Seifuddin F., Jang M.K., Yuen P.S., Kong H., Tunc I., Singh K., Charya A., Wilkins K., et al. Cell-free DNA maps COVID-19 tissue injury and risk of death and can cause tissue injury. JCI Insight. 2021;6 doi: 10.1172/JCI.INSIGHT.147610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scozzi D., Cano M., Ma L., Zhou D., Zhu J.H., O’Halloran J.A., Goss C., Rauseo A.M., Liu Z., Sahu S.K., et al. Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight. 2021;6 doi: 10.1172/jci.insight.143299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phetsouphanh C., Darley D.R., Wilson D.B., Howe A., Munier C.M.L., Patel S.K., Juno J.A., Burrell L.M., Kent S.J., Dore G.J., et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 2022;23:210–216. doi: 10.1038/S41590-021-01113-X. [DOI] [PubMed] [Google Scholar]
- 63.Fremed M.A., Farooqi K.M. Longitudinal outcomes and monitoring of patients with multisystem inflammatory syndrome in children. Front. Pediatr. 2022;10:820229. doi: 10.3389/FPED.2022.820229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Multisystem Inflammatory Syndrome in Children (MIS-C): Information for Healthcare Providers about Talking with Families and Caregivers https://www.cdc.gov/mis/mis-c/hcp/provider-families.html.
- 65.Ibarra A., Zhuang J., Zhao Y., Salathia N.S., Huang V., Acosta A.D., Aballi J., Toden S., Karns A.P., Purnajo I., et al. Non-invasive characterization of human bone marrow stimulation and reconstitution by cell-free messenger RNA sequencing. Nat. Commun. 2020;11:400. doi: 10.1038/S41467-019-14253-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stierman B., Abrams J.Y., Godfred-Cato S.E., Oster M.E., Meng L., Yip L., Patel P., Balachandran N., Prezzato E., Pierce T., et al. Racial and ethnic disparities in multisystem inflammatory syndrome in children in the United States, March 2020 to february 2021. Pediatr. Infect. Dis. J. 2021;40:e400–e406. doi: 10.1097/INF.0000000000003294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DeBiasi R.L., Harahsheh A.S., Srinivasalu H., Krishnan A., Sharron M.P., Parikh K., Smith K., Bell M., Michael D., Delaney M., et al. Multisystem inflammatory syndrome of children: subphenotypes, risk factors, biomarkers, cytokine profiles, and viral sequencing. J. Pediatr. 2021;237:125–135.e18. doi: 10.1016/j.jpeds.2021.06.002. [DOI] [PubMed] [Google Scholar]
- 68.Mölder F., Jablonski K.P., Letcher B., Hall M.B., Tomkins-Tinch C.H., Sochat V., Forster J., Lee S., Twardziok S.O., Kanitz A., et al. Sustainable data analysis with Snakemake. F1000Res. 2021;10:33. doi: 10.12688/f1000research.29032.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bushnell B., Rood J., Singer E. BBMerge - accurate paired shotgun read merging via overlap. PLoS One. 2017;12 doi: 10.1371/journal.pone.0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 72.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.García-Alcalde F., Okonechnikov K., Carbonell J., Cruz L.M., Götz S., Tarazona S., Dopazo J., Meyer T.F., Conesa A. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics. 2012;28:2678–2679. doi: 10.1093/bioinformatics/bts503. [DOI] [PubMed] [Google Scholar]
- 75.Chu T., Wang Z., Pe’er D., Danko C.G. Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat. Cancer. 2022;3:505–517. doi: 10.1038/s43018-022-00356-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oksanen J., Blanchet G.F., Friendly M., Kindt R., Legendre P., McGlinn D., Minchin P.R., O’Hara R.B., Simpsom G.L., Solymos P., et al. 2020. Vegan: Community Ecology PAckage. [Google Scholar]
- 77.Wickham H., Averick M., Bryan J., Chang W., McGowan L., François R., Grolemund G., Hayes A., Henry L., Hester J., et al. Welcome to the Tidyverse. J. Open Source Softw. 2019;4:1686. doi: 10.21105/joss.01686. [DOI] [Google Scholar]
- 78.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harris P.A., Taylor R., Minor B.L., Elliott V., Fernandez M., O’Neal L., McLeod L., Delacqua G., Delacqua F., Kirby J., et al. The REDCap consortium: building an international community of software platform partners. J. Biomed. Inform. 2019;95:103208. doi: 10.1016/J.JBI.2019.103208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Harris P.A., Taylor R., Thielke R., Payne J., Gonzalez N., Conde J.G. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 2009;42:377–381. doi: 10.1016/J.JBI.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tabula Sapiens Consortium∗, Jones R.C., Karkanias J., Krasnow M.A., Pisco A.O., Quake S.R., Salzman J., Yosef N., Bulthaup B., Brown P., et al. The Tabula Sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022;376 doi: 10.1126/science.abl4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The raw sequencing data reported in this study cannot be deposited in a public repository due to patient privacy concerns. De-identified RNA-seq count matrices and DNA methylation matrices have been uploaded to the NCBI (National Center for Biotechnology Information) GEO (Gene Expression Omnibus) database (GSE225223) and are publicly available. DOIs are listed in the key resources table.
-
•
Additional Supplemental Items are available from Mendeley Data at http://dx.doi.org/10.17632/5np8gym63j.1.
-
•
All original code has been depositied on GitHub: https://github.com/conorloy/Loy-etal_CellRepMed and is publicaly available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.