

Abstract
Immunotherapy is a powerful technique where immune cells are modified to improve cytotoxicity against cancerous cells to treat cancers that do not respond to surgery, chemotherapy, or radiotherapy. Expressing chimeric antigen receptor (CAR) in immune cells, typically T lymphocytes, is a practical modification that drives an immune response against cancerous tissue. CAR-T efficacy is suboptimal in solid tumors due to the tumor microenvironment (TME) that limits T lymphocyte cytotoxicity. In this study, we demonstrate that neutrophils differentiated from human pluripotent stem cells modified with AAVS1-inserted CAR constructs showed a robust cytotoxic effect against prostate-specific membrane antigen (PSMA) expressing LNCaP cells as a model for prostate cancer in vitro. Our results suggest that engineered CAR can significantly enhance the neutrophil anti-tumor effect, providing a new avenue in treating prostate cancers.
Keywords: Chimeric antigen receptor, Neutrophils, Cancer immunotherapy, Human pluripotent stem cells
Graphical Abstract
Introduction
Prostate cancer (PCa) is the 4th most common cancer worldwide, with 1.4 million cases in 2020 and an estimated number of deaths exceeding 320 thousand1. It is the most common cancer-associated death for men and commonly progresses into a castration-resistant variant with significant potential to metastasize. Metastatic, castration-resistant PCa (mCRPC) is the most advanced form of the disease, and patient outcomes and quality of life are low2. The prostate-specific membrane antigen (PSMA), an integral membrane protein expressed uniquely in prostate tissue, is upregulated in PCa tissue3,4. Monoclonal antibody treatment against PSMA can reduce PCa tumor advancement5.
T-lymphocytes expressing a chimeric antigen receptor (CAR) against tumor surface antigens have effectively targeted cancerous tissue. The receptor is a synthetic protein that combines a unique extracellular receptor domain with an invariable transmembrane and intracellular activation domain6–11. In theory, CARs can be designated to recognize any extracellular epitope and induce T-cell activation.
Despite promising to treat blood malignancy, CAR-T therapy suffers from several drawbacks, including allograft rejection response against the introduced cells, over-activity through cytokine storms, and potential graft versus host disease. Furthermore, solid tumors create a local anti-inflammatory zone using physical barriers, anti-inflammatory cytokines, small molecules, and tumor-recruited immune cells12–14. Inside the solid tumor microenvironment (TME), CAR-T response can be dampened below the threshold to eliminate the cancerous tissue. Several methods are attempted to remove these limitations, such as metabolic engineering15, adding inflammatory cytokines (TRUCKs)16, localized delivery of CAR-T, and blocking immunosuppressive signals from TME using antibody treatments14. An alternative approach to target solid tumors is to use other immune cells, specifically those with better ability to infiltrate solid tissue, such as macrophages (Mac) and natural killer (NK) cells. CAR-Mac has been shown in mouse models to reduce HER2+ breast cancer tumor burden and stimulate an immune response by T cell crosstalk17. Additionally, CAR-NK cells exhibit a strong cytotoxic effect on murine prostate cancer models18. The preclinical success of CAR-Mac/NK against solid tumors has led us to assess the capacity of neutrophils as a CAR-immune cell19.
From a clinical standpoint, using a patient’s peripheral immune cells for CAR therapy poses a significant time investment, and current viral genetic editing methods have limitations in immune cells. A potentially more clinically applicable solution is to produce “off the shelf” CAR-modified immune cells from specified progenitors or pluripotent stem cells. To provide “off the shelf” CAR-neutrophils, we edited H9 human pluripotent stem cell (hPSC) lines by inserting anti-PSMA CAR constructs into the AAVS1 safe harbor locus for constitutive and homogenous CAR expression in differentiated neutrophils. The resulting hPSC-derived anti-PSMA CAR-neutrophils (CAR-Neu) exhibited potent cytotoxic effects on PSMA+ LNCaP cells in vitro. Our results suggest that CAR-engineering can significantly enhance the neutrophil anti-tumor effect, providing a new direction in treating prostate cancers.
Results
Construction of anti-PSMA CAR knockin hPSCs
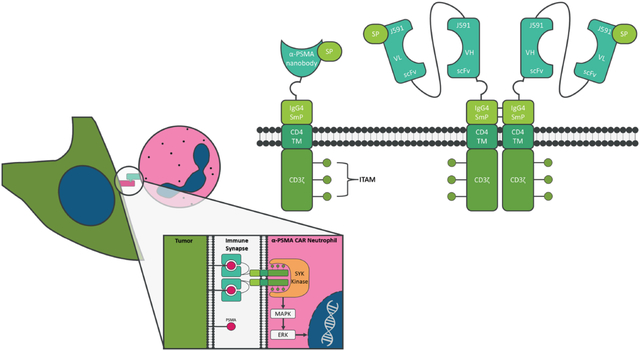
To investigate the anti-tumor effect of CAR-Neu on PCa cells, two CARs were designed (Fig. 1A). Both CARs target PSMA, using either a J591 single-chain fragment variable (scFv) minibody (MiB)20 or an anti-PSMA nanobody (NB)21, and share common domains including the GM-CSF-derived signal peptide (SP), IgG4 Fc domain (SmP), CD4 transmembrane domain, and intracellular CD3ζ signaling domain containing tyrosine-based activation motif (ITAM) binding sites. CAR constructs were inserted into the AAVS1 safe harbor locus via CRISPR/Cas9-mediated homologous recombination (Fig. 1B). After nucleofection, single cell-derived hPSC clones were isolated and genotyped by PCR (Fig. 1C). MiB CAR construct insertion resulted in 12 successfully targeted clones of which three were homozygous, and NB CAR construct insertion resulted in 8 successfully targeted clones, of which three were homozygous.
Figure 1:

CAR construct design and engineering of H9 hPSCs. (A) Schematic of anti-PSMA J591 minibody (MiB CAR) and nanobody CAR (NB CAR) donor plasmid containing a signal peptide (SP), an anti-PSMA binding domain (MiB or NB), IgG4 Fc domain (SmP), a CD4 transmembrane domain, and an intracellular CD3ζ signaling domain. (B) Schematic of Cas9-mediated knock-in strategy for targeting the AAVS1 safe harbor locus. The vertical arrow represents a targeted sgRNA, and the horizontal lines represent primer pairs for insertion efficiency and homozygosity assay. (C) PCR genotyping of single cell-derived hPSC clones for successful insertion of CAR constructs into the AAVS1 locus (AAVS1 positive) and confirmation of homozygosity (AAVS1 Homo) by lack of the ~240 bp band.
Differentiation of CAR-hPSCs into CAR-neutrophils
Neutrophils were differentiated from hPSCs following our previously published protocol19. Differentiation begins by the generation of hematopoietic stem and progenitor cells (HSPCs) (Fig. 2A). Activation of the canonical Wnt signaling pathway via CHIR9902122 and subsequent inhibition of the TGF-β signaling pathway with SB431542 differentiated hPSCs into hemogenic endothelium and induced endothelial to hematopoietic transition (EHT)23,24, respectively. Floating myeloid progenitors were differentiated from HSPCs with granulocyte/macrophage colony-stimulating factors (GM-CSF) and further specified into neutrophils with granulocyte colony-stimulating factor (G-CSF) along with the retinoic acid receptor agonist AM58025. Flow cytometry analysis confirmed the expression of key surface markers CD45, CD11b, and CD66b in hPSC-derived neutrophils (Fig. 2B). The expression of MPO in hPSC-derived neutrophils suggests their early-stage or immature phenotype, as MPO transcription occurs relatively early in a neutrophil’s lifespan and is found in primary, azurophilic granules26. The resulting cells also present a typical morphology of neutrophils by Wright-Giemsa stain with pink-violet cytosol, violet granules, and nucleus segmentation (Fig. 2C).
Figure 2:
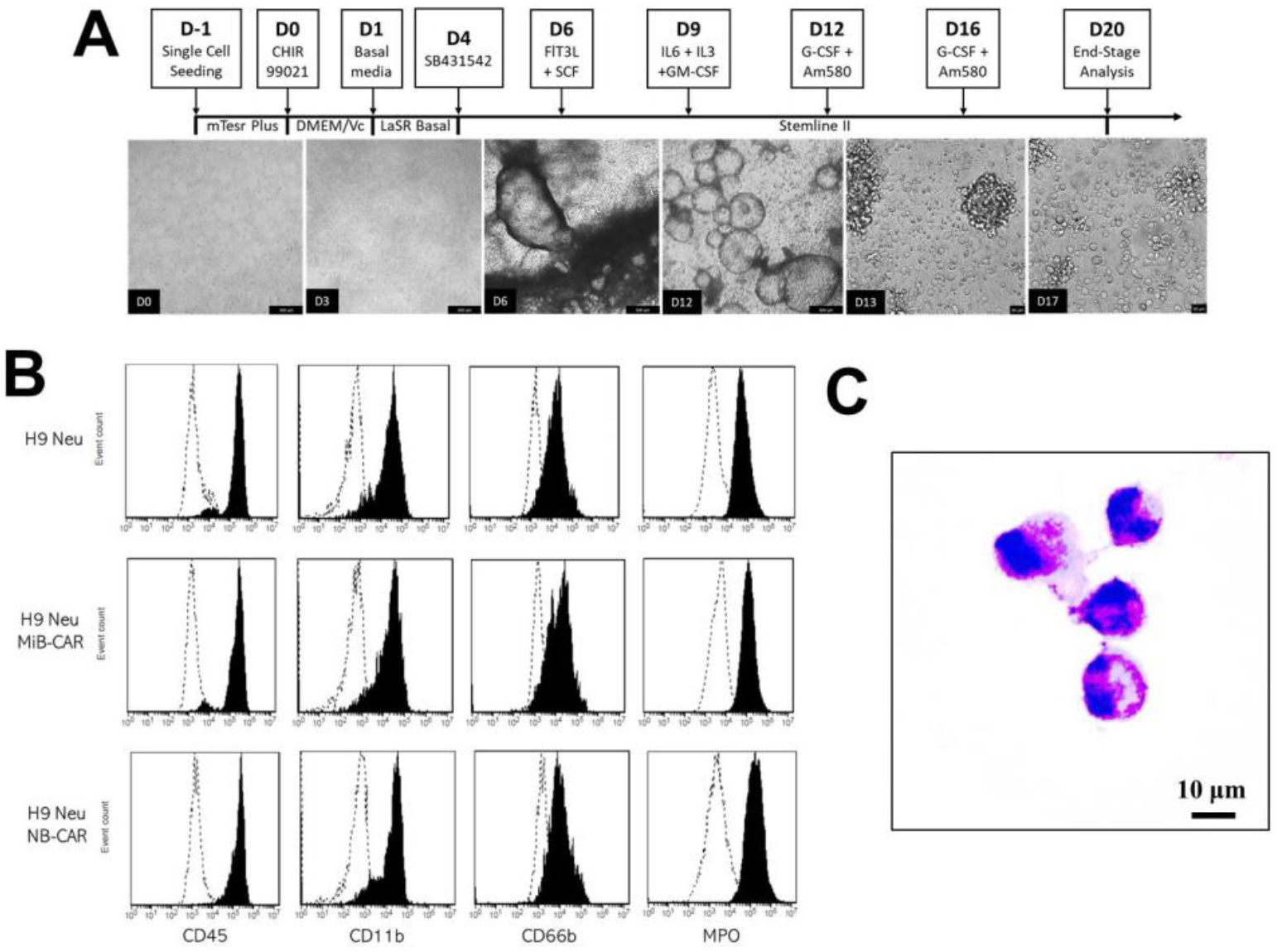
Differentiation of neutrophils from human pluripotent stem cells (hPSCs). (A) A Schematic of neutrophil differentiation with representative brightfield images is shown. Scale bars, 100 μm. (B) Flow cytometry analysis of indicated neutrophil surface markers in wildtype, MiB, and NB-CAR hPSC-derived neutrophils (Neu). (C) Wright Giemsa Stain of Day 20 hPSC-derived CAR-neutrophils. Scale bars, 10 μm.
Bulk RNA-seq analysis of hPSC-derived neutrophils
To further understand how differentiated neutrophils differ from their counterparts in peripheral blood (PB), bulk RNA sequencing (RNA-seq) was performed on hPSCs, hPSC-derived, and PB neutrophils. Hierarchical clustering of RNA-seq data demonstrated the similarity between hPSC-derived and PB neutrophils as they are clustered closely and distinct from undifferentiated hPSCs (Fig. 3A). The similarity among different neutrophil groups was confirmed by principal component analysis (PCA) (Fig. 3B). The distance between hPSC-derived and PB neutrophils indicated the heterogeneity and dynamics of neutrophils. As compared to undifferentiated hPSCs, the expression patterns of neutrophil-specific transcription factors and surface markers were identical in all neutrophil groups (Fig. 3C–D). Compared to PB neutrophils, lower expression of selected genes related to neutrophil phenotype, migration, and other functions, such as PTPRC (CD45), ITGAM (CD11b), MME, CXCR1, CXCR2, were observed, indicating the relative immaturity of hPSC-derived neutrophils. The relative immaturity was also confirmed by higher expression of neutrophil azurophilic granule markers, including PRTN3 and MPO26,27, in hPSC-derived neutrophils. We next performed gene set enrichment analysis (GSEA) to identify neutrophil-enriched signaling pathways (p < 0.05). The top 150 enriched gene sets are enriched in 20 pathways in all three neutrophil samples, including gene ontology (GO) related to immunoregulatory cytokine production, lipopolysaccharide, myeloid, granulocyte and neutrophil differentiation, neutrophil chemotaxis and migration (Fig. 3E–F).
Figure 3:

RNA-sequencing (RNA-seq) analysis of hPSCs, hPSC-derived and peripheral blood (PB) neutrophils. (A) Hierarchical clustering of RNA sequencing (RNA-seq) expression data of hPSCs, hPSC-derived, and PB neutrophils. (B) 3D score plot of the first three principal components (PCs) from principal component analysis (PCA) is shown. Each data point corresponds to different biological samples. (C-D) Heatmaps show selected ROS-generation-related genes, other neutrophil-function-related genes, transcription factors (C), general surface markers, chemokines (Chemos), chemoattractants (Chemoatt), Toll-like receptors (TLRs), Fc receptors, and adhesion molecules (D). (E) Venn diagram shows the number of gene sets that were enriched in different cell types (relative to hPSCs). The top 150 significantly enriched gene ontology (GO) (p<0.05), ranked by normalized enrichment score (NES) for each neutrophil group, were used for analysis. (F) Gene set enrichment analysis (GSEA) was performed, and heatmaps show 16 signaling pathways (p < 0.05) that were commonly enriched in each group relative to hPSCs. Commonly enriched gene sets related to myeloid, granulocyte, and neutrophil development and function are also listed.
In vitro functional analysis of hPSC-derived CAR-neutrophils
To determine if differentiated neutrophils retain key capabilities of PB neutrophils, several functional assays were performed in vitro. An initial assessment of hPSC-derived neutrophils (hPSC-Neu) to act as innate immune cells was performed using fluorescent marker-conjugated E. coli bioparticles to measure phagocytosis (Fig. 4A–B). Phagocytotic indices for differentiated neutrophils were measured from 80–100%, demonstrating that hPSC-derived neutrophils replicate the primary functionality of innate immune cells o recognize foreign targets for elimination. The formation of a tight hold between the target and effector cells is mediated by F-actin in response to the recognition of extracellular epitopes. Thus, the formation of this synapse is a critical measure of neutrophil performance. Differentiated neutrophils were co-cultured with PSMA+ human prostate adenocarcinoma line, LNCaP. Immunostaining analysis of F-actin confirmed the synapse formation between neutrophils and tumor cells (Fig. 4C).
Figure 4:

Functional analysis of hPSC-derived neutrophils. (A-B) Phagocytosis of pHrodo Green E. coli bioparticles. Representative flow cytometry analysis (A) and quantification (B) of phagocytosis of wildtype H9 (H9 w.t), minibody (MiB), and nanobody (NB) CAR-modified neutrophils were shown. (C) Immunological synapse formation between hPSC-derived neutrophils and LNCaP cells was stained and shown. (D) Cytotoxicity of H9 hPSC-derived neutrophils (wild type, MiB CAR, and NB CAR) against luciferase+ LNCaP for 24 hrs at effector: target ratios (Neutrophil: LNCaP) of 3:1, 5:1, and 10:1 was measured. Significance values were measured by Student’s T test. *: p < 0.05, **: p < 0.01, ***: p <0.001.
Finally, a cytotoxic assay was performed on hPSC-derived CAR-neutrophils in co-culture conditions with luciferase-expressing LNCaP (Fig. 4D). hPSC-derived CAR-neutrophils possess a marked increase in cytotoxicity than unmodified hPSC-Neu as indicated by a lower effector: target ratio necessary to achieve a similar cytotoxic effect. Neutrophils could release cytotoxic reactive oxygen species (ROS) to kill the target cells, and the kinetics of ROS production in different neutrophils coincided with their increased tumor-killing abilities (Fig. S1), indicating potential involvement of ROS in neutrophil-mediated tumor killing. In addition, neutrophils generated neutrophil extracellular traps (NETs) (Fig. S2), indicating the potential involvement of NETs in CAR-mediated tumor killing. As expected, anti-PSMA CAR-neutrophils showed similar anti-tumor ability against PSMA-negative MDA-MB-231 tumor cells as unmodified hPSC-derived neutrophils (Fig. S3), suggesting the specificity of hPSC-derived CAR-neutrophils. These functional assays demonstrate that both MiB and NB CAR-expressing hPSC-derived neutrophils can recognize LNCaP, form an immune synapse, and activate intracellular signaling cascades, which result in potent anti-tumor cytotoxicity against LNCaP in vitro.
Discussion
Unlike T or other immune cells, primary neutrophils are less commonly used in targeted cancer immunotherapy due to their short lifespan and resistance to genome editing. One potential solution to overcome this challenge is the engineering of hPSCs with synthetic CARs and their differentiation into “off-the-shelf” CAR neutrophils. We recently developed a chemically defined platform to produce neutrophils from multiple hPSCs, including induced pluripotent stem cell (iPSC) lines 19-9-7, 19-9-11, and 6-9-9, and human embryonic stem cell (hESC) lines H1, H9, and H1319,28. Although our study focused on engineering anti-PSMA CAR-neutrophils from H9 hESCs, we believe that our approach to producing CAR-neutrophils has the potential to be applied to patient-specific iPSCs for use in clinical settings.
Primary neutrophils exhibit antibody-dependent cellular cytotoxicity towards tumor cells via phagocytosis, which is facilitated by the Fcγ receptor and its downstream signaling pathways, including the tyrosine kinase Syk. In the case of our anti-PSMA CAR-neutrophils, they first form immune synapses with tumor cells, which may subsequently trigger the phosphorylation of Syk and activation of extracellular-signal-regulated kinase (ERK)1/219 (Fig. S4), reminiscent of signaling transduction in CAR natural killer cell-mediated cytotoxicity mechanism29.
Conclusion
The advent of CAR technology for immunotherapy has led to significant improvements in cancer treatments, especially those that do not respond to chemotherapy, surgery, or castration in the case of PCa. Improving the capacity of immune cells to recognize cancers is the critical function of CAR-based therapies. Solid cancers such as PCa still have many challenges before CAR treatments rival the efficacies in blood-borne tumors. One method to improve the efficacies in solid tumors is using alternatives for cytotoxic T lymphocytes. Several notable studies show improved tumor cytotoxicity using CAR-NK or CAR-macrophage. Our study presents a human pluripotent stem cell-derived CAR-neutrophil system that produces cytotoxic effects against a PCa model cancer cell LNCaP in vitro. Utilizing an hPSC differentiated cell allows for the precise insertion of the CAR construct into a clinically applicable locus (AAVS1). Furthermore, CAR-neutrophils can potentially be used in a cocktail with other CAR-immune cell systems or in combination with other treatments to produce synergistic effects. Further evaluation of PSMA-targeting hPSC-derived CAR neutrophils in an animal model of solid tumors will be critical to determine the therapeutic effects of this model in vivo.
Materials and Methods
hPSC Maintenance and Neutrophil Differentiation:
H9 hPSC line was obtained from WiCell and maintained on Matrigel-coated 6-well plates in mTeSR plus medium. To initiate neutrophil differentiation, 6 μM CHIR99021 (CHIR) was used to direct hPSCs into mesoendoderm in DMEM medium supplemented with 100 μg/mL ascorbic acid (DMEM/Vc) at day 0, followed by a medium change with LaSR basal medium on day 1. 50 ng/mL VEGF was added to the medium from day 2 to day 4. At day 4, 10 μM SB431542 was used to induce hematopoiesis in Stemline II medium (Sigma) supplemented with 25 ng/mL SCF and FLT3L. On day 6, cells were maintained in Stemline II medium with 50 ng/mL SCF and FLT3L, 25 ng/mL GM-CSF, 50 ng/mL IL-6, and 10 ng/mL IL-3. At day 9, the top half medium was aspirated and changed with 0.5 mL fresh Stemline II medium containing 50 ng/mL G-CSF, 50 ng/mL IL-6, and 10 ng/mL IL-3. On day 12, floating cells were gently harvested and filtered for terminal neutrophil differentiation in Stemline II medium supplemented with 2 mM GlutaMAX, 150 ng/mL G-CSF, and 2.5 μM retinoic acid agonist AM580. Half medium change was performed every 3 days, and mature neutrophils could be harvested for analysis starting from day 20.
Gene editing of hPSCs:
Cas9-mediated gene knockin were performed following our previously published manuscript19. Briefly, singularized 1–2.5 × 106 hPSCs were nucleofected with 6 μg SpCas9 AAVS1 gRNA T2 (Addgene; #79888) and 6 μg CAR donor plasmids in 100 μL human stem cell nucleofection solution (Lonza; #VAPH-5012) using program B-016 in a Nucleofector 2b. After cell seeding and recovery, drug selection was performed on nucleofected cells with 1 μg/mL puromycin (Puro) for 24 h. Individual drug-resistant clones were then picked using a microscope inside a tissue culture hood and expanded for 2–5 days in each well of a 96-well plate pre-coated with Matrigel, followed by a PCR genotyping.
Bulk RNA Sequencing:
Total RNA of sorted hPSC-derived CD16+ and peripheral blood neutrophils was prepared with Direct-zol RNA MiniPrep Plus kit (Zymo Research) and sequenced in Illumina HiSeq 2500 by the Center for Medical Genomics at Indiana University. The RefSeq transcript levels (RPKMs) HISAT2 program was employed to map the resulting sequencing reads to the human genome (hg 19), and the python script rpkmforgenes.py was used to quantify the. The original fastq files and processed RPKM text files were submitted to NCBI GEO (GSE188393). Principal component analysis (PCA) was performed in Perseus P2.0.6.0 and visualized in Mathematica 12.3. Gene expression data for each cell type were compared with that of hPSCs and significantly enriched gene ontology (p < 0.05) were considered for further analysis. MATLAB (Mathworks) and Microsoft Excel were used to identify the unique and common pathways in different cell types. Heatmaps and hierarchical clustering analysis of selected gene subsets after normalization were then plotted using Morpheus (Broad Institute).
Phagocytosis Assay:
Phagocytosis was assessed using pHrodo Green E.coli BioParticles conjugate according to the manufacturer’s protocol. In brief, pHrodo Green E. coli beads were resuspended in 2 mL of PBS and sonicated with an ultrasonicator 3 times. Beads per assay (100 mL) were opsonized by mixing with opsonizing reagent at a ratio of 1:1 and incubated at 37°C for 1 h. Beads were washed 3 times with mHBSS buffer by centrifugation at 4°C, 1,500 RCF for 15 min, and resuspended in mHBSS buffer. Differentiated neutrophils were resuspended in 100 μL of opsonized solution and incubated at 37°C for 1 h, followed by flow cytometry analysis using an Accuri C6 plus cytometer (Beckton Dickinson). Phagocytotic capacity was measured using the following index:
Where phagocytotic cells are positive for the fluorescent dye.
Immune Synapse Formation:
To visualize immunological synapses, 100 μL of LNCAP cells (1,000 cells/mL) were plated in a 96-well plate for 12 hours to adhere. Neutrophils (10,000 cells/mL) were added to LNCaP cells and incubated for 6 hours before fixation with 4% paraformaldehyde (in PBS). Cytoskeleton staining was then performed using an F-actin Visualization Biochem Kit (Cytoskeleton Inc.). Additionally, CD45 antibody and Hoechst reagent (DAPI) was additionally added to clarify neutrophils and LNCaP cells.
Cytotoxic Assay:
The viability of luciferase-expressing LNCaP was assessed by D-luciferin assay. 100 μL of tumor cells (50000 cells/mL) in RPMI medium containing 10% FBS were mixed with 100 μL of 150,000, 250,000 and 500,000 cells/mL neutrophils in 96 well plates, and then incubated at 37°C and 5% CO2 for 24 hr. After the incubation, the mixture was centrifuged at 1,000 rpm for 5 min, the suspension was removed and 100 μL of culture medium containing 150 μg mL-1 D-luciferin was added into each well for 30 min of incubation at 37°C in a humidified 5% CO2 incubator. The bioluminescence was measured by SpectraMax iD3. The cellular viability was calculated as following: Cellular viability = (Sample-blank) /(negative-blank)×100%.
ROS generation analysis:
100 μL of LNCaP cells (30,000 cells/mL) were seeded into wells of a 96-well plate 12 h before adding neutrophils at a neutrophil-to-tumor ratio of 10:1. After co-incubation for 12 h, the resulting cell mixture was treated with 10 mM H2DCFDA at 37°C for 50 min and then the fluorescence emission signal (480–600 nm) was collected in a SpectraMax iD3 microplate reader with an excitation wavelength of 475 nm.
Measurement of neutrophil extracellular trap formation:
100 μL of LNCaP cells (30,000 cells/mL) were seeded into wells of a 96-well plate 12 h before adding neutrophils at a neutrophil-to-tumor ratio of 10:1. After co-incubation for 12 h, the resulting cells were centrifuged for 5 min at 200 xg, and extracellular DNA in the supernatant samples was quantified using the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen) and characterized by the SpectraMax iD3 microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Supplementary Material
Highlights.
A chemically-defined protocol for massive production of neutrophils from hPSCs
CAR constructs significantly enhance antitumor activity of hPSC-derived neutrophils
CAR engineering does not alter the phenotype of hPSC-derived neutrophils
Anti-PSMA CAR-neutrophils display superior anti-prostate cancer activities
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Credit Author Statement
Jackson D. Harris: Conceptualization, Formal Analysis, Investigation, Writing- Original Draft Preparation. Yun Chang: Conceptualization, Formal Analysis, Investigation, Writing- Original Draft Preparation. Ramizah Syahirah: Validation, Writing- Reviewing and Editing. Xiaojun Lance Lian: Resources, Supervision, Funding Acquisition, Writing- Reviewing and Editing. Qing Deng: Resources, Supervision, Writing- Reviewing and Editing. Xiaoping Bao: Supervision, Funding Acquisition, Writing- Reviewing and Editing.
Conflict of Interest Statement
The authors declare no conflicts of interest.
References
- 1.Sung H et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 71, 209–249 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Henríquez I et al. Current and emerging therapies for metastatic castration-resistant prostate cancer (Mcrpc). Biomedicines 9, 1–13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang SS et al. Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res 59, 3192–3198 (1999). [PubMed] [Google Scholar]
- 4.Liu H et al. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res 57, 3629–3634 (1997). [PubMed] [Google Scholar]
- 5.Kuratsukuri K et al. Inhibition of prostate-specific membrane antigen (PSMA)-positive tumor growth by vaccination with either full-length or the C-terminal end of PSMA. Int J Cancer 102, 244–249 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Salter AI, Pont MJ & Riddell SR Chimeric antigen receptor–modified T cells: CD19 and the road beyond. Blood 131, 2621–2629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.June CH & Sadelain M Chimeric antigen receptor therapy. New England Journal of Medicine 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gill S, Maus M. v. & Porter DL Chimeric antigen receptor T cell therapy: 25 years in the making. Blood Rev 30, 157–167 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Barrett DM, Singh N, Porter DL, Grupp SA & June CH Chimeric antigen receptor therapy for cancer. Annu Rev Med 65, 333–347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu Y & Huang J The chimeric antigen receptor detection toolkit. Front Immunol 11, 1770 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadelain M, Brentjens R & Rivière I The basic principles of chimeric antigen receptor design. Cancer Discov 3, 388–398 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J & Bae JS Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediators Inflamm 2016, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klichinsky M et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nature Biotechnology 2020 38:8 38, 947–953 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuca G, Reppel L, Landoni E, Savoldo B & Dotti G Enhancing Chimeric Antigen Receptor T-Cell Efficacy in Solid Tumors. Clinical Cancer Research 26, 2444–2451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newick K, Moon E & Albelda SM Chimeric antigen receptor T-cell therapy for solid tumors. Molecular therapy-oncolytics 3, 16006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu S, Yi M, Qin S & Wu K Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol Cancer 18, 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y et al. CAR-macrophage: A new immunotherapy candidate against solid tumors. Biomedicine & Pharmacotherapy 139, 111605 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Montagner IM et al. Anti-PSMA CAR-engineered NK-92 Cells: An Off-the-shelf Cell Therapy for Prostate Cancer. Cells 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang Y et al. Engineering chimeric antigen receptor neutrophils from human pluripotent stem cells for targeted cancer immunotherapy. Cell Rep 40, 111128 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parker SA, Diaz ILC, Anderson KA & Batt CA Design, production, and characterization of a single-chain variable fragment (ScFv) derived from the prostate specific membrane antigen (PSMA) monoclonal antibody J591. Protein Expr Purif 89, 136–145 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Hassani M et al. Construction of a chimeric antigen receptor bearing a nanobody against prostate specific membrane antigen in prostate cancer. J Cell Biochem 120, 10787–10795 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Lian X et al. Efficient differentiation of human pluripotent stem cells to endothelial progenitors via small-molecule activation of WNT signaling. Stem Cell Reports 3, 804–816 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Armitage LH et al. High-Yield Monocyte, Macrophage, and Dendritic Cell Differentiation from Induced Pluripotent Stem Cells Running Title: High-Yield Monocyte Differentiation from hiPSC. doi: 10.1101/2021.04.29.441947. [DOI] [Google Scholar]
- 24.Yanagimachi MD et al. Robust and highly-efficient differentiation of functional monocytic cells from human pluripotent stem cells under serum- and feeder cell-free conditions. PLoS One 8, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brok-Volchanskaya VS et al. Effective and Rapid Generation of Functional Neutrophils from Induced Pluripotent Stem Cells Using ETV2-Modified mRNA. Stem Cell Reports 13, 1099–1110 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dalton JA, Higgins MK, Miller AH, Keefe FJ & Khuri FR Myeloperoxidase: A new player in autoimmunity.HHS Public Access. Cell Immunol 38, 457–464 (2016). [Google Scholar]
- 27.FALLOON J & GALLIN J Neutrophil granules in health and disease. Journal of Allergy and Clinical Immunology 77, 653–662 (1986). [DOI] [PubMed] [Google Scholar]
- 28.Chang Y, S.R. et al. Chemically-defined generation of human hemogenic endothelium and definitive hematopoietic progenitor cells. Biomaterials 285, 121569 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Hermanson DL, Moriarity BS, Kaufman DS Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 23 (2), 181–192 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.