

Vascular Ehlers-Danlos syndrome (vEDS), caused by damaging variants in the type III procollagen gene, COL3A1, has a median survival of 51 years.1 Dominant-negative (DN) variants, missense glycine substitutions within the triple helical domain and splicing variants altering in-phase exons, are considered more severe than haploinsufficiency (HI) variants, premature termination codons causing mRNA instability (i.e., frameshift, non-sense, large deletions).1,2 Penetrance, estimated from patients with vEDS, is high; ≥ 80% of genotype-positive individuals have ≥ one major complication.3 Increasingly, deleterious COL3A1 genotypes are identified through genomic screening programs, yet clinical risks in such unselected populations are not yet defined. We therefore used a genotype-first approach to minimize ascertainment bias, investigating vEDS-associated phenotypes among COL3A1+ biobank participants.
Mount Sinai’s BioMe biobank (median age 64, IQR 49–75 years; 58.4% Female; 22% African American (AA), 27% European American, 34% Hispanic/Latinx, 4% Asian), links 32,344 patients’ exome sequences to their longitudinal electronic health records (EHR), and the United Kingdom Biobank (UKBB), a prospective cohort study, contains phenotypic data and exome sequences for 200,643 participants (median age 57 ± 8 years; 54% Female; 94% white, 2% Asian, 2% Black). Exomes were queried for putatively damaging COL3A1 variants: rare (minor allele frequency < 0.1% in gnomAD) and (1) triple helical domain (exons 6–47)3 missense glycine substitutions; (2) stop-loss/start-gain; (3) frameshift insertion/deletion; or (4) canonical splice site variant within two base pairs of the exon. Variants with ≥ 1 likely benign/benign ClinVar designation were excluded. Mount Sinai’s IRB approved this study; informed consent was obtained at enrollment. Data supporting study findings are available from the corresponding author upon reasonable request.
Eighteen BioMe (11 females; 1 in 1,797, 95% C.I. 1:2,500–1:3,344) and 63 UKBB (33 females; 1 in 3,185, 95% C.I. 1:2,554–1:4,229) were COL3A1+ (Figure, A). vEDS-related phenotypes including major/minor diagnostic criteria,4 age/cause of death, and obstetric/surgical histories were assessed from EHRs (BioMe) or database entries (UKBB). Median age was 50 (BioMe) and 54 (UKBB) years, exceeding the median age when patients with vEDS are diagnosed following clinical events.3 No COL3A1+ individual was diagnosed with vEDS or a genetic arteriopathy.
Figure 1.
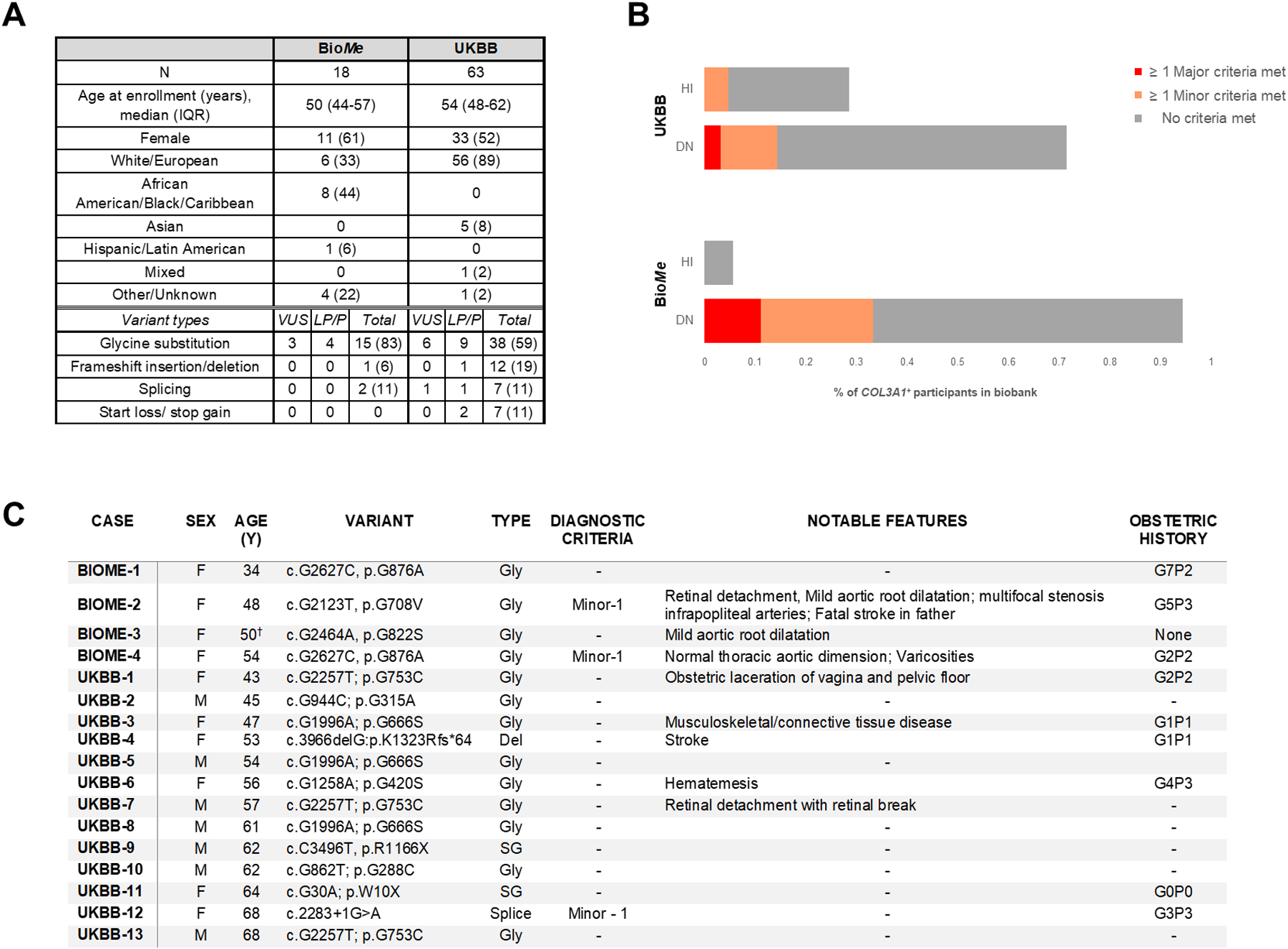
Demographic and clinical data, distribution of variant types and individuals meeting vEDS diagnostic criteria and selection of detailed cases among BioMe and UKBB participants with putatively deleterious COL3A1 variants. A, Demographic data and types of variants for BioMe and UKBB participants with putatively deleterious COL3A1+ genotypes. B, Distribution of vEDS major and minor criteria for BioMe and UKBB participants with putatively deleterious COL3A1 variants stratified according to haplo-insufficiency or dominant-negative variant types.. C, Table of biobank participants harboring COL3A1 variants previously reported in association with vEDS (likely pathogenic or pathogenic designations in ClinVar).A: Table entries are number (%) unless otherwise noted; Designations of variants in ClinVar are listed: VUS- variant of uncertain significance; LP- likely pathogenic; P- pathogenic; total indicates all variants including those not classified in ClinVar; B: HI- “haplo-insufficiency” variant; DN- “dominant negative” variant; C: Age listed is at last EHR entry (BioMe) or first assessment (UKBB); Gly- glycine substitution; SG- stop-gain; Spice- essential splice site mutation; Del- frameshift deletion; † - Participant died at age 50 of septic shock from sternal wound infection following mitral/aortic valve replacement
Although > 75% of included variants were DN (BioMe: 17, 94%; UKBB: 45, 71%) and predicted to cause severer phenotypes, no genotype-positive participant experienced organ rupture or fatal, unprovoked arterial rupture/dissection. Major vEDS criteria were met in two (11%) BioMe (aneurysm; family history) and two (3%) UKBB (aneurysm) participants. Minor criteria were met in four (22%) BioMe (varicose veins, talipes equinovarus) and six (10%) UKBB (varicose veins, easy bruising) participants (Figure, B). None had organ rupture, carotid-cavernous sinus arteriovenous fistula, tendon/muscle rupture, pneumothorax/hemopneumothorax, congenital hip dislocations, joint or craniofacial/skin features. Association with any vEDS criteria was not different between individuals harboring DN or HI variants (15, 24% vs. 3, 16%, p=0.4). Sixteen (20%) COL3A1+ individuals harbored variants previously reported as likely pathogenic or pathogenic (LP/P) in ClinVar (Figure, C); none met major and three (19%) met minor criteria. Amongst the remaining 65 with novel (n=55) or VUS (n=10) variants, 4 (6%) met major and 7 (11%) met minor criteria. Prevalence of any criteria was not different in those with LP/P versus novel/VUS variants (p=0.9).
84% of COL3A1+ participants had no documented vEDS features. Surgeries, including major surgeries, occurred in 89% of BioMe (with no major complications) and 90% of UKBB COL3A1+ participants. No pregnancy complications were noted for five BioMe (45%) and 30 UKBB (91%) COL3A1+ females with obstetric histories. Three participants were deceased, one (UKBB) from a subarachnoid hemorrhage secondary to an unspecified surgery.
The higher prevalence of relevant variants in BioMe versus UKBB may be due to health system rather than population level recruitment. Prevalence of deleterious COL3A1 variants in UKBB was still higher than previously cited for vEDS (~1:50,000–1:200,000).5 The four individuals who met major vEDS criteria, however, equate to a point prevalence between 1:16,172 and 1:100,322, consistent with previous estimates. Interestingly, 44% of COL3A1+ individuals in BioMe (none related) were AA, although vEDS has no known racial predilection.
Study limitations are those inherent to EHR/database phenotyping, including incompleteness. Outward criteria of vEDS may be subtle, missed even by clinical geneticists. Aneurysms are often asymptomatic preceding dissection or rupture. Phenotypic features may have been present in COL3A1+ participants but not suspected in routine clinical care. We do expect to have captured the severest manifestations of COL3A1+-related disease (i.e., arterial dissection/rupture, organ rupture) with our methods. Finally, COL3A1+ cases with fatalities at young ages would not be included in these biobanks, conferring a survival bias.
In summary: 1) relevant COL3A1 variants are commoner than previously understood; 2) COL3A1+ status, even for DN variants, does not reliably predict vEDS phenotypes or dangerous outcomes; 3) penetrance may be lower than previously predicted; and 4) assigning rare COL3A1 variants as putatively damaging based on the criteria used for this study is at least as predictive as established ClinVar classifications.
Acknowledgments:
We would like to thank the participants of the BioMe biobank and United Kingdom Biobank and Mount Sinai’s Charles Bronfman Institute for Personalized Medicine for their continued development of BioMe. We would also like to thank Nihir Patel, MS for his assistance with this study. This research was performed using the United Kingdom Biobank Resource (application number 16218).
Sources of Funding:
This study was supported by the grant support from the National Institute of Health / National Heart, Lung, and Blood Institute (NHLBI) to B.D.G. (HL135742) and A.R.K. (HL140083).
Nonstandard Abbreviations and Acronyms
- EHR
electronic health record
- DN
dominant negative
- HI
haplo-insufficiency
- UKBB
United Kingdom Biobank
- vEDS
vascular Ehlers-Danlos syndrome
Footnotes
Disclosures: None.
References:
- 1.Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med. 2014;16:881–888. [DOI] [PubMed] [Google Scholar]
- 2.Frank M, Adham S, Seigle S, Legrand A, Mirault T, Henneton P, Albuisson J, Denarié N, Mazzella JM, Mousseaux E, et al. Vascular Ehlers-Danlos syndrome: Long-term observational study. J Am Coll Cardiol. 2019;73:1948–1957. [DOI] [PubMed] [Google Scholar]
- 3.Frank M, Albuisson J, Ranque B, Golmard L, Mazzella JM, Bal-Theoleyre L, Fauret AL, Mirault T, Denarié N, Mousseaux E, et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur J Hum Genet. 2015;23:1657–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175:8–26. [DOI] [PubMed] [Google Scholar]
- 5.Byers PH, Belmont J, Black J, De Backer J, Frank M, Jeunemaitre X, Johnson D, Pepin M, Robert L, Sanders L, et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2017;175:40–47. [DOI] [PubMed] [Google Scholar]