

SUMMARY
Highly coordinated changes in gene expression underlie T cell activation and exhaustion. However, the mechanisms by which such programs are regulated and how these may be targeted for therapeutic benefit remain poorly understood. Here, we comprehensively profile the genomic occupancy of mSWI/SNF chromatin remodeling complexes throughout acute and chronic T cell stimulation, finding that stepwise changes in localization over transcription factor binding sites direct site-specific chromatin accessibility and gene activation leading to distinct phenotypes. Notably, perturbation of mSWI/SNF complexes using genetic and clinically relevant chemical strategies enhances the persistence of T cells with attenuated exhaustion hallmarks and increased memory features in vitro and in vivo. Finally, pharmacologic mSWI/SNF inhibition improves CAR-T expansion and results in improved anti-tumor control in vivo. These findings reveal the central role of mSWI/SNF complexes in the coordination of T cell activation and exhaustion and nominate small-molecule-based strategies for the improvement of current immunotherapy protocols.
Graphical abstract
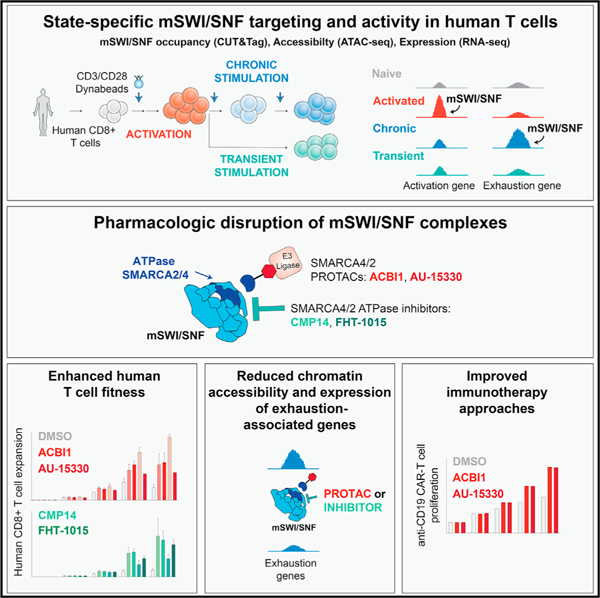
In brief
mSWI/SNF ATP-dependent chromatin remodeling complexes direct chromatin accessibility and gene expression during T cell activation and exhaustion. Targeting mSWI/SNF complexes with clinically relevant small-molecule inhibitors and degraders attenuates T cell exhaustion and increases T cell persistence and anti-tumor activity in cell culture systems and in vivo.
INTRODUCTION
T cells undergo dynamic morphologic and gene regulatory changes upon acute or sustained antigen exposure.1–5 Importantly, chronic antigen stimulation causes T cells to enter a dysfunctional state known as T cell exhaustion characterized by poor effector function, reduced proliferative capacity, expression of inhibitory receptors, and altered cytokine production.6 Targeting T cell exhaustion has formed the basis for numerous studies in the context of both chimeric antigen receptor (CAR)-T cell generation and checkpoint blockade.7–13 However, the molecular mechanisms governing T cell activation and exhaustion as well as the factors directing the expression of key state-specific biomarkers remain poorly understood. Uncovering such mechanisms may enable improvements to current immunotherapeutic approaches.
Studies in both mouse and human settings have defined chromatin changes during T cell activation and exhaustion, including linking locus-specific accessibility changes with anti-tumor responses.14–22 Species-level differences have also been identified owing to the fact that a number of gene regulatory networks differ between mouse and human cells, highlighting the need for increased understanding of human T cells.18 Studies aiming to identify strategies to prevent or reverse exhaustion and to define which T cell populations (i.e., terminally versus progenitor exhausted) should be targeted represent active areas of investigation.23–25
Several critical transcription factors (TFs) that contribute to distinct stages of T cell activation and exhaustion have been identified,3,24 including NFAT, NF-κB, AP-1, GATA3, and t-BET, as mediators of activation, and elevated T cell receptor (TCR)-responsive TFs such as TOX, NFATC1, IRF4, BATF, MYB, and others in exhausted states.6,25–33 Efforts to target such factors directly are challenged by high-affinity TF-DNA interactions and functional redundancy between multiple TFs, making the inhibition or depletion of a single TF often insufficient. Networks that encompass multiple TFs have been identified, yet strategies to prioritize those that play ‘‘master regulatory’’ roles and that are secondary remain challenging. With few exceptions, the role of chromatin regulatory complexes and epigenetic modifiers in T cell dysfunction remains less clear.8,15,34 Recently, several CRISPR screens in mouse T cells revealed chromatin regulatory proteins and protein complexes as mediators of exhaustion programs.35,36 In particular, genome-wide CRISPR screens have identified components of the mammalian SWI/SNF (mSWI/SNF) family of ATP-dependent chromatin remodeling complexes as mediators of regulatory T cell and exhausted states.37,38
Our group and others have shown that a wide range of human TFs interacts transiently with mSWI/SNF complexes resulting in their site-specific targeting genome wide.39–42 mSWI/SNF complexes are heterogeneous, multi-subunit entities that alter DNA-nucleosome contacts, generating chromatin accessibility and coordinating the timely binding of transcriptional machinery required for gene expression.43–47 mSWI/SNF complexes exist in three forms, termed canonical BAF (cBAF), polybromo-associated BAF (PBAF), and non-canonical BAF (ncBAF), each demarcated by the incorporation of distinct subunits and unique association with chromatin features.43,48,49The genes encoding the 29 total subunits are collectively mutated in over 20% of human cancers, in some cases representing hallmark drivers44,50 and have been implicated in both cellular differentiation and cell-state changes. Although the importance of mSWI/SNF complexes as regulators of chromatin accessibility in tumor-intrinsic settings represents an active area of investigation, their role in governing immune cell function remains less clear.51–59 Understanding the role for mSWI/SNF-directed chromatin remodeling across T cell states has uniquely high significance, given that such cells often need to undergo dynamic changes to carry out specific functions and to orchestrate anti-tumor immune responses.
RESULTS
Stepwise rewiring of mSWI/SNF complex occupancy and chromatin accessibility during acute and chronic human T cell stimulation
To study chromatin-level changes across T cell states, we developed a system for antigen-independent TCR stimulation of human CD8+ T cells (Figure 1A). T cells were profiled without stimulation (0 h) or at 3, 24, 48, and 72 h (3 days) following stimulation to capture different stages of early T cell activation. To profile changes related to chronic antigen stimulation, cells were replated in presence of new beads on days 3, 6, and 9 following initial activation (‘‘chronic,’’ [Ch] condition). In addition, a ‘‘transient’’ (Tr) stimulation condition in which beads were removed after the initial 3-day incubation was used to mimic a memory phenotype. We monitored T cell proliferation, immunophenotype, and functionality across these stages (Figures 1A, S1A, and S1B). Stimulated T cells displayed sustained proliferation during the first 6 days after activation and, as expected, lost proliferative capability upon transient stimulation. Chronically stimulated T cells also lost proliferative capacity by day 9, indicating the acquisition of an exhausted-like state (Figure S1A). Fluorescence-activated cell sorting (FACS) analyses performed at the early activation (3 and 24 h), activation (48 and 72 h), exhaustion-like (Day6-Day9-Ch), or memory (Day9-Tr) stages revealed the acquisition of markers consistent with these phenotypes (Figure S1B), such as the CD25 activation marker at 24 h, sustained the upregulation of the PD1 immune checkpoint and elevated terminal exhaustion markers, TIM-3 and CD39, at days 6 and 9 of the chronic but not of the transiently stimulated condition (Figures 1B and S1B). Chronically activated T cells expressed low levels of CCR7 and CD45RA and displayed reduced IFNγ, TNFα, and GZMb staining upon re-stimulation, indicating dysfunction (Figure S1B). In addition, we established a similar system in mouse CD8+ T cells, with highly consistent proliferation and immunophenotyping results (Figures S1C and S1D).
Figure 1. Stepwise changes in mSWI/SNF complex targeting and chromatin accessibility during CD8+ T cell activation and exhaustion.
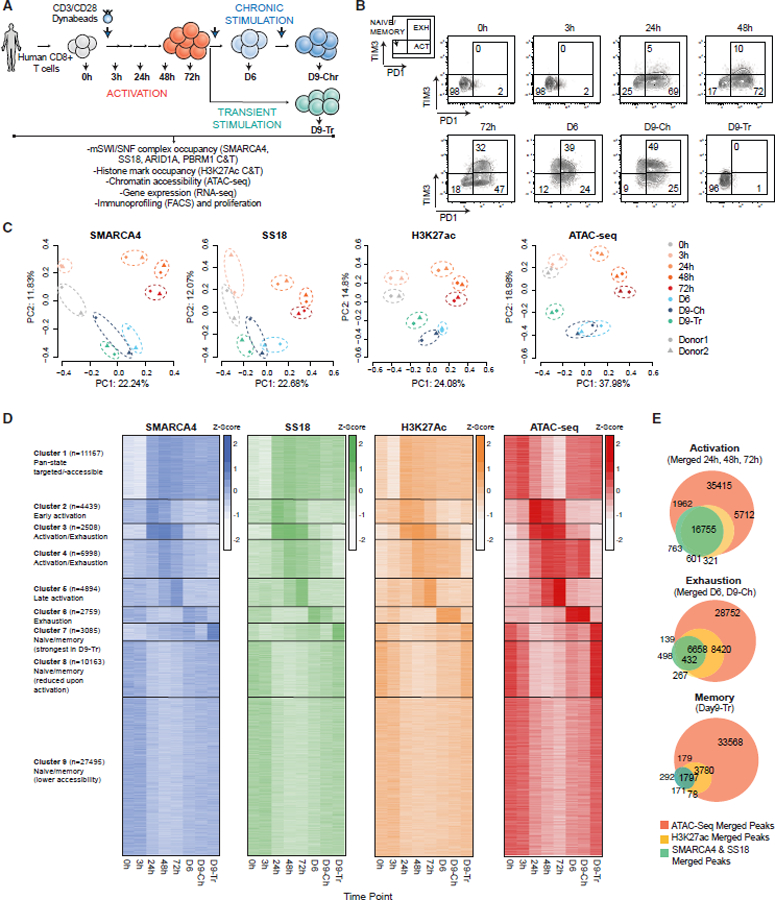
(A) Schematic for CD3/CD28 bead-based stimulation of human CD8+ T cells.
(B) FACS-based profiling of PD1 and TIM3 markers indicating naive/memory, activated, and exhausted populations.
(C) PCA for mSWI/SNF subunit and H3K27Ac CUT&Tag and ATAC-seq profiles across the time course (donors 1 and 2, see KRT for donor information).
(D) K-means clustering for SMARCA4, SS18, H3K37Ac, and ATAC-seq performed over merged SMARCA4, SS18, H3K27ac, and ATAC-seq peaks; heatmap intensity depicts quantile-normalized log2-transformed RPKM values transformed into Z scores.
(E) Venn diagrams showing the overlap between SMARCA4/SS18 merged, H3K27Ac CUT&Tag peaks with ATAC-seq peaks across time points shown.
See also Figures S1 and S2.
Owing to the long-held concept that T cells undergo major rearrangements in their nuclear architecture and chromatin accessibility during activation and exhaustion,60 we next sought to characterize the chromatin occupancy of the mSWI/SNF complexes, which are known to play directive roles in generating DNA accessibility.56,58,60–62 These complexes have been implicated in various T cell contexts, from dysfunction to the generation of T regulatory cells; however, comprehensive profiling of their genome-wide occupancy and activities across T cell populations has not been examined.35,37,60,63 To achieve this, we performed cleavage under targets and tagmentation (CUT&Tag) experiments using antibodies targeting the ATPase subunit, SMARCA4, which is present in all three subcomplexes of the mSWI/SNF family (cBAF, PBAF, and ncBAF).43,64,65 Further, we profiled SS18 (a member of cBAF and ncBAF), as well as ARID1A (cBAF-specific) and PBRM1 (PBAF-specific), in parallel with H3K27Ac, a marker of active chromatin. We observed striking increases in total mSWI/SNF and histone peak numbers at activation, consistently in T cells from two independent donors (Figures S2A–S2C). Principal-component analyses (PCAs) revealed concordant time point- and cell state-specific changes in mSWI/SNF as well as H3K27Ac occupancy from both human donors across the course (Figures 1C and S2D). In parallel, we characterized chromatin accessibility using assay for transposase-accessible chromatin using sequencing (ATAC-seq),66,67 which revealed similar changes and directionality upon PCA analysis (Figures 1C, S2E, and S2F).
We next merged the mSWI/SNF complex occupancy and accessibility data and performed k-means clustering analyses to reveal changes in complex localization and accessibility across the time course. The combination of SMARCA4-SS18-H3K27Ac-ATAC-seq-merged peaks (quantile-normalized, log2-transformed RPKM values transformed into Z scores) revealed 9 distinct clusters of mSWI/SNF occupancy and DNA accessibility (Figures 1D and S2G). Integrating these data with the immune profiling results (Figure S1B), we found that early activation was highlighted in cluster 2 (C2), activation and exhaustion in clusters 3 and 4 (C3 and C4), late activation in cluster 5 (C5), exhaustion in cluster 6 (C6), and memory phenotype in clusters 7 and 8 (C7 and C8) (Figures 1D, S1B, and S2G). Cluster 1 (C1) sites contained TSS-proximal targets of varied targeting and accessibility across conditions and cluster 9 (C9) sites encompassed TSS-distal sites of lower accessibility and with the highest mSWI/SNF signal prior to and at early stimulation (Figures 1D and S2H). Across all time points, mSWI/SNF complex occupancy, H3K27Ac signal, and accessibility overlapped substantially genome wide, with mSWI/SNF complex-bound sites representing a fraction of the total accessible sites (Figures 1E and S2I). Further, mSWI/SNF-bound sites represented increasingly lower fractions of total accessible sites across activation to exhaustion, suggesting an increasingly specific group of sites directing the chromatin accessibility and gene regulatory programs hallmark to these states (Figure 1E). Finally, we obtained similar results in mouse settings, albeit with fewer differences between Day9-Ch and Day9-Tr conditions (Figure S2J). Taken together, these studies establish mSWI/SNF complex binding and chromatin accessibility profiles throughout T cell activation and exhaustion, enabling the dissection of their roles in mediating state-specific transcriptional networks.
Differential, state-specific targeting and activity of mSWI/SNF complexes over TF binding sites during T cell activation and exhaustion
We next performed motif analyses using HOMER and archetype-based calling across the 9 clusters of mSWI/SNF-bound, accessible sites (Figures 2A, S3A, and S3B). Similarly, locus overlap analysis (LOLA) performed across the activation-exhaustion time course revealed state-specific TF binding enrichment over mSWI/SNF-occupied and accessible sites (Figure 2B). Sites corresponding to early activation were enriched in AP-1 (JUN/FOS), BATF, NF-κB, and NFAT motifs, corresponding to TFs known to play critical roles in early T cell activation (Figures 2A, 2B, S3A, and S3B).68–71 TF motifs enriched in sites at the middle to late activation stages included those for ATF3, BCL6, CREB1, JUND, and TBX1 (Figures 2A and 2B). Motifs in the exhaustion-associated C6 (Day 6 and Day9-Ch time points) included those for MYB/MYBL1, TCF7 (TCF1), which have been implicated in T cell stemness and/or exhaustion, as well as CUX2, POU5F1, and SOX3/10 TFs, which are less well characterized in T cells but have been suggested to interact with mSWI/SNF complexes (Figures 2A, 2B, and S3B).6,16,72–77 Intriguingly, we identified a particularly significant enrichment of mSWI/SNF complex occupancy over motifs corresponding to hepatocyte nuclear factor-1-beta (HNF1B) not previously implicated in T cell biology (Figures 2A, 2B, S3A, and S3B).
Figure 2. tate-specific transcription factor motif enrichment of mSWI/SNF complex occupancy and activity during T cell activation and exhaustion.
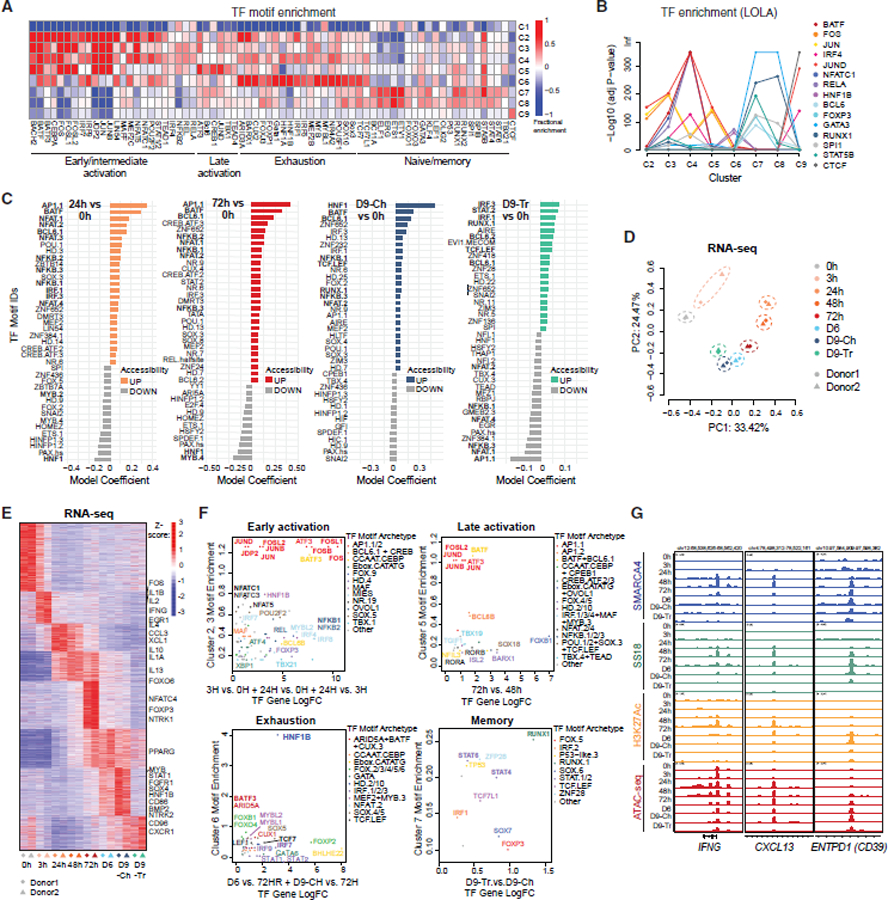
(A) Fractional motif enrichment in clusters C1–C9 (relative to all sites).
(B) LOLA enrichment of 15 selected TFs across C2–C9.
(C) Differential motif accessibility between time points indicated (top 40 coefficients of logistic regression models).
(D) PCA performed on RNA-seq datasets from T cells isolated from 2 independent donors at each time point.
(E) Z scored heatmap reflecting the top 25% most variable genes across the time course, partitioned into 8 groups by K-means clustering with selected genes labeled.
(F) Plots representing state (cluster(s)-specific) TF fractional motif enrichment (y axis) and gene expression (x axis).
(G) Representative SMARCA4, SS18, H3K27ac C&T, and ATAC-seq tracks over the IFNG, CXCL13, and ENTPD1 loci.
See also Figure S3.
Further, TF motifs identified under mSWI/SNF-occupied sites specifically at memory-like/naive clusters (C7/C8) included ETS factors (ETS1, ERG), RUNX1/2, GATA3, STAT factors, and others (Figures 2A and 2B). Several of these factors have been reported by our group and others to interact with mSWI/SNF complexes in other contexts.39,40,78,79 Finally, motifs enriched at mSWI/SNF target sites prior to and at early stimulation, as well as at Day 6, Day9-Tr, and Day9-Ch (C9) time points were nearly exclusively those for CTCF, targeted uniquely by ncBAF (Figures 2A and S3B).49,51
To define putative direct mSWI/SNF genomic targets as well as downstream accessible sites, we next used ATAC-seq data in isolation to identify TF motifs under mSWI/SNF-bound and -unbound accessible regions (Figures 2C and S3C). In addition to the strong enrichment of TF motifs shared with those identified at mSWI-SNF-bound sites (Figure 2C, bold), we also identified enrichment of TF motifs at sites of gained accessibility lacking mSWI/SNF occupancy, including ZNF and DMRT in early activation, and AIRE, MEF2, HLTF, and ZIM3 in exhaustion (Figure 2C). These data suggest that specific sites are made accessible following initial mSWI/SNF targeting, which in turn generates secondarily accessible regions that amplify state-specific programs.
We next integrated gene expression profiling by RNA sequencing (RNA-seq) with mSWI/SNF complex binding and accessibility. PCA analyses of RNA-seq data indicated stepwise changes in expression profiles, with greatest shifts between 0 h/3 h and 24 h early activation (PC1) and exhaustion (PC2) (Figures 2D, 2E, and S3D). Given that TFs are considered as the main directive factors governing transcriptional programs in T cells, we first focused on the impact of mSWI/SNF complex occupancy and accessibility generation over the expression of TF genes themselves. By integrating gene expression with fractional mSWI/SNF complex occupancy (i.e., the enrichment of mSWI/SNF binding over a given TF motif genome wide, relative to others), we identified AP-1 TF genes FOS, FOSB, FOSL1, ATF3, and BATF3 as upregulated mSWI/SNF targets during early activation and with reduced gene activation but retained fractional mSWI/SNF occupancy at late activation time points (Figures 2F and S3E). Interestingly, in the exhausted state, we identified uniquely high mSWI/SNF occupancy at HNF1B motifs and significant changes in expression of HNF1B (>3 log2FC). In the memory-like state, we identified more subtle changes in TF gene expression, of RUNX1, STAT4, STAT6, TCF7L1, and IRF1 genes coupled with slight changes in mSWI/SNF fractional enrichment (Figures 2F and S3F). Lastly, the visualization of mSWI/SNF complexes (SMARCA4, SS18), H3K27ac, and ATAC-seq signal over key genes for activation (IFNG) and early and late exhaustion (CXCL13 and ENTPD1, encoding CD39) confirmed concomitant regulatory region and/or promoter mSWI/SNF binding and chromatin opening (Figure 2G).
BAF complexes are bound and active over HNF1B TF binding sites genome wide in exhausted T cells
Notably, examining the top 10% differentially upregulated genes across activation, intermediate activation, late activation, exhaustion, and memory states, we found that 23%, 14%, 29%, 40%, and 24% of upregulated gene loci, respectively, were occupied by mSWI/SNF complexes (Figures 3A and S3F). Of note, mSWI/SNF occupancy was present over the greatest percentage (40%) of loci corresponding to genes upregulated at exhaustion (Figure 3A), suggesting a heightened role for mSWI/SNF complexes in the establishment and maintenance of the exhaustion transcriptional signature. Next, we monitored expression changes of key hallmark genes of naive, activated, and exhausted T cells (Figure 3B). Genes known to be expressed in naive T cells (IL7R, TCF7, SELL) had the highest expression at the no stimulation (0 h) time point, whereas levels of hallmark activation genes such as IFNG, IL2, PDCD1, CXCL13, GZMB, and LAG3 were most elevated at the 3–72 h time points (Figures 3B, S3D, and S3F). Importantly, key exhaustion hallmark genes (TOX, ENTPD1, ITGA2, and TIGIT) were upregulated in the exhaustion-like state (Day 6, Day9-Ch) and bound by mSWI/SNF. Additional key exhaustion hallmarks (HAVCR2, PRDM1, CTLA4) were upregulated as secondary (non-mSWI/SNF bound) target genes (Figure 3B). These studies inform the chromatin landscape and gene regulatory signatures across T cell states, highlighting potential mSWI/SNF-directed as well as downstream changes.
Figure 3. Exhaustion-associated gene expression and chromatin targeting is partially mediated by the HNF1B transcription factor.
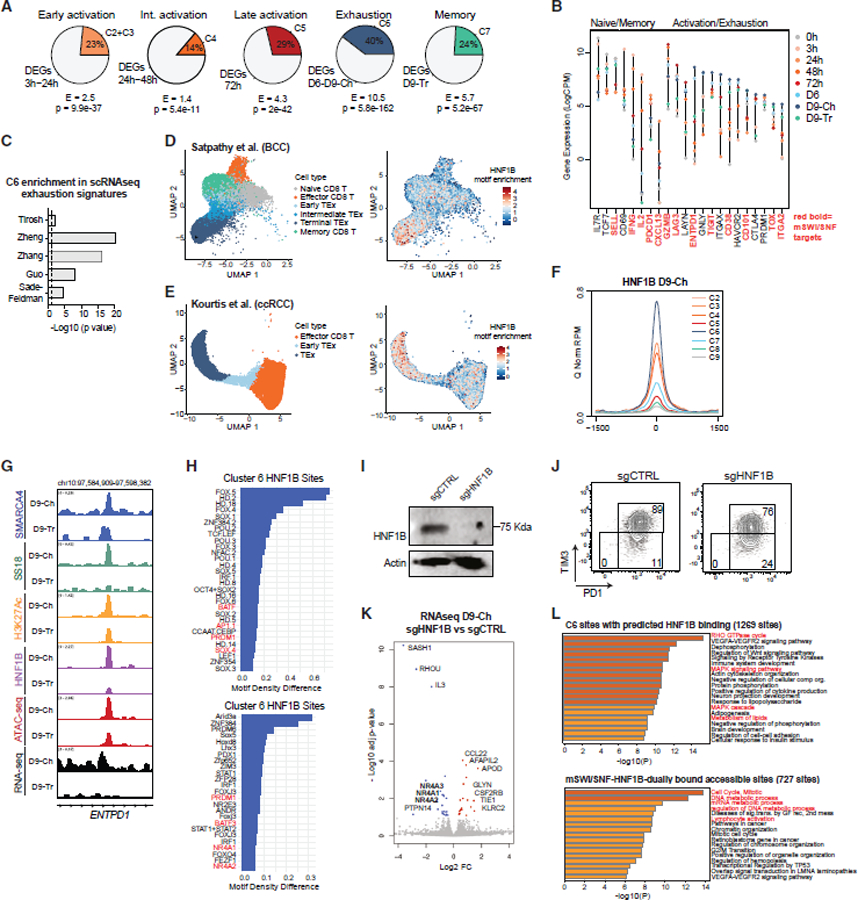
(A) Pie charts representing fractions of the top 10% differentially expressed genes near sites within clusters indicated.
(B) Lollipop plots representing the gene expression (LogCPM) of marker genes for the different states throughout the time course with mSWI/SNF-bound genes indicated.
(C) Enrichment of C6-associated genes across exhaustion signatures from published scRNA-seq datasets.
(D) UMAP projections of 12,643 CD8+ T cells from basal cell carcinoma (BCC) tumor biopsies, clustered by phenotype (left) or colored by HNF1B motif enrichment (right).
(E) UMAP projection of 13,613 CD8+ T cells from clear cell renal cell carcinoma (ccRCC) tumor biopsies, clustered by phenotype (left) or colored by HNF1B motif enrichment (right).
(F) Enrichment of HNF1B (CUT&Tag performed on Day9-Ch T cells) across clusters.
(G) Representative tracks over the ENTPD1 locus.
(H) Motif enrichment over HNF1B target sites in (top) cluster 6 HNF1B target sites and (bottom) all HNF1B target sites.
(I) Western blot for HNF1B and beta-actin in Day9-Ch sgCTRL and sgHNF1B T cells (donor 7).
(J) PD1 and TIM3 immunoprofiling on sgCTRL and sgHNF1B T cells at Day9-Ch.
(G) Volcano plot depicting differential gene expression (RNA-seq) in sgCTRL and sgHNF1B T cells.
(H) Metascape analysis performed over (top) C6 sites with predicted HNF1B binding (>2 motifs) and (bottom) C6 sites with CUT&Tag HNF1B binding, mSWI/SNF occupancy and accessibility.
See also Figure S3.
Given the uniquely abundant collection of mSWI/SNF target genes among exhaustion hallmarks (Figures 3A and 3B), we next compared the observed changes in gene expression with exhaustion and memory signatures derived from single-cell human tumor microenvironment transcriptomic datasets80–84 (Table S1). Across 5 scRNA-seq datasets evaluated, exhaustion signatures were the most highly enriched over our Day 6 and Day9-Ch time points, whereas memory signatures were enriched in the unstimulated and Day9-Tr time points (Figure S3G). Remarkably, cluster 6 genes, characterized by mSWI/SNF occupancy exhaustion time points and enriched in HNF1B motifs, displayed significant enrichment in all exhaustion signatures, indicating that mSWI/SNF directly regulate a collection of genes hallmarking exhaustion (Figures 3C, S3G, and S3H). Further, we extracted scATAC-seq profiles of intra-tumoral T cells from human basal cell carcinoma (BCC, Satpathy et al.) and clear cell renal cell carcinoma (ccRCC, Kourtis et al.) samples. UMAP projections identified distinct T cell subsets (Figures 3D and 3E) and ChromVar analyses revealed enrichment of the HNF1B motif specifically in exhausted T cell states (Figures 3D, 3E, S3I, and S3J).22,60
Owing to these findings, we next profiled the occupancy of HNF1B using CUT&Tag at Day9-Ch and Day9-Tr time points, which exhibit high and low expression of HNF1B, respectively (Figure S3K). Indeed, HNF1B occupancy from Day9-Ch T cells was most enriched over C6 (exhausted) BAF-bound accessible sites (Figures 3F and S3L), exemplified at the ENTPD1 locus (Figure 3G). Archetype and non-archetype motif enrichment analyses performed over HNF1B target sites and over HNF1B sites within C6 revealed the co-enrichment of other exhaustion-associated TFs such as BATF, NR4A1/2, SOX4, PRDM1, and others (Figures 3H and S3M). In line with this, sgRNA-mediated knockout of HNF1B in human CD8+ T cells resulted in reduced TIM3+PD1+ exhausted cells (Figures 3I and 3J). However, HNF1B KO failed to generate a proliferative advantage (Figure S3N). Further, RNA-seq analyses performed on Day9-Ch wild-type (WT) and HNF1B KO T cells revealed substantial downregulation of exhaustion-associated NR4A1-3 genes and the differential expression of cytokine genes such as GLNY (Figure 3K). Other genes less characterized in T cell biology such as SASH1 and RHOU were strongly downregulated. Finally, the analysis of genes near C6 sites with HNF1B motif density and those containing HNF1B binding exhibiting reduced expression included those involved in metabolism, MAPK signaling, and immune system development (Figures 3L and S3O).
Lastly, we identified TF motifs with accessibility changes across the time course in mouse CD8+ T cells. Although the majority of TF motifs enriched under sites of mSWI/SNF occupancy were enriched similarly in human and mouse settings, few notable exceptions included the exhaustion state (C6)-enriched HNF1B (homeodomain-containing Hd.10 factors) motifs, which were specific to human cells (Figures S3P and S3Q). Taken together, these data define the targeting specificities of mSWI/SNF complexes across different T cell states, suggesting their key roles in orchestrating the exhaustion expression signature.
Chromatin-focused CRISPR-Cas9 screens identify cBAF components regulators of T cell exhaustion
We next aimed to unbiasedly characterize the roles for chromatin regulatory factors in modulating the exhaustion hallmarks of chronically stimulated T cells.85–89 We generated lentiviral constructs expressing RFP and a custom sgRNA library targeting 310 known epigenetic regulators, containing 6 sgRNAs per gene, non-targeting sgRNAs as negative controls, and sgRNAs targeting PD1 and HAVCR2 (TIM3) as positive controls (total: 1,928 sgRNAs) (Table S2). We then performed a CRISPR-Cas9 screen in mouse CD8+ T cells purified from Rosa26Cas9-EGFP mice (Figure 4A). Cells were activated for 24 h, transduced with the sgRNAs library, ensuring a proper library expression by day 3 of the activation protocol, then chronically stimulated until day 9. At Day9-Ch, sgRNA-RFP+, PD1+TIM3+ T cells were sorted and sgRNA library representation was compared with the initial library representation. Quality control analyses of deep-sequenced sgRNAs libraries confirmed efficient capture of all sgRNAs (97.6%–99% of all sgRNAs) and an even distribution of sgRNA sequences (Gini indexes: 0.05–0.09) (Figure S4A). We then analyzed the genes whose sgRNAs were enriched or depleted in the PD1+TIM3+ population (Figure 4B; Table S3). Interestingly, most of the significant hits (abs log2FC > 1, false discover rate [FDR] < 0.05) were depleted (n = 27), while only 2 hits in total were enriched, highlighting the diverse chromatin-level contributions to T cell dysfunction. In addition to positive controls Pd1 and Havcr2, depleted hits included genes encoding the mSWI/SNF complexes (Arid1a, Dpf2, Smarcc1, Smarca4), genes involved in histone acetylation (Kat5, Kat8, Ep300, Hdac3), methylation (Kmt2d, Prmt5, Ezh2, Kdm1a, Kdm6a), and other processes, highlighting a range of epigenetic mechanisms playing potential roles in regulating exhaustion immune checkpoints (Figures 4B and S4B). Intriguingly, genes encoding mSWI/SNF complex subunits were the top-scoring genes depleted in the PD1+/TIM3+ population (Arid1a = rank 2, Dpf2 = rank 6, Smarcc1 = rank 10, and Smarca4 = rank 15), specifically, the cBAF complex43 (Figures 4B, 4C, and S4B; Table S3). Arid1b, the paralog for Arid1a, was moderately depleted but to a lesser extent given its lower expression and hence likely lower stoichiometric abundance in cBAF complexes (Figure S4C). PBAF- and ncBAF-specific subunits such as Arid2, Pbrm1, and Brd9 were also depleted but to a minimal extent. Taken together, these data highlight unbiasedly the role of the mSWI/SNF complexes, specifically, Arid1a- and Dpf2-containing cBAF complexes, as among the most significant determinants of exhausted state.
Figure 4. Chromatin-focused CRISPR-Cas9 screens identify cBAF components as regulators of T cell exhaustion.
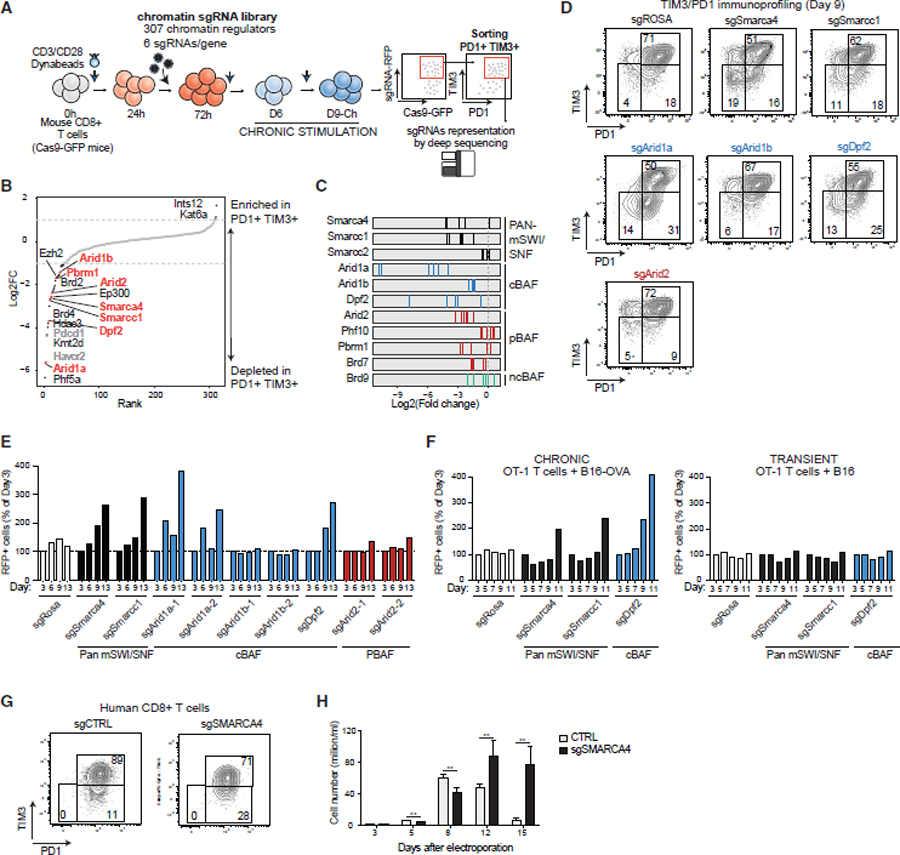
(A) Schematic for CD8+ PD1+/TIM3+ T cell screening using a custom sgRNA library of chromatin regulators.
(B) Rank plot depicting log2FC scores (average of n = 6 guides) targeting chromatin regulator genes and negative/positive controls. Depleted genes are highlighted in black; positive controls are highlighted in gray; mSWI/SNF complex genes are highlighted in orange.
(C) Log2FC values for n = 6 independent guides in PD1+TIM3+ cells.
(D) FACS plots depicting PD1+/TIM3+ T cell populations in control and mSWI/SNF subunit KO conditions.
(E) Bar graph depicting RFP+ cells (% cells of day 3) for control, pan-mSWI/SNF, cBAF, and PBAF genes.
(F) Bar graph depicting RFP+ cells (% cells of day 3) for chronic (+B16+OVA) and transient (+B16) stimulation of OT-1 T cells in each sgRNA condition.
(G) FACS plots depicting PD1+/TIM3+ T cell populations in control and sgSRMARCA4 KO conditions in human CD8+ T cells. Donor 7 was used for this experiment, which was conducted together with the experiment in Figure 3J, thus the same control was used.
(H) Bar graph depicting cell proliferation of human sgCTRL or sgSMARCA4 CD8+ T cells. Error bars represent mean ± SD of 3 technical replicates. **p < 0.01.
See also Figure S4.
To functionally validate top hits, we transduced two independent sgRNA-RFP plasmids into Cas9-EGFP T cells and evaluated for PD1+ and TIM3+ populations. We observed a reduction in the percentage of PD1+TIM3+ cells upon the depletion of all pan-mSWI/SNF and cBAF-specific components at day 9. By contrast, the depletion of Arid2 had a minimal effect (Figures 4D and S4D). Although no significant proliferation differences were identified before day 9, pan-mSWI/SNF and cBAF subunit knockout cells (but not PBAF or ncBAF KO cells) out-competed WT cells at day 9, indicating increased T cell persistence (Figure 4E). We also monitored a slight increase in the percentage of cells with a central memory (CM) phenotype (CD44+ CD62L+) (Figure S4E) and a decrease in the killing capacity in the B16-OVA/OT-1 system at increasing target-effector (T-E) ratios, confirming a memory-like phenotype (Figure S4F). Finally, to confirm these findings, we performed a second CRISPR screen in mouse CD8+ T cells in which we sorted for the top and bottom 15% TIM-3-expressing cells (Figures S4G and S4H), which revealed similar results (Figures S4I–S4K; Table S4).
We next sought to validate these findings using an independent system, based on the co-culture of mouse OT-1 CD8+ T cells with B16 cells expressing the model antigen OVA (chronic stimulation) or B16 WT cells as control (transient stimulation) (Figure S4L). Splenocytes from Rosa26Cas9-EGFP-OT-1 mice were incubated with the MHC-I-specific OVA peptide SIINFEKL for 48 h to achieve T cell activation, then CD8+ T cells were purified, infected with sgRNAs, and co-cultured with B16 or B16-OVA cells (Figure S4L). Here again, we observed a decrease in T cell proliferation at day 9 of stimulation (Figure S4M), upregulated PD1 and TIM3, and an effector phenotype (CD44+ CD62L−), indicative of exhaustion-like features (Figure S4N). Consistent with our previous findings, the depletion of Smarca4, Smarcc1, and Dpf2 led to a decreased percentage of PD1+TIM3+ cells and an increase in the CM population (Figure S4O). Also in this model, we detected an increased persistence of chronically, but not transiently, stimulated T cells upon the sgRNA-mediated depletion of Smarcc1, Smarca4, and Dpf2 (Figure 4F). Finally, to validate these findings in human CD8+ T cells, we electroporated the Cas9 ribonucleoprotein and either control (CTRL) or SMARCA4-targeting sgRNAs in human CD8+ T cells. In the setting of SMARCA4 KO (Figure S4P), we observed a decrease in the percentage of PD1+ TIM3+ T cells (Figure 4G) and increased persistence of human CD8+ T cells (Figure 4H). Taken together, these studies highlight the unique role of mSWI/SNF complexes, particularly cBAF, in T cell exhaustion in concordance with the high degree of mSWI/SNF targeting and activity over exhaustion genes.
Diverse mSWI/SNF ATPase-specific inhibitors and degraders attenuate T cell exhaustion and increase memory phenotypes
We next sought to evaluate the impact of pharmacologic mSWI/SNF perturbation, using both small-molecule allosteric inhibitors of SMARCA4/2 ATPase activity, CMP14 and FHT-1015, as well as degraders of the SMARCA4/2 ATPase protein subunits, ACBI1 and AU-15330, which result in the degradation of the entire 5-subunit ATPase module of mSWI/SNF complexes.43,56,90–93 Of note, an analog of FHT-1015, FHD-286, recently entered Phase I clinical trials for hematologic and solid tumors (NCT04891757 and NCT04879017).
We activated human CD8+ T cells with CD3/CD28 beads for 3 days and added either DMSO (control) or one of the four mSWI/SNF SMARCA4/2-targeting compounds at two different concentrations (50 and 100 nM) for an additional 6 days coupled with chronic stimulation (Figures 5A and S5A). At day 9, treatment with all compounds (ACBI1, AU-15330, CMP14, and FHT-1015) resulted in statistically significant reductions of PD1+/TIM3+ exhausted populations at 51.5%, 51.5%, 20%, and 50%, respectively, compared with DMSO-treated conditions, consistently across donors (Figures 5B, 5C, and S5B). This was coupled with statistically significant increases in activated/progenitor-exhausted T cells (PD1+/TIM3−) and lower levels of CD39, an additional marker of terminal exhaustion (Figures 5B, 5C, S5B, and S5C). The profiling of CD45RA and CCR7 indicated a decrease in the effector T cell pool and an increase in both effector memory (EM) and CM cells upon treatment (Figures S5D–S5F). We also observed decreases in the expression of cytokines (IFNγ, TNFα, or both) of T cells treated with all compounds (Figure S6A). Remarkably, although treatment did not significantly impact cell proliferation or viability until day 9, we observed an increase in persistence in treated cells from multiple donors of different ages and sex across further stimulations (fold differences: ACBI1: 6- to 20-fold; AU-15330: 5- to 13-fold; CMP14: 5- to 17-fold; FHT-1015: 2- to 5-fold) and a decrease in the percentage of Annexin-positive apoptotic cells in the treated conditions (Figures 5D, S6B, and S6C).
Figure 5. Pharmacologic disruption of mSWI/SNF complexes attenuates human T cell exhaustion.
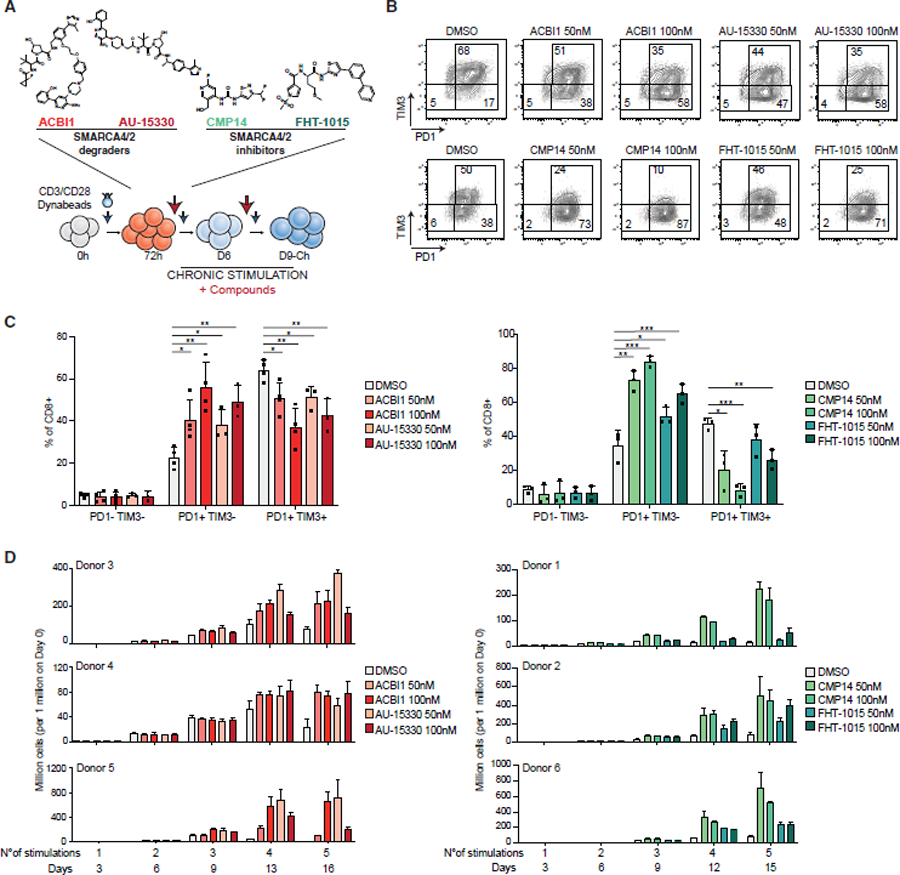
(A) Schematic for inhibitor and degrader experiments with compounds added at day 3 and refreshed (with stimulation) every 3 days.
(B) FACS plots depicting PD1/TIM3 populations in CD8+ T cells at day 9 treated with 50 nM and 100 nM of SMARCA4/2 degraders (donor 3) and inhibitors (donor 1).
(C) Bar graph depicting % of CD8+ T cells in PD1−/TIM3−, PD1+TIM3−, and PD1+TIM3+ populations in DMSO, ACBI1, and AU-15330 (Left) or DMSO, CMP14 and FHT-1015 (right) conditions. Error bars represent mean ± SD of 3 or 4 independent CD8+ T cell donors. Statistical analysis was performed using an unpaired t test.
(D) Bar graphs depicting cell number upon treatment with ACBI1/AU-15330 (left, donors 3, 4, and 5) or CMP14/FHT-1015 (right, donors 1, 2, and 6). See KRT for donor information. Error bars represent mean ± SD of 3 technical replicates per donor. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
See also Figures S5 and S6.
To test whether mSWI/SNF PROTACs or inhibitors could not only prevent the onset of exhaustion but also revert it, we activated human T cells and treated them with mSWI/SNF inhibitors or degraders starting at days 3, 6, or 9. Intriguingly, although treatment starting at day 3 attenuated the onset of exhaustion, as assessed by FACS and cell proliferation, the impact was reduced or absent if the treatment was initiated at day 6 or day 9, respectively (Figures S6D and S6E), suggesting a narrowing window amenable to chromatin landscape modification during the progenitor exhausted-like (Day 6) to terminal exhaustion-like (Day9-Ch) states.
Finally, we performed similar experiments using mouse CD8+ T cells. Also in the mouse T cell context, treatment with CMP14 and FHT-1015 SMARCA4/2 ATPase inhibitors led to a reduction of PD1+TIM3+ cells and in cytokines expression (Figure S6F). Taken together, these results indicate that pharmacologic targeting of the SWI/SNF complex attenuates the onset of exhaustion hallmarks and promotes increased persistence and memory phenotypic features of both human and mouse T cells.
Pharmacologic disruption of mSWI/SNF complex activity alters accessibility over TF motif sites and inhibits T cell exhaustion
To dissect the mechanistic basis for the observed phenotypes, we performed ATAC-seq upon treatment with SMARCA4/2 ATPase degraders and inhibitors during chronic stimulation in human CD8+ T cells. Notably, PCA analyses revealed dramatic differences in chromatin accessibility between control and treated cells at Day9-Ch, consistent between independent donors, with PC1 capturing the impact of SMARCA4/2 pharmacologic perturbation (Figures 6A and S7A). As expected from previous studies, treatment with these compounds resulted in genome-wide decreases in accessibility, with sites affected being highly consistently impacted across all four compound treatments relative to control (Figures 6B and S7A–S7C). Notably, sites decreased in accessibility were enriched for motifs corresponding to the HNF1B TF, underscoring the function of mSWI/SNF complexes in mediating accessibility over HNF1B binding motifs (Figure 6C). In addition, motifs corresponding to BATF, AP1, NF-κB, and FOX were also enriched over sites exhibiting decreased accessibility following treatment (Figure 6C).
Figure 6. mSWI/SNF pharmacological disruption alters chromatin accessibility and TFs recruitment at key T cell activation and exhaustion sites.
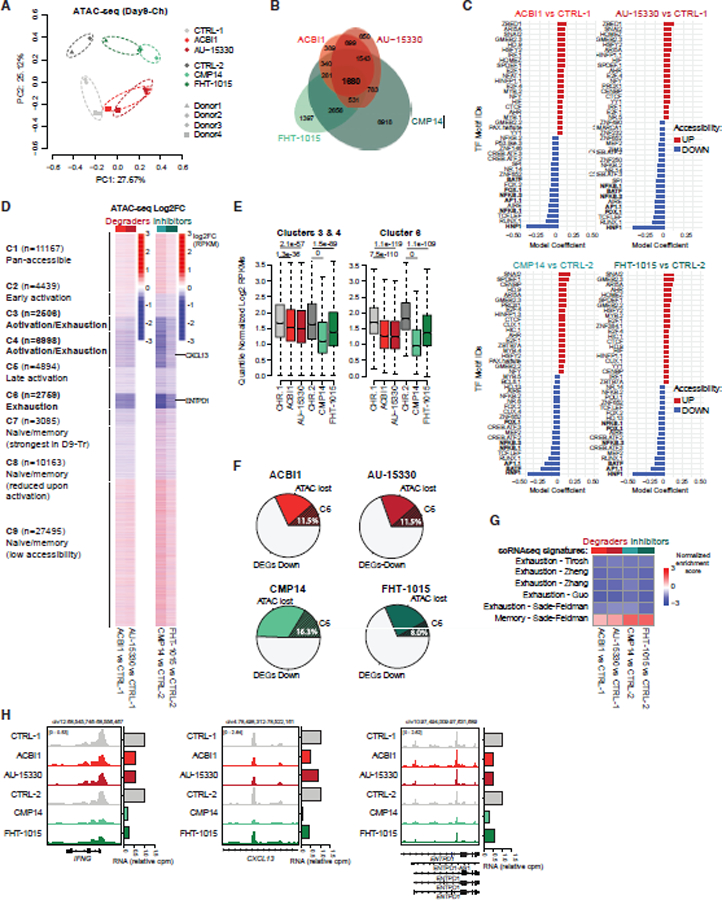
(A) PCA of ATAC-seq profiles of control (CTRL) and ACBI1, AU-15330, CMP14, or FHT-1015-treated human CD8+ T cells (100 nM), at day 9. CTRL-1 and CTRL-2 are the controls for the ACBI1/AU-15330 (donors 3 and 4) and CMP14/FHT-1015 (donors 1 and 2) experiments, respectively.
(B) Venn diagram showing the overlap in sites with decreased accessibility (logFC < −1) upon treatment with ACBI1, AU-15330, CMP14, or FHT-1015 (100 nM).
(C) Top 40 coefficients of logistic regression models fitting motif counts across all sites to changes in accessibility for indicated comparisons.
(D) Heatmap showing the log2 fold-change of accessibility upon ACBI1, AU-15330, CMP14, or FHT-1015 treatment compared with control in the 9 clusters identified in Figure 1.
(E) Quantification of chromatin accessibility (quantile-normalized log2 RPKM) at sites within clusters 3 and 4 and cluster 6. CTRL-1 and CTRL-2 are the controls for the ACBI1/AU-15330 and CMP14/FHT-1015 experiments, respectively. P values were computed using standard t tests.
(F) Pie charts representing percentages of downregulated genes after treatment near cluster 6 (C6) sites with strong decreases in accessibility (log2FC < −1).
(G) Gene set enrichment analysis (GSEA) analysis of exhaustion and memory signatures derived from scRNA-seq datasets in the selected comparisons.
(H) Representative SMARCA4, SS18, H3K27ac C&T, and ATAC-seq tracks over the IFNG, CXCL13, and ENTPD1 loci.
See also Figure S7.
Chromatin accessibility changes upon all treatments compared with control were highly concordant across the 9 clusters identified (Figures 1, 6D, and 6E). Importantly, C6 exhaustion-associated sites exhibited decreases in the accessibility of the highest significance and magnitude, whereas C3 and C4 sites, which included sites broadly accessible during activation and exhaustion, decreased with lower fold changes (Figures 6D and 6E), indicating that mSWI/SNF ATPase inhibition most strongly suppressed the accessibility over regions enriched for exhaustion-associated genes as well as selected activation-associated genes (Figure 6F). Interestingly, mSWI/SNF disruption moderately impacted accessibility (both increases and decreases) over naive/memory-associated sites (C7 and C8), consistent with the fact that these cells gain some but not all memory-like features. Reflecting this, genes near sites with increased accessibility included TCF7, ID3, and KLF2, and those near sites with reduced accessibility included SELL and BHLHE40 genes (Figures 6D–6F). Intriguingly, accessibility was markedly increased over genes within C1 and C9 clusters, in agreement with previous findings that cBAF complex perturbations result in enhanced abundance and function of ncBAF complexes over CTCF motifs, which we demonstrated were strongly enriched in the C9 cluster (Figures 2A, 2B, and S3B).56
To further investigate the impact of mSWI/SNF pharmacologic inhibition on the attenuation of exhaustion-like states, we next performed RNA-seq analyses to reveal changes in gene expression programs (Figures S7D and S7E). PCA analysis revealed similarly changed profiles and high concordance between differentially expressed genes across treatment conditions (Figures S7D, S7F, and S7G). Downregulated genes were enriched for those involved in immune-related pathways, key activation and exhaustion TFs, IFNγ response, and TNFα signaling via NF-κB (Figure S7H). A subset of genes downregulated upon mSWI/SNF inhibition was contained within genomic regions losing DNA accessibility upon compound treatment (30.5%–49.6%), a subset of which (8%–16.3% of total downregulated genes) were genes bound by mSWI/SNF in the exhausted state (C6) (Figure 6F). Examples included ENTPD1, TIGIT, IFNG, and GZMB, which demonstrated decreased accessibility and gene expression upon compound treatment (Figure S7I). Interestingly, we also observed chromatin opening and enhanced gene expression at a smaller set of sites (Figures S7A–S7C and S7E–S7I), exemplified over genes such as IRF1 (Figure S7J), which may indicate a set of factors that facilitate the increased T cell persistence.94
Finally, we applied the C6 mSWI/SNF perturbation signature to differentially accessible and differentially expressed genes with the exhaustion and memory signatures derived from human scRNA-seq datasets.80–84 Remarkably, treatment with all four distinct mSWI/SNF-disrupting compounds showed negative enrichment exhaustion signature genes, whereas memory genes increased in expression (Figures 6G and 6H). These data support a mechanism whereby mSWI/SNF inhibition or degradation attenuates the activation status of T cells, preventing them from undergoing exhaustion, and enables the maintenance of the memory-like phenotype with sustained proliferation capability over time.
Finally, we performed ATAC-seq in mouse CD8+ T cells treated with SMARCA4/2 ATPase inhibitors and found that sites reduced in accessibility were enriched for similar motifs, such as those corresponding to BATF, MYB, E2F, and NFY factors, but not for HNF1B, confirming its specificity to human cells. Importantly, examining the gene expression changes across clusters of mSWI/SNF occupancy and accessibility in mouse cells (Figure S2J), we identified the most strongly downregulated genes were again those in C6, corresponding to the exhaustion state (Figures S7K and S7L).
Pharmacologic disruption of mSWI/SNF increases in vitro persistence during CAR-T cell generation and enhances T cell-mediated anti-tumor efficacy in vivo
CAR-T infusion products displaying decreased exhaustion and increased memory hallmarks have been demonstrated to have increased efficacy in vivo.89,95–100 With the above results, we reasoned that pharmacologic mSWI/SNF complex perturbation may represent a viable approach to improve T cell fitness and prevent exhaustion during CAR-T cell manufacturing.101 CAR-Ts are routinely generated from both CD4+ and CD8+ T cells at variable ratios and expanded using beads similar to those used in our in vitro exhaustion experiments.102,103 As CD4+ T cells have been implicated to be longer-lasting relative to CD8+ cells, detected even decades after tumor remission, we sought to understand whether CD4+ T cells displayed similar increased persistence as CD8+ T cells in response to mSWI/SNF inhibition.101,104,105 Treatment with the mSWI/SNF ATPase inhibitor and degrader compounds led to a significant reduction in PD1+TIM3+ and CD39+ populations and increased CD4+ T cell persistence (Figures 7A–7C), indicating that these treatments have the same outcome in both CD8+ and CD4+ lineages.
Figure 7. mSWI/SNF targeting improves T cell-based cancer immunotherapy approaches.
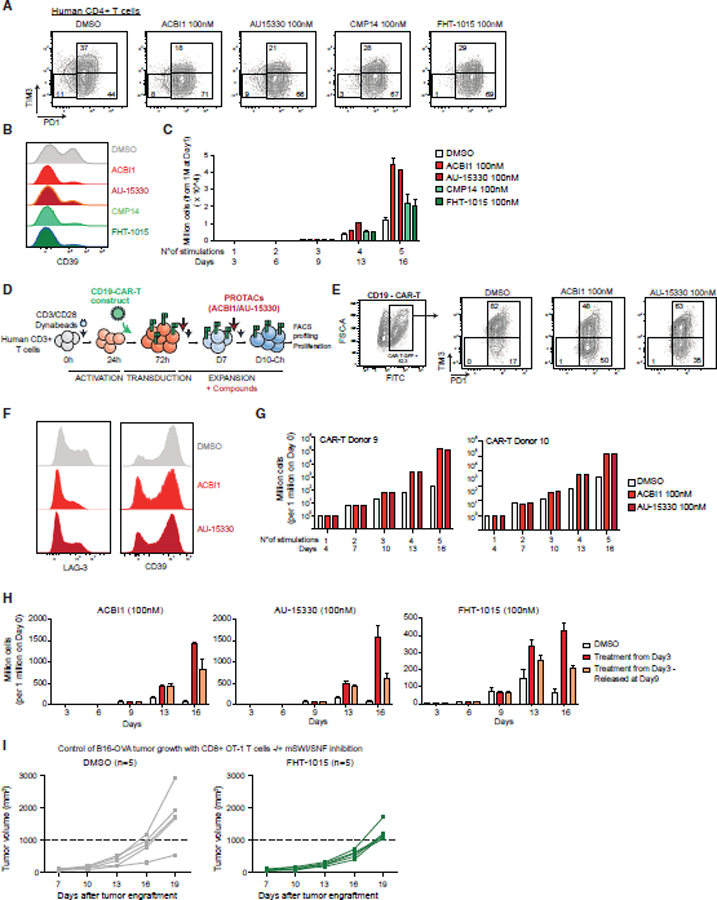
(A) FACS plots depicting PD1/TIM3 populations in DMSO, ACBI1, AU-15330, CMP14, and FHT-1015 conditions (100 nM), at day 9, for one human CD4+ T cell donor (donor 8).
(B) FACS plot depicting the profiling of CD39 in human CD4+ T cells treated with DMSO, ACBI1, AU-15330, CMP14, or FHT-1015 (100 nM), at day 9.
(C) Bar graph depicting human CD4+ T cell number upon treatment with DMSO, ACBI1, AU-15330, CMP14, or FHT-1015 (100 nM). Error bars represent the mean ± SD of 3 technical replicates of one donor.
(D) Schematic for CD19-CAR-T cell generation, stimulation, and treatments.
(E) FACS plots depicting CD19-CAR-T-GFP cells identification and PD1/TIM3 populations, in cells treated with ACBI1 or AU-15330 (donors 9 and 10).
(F) FACS histograms of LAG-3 and CD39 expression in CAR-T cells treated with DMSO, ACBI1, or AU-15330.
(G) Bar graphs depicting CAR-T cell number upon treatment with DMSO, ACBI1, or AU-15330 (100 nM). Error bars represent the mean for each donor.
(H) Bar graphs depicting cell number upon treatment with ACBI1, AU-15330, or FHT-1015 (all 100 nM) at day 3 onward or at day 3 with treatment washout at day 9. Error bars represent mean ± SD of 3 technical replicates of one donor.
(I) In vivo B16 melanoma tumor growth curves in mice injected with DMSO or FHT-1015-treated CD8+ OT-1 T cells. Two-way ANOVA generated p value for comparison between the two groups’ means at day 19: p < 0.05.
See also Figure S7.
We then generated CD19-CAR-T cells using total human CD3+ T cells and engineered to express a CAR-T construct targeting the CD19 antigen (Figures 7D and S7M). Cells were expanded for 2 additional days, then treated with either DMSO or SMARCA4/2 degraders, and analyzed at day 10 (Figures 7D–7F). Remarkably, the treatment of CAR-T cells with mSWI/SNF ATPase degraders resulted in the marked depletion of PD1+TIM3+, LAG3+, and CD39+ populations (Figures 7D–7F) and increased persistence across two independent human T cell donors (Figure 7G). These results suggest potential approaches in which CAR-T cells are expanded in the presence of SMARCA4/2 degraders or inhibitors, prior to infusion into patients. In an attempt to assess the potential duration of proliferative advantage of treated T cells once injected in vivo (where no inhibitor would be present), we released treatment on day 9 and monitored proliferation. Cells retained a significant proliferation advantage compared with DMSO-treated cells for at least 1 week after treatment release (Figure 7H).
Finally, to assess the anti-tumor functionality of mSWI/SNF-inhibited T cells, we implemented an OVA antigen-expressing melanoma model (B16) with a subsequent infusion of CD8+ T cells from OT-1 TCR transgenic mice. Intriguingly, the pre-treatment of CD8+ OT-1 T cells with the FHT-1015 SMARCA4/2 ATPase inhibitor resulted in decreased levels of day 9 in vitro cell killing at 24 and 48 h time points across a range of target:effector ratios relative to control T cells (Figure S7N), consistent with results using cBAF subunit genetic depletion experiments. Importantly, OT-1 T cells pretreated with FHT-1015 attenuated B16-OVA tumor growth in vivo relative to DMSO control-treated cells (Figure 7I). Collectively, these experiments demonstrate that T cells subjected to mSWI/SNF inhibition exhibit increased persistence resulting in enhanced anti-tumor activity.
DISCUSSION
In this study, we dissected the contribution of mSWI/SNF chromatin remodeling complexes targeting and activity to gene expression programs and functional phenotypes during T cell activation and exhaustion. Our efforts to probe the mechanistic contributions of mSWI/SNF complexes were inspired by the known extensive changes in chromatin accessibility during T cell activation and exhaustion and in CAR-T cell populations,14–22 coupled with work by our group and others establishing mSWI/SNF complexes as major mediators of the establishment and maintenance of tissue-specific chromatin accessibility.56,58,62,106,107 Further, recent CRISPR-based screening studies begun to reveal roles for mSWI/SNF complexes in T cell exhaustion,35,36 presenting opportunities to define their functional contributions.
Our studies describe the chromatin, gene regulatory, phenotypic, and in vitro and in vivo functional impact of four independent inhibitors and degraders that have been biochemically and structurally confirmed to specifically target the mSWI/SNF SMARCA2/4 ATPases. Underscoring the clinical relevance of our findings, the FHT-1015 compound is an analog of the phase I compound, FHD-286, currently being evaluated in human acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), and uveal melanoma.93 With these agents, we find highly consistent chromatin accessibility and gene regulatory impacts, suggesting that mSWI/SNF catalytic activity is equivalent to assembly and function of the entire ATPase module. The degradation of SMARCA4/2 prevents assembly of ACTL6A, beta-actin, SS18, and BCL7 family subunits on to mSWI/SNF family complexes.43,108,109
Based on a range of studies by our group and others, we suggest a model where the activities of mSWI/SNF complexes are controlled by the repertoire of TFs expressed in a given cell type, which collectively guides their positioning. Indeed, the pharmacologic inhibition of mSWI/SNF complexes results in the closing of previously accessible, complex-targeted regions enriched in DNA motifs corresponding to highly specific TFs (Figures 6C and 6D). In addition, small-molecule inhibition resulted in minimal cell viability impacts, underscoring the preferential role for mSWI/SNF complexes over sites important for the exhaustion program rather than those supporting cell homeostatic programs (Figures 5D, 7D–7H, S6B, and S6C). These data, coupled with immunophenotyping indicating decreased T cell exhaustion, suggest the favorable utility of mSWI/SNF inhibitors in the setting of ex vivo-manipulated CAR-T cells. The use of small-molecule inhibitors represents a more facile, lower-cost, and safer approach to improve CAR-T cells fitness, contrary to the challenges and safety concerns associated with genetic strategies such as DNMT3A deletion.8,110,111 Intriguingly, in both of our screens, Dnmt3a, which has been previously shown to contribute to T cell exhaustion, was not depleted relative to mSWI/SNF genes, indicating relative contributions of these distinct epigenetic regulators (Figures 4 and S4; Tables S3 and S4)8,34,112,113
Of note, our studies suggest that mSWI/SNF inhibition can prevent T cell exhaustion but may not be able to revert it (Figures S6D and S6E). This indicates that mSWI/SNF complexes may facilitate the stable binding of important TFs to exhaustion sites—once the exhaustion program has been triggered, mSWI/SNF inhibition may not be sufficient to displace those TFs. Additional studies employing different time points at which mSWI/SNF inhibitors are introduced will be needed to comprehensively address whether mSWI/SNF perturbation can affect already exhausted cells and reinvigorate tumor-infiltrating T cells in vivo.
We identify here several previously unknown genomic features of the exhausted T cell state as well as determinants of exhaustion-specific mSWI/SNF targeting, such as the connection between mSWI/SNF occupancy and the strong enrichment of HNF1B sites. HNF1B is a heterodimeric TF (heterodimerizes with HNF1A) and was originally characterized for its functions in the development of the pancreas and liver and in controlling insulin production.114 Of note, we found that HNF1B is not expressed in mouse T cells (Figure S3K), which may reconcile why HNF1B was not previously identified in any CRISPR screen, all of which have been performed in mouse T cell settings. For HNF1B in particular, mice with heterozygous mutations in HNF1B show no phenotype relative to that seen in humans.114 In solid tumors, HNF1B regulates glucose uptake, glucose metabolism, and mitochondrial function.102,103,115–118 Exhausted T cells are known to have rewired glucose metabolism, a higher rate of glycolysis and impaired oxidative phosphorylation.103 Gene ontology (GO) analysis of HNF1B targets identified the MAPK signaling pathway and metabolic pathways as enriched processes (Figure 3L), pointing toward a connection between HNF1B and T cell metabolism that could be further explored.
In summary, our study presents a comprehensive dissection of the mechanisms by which mSWI/SNF complexes regulate T cell activation and exhaustion, serving as a valuable resource for the field and advancing new clinically relevant strategies for immunotherapy improvement.
Limitations of the study
Although our results suggest that cBAF complexes specifically mediate T cell exhaustion, SMARCA4/2 inhibitors and degraders target all mSWI/SNF family sub-complexes, thus we cannot exclude the contributions of ncBAF and PBAF activities. Further, although we have accomplished an extensive repertoire of genomic studies in primary T cells, we were unable to biochemically identify and characterize TF-BAF complex protein-protein interactions owing to limitations in cell numbers. Additionally, the experiments presented here were performed across donors with varied naive:memory T cell ratios—further work will be required to compare the impact of cBAF disruption in human naive versus memory T cell populations. Finally, in vivo studies assessing the impacts of mSWI/SNF inhibitors and degraders may pose challenges in uncoupling tumor cell intrinsic and T cell microenvironmental changes.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Cigall Kadoch (cigall_kadoch@dfci.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Genomics data (CUT&Tag, ATAC-seq, and RNA-seq) have been deposited at GEO under accession number GSE212357 and are publicly available as of the date of publication. This paper also analyzes existing, publicly available data (GSE181062 and GSE181064 key resources table). Unprocessed blot images have been deposited in Mendeley (Mendeley data: https://doi.org/10.17632/7msw2d62r6.1). See Key Resources Table for details.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead, Cigall Kadoch, upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Brilliant Violet 605™ anti-human CD279 (PD-1) | BioLegend | Cat# 329923; RRID: AB_11124107 |
| APC/Cyanine7 anti-human CD366 (Tim-3) | BioLegend | Cat# 345025; RRID: AB_2565716 |
| Brilliant Violet 605™ anti-human CD197 (CCR7) | BioLegend | Cat# 353223; RRID: AB_11124325 |
| APC/Cyanine7 anti-human CD45RA | BioLegend | Cat# 304127; RRID: AB_10708419 |
| APC anti-human CD39 | BioLegend | Cat# 328209; RRID: AB_1953233 |
| APC/Cyanine7 anti-mouse CD279 (PD-1) | BioLegend | Cat# 135223; RRID: AB_2563522 |
| PerCP/Cyanine5.5 anti-mouse CD366 (Tim-3) | BioLegend | Cat# 119717; RRID: AB_2571934 |
| APC anti-mouse CD62L | BioLegend | Cat# 104411; RRID: AB_313098 |
| PerCP/Cyanine5.5 anti-mouse/human CD44 | BioLegend | Cat# 103032; RRID: AB_2076204 |
| PE anti-human TNF-α | BioLegend | Cat# 502908; RRID: AB_315260 |
| Alexa Fluor® 700 anti-human IFN-γ | BioLegend | Cat# 506515; RRID: AB_961351 |
| APC anti-mouse TNF-α | BioLegend | Cat# 506307; RRID: AB_315429 |
| PE/Cyanine7 anti-mouse IFN-γ | BioLegend | Cat# 505825; RRID: AB_1595591 |
| PE/Cyanine7 anti-human CD3 | BioLegend | Cat# 317333; RRID: AB_2561451 |
| APC/Cyanine7 anti-mouse CD8a | BioLegend | Cat# 100714; RRID: AB_312753 |
| FITC anti-human CD3 | ThermoFisher Scientific | Cat#11-0038-42; RRID: AB_2043831 |
| PE/Cy7 anti-human CD8 | Biolegend | Cat# 344712; RRID: AB_2044007 |
| Alexa Fluor® 700 anti-human CD4 | Biolegend | Cat# 317426; RRID: AB_571942 |
| Mouse Anti-Actin (clone C4) | Millipore | Cat# MAB1501; RRID: AB_2223041 |
| PE/Cyanine7 anti-human CD25 | BioLegend | Cat# 302612; RRID: AB_314282 |
| Rabbit Anti-IgG Donkey Polyclonal Antibody (HRP) | Cytiva | Cat# NA934-1ML; RRID: AB_772206 |
| Normal Rabbit IgG | Cell Signaling Technology | Ca# 2729S; RRID: AB_1031062 |
| Anti-Histone H3 (acetyl K27) | Abcam | Cat# ab4729; RRID: AB_2118291 |
| Rabbit Anti-SMARCA4 (Brg1) (D1Q7F) | Cell Signaling Technology | Cat# 49360S; RRID: AB_2728743 |
| Rabbit Anti-SS18 (D614Z) | Cell Signaling Technology | Cat# 21792S; RRID: AB_2728667 |
| Rabbit Anti-ARID1A (D2A8U) | Cell Signaling Technology | Cat# 12354S; RRID: AB_2637010 |
| Rabbit Anti-PBRM1 (91894) | Cell Signaling Technology | Cat# 91894S; RRID: AB_2800173 |
| Rabbit Anti-HNF1B | Proteintech | Cat# 12533-1-AP; RRID: AB_2116758 |
|
Bacterial and virus strains | ||
| MegaX DH10B electro-competent bacteria | Life Technologies | Cat# C640003 |
|
Biological samples | ||
| Primary Human CD8+ T cells isolated from PBMCs (Donor 1: 24y/o, Female; Donor 2: 35y/o, Male; Donor 5: 24y/o, Male) | StemCell Technologies Inc | Cat# 70027 |
| Primary Human CD8+/CD4+ T cells isolated from PBMCs (Donor 3: 28y/o, Female; Donor 4: 19y/o, Female; Donor 6: 28y/o, Female; Donor7: <30y/o, N/A; Donor8: <30y/o, Male) | New York Blood Center | https://www.nybc.org/ |
| Primary Human CD3+ T cells isolated from PBMCs: (Donor 9: 43y/o, Male; Donor 10: 42y/o, Female) | Indiana University | N/A |
|
Chemicals, peptides, and recombinant proteins | ||
| Zombie Aqua™ Fixable Viability Kit | BioLegend | Cat# 423101 |
| Zombie Violet™ Fixable Viability Kit | BioLegend | Cat# 423113 |
| eBioscience Foxp3/Transcription Factor Staining Buffer Set kit | Life Technologies | Cat# 00-5523-00 |
| BsmBI-v2 | New England Biolabs | Cat# R0739L |
| Anza Esp1 enzyme | Life Technologies | Cat# IVGN0136 |
| T4 DNA ligase | New England Biolabs | Cat# M0202L |
| ATP | New England Biolabs | Cat# PO756S |
| Plasmid safe ATP-dependent DNase | Thermo Fisher Scientific | Cat# NC9046399 |
| TaKaRa Ex Taq DNA Polymerase | Takara Bio | Cat# RR001B |
| Polyethylenimine (PEI) | Thermo Fisher Scientific | Cat# NC1014320; CAS: 9002-98-6 |
| Polybrene | Santa Cruz Biotechnology | Cat# SC-134220; CAS: 28728-55-4 |
| TaKaRa Ex Taq DNA Polymerase | Takara Bio | Cat# RR001B |
| 10mM 2-Mercaptoethanol | Life Technologies | Cat# 21985023; CAS: 60-24-2 |
| T4 PNK | New England Biolabs | Cat# VWR #101228-174 |
| T4 ligase | New England Biolabs | Cat# M0202M |
| SIINKEFL peptide (OVA 257-264) | Invivogen | Cat# vac-sin |
| ACBI1 | SelleckChem | Cat# S9612; CAS: 2375564-55-7 |
| AU-15330 | MedChem Express LLC | Cat #HY-145388; CAS: 2380274-50-8 |
| Cas9-GFP ribonucleoprotein | Integrated DNA Technologies | Cat#10008100 |
| CMP14 | Courtesy of Jun Qi; Papillon et al.90 | N/A |
| FHT-1015 | MedChemExpress® | Cat# HY-144896; CAS: 2368903-18-6 |
| Recombinant Human IL-2 | Peprotech, Inc. | Cat# 200-02-1MG |
| Recombinant Human IL-7 | Peprotech, Inc. | Cat# 200-07-50UG |
| Recombinant Murine IL-2 | Peprotech, Inc. | Cat# 212-12-50UG |
| Recombinant Murine IL-7 | Peprotech, Inc. | Cat# 212-17-50UG |
| Recombinant Murine IFNg | Peprotech, Inc. | Cat# 315-05 |
| APC Annexin V | BioLegend | Cat# 640920 |
|
Critical commercial assays | ||
| QIAquick Gel Extraction Kit | Qiagen | Cat# 28706X4 |
| Phusion High-Fidelity PCR Kit | Life Technologies | Cat# F553S |
| MinElute Reaction Cleanup Kit | Qiagen | Cat# 28204 |
| PureLink™ HiPure Plasmid Filter Maxiprep Kit | Thermo Fisher Scientific | Cat#K21001 |
| QIAamp DNA Mini Kit | Qiagen | Cat# 51304 |
| PureLink™ HiPure Plasmid Filter Maxiprep Kit | Thermo Fisher Scientific | Cat# K21001 |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74136 |
| Nebnext Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | Cat# E7490 |
| NEXTFLEX® Poly(A) Beads 2.0 | Perkin Elmer | Cat# NOVA-512991 |
| Nebnext Ultra II Directional RNA Library Prep Kit | New England Biolabs | Cat# E7760 |
| NEXTFLEX® Rapid Directional RNA-Seq Kit 2.0 | Perkin Elmer | Cat# NOVA-5198-01 |
| Qiagen MinElute Reaction clean up kit | Qiagen | Cat# 28206 |
| NextSeq™ 500/550 High output flow cell kit | Illumina | Cat# 20024906 |
| CellTiterGlo | Promega | Cat# G7571 |
|
Deposited data | ||
| Cut&Tag-seq, RNA-seq, and ATAC-seq | This study | GSE212357 |
| scATAC-seq | Sathpathy et al.60 | GSE181062 |
| scATAC-seq | Kourtis et al.22 | GSE181064 |
| Unprocessed blot images | This study | Mendeley data: https://doi.org/10.17632/7msw2d62r6.1 |
|
Experimental models: Organisms/strains | ||
| Mice (C57BL/6J) | The Jackson Laboratory | Jackson strain #000664; RRID: IMSR_JAX:000664 |
| OT-1 mice (C57BL/6-Tg(TcraTcrb)1100Mjb/J) | The Jackson Laboratory | Jackson strain #003831; RRID: IMSR_JAX:003831 |
| Cas9 Mice (Gt(ROSA)26Sortm1.1 (CAG-cas9*,-EGFP)Fezh/J) | The Jackson Laboratory | Jackson strain #024858; RRID: IMSR_JAX:024858 |
| Rag1KO mice B6.129S7-Rag1<tm1Mom>/J | The Jackson Laboratory | Jackson strain #002216; RRID: IMSR_JAX:002216 |
|
Oligonucleotides | ||
| Mouse sgRNAs oligo pool targeting epigenetic genes | Twist Bioscience | https://www.twistbioscience.com/products/oligopools |
| sgROSA (mouse) 5’-AACGGCTCCA CCACGCTCGG-3’ | This study | N/A |
| sgSMARCA4-1 (mouse, sg1553 in library) 5’- ATAATGGCCTACAAGATGT- 3’ | This study | N/A |
| sgSMARCA4-2 (mouse, sg1554 in library) 5’- ATTGCCCGACCACCTGCAGA- 3’ | This study | N/A |
| sgSMARCC1-1 (mouse, sg1563 in library) 5’- TGCCCCAAGAATGTGACACA- 3’ | This study | N/A |
| sgSMARCC1-2 (mouse, sg1564 in library) 5’- TTGGTGACATGCTTCCCAA- 3’ | This study | N/A |
| sgARID1A-1 (mouse, sg1844 in library) 5’- TGCACCACAAGCACCCAGAG- 3’ | This study | N/A |
| sgARID1A-2 (mouse, sg1845 in library) 5’- GTCACTGAGGAAGCGCACCA- 3’ | This study | N/A |
| sgARID1B-1 (mouse, sg1851 in library) 5’- GCTCAGCACCCCGTACCCCG- 3’ | This study | N/A |
| sgARID1B-2 (mouse, sg1853 in library) 5’- CAGCAGAGCAGCCCATACCC- 3’ | This study | N/A |
| sgDPF2-1 (mouse, sg0385 in library) 5’- TCAGCAGATCCAGACACAGG- 3’ | This study | N/A |
| sgDPF2-2 (mouse, sg0386 in library) 5’- AAGGAGTGAGACAGTACATG- 3’ | This study | N/A |
| sgARID2-1 (mouse, sg1854 in library) 5’- GCCGTTTAAGAAGATCCCTG- 3’ | This study | N/A |
| sgARID2-2 (mouse, sg1858 in library) 5’- ACCAGAGTCACTACTTTAGG- 3’ | This study | N/A |
| SMARCA4 sgRNAs cocktail (human) 5’-ACUCCAGACCCACCCCUGGG-3’5’- CCCUAGCCCGGGUCCCUCGC-3’5’- GUCCUGCUGAGGGCGGCCCU-3’ | Synthego | https://www.synthego.com/products/crispr-kits/gene-knockout-kit |
| HNF1B sgRNAs cocktail (human) 5’-AGCCCUCGUCGCCGGACAAG-3’5’- GGCCGAGCCCGACACCAAGC-3’5’- CGGGGUCACCAAGGAGGUGC-3’ | Synthego | https://www.synthego.com/products/crispr-kits/gene-knockout-kit |
|
Recombinant DNA | ||
| pLKO5.sgRNA.EFS.tRFP plasmid | Addgene | Cat #57823; RRID: Addgene_57823 |
| psPAX2 plasmid | Addgene | Cat#12260; RRID: Addgene_12260 |
| pMD2G plasmid | Addgene | Cat#12259; RRID: Addgene_12259 |
| Epigenetic mouse CRISPR library cloned in pLKO5.sgRNA.EFS.tRFP plasmid | This study | N/A |
| Anti-CD19-FMC63scFV-41BB-GFP | This study | N/A |
|
Software and algorithms | ||
| MAGECK | MAGeCKFlute package119 | https://sourceforge.net/p/mageck/wiki/Home/. |
| STAR v2.5.2b | Dobin et al.120 | https://github.com/alexdobin/STAR; RRID: SCR_004463 |
| deepTools v2.5.3 | Ramírez et al.121 | https://deeptools.readthedocs.io/en/develop/; RRID: SCR_016366 |
| Trimmomatic v0.36 | Bolger et al.122 | http://www.usadellab.org/cms/?page=trimmomatic; RRID: SCR_011848 |
| Bowtie2 v2.29 | Langmead and Salzberg.123 | https://bowtie-bio.sourceforge.net/index.shtml; RRID: SCR_016368 |
| Picard v2.8.0 | Picard.124 | https://github.com/broadinstitute/picard; RRID:SCR_006525 |
| SAMtools v 0.1.19 | Li et al.125 | https://samtools.sourceforge.net; RRID: SCR_002105 |
| MACS2 v2.1.1 software | Zhang et al.126 | https://pypi.org/project/MACS2/; RRID: SCR_013291 |
| UCSC utilities | Kuhn et al.127 | https://genome.ucsc.edu/util.html; RRID: SCR_005780 |
| CutRunTools | Zhu et al.128 | https://zenodo.org/record/3374112#.Y22KTy2B3xs |
| edgeR v3.12.1 | Robinson et al.129 | https://bioconductor.org/packages/release/bioc/html/edgeR.html; RRID: SCR_012802 |
| GSEA | Subramanian et al.130 | https://www.gsea-msigdb.org/gsea/index.jsp; RRID: SCR_003199 |
| DESeq2 v1.30.1 | Love et al.131 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html; RRID: SCR_015687 |
| corpcor | Opgen-Rhein and Strimmer132; Schäfer and Strimmer.133 | https://cran.r-project.org/web/packages/corpcor/index.html |
| Bedtools multiIntersectBed and mergeBed functions | Quinlan and Hall134 | https://bedtools.readthedocs.io/en/latest/ |
| ChIPpeakAnno v3.17.0 | Zhu et al.135 | https://bioconductor.org/packages/release/bioc/html/ChIPpeakAnno.html; RRID: SCR_012828 |
| LOLA v1.12.0 | Sheffield and Bock136 | https://bioconductor.org/packages/release/bioc/html/LOLA.html; RRID: SCR_006912 |
| HOMER v4.9 | Heinz et al.137 | http://homer.ucsd.edu/homer/; RRID: SCR_010881 |
| non-redundant archetype consensus motifs | Vierstra et al.138 | https://www.vierstra.org/resources/motif_clustering |
| GLMnet | Friedman et al.139 | https://glmnet.stanford.edu/index.html; RRID: SCR_015505 |
| ChromVAR | Schep et al.140 | https://greenleaflab.github.io/chromVAR/index.html |
|
Other | ||
| Lymphoprep™ | STEMCELL Technologies Inc | Cat# 07811 |
| Pan T Cell Isolation kit, Human | Miltenyi Biotec | Cat# 130-096-535 |
| CD8+ T Cell Isolation Kit, Human | Miltenyi Biotec | Cat# 130-096-495 |
| CD4+ T Cell Isolation Kit, Human | Miltenyi Biotec | Cat# 130-096-533 |
| CD8a+ T Cell Isolation Kit, Mouse | Miltenyi Biotech | Cat #130-104-075 |
| Human T-Activator CD3/CD28 Dynabeads | Thermo Fisher Scientific | Cat# 11132D |
| Mouse T-Activator CD3/CD28 Dynabeads | Thermo Fisher Scientific | Cat# 11453D |
| ACK lysis buffer | Quality Biological Inc | Cat# 118156101C |
| 1X GlutaMAX | Life Technologies | Cat# 35050061 |
| 1X Non-essential Amino Acids | Life Technologies | Cat# 11140050 |
| 1X Sodium Pyruvate | Thermo Fisher Scientific | Cat# MT25000CI |
| Cell Stimulation Cocktail | Affymetrix | Cat# 00-4970-93 |
| Human TruStain FcX™ | Human TruStain FcX™ | Cat# 422302 |
| Ampure beads | Thermo Fisher Scientific | Cat# NC9933872 |
| Concanavalin A | BioMag®Plus | Cat# 86057 |
| CUTANA pAG-Tn5 | Epicycpher | Cat# 15-1117 |
| 4%−12% Bis-Tris polyacrylamide gels | Life Technologies | Cat# NP0336BOX |
| Immobilon PVDF Transfer Membrane | Millipore | Cat# IPVH00010 |
| eBioscience Brefeldin A Solution | Life Technologies | Cat# 00-4506-51 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Life Technologies | Cat# 34095 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary T cells
Human peripheral blood mononuclear cells (PBMCs) isolated from 19- to 43-year-old male and female healthy donors were obtained through the New York Blood Center (NYBC). These de-identified human PBMC samples were collected under an IRB-exempt protocol with donors providing written consent for banking and research of their specimens. For selected experiments in which high cell numbers were needed (Figures 1, 5, and 6), CD8+ T cells isolated from healthy donors PBCMCs were purchased from StemCell (StemCell Technologies Inc, Cat# 70027). Detailed donor information is reported in the key resources table.
METHOD DETAILS
Human CD8+ and CD4+ T cell isolation
For T cell isolation, blood samples were diluted with 1 volume of PBS 2% FBS, then the diluted blood was added dropwise to 1 volume of Lymphoprep (Stemcell Technologies Inc, Cat# 07811) at room temperature. Samples were centrifuged at 800g for 20 minutes at 22 °C, then the PBMCs layer was harvested, washed two times in PBS 2% FBS, and resuspended in PBS 2% FBS. CD8+ T cells were then purified by two subsequent rounds of isolation: first, T cells were enriched using the Pan T Cell Isolation kit (Miltenyi Biotec, Cat# 130-096-535); then, negative CD8 T cell isolation was performed with the CD8+ T Cell Isolation Kit, human (Miltenyi Biotec, Cat# 130-096-495) or negative CD4 T cell isolation was performed with the CD4+ T Cell Isolation Kit, human (Miltenyi Biotec, Cat# 130-096-533). Both enrichment steps were performed using an AutoMACS machine. For both mouse and human cells, CD8 T cell purity was assessed by FACS staining using mouse or human anti-CD3 and anti-CD8 antibodies at 1:100 dilution (PE/Cyanine7 anti-human CD3, BioLegend, # Cat317333; APC/Cyanine7 anti-mouse CD8a, BioLegend, Cat# 100714; FITC anti-human CD3, ThermoFisher Scientific, Cat#11-0038-42; PE/Cy7 anti-human CD8, Biolegend, Cat# 344712). Human CD4+ T cell purity was assessed using the A700 anti-human CD4 antibody, Biolegend, Cat# 317426 at 1:100 dilution.
Mouse CD8+ T cell isolation
Mouse CD8+ T cells were isolated from spleens and lymph nodes of male and female 8–12 weeks old C57BL/6J mice (Jackson strain #000664), C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-1 mice) (Jackson strain #003831), Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J (Cas9 mice) (Jackson strain #024858). Cas9-OT-1 mice were obtained by breeding OT-1 and Cas9 strains, and both Cas9 homozygous and heterozygous mice were used for experiments. All animals used were bred and maintained at NYU School of Medicine and all experiments were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committees at NYU. For T cell isolation, organs were harvested and single cell suspensions were obtained by smashing and filtering through a 40μM strainer. In spleen samples, red blood cell lysis was performed by incubation in ACK lysis buffer (Quality Biological Inc, Cat# 118156101C) for 1 minute, followed by resuspension in FACS buffer (PBS 2% FBS). Negative CD8+ T cell isolation was then performed with the CD8a+ T Cell Isolation Kit, mouse (Miltenyi Biotech, Cat #130-104-075), following the manufacturer’s instructions, utilizing an AutoMACS machine.
In vitro T cell activation and exhaustion
Mouse or human CD8+ T cells were cultured in RPMI media supplemented with 10% FBS, 1% Pen/Strep, 1X GlutaMAX (Life Technologies Cat# 35050061), 1X Non-essential Amino Acids (Life Technologies Cat# 11140050), 1X Sodium Pyruvate (Thermo Fisher Scientific Cat# MT25000CI) and 10mM 2-Mercaptoethanol (Life Technologies Cat# 21985023). Non-activated cells were maintained in culture for maximum 3 days in presence of 1ng/μl mouse or human IL7 (Murine IL-7, Peprotech, Inc., Cat# 217-17-50UG; Recombinant Human IL-7, Peprotech, Inc., Cat# 200-07-50UG), while activated cells were supplemented with 30U/ml mouse or human IL2 (Recombinant Murine IL-2, Peprotech, Inc., Cat# 212-12-50UG; Recombinant Human IL-2, Peprotech, Inc., Cat# 200-02-1MG).
For in vitro T cell activation and exhaustion experiments, cells were thawed and plated at 1 million/ml in presence of Mouse T-Activator CD3/CD28 Dynabeads (Thermo Fisher Scientific, Cat# 11453D) or Human T-Activator CD3/CD28 Dynabeads (Thermo Fisher Scientific, Cat# 11132D) at 1:1 beads-to-cells ratio. After 2 days, cells were split 1:2 by adding fresh media. Beads were removed on day 3 and cells were replated at 0.5M/ml in presence of new beads at 1:1 ratio. Cells were split 1:3 on day 4 and 1:2 on day. 5. Beads were removed again on day 6 and cells were replated at 0.5 million/ml in presence of new beads at 1:1 ratio. Cells were split 1:2 on day 7 and collected on day 9. For RNA-seq, ATAC-seq and C&T profiling, cells were harvested at 0h, 3h, 24h, 48h, 72h, 6 Days and 9 Days along this protocol.
For long-term experiments in the presence of inhibitors or with genetic KO lines, cells were replated at 1M/ml on day 9 in presence of new beads at 1:1 ratio, then split 1:2 on day 10. Beads were removed on day 12 or 13 and cells were replated at 1M/ml in presence of new beads at 1:1 ratio. Cells were then harvested on day 15 or 16. At every time point, alive and dead cells were counted by diluting 10μl of cell suspension with 10ul of Trypan blue and analyzed on a Countess machine (Thermo Fisher Scientific).
FACS staining
For surface FACS staining, cells were harvested, washed in PBS 2% FBS (FACS buffer), incubated in Fc Block Solution (Human TruStain FcX™, BioLegend Cat# 422302) for 5 minutes, then incubated for 30 minutes at 4°C in FACS buffer with antibodies targeting the proteins of interest. Cells were washed two times in FACS buffer and diluted in FACS buffer supplemented with DAPI for live/dead cell exclusion. The following antibodies were used at 1:100 dilution unless stated otherwise: Brilliant Violet 605™ anti-human CD279 (PD-1) (BioLegend, Cat# 329923), APC/Cyanine7 anti-human CD366 (Tim-3) (BioLegend, Cat# 345025), Brilliant Violet 605™ anti-human CD197 (CCR7) (BioLegend, Cat# 353223), APC/Cyanine7 anti-human CD45RA (BioLegend, Cat# 304127), APC anti-human CD39 (BioLegend, Cat# 328209), APC/Cyanine7 anti-mouse CD279 (PD-1) (BioLegend, Cat# 135223, 1:200 dilution), PerCP/Cyanine5.5 anti-mouse CD366 (Tim-3) (BioLegend, Cat# 119717), APC anti-mouse CD62L (BioLegend, Cat# 104411, 1:400 dilution), PerCP/Cyanine5.5 anti-mouse/human CD44 (BioLegend, Cat# 103032).
For intracellular cytokine profiling, cells were stimulated with Cell Stimulation Cocktail (Affymetrix, Cat# 00-4970-93) and supplemented with Brefeldin A (eBioscience Brefeldin A Solution, Life Technologies, Cat# 00-4506-51) for 3h at 37°C to block cytokine secretion. Cells were then harvested and stained with Zombie dyes for live/dead cell discrimination (Zombie Aqua™ Fixable Viability Kit, BioLegend, Cat# 423101 or Zombie Violet™ Fixable Viability Kit, BioLegend, Cat# 423113), following the manufacturer’s instructions. Fixation and permeabilization were then performed using the eBioscience Foxp3/Transcription Factor Staining Buffer Set kit (Life Technologies, Cat# 00-5523-00), according to the manufacturer’s instructions. The following antibodies for intracellular FACS analyses were used at 1:100 dilution: PE anti-human TNF-α (BioLegend, Cat# 502908), Alexa Fluor® 700 anti-human IFN-γ (BioLegend, Cat# 506515), APC anti-mouse TNF-α (BioLegend, Cat# 506307), PE/Cyanine7 anti-mouse IFN-γ (BioLegend, Cat# 505825). Annexin staining was performed with APC Annexin V (BioLegend, Cat# 640920). Samples were analyzed using a Fortessa cytometer.
Chromatin-focused CRISPR screen
Library design
For designing a chromatin-focused CRISPR library, a list of epigenetic modifiers was first compiled based on literature search.141,142 Domain-focused sgRNA sequences were then designed using the Sanjana lab software, accessible through http://guides.sanjanalab.org/#/, with the option to target protein domains selected, and expression data and average data from all tissues were used to pick and define exons. A target of 6 sgRNAs were generated per gene. 60 non-targeting sgRNAs, as well as Pdcd1 and Havcr2 sgRNAs were added as controls. A ‘G’ was added at the 5’ of every sgRNA, if not already present. The following overhangs were then added: AGGCACTTGCTCGTACGACGCGTCTCACACC – (sgRNA 20 nt) – GTTTCGAGACGATGTGGGCCCGGCACCTTAA. The final library consisted of 1928 sgRNAs targeting 310 protein-coding genes.
Library cloning
sgRNA sequences were cloned in the pLKO5.sgRNA.EFS.tRFP plasmid (Addgene, Cat #57823) The plasmid was a gift from Bejamin Ebert. Briefly, plasmid restriction was performed with BsmBI-v2 (NEB, Cat# R0739L) for 2h at 55°C, then the digested plasmid was run on a 1% agarose gel and purified using the QIAquick Gel Extraction Kit (Qiagen, Cat# 28706X4). The sgRNA library was diluted to 1ng/μl in H20, then PCR amplified using the Phusion High-Fidelity PCR Kit (Life Technologies, Cat# F553S), with the following primers: Forward primer: AGGCACTTGCTCGTACGACG, Reverse primer: ATGTGGGCCCGGCACCTTAA. Two ng per reaction were used and five total (50μl) reactions were performed to ensure the maintenance of library representation. PCR conditions were the following: 30 seconds at 98 °C, then 10 seconds at 98 °C, 30 seconds at 53 °C, 30 seconds at 72 °C, for 24 cycles, then 5 minutes at 72 °C. The PCR product was then run on a 1% agarose gel and purified using the QIAquick Gel Extraction Kit (Qiagen, Cat# 28706X4). Cloning into the library vector was then performed using Golden Gate cloning, with the following protocol: 5μg digested vector, 500 ng PCR insert, 5μl Anza Esp1 enzyme (Life Technologies, Cat# IVGN0136), 5μl T4 DNA ligase (New England Biolabs, Cat# M0202L), 20ul Anza Buffer, 20 μl 10mM ATP (New England Biolabs, Cat# PO756S), and H20 to 200ul final volume. The reaction was incubated for 30 minutes at 37 °C, then 30 minutes at 16 °C for 25 cycles. Samples were incubated with 1μl of Plasmid safe ATP-dependent Dnase (Thermo Fisher Scientific, Cat# E3101K) and incubated at 37 °C for 15 minutes. Reaction cleanup was then performed using the MinElute Reaction Cleanup Kit (Qiagen, Cat# 28204), and the elution product was electroporated into MegaX DH10B electro-competent bacteria (Life Technologies, Cat# C640003) using a BioRad Gene Pulser II Electroporation system. Following incubation at 37 °C for 1h, bacteria were plated in 4×24cm square LB plates containing Ampicillin and grown at 30°C for ~20h. The next day, bacteria were harvested from the plates and grown for 2 hours in 500ml liquid LB media with Ampicillin. Plasmid DNA was harvested using the PureLink™ HiPure Plasmid Filter Maxiprep Kit (Thermo Fisher Scientific, Cat#K21001). Library representation was checked by amplifying 200ng of library using the TaKaRa Ex Taq DNA Polymerase (Takara Bio, Cat# RR001B) for 15 cycles, and sequencing 10 million reads on a MiSeq 2 instrument, followed by alignment and QC using MAGECK.
CRISPR screen
For lentivirus production, HEK293T cells were plated in five 15 cm dishes (9 million cells each), in DMEM media supplemented with 10% FBS, 1% Pen/Strep and 1X Glutamax (Life Technologies, Cat# 35050061). The next day, cells were transfected using PEI (Polyethylenimine, Linear, Thermo Fisher Scientific, Cat# NC1014320) with the following plasmids (ug per plate): 15μg psPAX2 (Addgene, Cat#12260), 10μg pMD2G (Addgene, Cat#12259), and 20μg library plasmid. Media was changed 6h after transfection with HEK293T media, and again 24h after transfection with T cell media. 48h after transfection, the viral supernatant was filtered through a 0.45μM filter and added dropwise to mouse CD8+ T cells pre-activated for 24h, in the presence of 5 μg/ml Polybrene (Santa Cruz Biotechnology, Cat# SC-134220). The viral supernatant from each 15 cm dish was pooled and used to transduce 16 million T cells. Spin infection was performed by centrifuging at 1500g, for 60 minutes at 32°C. A second viral collection was performed 72h after transfection and a second round of spin infection was performed. After 2 days, cells were harvested, beads were removed, cells were washed two times in PBS and resuspended in media supplemented with DAPI. RFP+ cells were sorted using a SY3200 Cell Sorter. One million cells were then harvested to assess initial library representation (coverage = ~500x). Cells were then cultured as described in the ‘In vitro T cell activation and exhaustion’ section. At Day 9, cells were harvested and stained with PD1 and TIM3 antibodies and PD1 high TIM3 high or TIM3 low/high populations were sorted (1 to 2 million cells = coverage ~500–1000x). Genomic DNA was purified using the QIAamp DNA Mini Kit (Qiagen, Cat# 51304) and sgRNA sequences were amplified from genomic DNA using the TaKaRa Ex Taq DNA Polymerase (Takara Bio, Cat# RR001B). Five reactions per condition, each containing 1μg of genomic DNA, were performed to maintain library representation. The primers used were:
P5 primers: equimolar mix of: For_01:AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTCTTGTGGAAAGGACGAAACACC, For_02:AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTACTTGTGGAAAGGACGAAACACC, For_03:AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGACTTGTGGAAAGGACGAAACACC, For_04:AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTCGACTTGTGGAAAGGACGAAACACC, For_05:AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTACGACTTGTGGAAAGGACGAAACACC;
P7 primers (indexed):
IDX1:CAAGCAGAAGACGGCATACGAGATTCGCCTTGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTCTACTATTCTTTCCCCTGCACTGT,
IDX2:CAAGCAGAAGACGGCATACGAGATATAGCGTCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTCTACTATTCTTTCCCCTGCACTGT,
IDX4:CAAGCAGAAGACGGCATACGAGATATTCTAGGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTCTACTATTCTTTCCCCTGCACTGT,
IDX8:CAAGCAGAAGACGGCATACGAGATTTGAATAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTCTACTATTCTTTCCCCTGCACTGT.
PCR conditions were the following: 1 minute at 95 °C, then 30 seconds at 95 °C, 30 seconds at 52 °C, 10 minutes at 72 °C, for 22 cycles, then 10 minutes at 72 °C. PCR product purification and size selection were performed using Ampure beads (Thermo Fisher Scientific, Cat# NC9933872), with right selection using beads at 0.4x ratio and left selection with beads at 0.6x ratio. Samples were sequenced at 10 million single-end reads each on a NextSeq500 instrument.
Mouse CRISPR sgRNAs knock-out experiments
For single gRNAs validations, two sgRNA sequences with the greatest efficiency per gene were identified within the CRISPR library, and individually cloned in the pLKO5.sgRNA.EFS.tRFP plasmid (Addgene, Cat #57823). Briefly, the plasmid was digested with BsmBI-v2 (NEB, Cat# R0739L) for 2h at 55°C, then run on a 1% agarose gel, followed by isolation from the gel band using the QIA-quick Gel Extraction Kit (Qiagen, Cat# 28706X4). sgRNA oligos were phosphorylated and annealed by incubation in T4 PNK (New England Biolabs, Cat# VWR #101228-174) and T4 ligase buffer at 37°C for 30 minutes. Temperature was then gradually decreased (−1°C/minute) until room temperature. Oligos were diluted 1:200 in H20 and 1μl of diluted oligos were ligated with 25ng of digested vector for 1h at room temperature with T4 ligase (New England Biolabs, Cat# M0202M). Following transformation in Stbl3 cells, single colonies were analyzed by Sanger sequencing using a primer targeting the U6 promoter (sequence: GACTATCATATGCTTACCGT), expanded and purified using the PureLink™ HiPure Plasmid Filter Maxiprep Kit (Thermo Fisher Scientific, Cat#K21001). Lentiviral transduction was performed by transfecting HEK293T cells with 3.75μg psPAX2 (Addgene, Cat#12260), 2μg pMD2G (Addgene, Cat#12259), and 5μg library plasmid. Viral supernatant was collected 48h and 72h after transfection, filtered through a 0.45μM filter and used to spin-infect pre-activated mouse CD8+ T cells at 1500g, for 60 minutes at 32°C. RFP% was assessed using Fortessa machine at 3 day intervals post-activation in parallel with the in vitro exhaustion protocol.
Human CRISPR sgRNAs knock-out experiments
For sgSMARCA4 and sgHNF1B KO experiments in human cells, three CTRL or SMARCA4- or HNF1B-targeting sgRNAs were designed and synthetized by Synthego (https://www.synthego.com/products/crispr-kits/gene-knockout-kit). The Synthego-optimized multi-sgRNA approach was used, where the three different sgRNAs were co-electroporated in one reaction to increase knock-out efficiency. The SMARCA4-targeting sgRNA sequences were the following: ACUCCAGACCCACCCCUGGG, CCCUAGCCCGGGUCCCUCGC, GUCCUGCUGAGGGCGGCCCU. The HNF1B-targeting sgRNA sequences were the following: AGCCCUCGUCGCCGGACAAG, GGCCGAGCCCGACACCAAGC, CGGGGUCACCAAGGAGGUGC.
Human CD8+ T cells were activated for 48h, then beads were removed, and 200,000 T cells were electroporated with a mix consisting of 1.5 ug of Cas9-GFP ribonucleoprotein (Integrated DNA Technologies, Cat#10008100) and 1ug of sgRNAs, using the Neon transfection system (1,200V, Width=40, 1 pulse). After electroporation cells were plated at 1 million/ml in antibiotics-free media. After 4 hours, cells were harvested and GFP+ cells were sorted. Cells were then replated at 1 million/ml with activation beads, and expanded through the in vitro exhaustion protocol.
B16-OVA in vitro exhaustion model
B16-F10 and B16-F10-OVA cell lines used for co-culture mediated T cell exhaustion were a gift of Dr. Weber’s lab. For B16-T cells cocultures, splenocytes were harvested from OT-1/Cas9 mice and cultured at a concentration of 10 million/ml in T cell media in the presence of 1μM SIINKEFL peptide (OVA 257–264, Invivogen # vac-sin). After 48h, CD8+ T cells were purified with the CD8a+ T Cell Isolation Kit, mouse (Miltenyi Biotech, Cat #130-104-075), following the manufacturer’s instructions. Purified T cells were plated on 6-well plates containing B16 or B16-OVA cells, pretreated for 24h with 1ng/μl IFNg to promote MHCI expression. T cells were passaged on new B16 or B16-OVA plates, pre-treated with IFNg, every 48 hours. For CRISPR KO experiments in this model, cells were transduced as previously described following T cell purification, then cultured on B16 or B16-OVA plates and profiled 9 days after activation.
Western blots
Western blots were performed as described previously.143 Briefly, proteins were isolated in RIPA buffer, quantified and loaded on 4%−12% Bis-Tris polyacrylamide gels (Thermo Fisher Scientific). Proteins were then transferred onto PVDF membranes (Millipore) and probed using the SMARCA4 antibody (Cell Signaling Technology Cat# 49360T, 1:1000 dilution), the anti-HNF1B antibody Proteintech Cat# 12533-1-AP, 1:1000 dilution) or the anti-Actin (Millipore Cat# MAB1501, 1:10000 dilution). Following incubation with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare), chemiluminescence was assessed with ECL (Life Technologies).
Cell killing assays
Killing assays were performed in 96-well plates, by mixing 50000 B16 or B16-OVA cells (Target) and serial dilutions of OT-1 T cells (Effector) at Day9 of the chronic stimulation protocol, in 200 ul of RPMI media supplemented with 10% FBS, 1% Pen/Strep, 1X GlutaMAX (Life Technologies Cat# 35050061), 1X Non-essential Amino Acids (Life Technologies Cat# 11140050), 1X Sodium Pyruvate (Thermo Fisher Scientific Cat# MT25000CI) and 10mM 2-Mercaptoethanol (Life Technologies Cat# 21985023). Four replicates were seeded per condition, and controls containing B16 or B16-OVA without T cells were included. After 24h or 48h incubation at 37°C, media was removed, wells were washed twice in 200μl of PBS, then 100ul of PBS per well were added. CellTiterGlo (Promega Cat# G7571) was then added (100μl/well), plates were incubated for 10 minutes protected from light, and measured luminescence.
CD19-CAR-T cell experiments
For CAR-T experiments, we generated an anti-CD19 CAR lentiviral vector incorporating a 41BB co-stimulatory domain and CD3ζ activation domain. The single-chain variable fragment (scFv) was derived from the murine FMC63 anti-human antibody, that has high affinity and specificity for CD19 and it is utilized in clinical trials. The complete CAR construct is driven by an EF1α promoter and contains the internal ribosome entry site (IRES)-GFP signal for cell selection. The CAR-T vector was cloned in house in the Perna Lab (Indiana University). For CAR-Ts preparation, peripheral blood was obtained from de-identified healthy human volunteers under IRB-exempt protocol with written consent for banking and research of their specimens given for each donor. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation, purified using the Human Pan T Cell Isolation Kit (Miltenyi Biotec, Cat#130-096-535), stimulated with CD3/CD28 T cell activator Dynabeads (Thermo Fisher Scientific, Cat# 11132D) for 2 days and cultured in X-VIVO-15 media (Lonza Cat#BE02-060Q) supplemented with human serum (5%) and IL-2 (200U/ml). After 24 hours of activation, cells were transduced with lentiviruses encoding anti-CD19 CAR and GFP genes, in presence of polybrene. Transduction efficiencies were assessed by FACS and were ranging from 20 to 50%. Then, cells were counted and plated (0.5 million/ml) in the presence of beads, and DMSO or PROTACS, ACBI1 or AU-15330 (100nM). The same process was repeated at 3–4 days intervals. At Day 10, beads were removed and immunophenotype was analyzed by flow-cytometry using the following markers: CCR7, CD45RA, PD1, TIM3, LAG-3, CD39 (vendors and catalogs previously stated in methods). To assess in vitro persistence, cells were incubated with fresh beads at 3-days intervals and counted at Day 13 and 16.
In vivo B16-OVA killing assays
For in vivo experiments, 8–12 weeks-old Rag1KO mice B6.129S7-Rag1<tm1Mom>/J (Jackson # 002216), were injected subcutaneously with 0.5 million B16-OVA cells in 100ml PBS. The same day, mouse CD8+ OT-1 T cells were purified from 8–12 week old OT-1 mice, and activated in vitro with CD3/CD28 Dynabeads at 1:1 ratio. After 72h, beads were removed, cells were counted and plated with fresh beads at 1:1 ratio in presence of DMSO or FHT-1015 at 100nM. Cells were split 1:2 or 1:3 every day by adding fresh media and DMSO or FHT-1015. At Day 7, T cells were washed in PBS, counted and 2 million cells were injected intravenously into tumor bearing mice. Tumor growth was assessed by caliper measurement every 3 days.
SMARCA4/SMARCA2 Inhibitor and Degrader Small Molecule Treatment Studies
For inhibitor and small-molecule PROTAC treatments, mouse or human T cells were activated for 3 days as previously described. At Day 3, cells were counted and plated at 0.5 million /ml in presence of DMSO, 50nM or 100nM of inhibitor or PROTAC. At every subsequent time point (Days 6, 9, 12, 16), beads were removed and cells were replated at 0.5M/ml in presence of inhibitors or PROTACs and fresh beads at 1:1 ratio. Cells were diluted 1:2 or 1:3 in between time points by adding media containing the corresponding concentration of inhibitor or PROTAC. The drugs used were: ACBI1 (SelleckChem, Cat#S9612), AU-15330 (MedChem Express Llc, Cat #HY-145388), CMP14 (synthesized), FHT-1015 (synthesized).
RNA-seq
For RNAseq experiments, 50.000 to 0.5 million cells were harvested on ice and washed in cold PBS. RNA extraction was then performed using the Rneasy Plus Mini Kit (Qiagen Cat#74136), following the manufacturer’s instructions. Poly-A selection was performed using the the Nebnext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs Cat#E7490) for all RNAseq experiments, except the RNAseq upon PROTAC treatment experiment, where NEXTFLEX® Poly(A) Beads 2.0, (Perkin Elmer Cat#NOVA-512991) were used. Library preparation was performed using the Nebnext Ultra II Directional RNA Library Prep Kit (New England Biolabs Cat# E7760), or NEXTFLEX® Rapid Directional RNA-Seq Kit 2.0 (Perkin Elmer Cat#NOVA-5198-01) for PROTAC experiments. For all libraries, quality was assessed by Tapestation. Samples were sequenced on NovaSeq6000 and NextSeq500 machines (Illumina) at sequencing depth of 30 million reads per sample.
ATAC-seq
Cells were harvested at 0h, 3h, 24h, 48h, 72h, Day6 and Day9. ATAC-seq experiments were completed and samples were prepared into libraries using the previously described methodology.66,67,144 Cells (50,000) were collected in media and washed in cold PBS. Cells were spun at 500rcf for 5 minutes to form a pellet and PBS was removed. Cold lysis buffer was added and cells were gently resuspended by pipetting. Resuspended cells were incubated on ice for three minutes. Lysis was quenched by adding wash buffer and mixing by inverting the tube three times. Lysed material was pelleted at 400 rcf for 10 minutes, and supernatant was discarded. The pelleted DNA was resuspended in transposition reaction buffer and the transposition reaction was carried out for 30 minutes at 37°C with gentle shaking at 1,000 rpm on a thermomixer. The resultant tagmented DNA was purified using Qiagen MinElute Reaction clean up kit (Qiagen Cat# 28206) and eluted in dH20. Tagmented DNA libraries were amplified with 7 total cycles using a standard ATAC-seq amplification protocol and custom PCR primers. ATAC-seq libraries were sequenced on the Illumina NextSeq500 with 35 base-pair paired end sequencing parameters and using the NextSeq™ 500/550 High output flow cell kit (Illumina Cat# 20024906).
Cut & Tag
The epicypher protocol for Cleavage under targets and tagmentation was used with slight modifications.65 Concanavalin A (ConA, BioMag®Plus Cat# 86057) beads were activated with bead activation buffer and stored on ice until further use. Cells (100,000) were collected and washed with cold PBS. Cells were spun at 300rcf for 5 minutes at 4°C and PBS supernatant was removed from the cell pellet. Nuclear extraction buffer was added to the tube and the pellet was gently resuspended by pipetting to lyse cells and extract nuclei. Activated ConA beads and nuceli were incubated were mixed and incubated at room temperature for 10 minutes. The nuclei-conjugated bead complexes were resuspended in antibody binding buffer and add primary antibody rotating on a nutator overnight at 4°C (Added 2.5, 0.5, 0.25, 0.25, 0.67 and 0.25 ug of IgG, kH3K27Ac, Brg1, SS18, ARID1A, and PBRM1 respectively). Primary antibody mixture was removed, and nuclei-bead complexes were incubated with 0.5μg secondary antibody in digitonin 150 buffer for 1 hour at room temperature on the nutator. After secondary antibody incubation, samples were washed with digitonin 150 buffer and resuspended in digitonin 300 buffer supplemented with 2 microliters of CUTANA pAG-Tn5 (Epicycpher Cat#15-1117) added per sample. Samples were incubated with Tn5 for 1 hour at room temperature on the nutator. Digitonin 300 buffer was added two times to remove excess enzyme from samples. Targeted chromatin tagmentation was completed following the epicypher protocol. Libraries were amplified with 14 PCR cycles and purified by single sided 1.3x AMPure bead purification. The NextSeq500 and 35 base-pair paired end sequencing parameters were used for library sequencing.
QUANTIFICATION AND STATISTICAL ANALYSIS
NGS Data Processing
Cut and Tag, ATAC-Seq, and RNA-Seq samples were sequenced with the Illumina technology, and output data were demultiplexed using the bcl2fastq software tool. RNA-Seq reads were aligned to the hg19 genome with STAR v2.5.2b,120 and tracks were generated using the deepTools v2.5.3 bamCoverage function.121 For ATAC-Seq data, quality read trimming was carried out by Trimmomatic v0.36,122 followed by alignment, duplicate read removal, and read quality filtering using Bowtie2 v2.29,123 Picard v2.8.0,124 and SAM-tools v 0.1.19,125 respectively, and ATAC-seq broad peaks were called with the MACS2 v2.1.1 software126 using the BAMPE option and a broad peak cutoff of 0.001. For ATAC-Seq track generation, output BAM files were converted into BigWig files using MACS2 and UCSC utilities127 in order to display coverage throughout the genome in RPM values. For Cut and Tag libraries, the CutRunTools pipeline was leveraged to perform read trimming, quality filtering, alignment, peak calling, and track building using default parameters.128 All sequencing data analyzed in this study have been deposited at NCBI’s Gene Expression Omnibus under accession number GSE212357.
RNA-seq data analysis
For RNA-seq data, output gene count tables from STAR based on alignments to the hg19 reflat annotation were used as input into edgeR v3.12.1129 to obtain normalized log CPM values and to evaluate differential gene expression. Log2 fold change values from edgeR were used as input into GSEA,130 and the GseaPreranked tool was run with default settings to measure gene set enrichment. For day 9 treatment comparisons, the DESeq2 v1.30.1 R software package was used to evaluate differential gene expression.131 For heatmaps displaying differentially expressed genes, log CPMs were transformed into Z-scores followed by hierarchical or K-means clustering. In order analyze gene set enrichment for select subsets of genes, hypergeometric tests were performed on overlaps with various MSIGDB gene sets and select enriched gene sets were displayed. Principal component analysis was performed using the wt.scale and fast.svd functions from the corpcor R package on RPKM values,132,133 which were quantified using median length isoforms and total mapped read counts computed by the Samtools idxstats function.
Cut & Tag and ATAC-seq data analysis and integration
The Bedtools multiIntersectBed and mergeBed functions were used for peak merging,134 and the R package, ChIPpeakAnno v3.17.0,135 was used to visualize peak overlaps. Distance-to-TSS peak distributions were computed utilizing Ensembl protein-coding gene coordinates. To generate the heatmap in Figure 1D, which served as a platform for several downstream analyses, first, 32 sets of peaks derived from the SS18 and SMARCA4 samples (called by the CutRunTools pipeline) from both donors and from all time points were merged with the Bedtools multiIntersectBed function. Since there wsere two donors for every time point, we removed any peaks or parts of peaks that did not overlap with at least one peak to remove outlier peaks and outlier peak segments. This overlap information from the 32 sets of peaks was provided by the output of the Bedtools multiIntersectBed function. After the outlier peaks and outlier peak segments were removed, the Bedtools mergeBed function was used to merge the filtered peaks. In an identical manner, the peaks of the H3K27ac and ATAC-seq samples were separately merged. Second, these three sets of merged peaks from all time points were overlapped and merged to generate the Venn Diagram in Figure S2I, which represent the sites in the Figure 1D heatmap. Third, the Cut & Tag and ATAC-seq RPKM data from the merged peaks were log2 transformed, followed by the separate quantile normalization of each BAF subunit, H3K27ac, and ATAC-seq data across time points. Finally, K-means clustering was applied in a semi-unsupervised manor to partition the SMARCA4, SS18, H3K27ac and ATAC-seq data into the 9 groups or clusters, which are exhibited in Figure 1D, followed by transformation into Z-scores across timepoints to highlight the differences within clusters among time points.
IgG control subtraction and data normalization
CUT&TAG datasets from two independent human T cell donors were merged and RPKM values were computed for SMARCA4, SS18, H3K27Ac and IgG samples for each timepoint across the T cell activation and exhaustion time course (as above). The RPKM values for each mark were log2-transformed and individually subjected to quantile normalization. Quantile-normalized IgG signals were then subtracted from the SMARCA4, SS18, and H3K27Ac quantile-normalized signals. From here, for selected figure panels, quantile-normalized, IgG-subtracted signal values were separately transformed into Z-scores for all timepoints. Finally, IgG control peaks that were present and overlapped SMARCA4, SS18 and H3K27Ac merged sites were removed, while maintaining the order of the non-overlapping sites. Heatmaps were generated from the resultant non-overlapping and IgG-subtracted sites.
Principal component analyses (PCA) were also performed on these quantile-normalized log2-transformed RPKM values. For day 9 treatment analyses, DESeq2 was used to evaluate differential accessibility, and quantile-normalized log2 RPKM values were transformed into Z-scores following by hierarchical clustering to display differentially accessible sites.
Transcription factor motif and archetype analyses
Transcription factor enrichment and motif analyses were carried out by the LOLA v1.12.0136 and HOMER v4.9137 software packages, respectively. In addition to using HOMER to analyze motif enrichment, for several motif enrichment analyses conducted in this study, we determined the number of motif occurrences for 286 non-redundant archetype consensus motifs138 within +/− 250 base pairs of peak centers for each peak within given peak sets. The coordinates of these archetype motifs as well as non-archetype motifs across the entire human and mouse genomes can be downloaded from the following resource https://www.vierstra.org/resources/motif_clustering#downloads.
Average archetype and non-archetype motif occurrences and densities within all sites and within clusters of sites were also determined, and fractional enrichment values relative to all sites were displayed for select motifs with high occurrence and variability. The following formulas were used to compute motif densities and enrichment.
| 1. |
| 2. |
| 3. |
| 4. |
| 5. |
These archetype motif fraction enrichment values in clusters were also plotted against corresponding TF gene log fold change values for several stepwise comparisons across the T-cell activation and exhaustion time course. For logistic regression analyses on archetype motifs, matrices of motif counts for given merged peaks were generated, and the R software program GLMnet139 was used to produce logistic regression models to fit the motif counts to binomial vectors where 1’s represented sites with BAF or ATACseq log2 fold changes > 0. Divergent barplots were used to display large coefficients in output models to estimate the influence of motifs on BAF occupancy or accessibility in terms of magnitude, directionality and predictability.
CRISPR screen data analysis
Data analysis was performed using MAGECK,119 following the standard analysis pipeline reported in https://sourceforge.net/p/mageck/wiki/Home/. Data visualization was performed using the Bioconductor MAGeCKFlute package.
scRNAseq and scATACseq datasets and analyses
scRNA-seq
scRNAseq signatures consisted of the marker genes identified in exhausted or memory cells from the several literature sources.80–84 These gene lists were used as ‘‘gene set’’ inputs into GSEA along with log2 fold change values from edgeR for all expressed genes for several given comparisons, and the GseaPreranked tool was run with default settings to measure gene set enrichment.130 A positive score indicates an enrichment of genes within a given gene set that have increasing expression, while a negative score indicates an enrichment of genes within a given gene set that have decreasing expression. GSEA output normalized enrichment scores or regular enrichment scores were displayed in heatmaps. Negative log base 10 p-values from select the indicated gene sets were displayed in barplots.
scATAC-seq
To assess the TF activity toward SWI/SNF bound regions in tumor infiltrating human CD8+ T cells, we used publicly available scATAC-seq datasets (GSE181062, GSE181064).22,60 From the raw data, we used read counts that fall into the peak coordinate defined as SWI/SNF bound regions in this study. We then used the Viestra et al. motif database as an input of the ‘motifmatchr’ function of ChromVAR and followed the ChromVAR workflow with default settings.140 ‘deviationScores’ is used for visualization of TF activity and cell type annotations in the original papers were used.
Supplementary Material
Highlights.
mSWI/SNF targeting and activity is specific to T cell activation and exhaustion states
Genetic and chemical disruption of cBAF complexes enhances T cell persistence
cBAF complex activities facilitate T cell exhaustion and prevent memory features
mSWI/SNF pharmacologic disruption improves CAR-T expansion and anti-tumor control
ACKNOWLEDGMENTS
We are grateful to all members of the Kadoch and Aifantis laboratories for their helpful discussions during the preparation of this study. We are also grateful to Drs. Arlene Sharpe, Sarah Weiss, Debamatta Sen, and Hsaio-Wei Tsao for their helpful insights and for providing mouse T cell protocols and reagents, as well as Z. Herbert and M. Sullivan of the Molecular Biology Core Facility (MBCF) at the Dana-Farber Cancer Institute. We would also like to thank the NYU Genome Technology Center for sequencing experiments. This work was supported in part by NIH 1F31CA271427-01 (K.H.), NIH 5F30CA239317 (D.E.C.), T32GM007753 and T32GM144273 (D.E.C.), and 1DP2CA195762 (C.K.). C.K. was also supported by the Mark Foundation for Cancer Research Emerging Leader Award. E.B. was supported by the Swiss National Science Foundation (SNSF) and the Lymphoma Research Foundation (LRF). I.A. was supported by the NIH/NCI (5R01CA173636, 5R01CA228135, 5P01CA229086, 5R01CA242020, 1R01CA243001, and 1R01CA252239) and the Vogelstein Foundation. NYU Langone’s Genome Technology Center (RRID: SCR_017929) is partially supported by the Cancer Center Support grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center.
Footnotes
DECLARATION OF INTERESTS
C.K. is the scientific founder, scientific advisor to the board of directors, scientific advisory board member, shareholder, and consultant for Foghorn Therapeutics, Inc. (Cambridge, MA). C.K. is also a member of the scientific advisory board and is a shareholder of Nested Therapeutics and Nereid Therapeutics, serves on the scientific advisory board for Fibrogen, Inc. and on the Molecular Cell editorial board, and is a consultant for Cell Signaling Technologies and Google Ventures. I.A. is a scientific consultant for Foresite Labs and receives research funding from AstraZeneca Inc. F.P. is an inventor on patents related to adoptive cell therapies, held by MSKCC (some licensed to Takeda), serves as a consultant for AstraZeneca, and receives research support from Lonza and NGMBio. A.T. is a scientific advisor to Intelligencia AI.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2023.02.026.
REFERENCES
- 1.Kaech SM, and Cui W (2012). Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol 12, 749–761. 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hämmerling GJ, et al. (2016). Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45, 389–401. 10.1016/j.immuni.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seo W, Jerin C, and Nishikawa H (2021). Transcriptional regulatory network for the establishment of CD8+ T cell exhaustion. Exp. Mol. Med 53, 202–209. 10.1038/s12276-021-00568-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang N, and Bevan MJ (2011). CD8+ T cells: foot soldiers of the immune system. Immunity 35, 161–168. 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJW, et al. (2015). Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241. 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wherry EJ, and Kurachi M (2015). Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499. 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pardoll DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264. 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prinzing B, Zebley CC, Petersen CT, Fan Y, Anido AA, Yi Z, Nguyen P, Houke H, Bell M, Haydar D, et al. (2021). Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci. Transl. Med 13, eabh0272. 10.1126/scitranslmed.abh0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pauken KE, and Wherry EJ (2015). Overcoming T cell exhaustion in infection and cancer. Trends Immunol 36, 265–276. 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang B, Zhang W, Jankovic V, Golubov J, Poon P, Oswald EM, Gurer C, Wei J, Ramos I, Wu Q, et al. (2018). Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8+ T cell dysfunction and maintain memory phenotype. Sci. Immunol 3, eaat7061. 10.1126/sciimmunol.aat7061. [DOI] [PubMed] [Google Scholar]
- 11.Klein Geltink RI, Edwards-Hicks J, Apostolova P, O’Sullivan D, Sanin DE, Patterson AE, Puleston DJ, Ligthart NAM, Buescher JM, Grzes KM, et al. (2020). Metabolic conditioning of CD8+ effector T cells for adoptive cell therapy. Nat. Metab 2, 703–716. 10.1038/s42255-020-0256-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chowell D, Yoo S-K, Valero C, Pastore A, Krishna C, Lee M, Hoen D, Shi H, Kelly DW, Patel N, et al. (2022). Improved prediction of immune checkpoint blockade efficacy across multiple cancer types. Nat. Biotechnol 40, 499–506. 10.1038/s41587-021-01070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. (2014). Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med 371, 2189–2199. 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li P, and Leonard WJ (2018). Chromatin accessibility and interactions in the transcriptional regulation of T cells. Front. Immunol 9, 2738. 10.3389/fimmu.2018.02738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henning AN, Roychoudhuri R, and Restifo NP (2018). Epigenetic control of CD8+ T cell differentiation. Nat. Rev. Immunol 18, 340–356. 10.1038/nri.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao H-W, Godec J, LaFleur MW, Brown FD, et al. (2016). The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169. 10.1126/science.aae0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gennert DG, Lynn RC, Granja JM, Weber EW, Mumbach MR, Zhao Y, Duren Z, Sotillo E, Greenleaf WJ, Wong WH, et al. (2021). Dynamic chromatin regulatory landscape of human CAR T cell exhaustion. Proc. Natl. Acad. Sci. USA 118. e2104758118. 10.1073/pnas.2104758118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang P, Zhang Z, Hu Y, Liang Z, Han Y, Li X, Zeng X, Zhang H, Zhu M, Dong J, et al. (2022). Single-cell ATAC-seq maps the comprehensive and dynamic chromatin accessibility landscape of CAR-T cell dysfunction. Leukemia 36, 2656–2668. 10.1038/s41375-022-01676-0. [DOI] [PubMed] [Google Scholar]
- 20.Wang W, Fasolino M, Cattau B, Goldman N, Kong W, Frederick MA, McCright SJ, Kiani K, Fraietta JA, and Vahedi G (2020). Joint profiling of chromatin accessibility and CAR-T integration site analysis at population and single-cell levels. Proc. Natl. Acad. Sci. USA 117, 5442–5452. 10.1073/pnas.1919259117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen GM, Chen C, Das RK, Gao P, Chen C-H, Bandyopadhyay S, Ding Y-Y, Uzun Y, Yu W, Zhu Q, et al. (2021). Integrative bulk and single-cell profiling of premanufacture T-cell populations reveals factors mediating long-term persistence of CAR T-cell therapy. Cancer Discov 11, 2186–2199. 10.1158/2159-8290.CD-20-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kourtis N, Wang Q, Wang B, Oswald E, Adler C, Cherravuru S, Malahias E, Zhang L, Golubov J, Wei Q, et al. (2022). A single-cell map of dynamic chromatin landscapes of immune cells in renal cell carcinoma. Nat. Cancer 3, 885–898. 10.1038/s43018-022-00391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Leun AM, Thommen DS, and Schumacher TN (2020). CD8+ T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer 20, 218–232. 10.1038/s41568-019-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Utzschneider DT, Gabriel SS, Chisanga D, Gloury R, Gubser PM, Vasanthakumar A, Shi W, and Kallies A (2020). Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat. Immunol. 21, 1256–1266. 10.1038/s41590-020-0760-z. [DOI] [PubMed] [Google Scholar]
- 25.Tsui C, Kretschmer L, Rapelius S, Gabriel SS, Chisanga D, Knöpper K, Utzschneider DT, Nüssing S, Liao Y, Mason T, et al. (2022). MYB orchestrates T cell exhaustion and response to checkpoint inhibition. Nature 609, 354–360. 10.1038/s41586-022-05105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macian F (2005). NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol 5, 472–484. 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- 27.Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, Utzschneider DT, von Hoesslin M, Cullen JG, Fan Y, et al. (2019). TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269. 10.1038/s41586-019-1326-9. [DOI] [PubMed] [Google Scholar]
- 28.Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, et al. (2019). TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274. 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, et al. (2019). TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218. 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yao C, Sun H-W, Lacey NE, Ji Y, Moseman EA, Shih H-Y, Heuston EF, Kirby M, Anderson S, Cheng J, et al. (2019). Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat. Immunol 20, 890–901. 10.1038/s41590-019-0403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, López-Moyado IF, Georges RO, Zhang W, Onodera A, et al. (2019). TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 116, 12410–12415. 10.1073/pnas.1905675116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, Pellegrini M, Zehn D, Berberich-Siebelt F, Febbraio MA, et al. (2017). Transcription factor IRF4 promotes CD8+ T cell exhaustion and limits the development of memory-like T cells during chronic Infection. Immunity 47, 1129–1141.e5. 10.1016/j.immuni.2017.11.021. [DOI] [PubMed] [Google Scholar]
- 33.Martinez GJ, Pereira RM, Äijö T, Kim EY, Marangoni F, Pipkin ME, Togher S, Heissmeyer V, Zhang YC, Crotty S, et al. (2015). The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 42, 265–278. 10.1016/j.immuni.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, Carter R, Awad W, Neale G, Thomas PG, et al. (2017). De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell 170, 142–157.e19. 10.1016/j.cell.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo A, Huang H, Zhu Z, Chen MJ, Shi H, Yuan S, Sharma P, Connelly JP, Liedmann S, Dhungana Y, et al. (2022). cBAF complex components and MYC cooperate early in CD8+ T cell fate. Nature 607, 135–141. 10.1038/s41586-022-04849-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belk JA, Yao W, Ly N, Freitas KA, Chen Y-T, Shi Q, Valencia AM, Shifrut E, Kale N, Yost KE, et al. (2022). Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell 40, 768–786.e7. 10.1016/j.ccell.2022.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loo C-S, Gatchalian J, Liang Y, Leblanc M, Xie M, Ho J, Venkatraghavan B, Hargreaves DC, and Zheng Y (2020). A genome-wide CRISPR screen reveals a role for the non-canonical nucleosome-remodeling BAF complex in Foxp3 expression and regulatory T cell function. Immunity 53, 143–157.e8. 10.1016/j.immuni.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou M, Yuan J, Deng Y, Fan X, and Shen J (2021). Emerging role of SWI/SNF complex deficiency as a target of immune checkpoint blockade in human cancers. Oncogenesis 10, 3. 10.1038/s41389-020-00296-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sandoval GJ, Pulice JL, Pakula H, Schenone M, Takeda DY, Pop M, Boulay G, Williamson KE, McBride MJ, Pan J, et al. (2018). Binding of TMPRSS2-ERG to BAF chromatin remodeling complexes mediates prostate oncogenesis. Mol. Cell 71, 554–566.e7. 10.1016/j.molcel.2018.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boulay G, Sandoval GJ, Riggi N, Iyer S, Buisson R, Naigles B, Awad ME, Rengarajan S, Volorio A, McBride MJ, et al. (2017). Cancer-specific retargeting of BAF complexes by a prion-like domain. Cell 171, 163–178.e19. 10.1016/j.cell.2017.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W, Haswell JR, Park PJ, and Roberts CW (2017). The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 8, 14648. 10.1038/ncomms14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barisic D, Stadler MB, Iurlaro M, and Schübeler D (2019). Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature 569, 136–140. 10.1038/s41586-019-1115-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mashtalir N, D’Avino AR, Michel BC, Luo J, Pan J, Otto JE, Zullow HJ, McKenzie ZM, Kubiak RL, St Pierre R, et al. (2018). Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell 175, 1272–1288.e20. 10.1016/j.cell.2018.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mittal P, and Roberts CWM (2020). The SWI/SNF complex in cancer — biology, biomarkers and therapy. Nat. Rev. Clin. Oncol 17, 435–448. 10.1038/s41571-020-0357-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Centore RC, Sandoval GJ, Soares LMM, Kadoch C, and Chan HM (2020). Mammalian SWI/SNF chromatin remodeling complexes: emerging mechanisms and therapeutic strategies. Trends Genet 36, 936–950. 10.1016/j.tig.2020.07.011. [DOI] [PubMed] [Google Scholar]
- 46.Kadoch C, and Crabtree GR (2015). Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci. Adv. 1, e1500447. 10.1126/sciadv.1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cenik BK, and Shilatifard A (2021). COMPASS and SWI/SNF complexes in development and disease. Nat. Rev. Genet 22, 38–58. 10.1038/s41576-020-0278-0. [DOI] [PubMed] [Google Scholar]
- 48.Mashtalir N, Dao HT, Sankar A, Liu H, Corin AJ, Bagert JD, Ge EJ, D’Avino AR, Filipovski M, Michel BC, et al. (2021). Chromatin landscape signals differentially dictate the activities of mSWI/SNF family complexes. Science 373, 306–315. 10.1126/science.abf8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michel BC, D’Avino AR, Cassel SH, Mashtalir N, McKenzie ZM, McBride MJ, Valencia AM, Zhou Q, Bocker M, Soares LMM, et al. (2018). A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol 20, 1410–1420. 10.1038/s41556-018-0221-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, and Crabtree GR (2013). Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet 45, 592–601. 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.St Pierre R, Collings CK, Samé Guerra DD, Widmer CJ, Bolonduro O, Mashtalir N, Sankar A, Liang Y, Bi WL, Gerkes EH, et al. (2022). SMARCE1 deficiency generates a targetable mSWI/SNF dependency in clear cell meningioma. Nat. Genet 54, 861–873. 10.1038/s41588-022-01077-0. [DOI] [PubMed] [Google Scholar]
- 52.Zullow HJ, Sankar A, Ingram DR, Samé Guerra DD, D’Avino AR, Collings CK, Lazcano R, Wang W-L, Liang Y, Qi J, et al. (2022). The FUS::DDIT3 fusion oncoprotein inhibits BAF complex targeting and activity in myxoid liposarcoma. Mol. Cell 82, 1737–1750.e8. 10.1016/j.molcel.2022.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mathur R, Alver BH, San Roman AK, Wilson BG, Wang X, Agoston AT, Park PJ, Shivdasani RA, and Roberts CWM (2017). ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet 49, 296–302. 10.1038/ng.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, Kim Y, Kryukov GV, Ghandi M, Aguirre AJ, et al. (2014). ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med 20, 251–254. 10.1038/nm.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu G, Chhangawala S, Cocco E, Razavi P, Cai Y, Otto JE, Ferrando L, Selenica P, Ladewig E, Chan C, et al. (2020). ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat. Genet 52, 198–207. 10.1038/s41588-019-0554-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pan J, McKenzie ZM, D’Avino AR, Mashtalir N, Lareau CA, St Pierre R, Wang L, Shilatifard A, and Kadoch C (2019). The ATPase module of mammalian SWI/SNF family complexes mediates subcomplex identity and catalytic activity–independent genomic targeting. Nat. Genet 51, 618–626. 10.1038/s41588-019-0363-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McBride MJ, Pulice JL, Beird HC, Ingram DR, D’Avino AR, Shern JF, Charville GW, Hornick JL, Nakayama RT, Garcia-Rivera EM, et al. (2018). The SS18-SSX fusion oncoprotein hijacks BAF complex targeting and function to drive synovial sarcoma. Cancer Cell 33, 1128–1141.e7. 10.1016/j.ccell.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakayama RT, Pulice JL, Valencia AM, McBride MJ, McKenzie ZM, Gillespie MA, Ku WL, Teng M, Cui K, Williams RT, et al. (2017). SMARCB1 is required for widespread BAF complex–mediated activation of enhancers and bivalent promoters. Nat. Genet 49, 1613–1623. 10.1038/ng.3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Agarwal R, Chan Y-C, Tam CS, Hunter T, Vassiliadis D, Teh CE, Thijssen R, Yeh P, Wong SQ, Ftouni S, et al. (2019). Dynamic molecular monitoring reveals that SWI–SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat. Med. 25, 119–129. 10.1038/s41591-018-0243-z. [DOI] [PubMed] [Google Scholar]
- 60.Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 37, 925–936. 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Valencia AM, Collings CK, Dao HT, St Pierre R, Cheng Y-C, Huang J, Sun Z-Y, Seo H-S, Mashtalir N, Comstock DE, et al. (2019). Recurrent SMARCB1 mutations reveal a nucleosome acidic patch interaction site that potentiates mSWI/SNF complex chromatin remodeling. Cell 179, 1342–1356.e23. 10.1016/j.cell.2019.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iurlaro M, Stadler MB, Masoni F, Jagani Z, Galli GG, and Schübeler D (2021). Mammalian SWI/SNF continuously restores local accessibility to chromatin. Nat. Genet 53, 279–287. 10.1038/s41588-020-00768-w. [DOI] [PubMed] [Google Scholar]
- 63.Cortez JT, Montauti E, Shifrut E, Gatchalian J, Zhang Y, Shaked O, Xu Y, Roth TL, Simeonov DR, Zhang Y, et al. (2020). CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature 582, 416–420. 10.1038/s41586-020-2246-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Henikoff S, Henikoff JG, Kaya-Okur HS, and Ahmad K (2020). Efficient chromatin accessibility mapping in situ by nucleosome-tethered tagmentation. eLife 9, e63274. 10.7554/eLife.63274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, and Henikoff S (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10, 1930. 10.1038/s41467-019-09982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, Yates K, Godec J, Klatt MG, Regev A, et al. (2014). The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol 15, 373–383. 10.1038/ni.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seo H, González-Avalos E, Zhang W, Ramchandani P, Yang C, Lio C-WJ, Rao A, and Hogan PG (2021). BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat. Immunol 22, 983–995. 10.1038/s41590-021-00964-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kane LP, Lin J, and Weiss A (2002). It’s all Rel-ative: NF-κB and CD28 costimulation of T-cell activation. Trends Immunol 23, 413–420. 10.1016/S1471-4906(02)02264-0. [DOI] [PubMed] [Google Scholar]
- 71.Klein-Hessling S, Muhammad K, Klein M, Pusch T, Rudolf R, Flöter J, Qureischi M, Beilhack A, Vaeth M, Kummerow C, et al. (2017). NFATc1 controls the cytotoxicity of CD8+ T cells. Nat. Commun 8, 511. 10.1038/s41467-017-00612-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gautam S, Fioravanti J, Zhu W, Le Gall JB, Brohawn P, Lacey NE, Hu J, Hocker JD, Hawk NV, Kapoor V, et al. (2019). The transcription factor c-Myb regulates CD8+ T cell stemness and antitumor immunity. Nat. Immunol 20, 337–349. 10.1038/s41590-018-0311-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336. 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abdelsamed HA, Moustaki A, Fan Y, Dogra P, Ghoneim HE, Zebley CC, Triplett BM, Sekaly R-P, and Youngblood B (2017). Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J. Exp. Med 214, 1593–1606. 10.1084/jem.20161760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.King HW, and Klose RJ (2017). The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. eLife 6, e22631. 10.7554/eLife.22631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang X, Park KM, Gontarz P, Zhang B, Pan J, McKenzie Z, Fischer LA, Dong C, Dietmann S, Xing X, et al. (2021). OCT4 cooperates with distinct ATP-dependent chromatin remodelers in naïve and primed pluripotent states in human. Nat. Commun. 12, 5123. 10.1038/s41467-021-25107-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weider M, Küspert M, Bischof M, Vogl MR, Hornig J, Loy K, Kosian T, Müller J, Hillgärtner S, Tamm ER, et al. (2012). Chromatin-remodeling factor Brg1 is required for Schwann cell differentiation and myelination. Dev. Cell 23, 193–201. 10.1016/j.devcel.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 78.Wang Z, Wang P, Li Y, Peng H, Zhu Y, Mohandas N, and Liu J (2021). Interplay between cofactors and transcription factors in hematopoiesis and hematological malignancies. Signal Transduct. Target. Ther 6, 24. 10.1038/s41392-020-00422-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takaku M, Grimm SA, Shimbo T, Perera L, Menafra R, Stunnenberg HG, Archer TK, Machida S, Kurumizaka H, and Wade PA (2016). GATA3-dependent cellular reprogramming requires activation-domain dependent recruitment of a chromatin remodeler. Genome Biol 17, 36. 10.1186/s13059-016-0897-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zheng C, Zheng L, Yoo J-K, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, et al. (2017). Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356.e16. 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 82.Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, Gao R, Kang B, Zhang Q, Huang JY, et al. (2018). Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272. 10.1038/s41586-018-0694-x. [DOI] [PubMed] [Google Scholar]
- 83.Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, et al. (2018). Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 24, 978–985. 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 84.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013.e20. 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, Boesteanu AC, Wang Y, O’Connor RS, Hwang W-T, et al. (2018). Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med 24, 563–571. 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, and Rosenberg SA (2009). Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 114, 1537–1544. 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, Good Z, Belk JA, Daniel B, Klysz D, et al. (2021). Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 372, eaba1786. 10.1126/science.aba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, et al. (2016). Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165. 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Deng Q, Han G, Puebla-Osorio N, Ma MCJ, Strati P, Chasen B, Dai E, Dang M, Jain N, Yang H, et al. (2020). Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med 26, 1878–1887. 10.1038/s41591-020-1061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Papillon JPN, Nakajima K, Adair CD, Hempel J, Jouk AO, Karki RG, Mathieu S, Möbitz H, Ntaganda R, Smith T, et al. (2018). Discovery of orally active inhibitors of Brahma homolog (BRM)/SMARCA2 ATPase activity for the treatment of Brahma related gene 1 (BRG1)/SMARCA4-mutant cancers. J. Med. Chem 61, 10155–10172. 10.1021/acs.jmedchem.8b01318. [DOI] [PubMed] [Google Scholar]
- 91.Farnaby W, Koegl M, Roy MJ, Whitworth C, Diers E, Trainor N, Zollman D, Steurer S, Karolyi-Oezguer J, Riedmueller C, et al. (2019). BAF complex vulnerabilities in cancer demonstrated via structure-based Protac design. Nat. Chem. Biol 15, 672–680. 10.1038/s41589-019-0294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xiao L, Parolia A, Qiao Y, Bawa P, Eyunni S, Mannan R, Carson SE, Chang Y, Wang X, Zhang Y, et al. (2022). Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature 601, 434–439. 10.1038/s41586-021-04246-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vaswani RG, J.A., N., Millan DS, Schiller SER, Wilson KJ, and Huang DS (2020). Compounds and Uses Thereof [Google Scholar]
- 94.Huang Z, Zak J, Pratumchai I, Shaabani N, Vartabedian VF, Nguyen N, Wu T, Xiao C, and Teijaro JR (2019). IL-27 promotes the expansion of self-renewing CD8(+) T cells in persistent viral infection. J. Exp. Med 216, 1791–1808. 10.1084/jem.20190173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yee C (2014). The use of endogenous T cells for adoptive transfer. Immunol. Rev 257, 250–263. 10.1111/imr.12134. [DOI] [PubMed] [Google Scholar]
- 96.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, et al. (2013). CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med 5, 177ra38. 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sadelain M, Brentjens R, and Rivière I (2013). The basic principles of chimeric antigen receptor design. Cancer Discov 3, 388–398. 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, Klebanoff CA, Johnson LA, Kerkar SP, Yang S, et al. (2011). Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood 117, 808–814. 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, and Riddell SR (2008). Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Invest 118, 294–305. 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. (2011). A human memory T cell subset with stem cell–like properties. Nat. Med 17, 1290–1297. 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, Gao P, Bandyopadhyay S, Sun H, Zhao Z, et al. (2022). Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 602, 503–509. 10.1038/s41586-021-04390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, Peralta R, Wang Y, Wang Y, DePeaux K, et al. (2021). Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol 22, 205–215. 10.1038/s41590-020-00834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vardhana SA, Hwee MA, Berisa M, Wells DK, Yost KE, King B, Smith M, Herrera PS, Chang HY, Satpathy AT, et al. (2020). Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol 21, 1022–1033. 10.1038/s41590-020-0725-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, and Wherry EJ (2014). Molecular and transcriptional basis of CD4+ T cell dysfunction during chronic. Immunity 40, 289–302. 10.1016/j.immuni.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Miggelbrink AM, Jackson JD, Lorrey SJ, Srinivasan ES, Waibl-Polania J, Wilkinson DS, and Fecci PE (2021). CD4 T-cell exhaustion: does it exist and what are its roles in cancer? Clin. Cancer Res 27, 5742–5752. 10.1158/1078-0432.CCR-21-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kelso TWR, Porter DK, Amaral ML, Shokhirev MN, Benner C, and Hargreaves DC (2017). Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. eLife 6, e30506. 10.7554/eLife.30506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang X, Lee RS, Alver BH, Haswell JR, Wang S, Mieczkowski J, Drier Y, Gillespie SM, Archer TC, Wu JN, et al. (2017). SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet 49, 289–295. 10.1038/ng.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mashtalir N, Suzuki H, Farrell DP, Sankar A, Luo J, Filipovski M, D’Avino AR, St Pierre R, Valencia AM, Onikubo T, et al. (2020). A structural model of the endogenous human BAF complex informs disease mechanisms. Cell 183, 802–817.e24. 10.1016/j.cell.2020.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pan J, Meyers RM, Michel BC, Mashtalir N, Sizemore AE, Wells JN, Cassel SH, Vazquez F, Weir BA, Hahn WC, et al. (2018). Interrogation of mammalian protein complex structure, function, and membership using genome-scale fitness screens. Cell Syst 6, 555–568.e7. 10.1016/j.cels.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang Q, Hresko ME, Picton LK, Su L, Hollander MJ, Nunez-Cruz S, Zhang Z, Assenmacher CA, Sockolosky JT, Garcia KC, et al. (2021). A human orthogonal IL-2 and IL-2Rb system enhances CAR T cell expansion and antitumor activity in a murine model of leukemia. Sci. Transl. Med 13, eabg6986. 10.1126/scitranslmed.abg6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aspuria P-J, Vivona S, Bauer M, Semana M, Ratti N, McCauley S, Riener R, de Waal Malefyt R, Rokkam D, Emmerich J, et al. (2021). An orthogonal IL-2 and IL-2Rb system drives persistence and activation of CAR T cells and clearance of bulky lymphoma. Sci. Transl. Med 13, eabg7565. 10.1126/scitranslmed.abg7565. [DOI] [PubMed] [Google Scholar]
- 112.Zebley CC, Brown C, Mi T, Fan Y, Alli S, Boi S, Galletti G, Lugli E, Langfitt D, Metais J-Y, et al. (2021). CD19-CAR T cells undergo exhaustion DNA methylation programming in patients with acute lymphoblastic leukemia. Cell Rep 37, 110079. 10.1016/j.celrep.2021.110079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y, and Zhang Y (2020). T cell dysfunction and exhaustion in cancer. Front. Cell Dev. Biol 8, 17. 10.3389/fcell.2020.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.El-Khairi R, and Vallier L (2016). The role of hepatocyte nuclear factor 1b in disease and development. Diabetes Obes. Metab 18, 23–32. 10.1111/dom.12715. [DOI] [PubMed] [Google Scholar]
- 115.Casemayou A, Fournel A, Bagattin A, Schanstra J, Belliere J, Decramer S, Marsal D, Gillet M, Chassaing N, Huart A, et al. (2017). Hepatocyte nuclear Factor-1B controls mitochondrial respiration in renal tubular cells. J. Am. Soc. Nephrol 28, 3205–3217. 10.1681/ASN.2016050508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Okamoto T, Mandai M, Matsumura N, Yamaguchi K, Kondoh H, Amano Y, Baba T, Hamanishi J, Abiko K, Kosaka K, et al. (2015). Hepatocyte nuclear factor-1β (HNF-1 β) promotes glucose uptake and glycolytic activity in ovarian clear cell carcinoma. Mol. Carcinog 54, 35–49. 10.1002/mc.22072. [DOI] [PubMed] [Google Scholar]
- 117.Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, and Delgoffe GM (2016). The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45, 374–388. 10.1016/j.immuni.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yu YR, Imrichova H, Wang H, Chao T, Xiao Z, Gao M, Rincon-Restrepo M, Franco F, Genolet R, Cheng WC, et al. (2020). Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat. Immunol 21, 1540–1551. 10.1038/s41590-020-0793-3. [DOI] [PubMed] [Google Scholar]
- 119.Wang B, Wang M, Zhang W, Xiao T, Chen CH, Wu A, Wu F, Traugh N, Wang X, Li Z, et al. (2019). Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc 14, 756–780. 10.1038/s41596-018-0113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Broad Institute. Picard Toolkit. Broad Institute, GitHub repository https://github.com/broadinstitute/picard [Google Scholar]
- 125.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kuhn RM, Haussler D, and Kent WJ (2013). The UCSC genome browser and associated tools. Brief. Bioinform 14, 144–161. 10.1093/bib/bbs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhu Q, Liu N, Orkin SH, and Yuan G-C (2019). CUT&RUNTools: a flexible pipeline for CUT&RUN processing and footprint analysis. Genome Biol 20, 192. 10.1186/s13059-019-1802-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Opgen-Rhein R, and Strimmer K (2007). Accurate ranking of differentially expressed genes by a distribution-free shrinkage approach. Stat. Appl. Genet. Mol. Biol 6, Article9. 10.2202/1544-6115.1252. [DOI] [PubMed] [Google Scholar]
- 133.Schäfer J, and Strimmer K (2005). A shrinkage approach to large-scale covariance matrix estimation and implications for functional genomics. Stat. Appl. Genet. Mol. Biol 4, Article32. 10.2202/1544-6115.1175. [DOI] [PubMed] [Google Scholar]
- 134.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin SM, Lapointe DS, and Green MR (2010). ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics 11, 237. 10.1186/1471-2105-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sheffield NC, and Bock C (2016). LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics 32, 587–589. 10.1093/bioinformatics/btv612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vierstra J, Lazar J, Sandstrom R, Halow J, Lee K, Bates D, Diegel M, Dunn D, Neri F, Haugen E, et al. (2020). Global reference mapping of human transcription factor footprints. Nature 583, 729–736. 10.1038/s41586-020-2528-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Friedman J, Hastie T, and Tibshirani R (2010). Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- 140.Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978. 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Feinberg AP, Koldobskiy MA, and Göndör A (2016). Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet 17, 284–299. 10.1038/nrg.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hogg SJ, Beavis PA, Dawson MA, and Johnstone RW (2020). Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Discov 19, 776–800. 10.1038/s41573-020-0077-5. [DOI] [PubMed] [Google Scholar]
- 143.Chen X, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A, Sakellaropoulos T, Gong Y, Kloetgen A, Yap YS, et al. (2019). Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment. Cancer Discov 9, 890–909. 10.1158/2159-8290.CD-19-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, and Greenleaf WJ (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomics data (CUT&Tag, ATAC-seq, and RNA-seq) have been deposited at GEO under accession number GSE212357 and are publicly available as of the date of publication. This paper also analyzes existing, publicly available data (GSE181062 and GSE181064 key resources table). Unprocessed blot images have been deposited in Mendeley (Mendeley data: https://doi.org/10.17632/7msw2d62r6.1). See Key Resources Table for details.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead, Cigall Kadoch, upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Brilliant Violet 605™ anti-human CD279 (PD-1) | BioLegend | Cat# 329923; RRID: AB_11124107 |
| APC/Cyanine7 anti-human CD366 (Tim-3) | BioLegend | Cat# 345025; RRID: AB_2565716 |
| Brilliant Violet 605™ anti-human CD197 (CCR7) | BioLegend | Cat# 353223; RRID: AB_11124325 |
| APC/Cyanine7 anti-human CD45RA | BioLegend | Cat# 304127; RRID: AB_10708419 |
| APC anti-human CD39 | BioLegend | Cat# 328209; RRID: AB_1953233 |
| APC/Cyanine7 anti-mouse CD279 (PD-1) | BioLegend | Cat# 135223; RRID: AB_2563522 |
| PerCP/Cyanine5.5 anti-mouse CD366 (Tim-3) | BioLegend | Cat# 119717; RRID: AB_2571934 |
| APC anti-mouse CD62L | BioLegend | Cat# 104411; RRID: AB_313098 |
| PerCP/Cyanine5.5 anti-mouse/human CD44 | BioLegend | Cat# 103032; RRID: AB_2076204 |
| PE anti-human TNF-α | BioLegend | Cat# 502908; RRID: AB_315260 |
| Alexa Fluor® 700 anti-human IFN-γ | BioLegend | Cat# 506515; RRID: AB_961351 |
| APC anti-mouse TNF-α | BioLegend | Cat# 506307; RRID: AB_315429 |
| PE/Cyanine7 anti-mouse IFN-γ | BioLegend | Cat# 505825; RRID: AB_1595591 |
| PE/Cyanine7 anti-human CD3 | BioLegend | Cat# 317333; RRID: AB_2561451 |
| APC/Cyanine7 anti-mouse CD8a | BioLegend | Cat# 100714; RRID: AB_312753 |
| FITC anti-human CD3 | ThermoFisher Scientific | Cat#11-0038-42; RRID: AB_2043831 |
| PE/Cy7 anti-human CD8 | Biolegend | Cat# 344712; RRID: AB_2044007 |
| Alexa Fluor® 700 anti-human CD4 | Biolegend | Cat# 317426; RRID: AB_571942 |
| Mouse Anti-Actin (clone C4) | Millipore | Cat# MAB1501; RRID: AB_2223041 |
| PE/Cyanine7 anti-human CD25 | BioLegend | Cat# 302612; RRID: AB_314282 |
| Rabbit Anti-IgG Donkey Polyclonal Antibody (HRP) | Cytiva | Cat# NA934-1ML; RRID: AB_772206 |
| Normal Rabbit IgG | Cell Signaling Technology | Ca# 2729S; RRID: AB_1031062 |
| Anti-Histone H3 (acetyl K27) | Abcam | Cat# ab4729; RRID: AB_2118291 |
| Rabbit Anti-SMARCA4 (Brg1) (D1Q7F) | Cell Signaling Technology | Cat# 49360S; RRID: AB_2728743 |
| Rabbit Anti-SS18 (D614Z) | Cell Signaling Technology | Cat# 21792S; RRID: AB_2728667 |
| Rabbit Anti-ARID1A (D2A8U) | Cell Signaling Technology | Cat# 12354S; RRID: AB_2637010 |
| Rabbit Anti-PBRM1 (91894) | Cell Signaling Technology | Cat# 91894S; RRID: AB_2800173 |
| Rabbit Anti-HNF1B | Proteintech | Cat# 12533-1-AP; RRID: AB_2116758 |
|
Bacterial and virus strains | ||
| MegaX DH10B electro-competent bacteria | Life Technologies | Cat# C640003 |
|
Biological samples | ||
| Primary Human CD8+ T cells isolated from PBMCs (Donor 1: 24y/o, Female; Donor 2: 35y/o, Male; Donor 5: 24y/o, Male) | StemCell Technologies Inc | Cat# 70027 |
| Primary Human CD8+/CD4+ T cells isolated from PBMCs (Donor 3: 28y/o, Female; Donor 4: 19y/o, Female; Donor 6: 28y/o, Female; Donor7: <30y/o, N/A; Donor8: <30y/o, Male) | New York Blood Center | https://www.nybc.org/ |
| Primary Human CD3+ T cells isolated from PBMCs: (Donor 9: 43y/o, Male; Donor 10: 42y/o, Female) | Indiana University | N/A |
|
Chemicals, peptides, and recombinant proteins | ||
| Zombie Aqua™ Fixable Viability Kit | BioLegend | Cat# 423101 |
| Zombie Violet™ Fixable Viability Kit | BioLegend | Cat# 423113 |
| eBioscience Foxp3/Transcription Factor Staining Buffer Set kit | Life Technologies | Cat# 00-5523-00 |
| BsmBI-v2 | New England Biolabs | Cat# R0739L |
| Anza Esp1 enzyme | Life Technologies | Cat# IVGN0136 |
| T4 DNA ligase | New England Biolabs | Cat# M0202L |
| ATP | New England Biolabs | Cat# PO756S |
| Plasmid safe ATP-dependent DNase | Thermo Fisher Scientific | Cat# NC9046399 |
| TaKaRa Ex Taq DNA Polymerase | Takara Bio | Cat# RR001B |
| Polyethylenimine (PEI) | Thermo Fisher Scientific | Cat# NC1014320; CAS: 9002-98-6 |
| Polybrene | Santa Cruz Biotechnology | Cat# SC-134220; CAS: 28728-55-4 |
| TaKaRa Ex Taq DNA Polymerase | Takara Bio | Cat# RR001B |
| 10mM 2-Mercaptoethanol | Life Technologies | Cat# 21985023; CAS: 60-24-2 |
| T4 PNK | New England Biolabs | Cat# VWR #101228-174 |
| T4 ligase | New England Biolabs | Cat# M0202M |
| SIINKEFL peptide (OVA 257-264) | Invivogen | Cat# vac-sin |
| ACBI1 | SelleckChem | Cat# S9612; CAS: 2375564-55-7 |
| AU-15330 | MedChem Express LLC | Cat #HY-145388; CAS: 2380274-50-8 |
| Cas9-GFP ribonucleoprotein | Integrated DNA Technologies | Cat#10008100 |
| CMP14 | Courtesy of Jun Qi; Papillon et al.90 | N/A |
| FHT-1015 | MedChemExpress® | Cat# HY-144896; CAS: 2368903-18-6 |
| Recombinant Human IL-2 | Peprotech, Inc. | Cat# 200-02-1MG |
| Recombinant Human IL-7 | Peprotech, Inc. | Cat# 200-07-50UG |
| Recombinant Murine IL-2 | Peprotech, Inc. | Cat# 212-12-50UG |
| Recombinant Murine IL-7 | Peprotech, Inc. | Cat# 212-17-50UG |
| Recombinant Murine IFNg | Peprotech, Inc. | Cat# 315-05 |
| APC Annexin V | BioLegend | Cat# 640920 |
|
Critical commercial assays | ||
| QIAquick Gel Extraction Kit | Qiagen | Cat# 28706X4 |
| Phusion High-Fidelity PCR Kit | Life Technologies | Cat# F553S |
| MinElute Reaction Cleanup Kit | Qiagen | Cat# 28204 |
| PureLink™ HiPure Plasmid Filter Maxiprep Kit | Thermo Fisher Scientific | Cat#K21001 |
| QIAamp DNA Mini Kit | Qiagen | Cat# 51304 |
| PureLink™ HiPure Plasmid Filter Maxiprep Kit | Thermo Fisher Scientific | Cat# K21001 |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74136 |
| Nebnext Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | Cat# E7490 |
| NEXTFLEX® Poly(A) Beads 2.0 | Perkin Elmer | Cat# NOVA-512991 |
| Nebnext Ultra II Directional RNA Library Prep Kit | New England Biolabs | Cat# E7760 |
| NEXTFLEX® Rapid Directional RNA-Seq Kit 2.0 | Perkin Elmer | Cat# NOVA-5198-01 |
| Qiagen MinElute Reaction clean up kit | Qiagen | Cat# 28206 |
| NextSeq™ 500/550 High output flow cell kit | Illumina | Cat# 20024906 |
| CellTiterGlo | Promega | Cat# G7571 |
|
Deposited data | ||
| Cut&Tag-seq, RNA-seq, and ATAC-seq | This study | GSE212357 |
| scATAC-seq | Sathpathy et al.60 | GSE181062 |
| scATAC-seq | Kourtis et al.22 | GSE181064 |
| Unprocessed blot images | This study | Mendeley data: https://doi.org/10.17632/7msw2d62r6.1 |
|
Experimental models: Organisms/strains | ||
| Mice (C57BL/6J) | The Jackson Laboratory | Jackson strain #000664; RRID: IMSR_JAX:000664 |
| OT-1 mice (C57BL/6-Tg(TcraTcrb)1100Mjb/J) | The Jackson Laboratory | Jackson strain #003831; RRID: IMSR_JAX:003831 |
| Cas9 Mice (Gt(ROSA)26Sortm1.1 (CAG-cas9*,-EGFP)Fezh/J) | The Jackson Laboratory | Jackson strain #024858; RRID: IMSR_JAX:024858 |
| Rag1KO mice B6.129S7-Rag1<tm1Mom>/J | The Jackson Laboratory | Jackson strain #002216; RRID: IMSR_JAX:002216 |
|
Oligonucleotides | ||
| Mouse sgRNAs oligo pool targeting epigenetic genes | Twist Bioscience | https://www.twistbioscience.com/products/oligopools |
| sgROSA (mouse) 5’-AACGGCTCCA CCACGCTCGG-3’ | This study | N/A |
| sgSMARCA4-1 (mouse, sg1553 in library) 5’- ATAATGGCCTACAAGATGT- 3’ | This study | N/A |
| sgSMARCA4-2 (mouse, sg1554 in library) 5’- ATTGCCCGACCACCTGCAGA- 3’ | This study | N/A |
| sgSMARCC1-1 (mouse, sg1563 in library) 5’- TGCCCCAAGAATGTGACACA- 3’ | This study | N/A |
| sgSMARCC1-2 (mouse, sg1564 in library) 5’- TTGGTGACATGCTTCCCAA- 3’ | This study | N/A |
| sgARID1A-1 (mouse, sg1844 in library) 5’- TGCACCACAAGCACCCAGAG- 3’ | This study | N/A |
| sgARID1A-2 (mouse, sg1845 in library) 5’- GTCACTGAGGAAGCGCACCA- 3’ | This study | N/A |
| sgARID1B-1 (mouse, sg1851 in library) 5’- GCTCAGCACCCCGTACCCCG- 3’ | This study | N/A |
| sgARID1B-2 (mouse, sg1853 in library) 5’- CAGCAGAGCAGCCCATACCC- 3’ | This study | N/A |
| sgDPF2-1 (mouse, sg0385 in library) 5’- TCAGCAGATCCAGACACAGG- 3’ | This study | N/A |
| sgDPF2-2 (mouse, sg0386 in library) 5’- AAGGAGTGAGACAGTACATG- 3’ | This study | N/A |
| sgARID2-1 (mouse, sg1854 in library) 5’- GCCGTTTAAGAAGATCCCTG- 3’ | This study | N/A |
| sgARID2-2 (mouse, sg1858 in library) 5’- ACCAGAGTCACTACTTTAGG- 3’ | This study | N/A |
| SMARCA4 sgRNAs cocktail (human) 5’-ACUCCAGACCCACCCCUGGG-3’5’- CCCUAGCCCGGGUCCCUCGC-3’5’- GUCCUGCUGAGGGCGGCCCU-3’ | Synthego | https://www.synthego.com/products/crispr-kits/gene-knockout-kit |
| HNF1B sgRNAs cocktail (human) 5’-AGCCCUCGUCGCCGGACAAG-3’5’- GGCCGAGCCCGACACCAAGC-3’5’- CGGGGUCACCAAGGAGGUGC-3’ | Synthego | https://www.synthego.com/products/crispr-kits/gene-knockout-kit |
|
Recombinant DNA | ||
| pLKO5.sgRNA.EFS.tRFP plasmid | Addgene | Cat #57823; RRID: Addgene_57823 |
| psPAX2 plasmid | Addgene | Cat#12260; RRID: Addgene_12260 |
| pMD2G plasmid | Addgene | Cat#12259; RRID: Addgene_12259 |
| Epigenetic mouse CRISPR library cloned in pLKO5.sgRNA.EFS.tRFP plasmid | This study | N/A |
| Anti-CD19-FMC63scFV-41BB-GFP | This study | N/A |
|
Software and algorithms | ||
| MAGECK | MAGeCKFlute package119 | https://sourceforge.net/p/mageck/wiki/Home/. |
| STAR v2.5.2b | Dobin et al.120 | https://github.com/alexdobin/STAR; RRID: SCR_004463 |
| deepTools v2.5.3 | Ramírez et al.121 | https://deeptools.readthedocs.io/en/develop/; RRID: SCR_016366 |
| Trimmomatic v0.36 | Bolger et al.122 | http://www.usadellab.org/cms/?page=trimmomatic; RRID: SCR_011848 |
| Bowtie2 v2.29 | Langmead and Salzberg.123 | https://bowtie-bio.sourceforge.net/index.shtml; RRID: SCR_016368 |
| Picard v2.8.0 | Picard.124 | https://github.com/broadinstitute/picard; RRID:SCR_006525 |
| SAMtools v 0.1.19 | Li et al.125 | https://samtools.sourceforge.net; RRID: SCR_002105 |
| MACS2 v2.1.1 software | Zhang et al.126 | https://pypi.org/project/MACS2/; RRID: SCR_013291 |
| UCSC utilities | Kuhn et al.127 | https://genome.ucsc.edu/util.html; RRID: SCR_005780 |
| CutRunTools | Zhu et al.128 | https://zenodo.org/record/3374112#.Y22KTy2B3xs |
| edgeR v3.12.1 | Robinson et al.129 | https://bioconductor.org/packages/release/bioc/html/edgeR.html; RRID: SCR_012802 |
| GSEA | Subramanian et al.130 | https://www.gsea-msigdb.org/gsea/index.jsp; RRID: SCR_003199 |
| DESeq2 v1.30.1 | Love et al.131 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html; RRID: SCR_015687 |
| corpcor | Opgen-Rhein and Strimmer132; Schäfer and Strimmer.133 | https://cran.r-project.org/web/packages/corpcor/index.html |
| Bedtools multiIntersectBed and mergeBed functions | Quinlan and Hall134 | https://bedtools.readthedocs.io/en/latest/ |
| ChIPpeakAnno v3.17.0 | Zhu et al.135 | https://bioconductor.org/packages/release/bioc/html/ChIPpeakAnno.html; RRID: SCR_012828 |
| LOLA v1.12.0 | Sheffield and Bock136 | https://bioconductor.org/packages/release/bioc/html/LOLA.html; RRID: SCR_006912 |
| HOMER v4.9 | Heinz et al.137 | http://homer.ucsd.edu/homer/; RRID: SCR_010881 |
| non-redundant archetype consensus motifs | Vierstra et al.138 | https://www.vierstra.org/resources/motif_clustering |
| GLMnet | Friedman et al.139 | https://glmnet.stanford.edu/index.html; RRID: SCR_015505 |
| ChromVAR | Schep et al.140 | https://greenleaflab.github.io/chromVAR/index.html |
|
Other | ||
| Lymphoprep™ | STEMCELL Technologies Inc | Cat# 07811 |
| Pan T Cell Isolation kit, Human | Miltenyi Biotec | Cat# 130-096-535 |
| CD8+ T Cell Isolation Kit, Human | Miltenyi Biotec | Cat# 130-096-495 |
| CD4+ T Cell Isolation Kit, Human | Miltenyi Biotec | Cat# 130-096-533 |
| CD8a+ T Cell Isolation Kit, Mouse | Miltenyi Biotech | Cat #130-104-075 |
| Human T-Activator CD3/CD28 Dynabeads | Thermo Fisher Scientific | Cat# 11132D |
| Mouse T-Activator CD3/CD28 Dynabeads | Thermo Fisher Scientific | Cat# 11453D |
| ACK lysis buffer | Quality Biological Inc | Cat# 118156101C |
| 1X GlutaMAX | Life Technologies | Cat# 35050061 |
| 1X Non-essential Amino Acids | Life Technologies | Cat# 11140050 |
| 1X Sodium Pyruvate | Thermo Fisher Scientific | Cat# MT25000CI |
| Cell Stimulation Cocktail | Affymetrix | Cat# 00-4970-93 |
| Human TruStain FcX™ | Human TruStain FcX™ | Cat# 422302 |
| Ampure beads | Thermo Fisher Scientific | Cat# NC9933872 |
| Concanavalin A | BioMag®Plus | Cat# 86057 |
| CUTANA pAG-Tn5 | Epicycpher | Cat# 15-1117 |
| 4%−12% Bis-Tris polyacrylamide gels | Life Technologies | Cat# NP0336BOX |
| Immobilon PVDF Transfer Membrane | Millipore | Cat# IPVH00010 |
| eBioscience Brefeldin A Solution | Life Technologies | Cat# 00-4506-51 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Life Technologies | Cat# 34095 |