

Abstract
Induction of cell death is inexorably linked with cancer therapy, but this can also initiate wound healing processes that have been linked to cancer progression and therapeutic resistance. Here we describe the contribution of apoptosis and the lytic cell death pathways in the response to therapy, including chemotherapy and immunotherapy. We also discuss how necroptosis, pyroptosis and ferroptosis function to promote tumor immunogenicity, along with emerging findings that these same forms of death can paradoxically contribute to immune suppression and tumor progression. Understanding the duality of cell death in cancer may allow for the development of therapeutics that shift the balance towards regression.
Keywords: cell death, apoptosis, necroptosis, pyroptosis, ferroptosis, immunogenicity, immunogenic cell death, tumor microenvironment, immunotherapy
Cell death pathways and the tumor immune microenvironment
Neoplastic cell death is the primary goal of most cancer therapeutics, and the myriad of pathways through which regulated cell death (RCD) can occur are becoming increasingly clear. Forms of RCD are classified according to their morphotypes into: (1) type I cell death or apoptosis; (2) type II cell death or autophagy; and type III cell death or necrosis [1]. Apoptosis is the best understood and is defined by clear morphological changes that include the rounding-up of the cell, retraction of pseudopods, decrease in cellular volume (pyknosis), chromatin condensation, nuclear fragmentation (karyorrhexis), plasma membrane blebbing, and the activation of caspases [2]. Autophagy is characterized by engaging the autophagosome machinery for the degradation and the removal of damaged proteins and organelles within a cell. However, while autophagy often accompanies cell death, it is not a mechanism of death per se [3]. In contrast, type III necrotic cell death does not display distinctive features of type I or II cell death and is characterized by the loss of membrane integrity and swelling of subcellular organelles (oncosis) with disposal of cell corpses in the absence of lysosomal involvement and phagocytic activity [1] (Figure 1). Although the various cell death modalities have distinct features, there is a substantial amount redundancy and crosstalk between the signaling pathways. Thus, blocking one form of cell death may engage another, with necroptosis occurring mostly in the absence of apoptotic machinery being illustrative of this process. The various forms of cell death have been expertly reviewed elsewhere [4–6], with a detailed description also found in Box 1.
Figure 1. Overview of cell death modalities.
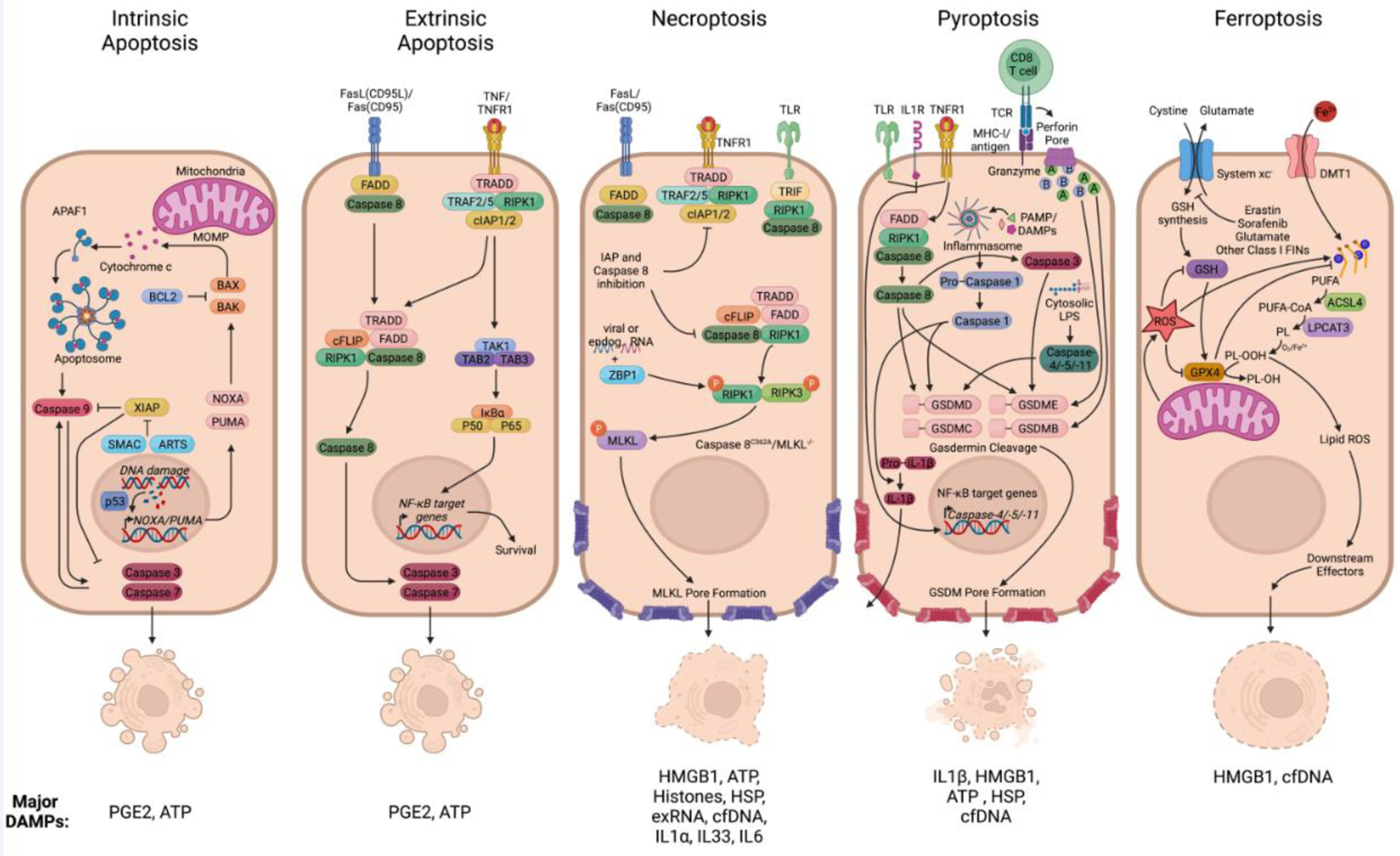
Active cell death processes are classified into sub forms according to the genes regulating each modality. (Panel 1) Caspase-dependent forms of non-lytic programmed cell death include intrinsic apoptosis that is initiated by MOMP and subsequent release of cytochrome c that can be controlled by BAX/BAK. This triggers the formation of the multiprotein complex termed the ‘apoptosome’ and activation of caspase-9, leading to downstream activation of executioner caspases-3 and -7. PGE2 and ATP are among the most common DAMPs released from apoptotic cells. While ATP is considered as a find-me signal for phagocytic cells, PGE2 negatively regulating death induced inflammation, promotes survival and angiogenesis and can act on surrounding stroma and the tumor microenvironment. (Panel 2) A variety of cell surface receptors, mainly from the TNF receptor superfamily such as CD95 or TNFR1, can lead to extrinsic apoptosis signaling via caspase-8 and RIPK1. Alternatively, these can lead to the induction of survival signaling via TAK1 mediated NF-kB activation driving survival gene expression. (Panel 3) Under apoptosis compromised conditions, such as depletion or pharmacological inhibition of caspase-8 and the IAPs, activation of RIPK1 can lead to necroptosis via RIPK3 and MLKL. The DAMPs described for necroptosis are HMGB1, ATP, Histones, HSP, exRNA, cfDNA, IL1α, IL33, and IL6. These generally have pro-inflammatory features; however, dose and context are critical in determining outcome. For example, IL-6 can promote and inhibit anti-tumor immunity. (Panel 4) Pyroptosis is induced through different cell surface receptors including TLRs, IL1R, and TNFR1. Essential for the execution of pyroptosis is the cleavage of GSDMs, leading to pore-formation, release of mature IL-1β, and the leakage of cellular content. This can be triggered by inflammasome activation and caspase-1, activated caspase-8 and caspase-3, or via granzymes from cytotoxic lymphocytes. Among the DAMPs release during pyroptosis (IL1β, HMGB1, HSP, cfDNA), IL1β is the strongest inducer of inflammation. (Panel 5) Ferroptosis occurs as a consequence of iron overload and accumulation of peroxidized lipids, caused either by GPX4 inhibition or glutathione deprivation. The few DAMPs described during ferroptotic cells have all been found to have pro-inflammatory functions. Figure created with BioRender.
Figure 1 Abbreviations:
BCL2 B-Cell Lymphoma 2
BAX BCL2 Associated X
BAK BCL2 Antagonist/Killer 1
XIAP X-Linked Inhibitor Of Apoptosis
SMAC Second Mitochondria-Derived Activator Of Caspase
ARTS Apoptosis-Related Protein In The TGF-Beta Signaling
PUMA P53-Upregulated Modulator Of Apoptosis
p53 Tumor protein P53
PGE2 Prostaglandin E2
ATP adenosine 5′-triphosphate
TNFR1 TNF receptor superfamily member 1A
FADD Fas associated via death domain
TRADD TNFRSF1A Associated Via Death Domain
TRAF2/5 TNF Receptor Associated Factor 2/5
RIPK1 Receptor Interacting Serine/Threonine Kinase 1
cIAP1/2 Cellular Inhibitor Of Apoptosis 1/2
cFLIP CASP8 And FADD Like Apoptosis Regulator
ZBP1 Z-DNA Binding Protein 1
RIPK3 Receptor Interacting Serine/Threonine Kinase 3
MLKL Mixed Lineage Kinase Domain Like Pseudokinase
HMGB1 High Mobility Group Box 1
HSP heat shock proteins
exRNA extracellular RNA
cfDNA cell-free DNA
IL1a Interleukine 1 alpha
IL33 Interleukin-33
IL6 Interleukin-6
TLR Toll-like receptor
MHC-I Major histocompatibility complex 1
TCR T cell receptor
PAMP Pattern associated molecular patterns
DAMP Danger associated molecular patterns
LPS Lipopolysaccharides
GSDMD/E/C/B Gasdermin D/E/C/B
NF-kB Nuclear factor kappa B
IL1b Interleukine 1 beta
ROS Reactive oxigen species
GSH Glutathione synthetase
GPX4 Glutathione Peroxidase 4
PUFA Polyunsaturated fatty acids
ACSL4 acyl-CoA synthetase long chain family member 4
LPCAT3 Lysophosphatidylcholine acyltransferase 3
CoA Coenzyme A
PL Phospholipid
Box 1: Regulated Cell Death Signaling Pathways.
Intrinsic apoptosis serves as a response to internal stresses such as hypoxia, cell cycle checkpoint arrest, metabolic stresses, ER stress, cytokine deprivation, genomic stress and oncogenic signaling. Intrinsic apoptosis occurs through tightly controlled release of mitochondrial proapoptotic factors by pro- and anti-apoptotic members of the B-cell lymphoma-2 (BCL2) family [5]. Outer membrane permeabilization by the pore forming molecules BAX and BAK permits the release of cytochrome C and the proapoptotic factors Smac and Omi. As a result, caspase-9 mediates downstream activation of executioner caspase-3, -6 and -7 upon forming a dimer with the adapter protein apoptotic protease-activating factor-1 (APAF1), also termed the “apoptosome complex”. Caspases cleave a range of essential substrates including actin and laminin, activate nucleases to cleave DNA, and engage in self cleavage to perpetuate an irreversible positive feedback loop.
The extrinsic apoptosis pathway is induced by extracellular stimuli activating the death receptors (DRs), members of the TNF receptor (TNFR) superfamily such as TNFR1, CD95/Fas and TNF-related apoptosis-inducing ligand receptor-1 (TRAIL). The activation of certain internal pattern recognition receptors by viral DNA, such as RIG-I-like and Toll-like receptors (TLRs), can also trigger extrinsic apoptosis signaling. Stimulation of DRs cause procaspase-8 monomers to be recruited to the cytoplasmic region of the DR, leading to a complex formation and caspase-8 dimerization. In the best understood example, TNFα ligation to TNFR1 triggers the recruitment of receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and TRADD, leading to binding of TNFR-associated factors 2/5 (TRAF2/5) and cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1/2) to form “complex I”. In this complex, cIAP1/2 and the linear ubiquitin chain assembly complex (LUBAC)-mediated ubiquitylation of RIPK1 and other proteins within the complex leads to potent NFκB activation and cell survival. However, when ubiquitylation of RIPK1 is compromised, RIPK1 and TRADD are released from TNFR1 to form a series of dynamic cytosolic signaling platforms known as complex II, including the death-induced signaling complex (DISC) consisting of TRADD, FADD, caspase-8, and/or cFLIP. As will be discussed below, this complex is also capable of inducing necroptosis.
Necroptosis was originally identified as a backup mechanism for apoptosis under conditions of caspase inhibition [6]. As such, a variety of stimuli can promote necroptosis, including TNF ligand family members, interferons (IFNs), ligands for TLRs that utilize the adapter TRIF (TIR-domain-containing adapter-inducing interferon-β), and viral DNA through Z-DNA-Binding Protein 1 (ZBP1). Necroptosis occurs if complex II forms in the absence of caspase 8 or the presence of the short cFLIP isoform, which causes RIPK1 to interact with RIPK3 via the RHIM domain. Alternatively, RHIM domain containing molecules such as ZBP1 can directly interact with RIPK3. Phosphorylated RIPK3 recruits and phosphorylates MLKL, which acts as an executioner by oligomerizing into a pore complex that disrupts membrane integrity. This induces swelling of intracellular organelles and the release of multiple DAMPs that promote an inflammatory response.
Pyroptosis was identified following necrotic cell death in macrophages upon Salmonella or shigella infection [6]. Pyroptosis displays specific morphological changes that overlap with apoptosis and necroptosis, including chromatin condensation, cellular swelling, and plasma membrane rupture. Depending on the initiating stimulus, pyroptosis is mediated by activation of one or more caspases, including caspase-1, caspase-3, caspase-4 and caspase-5 (as well as caspase-11 in mice), resulting in cleavage and activation of the gasdermin (GSDM) superfamily and the formation of pores in the plasma membrane. A unique feature of pyroptosis is its association with inflammasome formation and canonical caspase-1 activation, cleaving IL1β and IL18 into mature forms for release through GSDMD pores even in the absence of full membrane permeabilization.
Ferroptosis was first observed in 2003, with the term itself coined in 2012. Ferroptotic cells are characterized by dysmorphic small mitochondria with shrunken cristae and condensed or ruptured cell membranes [6]. Ferroptosis can be triggered when hydrogen peroxide reacts with free iron atoms in the cytosol, causing lipid peroxidation and generation of toxic by-products. Ferroptosis is prevented by the phospholipid hydroperoxidase GPX4 and the cofactor glutathione (GSH), and conversely can be triggered in a canonical manner by compromising GPX4 activity or GSH deprivation. In addition, ferroptosis can be induced non-canonically by treatment of cells with hemoglobin, ferrous ammonium sulfate or other factors that cause iron overload. Ferroptosis may overlap and/or crosstalk with other cell death pathways, as it has been shown that GPX4 inactivation sensitizes to apoptosis, pyroptosis and necroptosis. The role of ferroptosis has been mainly defined during pharmacological interventions using anticancer drugs or other pathological settings such as tissue injury, but whether it plays a role during physiological settings such as development or tissue homeostasis remains to be elucidated.
Beyond the obvious goal of reducing the number of neoplastic cells, cancer cell death is the first step in the cancer-immunity cycle [7]. Tumor destruction allows for the acquisition of antigens by conventional dendritic cells (cDCs), while the release of damage-associated molecular patterns (DAMPs) and inflammatory cytokines promotes cDC migration to draining lymph nodes and the induction of an adaptive immune response. For this reason, there is intense interest in identifying and inducing immunogenic forms of cell death that optimize the immune response against cancer, especially in the era of immune checkpoint inhibitors. However, cell death engagement can also be counterproductive [8,9] and certain cancers with high rates of spontaneous apoptosis or necrotic lesions demonstrate increased aggressiveness [8,10]. These observations may be due to compensatory proliferation, in which treatment-resistant cells gain access to space in which to repopulate [9,11], reminiscent of the rapid regrowth observed following treatment with angiogenic inhibitors [11]. There is also evidence that cell death can promote angiogenesis and metastasis, both directly and indirectly through the recruitment of myeloid cells. Similarly, myeloid cell recruitment has been linked to the creation of an immunosuppressive tumor microenvironment that limits T cell cytotoxic activity. As cancer cell death is a necessary component of therapy, understanding these dissonant properties is critical for optimizing therapeutic interventions. Here we will discuss the types of RCD induced by anti-cancer therapies and how these impact tumor progression and the immune microenvironment, with a focus on the underappreciated pro-tumor properties.
Cancer therapy and cell death modalities
The goal of non-invasive cancer therapy is to maximize tumor cell death while minimizing healthy tissue toxicity through the targeting of cancer-selective properties, including higher proliferation rates and activation of pro-survival pathways. These approaches primarily induce intrinsic apoptosis and historically there has been limited interest in understanding how this may impact the tumor microenvironment. The first major change in this area was the discovery of immunogenic forms of apoptosis that are induced by treatment with specific chemotherapies [12]. The second major change has been the approval of immune checkpoint inhibitors such as anti-programmed death (PD)-1, which has led to studies investigating combination therapy with small molecule inhibitors [13]. As the number of approved immune and targeted therapies expands, it is increasingly important to understand how these can be combined or sequenced appropriately to maximize efficacy [14,15]. Below we detail our understanding of how therapies induce the different forms of cell death, while recognizing that cancer type, stage, and mutational status may regulate outcomes in ways that have not been explored.
Radiation and chemotherapy
Ionizing radiation is widely used in cancer therapy, including the use of fractionated radiotherapy to limit tissue toxicity [16]. Radiotherapy induces DNA damage directly as well as indirectly through the generation of free radicals from water, thereby inducing single- and double-strand breaks. While most breaks are repaired, around 5% of double-strand break repairs fail and drive senescence or intrinsic apoptosis through p53 activation or mitotic catastrophe [17,18]. Some reports have also found a role for extrinsic apoptosis due to upregulation of death receptors such as DR5 and Fas [19–21]. Many of the immunogenic properties associated with radiation-induced cell death, such as the release of DAMPs, occur in a delayed fashion consistent with secondary necrosis. However, in addition to the canonical apoptotic death induced by caspase activation and mitochondrial outer membrane permeabilization by BAX and BAK, multiple different cellular outcomes have been described following irradiation, such as necroptosis and iron-dependent cell death [22–25]. These pathways are not necessary for cell death to occur, but could feasibly impact how the immune system responds to irradiated tumor cells in a cell line and dose-dependent manner.
Chemotherapeutics work broadly by inducing DNA damage (e.g. cyclophosphamide, carboplatin), interrupting DNA replication or RNA synthesis (e.g. gemcitabine, doxorubicin, 5-fluoruricil, mitoxantrone), or by targeting microtubules to directly induce chromosomal missegregation (e.g. paclitaxel). As with radiation, these events are mostly associated with induction of intrinsic apoptosis [26–29]. However, there are numerous reports of ferroptosis being induced by cisplatin or paclitaxel in lung, head and neck, colorectal, gastric and pancreatic ductal carcinoma cells [30–37]. Necroptosis has also been noted using etoposide on human breast cancer and sarcoma cells [38], as well as with the alkylating agent Methyl methanesulfonate in human A549 lung adenocarcinoma cells [39], and another alkylating agent 3-Bromopyruvate in T24 bladder carcinoma cells [40]. Finally, pyroptosis is frequently observed during response to various chemotherapies, including for paclitaxel, cisplatin, etoposide, and doxorubicin [41–45]. In this case, pyroptosis occurs as a result of the executioner caspase-3, which can cleave GSDMD and GSDME in cells that express these molecules, including epithelial cells [42,46]. This has been suggested to represent secondary necrosis, as GSDM pores cause loss of membrane integrity and the release of DAMPs [47]. Thus, inflammasome-independent pyroptosis may be a general property of many cytotoxic therapies, with cell type selectivity driven by GSDMD and GSDME expression, which are reduced in some cancers [48]. As we will discuss below, it is also tempting to speculate that GSDMD/E activation explains the observation that irradiation and certain chemotherapies induce an immunogenic form of cell death that is partially defined by the release of intracellular DAMPs. The molecular underpinnings that explain how chemotherapies induce other forms of necrotic-like cell death remain unclear.
Targeted therapeutics
Antagonist antibodies and tyrosine kinase inhibitors (TKI) that target growth factor receptors or their downstream proliferation pathways induce expression of pro-apoptotic genes and downregulate expression of anti-apoptotic proteins, thereby increasing the frequency of intrinsic apoptosis in tumors. As described above for chemotherapy, this implies that target therapies will also induce a degree of GSDMD/E activation and pyroptosis. Indeed, specific inhibitors targeting KRAS-, ALK-, or EGFR driven lung cancer cells induced some degree of pyroptosis, and this contributed to efficacy in xenograft models [49]. In addition, a more recent study showed in GSDMD expressing melanoma that combining the BRAF- and MEK inhibitor evoked pyroptosis and this was important for enhanced cytotoxic T-cell infiltration and antitumor immunity [43]. Presumably there will be a major expansion of studies investigating the potential of inhibitors to induce pyroptosis and work in combination with immune checkpoint blockade. Whether other forms of cell death are induced by inhibitors, especially inhibitors against molecules other than kinases, also needs to be determined. For now, it has been described that PARP inhibition using Olaparib promotes ferroptosis in BRCA-proficient ovarian cancer by downregulating the cystine transporter SLC7A11 [50], and that combining the HDAC inhibitor Givinostat with the TKI Sorafenib induces necroptosis in Hodgkin lymphoma cells [51].
Cell death and survival pathways can also be directly targeted in cancer, with the prime example being the anti-apoptotic BCL-2 family members (BCL-2, MCL-1, BCL-XL). BH-3 mimetic drugs bind to different BCL-2 family members by mimicking BH3 domains of proapoptotic BCL-2 (e.g. BAD, BIM, NOXA, PUMA) family proteins [52], resulting in cell death. This approach has found success in the treatment of chronic lymphocytic leukemia (CLL) and acute myeloid leukemia (AML) with the approval of the BCL-2 inhibitor, venetoclax [53]. Interestingly, it has also been described that sublethal intrinsic apoptotic signaling and the release of cytochrome c by BH-3 mimetic drugs can promote drug resistant persister cells [54], highlighting that even directly targeting cell death pathways with therapeutics results in complex outcomes.
Oncolytic Viruses
Oncolytic viruses (OV) preferentially infect and replicate in cancer cells over normal cells and have shown clinical efficacy through their ability to lyse cells, in addition to their enhancement of anti-tumor immunity [55,56]. Obviously, through their ability to lyse infected cells, OV eventually produce a necrotic form of cell death during the final stages of infection. However, multiple pattern recognition receptors are activated within cells that can initiate cell death pathways as a protective response against infection, including apoptosis, necroptosis, pyroptosis, and autophagic cell death [56,57].
A recent study conducted a systematic comparison of cell death induced by the most common pre-clinical and clinical OV wild-type strains including Adenovirus, Semliki Forest virus, and Vaccinia virus [58]. All viruses showed similar levels of lysis upon infection, but dramatically different forms of RCD. Adenovirus infection induced autophagic cell death, Vaccinia caused necroptosis, while Semliki promoted immunogenic apoptosis. All methods of cell death produced classic signs of immunogenic cell death, including release of HMGB1 and surface expression of calreticulin, as well as efferocytosis and expression of maturation markers by DCs. Surprisingly however, vaccinia virus infection of tumor cells reduced the ability of monocyte-derived DCs to induce IFN-γ expression by antigen-specific human T cells [58]. Whether this was due to vaccinia virus infection or necroptosis itself remain unclear, especially as using an adenovirus bearing an autoactivated RIPK3 construct produced superior immune responses against B16 melanoma tumors as compared to those inducing caspase-dependent cell death [59]. Determining the optimal type of cell death to induce with OVs may be one path towards improving therapeutic efficacy, both alone and in combination with immune checkpoint blockade.
Immunotherapy
CD8+ T cells and natural killer (NK) cell initiate cell death programs in target cells through two distinct mechanisms. One pathway involves the expression of FAS-ligand (CD96L) or TNF-related apoptosis-inducing ligand (TRAIL), which binds to FAS-receptor (CD95) or TRAIL-R1-R2 respectively, and trigger extrinsic apoptosis via FADD/Caspase-8 in the target cell [60,61]. The other pathway involves the secretion of granzyme B and perforin through granule exocytosis, with perforin pores allowing granzyme B to enter and mediate cleavage and activation of caspase-3 in target cells. [62]. Granzyme and perforin-mediated killing is the primary method employed by cytotoxic lymphocytes, with protection against cancer lost in the absence of either of these molecules. However, there does appear to be a secondary role for the extrinsic pathway [63]. Using live cell imaging and fluorescent localization reporters to detect caspase-8 and Granzyme B activity, it was found that NK cells first mediated killing through fast-acting granules. While granule content gradually decreased over time, this was compensated for by increasing expression of FAS-ligand and a transition to slower acting extrinsic apoptosis [64].
Interestingly, the same study noted that while FAS and caspase-8 mediated cell death clearly consisted of apoptotic morphology, a fraction of cells killed via granzyme B displayed morphological features of the rapid membrane rupture seen in necrotic RCD programs [64]. This was likely due to the aforementioned cleavage and activation of GSDMD or GSDME by caspase-3 [46,65]. GSDME and GSDMB can also be directly cleaved by granzyme B and granzyme A, respectively [66,67]. Whether through primary or secondary cleavage by granzymes, this activation of GSDMs is critical for anti-tumor immunity to occur in tumors expressing these molecules. Beyond direct cytotoxic activity, expression of IFN-γ by CD8+ T cells has been shown to sensitize tumors to ferroptosis [68,69]. This occurs through downregulation of subunits of the glutamate-cystine antiporter system xc− (SLC3A2, SLC7A11) and upregulation of acyl-CoA synthetase long chain family member 4 (ACSL4), both of which promote lipid peroxidation and ferroptosis. Critically, these ferroptosis-related genes regulate T cell immunity in murine models and correlate with negatively (SLC3A2, SLC7A11) or positively (ACSL4) with patient outcome in response to immune checkpoint blockade [68,69]. Taken together, it is evident that non-apoptotic forms of cell death are necessary to initiate and sustain anti-tumor immunity, with cytotoxic T cell activity having the potential to engage these cell death modalities to promote a virtuous cycle. Below we will discuss what is known about the mechanisms by which the various forms of non-apoptotic cell death promote an immune response against cancer.
Cell death and the immune response
Apoptosis is well established as a silent form of cell death, due to the clearance of cell corpses prior to loss of membrane stability and the “eat-me” signaling pathways that actively suppress inflammation and prevent the adaptive immune system from responding [70,71]. This suggests a critical role for secondary necrosis and/or RCD pathways that lead to loss of membrane integrity as the modalities integral for the generation of an anti-tumor immune response. However, much of the work investigating the mechanisms by which dying cells induce an adaptive immune response has been done in the context of chemotherapies that induce “immunogenic cell death” or “immunogenic apoptosis”, and it is not clear the extent to which this overlaps with other forms of RCD. The same DAMPs released during immunogenic apoptosis are found in the debris of cells undergoing necrosis, necroptosis, pyroptosis, and ferroptosis (Figure 2), but the kinetics and mechanism of release are poorly defined. In some cases it is also unclear the extent to which these DAMPs drive local or systemic adaptive immunity remains. For example, immune activation during necroptosis was found to depend upon active cytokine production mediated by RIPK3, with viable MLKL-deficient cells capable of promoting anti-tumor immunity in the absence of DAMP release [59,72].
Figure 2. Cell death and release of immune regulatory factors.
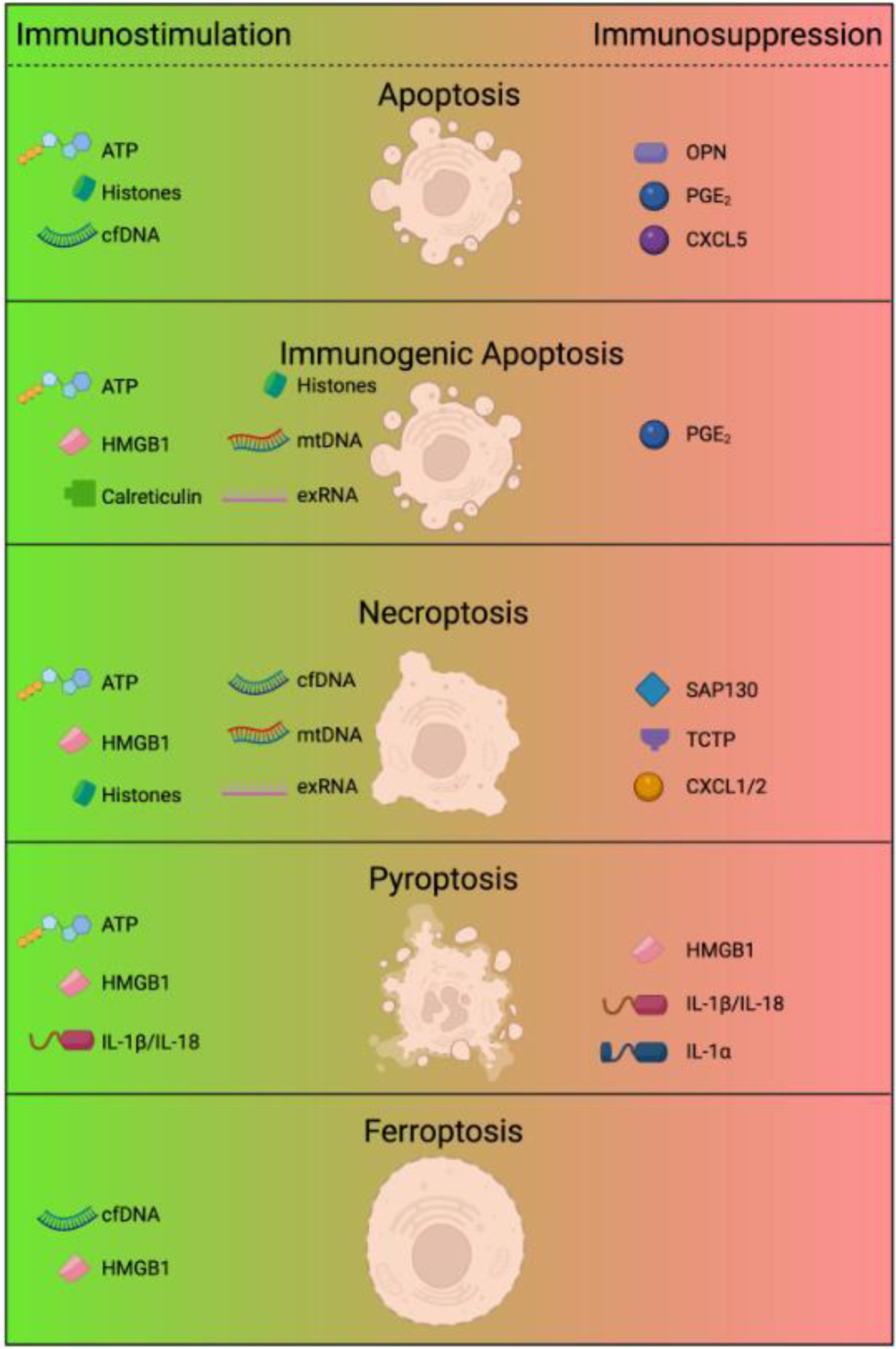
The form of cell death determines the type of DAMPs, cytokines, or other factors released by cells. Depending on the cell death modality and the DAMPs released, this can have either immunostimulatory or immunosuppressive effects on the tumor microenvironment. Apoptotic death is often considered to be immunologically silent despite the release of ATP and other DAMPs, due to the balance with immunosuppressive factors such as OPN and PGE2, as well a consequence of macrophage efferocytosis. In contrast, the immunogenic potential and antitumor features of immunogenic apoptosis is attributed to the combined release of HMGB1, mtDNA, exRNA, and histones, along with calreticulin exposure on the cell surface. Necrotic-like forms of lytic death generally include the release of intracellular DAMPs, including ATP, TCTP, SAP130, Histones, HMGB1, mtDNA, cfDNA, and exRNA. However, different forms of cell necrotic death are not necessarily equivalent and the form, type, and quantity of DAMPs may vary. Just as importantly, different forms of cell death can promote the release of unique cytokines with immune regulatory function. Beyond the connection of IL-1β with pyroptosis, necroptosis has been shown to induce CXCL1 and CXCL2 expression, major chemokines involved in the recruitment of neutrophils. These factors could promote or inhibit anti-tumor immunity in a context-specific manner. Figure created with BioRender.
Figure 2 Abbreviation list:
ATP Adenosine 5′-triphosphate
cfDNA Cell free DNA
OPN Osteopontin
PGE2 Prostaglandin E2
CXCL5 C-X-C motif chemokine ligand 5
mtDNA Mitochondrial DNA
exRNA Extracellular RNA
TCTP Translationally-Controlled Tumor Protein
SAP130 Sin3A Associated Protein 130
CXCL1/2 C-X-C motif chemokine ligand 1/2
IL1α Interleukine-1 alpha
IL1β Interleukine-1 beta
Immunogenic cell death
For cell death to be immunogenic it must allow for antigen acquisition and presentation by cDCs, as well as promote cDC maturation to induce migration to draining lymph nodes and expression of the costimulatory molecules and cytokines necessary to promote a T cell response. As mentioned, immunogenic apoptosis has been described in detail following irradiation or administration of select chemotherapies (e.g. anthracyclines). Because of the direct cytotoxic effect of these therapies, cells undergo apoptosis in a manner that involves the release of intracellular DAMPs such as ATP and the chromatin-binding protein high-mobility group box 1 (HMGB1). Release of ATP and annexin-A1 recruit cDCs and progenitor populations through P2Y2- and formyl peptide receptor 1-dependent pathways, respectively [73–75]. Recruited cDCs can then recognize the translocation of the endoplasmic reticulum (ER) protein calreticulin to the cell surface as an “eat me signal” [76,77]. Finally, the maturation of cDCs occurs through various pattern recognition receptors (PRRs) binding to DAMPs, with TLR4 recognition of HMGB1 most commonly described as critical for therapy induced immunity [78,79].
The molecular pathways that drive immunogenic apoptosis are not known, but two lines of evidence converge to suggest a partial explanation for the defining features of the phenotype. Namely, that caspase-3 can cleave GSDMD and GSDME in response to chemotherapy [42,46] and that cleavage of either GSDMD or GSDME is important for a T cell response against solid tumors [66]. Thus, it is possible that immunogenic cell death/apoptosis reflects secondary pyroptosis and the release of DAMPs through GSDMD/E pores. At least HMGB1 release has been described as a consequence of GSDME cleavage by caspase-3 in melanoma cells undergoing intrinsic apoptosis [43]. Pyroptosis would also explain the ability of certain chemotherapies to induce the release of IL-1β, although this would also depend upon constitutive or induced expression of Il1b by the cells. One phenotype not explained by pyroptosis is the need for phosphorylation of eIF2a during an ER stress response and the subsequent activation of exocytosis, which exposes intracellular contents such as calreticulin prior to loss of plasma membrane integrity [80,81]. While chemotherapies such as paclitaxel induce ER stress [82,83], this does not necessarily correlate with their proposed ability to induce immunogenic apoptosis [84].
At the same time, other studies have described a distinct mechanism by which cancer cells become immunogenic during therapy, in this case through the release of DNA and activation of the host cGAS-STING pathway following irradiation [85–87], paclitaxel chemotherapy [88], PARP inhibitors [89], the BH-3 mimetic ABT-737 [90], and in models of spontaneous tumor regression [91]. In each of these cases, TLR4 signalling through MyD88 was dispensable for T cell activity. Interestingly, HMGB1 release is still required for activation of STING in cDCs following paclitaxel therapy, as this is necessary for tumor DNA to enter host cells [88]. What might explain these discrepancies? Each of these studies utilized different tumor models, suggesting the possibility of intrinsic differences between the cell lines. For example, TLR3 in tumor cells can drive intrinsic expression of type I interferons (IFN-I) following anthracycline chemotherapy, while tumor cells with deficient autophagy pathways stimulate their own IFN-I production via cGAS-STING following irradiation [92–94]. As IFN-I are critical for cDC migration and the development of anti-tumor immunity [95], it may be that there are multiple routes to engage the same pathway depending on the cancer cell, the tissue, and the type of therapy being examined. Even calreticulin surface exposure is dispensable for type I cDCs to acquire tumor antigen, as CLEC9A can bind to exposed F-actin on necrotic corpses provided that the inhibitor gelsolin is not present [96,97].
Spontaneous necroptosis has not been described to promote anti-tumor immunity in murine models. Instead, studies have relied upon genetic systems to induce a clean form of cell death. Injecting cells with activated RIPK3, either in a prophylactic vaccine setting or directly into tumors, promoted CD8+ T cell responses and reduced tumor growth [59,72]. However, no impact on tumor growth was observed upon the injection of cells undergoing MLKL-induced necroptosis. Thus, the release of ATP and HMGB1 was insufficient to promote a T cell response. Instead, it was found that expression of NF-κB-dependent cytokines via the RIPK1/RIPK3 interaction was required, with no role for host PRR such as STING or TLRs. However, these results are in direct contrast to the finding that expressing MLKL through electroporation into mouse tumors leads to tumor regression, as this would not induce cytokine production via RIPK3 [98]. As before, this may reflect the use of different tumor models or treatment methods. Regardless, more work is required to define the immunogenic potential of necroptosis, including whether it can be induced therapeutically through non-genetic means.
The immunogenic properties of ferroptosis are even less understood. Inducing ferroptosis in a model of ovarian cancer promotes response to immune checkpoint blockade, and production of IFN-γ by CD8+ T cells further increases the sensitivity of tumor cells to ferroptosis [68]. However, using ferroptotic cells as a vaccine fails to induce a protective immune response against subsequent tumor inoculation. Surprisingly, the presence of ferroptotic cells even inhibits the anti-tumor immunity induced by the injection of cells undergoing immunogenic apoptosis, despite the release of ATP and HMGB1 [99]. The reason for this is unclear, but could relate to changes in the lipid profile of the tumor cells and the ability of certain lipids to regulate myeloid cell function by inducing ER stress [100,101].
Immunosuppressive cell death
As with all aspects of immunology, context is key to determining outcome. In addition to classical features of apoptosis inducing an immunosuppressive phenotype in macrophages (e.g. PGE2, phosphatidylserine), chemotherapy and/or chemotherapy-induced cell death can induce the expression of cytokines and chemokines that shape the tumor microenvironment. This has been most extensively described for paclitaxel chemotherapy, which induces macrophage recruitment in mammary carcinomas and breast tumors [102], likely due to increased expression of factors such as colony stimulating factor (CSF)-1 [103]. Beyond bulk changes in macrophage infiltration, single cell RNA sequencing of on-treatment biopsies has revealed that paclitaxel increases infiltration of immunosuppressive macrophage subsets while reducing infiltration of immunostimulatory populations associated with T cell function and therapeutic efficacy [104]. The extent to which these observations are due to death and cellular debris, as opposed to stress responses in the cells, is not clear. One example of the former is the ability of cellular debris from 5-fluorouracil treated cells to induce osteopontin expression by live tumor cells and macrophages, resulting in increased tumor growth [105]. Similar observations have been made for necrotic cells, with the release of the translationally controlled tumor protein (TCTP) inducing NF-κB signaling via TLR2, increasing expression of CXCL1 and CXCL2, and recruiting suppressive neutrophils that promote tumor growth [106,107].
Very little is known about the potential for other forms of RCD to promote the recruitment of macrophages, neutrophils, or other immune populations with suppressive functions. As mentioned, ferroptotic cells suppress the efficacy of therapeutic vaccination through an unidentified mechanism [99]. High levels of IL-1β in experimental tumor models and body fluids of patients often correlated with poor prognosis, carcinogenesis and invasiveness [108–110], consistent with the ability of these cytokines to recruit immunosuppressive myeloid cells and directly limit the function of cytotoxic lymphocytes [111,112]. However, inflammasome activation is not synonymous with pyroptosis, and only one study has implicated the release of DAMPs following activation of AIM2 [113]. Specifically, HMGB1 was found to promote PD-L1 expression in cancer cells via signaling through the receptor for advanced glycation endproducts (RAGE), whereas neutralizing IL-1β or IL-18 had no impact on animal survival in the KrasG12D model of pancreatic adenocarcinoma.
In the same model of murine pancreatic adenocarcinoma, another group found that RIPK3 activity promotes neutrophil recruitment via increased CXCL1 expression, as well as macrophage infiltration through the release of the histone deacetylase complex subunit 130, which binds to the C-type lectin receptor Mincle [114]. Loss of RIPK3 reversed this immunosuppressive phenotype and restricted tumor growth in a T cell-dependent manner [114]. Interestingly, gemcitabine upregulates expression of RIPK1 and RIPK3, and gemcitabine-induced cell death is reversed by the use of the MLKL inhibitor necrosulfonamide, suggesting single-agent induced necroptosis in human pancreatic adenocarcinomas [114]. As CXCL1 and CXCL2 expression is also observed during synthetic activation of RIPK3 [72], it is possible that recruitment of immunosuppressive myeloid cells is a general property of necroptosis that could complicate targeting this pathway as a therapeutic modality in vivo.
Cell death and tumor progression
Apoptotic cells discharge mitogenic signals to their surrounding cells, orchestrating their replacement during normal tissue homeostasis or during wound healing [115–117]. It is perhaps not surprising then that these same pathways have been shown to play a role in tumors. Wound healing also involves a number of other processes, including cell migration, extracellular matrix remodeling, angiogenesis, lymphangiogenesis, and innervation, each of which can contribute to tumor progression, growth, and metastasis. This is consistent with high rates of spontaneous apoptosis correlating with poor outcome, and that necrotic lesions in some tumors are a feature of aggressiveness [118–122]. Here we will discuss the available evidence that cell death—whether due to starvation, inflammation, or cytotoxic therapies—can impact the tumor microenvironment in ways that promote therapeutic resistance.
Survival and proliferation
Numerous mechanisms have been described through which apoptosis promotes compensatory proliferation. Several studies demonstrated an important role for prostaglandin E2 (PGE2) in promoting therapy-induced proliferation, including following irradiation of breast cancer and glioblastoma, and chemoresistance in bladder cancer [123–125]. In human and murine models of breast cancer, irradiation was found to promote PGE2 release via active caspase-3, which cleaved phospholipase A2 and increased production of arachidonic acid, a precursor of PGE2 [123]. Extracellular vesicles from apoptotic cells have also been found to permit intercellular transfer of splicing factors to surviving cells, resulting in glioblastoma invasion and therapy resistance [126]. Another study showed that tumor cells under apoptotic stress secrete FGF2, thereby increasing expression of the pro-survival BCL2 molecule in neighboring cells[127]. Interestingly, the production of mitogenic cytokines is not limited to the tumor cells themselves, as dying cancer-associated fibroblasts can promote the survival of glioblastomas during irradiation through induced IGF-1 production [128].
Caspase-8 activity has even been found to promote tumorigenesis in a model of inflammatory hepatocarcinogenesis via the generation of chromosomal aberrations [129]. This process is inhibited by necroptosis in apoptotic-resistance hepatocytes, consistent with another study describing how p53 mutated “unfit” cells undergo necroptosis and are extruded from monolayers of healthy cells in vitro [130]. These studies suggest an important role for necroptosis in restraining tumor formation. Whether necroptosis can promote cancer cell survival in established tumors has not been reported. However, several studies have found that the release of HMGB1 by necrotic cells can induce signalling through TLR4 and/or RAGE, leading to proliferation through ERK and p38, as well as survival through reduced Bax mitochondrial translocation [131,132].
Angiogenesis and metastasis
Angiogenesis as a consequence of cell death has not been described, but several lines of evidence suggest there may be a link. In addition to promoting cell survival, PGE2 increases angiogenesis through its ability to enhance expression of VEGF-A by neoplastic or endothelial cells [133], with a VEGF-A-independent pathway also described [134]. That said, deficiency in COX-2 had no impact on tumor growth in immunodeficient mice [135], and the pathway may only be important in certain cancer types or during specific forms of therapy. Since apoptosis, necrosis, and necroptosis increase myeloid cell invasion, and myeloid cells regulate vascular density and structure through VEGF-A production and bioavailability [136–138], it is reasonable to infer a causal link here as well. Notably, PGE2 can increase expression of CXCL1, which in addition to promoting myeloid cell recruitment, directly increases endothelial cell migration and tube formation [139]. Similarly, CX3CL1 is released from apoptotic cells and acts as a “find-me” signal to macrophages, in addition to promoting angiogenesis through its receptor CX3CR1 in multiple malignancies [140–142].
Interestingly, there is direct evidence that cell death can promote lymphangiogenesis. Apoptotic cells release sphingosine-1-phosphate, recruiting lipocalin 2-expressing macrophages that in turn drive VEGF-C expression, lymphangiogenesis, and increased metastasis in a syngeneic model of mammary carcinoma [143,144]. In this same model, the cell death associated with postpartum involution was found to promote metastasis through MerTK-dependent efferocytosis and macrophage polarization [145]. In addition, cancer cell autonomous effects of cell death have been described following doxorubicin treatment, with the release of the nucleotide uridine diphosphate signaling through the pyrimidinergic receptor P2Y6 to promote invasion and metastasis of the MDA-MB-231 human triple negative breast cancer cell line [146]. It should also be noted that extracellular vesicles such as exosomes, microvesicles or apoptotic bodies promote metastasis through several distinct mechanisms, and the production of these vesicles is substantially increased by chemotherapy or irradiation [147].
Beyond cell death promoting escape from the primary tumor via the vasculature or lymphatics, two studies have shown that the necroptotic pathways are involved in extravasation and the establishment of metastatic lesions in ectopic organs. Specifically, loss of RIPK3 in the endothelial compartment reduced the ability of B16 tumor cells to transmigrate through the endothelial barrier and form pulmonary foci following intravenous injection [148,149]. In one study this was due to tumor cells inducing necroptosis in endothelial cells via DR6 and its cognate ligand amyloid precursor protein [148]. In the other study this was mediated by a non-canonical role of RIPK3 promoting VEGF-dependent vascular permeability [149]. Nevertheless, the same phenotype demonstrates that RIPK3 in endothelial cells regulates intravasation into the lungs. Cancer cell expression of MLKL has also been shown to promote extravasation—but not intravasation—of the MVT-1 mammary carcinoma cell line, although the mechanism by which this occurs is unknown [150]. Critically, this is not mediated by signalling through RIPK1, but rather direct activation of RIPK3 by ZPB1 [151].
Concluding remarks
Considering the vast amount of cellular content released and the active pre-death upregulation of inflammatory signaling (e.g. NF-κB induction during necroptosis) [72], we know relatively little about how DAMPs function in tumors [152–154]. This may also reflect limitations in our understanding of the factors necessary for cDC maturation, migration, and T cell stimulation, especially within an immunosuppressive tumor microenvironment that can itself be established by dying cells. Moreover, despite the increased delineation of the molecular pathways that underly different cell death cascades, their physiological roles in regulating the tumor microenvironment are poorly understood. One challenge is linking a specific form of cell death with a particular outcome. Outside of experimental systems using ‘pure’ forms of cell death, tumor cells undergo different and sometimes overlapping forms of death, and inhibiting one form can simply shift the balance towards another, making data interpretation challenging and leaving a number of open questions (see outstanding questions).
Outstanding Questions.
Are secondary cell death pathways important for therapeutic outcome?
What properties of tumors regulate the type of therapy-induced cell death?
With overlapping pathways, how can the type of cell death be controlled?
How do dying cancer cells communicate with neighboring malignant cells?
How can cytotoxic therapies be rationally combined with immunotherapy?
Can oncolytic viruses or CAR-T cell therapies be used to engage specific death programs?
How tumor intrinsic and extrinsic properties regulate the outcome of cell death on the tumor microenvironment is also relatively unclear. At an intrinsic level, cancer cells can restrict expression of genes necessary for apoptosis, necroptosis and pyroptosis. Similarly, activation of the cGAS-STING pathway in tumor cells can have diverse consequences depending upon expression of additional regulators [155]. It is not hard to imagine that the mutational status of a tumor would regulate the physiological outcome of cell death by establishing unique microenvironments [156,157]. For example, mutation of Kras promotes neutrophil infiltration, limiting the ability of macrophages to clear apoptotic bodies, thereby leading to increased secondary necrosis. At an extrinsic level, dying tumor cells communicate through paracrine signaling with neighboring live tumor and non-immune stromal populations. For example, during a wound-healing response fibroblasts are pivotal in orchestrating various processes including immune cell behavior, survival and retention at wound sites, and similar pathways are at play within tumors [158]. The degree and type of fibrosis within tumors, the extent to which tumors are vascularized, and the presence of functional lymphatics would all be expected to impact how the immune system responds to cell death. How this process plays out in response to apoptosis is poorly understood, and even less is known regarding the impact of other forms of RCD. This is particularly important given the need to determine the best way to combine cytotoxic, targeted, and immune therapies to achieve optimal patient outcomes. Recent studies have shown that immune therapies in melanoma work best when given before targeted therapeutics, but the best way to combine cytotoxic therapies with immune checkpoint blockade remains to be determined.
Highlights:
Cancer therapies induce different forms of programmed cell death
Cell death modalities have differing impacts on anti-tumor immunity
Aspects of cell death can promote tumor growth and therapy resistance
Potential to take advantage cell death pathways for cancer therapy
Acknowledgments
BR is supported by the Department of Defense Breast Cancer Research Program (W81XWH-20-1-0012 and W81XWH-22-1-0406), the NIH/NCI (R01CA230610), and a Cancer Research Institute Lloyd J. Old STAR Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
B.R. is a member of the scientific advisor board for Omios Biologics, LLC. B.R. has a secondary faculty appointment at the University of South Florida.
References
- 1.Galluzzi L et al. (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death & Differentiation 25, 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Green DR and Llambi F (2015) Cell Death Signaling. Cold Spring Harb Perspect Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kroemer G and Levine B (2008) Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 9, 1004–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Napoletano F et al. (2019) Intersections between Regulated Cell Death and Autophagy. Trends Cell Biol 29, 323–338 [DOI] [PubMed] [Google Scholar]
- 5.Fuchs Y and Steller H (2015) Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nature Reviews Molecular Cell Biology 16, 329–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang D et al. (2019) The molecular machinery of regulated cell death. Cell Res 29, 347–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen DS and Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10. [DOI] [PubMed] [Google Scholar]
- 8.Ichim G and Tait SW (2016) A fate worse than death: apoptosis as an oncogenic process. Nat Rev Cancer 16, 539–548 [DOI] [PubMed] [Google Scholar]
- 9.Jiang M. j. et al. (2020) Dark Side of Cytotoxic Therapy: Chemoradiation-Induced Cell Death and Tumor Repopulation. Trends in Cancer 6, 419–431. [DOI] [PubMed] [Google Scholar]
- 10.Castillo Ferrer C et al. (2021) Apoptosis - Fueling the oncogenic fire. FEBS J 288, 4445–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sennino B and McDonald DM (2012) Controlling escape from angiogenesis inhibitors. Nat Rev Cancer 12, 699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kroemer G et al. (2013) Immunogenic cell death in cancer therapy. Annual Review of Immunology 31, 51–72. [DOI] [PubMed] [Google Scholar]
- 13.Goel S et al. (2017) CDK4/6 inhibition triggers anti-tumour immunity. Nature 548, 471–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haas L et al. (2021) Acquired resistance to anti-MAPK targeted therapy confers an immune-evasive tumor microenvironment and cross-resistance to immunotherapy in melanoma. Nat Cancer 2, 693–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phadke MS et al. (2021) Targeted Therapy Given after Anti-PD-1 Leads to Prolonged Responses in Mouse Melanoma Models through Sustained Antitumor Immunity. Cancer Immunol Res 9, 554–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prasanna PG et al. (2012) Normal tissue protection for improving radiotherapy: Where are the Gaps? Transl Cancer Res 1, 35–48 [PMC free article] [PubMed] [Google Scholar]
- 17.Minchom A et al. (2018) Dancing with the DNA damage response: next-generation anti-cancer therapeutic strategies. Ther Adv Med Oncol 10, 1758835918786658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sia J et al. (2020) Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Frontiers in cell and developmental biology 8, 41–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu GS et al. (1997) KILLER/DR5 is a DNA damage–inducible p53–regulated death receptor gene. Nature genetics 17, 141–143 [DOI] [PubMed] [Google Scholar]
- 20.Chakraborty M et al. (2003) Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. Journal of immunology (Baltimore, Md. : 1950) 170, 6338–6347 [DOI] [PubMed] [Google Scholar]
- 21.Sheard MA et al. (2003) Role of p53 in regulating constitutive and X-radiation-inducible CD95 expression and function in carcinoma cells. Cancer research 63, 7176–7184. [PubMed] [Google Scholar]
- 22.Adjemian S et al. (2020) Ionizing radiation results in a mixture of cellular outcomes including mitotic catastrophe, senescence, methuosis, and iron-dependent cell death. Cell Death & Disease 11, 1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vakifahmetoglu H et al. (2008) Death through a tragedy: mitotic catastrophe. Cell Death Differ 15, 1153–1162 [DOI] [PubMed] [Google Scholar]
- 24.Brown JM and Attardi LD (2005) The role of apoptosis in cancer development and treatment response. Nat Rev Cancer 5, 231–237 [DOI] [PubMed] [Google Scholar]
- 25.Wang HH et al. (2018) Ablative Hypofractionated Radiation Therapy Enhances Non-Small Cell Lung Cancer Cell Killing via Preferential Stimulation of Necroptosis In Vitro and In Vivo. Int J Radiat Oncol Biol Phys 101, 49–62 [DOI] [PubMed] [Google Scholar]
- 26.Kaufmann SH and Earnshaw WC (2000) Induction of Apoptosis by Cancer Chemotherapy. Experimental Cell Research 256, 42–49. [DOI] [PubMed] [Google Scholar]
- 27.van Schaik TA et al. (2021) Therapy-Induced Tumor Cell Death: Friend or Foe of Immunotherapy? Front Oncol 11, 678562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seitz SJ et al. (2010) Chemotherapy-induced apoptosis in hepatocellular carcinoma involves the p53 family and is mediated via the extrinsic and the intrinsic pathway. International journal of cancer 126, 2049–2066 [DOI] [PubMed] [Google Scholar]
- 29.Friesen C et al. (1996) Involvement of the CD95 (APO-1/FAS) receptor/ligand system in drug-induced apoptosis in leukemia cells. Nature medicine 2, 574–577 [DOI] [PubMed] [Google Scholar]
- 30.Zhang X et al. (2020) Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. Journal of cellular physiology 235, 3425–3437 [DOI] [PubMed] [Google Scholar]
- 31.Guo J et al. (2018) Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer Res Treat 50, 445–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lv C et al. (2017) Low-Dose Paclitaxel Inhibits Tumor Cell Growth by Regulating Glutaminolysis in Colorectal Carcinoma Cells. Front Pharmacol 8, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giannakakou P et al. (2001) Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel-induced cytotoxicity. Oncogene 20, 3806–3813 [DOI] [PubMed] [Google Scholar]
- 34.Sun Y et al. (2021) ent-Kaurane diterpenoids induce apoptosis and ferroptosis through targeting redox resetting to overcome cisplatin resistance. Redox biology 43, 101977–101977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roh JL et al. (2016) Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett 381, 96–103 [DOI] [PubMed] [Google Scholar]
- 36.Fu D et al. (2021) Induction of ferroptosis by ATF3 elevation alleviates cisplatin resistance in gastric cancer by restraining Nrf2/Keap1/xCT signaling. Cellular & molecular biology letters 26, 26–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du J et al. (2021) DHA exhibits synergistic therapeutic efficacy with cisplatin to induce ferroptosis in pancreatic ductal adenocarcinoma via modulation of iron metabolism. Cell death & disease 12, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tenev T et al. (2011) The Ripoptosome, a Signaling Platform that Assembles in Response to Genotoxic Stress and Loss of IAPs. Molecular Cell 43, 432–448. [DOI] [PubMed] [Google Scholar]
- 39.Jiang Y et al. (2016) Methyl methanesulfonate induces necroptosis in human lung adenoma A549 cells through the PIG-3-reactive oxygen species pathway. Tumour Biol 37, 3785–3795 [DOI] [PubMed] [Google Scholar]
- 40.Konstantakou EG et al. (2015) 3-BrPA eliminates human bladder cancer cells with highly oncogenic signatures via engagement of specific death programs and perturbation of multiple signaling and metabolic determinants. Mol Cancer 14, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X et al. (2020) The role of Caspase-1/GSDMD-mediated pyroptosis in Taxol-induced cell death and a Taxol-resistant phenotype in nasopharyngeal carcinoma regulated by autophagy. Cell Biol Toxicol 36, 437–457 [DOI] [PubMed] [Google Scholar]
- 42.Zhang CC et al. (2019) Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis 24, 312–325 [DOI] [PubMed] [Google Scholar]
- 43.Erkes DA et al. (2020) Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov 10, 254–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meng L et al. (2019) Doxorubicin induces cardiomyocyte pyroptosis via the TINCR-mediated posttranscriptional stabilization of NLR family pyrin domain containing 3. J Mol Cell Cardiol 136, 15–26 [DOI] [PubMed] [Google Scholar]
- 45.Shen X et al. (2021) Caspase 3/GSDME-dependent pyroptosis contributes to chemotherapy drug-induced nephrotoxicity. Cell Death Dis 12, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y et al. (2017) Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 [DOI] [PubMed] [Google Scholar]
- 47.Rogers C et al. (2017) Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 8, 14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia X et al. (2019) The role of pyroptosis in cancer: pro-cancer or pro-”host”? Cell Death Dis 10, 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu H et al. (2018) Molecular Targeted Therapies Elicit Concurrent Apoptotic and GSDME-Dependent Pyroptotic Tumor Cell Death. Clinical cancer research : an official journal of the American Association for Cancer Research 24, 6066–6077 [DOI] [PubMed] [Google Scholar]
- 50.Hong T et al. (2021) PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol 42, 101928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Locatelli SL et al. (2014) BIM upregulation and ROS-dependent necroptosis mediate the antitumor effects of the HDACi Givinostat and Sorafenib in Hodgkin lymphoma cell line xenografts. Leukemia 28, 1861–1871 [DOI] [PubMed] [Google Scholar]
- 52.Merino D et al. (2018) BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 34, 879–891 [DOI] [PubMed] [Google Scholar]
- 53.Diepstraten ST et al. (2022) The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer 22, 45–64 [DOI] [PubMed] [Google Scholar]
- 54.Kalkavan H et al. (2022) Sublethal cytochrome c release generates drug-tolerant persister cells. Cell 185, 3356–3374.e3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaufman HL and Maciorowski D (2021) Advancing oncolytic virus therapy by understanding the biology. Nat Rev Clin Oncol 18, 197–198 [DOI] [PubMed] [Google Scholar]
- 56.Santos Apolonio J et al. (2021) Oncolytic virus therapy in cancer: A current review. World J Virol 10, 229–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kabiljo J et al. (2020) From threat to cure: understanding of virus-induced cell death leads to highly immunogenic oncolytic influenza viruses. Cell Death Discovery 6, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma J et al. (2020) Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis 11, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Snyder AG et al. (2019) Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Science immunology 4, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee J et al. (2018) Fas Ligand localizes to intraluminal vesicles within NK cell cytolytic granules and is enriched at the immune synapse. Immunity, inflammation and disease 6, 312–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kägi D et al. (1994) Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369, 31–37 [DOI] [PubMed] [Google Scholar]
- 62.Schmidt H et al. (2011) Effector granules in human T lymphocytes: proteomic evidence for two distinct species of cytotoxic effector vesicles. Journal of proteome research 10, 1603–1620 [DOI] [PubMed] [Google Scholar]
- 63.Hassin D et al. (2011) Cytotoxic T lymphocyte perforin and Fas ligand working in concert even when Fas ligand lytic action is still not detectable. Immunology 133, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prager I et al. (2019) NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. The Journal of experimental medicine 216, 2113–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rogers C et al. (2017) Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nature communications 8, 14128–14128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Z et al. (2020) Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou Z et al. (2020) Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science (New York, N.Y.) 368, [DOI] [PubMed] [Google Scholar]
- 68.Wang W et al. (2019) CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liao P et al. (2022) CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 40, 365–378.e366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Merad M et al. (2013) The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annual Review of Immunology 31, 563–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Green DR et al. (2009) IMMUNOGENIC AND TOLEROGENIC CELL DEATH. Nature reviews. Immunology 9, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yatim N et al. (2015) RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science 350, 328–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Michaud M et al. (2011) Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science (New York, N.Y.) 334, 1573–1577. [DOI] [PubMed] [Google Scholar]
- 74.Kepp O et al. (2021) ATP and cancer immunosurveillance. Embo j 40, e108130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vacchelli E et al. (2015) Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science (New York, N.Y.) 350, 972–978 [DOI] [PubMed] [Google Scholar]
- 76.Obeid M et al. (2007) Calreticulin exposure dictates the immunogenicity of cancer cell death. Nature medicine 13, 54–61 [DOI] [PubMed] [Google Scholar]
- 77.Sprooten J et al. (2020) Necroptosis in Immuno-Oncology and Cancer Immunotherapy. Cells 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Garg AD et al. (2015) Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Frontiers in immunology 6, 588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yamazaki T et al. (2014) Defective immunogenic cell death of HMGB1-deficient tumors: compensatory therapy with TLR4 agonists. Cell Death and Differentiation 21, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bezu L et al. (2018) eIF2α phosphorylation is pathognomonic for immunogenic cell death. Cell Death Differ 25, 1375–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kepp O et al. (2013) Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev 24, 311–318 [DOI] [PubMed] [Google Scholar]
- 82.Tanimukai H et al. (2013) Paclitaxel induces neurotoxicity through endoplasmic reticulum stress. Biochem Biophys Res Commun 437, 151–155 [DOI] [PubMed] [Google Scholar]
- 83.Chen L et al. (2019) EIF2A promotes cell survival during paclitaxel treatment in vitro and in vivo. J Cell Mol Med 23, 6060–6071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fucikova J et al. (2020) Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis 11, 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Deng L et al. (2014) STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han C et al. (2020) Tumor cells suppress radiation-induced immunity by hijacking caspase 9 signaling. Nat Immunol 21, 546–554 [DOI] [PubMed] [Google Scholar]
- 87.Yamazaki T et al. (2020) Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol 21, 1160–1171 [DOI] [PubMed] [Google Scholar]
- 88.de Mingo Pulido Á et al. (2021) The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity 54, 1154–1167.e1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pantelidou C et al. (2019) PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov 9, 722–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Giampazolias E et al. (2017) Mitochondrial permeabilization engages NF-κB-dependent anti-tumour activity under caspase deficiency. Nat Cell Biol 19, 1116–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Woo S-R et al. (2014) STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sistigu A et al. (2014) Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 20, 1301–1309 [DOI] [PubMed] [Google Scholar]
- 93.Harding SM et al. (2017) Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yamazaki T et al. (2020) Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nature immunology 21, 1160–1171 [DOI] [PubMed] [Google Scholar]
- 95.Fuertes MB et al. (2011) Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. Journal of Experimental Medicine 208, 2005–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ahrens S et al. (2012) F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 36, 635–645. [DOI] [PubMed] [Google Scholar]
- 97.Giampazolias E et al. (2021) Secreted gelsolin inhibits DNGR-1-dependent cross-presentation and cancer immunity. Cell 184, 4016–4031.e4022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Van Hoecke L et al. (2018) Treatment with mRNA coding for the necroptosis mediator MLKL induces antitumor immunity directed against neo-epitopes. Nat Commun 9, 3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wiernicki B et al. (2022) Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun 13, 3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Di Conza G et al. (2021) Tumor-induced reshuffling of lipid composition on the endoplasmic reticulum membrane sustains macrophage survival and pro-tumorigenic activity. Nat Immunol 22, 1403–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cubillos-Ruiz JR et al. (2015) ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 161, 1527–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.DeNardo DG et al. (2011) Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 1, 54–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ruffell B et al. (2012) Leukocyte composition of human breast cancer. Proc Natl Acad Sci U S A 109, 2796–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang Y et al. (2021) Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 39, 1578–1593.e1578 [DOI] [PubMed] [Google Scholar]
- 105.Chang J et al. (2019) Chemotherapy-generated cell debris stimulates colon carcinoma tumor growth via osteopontin. FASEB J 33, 114–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li M et al. (2001) An essential role of the NF-kappa B/Toll-like receptor pathway in induction of inflammatory and tissue-repair gene expression by necrotic cells. J Immunol 166, 7128–7135 [DOI] [PubMed] [Google Scholar]
- 107.Hangai S et al. (2021) Orchestration of myeloid-derived suppressor cells in the tumor microenvironment by ubiquitous cellular protein TCTP released by tumor cells. Nat Immunol 22, 947–957 [DOI] [PubMed] [Google Scholar]
- 108.Apte RN et al. (2006) Effects of micro-environment- and malignant cell-derived interleukin-1 in carcinogenesis, tumour invasiveness and tumour-host interactions. Eur J Cancer 42, 751–759 [DOI] [PubMed] [Google Scholar]
- 109.Apte RN et al. (2006) The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev 25, 387–408. [DOI] [PubMed] [Google Scholar]
- 110.Maker AV et al. (2011) Cyst fluid interleukin-1beta (IL1beta) levels predict the risk of carcinoma in intraductal papillary mucinous neoplasms of the pancreas. Clin Cancer Res 17, 1502–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lillo S and Saleh M (2022) Inflammasomes in Cancer Progression and Anti-Tumor Immunity. Front Cell Dev Biol 10, 839041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rébé C and Ghiringhelli F (2020) Interleukin-1β and Cancer. Cancers (Basel) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li C et al. (2018) PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev Cell 46, 441–455.e448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Seifert L et al. (2016) The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 10.1038/nature17403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ryoo HD et al. (2004) Apoptotic Cells Can Induce Compensatory Cell Proliferation through the JNK and the Wingless Signaling Pathways. Developmental cell 7, 491–501. [DOI] [PubMed] [Google Scholar]
- 116.Huh JR et al. (2004) Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Current biology : CB 14, 1262–1266 [DOI] [PubMed] [Google Scholar]
- 117.Pérez-Garijo A et al. (2004) Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development (Cambridge, England) 131, 5591–5598 [DOI] [PubMed] [Google Scholar]
- 118.Caruso R et al. (2011) Histologic coagulative tumour necrosis as a prognostic indicator of aggressiveness in renal, lung, thyroid and colorectal carcinomas: A brief review. Oncology letters 3, 16–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Naresh KN et al. (2001) Apoptosis index is a predictor of metastatic phenotype in patients with early stage squamous carcinoma of the tongue: a hypothesis to support this paradoxical association. Cancer 91, 578–584 [PubMed] [Google Scholar]
- 120.Sun B et al. (2006) Extent, relationship and prognostic significance of apoptosis and cell proliferation in synovial sarcoma. European journal of cancer prevention : the official journal of the European Cancer Prevention Organisation (ECP) 15, 258–265. [DOI] [PubMed] [Google Scholar]
- 121.Alcaide J et al. (2013) The role and prognostic value of apoptosis in colorectal carcinoma. BMC clinical pathology 13, 24–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.De Jong JS et al. (2000) Number of apoptotic cells as a prognostic marker in invasive breast cancer. British journal of cancer 82, 368–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Huang Q et al. (2011) Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nature medicine 17, 860–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kurtova AV et al. (2014) Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 517, 209–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brocard E et al. (2015) Radiation-induced PGE2 sustains human glioma cells growth and survival through EGF signaling. Oncotarget 6, 6840–6849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pavlyukov MS et al. (2018) Apoptotic Cell-Derived Extracellular Vesicles Promote Malignancy of Glioblastoma Via Intercellular Transfer of Splicing Factors. Cancer cell 34, 119–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bock FJ et al. (2021) Apoptotic stress-induced FGF signalling promotes non-cell autonomous resistance to cell death. Nat Commun 12, 6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tommelein J et al. (2017) Radiotherapy-Activated Cancer-Associated Fibroblasts Promote Tumor Progression through Paracrine IGF1R Activation. Cancer research 78, 659–670 [DOI] [PubMed] [Google Scholar]
- 129.Vucur M et al. (2013) RIP3 Inhibits Inflammatory Hepatocarcinogenesis but Promotes Cholestasis by Controlling Caspase-8- and JNK-Dependent Compensatory Cell Proliferation. CellReports 4, 776–790 [DOI] [PubMed] [Google Scholar]
- 130.Watanabe H et al. (2018) Mutant p53-Expressing Cells Undergo Necroptosis via Cell Competition with the Neighboring Normal Epithelial Cells. Cell Rep 23, 3721–3729. [DOI] [PubMed] [Google Scholar]
- 131.Zhou J et al. (2015) HMGB1 induction of clusterin creates a chemoresistant niche in human prostate tumor cells. Scientific reports 5, 15085–15085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.He S et al. (2018) HMGB1 released by irradiated tumor cells promotes living tumor cell proliferation via paracrine effect. Cell death & disease 9, 648–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Finetti F et al. (2020) Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology (Basel) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xu L et al. (2014) COX-2 inhibition potentiates antiangiogenic cancer therapy and prevents metastasis in preclinical models. Sci Transl Med 6, 242ra284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zelenay S et al. (2015) Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 162, 1257–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lin EY et al. (2007) Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Molecular oncology 1, 288–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stockmann C et al. (2008) Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature 456, 814–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bergers G et al. (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biology 2, 737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang D et al. (2006) CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J Exp Med 203, 941–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li F et al. (2010) Down-regulation of fractalkine inhibits the in vitro and in vivo angiogenesis of the hepatocellular carcinoma HepG2 cells. Oncol Rep 24, 669–675 [PubMed] [Google Scholar]
- 141.Tang J et al. (2015) Upregulation of fractalkine contributes to the proliferative response of prostate cancer cells to hypoxia via promoting the G1/S phase transition. Mol Med Rep 12, 7907–7914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tardaguila M and Mañes S (2013) CX3CL1 at the crossroad of EGF signals: Relevance for the progression of ERBB2+ breast carcinoma. Oncoimmunology 2, e25669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yang J et al. (2013) Lipocalin 2 is a novel regulator of angiogenesis in human breast cancer. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 27, 45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jung M et al. (2016) Lipocalin 2 from macrophages stimulated by tumor cell–derived sphingosine 1-phosphate promotes lymphangiogenesis and tumor metastasis. Science signaling 9, ra64-NA. [DOI] [PubMed] [Google Scholar]
- 145.Stanford JC et al. (2014) Efferocytosis produces a prometastatic landscape during postpartum mammary gland involution. The Journal of clinical investigation 124, 4737–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ma X et al. (2016) Chemotherapy-induced uridine diphosphate release promotes breast cancer metastasis through P2Y6 activation. Oncotarget 7, 29036–29050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Abhange K et al. (2021) Small extracellular vesicles in cancer. Bioact Mater 6, 3705–3743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Strilic B et al. (2016) Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 536, 215–218 [DOI] [PubMed] [Google Scholar]
- 149.Hänggi K et al. (2017) RIPK1/RIPK3 promotes vascular permeability to allow tumor cell extravasation independent of its necroptotic function. Cell death & disease 8, e2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jiao D et al. (2018) Necroptosis of tumor cells leads to tumor necrosis and promotes tumor metastasis. Cell research 28, 868–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Baik JY et al. (2021) ZBP1 not RIPK1 mediates tumor necroptosis in breast cancer. Nat Commun 12, 2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Krysko DV et al. (2012) Immunogenic cell death and DAMPs in cancer therapy. Nature reviews. Cancer 12, 860–875 [DOI] [PubMed] [Google Scholar]
- 153.Yatim N et al. (2017) Dying cells actively regulate adaptive immune responses. Nature Reviews Immunology. 10.1038/nri.2017.9 [DOI] [PubMed] [Google Scholar]
- 154.Gong T et al. (2020) DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nature Reviews Immunology 20, 95–112. [DOI] [PubMed] [Google Scholar]
- 155.Li J et al. (2021) Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov 11, 1212–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wellenstein MD and de Visser KE (2018) Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 48, 399–416 [DOI] [PubMed] [Google Scholar]
- 157.Duits DEM and de Visser KE (2021) Impact of cancer cell-intrinsic features on neutrophil behavior. Semin Immunol 57, 101546. [DOI] [PubMed] [Google Scholar]
- 158.Barrett RL and Puré E (2020) Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. Elife 9 [DOI] [PMC free article] [PubMed] [Google Scholar]