

Abstract
Increasing evidence suggests that the gut microbiome may modulate the efficacy of cancer immunotherapy. In a B cell-lymphoma patient cohort from five centers in Germany and the United States (Germany, N=66; US, N=106, Total, N=172), we demonstrate that wide-spectrum antibiotics treatment (‘high-risk antibiotics’) prior to CD19-targeted chimeric antigen receptor (CAR)-T cell therapy is associated with adverse outcomes, but this effect is likely confounded by an increased pre-treatment tumor burden and systemic inflammation in high-risk antibiotics-pre-treated patients. To resolve this confounding effect and gain insights into antibiotics-masked microbiome signals impacting CAR-T efficacy, we focused on the high-risk antibiotics non-exposed patient population. Indeed, in these patients, significant correlations were uncovered between pre-CAR-T-infusion Bifidobacterium longum and microbiome-encoded peptidoglycan biosynthesis, and CAR-T treatment-associated 6-months survival or lymphoma progression. Furthermore, predictive pre-CAR-T treatment microbiome-based machine-learning algorithms trained on the high-risk antibiotics non-exposed German cohort and validated by the respective US cohort robustly segregated long-term responders from non-responders. Bacteroides, Ruminococcus, Eubacterium and Akkermansia were most important in determining CAR-T responsiveness, with Akkermansia also associating with pre-infusion peripheral T cell levels in these patients. Collectively, we uncover conserved microbiome features across clinical and geographical variations, which may enable cross-cohort microbiome-based predictions of outcomes in CAR-T cell immunotherapy.
Keywords: CAR-T cell therapy, lymphoma, microbiome, cancer immunotherapy, response prediction, cross-cohort study
Introduction
Compelling evidence from human studies and preclinical experiments have identified the gut microbiome and its effectors as potential major biological variables impacting the efficacy of T cell driven cancer immunotherapies and their toxicities. Gut microbes such as Akkermansia muciniphila, Faecalibacterium prausnitzii and Bifidobacterium longum have been associated with improved responses to immune checkpoint blockade (ICB)1–3, Eubacterium limosum has been associated with reduced relapse after allogeneic hematopoietic cell transplantation (allo-HCT)4, and Enterococcus has been associated with the development of post-HCT graft-versus-host disease5. Gut microbes produce metabolites that can modulate T cell function, as has been shown for inosine produced by bifidobacteria, which can enhance ICB efficacy through Th1 cell activation6. Exploratory clinical trials suggest that fecal microbiome transfers from ICB responders to previously ICB refractory melanoma patients can lead to antitumor ICB responses in a subset of patients7,8. Conversely, depletion of gut microbes by broad-spectrum antibiotics prior to T cell therapy can reduce ICB efficacy9 or increase GVHD-related mortality10.
Adoptive T cell transfer therapy with CAR-T cells represents a breakthrough immunotherapy in the treatment of hematologic malignancies. CARs are genetically engineered hybrid receptors with antigen-specificity of monoclonal antibodies that are overexpressed in patient autologous T cells to redirect these cells towards tumor surface antigens and kill malignant cells without the need for MHC-restricted antigen presentation11. Four CD19-targeted CAR-T cell products have received approval for the treatment of refractory and relapsing (r/r) B cell malignanices12. However, despite high initial response rates, complete and long-term remission is only achieved in up to 40% of patients13–16. The loss of antitumor efficacy has been attributed to CAR-T cell exhaustion in the tumor microenvironment17,18, or, to a lesser extent, immune evasion of malignant cells with loss of tumor surface antigen expression19. Additionally, lymphoma-intrinsic genomic alterations are suggested to confer resistance of malignant cells against CD19 CAR-T cells20. Other clinical variables with negative impact on CAR-T responsiveness include high metabolic tumor volume, elevated serum lactate dehydrogenase (LDH) levels, or elevated C-reactive protein (CRP) as a marker of systemic inflammation21–23, but are overlapping between responders and non-responders and have therefore not been included in standardized treatment decision algorithms. Interestingly, treatment of B cell lymphoma and leukemia patients with broad-spectrum antibiotics prior to CD19-targeted chimeric antigen receptor (CAR)-T cell therapy was recently associated with reduced survival and increased neurotoxicity24. Understanding the causal nature of possible microbiome contributions to CAR-T therapy effectiveness and adverse effects may enable better understanding of differential CAR-T cell activation, persistence, and clinical efficacy, and ultimately the prediction of response to CAR-T cell immunotherapy even prior to treatment. To address this question in a prospective clinical study, we have built an international consortium of German and US lymphoma patient cohorts receiving CD19-targeted CAR-T cell immunotherapies and studied the role of the gut microbiome and its potential confounders in clinical outcomes of CAR-T therapy.
Results
Antibiotics and disease severity associated with CAR-T outcomes.
Following recent reports implicating gut microbiome disruption by broad-spectrum antibiotics as a potential contributor to reduced antitumor efficacy of immunotherapies1,9,24, we conducted a clinical study in a cohort of r/r lymphoma patients to dissect gut microbiome features associated with clinical outcomes, as potential predictors of response to CAR-T cell therapy. To minimize confounding factors related to sample collection, processing protocols, statistical power, and geographic or disease-related variables25, we established an international consortium of five tertiary medical centers across Germany and the USA, engaged in CAR-T cell treatment of patients with lymphoma.
We began our investigation by exploring the associations between antibiotic treatments prior to commercial CD19-directed CAR T-cell treatment and outcomes in r/r lymphoma patients that included German (n=66) and US (n=106) patients from three German and two US-based tertiary hospitals who were prospectively recruited between the years 2018 and 2021 (see Table 1 for demographic and disease characteristics). Of 172 patients, 122 were treated with axicabtagene ciloleucel (axi-cel), 49 received tisagenlecleucel (tisa-cel) and 1 received lisocabtagene maraleucel (liso-cel). The median age of participants was 62.0 years (range: 22.0 – 88.0 years) with 55 being females and 117 males, and they received a median of three lines of systemic therapy prior to CAR-T cell therapy. Histological diagnoses included diffuse large B-cell lymphoma (DLBCL) or transformed follicular lymphoma (TFL) (n=162), primary mediastinal lymphoma (PMBCL) (n=4), Richter’s transformed chronic lymphocytic leukemia (TCLL) (n=3) and transformed marginal zone lymphoma (TMZL) (n=3). The median duration of follow-up among survivors in the combined cohort was 13.2 (range: 2.0-32.4) months. For all patients, the best overall response rate (ORR) and complete response (CR) rate at day 90 after CAR-T cell administration was 58.1% (100/172) and 47.6% (82/172), respectively (see also Table 2 for outcome characteristics).
Table 1.
Patient characteristics by country prior to CAR-T cell treatment
| Cohorts by country | |||
|---|---|---|---|
| Characteristic | Total cohort (N=172) | Germany (N=66) | US (N=106) |
| Age | |||
| Median (range) - yr | 62 (24-88) | 63 (31-82) | 62 (24-88) |
| ≥65 yr - no(%) | 75 (43.6) | 29 (43.9) | 46 (43.4) |
| Gender - no (%) | |||
| Female | 55 (32.0) | 20 (30.3) | 35 (33.0) |
| Male | 117 (68) | 46 (60.7) | 71 (67.0) |
| Diagnosis - no (%) | |||
| DLBCL/TFL | 162 (94.2) | 63 (95.4) | 99 (93.4) |
| PMBCL | 4 (2.3) | 1 (1.5) | 3 (2.8) |
| Richter TCLL | 3 (1.7) | 1 (1.5) | 2 (1.9) |
| TMZL | 3 (1.7) | 1 (1.5) | 2 (1.9) |
| CAR-T product - no (%) | |||
| Axi-cel | 122 (70.9) | 30 (47.0) | 91 (85.8) |
| Tisa-cel | 49 (28.5) | 34 (51.5) | 15 (14.2) |
| Liso-cel | 1 (0.6) | 1 (1.5) | 0 |
| Center - no (%) | |||
| Germany | 66 (38.4) | Heidelberg | MDACC |
| United States | 106 (61.6) | 21 (31.8) | 59 (55.7) |
| Munich | Moffitt | ||
| 41 (62.1) | 47 (44.3) | ||
| Regensburg | |||
| 4 (6.1) | |||
| ECOG performance-status score - no (%) | |||
| 0 | 52 (30.2) | 30 (45.5) | 22 (20.8) |
| 1 | 97 (56.4) | 27 (40.9) | 70 (66.0) |
| 2 | 18 (10.5) | 6 (9.1) | 12 (11.3) |
| 3 | 5 (2.9) | (4.5) | 2 (1.9) |
| Ann Arbor lymphoma staging system - no (%) | |||
| Stage I/II | 41 (23.8) | 22 (33.3) | 19 (17.9) |
| Stage III/IV | 131 (76.2) | 44 (66.7) | 87 (82.1) |
| Bulky disease - no (%) | |||
| ≥10 cm | 20 (12.5) | 9 (13.6) | 11 (11.7) |
| Missing (no) | 12 | 12 | |
| Bridging therapy - no. of pat. (%) | 109 (65.3) | 54 (81.8) | 55 (54.5) |
| Missing (no) | 5 | 5 | |
| Prior therapies - no (%) | |||
| 1 - 3 | 101 (61.2) | 38 (61.3) | 63 (61.2) |
| 4 - 6 | 61 (37.0) | 23 (37.1) | 38 (36.9) |
| 7 - 9 | 3 (1.9) | 1 (1.6) | 8 (7.8) |
| Missing (no) | 7 | 4 | 3 |
Legend: DLBCL, diffuse large B cell lymphoma; TFL, transformed follicular lymphoma; PBMCL: primary mediastinal B cell lymphoma; TCLL; transformed chronic lymphocytic leukaemia; TMZL: transformed marginal zone lymphoma.
Table 2.
Patient outcomes by country after CAR-T cell treatment
| Cohorts by country | |||
|---|---|---|---|
| Outcome | Total cohort (N=172) | Germany (N=66) | US (N=106) |
| Response day 90 - no (%) | |||
| CR | 82 (47.7) | 30 (45.5) | 52 (49.1) |
| PR | 17 (9.9) | 11 (16.7) | 6 (5.7) |
| SD | 1 (0.6) | 1 (1.5) | 0 |
| PD | 53 (30.8) | 16 (24.2) | 37 (34.9) |
| Death | 19 (11.1) | 8 (12.1) | 11 (10.4) |
| Response day 180 - no (%) | |||
| CR | 72 (41.9) | 27 (40.9) | 45 (42.4) |
| PR | 8 (4.7) | 5 (7.6) | 3 (2.8) |
| PD | 56 (32.5) | 21 (31.8) | 35 (33.0) |
| Death | 36 (20.9) | 13 (19.7) | 23 (21.7) |
| Progression early/late - no (%) | |||
| Early | 79 (47.6) | 33 (51.6) | 54 (52.9) |
| Late or none | 87 (52.4) | 31 (48.4) | 48 (47.1) |
| Cytokine release syndrome - no (%) | |||
| 0 | 18 (10.5) | 7 (10.6) | 11 (10.4) |
| 1 | 86 (50.0) | 35 (53.0) | 51 (48.1) |
| 2 | 57 (33.1) | 19 (28.8) | 38 (35.9) |
| 3 | 8 (4.7) | 5 (7.6) | 3 (2.8) |
| 4 | 3 (1.7) | 0 | 3 (2.8) |
| Neurotoxicity (ICANS) - no (%) | |||
| 0 | 90 (52.3) | 41 (62.1) | 49 (46.2) |
| 1 | 29 (16.9) | 11 (16.7) | 18 (17.0) |
| 2 | 23 (13.4) | 10 (15.2) | 13 (12.3) |
| 3 | 22 (12.9) | 3 (4.5) | 19 (17.9) |
| 4 | 8 (4.7) | 1 (1.5) | 7 (6.6) |
Legend: CR, complete remission: PR, partial remission; SD, stable disease; PD, progressive disease.
Antibiotics exposure within a three-week period before CAR-T cell infusion was associated with significantly decreased progression-free survival (PFS) (Fig. 1A, hazard ratio = 2.04) in antibiotics pre-treated patients in both the US (hazard ratio = 2.02; 41.5% of 106 received antibiotics) and the German cohorts (hazard ratio = 2.20; 27% of 66 with antibiotics) (Extended Data Fig. 1A). Likewise, antibiotic exposures were associated with a significantly higher cumulative incidence of progression (odds ratio = 1.58; Extended Data Fig. 1B) and a significantly reduced overall survival (OS) (hazard ratio = 2.39; Extended Data Fig. 1C). Various antibiotic classes were administered to patients during the pre-CAR-T cell infusion period, with piperacillin-tazobactam, cefepime or meropenem being most frequently administered before CAR-T cell infusion and thereafter (Extended Data Fig. 2A–D). Assessment of the impact of individual antibiotic classes prior to CAR-T cell infusion (i.e., days −21 to 0 relative to infusion) on survival outcomes demonstrated disparate association strengths of individual drug classes on progression to CAR-T immunotherapy (Fig. 1B). Based on these results, we defined ‘high-risk antibiotics’ including meropenem, cefepime, ceftazidime, and piperacillin-tazobactam antibiotics that were associated with significantly reduced responses to CAR-T cell therapy. These antibiotics exert a considerable broad-spectrum, anti-pseudomonal activity, and are also known to severely disrupt the gut microbiome by targeting many anaerobic commensals10,26. All other antibiotics administered to the patients in either geographical location were defined as ‘low-risk antibiotics’. Recipients of high-risk antibiotics displayed significantly higher rates of disease progression (odds ratio = 2.60; Fig. 1C), whereas recipients of low-risk antibiotics had a progression incidence similar to that of patients who avoided antibiotic exposure prior to CAR-T infusion (Fig. 1C). This translated to a significantly reduced PFS (hazard ratio = 3.05) and OS (hazard ratio = 2.99; Fig. 1D and 1E) in patients receiving high-risk antibiotics. These findings were observed independently in both the US and German cohorts (Extended Data Fig. 1D).
Figure 1. Associations of antibiotic drug exposure prior to CD19-directed CAR-T cell therapy with survival outcomes, tumor burden and inflammation.
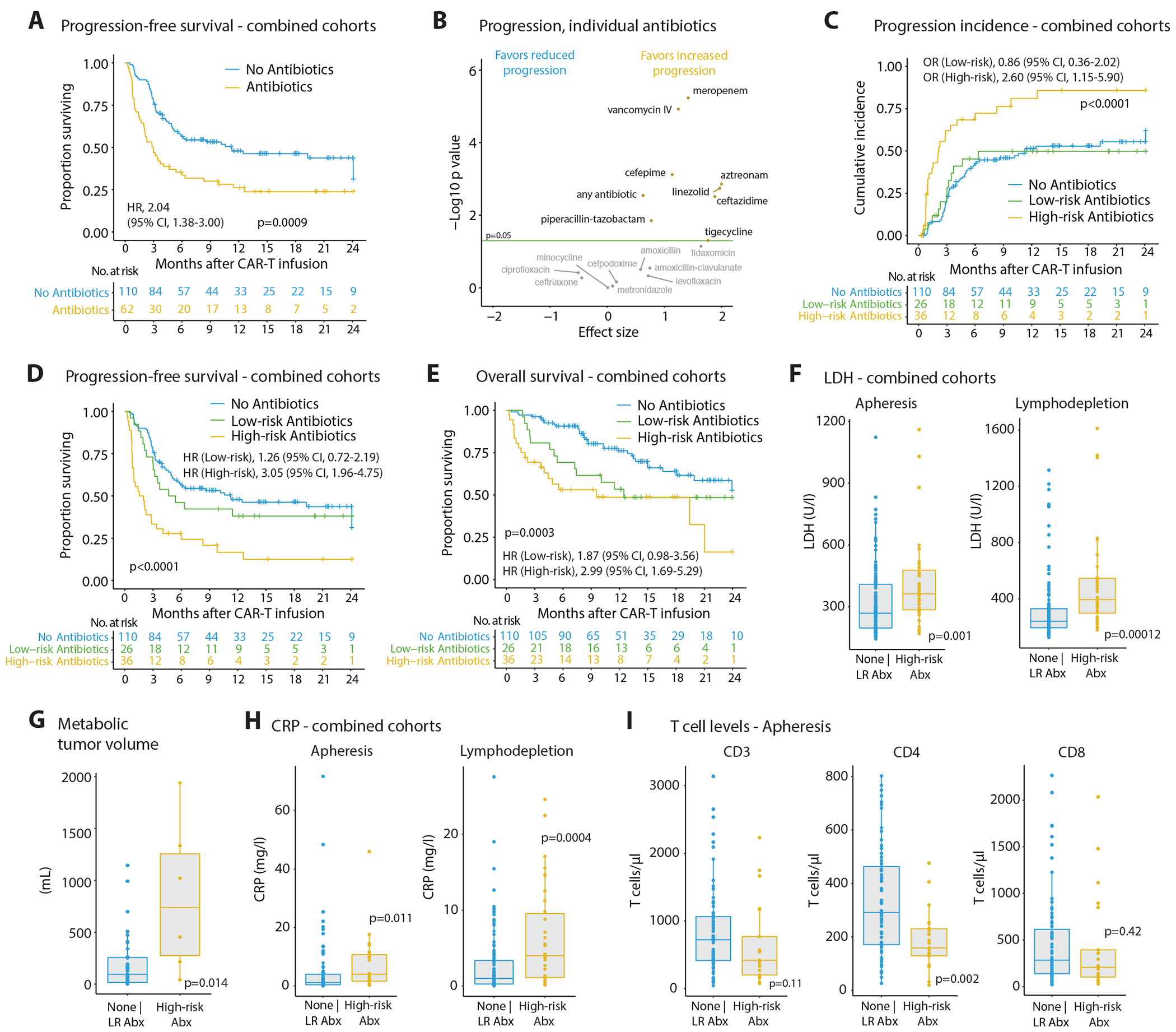
(A) The international cohort of non-Hodgkin’s lymphoma (NHL) patients consisted of German (n = 66) and US (n = 106) participants prospectively recruited between 2018 and 2021 and assessed for antibiotic exposures within 3 weeks prior to CAR-T cell infusion and survival outcomes up to 24 months following CAR-T cell treatment; progression-free survival (PFS) was significantly reduced in patients receiving any antibiotic treatment within the baseline time period. (B) Effects of individual antibiotic classes administered in the 3-weeks pre-CAR-T period on progression incidence in our international patient cohort. (C) Incidence of lymphoma progression, (D) Kaplan-Meier curves on PFS and (E) overall survival (OS) in our international patient cohort after CAR-T cell therapy stratified by high-risk (i.e., broad-spectrum carbapenem, cephalosporin or piperacillin/tazobactam antibiotics) vs. low-risk (any other antibiotic) or no antibiotic exposure in the 3-weeks period prior to CAR-T cell infusion (logrank test: for (C) p = 2.26e-05, for (D) p = 3.8e-5). (F) Serum levels of LDH as a marker for tumor burden at the time point of leukocyte apheresis (left) or during lymphodepleting chemotherapy (right) stratified into patients receiving no or low-risk (LR) antibiotics (LDH: n = 132, CRP: n = 127) vs. high-risk (HR) antibiotics (LDH: n = 36, CRP: n = 32) during the pre-CAR-T cell therapy time window. (G) Assessment of metabolic tumor volume by FDG-PET-CT scans in a subset of German and US patients combined and stratified by the type of baseline antibiotic exposure (none | LR Abx: n = 33; HR Abx: n = 8). (H) Serum levels of CRP as a marker for systemic inflammation measured at the time point of leukocyte apheresis (left) or during lymphodepleting chemotherapy (right) stratified into patients receiving no or low-risk (LR) antibiotics (LDH: n = 132, CRP: n = 127) vs. high-risk (HR) antibiotics (LDH: n = 36, CRP: n = 32) during the pre-CAR-T cell therapy time window. (I) Peripheral blood counts of CD3, CD4 and CD8 T cells in a subset of German and US patients combined and stratified by the type of antibiotic exposure in the 3-weeks pre-CAR-T cell period (none | LR Abx: n = 36; HR Abx: n = 15). Statistics by logrank and Mann-Whitney tests.
Immune-related toxicities constitute important factors contributing to morbidity and mortality in CAR-T cell immunotherapy14,27. We therefore investigated whether exposure to antibiotics in the pre-infusion time window was associated with subsequent development of CAR-T cell-mediated toxicities. Indeed, high-risk antibiotic treatments were not associated with cytokine-release syndrome (CRS; Extended Data Fig. 1E), but were correlated with a significantly higher rate of immune-effector cell-associated neurologic syndromes (ICANs) of any grade (high-risk antibiotics = 23/36 (63.8%) versus 42/110 (38.2%); Extended Data Fig. 1E) in the combined cohort, which was in line with a previous study24.
We reasoned that increased disease progression in antibiotics pre-treated patients could be explained by one of two scenarios: In the first ‘causality’ scenario, antibiotics-induced microbiome disruptions would adversely impact CAR-T therapy efficacy. In the second ‘reverse causality’ scenario high-risk antibiotics-treated patients could feature an a priori more advanced disease and/or deteriorated clinical status, which would require an intensive bridging therapy and antibiotics to treat infectious complications. This altered clinical status would render subsequent CAR-T cell treatment less effective in antibiotics-pretreated patients, independently of the associated changes in the microbiome, as was suggested in other forms of immunotherapy28. To assess these possibilities, we investigated tumor burden and systemic inflammation in our lymphoma cohort prior to CAR-T therapy initiation. Indeed, serum LDH, representing a surrogate marker for tumor burden, was significantly higher in patients receiving high-risk antibiotic treatments at the time point of leukocyte apheresis and lymphodepletion, compared to patients not receiving antibiotics or treated with low-risk antibiotics (Fig. 1F), a finding that was observed independently in the German and the US cohort (Extended Data Fig. 3A). In corroboration, the metabolic tumor volume (assessed by FDG-PET-CT scans prior to CAR-T cell infusion) was significantly higher in high-risk antibiotics-administered patients (Fig. 1G; N = 41 patients; scan data available from LMU Munich and Moffitt Cancer Center). Likewise, serum CRP levels were significantly elevated in high-risk antibiotic treated patients at both time points (Fig. 1H and Extended Data Fig. 3B). This increased level of inflammation in these patients was also mirrored by significantly increased serum interleukin (IL)-6 at the day of CAR-T cell infusion (i.e., day 0; Extended Data Fig. 3C). No associations were observed between antibiotics administration and the presence of bulky tumors (Extended Data Fig. 3D), but significantly more high-risk antibiotic pretreated patients also presented with a decreased physical status according to the ECOG performance (Extended Data Fig. 3E). There was no correlation between the two major CAR-T cell products (axi-cel or tisa-cel) administered to the patients and antibiotic risk stratification or performance status (Extended Data Fig. 3F and 3G, left). However, the toxicity profiles differed between the two products, with axi-cel associated with increased ICANS frequency (Extended Data Figure 3G, right), a finding in line with previous observations29.
In addition to tumor burden and physical status, the quality of the autologous CAR-T cell product could affect survival outcomes30. This feature heavily depends on the quality of T cells harvested from each patient. Intriguingly, patients receiving high-risk antibiotics displayed significantly lower peripheral CD4 T cell counts and a trend towards lower CD3 and CD8 T cells in the combined cohort (Fig. 1I), a result seen separately in the US and the German cohorts (Extended Data Fig. 3H). Furthermore, the number of prior lines of therapy was associated with differences in peripheral T cell levels (Extended Data Fig. 3I), which was also recently reported in a pediatric CAR-T cell cohort31. In a subset of US patients, we also performed an immunological characterization of patient T cells at apheresis and found a significant reduced number of effector/effector memory CD 4 and CD8 T cells in high-risk antibiotics-treated patients (Extended Data Fig. 3J and 3K). A multivariate testing by Cox proportional hazards analysis for PFS and OS that included age, performance status (ECOG), country, LDH, CRP, presence of bulky disease and the number of therapies that a patient received prior to CAR-T cell infusion demonstrated that exposure to high-risk antibiotics remained significantly associated with PFS (HR = 2.19; Extended Data Fig. 4A) but lost its association with OS (HR= 1.96; Extended Data Fig. 4B). Importantly, serum LDH levels above the upper limit of normal were significantly associated with poorer survival outcomes (Extended Data Fig. 4A and 4B).
Together, exposure to broad-spectrum, ‘high-risk’ antibiotics such as meropenem, piperacillin-tazobactam or cefepime within three weeks prior to CAR-T cell therapy was associated with worse PFS and OS in patients across cohorts. Clinically, administration of these antibiotics was related to a higher pre-CAR-T treatment tumor burden, increased systemic inflammation, and a reduced peripheral T cell pool and immunophenotype, which likely drive the noted differences in CAR-T therapy effectiveness.
Microbiome profiles linked to clinical covariates and outcomes.
To circumvent the above potential confounders, we next assessed the changes of the gut microbiome observed over the course of CAR-T cell therapy in the entire CAR-T-treated cohort, or in segregated high-risk-antibiotics-treated, or non-high risk-antibiotics-treated patients. In these analyses, we aimed to uncover associations between microbiome features and major clinical and immunotherapy readouts, and their links to high-risk antibiotics exposure. To this aim, we prospectively collected 351 stool samples from 116 of 172 patients at all five CAR-T cell centers between days −62 to 208 relative to CAR-T cell infusion and performed shotgun metagenome sequencing to investigate bacterial species composition, abundance of metabolic pathways encoded in the microbiome and its gene contents (Extended Data Fig. 5A). All included samples contained at least 500,000 metagenomic reads after removing host-related reads (see Methods). We observed a considerable variance in alpha diversity between samples over time (Fig. 2A), with exposures to high-risk antibiotics associated with significantly reduced microbial diversity (Fig. 2B). Baseline alpha diversity (or averaged alpha diversity in case of multiple baseline samples obtained) was not associated with responses to CAR-T immunotherapy and progression (both assessed at day 180 after infusion) irrespective of antibiotic exposures (Fig. 2C, Extended Data Fig. 5B). Similarly, no significant associations were observed between baseline alpha diversity and CRS or with ICANS as major immune-related toxicities (Extended Data Fig. 5C and 5D).
Figure 2. Gut microbiome characteristics and associations with clinical variables in CAR-T cell treated patients.

(A) Variation of microbial alpha diversity assessed by Shannon index over the course of CAR-T cell therapy in the subset of German and US patients; microbiome sequencing performed by shotgun metagenome sequencing (n = 116 patients, n = 351 fecal samples); diversity metrics shown between day −20 and 70 relative to CAR-T cell therapy. The red line displays the 7-days-window moving median of the Shannon indices with spline interpolation smoothing. (B) Left, Shannon’s alpha diversity as in (A), but stratified by the type of antibiotic exposure during specimen collection over the course of CAR-T cell therapy; right, analysis of Shannon’s diversity of paired patient samples before (i.e., during none or LR Abx administration) and during HR Abx exposures (n = 39 patients); in case of repeated samples per patient before or during HR Abx exposures sample means were used for plotting and statistics. (C) Mean-per-patient alpha diversity for patient samples collected between day 0 (day of CAR-T cell infusion) and day −21 grouped by response at day 180 (CR, complete remission [n = 42] vs. no CR [n = 53 patients]) or early progression (≤ 180 days: progression within 180 days after infusion [n = 47] vs. no progression within follow-up or progression after day 180 [n = 48 patients)]; HR antibiotic samples were included. (D) PERMANOVA analyses of the variance in microbiome features by analyzing the composition of species, metabolic pathways and microbial genes in samples collected within the 3-weeks period before CAR-T cell infusion and calculated as mean per patient (in case of multiple samples per patient; n = 95 patients, n = 124 samples included) and related to the following clinical variables: age (< 65 vs. >= 65 years), ECOG (grade 0-1 vs. 2-3), LDH (< 280 vs. >=280 U/ml assessed during lymphodepletion chemotherapy), presence of bulky disease (>10 cm: present vs. absent), number of lymphoma therapies prior to CAR-T cell infusion (0-3/4-6/7-10), antibiotic administration during baseline time period (none or only low-risk antibiotic exposure vs. high-risk antibiotic treatment given on or within the last two-week before the sampling day). (E) Principal coordinate analysis (PCoA) based on Bray Curtis dissimilarity metrics of the microbiome taxa and pathways beta-diversity in all longitudinally collected samples, color-coded for the antibiotic risk exposure at the sampling day. (F) Left, volcano plot showing the log2 fold-change in the relative abundance of microbial species in none and/or low-risk samples vs. those in high-risk antibiotic exposed samples against the significance of the abundance differences by Mann–Whitney tests. Samples with both p-value < 0.05 and an absolute log2 fold change > 1 are highlighted in red. Right, bar-plot showing the fold-change of the species found to be significantly different between antibiotic risk groups; here only stool microbiomes collected prior to CAR-T cell infusion were analyzed (n = 96 patients). (G) PERMANOVA analyses of the variance in microbiome features of baseline samples (mean-per-patient) by species, pathways and genes composition explained by the following therapy outcomes: response (see C), survival (alive vs. dead at day 180), early progression (see C), CRS (grade 0-1 vs. >= 2), ICANS (grade 0 vs. >= 1). (H) Heatmap of the PERMANOVA results as in (G), but excluding samples collected while or less than 14 days after exposure to high-risk antibiotics. Number of patients included in PERMANOVA tests: n = 95 in (G), n = 79 in (H). P-values (PERMANOVA): * p < 0.05, ** p < 0.01. For (B) and (C) Wilcoxon signed-rank test and Mann-Whitney test were used, respectively.
Next, we analyzed the effects of various clinical covariates on the compositional variance of the pre-infusion gut microbiome in our combined patient cohort by testing the beta-diversity differences between groups based on Bray-Curtis distances. These analyses revealed significant contributions of country (Germany vs. US), bulky disease, the number of prior lines of lymphoma therapies, and, notably, the exposure to high-risk antibiotics on pre-infusion variances in species composition, metabolic pathways, or the microbiome gene reservoir (Fig. 2D). Importantly, taxonomic and pathway compositions featured separation according to antibiotic risk strata (Fig. 2E, including all longitudinally collected stool samples; see also Extended Data Fig. 5E–H displaying the same data colored according to country, ECOG performance, prior therapies, and days relative to CAR-T cell infusion). Roseburia, Bifidobacterium, Lactobacillus and Eubacterium spp. were the most abundant commensals noted in non-antibiotic or low-risk antibiotic exposed baseline samples, whereas Prevotella, Veillonella, or Enterococcus spp. were among the major bacteria significantly more abundant in high-risk antibiotic exposed specimens (Fig. 2F). Metabolically, pathways involved in peptidoglycan biosynthesis, or lactose and galactose degradation dominated in high-risk antibiotic-exposed samples (Extended Data Fig. 6A and 6B). Klebsiella pneumoniae, Streptococcus thermophilus, Lactobacillus rhamnosus or D-galactose biosynthesis and mycolate biosynthesis were among the top microbiome features differing between German and US patients (Extended Data Fig. 6C and 6D).
We further analyzed the associations between pre-infusion microbiome compositions and major clinical outcomes of CAR-T cell therapy, by comparing the compositions of microbiomes between outcome groups. We began by assessing the correlations between microbiome feature variability and clinical disease readouts. When using all baseline samples (including antibiotic exposed microbiomes; n = 95 patients), we found that species and pathway or gene variability were only weakly associated with any of the major outcomes studied (Fig. 2G). In contrast, exclusion of high-risk antibiotics-exposed specimens resulted in a higher portion of species, pathway and gene variance explaining treatment response and early disease progression (Fig. 2H; n = 79 patients). Next, we considered the differences in individual microbiome features between different outcomes. We found Bifidobacterium longum, Eubacterium eligens and Parabacteroides merdae to be more abundant in patients alive at 6 months after infusion when including all samples (Extended Data Fig. 7A), and Bifidobacterium longum after excluding high-risk antibiotic treated patients (Extended Data Fig. 7B). In contrast, peptidoglycan biosynthesis or D-galactose biosynthesis pathways were more prevalent in the gut microbiome of patients that did not survive the first 6 months after infusion or experienced early disease progression (Extended Data Fig. 7C and 7D, respectively). We did not detect significant species associations with CR as a response variable (Extended Data Fig. 8A).
Our analysis of global microbiome compositions revealed several patient characteristics, clinical covariates and immunotherapy outcome variables that significantly explained microbial feature variability in our patient cohort. However, these environmental and patient-intrinsic factors can interact with each other and, thus, may represent overlapping associations with the microbiome architecture32. To further explore these interdependences, we correlated patients’ pre- treatment species and pathway abundances (i.e., from stool specimens collected in the first 21 days before CAR-T cell infusion, or averaged abundances in cases in which multiple samples per patient were collected during this period) with major demographic, clinical and outcome variables. Importantly, when considering all baseline samples (including antibiotic-exposed microbiomes), species abundances differing between antibiotic exposure groups were also different for age, country, patient performance status (ECOG), and 6-months survival or CRS (Extended Data Fig. 8A). Intriguingly, country of residence had a distinct effect on metabolic pathway composition with a small overlap with significantly different pathways for response (Extended Data Fig. 8B). After exclusion of high-risk antibiotic samples, Bifidobacterium longum was significantly associated with 6-months survival (Extended Data Fig. 9A), and this microbial association was independent of associations with other demographic or clinical variables. Likewise, after high-risk antibiotics exclusion, peptidoglycan biosynthesis or geranyl-geranyl-diphosphate biosynthesis pathways were significantly associated with 6-months survival or response, without overlaps with age, country, performance status or tumor burden (Extended Data Fig. 9B). In a separate approach, we studied the effects of gender or age per se on survival outcomes, but did not observe significant impacts on PFS (gender: hazard ratio = 1.2 [95% CI, 0.8-1.8], p=0.5; age [<65/>=65 years]: hazard ratio = 0.9 [95% CI, 0.6-1.3], p=0.6) or OS (gender: HR = 1.4 [95% CI, 0.8-2.4], p=0.3; age [<65/>=65 years]: HR = 0.9 [95% CI, 0.6-1.6], p=0.8; all carried out in univariate analyses).
Collectively, these results suggest that antibiotic drug exposures, country of residence (i.e., Germany vs. US), patients’ pre-treatment health status and tumor burden constitute major determinants of the diversity, composition, and functional repertoire of the gut microbiome in our patient cohorts. These variables are important covariates that were found to overlap in most cases in their effects on major CAR-T cell outcome parameters, as demonstrated by significant associations between antibiotic exposures with beta-lactam resistance encoding peptidoglycan biosynthesis pathway regarding PFS (Extended Data Fig. 10A). Notably, exclusion of high-risk antibiotic-exposed patients strengthened the associations between microbiome features with outcomes including survival and progression, with multiple features, including Bifidobacterium longum and peptidoglycan biosynthesis, being strongly correlated with long-term survival or disease progression following CAR-T cell therapy, independently of other demographic or clinical variables.
Microbiome-based modeling of response prediction across cohorts.
The above results suggested that distinct microbiome features may be associated with the antitumor efficacy of CAR-T cell therapy after accounting for antibiotics perturbation. To assess the predictive power of baseline taxonomic microbiome features in predicting long-term response to CAR-T cell therapy (CR vs. no-CR on day 180 after CAR-T cell infusion), we utilized two types of supervised machine learning frameworks, in which the German cohort was used for training the models and the US cohort was used as an external validation cohort. In a deterministic logistic-regression model algorithm, the input data contained clinical data (age and gender) and microbiome data on species abundances at baseline (3-weeks pre-CAR-T cell infusion period). Inclusion of all patients in the training process demonstrated limited performance in predicting response to CAR-T therapy as the endpoint (area under the receiver operating curve (AUROC) for the US validation cohort: 0.64; Extended Data Fig. 10B). Similarly, a PCA of the microbial features selected during the training for the machine-learning model including all samples did not show a clear segregation of CR vs. non-CR at 6 months (see Extended Data Fig. 10C).
To further explore these results, we fitted a second, probabilistic prediction model, which provides quantification of uncertainty in its predictions. Like the previous model, predictions trained on the entire cohort (including antibiotics-treated patients) by this probabilistic model and corroborated by the validation (US) dataset were not significantly better than those obtained by a random guess (Fig. 3A; AUROC=0.52±0.05 for the US validation cohort). Collectively, these poor predictions likely reflect our initial observations that prior high-risk antibiotics treatment may obscure microbiome associations with survival outcomes. Indeed, using the probabilistic model, training set predictions of high-risk antibiotics-treated individuals were characterized by a high variability in contrast to the non-high-risk antibiotics-treated group (Fig. 3B), suggesting that high-risk antibiotic exposure constitutes a major source of uncertainty for the predictive model. To validate the confounding effects of high-risk antibiotics exposure on predictability of CAR-T responses, we repeated our analysis using both probabilistic and deterministic models, after excluding patients exposed to high-risk antibiotics prior to CAR-T cell infusion. Importantly, predictability using both the probabilistic (Fig. 3C; AUROC=0.67±0.04 for the validation cohort; Extended data Fig. 10D for training data) and deterministic models (Extended Data Fig. 10E; AUROC=0.69 for the validation cohort) was markedly improved.
Figure 3. Prediction of response to CAR-T cell therapy by specific gut commensals.

Bayesian logistic regression models were trained on clinical variables and pre-CAR-T cell infusion gut microbial species composition (mean per patient) of the German cohort (with all baseline samples: n = 50 patients; after excluding high-risk antibiotic samples: n = 43 patients) and their performance was assessed on the US cohort (with all baseline samples: n = 45 patients; after excluding high-risk antibiotic samples: n = 36 patients). (A) Area under the curve (AUC) receiver operating characteristic (ROC) curves indicating the false-positive and true-positive rates for predicting complete remission (CR) vs. non-CR at day 180 based on an analysis including all patient samples; data for the US cohort as validation dataset are presented. (B) Bar plots summarizing individual predictions of patients in the training dataset based on 500 random draws from the guide function; the y-axis shows the predicted probability of the day 180 - response (CR vs. no CR; n = 16 HR abx; 79 none | LR abx patients). (C) AUROC curves indicating the false-positive and true-positive rates for predicting complete remission (CR) vs. non-CR at day 180 based on an analysis that excluded samples collected on or less than two weeks after exposure to high-risk antibiotics (data for the US cohort as validation dataset are presented). (D) The regression coefficients of the gut microbiome species that were found to be important for the prediction of CR vs. non-CR on day 180; species with the top 25% absolute coefficient values are presented. (E) Kaplan-Meier curve for PFS of patients after CAR-T cell infusion und stratified by the median baseline relative abundance [low (<= median) or high (> median)] of Akkermansia muciniphila as one of the top features for CR in 3C; only pre-infusion samples without HR antibiotic exposure were included (n = 79). (F) Differences in relative abundances for five among the most contributing species with highest coefficients for predicting response are shown with grouping by response and high-risk antibiotic exposures (n = 38 / 41 / 4 / 12 patients [from left to right]); FDR corrected p-values for B. eggerthii are 0.0066 (for both comparisons) and for E. ramosum are 0.0476 (for all three comparisons). Group comparisons by pair-wise Mann-Whitney tests with FDR correction of p-values.
We next assessed which species potentially drive the discrimination of response to CAR-T cell therapy and analyzed their importance for response prediction (based on the deterministic machine learning model). Based on regression coefficients, we observed that a higher relative abundance of Bacteroides eggerthii, Ruminococcus lactaris, Eubacterium spp. CAG 180, Akkermansia muciniphila and Erysipelatoclostridium ramosum increased the probability for CR prediction, whereas higher relative abundances of Bacteroides stercoris and others increased the probability for non-response prediction (Fig. 3D). Age and gender, co-considered with gut species abundances in the models, were not among the top 25% of the features contributing to the prediction of clinical response.
We further assessed the contribution of gut commensals featuring the top regression coefficients for response and non-response on survival outcomes features (high vs. low median abundance of bacteria in the baseline microbiome; after exclusion of high-risk antibiotics samples). We observed that high abundances of Akkermansia muciniphila or Ruminococcus lactaris were associated with increased PFS (Akkermansia muciniphila: hazard ratio = 2.2 [Fig. 3E]; Ruminococcus lactaris: hazard ratio = 1.83 [p=0.12]), whereas high abundances of Bacteroides stercoris were linked to decreased PFS (Extended Data Fig. 10F). A borderline significance was noted when using the Cox proportional hazards regression analyses, likely due to small sample sizes per groups. As species predictive of immunotherapy response in our model were mostly anaerobes that are targeted by broad-spectrum antibiotics, we looked into abundance differences of these species following response and antibiotics exposure stratification. Indeed, high-risk antibiotic exposure was commonly associated with lower abundances of these species, similar to the non-response setting (Fig. 3F). Similarly, species predicting non-response after CAR-T cell administration were also downregulated in high-risk antibiotics-exposed specimens (Extended Data Fig. 10G; presented are statistical differences after FDR correction). Overall, these results provide putative candidates contributing to the reduced predictive power of the gut microbiome upon broad-spectrum antibiotic treatment.
Finally, given these findings and the recently suggested roles of gut commensal microbes in modulating human T cell responses33,34, we explored associations of baseline species abundances with peripheral blood T cell counts around the time point of apheresis in the combined cohort. When including all specimens, we found significant positive correlations of relative abundances of Lachnospira pectinoschiza with CD3 and CD4 T cell levels (Fig. 4A). Bacteroides uniformis, Bacteroides ovatus, Blautia spp. and Faecalibacterium prausnitzii abundances were negatively correlated with CD3 and CD8 T cell levels (Fig. 4A). After excluding high-risk antibiotics samples as potential confounders, we observed significant positive associations of Lachnospira pectinoschiza and Akkermansia muciniphila with CD3 and CD4 T cell counts, whereas Firmicutes bacterium CAG 424 was linked to low CD4 T cell counts in peripheral blood (Fig. 4B and 4C). In addition, higher Lachnospira, Bacteroides eggerthii or Ruminococcus lactaris abundances featured a trend towards reduced systemic inflammation, as assessed by serum CRP or IL-6 levels during lymphodepletion chemotherapy (Fig. 4D and 4E). These microbe-T cell associations suggest that gut microbes such as Akkermansia muciniphila, identified as classifiers of response to CD19-directed CAR-T cell therapy, may also associate with a homeostatic microbiome-immune cell interactions, thereby meriting future studies.
Figure 4. Associations of gut microbes with patient T cells before CAR-T cell infusion.
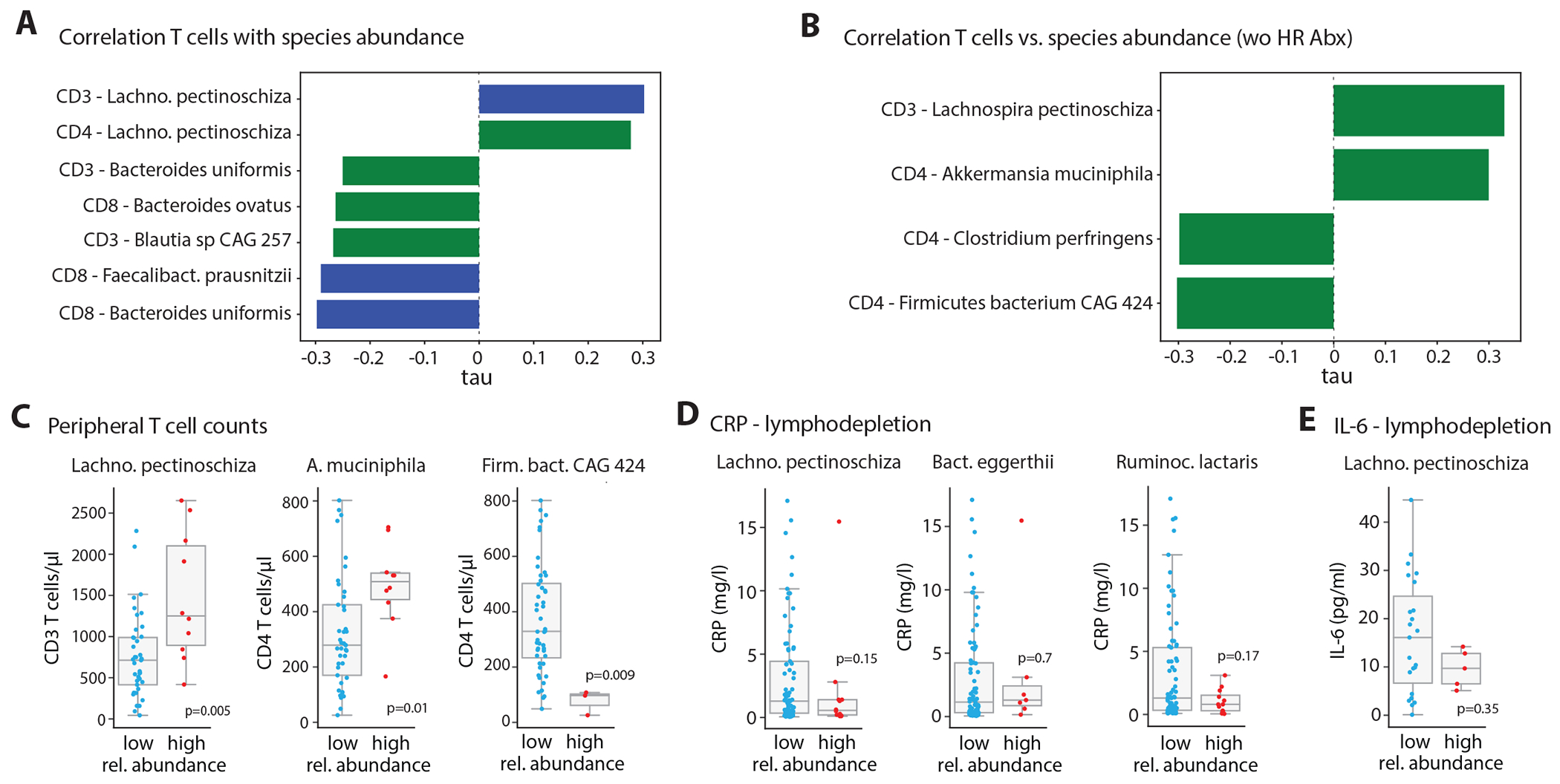
(A) Kendall’s tau coefficient of significant correlations (green bar: p-value < 0.01; blue bar: p-value < 0.01 and FDR-corrected p-value < 0.05) between species in the microbiome of patients in the 3-weeks pre-infusion period and blood T cells counts at apheresis with HR antibiotic exposed samples included. (B) As in (A), but excluding samples collected while or less than two weeks after exposure to high-risk antibiotics. (C) Blood T cells counts at apheresis compared between patients with low (<= median) or high (> median) abundance of the indicated species; samples collected while or less than two weeks after exposure to high-risk antibiotics were excluded (number of patients L. p.: low / high abundance = 41 / 10; A.m.: low / high abundance = 41 / 10; F.b.: low / high abundance = 48 / 3). (D) Serum CRP (number of patients: L. p.: low / high abundance = 77 / 12; B.e.: low / high abundance = 82 / 7; F.b.: low / high abundance = 77 / 12) and (E) interleukin (IL)-6 levels (number of patients: L. p.: low / high abundance = 23 / 5) at the time point of lymphodepletion chemotherapy compared between patients with low (<= median) or high (> median) abundance of the indicated species using all baseline samples. Group comparisons carried out by Mann-Whitney tests; for (C)-(E) no abundance filtration was applied.
DISCUSSION
We report in B cell lymphoma patients receiving CAR-T cell therapy that high-risk antibiotic treatment (i.e., broad-spectrum antibiotics such as piperacillin/tazobactam, meropenem, or cefepime) administered in a 3-weeks period prior to T cell infusion is associated with increased progression of the underlying disease and reduced overall survival. These findings were replicated independently in a German and a US cohorts and are in line with data of a recent US study of lymphoma and acute lymphoblastic leukemia patients24. However, our results suggest that administration of these classes of antibiotics constitutes a likely surrogate for poor disease status and a state of increased systemic inflammation prior to initiation of CAR-T cell therapy, as was recently suggested in cancer patients treated with other ICBs28,35. As such, rather than causally driving altered CAR-T responsiveness, antibiotic treatments may likely disrupt microbiome features that otherwise would be clearly associated with immunotherapy outcomes. For instance, broad-spectrum antibiotic treatment reduced the intestinal abundances of microbes in our patients such as Roseburia, Bifidobacterium or Ruminococcus spp., which were previously associated with beneficial outcomes of ICB cancer immunotherapies1,36.
Exposures to high-risk antibiotics in our study, in turn, led to increased intestinal abundances of Enterococcus, Streptococcus or Klebsiella spp., and also led to an enrichment of microbial pathways for peptidoglycan biosynthesis and related metagenomes that are involved in antibiotic-resistance. Exclusion of patients featuring such antibiotics-induced dysbiosis already present before CAR-T cell infusion unmasked potent associations between baseline microbiome configurations at the taxonomic and metagenome level with CAR-T treatment response, survival, and toxicity. Even more importantly, such a careful patient inclusion enabled a logistic regression-based machine learning prediction of immunotherapy response. Machine learning enables to explore and identify inter-cohort-generic microbial signatures in an unsupervised manner associated with the state of cancer disease in patients37, and has recently been applied to estimate microbiome-based prediction of ICB efficacy36.
Studying the non-antibiotic disrupted baseline microbiomes, we developed a model that enabled prediction of long-term response to CAR-T cell therapy. As treatment responses may be inherently limited by population and microbiome heterogeneity, we performed a cross-cohort evaluation utilizing our German and US cohorts to train, optimize and validate the machine learning model across geography and varying treatment protocols, which enabled a prediction ability based on the gut microbiome as a major biological variable. Within our model, we identified Bacteroides, Ruminococcus, Eubacterium and Akkermansia spp. as major taxa that contribute to prediction of CAR-T response. These microbes were also previously reported to be associated with better response to ICB immunotherapy2,3,38,39. Among those, Akkermansia abundances were also associated with significantly higher baseline peripheral T cell levels which, in turn, likely improves the quality and performance of the manufactured CAR-T cell product. Akkermansia muciniphila degrades mucin with a subsequent release of the short-chain fatty acids acetate and propionate40, which can enhance effector cytokines in T cells41. In vitro stimulation with Akkermansia has been shown to stimulate the release of the effector cytokine IFN-γ from CD4+ an CD8+ memory T cells2, while a lipid from Akkermansia’s cell membrane can activate a non-canonical TLR2-TLR1 pathway in immune cells42. A mechanistic elucidation of the effects of Akkermansia muciniphila and the other response-related species to favorable antitumor CAR-T cell efficacy merits future studies.
Of note, limitations of our study include differences and constraints in sample size of the German and US cohorts, limiting our Cox proportional hazards regressions for PFS. Another limitation that is inherent to observational studies is that the associations observed do not demonstrate causality, meriting future microbiome causative studies, such as ones featuring microbiome transfers into CAR-T treated mice. Of note in our study, we observed center-specific differences in the use of broad-spectrum antibiotics. Specifically, cefepime was only administered in US patients, which could confound microbiome signatures given its narrower anti-microbial activity spectrum as compared to other high-risk antibiotics43. These limitations notwithstanding, our clinical and analytical design enabled the uncovering a novel set of microbiome-based biomarkers that enable data-driven stratification in non-antibiotics pretreated patients. These may be potentially clinically harnessed to the individual and utilized for optimizing patient selection or for tailored management and follow-up of CAR-T cell treated patients.
METHODS
Patient cohorts.
The present study included international patient cohorts from Germany and the United States: patients receiving CD-19 targeted CAR-T cell therapy at the Department of Medicine III, University Hospital of the Ludwig Maximilian University (LMU) Munich, at the Department of Internal Medicine V, University Hospital Heidelberg, at the Department of Internal Medicine III, University Hospital Regensburg (i.e., German cohort; n=66) and at the Department of Blood and Marrow Transplant and Cellular Immunotherapy, Moffitt Cancer Center, Tampa, FL and the Department of Lymphoma/Myeloma, MD Anderson Cancer Center, Houston, TX (i.e., US cohort; n=106). These patients received CAR-T cell infusions between 2019 – 2021 (German cohort) and 2018 – 2021 (US cohort) with axicabtagene ciloleucel (axi-cel; n=122), tisagenlecleucel (tisa-cel; n=49) or lisocabtagene maraleucel (liso-cel; n=1). NHL - Patients with DLBCL/TFL, PMBCL, Richter’s TCLL and TMZL were included in the analysis; for more details see Tables 1 and 2.
Regulatory compliance.
Collection and analyses of clinical data and patient biomaterial were approved by the Ethics Committees of the University of Heidelberg (protocol no. S-877/2019; German Clinical Trials Register ID DRKS00020506), of LMU Munich (protocol # 19-817) and of University of Regensburg (protocol # 14-47 2-101) according to the principles of the Declaration of Helsinki. At Moffitt Cancer Center, data and specimen collection was approved under Advara IRB Pro00029290, and at MDACC under IRB protocol LAB04-717. Written informed consent was obtained from all the participants of the study. Patients did not receive compensation for participating in the study.
Clinical data collection.
In addition to general epidemiological patient characteristics such as age or gender, we recorded indices of tumor burden at leukocyte apheresis and/or lymphodepletion by serum LDH levels, presence of bulky disease (tumor diameter > 10cm), metabolic tumor volume assessed by fluorine-18-fluorodeoxyglucose (FDG) positron emission tomography (PET) computed tomography (CT) scans (data available on a subset of 10 German and 31 US patients), Ann Arbor stage and ECOG. We collected the number of prior oncological therapies, and whether or not patients underwent bridging chemo- and/or radiotherapy from patient records. Serum CRP levels were recorded at apheresis and lymphodepletion, and blood T cell counts (CD3, CD4, CD8) were assessed at apheresis through routine clinical FACS applications. Exposure to antibiotic drugs was recorded from patient medical records from each center. Baseline antibiotic exposure was considered for any antibiotic administration between the day of CAR-T cell infusion (day 0) and three weeks prior to infusion of the engineered T cells.
Assessment of clinical outcomes.
Tumor response to CAR-T cell immunotherapy was classified as either complete remission (CR) or non-CR (partial remission, stable disease, progressive disease or death) 90 and 180 days after CAR-T cell infusion by the treating physician; here, imaging results through CT-/MRI-scans were used for tumor response assessment. Overall survival (OS) and progression-free survival (PFS) were prospectively recorded until death, to the date of progression or date of last contact alive. The development of cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity (ICANS) were recorded and graded according to the consensus guidelines of the American Society of Transplantation and Cellular Therapy (ASTCT)44.
Fecal sample collection, DNA extraction and microbiome sequencing.
Prospective fecal sample collection was performed according to institutional practices. In Heidelberg, Munich and Regensburg samples were collected at the hospital facilities immediately after donation, aliquoted and frozen (−80°C) until DNA extraction. At MDACC, patients were provided an OMNIgene GUT kit for in- and outpatient sample collection and were frozen (−80°C) prior to extraction. At Moffitt Cancer Center, samples were collected on fecal occult blood test cards stored at −80C as previously described45. At all centers, fecal specimens were obtained serially from patients at different time points before, during and early after CAR-T cell infusion; see Extended Data Figure 6A for samples obtained per time point relative to CAR-T treatment.
DNA extraction for microbiome analyses:
DNA was extracted centrally for samples of the German cohort at DKFZ from 200mg collected stool material using the PureLink DNA isolation kit (Invitrogen) as per manufacturer protocol. For MDACC samples, bacterial genomic DNA was extracted using QIAamp Fast DNA stool mini kit (Qiagen), following the manufacturer’s protocol, modified to include an intensive bead-beating lysis step. A 3.2mm steel bead and zirconium beads were added to DNA bead-beading steps for intensive lysis. For Moffitt samples, the Mo Bio Powersoil DNA extraction kit was used to extract DNA from window cutouts from FOBT cards with a bead-beating step added to facilitate lysis.
Microbiome sequencing.
For German samples, DNA concentrations were measured using a Qubit 4.0 fluorometer with a Qubit™ 1X dsDNA HS Assay-Kit (Life Technologies, Grand Island, NY, USA, Cat. No Q33230). For shotgun metagenome sequencing whole-genome libraries were prepared from DNA extracted as above from stool samples and further normalized to a concentration of 0.2 ng/μl. Illumina sequencing libraries were prepared from 1 ng DNA using the Nextera XT DNA Library Preparation kit (Illumina, San Diego, CA, USA, FC-131-1096) according to the manufacturer’s recommended protocol, with reaction volumes scaled accordingly. Concentrations for each pooled library were determined using Qubit™ 1X dsDNA HS Assay-Kit. Prior to sequencing, libraries were pooled by collecting equal volumes of normalized libraries (2 μl) from batches with up to 16 samples. The resulting multiplexes were sequenced on Illumina NextSeq 550 to obtain 100 bp paired-ends reads with Unique Dual Barcodes to yield ~30-80 million paired end reads per sample. Post-sequencing demultiplexing and generation of FASTQ files was carried out using the bcl2fastq Conversion Software (Illumina, San Diego, CA, USA). For MDACC samples, libraries were prepared using the Illumina DNA prep kit (Illumina, Cat# 20060060) following the manufacturer’s protocol. Library quality was determined by Agilent high sensitivity D1000 screen tape assay (Agilent, Cat# 5067-5584) and pooled with an equal molar ratio. The final libraries were sequenced on Illumina NovaSeq 6000 platform with a 2x150 bp paired-end read protocol, resulting in ~5 Gb per sample. Moffitt samples were sequenced at Microbiome Insights (Vancouver, Canada) in a CAP-accredited laboratory, and paired-end sequencing (2x150 bp) was done on an Illumina NextSeq 500 platform.
Analyses of sequencing data.
Sequencing data processing.
Sequencing reads from each sample in a pool were de-multiplexed based on their associated barcode sequence. All down-stream sequencing data analyses were performed centrally at DKFZ. In order to remove host contaminant reads, sequencing reads mapping to the human genome were filtered out using KneadData (kneaddata version v0.10.0; Huttenhower Lab https://github.com/biobakery/kneaddata; human database hg37dec_v0.1). The remaining reads were then rarified by random subsampling of 5e5 forward reads and the corresponding 5e5 reverse reads per sample, using seqtk (https://github.com/lh3/seqtk [version 1.3-r116-dirty]) with random seed 1. Taxonomic and functional profiling of the metagenomes was performed by MetaPhlAn (version 3.0)46 and HUMAnN3 (version v3.0.0.alpha.4)46.
Data partitioning and aggregation.
Samples taken between 0-21 days before the infusion day were considered as basal samples. For these samples, those that were sampled less than 14 days after a treatment of the patient with a high-risk antibiotic were considered as high-risk-antibiotics baseline samples, and accordingly the other basal samples as no-high-risk-antibiotics basal samples. Following these partitions, in order to aggregate per-patient multiple basal samples, the mean value for each microbiome feature in these samples was derived. Unless otherwise indicated, species, metabolic pathways and genes features were filtered by the condition of having among the samples a maximal fractional abundance bigger than 1e-3, 1e-4 and 1e-4, respectively. Fractional abundance smaller than the corresponding threshold were re-assigned to the threshold value. Log transformation was applied in the logistic regression modelling of clinical outcomes.
Microbiome diversity indices.
Alpha-diversity parameters summarize a microbial community according to the number, or the relative abundance, of the different taxonomic units composing it. Here, we calculated alpha-diversity using the Shannon index at the species level, a measure which weighs the relative abundance of the species. Baseline alpha diversity was assessed as diversity of a sample collected in the first 21 days before CAR-T cell infusion, or averaged diversity in case of multiple samples per patient were collected during this period. To analyze differences in the microbiome composition (i.e., beta-diversity) between samples, we performed a principal coordinate analysis (PCoA) based on species-level Bray-Curtis dissimilarity. Shannon indices and Bray-Curtis dissimilarities were calculated in Python using the skbio.diversity.alpha.shannon and scipy.spatial.distance.pdist functions. Python versions (3.7.3, 3.9.7 and 3.6.8) were used in the microbiome data analyses.
Quantification and statistical analyses.
Group comparisons.
We assessed statistical differences by Mann-Whitney tests for two-group pair-wise comparisons (two-sided tests were performed) and Kruskal-Wallis rank tests for non-parametric comparisons of more than two groups; FDR correction for multiple testing was performed as indicated in the figure legends. For contingency analyses on clinical features, we applied Fisher’s exact tests as indicated in the figure legends. We also performed clinical covariate analyses computing associations using Kendall’s tau as indicated in the text. PERMANOVAs were performed for testing the differences between groups based on Bray-Curtis distances (implemented in Python, using the functions scipy.spatial.distance.pdist and DistanceMatrix of the SciPy [version 0.5.6] and the Skbio [version 1.4.1] packages, respectively). Statistical significance was set at p < 0.05.
Survival analyses.
OS, PFS and progression incidence were visualized by Kaplan-Meier curves. Cox proportional-hazards regression models were used to investigate the association between antibiotic treatments, clinical co-variates and microbiome features after binarizing the predictor variables (taxa abundance, age, ECOG, LDH, CRP, number of prior therapies). Univariate testing of individual features vs. multivariate testing by logrank and Wald tests to assess the effects of antibiotic treatments or microbiome features and clinical metadata on overall survival, progression-free survival and progression incidence were performed as indicated in the text; the survival (version 3.3-1) and survminer (version 0.4.9) R packages (R version 4.2.1) were used to compute the models.
Machine learning.
Datasets for this part included either all the samples collected in the 3-weeks pre-CAR-T cell infusion time period, i.e., baseline samples, with a mean rel. abundance calculated in case of multiple samples per patient, or only the non-high-risk-antibiotics baseline samples. The German and US cohorts were used as the training and external validation test cohorts, respectively. The initial set of input features included species relative abundance as well as patients age and gender. The prediction workflow consisted of capping and filtration of species relative abundance and normalization of all features, followed by classification layer that is a logistic regression (sklearn.linear_model.LogisticRegression) with elastic net regularization penalty 47. Accordingly, the hyper parameters of the workflow were capping threshold, regularization strength and the L1 ratio. First, we tuned the hyperparameters of the model using grid search cross validation applied to the training (German) cohort, and re-fit the model using the optimal set of hyperparameters to the entire training set. Then, we trained the final model using the set of up to 30 features corresponding to highest magnitude regression coefficients. The same set of features was used for training Bayesian logistic regression models 48 specified as following:
where is the input data, denotes the outcome label and is the sigmoid function. The model was implemented using the Pyro probabilistic programming language framework 49. We approximate a joint posterior distribution over the parameters given the training data: , using a reparametrized variational distribution (guide function) implemented by pyro’s AutoLowRankMultivariateNormal class. The parameters of the guide were then found via stochastic variational inference (SVI) for maximizing the evidence lower bound (ELBO) 50. Optimization was performed for 15000 iterations with SGD optimizer setting the learning rate at 0.01. Predictions were conducted by applying 500 random samples of the parameters from the guide function to the data according to the logistic model: .
Data presentation.
Grouped data are presented as scatter – box plots with the following definitions: whisker min: Q1 - 1.5*ICR, whisker max: Q1 + 1.5*ICR, line within the box = median, lower box bound = Q1 (i.e., 25th percentile), upper box bound = Q3 (i.e., 75th percentile) and IQR is defined as Q3 - Q1.
Extended Data
Extended Data Figure 1. Effects of antibiotic exposure on survival outcomes and on development of toxicities in CD19-targeted CAR-T cell treated lymphoma patients.

(A) Kaplan-Meier curves for progression-free survival (PFS) in the German and US cohorts displayed separately according to exposure to any antibiotic within 3 weeks before CAR-T cell infusion. (B) Incidence of progression and (C) overall survival in the combined cohort stratified by pre-infusion antibiotic exposure. (D) PFS for the US and the German patient cohorts displayed according to antibiotic risk strata (no antibiotic exposure vs. exposures to low- or to high-risk antibiotics in the three-weeks pre-infusion time window). (E) Histograms of the frequencies of CRS or ICANS grades according to antibiotic risk stratification; statistics by logrank tests (for survival analyses) and Fisher’s exact tests.
Extended Data Figure 2. Antibiotic classes administered to CAR-T cell treated lymphoma patients.

Frequency of individual antibiotic drugs administered to patients within three weeks before (A) and four weeks after (B) CAR-T cell infusion. Histogram of patients receiving individual antibiotics over the course of CAR-T cell therapy for the 8 most frequently prescribed antibiotics in the combined cohort (C) and stratified by country (D). Note that cefepime was only administered to US patients and is therefore not shown in D.
Extended Data Figure 3. Associations of pre-CAR-T cell infusion antibiotic exposure, tumor burden, performance status, CAR-T cell product and peripheral blood phenotypes.
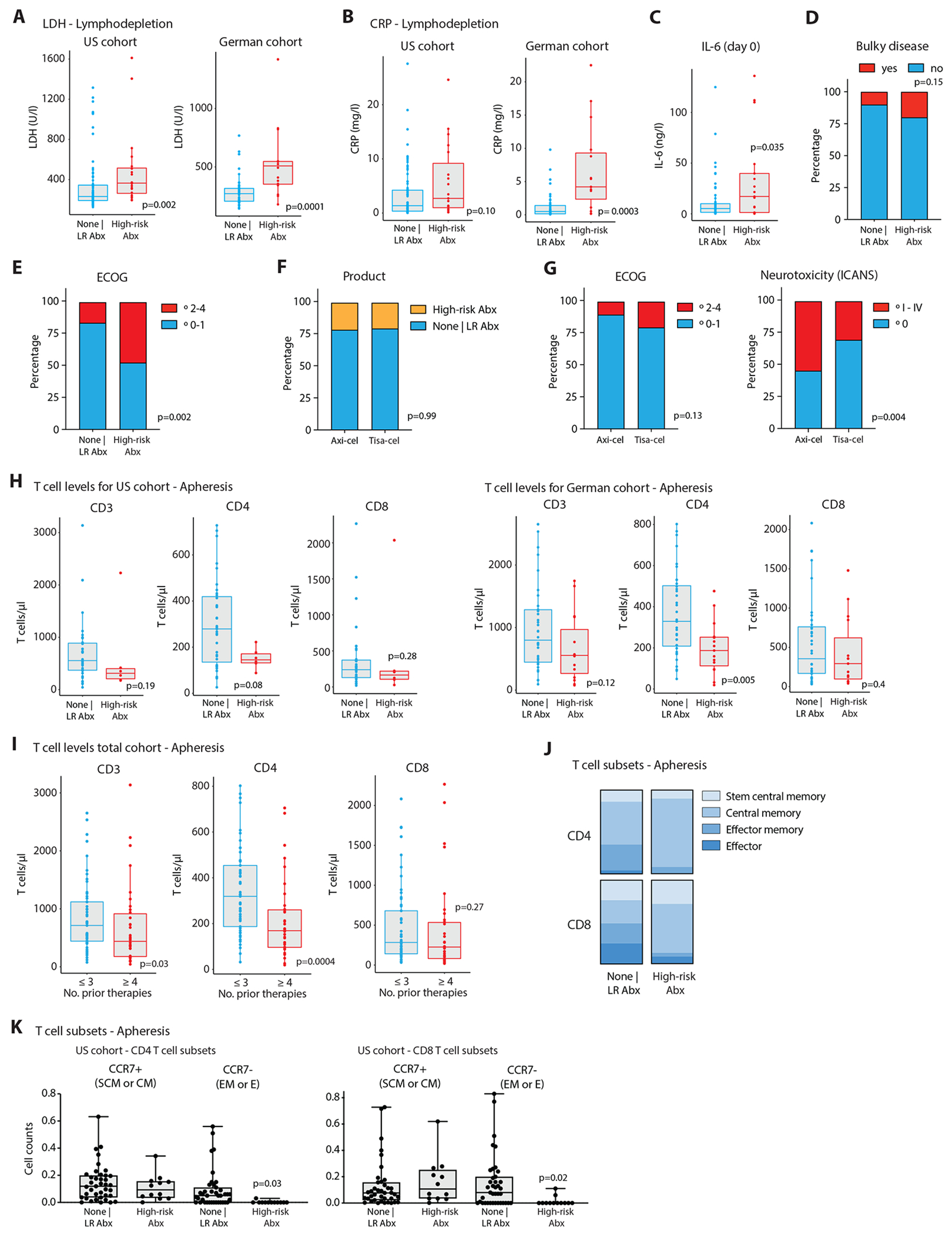
Serum levels of LDH (A) in German and US patients during the time point of lymphodepletion chemotherapy stratified by whether patients received none or low-risk antibiotics (US: n = 83, Germany: n= 20) versus high-risk antibiotics (US: n = 20: Germany: n= 16). (B) Serum levels of CRP in German and US patients during the time point of lymphodepletion chemotherapy stratified by whether patients received none or low-risk antibiotics versus high-risk antibiotics. (C) IL-6 serum levels in patients at the day of CAR-T cell infusion (none | LR antibiotics: n = 67; HR antibiotics: n = 18). (D) Histograms of the frequencies of bulky lymphomas (>10cm by radiological measurement), or (E) ECOG performance status according to antibiotic risk stratification. (F) Histograms of the frequencies of antibiotic exposures, or (G) ECOG performance status (left), or ICANS grades (right) according to type of CAR-T cell product administered to the patients. (H) Peripheral blood CD3, CD4 and CD8 T cell counts at the time point of leukapheresis displayed separately for US and German patients. (I) Blood T cell counts stratified by the number of treatment lines prior to CD19-directed CAR-T cell therapy (≤ 3: n = 32; ≥ 4: n = 19). (J and K) FACS analyses of peripheral blood T cell subsets of CAR-T cell treated patients from Moffitt Cancer Center at the time of leukocyte apheresis and stratified by antibiotic exposure (none | LR antibiotics: n = 39; HR antibiotics: n = 12). CD4 and CD8 stem central memory (SCM; CCR7+, CD45RO−), central memory (CM; CCR7+, CD45RO+), effector memory (EM; CCR7−, CD45RO+), and effector (E; CCR7−CD45RO−) subsets are shown. Statistics for A-C, H, I, K by Mann-Whitney tests, for E-G by Fisher’s exact tests.
Extended Data Figure 4. Multivariate analysis of antibiotic exposure prior to CD19-targeted CAR-T cell infusion and PFS or OS.
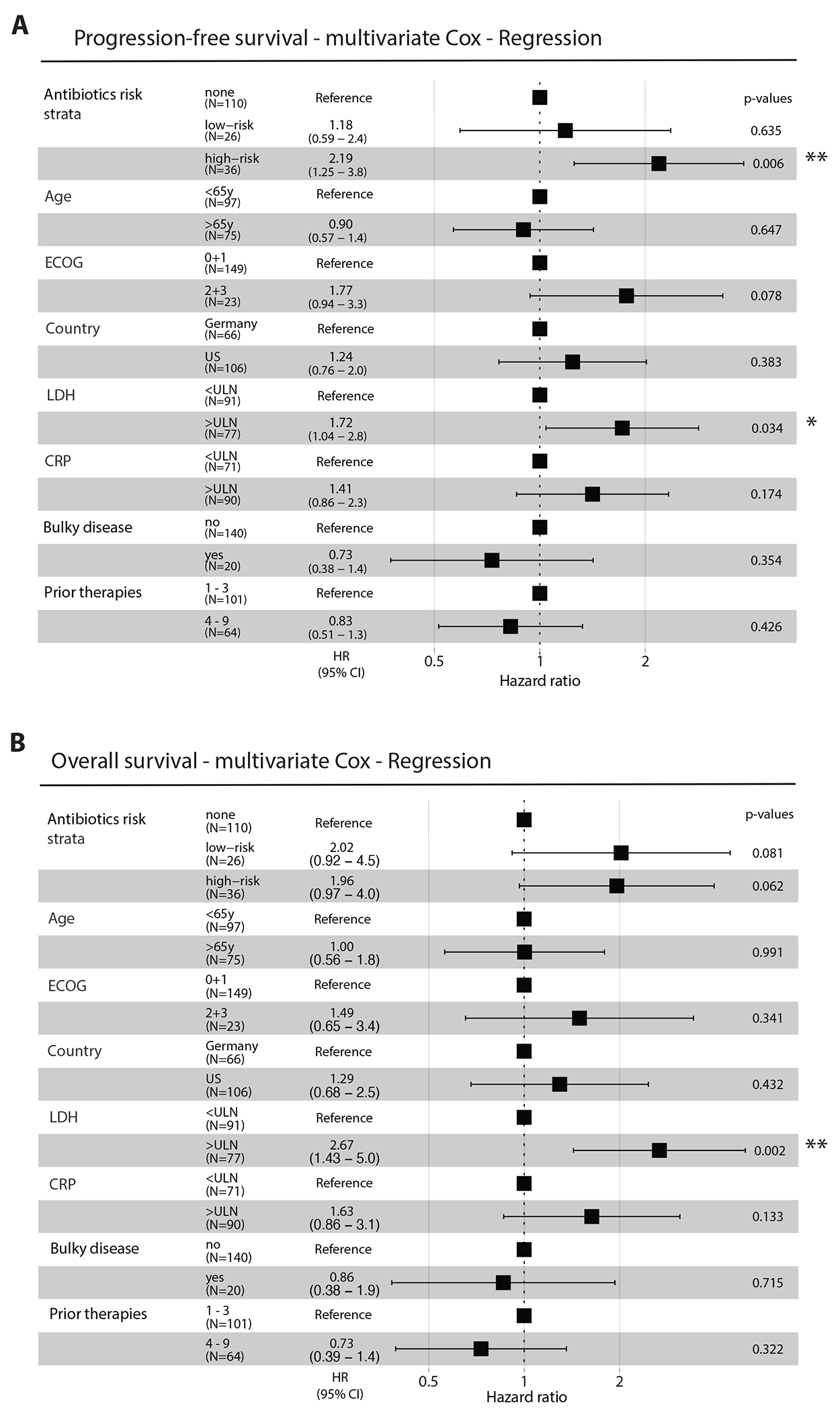
(A) The multivariate Cox model was adjusted for age (stratified by 65 years of age), ECOG (grade 0 and 1 vs. higher), country (Germany vs. US), LDH (normal vs. higher than ULN), CRP (normal vs. higher than 5mg/dl), bulky disease (> 10cm) and number of prior therapies (1-3 vs. 4-9). (B) The multivariate Cox model was adjusted for age (stratified by 65 years of age), ECOG (grade 0 and 1 vs. higher), country (Germany vs. US), LDH (normal vs. higher than ULN), CRP (normal vs. higher than 5mg/dl), bulky disease (> 10cm) and number of prior therapies (1-3 vs. 4-9). Error bars represent the low and high 95% confidence intervals (CI) of HR; *p<0.05; **p<0.01
Extended Data Figure 5. Gut microbiome diversity metrics grouped by clinical variables and outcomes.

(A) Number of stool samples collected over the course of CAR-T cell therapy by center and country (n = 351 in total). (B) Shannon indices for alpha diversity of the basal gut microbiome (i.e., samples collected between days −21 and 0 relative to CAR-T cell infusion and species composition averaged by mean in case of multiple samples per patient) and grouped for CR vs. no CR at day 180 (left; CR: n = 38, no CR: n = 41 patients), or early progression at day 180 (right; ≤ 180 days: progression within 6 months after infusion (n = 37); no | > 180 days: no progression within follow-up or progression after 6 months after infusion (n = 42 patients) after excluding specimens collected while or less than two weeks after high-risk antibiotics exposure. (C and D) Shannon’s diversity of the basal gut microbiome, after the same exclusion of HR antibiotic samples, grouped for CRS (n = 7, 40, 28, 4 patients [grade 0, 1, 2, 3+4]) and ICANS grades (n = 45, 12, 11, 11 patients [grade 0, 1, 2, 3+4]). (E) PCoA plots based on Bray-Curtis dissimilarity metrics of the microbiome species and metabolic pathways beta-diversity in all samples color-coded for German vs. US patients. (F – H) PCoA plots as in (E) for species composition color-coded for ECOG levels (F), number of prior therapy lines (PT; prior to CAR-T cell therapy) (G), and day of specimen collection relative to the day of CAR-T cell infusion (i.e., day 0) (H). Group comparisons carried out by Mann-Whitney tests or Kruskal-Wallis rank tests.
Extended Data Figure 6. Analyzing microbiome features associated with major cohort characteristics.

(A) Differential relative abundance analyses by log-fold changes and statistical testing by Mann-Whitney tests illustrated with volcano plots for species and metabolic pathways encoded in the fecal metagenomes: the log2 abundance in none | LR antibiotic samples (n = 79) / abundance in HR antibiotic samples (n = 16), versus the significance of the abundance difference. Metabolic pathways that have both p-value < 0.05 and absolute log2 fold change > 1 are shown in red. (B) Bar-plots indicating the fold-change of the top features revealed by the differential abundance analyses described in (A). (C and D) Differential abundance analyses for species and metabolic pathway composition comparing the German (n = 50 patients) and the US (n = 45 patients) cohort. Here, comparisons of fecal metagenomes for samples collected in the 3-weeks pre-CAR-T cell infusion window are shown. Group comparisons carried out by Mann-Whitney tests.
Extended Data Figure 7. Analyzing microbiome features associated with major outcomes of CAR-T cell therapy.
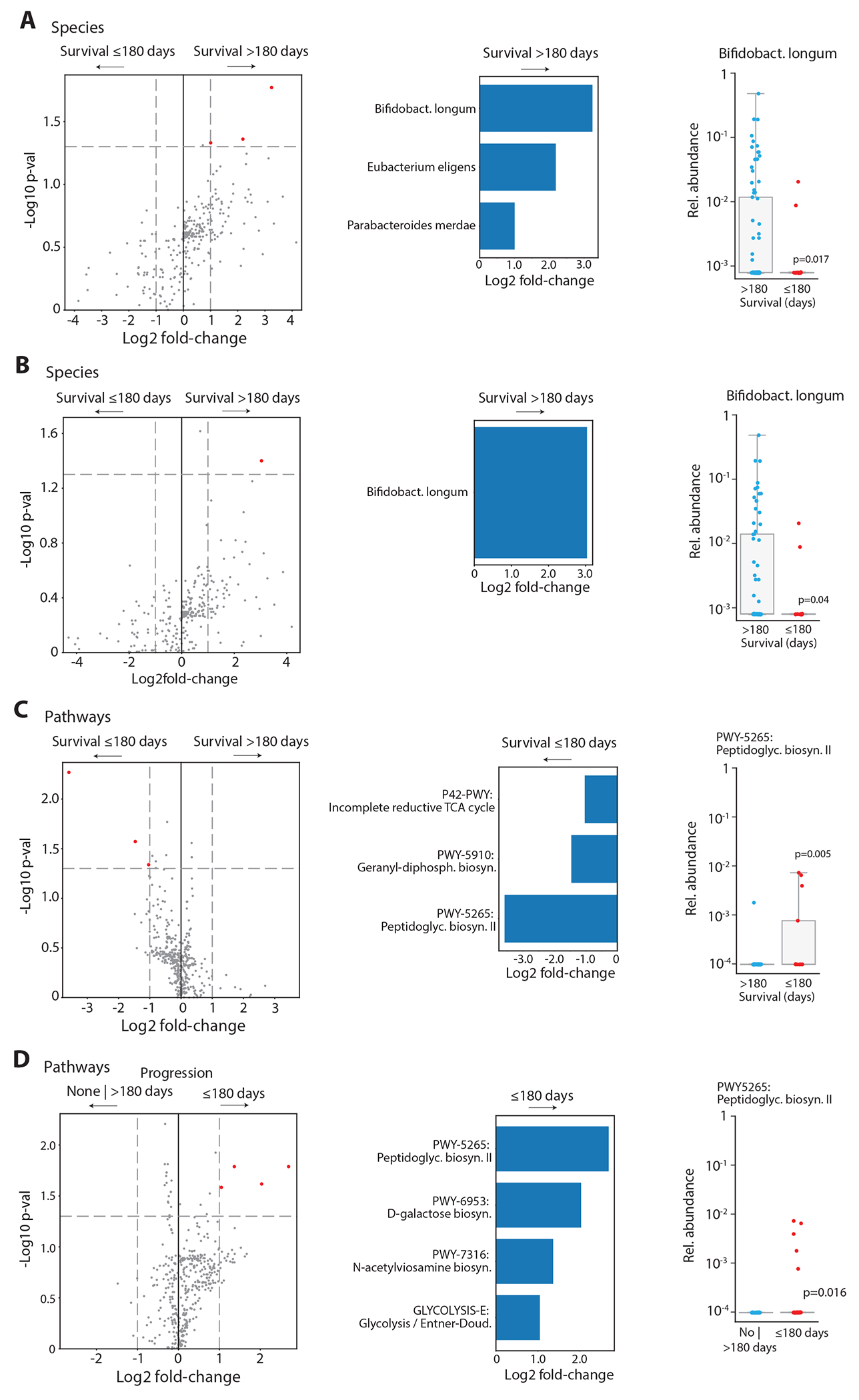
Differential abundance analyses were done as described in Extended Data Figure 7 with different grouping variables. (A) Left, volcano plots showing species and metabolic pathway compositions of the abundance in patients with survival > 180 day (n = 77) / abundance in patients with survival ≤ 180 day (n= 18) versus the significance of the abundance difference by Mann–Whitney tests. All samples collected in the pre-infusion period were included here. Features with a p-value < 0.05 and absolute log2 change > 1 are shown in red. Middle, bar-plots indicating the fold-change of the top species found in (A). Right, comparison of the relative abundances of the indicated species between patients with survival shorter or longer than 180 days. (B) As in (A), but samples collected during and less than two weeks after exposure to high-risk antibiotics were excluded (alive: n = 66; dead: n = 13). (C and D) As in (B), for metabolic pathways encoded in fecal metagenomes comparing 6-months survival (alive vs. dead at day 180) and early progression (≤ 180 days: progression within 6 months after infusion vs. no progression within follow-up or progression after day 180; n = 37 vs. n = 42). Group comparisons were computed by Mann-Whitney tests.
Extended Data Figure 8. Summary of differentially abundant microbiome features by clinical variables and outcomes including all specimens collected in the 3-weeks pre-infusion period.
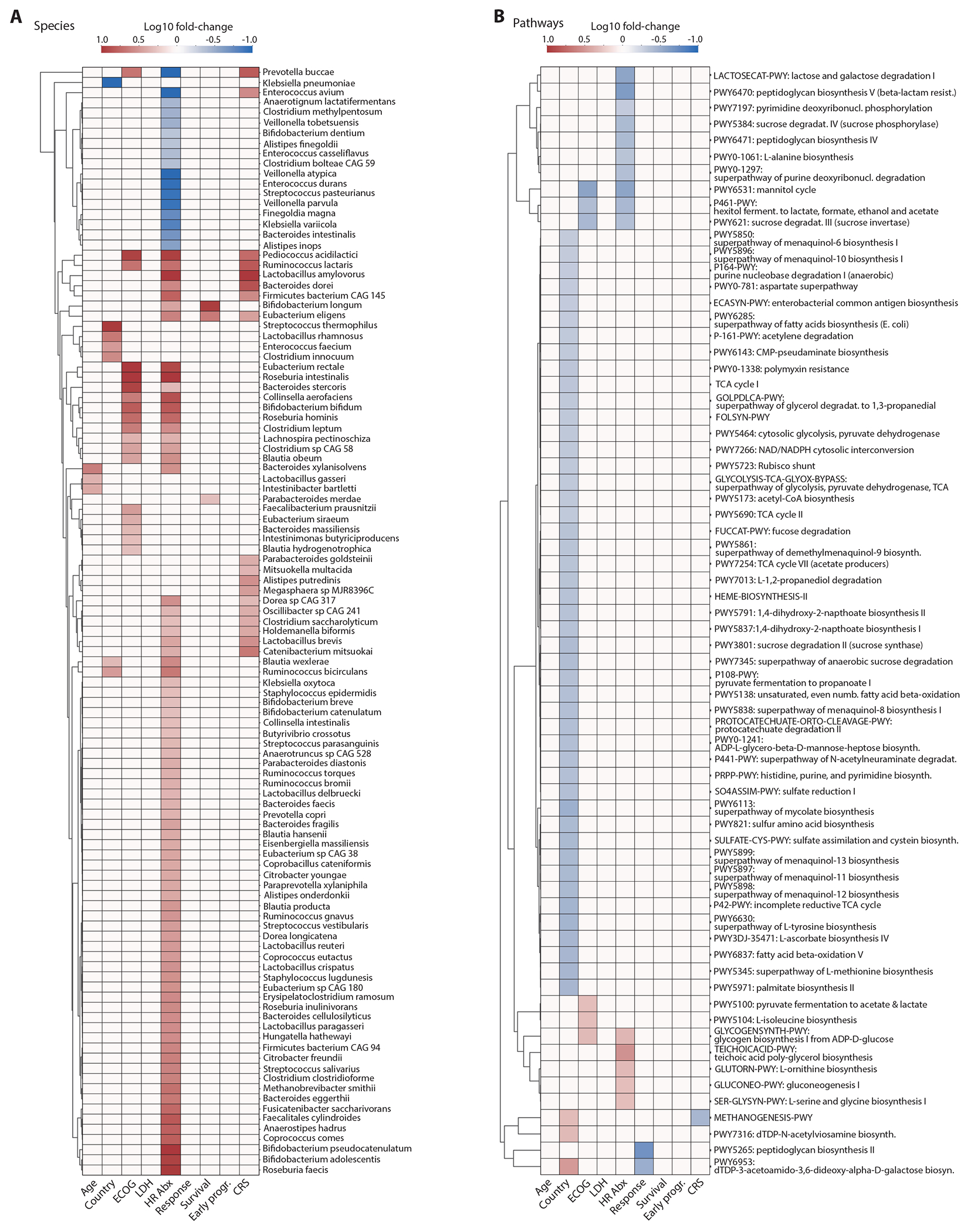
Heatmaps showing the associations between the species (A) and metabolic pathways (B) of the baseline gut microbiome and clinical parameters and major outcomes (Mann–Whitney tests, p-value < 0.05). Color indicates the Log10 of the following ratios: age (>= 65 / < 65 years), country (Germany / US), ECOG (grades 0-1 / 2-3), LDH (LDH at lymphodepletion chemotherapy: >= 280 / < 280 U/ml), HR antibiotic exposures (no / yes), response (CR / no CR at day 180), survival (alive / dead at day 180), early progression (lymphoma progression until day 180 / progression > 180 days or no progression during follow-up) and CRS (grades <=1 / >1). Associations with p-value > 0.05 or with absolute log2 fold change < 1 were set on the heatmap to have a log10 fold change of zero.
Extended Data Figure 9. Summary of differentially abundant microbiome features by clinical variables and outcomes excluding baseline specimens collected while or within 2 weeks after exposure to high-risk antibiotics.
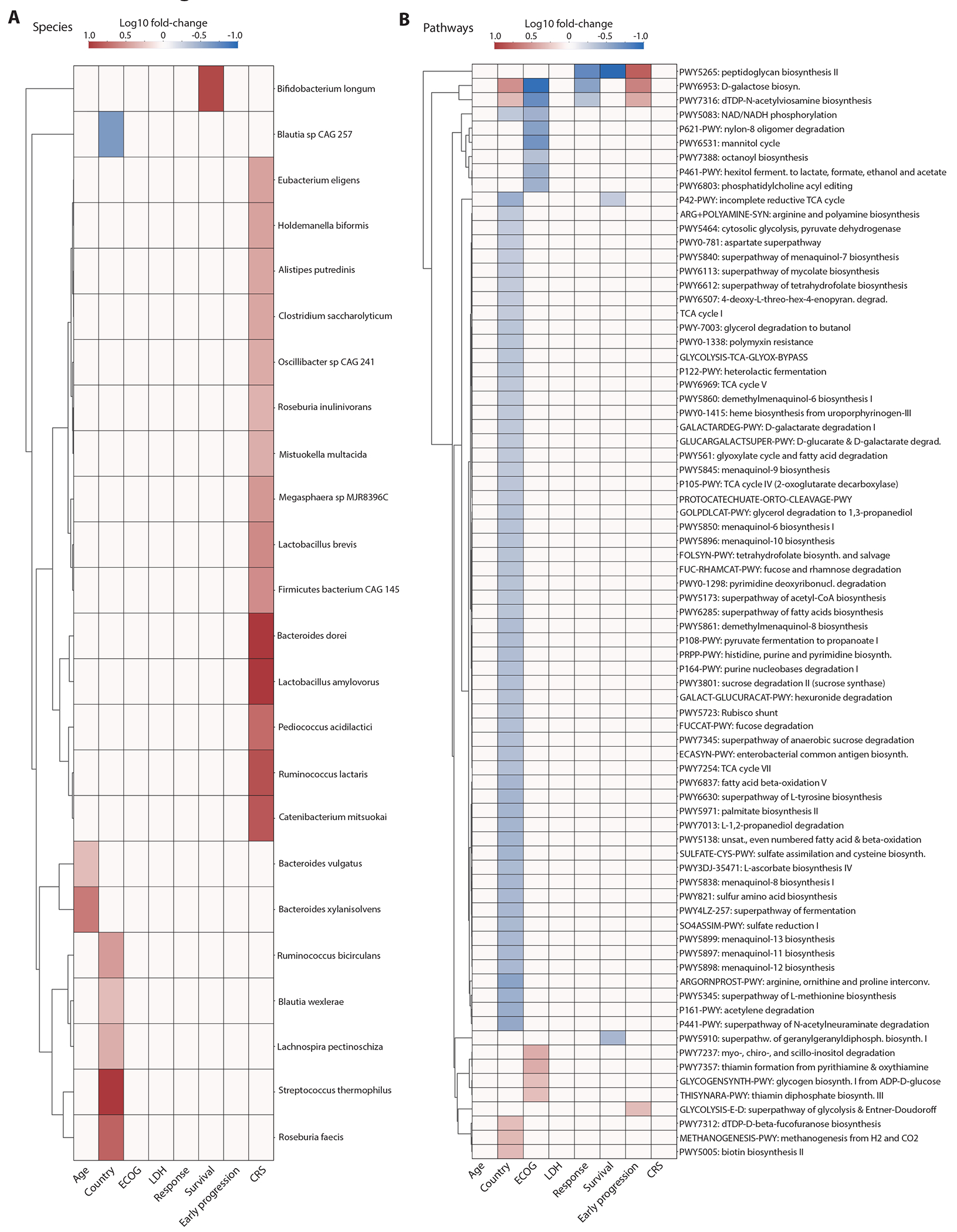
Heatmaps showing the associations between the species (A) and metabolic pathways (B) of the baseline gut microbiome and clinical parameters and major outcomes (Mann–Whitney tests, p-value < 0.05). Color indicates the Log10 of the following ratios: age (>= 65 / < 65 years), country (Germany / US), ECOG (grades 0-1 / 2-3), LDH (LDH at lymphodepletion chemotherapy: >= 280 / < 280 U/ml), response (CR / no CR at day 180), survival (alive / dead at day 180), early progression (lymphoma progression until day 180 / progression > 180 days or no progression during follow-up) and CRS (grades <=1 / >1). Associations with p-value > 0.05 or with absolute log2 fold change < 1 were set on the heatmap to have a log10 fold change of zero.
Extended Data Figure 10. Interaction of antibiotic exposures, survival outcomes and response prediction modeling.
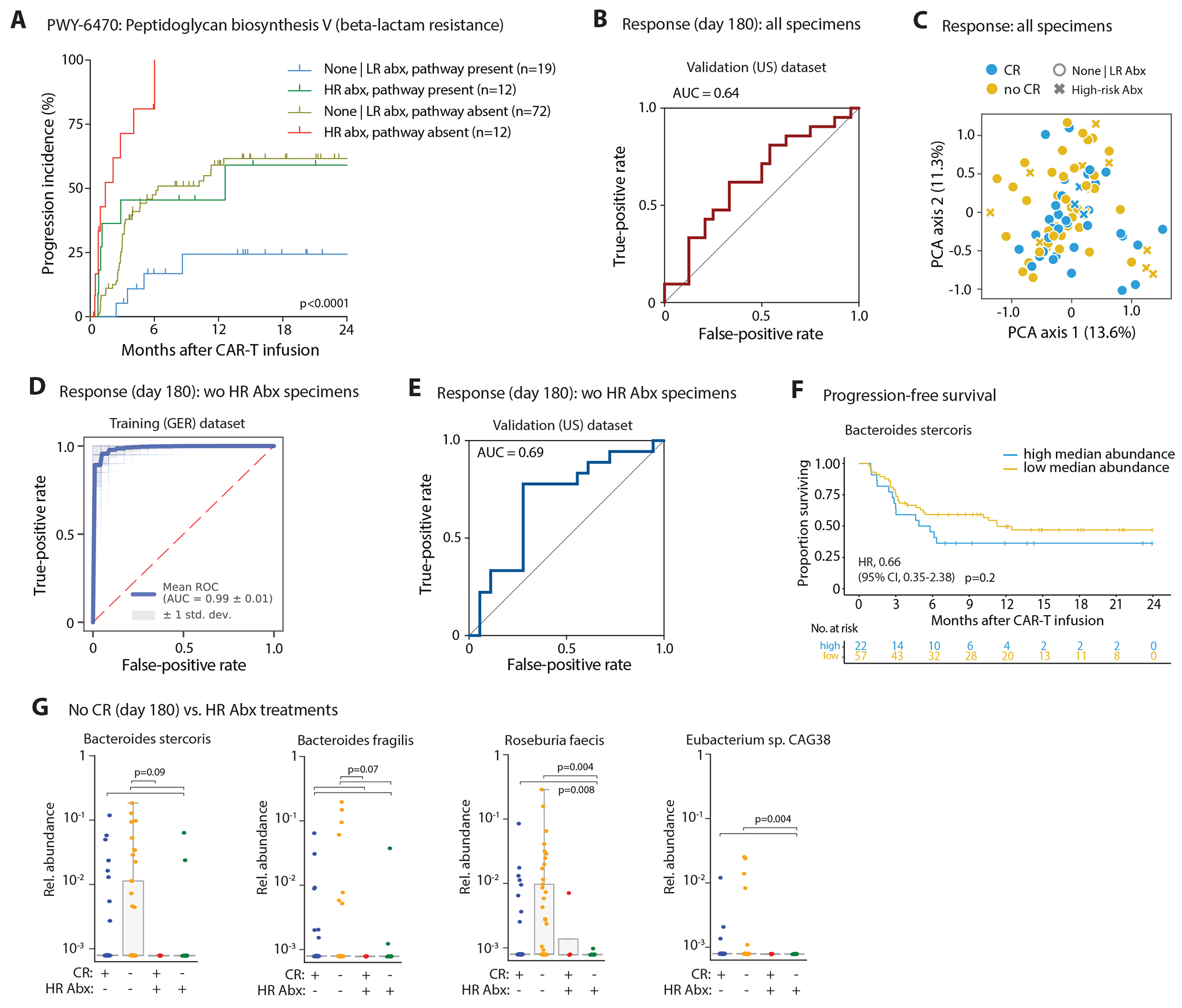
(A) Incidence curves for disease progression in patients were stratified according to high-risk antibiotics exposure in the 3-weeks pre-CAR-T cell infusion time window and according to presence of the peptidoglycan biosynthesis V - pathway in the patients’ stool metagenomes. (B) Deterministic logistic regression models were trained on clinical variables and pre-CAR-T cell infusion gut microbial species composition (mean per patient) of the German cohort and their performance was assessed on the US cohort (with all baseline samples: n = 45 patients; after excluding high-risk antibiotic samples: n = 36 patients). Area under the curve (AUC) receiver operating characteristic (ROC) curves indicating the false-positive and true-positive rates for predicting complete remission (CR) vs. non-CR at day 180 based on an analysis including all patient samples. (C) PCA computed for the whole dataset (i.e., training and validation data including high-risk antibiotic samples) based on the features selected during the training process of response at day 180 after CAR-T cell infusion. (D) AUROC curves indicating the false-positive and true-positive rates for predicting complete remission (CR) vs. non-CR at day 180 after training the probabilistic model on the German dataset that excluded high-risk antibiotics exposed samples. (E) AUROC curves indicating the false-positive and true-positive rates for predicting complete remission (CR) vs. non-CR at day 180 based on a deterministic model analysis that excluded samples collected on or less than two weeks after exposure to high-risk antibiotics; data displayed for the US validation cohort. (F) PFS curve of patients after CAR-T cell infusion stratified by the median baseline relative abundance of Bacteroides stercoris as top feature for non-CR as revealed in the machine learning model for CR; only pre-infusion samples without HR antibiotic exposure were included. (G) Differences in relative abundances for the four most important species predicting non-response (no CR) grouped by response and high-risk antibiotic exposures; group comparisons carried out by pairwise Mann-Whitney tests with FDR correction of p-values (n = 38 / 41 / 4 / 12 patients [from left to right]); FDR corrected p-values for B. stercoris are 0.089 (for all three comparisons), for B. fragilis are 0.0725 (for all four comparisons), and for E. sp. CAG38 are 0.0037 (for both comparisons). Statistics in (A) and (F) performed by logrank tests.
ACKNOWLEDGMENTS
We thank the members of the DKFZ Microbiome & Cancer Division and Elinav lab, Weizmann Institute of Science for insightful discussions; participants, investigators, collaborators, and study site personnel engaging in the trials. The authors thank the REG allo-SCT/CAR team, especially Heike Bremm, Tatjana Schifferstein and Yvonne Schumann for their help in collecting and cryopreserving stool samples, and Sigrun Gleich for data management. We are grateful for our partnership with the Mark Foundation (Endeavor Award 2021 to C.S-T., E.E., R.J., M.J., M.D.), who funded key aspects of this study. C.S-T. is supported by the German José Carreras Leukemia Foundation (01 R/2020), the German Research Foundation (DFG) (STE 2964/5-1), the Baden-Württemberg Stiftung, the DFG Excellence Cluster EXC-2124 (Controlling Microbes to Fight Infections, CMFI) and NCT Heidelberg Funds against Cancer. V.B. is supported by the Else-Kröner Fresenius-Stiftung and the German Cancer Consortium (DKTK) and the Bavarian Cancer for Cancer Research (BZKF). M.-L.S. is supported by the Olympia Morata Program of the University of Heidelberg (OM 09/2019). H.P. is supported by the DFG (Projektnummer 360372040 [SFB1335], 395357507 [SFB1371], 324392634 [TRR221]), the German Cancer Aid (70114547), the Wilhelm Sander Foundation (2021.040.1); H.P. is supported by the EMBO Young Investigator Program. This work was supported in part by Cancer Center Support Grant (P30CA016672) from the National Cancer Institute and the Microbiome Core Facility at MDACC. This work was supported in part by Cancer Center Support Grant (P30 CA076292) from the National Cancer Institute to Moffitt Cancer Center and the Moffitt Flow Cytometry Core Facility. E.E. is supported by the European Research Council, Israel Science Foundation, Israel Ministry of Science and Technology, Israel Ministry of Health, Helmholtz Foundation, Garvan Institute of Medical Research, European Crohn’s and Colitis Organization, Deutsch-Israelische Projektkooperation, IDSA Foundation and Wellcome Trust, Charlie Teo Foundation. E.E. is the incumbent of the Sir Marc and Lady Tania Feldmann Professorial Chair, a senior fellow of the Canadian Institute of Advanced Research (CIFAR) and an international scholar of the Bill & Melinda Gates Foundation and Howard Hughes Medical Institute (HHMI).
DECLARATION OF INTERESTS
V.B. received research funding from Bristol Myers-Sqibb (BMS)/Celgene, Gilead, Janssen, Novartis, Roche and Takeda; V.B. received honoraria from Gilead, Janssen and Novartis. M.-L.S. is a consultant for Novartis, Gilead and Janssen. H.P. is a consultant for Gilead, Abbvie, Pfizer, Novartis, Servier, and BMS. H.P. received research funding from BMS. H.P. received honoraria from Novartis, Gilead, Abbvie, BMS, Servier and Janssen-Cilag. M.L.D. reports consultancy/advisory/honoraria for Kite/Gilead, Novartis, Atara, Precision Biosciences, Celyad, Bellicum, GSK, Adaptive Biotech, and Anixa Biosciences, and research funding from Kite/Gilead, Novartis, and Atara. FLL reports consultancy/advisory for Allogene, Amgen, Bluebird Bio, BMS/Celgene, Calibr, Cellular Biomedicine Group, Cowen, EcoR1, Emerging Therapy Solutions, GammaDelta Therapeutics, Gerson Lehrman Groupt, Iovance, Kite Pharma, Janssen, Legend Biotech, Novartis, Sana, Takeda, Wugen, and Umoja, and research funding from Kite/Gilead, Allogene, Novartis, BlueBird Bio, BMS, NCI, Leukemia and Lymphoma Society, and education or editorial activity for Aptitude Health, ASH, BioPharma Communications CARE Education, Clinical Care Options Oncology, Imedex, Society for Immunotherapy of Cancer. M.D.J. reports consultancy/advisory for Kite/Gilead, Novartis, BMS, MyeloidTx, and research funding from Incyte and Kite/Gilead. The remaining authors declare no competing interests. E.E. is a scientific cofounder of DayTwo and BiomX, and an advisor to Hello Inside, Igen, and Aposense in topics unrelated to this work.
Footnotes
Code Availability.
Code used in the present study is available under https://github.com/Elinav-Lab-DKFZ/CART-Microbiome and upon request.
Data Availability.
All data are available in the main text or the extended materials. The sequencing data generated during this study are available at European Nucleotide Archive (ENA) [project accession no. PRJEB54704] including basic phenotypes.
REFERENCES
- 1.Gopalakrishnan V et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103, doi: 10.1126/science.aan4236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Routy B et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97, doi: 10.1126/science.aan3706 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Matson V et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108, doi: 10.1126/science.aao3290 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peled JU et al. Intestinal Microbiota and Relapse After Hematopoietic-Cell Transplantation. J Clin Oncol 35, 1650–1659, doi: 10.1200/JCO.2016.70.3348 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stein-Thoeringer CK et al. Lactose drives Enterococcus expansion to promote graft-versus-host disease. Science 366, 1143–1149, doi: 10.1126/science.aax3760 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mager LF et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369, 1481–1489, doi: 10.1126/science.abc3421 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Davar D et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 371, 595–602, doi: 10.1126/science.abf3363 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baruch EN et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 371, 602–609, doi: 10.1126/science.abb5920 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Pinato DJ et al. Association of Prior Antibiotic Treatment With Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients With Cancer. JAMA Oncol, doi: 10.1001/jamaoncol.2019.2785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shono Y et al. Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patients and mice. Sci Transl Med 8, 339ra371, doi: 10.1126/scitranslmed.aaf2311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guedan S, Ruella M & June CH Emerging Cellular Therapies for Cancer. Annu Rev Immunol 37, 145–171, doi: 10.1146/annurev-immunol-042718-041407 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amini L et al. Preparing for CAR T cell therapy: patient selection, bridging therapies and lymphodepletion. Nat Rev Clin Oncol 19, 342–355, doi: 10.1038/s41571-022-00607-3 (2022). [DOI] [PubMed] [Google Scholar]
- 13.Park JH et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med 378, 449–459, doi: 10.1056/NEJMoa1709919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Locke FL et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol 20, 31–42, doi: 10.1016/S1470-2045(18)30864-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nastoupil LJ et al. Standard-of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results From the US Lymphoma CAR T Consortium. J Clin Oncol 38, 3119–3128, doi: 10.1200/JCO.19.02104 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schuster SJ et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 380, 45–56, doi: 10.1056/NEJMoa1804980 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Jain MD et al. Tumor interferon signaling and suppressive myeloid cells are associated with CAR T-cell failure in large B-cell lymphoma. Blood 137, 2621–2633, doi: 10.1182/blood.2020007445 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng Q et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat Med 26, 1878–1887, doi: 10.1038/s41591-020-1061-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plaks V et al. CD19 target evasion as a mechanism of relapse in large B-cell lymphoma treated with axicabtagene ciloleucel. Blood 138, 1081–1085, doi: 10.1182/blood.2021010930 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jain MD et al. Whole-genome sequencing reveals complex genomic features underlying anti-CD19 CAR T-cell treatment failures in lymphoma. Blood, doi: 10.1182/blood.2021015008 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dean EA et al. High metabolic tumor volume is associated with decreased efficacy of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv 4, 3268–3276, doi: 10.1182/bloodadvances.2020001900 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karschnia P et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood 133, 2212–2221, doi: 10.1182/blood-2018-12-893396 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Jacobson CA et al. Axicabtagene Ciloleucel in the Non-Trial Setting: Outcomes and Correlates of Response, Resistance, and Toxicity. J Clin Oncol 38, 3095–3106, doi: 10.1200/JCO.19.02103 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith M et al. Gut microbiome correlates of response and toxicity following anti-CD19 CAR T cell therapy. Nat Med 28, 713–723, doi: 10.1038/s41591-022-01702-9 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cullin N, Azevedo Antunes C, Straussman R, Stein-Thoeringer CK & Elinav E Microbiome and cancer. Cancer Cell 39, 1317–1341, doi: 10.1016/j.ccell.2021.08.006 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Galloway-Pena JR et al. Gut Microbiome Signatures Are Predictive of Infectious Risk Following Induction Therapy for Acute Myeloid Leukemia. Clin Infect Dis 71, 63–71, doi: 10.1093/cid/ciz777 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santomasso B, Bachier C, Westin J, Rezvani K & Shpall EJ The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am Soc Clin Oncol Educ Book 39, 433–444, doi: 10.1200/EDBK_238691 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Derosa L et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann Oncol 29, 1437–1444, doi: 10.1093/annonc/mdy103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bachy E et al. A real-world comparison of tisagenlecleucel and axicabtagene ciloleucel CAR T cells in relapsed or refractory diffuse large B cell lymphoma. Nat Med 28, 2145–2154, doi: 10.1038/s41591-022-01969-y (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finney OC et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest 129, 2123–2132, doi: 10.1172/JCI125423 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das RK, Vernau L, Grupp SA & Barrett DM Naive T-cell Deficits at Diagnosis and after Chemotherapy Impair Cell Therapy Potential in Pediatric Cancers. Cancer Discov 9, 492–499, doi: 10.1158/2159-8290.CD-18-1314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gacesa R et al. Environmental factors shaping the gut microbiome in a Dutch population. Nature 604, 732–739, doi: 10.1038/s41586-022-04567-7 (2022). [DOI] [PubMed] [Google Scholar]
- 33.Schluter J et al. The gut microbiota is associated with immune cell dynamics in humans. Nature 588, 303–307, doi: 10.1038/s41586-020-2971-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wastyk HC et al. Gut-microbiota-targeted diets modulate human immune status. Cell 184, 4137–4153 e4114, doi: 10.1016/j.cell.2021.06.019 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castello A, Rossi S, Toschi L & Lopci E Impact of Antibiotic Therapy and Metabolic Parameters in Non-Small Cell Lung Cancer Patients Receiving Checkpoint Inhibitors. J Clin Med 10, doi: 10.3390/jcm10061251 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee KA et al. Cross-cohort gut microbiome associations with immune checkpoint inhibitor response in advanced melanoma. Nat Med 28, 535–544, doi: 10.1038/s41591-022-01695-5 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cammarota G et al. Gut microbiome, big data and machine learning to promote precision medicine for cancer. Nat Rev Gastroenterol Hepatol 17, 635–648, doi: 10.1038/s41575-020-0327-3 (2020). [DOI] [PubMed] [Google Scholar]
- 38.McCulloch JA et al. Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1. Nat Med 28, 545–556, doi: 10.1038/s41591-022-01698-2 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dizman N et al. Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carcinoma: a randomized phase 1 trial. Nat Med 28, 704–712, doi: 10.1038/s41591-022-01694-6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Derrien M, Vaughan EE, Plugge CM & de Vos WM Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol 54, 1469–1476, doi: 10.1099/ijs.0.02873-0 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Luu M et al. Microbial short-chain fatty acids modulate CD8(+) T cell responses and improve adoptive immunotherapy for cancer. Nat Commun 12, 4077, doi: 10.1038/s41467-021-24331-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bae M et al. Akkermansia muciniphila phospholipid induces homeostatic immune responses. Nature 608, 168–173, doi: 10.1038/s41586-022-04985-7 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gamage H et al. Third generation cephalosporins and piperacillin/tazobactam have distinct impacts on the microbiota of critically ill patients. Sci Rep 11, 7252, doi: 10.1038/s41598-021-85946-4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS-ONLY REFERENCES:
- 44.Lee DW et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant 25, 625–638, doi: 10.1016/j.bbmt.2018.12.758 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Hogue SR, Gomez MF, da Silva WV & Pierce CM A Customized At-Home Stool Collection Protocol for Use in Microbiome Studies Conducted in Cancer Patient Populations. Microb Ecol 78, 1030–1034, doi: 10.1007/s00248-019-01346-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beghini F et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 10, doi: 10.7554/eLife.65088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hastie T TR, Friedman J The Elements of Statistical Learning: Data Mining, Inference, and Prediction. Second edn, 767 (Springer, 2009). [Google Scholar]
- 48.McElreath R A Bayesian Course with Examples in R and STAN. 2nd edn, 612 (Chapman & Hall, 2020). [Google Scholar]
- 49.Bingham E et al. Pyro: Deep Universal Probabilistic Programming. J Mach Learn Res 20 (2019). [Google Scholar]
- 50.Wingate D & Weber T Automated variational inference in probabilistic programming. arXiv 1301.1299, 1–7 (2013). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available in the main text or the extended materials. The sequencing data generated during this study are available at European Nucleotide Archive (ENA) [project accession no. PRJEB54704] including basic phenotypes.