

Abstract
Endothelial activation and sickle red blood cell (RBC) adhesion are central to the pathogenesis of sickle cell disease (SCD). Quantitatively, RBC-derived extracellular vesicles, REVs, are more abundant from SS RBCs compared with healthy RBCs (AA RBCs). Sickle RBC-derived REVs (SS REVs) are known to promote endothelial cell (EC) activation through cell signaling and transcriptional regulation at longer terms. However, the SS REV-mediated short term non transcriptional response of EC is unclear. Here, we examined the impact of SS REVs on acute microvascular EC activation and RBC adhesion at 2 hours. Compared with AA REVs, SS REVs promoted human pulmonary microvascular endothelial cells (HPMEC) activation indicated by increased von Willebrand Factor (vWF) expression. Under microfluidic conditions, we found abnormal SS RBC adhesion to HPMECs exposed to SS REVs. This enhanced SS RBC adhesion was reduced by heme binding protein hemopexin or vWF cleaving protease ADAMTS13 to a level similar to HPMECs treated with AA REVs. Consistent with these observations, hemin- or SS REV-induced microvascular stasis in SS mice with implanted dorsal skin-fold chambers that was inhibited by ADAMTS13. The adhesion induced by SS REVs was variable, and was higher with SS RBCs from patients with increased markers of hemolysis (LDH and reticulocyte count) or a concomitant clinical diagnosis of deep vein thrombosis. Our results emphasize the critical contribution made by REVs to the pathophysiology of SCD by triggering acute microvascular EC activation and abnormal RBC adhesion. These findings may help to better understand acute pathophysiological mechanism of SCD and thereby the development of new treatment strategies using vWF as a potential target.
1. INTRODUCTION
Sickle cell disease (SCD) is a genetically inherited blood disorder, in which a point mutation in the beta globin chain gene replaces A with T at codon 6. This causes a switch from a hydrophilic glutamic acid to a hydrophobic valine, producing abnormal sickle hemoglobin (HbS). HbS polymerizes into long and stiff intracellular structures under dehydration and deoxygenation, leading to sickle-shaped, excessively stiff, and adhesive red blood cells (RBCs). Sickle RBCs (SS RBCs) are prone to extra- and intravascular hemolysis [1]. During hemolysis, SS RBCs release damage-associated molecular patterns (DAMPs) including hemoglobin/ heme and extracellular vesicles (EVs) [2–6]. The DAMPs cumulatively promote a pro-inflammatory milieu with decreased in levels of natural occurring anticoagulant proteins (i.e., protein C and protein S), nitric oxide (NO) bioavailability, and ADAMTS13, and with increased levels of von Willebrand factor (vWF) [7]. The decreased of levels of natural anticoagulant proteins may promote endothelial damage [8, 9], the decreased NO bioavailability decreases its effect in impairing blood-endothelial cell adhesion [10], and the increased levels of vWF favors RBC adhesion on endothelium [11]. Consequently, these DAMP associated endothelial damages intermittent vaso-occlusion of the microvasculature in SCD [2, 12–14].
EVs are membrane-bound sub-micron particles (0.1–1.0 μm) generated from cells under normal and activated conditions with a broad spectrum of size, density, biochemical composition, and cellular origins [15, 16]. EVs contain various cargos including proteins, lipids, and nucleic acids, which may reflect the state of activation of the cells from which they originate [17] and can serve as vehicles for cellular communication [15]. EV biogenesis were associated with both ESCRT-dependent and independent pathways. Specifically, RBC-derived EVs (REVs) biogenesis involves alterations in RBC membrane proteins such as band 3 [6] and sphingomyelin [16, 18]. The level of total circulating EVs (including EVs from platelets, leukocytes, endothelial cells, and RBCs) have been reported to be higher in the bloodstream of subjects with SCD at steady state than in healthy controls and increased during SCD vaso-occlusive crises [19, 20]. REVs made up the most prevalent subtype, and correlated with hemolysis, oxygen saturation, pulmonary systolic pressure, and mortality [21]. REVs in SCD (SS REVs) may be produced through accelerated aging of RBCs during oxygenation/deoxygenation cycles [16], under the influence of stress factors [4, 22–24], or during hemolysis in capillary beds. SS REVs are reported to have increased phosphatidylserine (PS) expression on their surface [12], as well as to contain heme, Hb, oxidized Hb, ferryl Hb, and microRNAs [3, 25, 26]. EVs in SCD are known to have a range of pathophysiologic impacts on blood-vascular interactions including nitric oxide scavenging [10], immune modulation [18], coagulation [27], decrease endothelial monolayer integrity [28–30], and adhesion of blood cells to the endothelium [5, 28]. Garnier et al. showed plasma EV-treated ECs upregulated mRNA and protein expression of intercellular adhesion molecule-1 (ICAM-1) at longer terms (4 or 6 hours), which led to significantly enhanced neutrophil recruitment to human endothelium in SCD [5]. Gemel and Lapping-Carr et al. reported that plasma EVs isolated from subjects with SCD during acute chest syndrome significantly decreased levels of endothelial VE-cadherin protein and disrupted junctions at 48 hours [29, 30]. Although these works mainly utilized plasma EVs, the roles of REVs within the plasma EVs were emphasized. Camus et al. demonstrated that in transgenic SAD mice, SS REVs are capable of transferring heme to endothelial cells (ECs), triggering microvascular-occlusions, likely through toll-like receptor-4 (TLR-4) dependent pathways [3]. Importantly, ~45% of the cell-free heme in plasma is bound to REVs [3] therefore endothelial activation via cell-free heme is likely mediated by the transfer of heme through REVs [3]. However, most of the existing work indicate SS REVs promote endothelial activation at longer terms (>4 hours) through transcriptional responses, while the acute (≤2 hours) impact of REVs on endothelial activation has not been fully elucidated.
In this study, we utilized our previously developed endothelium-on-a-chip platform [31] to analyze RBC adhesion to REV-activated human pulmonary microvascular endothelial cells (HPMECs) under physiologic flow conditions. SS REVs were generated by increasing the intracellular calcium concentration in SS RBCs. Following short-term (2 hours) REV activation of microvascular ECs, which mimics the proinflammatory phenotype of microvascular endothelium during acute hemolysis in SCD, we analyzed RBC adhesion to the HPMECs using clinical blood samples from subjects with homogeneous SCD followed in our clinic. Here, we report SS RBC adhesion on REV-activated HPMECs and its clinical associations in SCD. The aim of the present study is to demonstrate the acute impact of REVs generated from SS RBCs on microvascular EC phenotype, focusing on abnormal RBC adhesion.
2. MATERIALS AND METHODS
2.1. Blood sample collection
All experiments were performed in accordance with approved study protocol by the Institutional Review Board (IRB) committee (IRB 05-14-07C). Blood samples from de-identified adult subjects with HbSS were collected in EDTA-containing vacutainer tubes at the outpatient clinic at University Hospitals Cleveland Medical Center (UHCMC) in Cleveland, Ohio. Informed consent was obtained from all study participants, and all blood samples were collected from participants with HbSS at clinical baseline (steady-state). Baseline was defined as steady state for at least two weeks before blood collection (no crises as identified by day hospital or emergency department visit, or hospital admission for pain). Healthy donor (HbAA) blood samples were collected in EDTA-containing vacutainer tubes from biorepository at Case Western Reserve University. There were no known medical issues with any of the healthy donors in this study. Hb composition of SCD samples was identified by high-performance liquid chromatography (HPLC) at the Core Laboratory of UHCMC. Clinical variables of the study subjects with SCD were obtained from the medical record.
2.2. Microfluidic channel endothelialization
Microfluidic channels were fabricated as previously described [34–38]. Briefly, the microchannels were fabricated by assembling three layers, including a glass slide as bottom layer, double-sided adhesive (DSA) film as middle layer, and polymethyl methacrylate (PMMA) as top layer. Microchannel geometry was determined by the middle DSA layer and channel inlet and outlet was determined on the top PMMA layer (Supplemental Fig. 1). The microchannels were incubated with 0.2 mg/mL fibronectin for 1 hour at 37°C. HPMECs were seeded into the microchannels and cultured with 5% CO2 at 37°C under 100 μL/min flow for 48–72 hours until a confluent monolayer over the microchannel bottom surface was formed (Supplemental Fig. 1).
2.3. Red blood cell derived extracellular vesicle (REV) generation and characterization
Pooled blood samples from 10 subjects with HbSS and 5 healthy donors were used to derive HbSS REV and HbAA REV, respectively. RBCs were isolated from either individual or pooled blood samples by centrifuging for 5 minutes at 500×g at room temperature. Plasma, buffy coat, and the near-plasma portion of the RBC layer were carefully removed. The isolated RBCs were washed twice with PBS, and were re-suspended in Hank’s balanced salt solution modified with calcium and magnesium (Hank’s buffer) at 20% hematocrit. The RBC suspensions were then stimulated with 2 μM calcium ionophore (A23187) for 1 hour at 37°C. To isolate generated REVs, cell suspensions were subject to differential centrifugation, initially at 1,500 ×g for 15 min followed by the second-step centrifugation at 3000×g for 15 min. Finally, the supernatants were centrifuged at 25,000×g for 2 hours at 4°C to harvest REV pellets. REVs were re-suspended in either cell culture media (for endothelial cell activation) or PBS (for REV characterization), split into 20 μL aliquots and stored in −80°C before using. The size distribution and concentration of the generated REVs were measured using ZetaView® particle tracking analyzer (Particle-Metrix, Germany). For measuring heme content, REVs were lysed using 0.5% Triton x-100 in deionized water. The lysed solutions were then characterized using Heme Assay Kit (Sigma-Aldrich, US). Equal total number of SS REVs and AA REVs were used for heme content measurement.
2.4. REV endothelial cell activation
For all endothelial activation experiments without hemopexin, stock REVs were thawed at 4 °C and brought to 37 °C in the cell culture incubator. To test the impact of hemopexin on REV-mediated HPMEC activation, REVs were incubated with 2 μM human hemopexin (Sigma-Aldrich) for 1 hour at room temperature. For all experiments, cultured HPMECs (LONZA) were washed with fresh culture medium and were then incubated with HbSS REVs or HbAA REVs for 2 hours at 37°C. The activated HPMECs were then washed with fresh culture medium and used for adhesion experiments. In control experiments, SS REVs and AA REVs were reconstituted to the same concentration.
2.5. Heme endothelial cell activation
Heme solution was used to activation endothelial cells in order to compare the activation effect of REVs and purified heme. Briefly, heme stock solution was prepared by dissolving bovine hemin in 0.1 M NaOH solution to obtain a final heme concentration of 40 mM. Next, the stock solution was diluted using RPMI-1640 (FBS and antibioticsfree) to the final working concentrations equivalent to the heme content in REVs determined using the protocol described in Section 2.3.
2.6. Fluorescent labeling of von Willebrand factor (vWF) on endothelial cells
Following REV activation, HPMECs were rinsed with fresh culture medium and fixed with 4% paraformaldehyde (PFA) for 15 minutes at room temperature. Fixed HPMECs were rinsed with PBS twice and blocked with 2% BSA for 1-hour at room temperature. After washing with PBS, HPMECs were incubated with sheep polyclonal anti-human vWF antibody (Abcam) conjugated with fluorescein isothiocyanate (FITC, 1:100 v/v dilution) for 1 hour at room temperature in dark. Fluorescent images were then acquired at multiple locations throughout the microchannel of REV activated HPMECs at 10X.
2.7. Adhesion Experiments
For all RBC adhesion experiments without VWF cleaving protease ADAMTS13, RBCs were isolated from individual blood samples by centrifugation for 5 minutes at 500×g at room temperature. The isolated RBCs were washed twice with PBS and were re-suspended in fresh basal medium supplemented with 10 mM HEPES at 20% hematocrit. To test the impact of ADAMTS13 on RBC adhesion, freshly isolated RBCs in basal medium supplemented with 10 mM HEPES at 20% hematocrit were incubated for 30 minutes under 37 °C with gentle shaking with 5 μg/mL ADAMTS13 (R&D Systems), and this RBC suspension in HEPES-ADAMTS13 was used for adhesion experiment [32, 33].
For all RBC adhesion experiment, a total sample volume of 15 μL was perfused into the microchannel at the shear rate of 1 dyne/cm2, corresponding to a typical value observed in human post-capillary venules. After blood perfusion, the microchannels were rinsed with fresh basal medium supplemented with 10 mM HEPES to remove non-adherent RBCs. Visual confirmation of adhesion results confirmed negligible contamination by platelets or white blood cells.
2.8. Mice
All animal experiments were approved by the University of Minnesota’s Institutional Animal Care and Use Committee. In this study, we utilized male and female HbSS-Townes on a 129/B6 mixed genetic background. The SS mice were created by knocking in human α and AγβS globins into the sites where murine α-globin and β-globins were knocked out [34]. SS mice have severe anemia and an SS RBC half-life of 2.5 days (normal mice half-life span ~22.6 days) [34]. All animals were monitored daily including weekends and holidays for health problems, food and water levels and cage conditions. Littermates were randomly assigned to different treatment groups. All animals were included in each endpoint analysis and there were no unexpected adverse events that required modification of the protocol. Mice were aged 11–14 weeks.
2.9. Statistical Methods
Mean ± standard deviation (SD) of the mean were reported for acquired data in this study. Minitab 18 Software (Minitab Inc., State College, PA) was used to perform all statistical analyses. Data normality was initially analyzed. For comparison, normally distributed data were analyzed by 2 groups t test. Statistical significance was set at 95% confidence level for all tests (P < 0.05).
3. RESULTS
3.1. Characterization of red blood cell derived extracellular vesicles (REVs)
We characterized and compared calcium ionophore-derived AA REVs, generated from the pooled blood samples from 5 healthy donors, and calcium ionophore-derived SS REVs, generated from the pooled blood samples from 5 people with SCD (Fig. 1). The nanoparticle tracking analysis (NTA) results demonstrated that derived AA and SS REVs had similar diameters (10 to 700 nm with >95% of them between 80–400 nm, Fig. 1A&B, mean size ± SD: AA REV = 205.4 ± 223.7 nm vs. SS REV = 181.7 ± 200.2 nm). However, per RBC volume, SS RBCs generated significantly more REVs than did AA RBCs (Fig. 1C, mean concentration ± SD: SS REV = 3.17 × 107 ± 3.91× 106/mL vs. AA REV = 6.35 × 106 ± 5.68× 105/mL, p = 0.011, 2-Sample t test). Heme Assay Kit determined that the generated SS REVs contain significantly higher concentration of heme than did same number of AA REVs (Fig. 2D, mean concentration ± SD: SS REV heme = 17.3 ± 1.1 μM vs. AA REV heme = 6.2 ± 0.3 μM).
Figure 1. SS RBCs generate increased level of REVs than AA RBCs and SS REVs carry higher concentration of heme than AA REVs. (A&B).
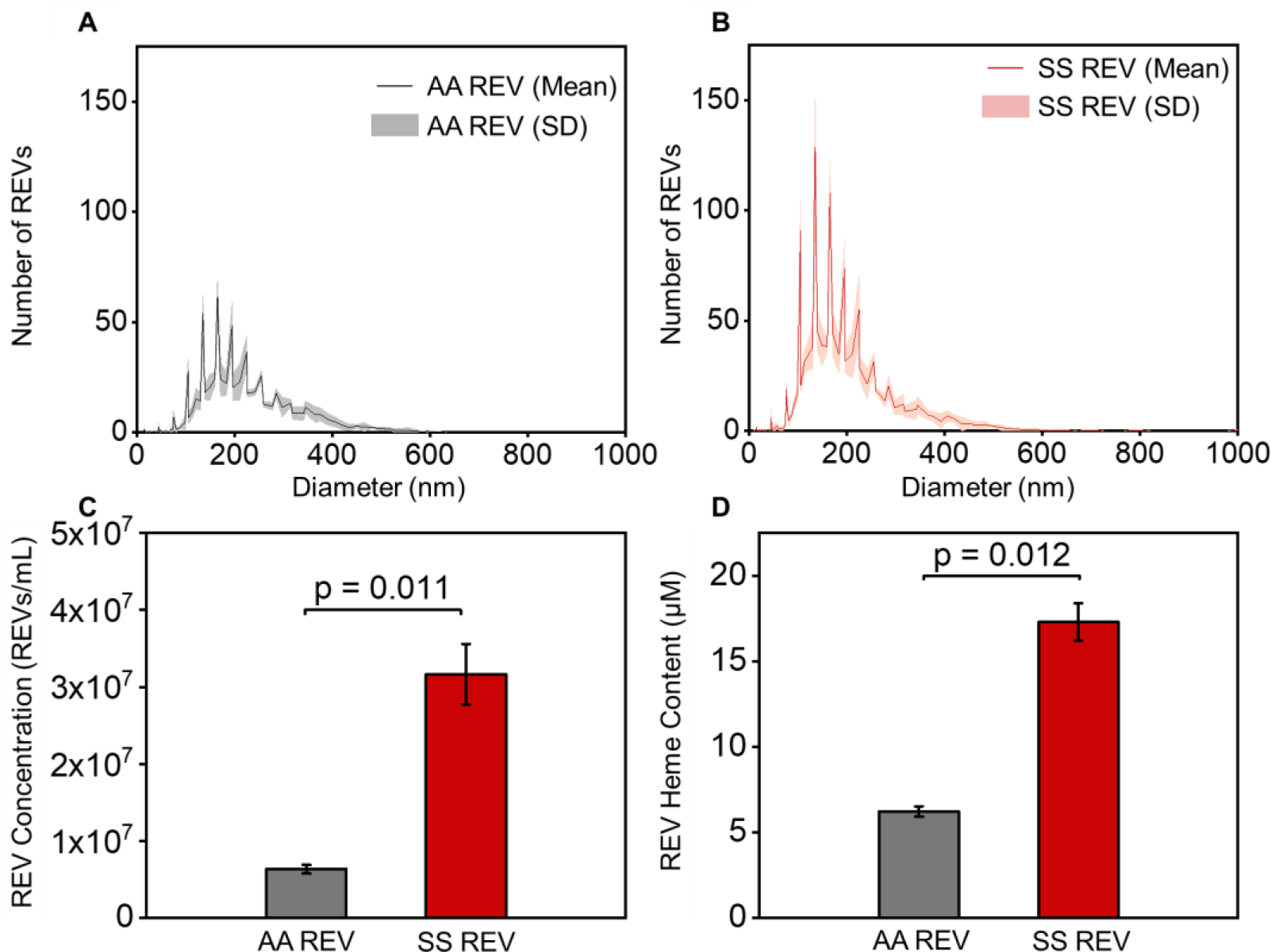
Size distribution of AA REVs and SS REVs generated from RBCs in pooled-samples from healthy donors or pooled-samples from patients with SCD. Black and red lines indicate the mean number of AA and SS REVs from 5 individual tests and gray and light-red shaded area indicate the standard deviation. AA REV and SS REV share similar size distribution (Mean size ± SD: AA REV = 205.4 ± 223.7 nm vs. SS REV = 181.7 ± 200.2 nm). (C) Concentration of AA REV and SS REV generated in vitro per microliter of purified RBCs in samples from either healthy donors (gray) and patients with SCD (red). SS RBCs generated statistically significantly higher quantity of REVs than AA RBCs (Mean concentration ± SD: SS REV = 3.17 × 107 ± 3.91× 106/mL vs. AA REV = 6.35 × 106 ± 5.68× 105/mL, p = 0.011, 2-Group t test). (D) Heme concentration carried by equal number of AA REVs (gray) and SS REVs (red). SS REVs carried statistically significantly higher concentration of heme than AA REVs (Mean concentration ± SD: SS REV = 17.3 ± 1.1 μM vs. AA REV = 6.2 ± 0.3 μM, p = 0.012, 2-Group t test).
Figure 2. SS REVs activate HPMECs, increase vWF expression and mediates enhanced adhesion of SS RBCs, which can be reduced by hemopexin and ADAMTS13.
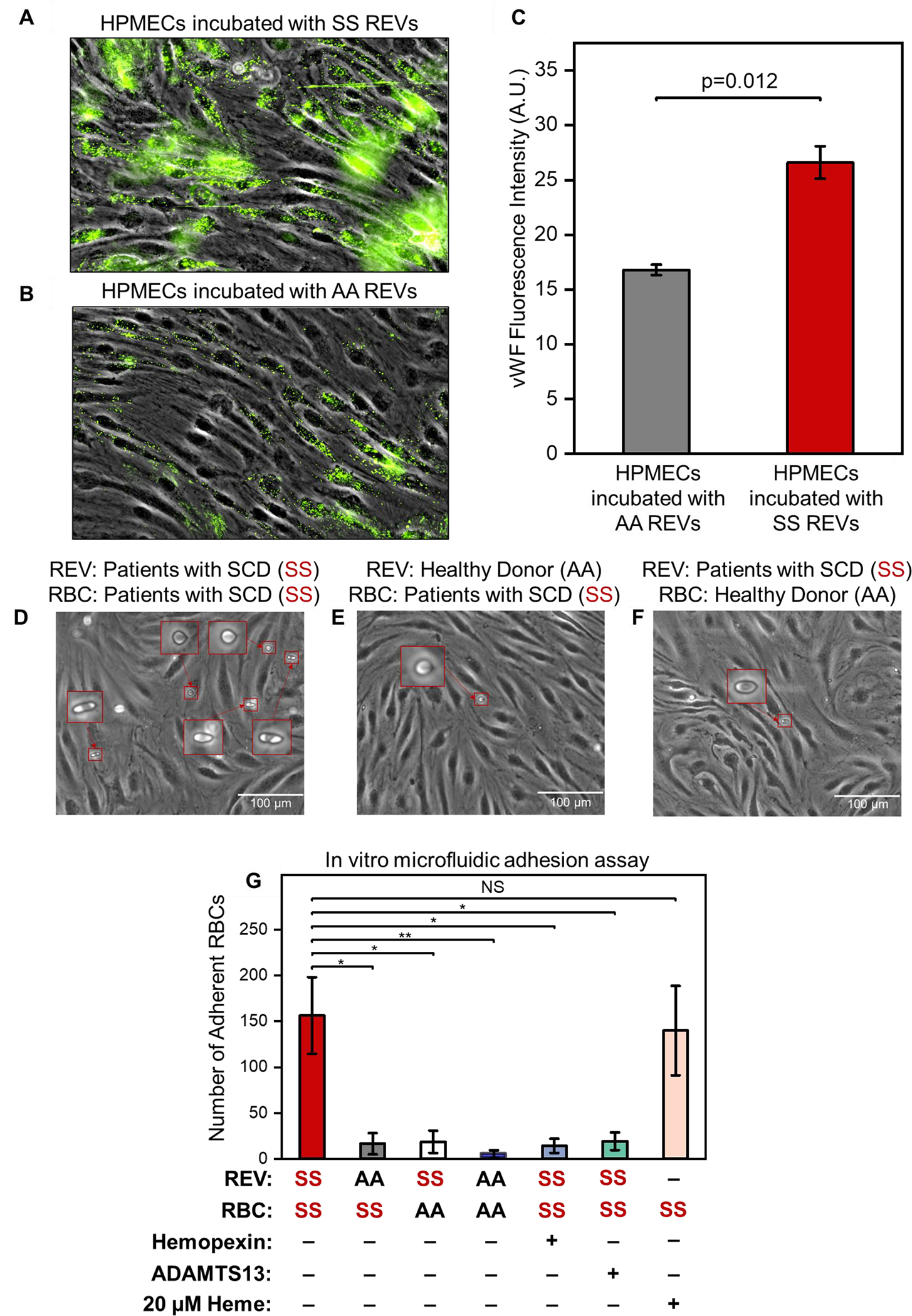
(A, B): HPMECs were treated with SS REVs (A) and AA REVs (B) for 2 hours in 37 °C and incubated with fluorescently labeled antibodies against vWF following a fixing step with 4% PFA. The SS REV treated HPMECs demonstrated significantly increased vWF expression comparing to HPMECs incubated with AA REVs (C, n=5, p=0.012, 2 Sample t test). (D) SS RBC adhesion on HPMECs activated with SS REVs. (E) SS RBCs adhesion on HPMECs treated with AA REVs. (F) AA RBCs adhesion on HPMECs treated with SS REVs. Insets are closer view of RBCs adhered to HPMECs. (G) Interaction between SS RBC and SS REV activated HPMECs is significantly stronger than the other test groups, and this interaction is reduced by hemopexin and ADAMTS13 (mean ± SD: SS REV-SS RBC = 157 ± 42, AA REV-SS RBC = 16 ± 12, AA REV-SS RBC = 19 ± 12, AA REV-AA RBC (5 ± 4), SS REV-SS RBC with hemopexin = 14 ± 8, SS REV-SS RBC with ADAMTS13 = 19 ± 10, and heme-SS RBC = 140 ± 49, p < 0.05 for all groups except for heme-SS RBC, n = 5 in each group, 2-Group t test).
*: p = 0.002
**: p = 0.001
NS: p = 0.058
3.2. SS REVs increase endothelial von Willebrand factor (vWF) expression
We incubated HPMECs with derived SS or AA REVs for 2 hours. vWF expression was significantly increased on HPMECs incubated with derived SS REVs (Fig. 2A, B) compared to vWF expression on HPMECs incubated with derived AA REVs (Fig. 2C, p = 0.012, 2-Sample t test).
3.3. HbSS RBC adhere specifically to HPMECs incubated with HbSS REV
We first verified the in vitro approach by examining the interactions between SS RBC, AA RBC, and HPMECs treated with SS REVs and AA REVs. HPMEC laminated microchannels were treated with derived SS REV (Fig. 2D and 2F) or derived AA REV (Fig. 2E). SS RBC suspensions were then perfused into the first 2 channels (Fig 2D & E), and AA RBC suspension was perfused into the third (Fig 2F, per Section 2.7). No visible injury or morphological changes were observed on HPMECs treated with SS REV or AA REV. More adherent SS RBCs were observed on HPMECs activated with SS REVs than with AA REVs (Fig. 2G, 157 ± 42 vs. 19 ± 12, p < 0.05) and AA RBC on HPMECs treated with SS REVs (Fig. 2G, 16 ± 12, p < 0.05), as well as AA RBC on HPMECs treated with AA REVs (Fig. 2G, 5 ± 4, p < 0.05).
Figure 5. Impact of REV on microvascular endothelial cell response.
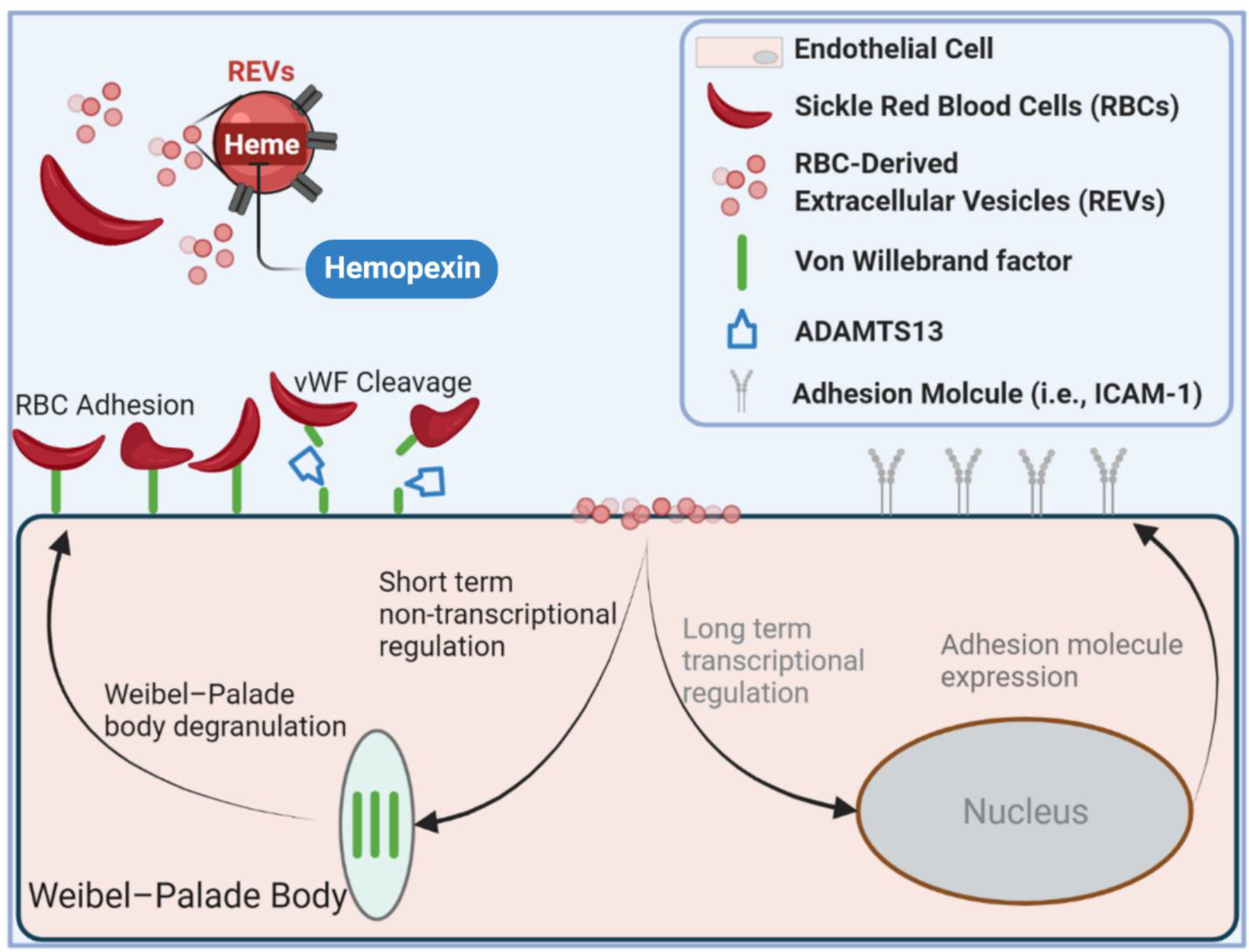
SS REV are capable of promoting HPMEC VWF expression within 2 hours, likely through the acute stress sentinel pathway of Weibel-Palade body degranulation. SS RBC adhere specifically to SS REV-activated HPMECs, likely mediated by endothelial VWF. The adhesion is decreased with VWF cleaving protease ADAMTS13, and with heme-binding protein hemopexin. Enhanced SS RBC adhesivity was observed in patients with elevated biomarkers of hemolysis and inflammation and thrombophilia.
3.4. REV-mediated RBC adhesion to HPMECs is attenuated by hemopexin
To demonstrate that the observed RBC adhesion was induced by REV-mediated HPMEC activation, we investigated the impact of the heme-binding protein hemopexin on REV-mediated RBC adhesion. Pre-incubating REVs with hemopexin reduced the HPMEC activation indicated by reduced number of adherent SS RBCs (Fig. 2G, 157 ± 42 vs. 14 ± 8, p < 0.05).
3.5. REV-mediated RBC adhesion to HPMECs is attenuated by ADAMTS13
To demonstrate that the observed RBC adhesion were mediated by vWF on REV-activated HPMECs, we investigated the impact of the vWF-specific protease ADAMTS13 on REV-mediated RBC adhesion. The addition of ADAMTS13 significantly reduced the adhesion of SS RBCs to SS REV-activated HPMECs (Fig. 2G, 157 ± 42 vs. 19 ± 10, p < 0.05). These results indicate that RBC adhesion was dependent on HPMEC vWF, because ADAMTS13 dramatically inhibited RBC adhesion, reaching a level that was close to SS RBC on HPMECs treated with AA REVs.
3.6. Heme is comparable to REVs in mediating RBC adhesion to HPMECs
To compare the impact of REVs and heme on activating HPMECs, we examined the SS RBC adhesion on HPMECs that are activated with 20 μM heme, which is comparable to the determined SS REV heme concentration of 17.3 ± 1.1 μM. The numbers of adherent SS RBCs on SS REV activated HPMECs and on 20 μM heme activated HPMECs demonstrated no statistical difference, although a slightly higher adhesion profile was observed on HPMECs activated with SS REVs (Fig. 2G, 157 ± 42 vs. 140 ± 49, p = 0.058).
3.7. Heme- or SS REV-induced vaso-occlusion in SS Towns mice that was inhibited by ADAMTS13
Since ADAMTS13 reduced RBC-endothelial cell adhesion and RBC adhesion to endothelium is believed to be involved in vaso-occlusion in SCD, we used Townes SS mice and a dorsal skin-fold chamber model to determine if ADAMTS13 would reduce microvascular stasis in response to hemin or SS REVs. Townes SS mice were surgically implanted with a dorsal skin-fold chamber. After chamber implantation, 20–21 subcutaneous flowing venules were selected at baseline using intravital microscopy. After selection and mapping of flowing venules, SS mice were infused via the tail vein with hemin (3.2 μmol/kg) or SS REVs at time zero to induce vaso-occlusion [3]. Twenty minutes later, mice were infused with ADAMTS13 (1 mg/kg) or sterile saline (10 ml/kg). The same venules selected at baseline were re-examined for stasis (no flow) at 1, 2, 3, and 4 hours after hemin or SS REV infusion. In SS mice, the levels of heme- and REV-mediated vaso-occlusion (stasis) at 1, 2, 3, and 4 hours were similar (Fig. 3A&B). Additionally, ADAMTS13 administered intravenously 20 minutes after hemin or SS REV infusion significantly stasis inhibited at 1, 2, 3, and 4 hours post-hemin or post-REV as compared to SS mice that received saline at 20 minutes (Fig. 3A&B).
Figure 3. Heme- and REV-induced vaso-occlusion in SS Towns mice are inhibited by ADAMTS13.
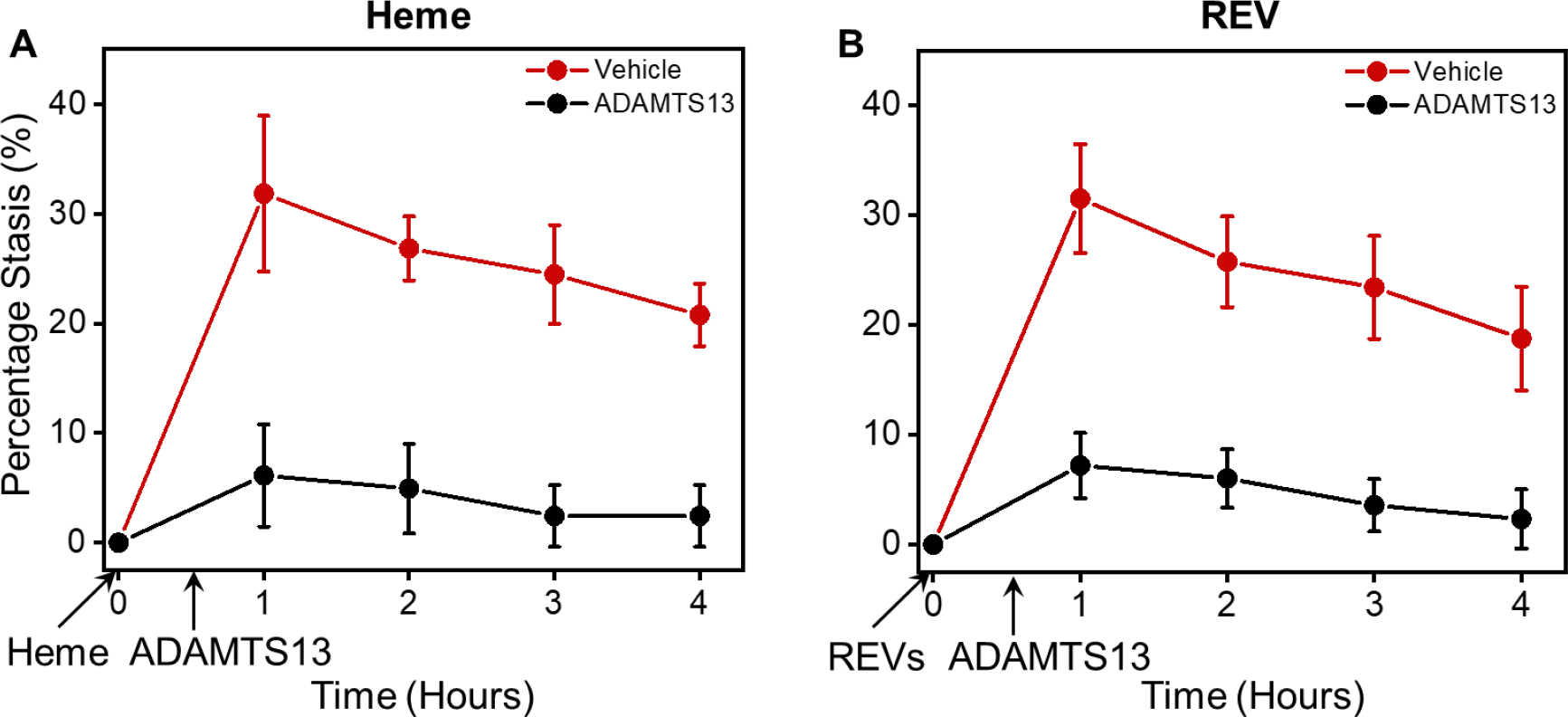
(A) Heme-induced vaso-occlusion (measured in percentage of stasis) in vivo in SS Towns mice is inhibited by ADAMTS13 (mean ± SD for 1, 2, 3, and 4 hours: Vehicle: 31.8% ± 7.1%, 26.8% ± 2.9%, 24.5% ± 4.5%, 20.8% ± 2.9% vs. ADAMTS13: 6.1% ± 4.7%, 4.9% ± 4.1%, 2.4% ± 2.8%, 2.4% ± 2.8%, p < 0.05 for all time points, n = 4 for each group at each time point). (B) REV-induced vaso-occlusion in vivo in SS Towns mice is inhibited by ADAMTS13 (mean ± SD for 1, 2, 3, and 4 hours: Vehicle: 31.5% ± 5.0%, 25.8% ± 4.1%, 23.4% ± 4.7%, 18.7% ± 4.7% vs. ADAMTS13: 7.2% ± 3.0%, 6.0% ± 2.7%, 3.6% ± 2.4%, 2.3% ± 2.7%, p < 0.05 for all time points, n = 4 for each group at each time point).
3.8. Clinical implications of REV-mediated RBC adhesion to HPMECs
To determine the association between clinical phenotypes and adhesion profiles, we examined a hemolytic subpopulation (Group 1, N = 6) with distinctly lower lactate dehydrogenase (LDH) levels and absolute reticulocyte counts (ARCs) compared with the 2nd subpopulation (Group 2, N = 9, Fig. 4A) [42]. Subjects in Group 1 had significantly lower LDH levels and ARCs (N = 8, LDH mean ± standard deviation: Group 1: 186 ± 99 IU vs. Group 2: 305 ± 55 IU, p = 0.031, 2-Group t test; ARCs mean ± standard deviation: Group 1: 162 ± 74 vs. Group 2: 401 ± 122 × 1010, p < 0.001, 2-Group t test). We then compared the RBC adhesion profile between Group 1 subjects and Group 2 subjects, and found significantly lower RBC adhesion to HbS REV activated HPMECs in Group 1 subjects than in Group 2 subjects (Fig. 4B, 117 ± 72 vs. 267± 125, p = 0.012, 2-Group t test). Subjects in Group 1 also had lower WBCs than did subjects in group 2 (Fig. 4C, 8.5 ± 1.2 × 109 vs. 11.4 ± 3.6 × 109 / L, p = 0.047, 2-Group t test). Group 1 also had a suggestion of having lower ferritin values (Fig. 4D, 876 ± 1527 ug/L vs. 2841 ± 2782 μg/L, p = 0.106, 2-Group t test). Similar differences of RBC adhesion profiles between two groups of subjects were also observed within the groups distinguished using single cutoff level of ARC level at 300 × 109/L or using single cutoff level of LDH level at 250 U/L (Supplemental Fig. 2).
Figure 4. RBC adhesion to REV-activated HPMECs correlates with subject clinical phenotype including hemolytic and inflammatory biomarkers.
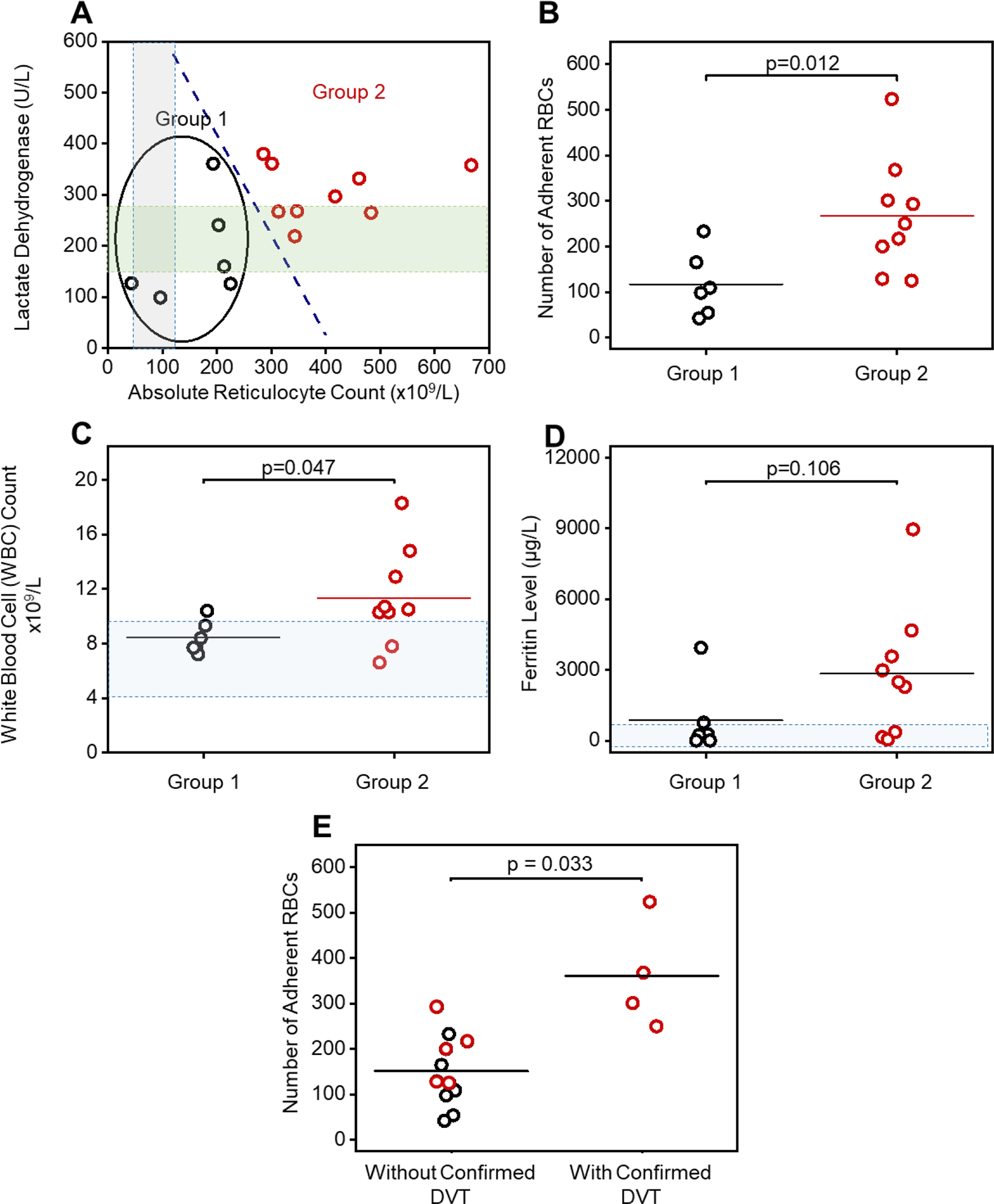
(A): A subpopulation (Group 1, N = 6) with distinct hemolysis markers of lactate dehydrogenase (LDH) levels and absolute reticulocyte count (ARCs) comparing to the rest (Group 2, N = 9) via k-means clustering analysis. RBCs from subjects in Group 2, with significantly higher LDH levels and ARCs, have greater adhesion to REV-activated HPMECs compared to the RBCs from subjects in Group 1 (B, Mean ± SD: 267 ± 125 vs. 117 ± 72, p = 0.012, 2-Group t test). The gray and green shaded areas indicate normal ranges for ARC and LDH, respectively. (C): Subjects in Group 2 with higher LDH and ARC and enhanced RBC adhesion have significantly higher WBC counts, than subjects in Group 1 (Mean ± SD: 11.4 ± 3.6 vs. 8.5 ± 1.2, p = 0.47, 2-Group t test). Shadowed area: WBC count range from 4.5 to 10 × 109. (D): Subjects in Group 2 with higher LDH and ARC and enhanced RBC adhesion have higher ferritin levels, although not statistically significant, than subjects in Group 1 (2841 ± 2804 vs. 876 ± 1527, p = 0.106, 2-Group t test). Shadowed area: normal range of ferritin level (2 to 1000 μg/L) [71, 72]. (E) Subjects with confirmed deep vein thrombosis (DVT) had significantly higher RBC adhesion to REV activated HPMECs than the ones without confirmed DVT (Mean ± SD: 361 ± 119 vs. 151 ± 78, p = 0.033, 2-Group t test). Six out of six subjects in Group 1 patient with lower RBC adhesion, and three out of six subjects in Group 2 did not have DVT. Four out of eight subjects in group 2 with higher RBC adhesion had DVT. DVT status of two out of eight subjects in Group 2 were not available (1 patient diagnosed as ‘unclear’, 1 patient record not accessible).
*In Fig. 4C 2 patients had the exact same WBC count at 7.1×109/L
Finally, subjects with a documented history of deep vein thromboses (DVT, N = 4) had higher RBC adhesion to REV-activated HPMECs compared to subjects without confirmed DVT (N = 11, Fig. 4E, 361 ± 119 vs. 151 ± 78, p = 0.033, 2-Group t test). Zero out 6 Group 1 subjects and 4 out 7 Group 2 subjects (with definitive DVT record) were diagnosed with DVT. DVT record of 2 subjects in Group 2 were not available (DVT diagnosis for one subject was not clear and DVT record was not accessible for the other subject). Similarly, Group 2 subjects with higher RBC adhesion profile had significantly greater possibility of having DVT than subjects in Group 1 (Table S1).
DISCUSSION
Two dominant pathophysiological events have been characterized with SCD: abnormal RBC adhesion and intravascular hemolysis. Intravascular hemolysis promotes endothelial activation and causes endothelial dysfunction; and SS RBCs are known to have reduced deformability [35, 36] and increased adhesivity to the activated endothelium, leading to vaso-occlusion. These mechanisms likely intersect and contribute to acute painful episodes and chronic organ damage [37].
SCD RBC membrane abnormalities include aberrant timing or increased rigidity during maturation, and abnormal activation by ‘stress signals’, of surface molecules such as VLA-4, CD36, LW and BCAM/LU [38–43]. Cumulative oxidative damage, resulting in excessive PS externalization on the SCD RBC membrane, causes abnormal adhesion [44, 45]. RBC adhesion to activated endothelium has been associated with elevated endothelium adhesion molecules including P-selectin [46], ICAM-1 [47], Vascular Cell adhesion molecule-1 (VCAM-1) [48], E-selectin [49], and vWF [50], as well as sub-endothelial proteins such as laminin (LN) under various physiological conditions. These abnormal interactions between SCD RBC and activated endothelium plays a pivotal role in the initiation and propagation of unpredictable VOC episodes, thereby contributing to the pathophysiology of SCD.
Intravascular hemolysis releases free heme, Hb, and REVs. Heme induces endothelial activation through TLR-4, resulting in increased expression of VCAM-1, ICAM-1, E-selectin, P-selectin, IL-1, IL-6, IL-8, and tissue factor via a pathway involving the activation of NF-kB phospho-p65 which are known to mediate RBC adhesion [2, 47, 49]. Importantly, up to one-third of cell-free heme in plasma is sequestered in circulating REVs [51]. Therefore, we set out to reproduce this important physiological mechanism in vitro. SS REVs can transfer heme to ECs in annexin-a5-sensitive fashion and have been reported to trigger transcriptional responses from middle- to longer-time periods (4–48 hours) [5, 29, 30, 51, 52] causing endothelial injury, linking hemolysis to chronic vascular injury in SCD mouse [4, 51, 52]. In addition to these long term transcriptional responses, heme is also known to activate acute stress through sentinel pathway mediated by TLR-4 signaling that leads to production of ROS, triggers WPB degranulation and rapid release of vWF to the cell surface, which eventually causes vaso-occlusion [2, 53]. It has been shown that vWF multimers released from endothelial cells results in a significant increase in adhesion of SS RBCs to endothelium ex vivo [11], suggesting an important role in the pathophysiology of sickle cell-induced microvascular occlusion [11, 50]. However, the relationship of this finding and SS REVs has not been well-described.
Leveraging the Endothelium-on-a-chip technology, we treated HPMECs with calcium ionophore-generated REVs in order to mimic the highly hemolytic plasma milieu that is often typical in people with HbSS [14]. We demonstrated enhanced vWF expression on HPMECs treated with SS REVs comparing to those treated with AA REVs within 2 hours. This demonstrates that REVs are capable of inducing acute endothelial response in the short term, in addition to known long-term effects. Further, vWF is known to mediate SS RBC adhesion independent of platelets [50, 54], and plasma levels of vWF may correlate with hemolysis in SCD [55]. We found that SS RBCs demonstrate increased adherence to SS REV-activated HPMECs, compared to the adhesion of SS RBCs to AA REV-treated HPMECs, and to the adhesion of AA RBCs to SS REV-activated HPMECs, and to the adhesion of AA RBCs to AA REV-treated HPMECs. These adhesion events were dramatically mitigated by heme-binding protein hemopexin or by vWF-specific protease ADAMTS13, reaching levels that were close to SS RBC on HPMECs treated with AA REVs.
Circulating EVs in SCD are known to have pro-thrombotic effect due to their surface tissue factor and PS expression. Circulating REVs in SCD are particularly known to contain significant amounts of cell-free heme and facilitate heme delivery to ECs [5, 56]. Here, we demonstrated the effect of hemopexin in mitigating REV-mediated acute endothelial activation. These results agreed with our previous work regarding the effect of hemopexin in mitigating heme-mediated vaso-occlusion in SS mice [2]. Furthermore, we compared the adhesion profiles of SS RBCs on HPMECs activated using SS REVs and on HPMECs activated using heme at the concentration carried by the SS REVs. The adhesion profiles between these two groups were statistically non-significant, despite the fact that slightly higher adhesion profile was observed on HPMECs activated by SS REVs compared to the ones activated by heme. These results agreed with previous publication by Camus et al. [51] that the effect of REVs in activating endothelium is largely carried via heme.
Heme is known to rapidly mobilize Weibel-Palade body (WPB) vWF and P-selectin onto EC surface and cause vaso-occlusion SS mice [2]. Belcher et al. previously demonstrated that polyclonal antibodies to vWF blocked stasis induced by heme in SCD mice with dorsal skin-fold chambers [2]. Here, in addition to the in vitro experimental results, we confirmed the effect of ADAMTS13 in mitigating both heme- and REV-mediated vaso-occlusion measured in percent of stasis in vivo using Townes SS mice. Together, both in vitro and in vivo results suggest a specific interaction between these cellular elements that is potentially mediated by HPMEC vWF expression induced by the heme content in SS REVs (Fig. 5). Despite the in vivo and in vitro studies demonstrating comparable impacts of REVs and heme or heme-loaded particles by this study and by others [51], the role of SS REVs in regulating vWF expression were investigated in vitro in this study. Future work will be conducted to confirm this SS REV regulated mechanism in vivo using mouse model. Additionally, the ligand vWF on SS RBCs mediating the adhesion events remains unclear therefore future work will be conducted in identifying potential ligands and to correlate the RBC surface expression of these ligands with subject phenotypes.
To analyze the heterogeneity of patient RBC adhesion to REV-activated endothelial cells, we uniformly activated HPMECs with SS REVs generated from pooled blood samples. Therefore, the RBC adhesion quantified in this study only depends on the adhesivity of RBCs in individual patients. Accordingly, we describe 2 groups, one of which is characterized by elevated LDH levels and ARCs, and had significantly greater RBC adhesion as compared to the other group (Fig. 4A&B). LDH and ARC are important in vivo hemolytic biomarkers which have been linked to disease severity [57, 58]. Therefore, these results suggest that RBCs are more adhesive in subjects with a more severe hemolytic phenotype in SCD. Additionally, we found that subjects with a documented history of confirmed deep vein thromboses (DVT, N = 4) had higher RBC adhesion to REV-activated HPMECs compared to subjects without confirmed DVT. We postulate that this is because the higher adhesivity indicates more severe sickling of RBCs, which has been reported to mediate entrapment of these cells within venous clot [59]. Future work will test the associations between REV-mediated RBC adhesion and patient clinical outcomes including hemolysis and record of DVT, and ongoing treatment, in increased number of patients.
In conclusion, in this study, we have improved our Endothelium-on-a-chip approach by activating the endothelial layer with a biologically complex in vitro generated REVs obtained from isolated RBC. We believe that this is physiologically relevant in comparison to a simpler biochemical signal such as TNF-α or heme. Our findings indicate that both pathological RBCs with higher adhesivity, and an activated endothelium are required for the observed abnormal interaction. The SS RBCs demonstrate enhanced adhesion to HPMECs activated with SS REVs in a subject specific fashion. This heterogeneity in adhesion solely reflects heterogeneous RBC adhesion to uniformly activated HPMECs (using pooled derived REV). The RBC adhesion profiles associated with hemolytic and inflammatory biomarkers including LDH, ARC, WBC, and ferritin levels. Future investigations will re-examine these clinical associations in expanded patient population, as well as examine RBC adhesion on endothelial cells that are activated with patient-specific REVs, which may better reflect the overall disease process in an individual patient. Understanding the important collective interplay between RBCs and REV-activated microvascular endothelial cells will better characterize the multicellular adhesion paradigm for acute and chronic vaso-occlusion in SCD and may enable us to develop more effective treatment paradigms.
Supplementary Material
ACKNOWLEDGEMENTS
This work is supported by National Heart, Lung, and Blood Institute (NHLBI) R01HL133574 (Gurkan), OT2HL152643 (Gurkan), U01HL117659 (Gurkan), R01HL114567 (Vercellotti/Belcher), K25HL159358 (An), and T32HL134622 (An). This article’s contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
Footnotes
CONFLICT OF INTEREST
RA, JAL, UAG, and Case Western Reserve University have financial interests in Hemex Health Inc. JAL, EK, UAG, and Case Western Reserve University have financial interests in BioChip Labs Inc. UAG and Case Western Reserve University have financial interests in Xatek Inc. UAG has financial interests in DxNow Inc. Financial interests include licensed intellectual property, stock ownership, research funding, employment, and consulting. Hemex Health Inc. offers point-of-care diagnostics for hemoglobin disorders, anemia, and malaria. BioChip Labs Inc. offers commercial clinical microfluidic biomarker assays for inherited or acquired blood disorders. Xatek Inc. offers point-of-care global assays to evaluate the hemostatic process. DxNow Inc. offers microfluidic and bio-imaging technologies for in vitro fertilization, forensics, and diagnostics. Competing interests of Case Western Reserve University employees are overseen and managed by the Conflict of Interests Committee according to a Conflict-of-Interest Management Plan. GMV and JDB receive research funding for CSL Behring and Astellas/Mitobridge.
REFERENCES
- 1.Kato Gregory J., Piel Frédéric B., Reid Clarice D., Gaston Marilyn H., Kwaku Ohene-Frempong Lakshmanan Krishnamurti, Smith Wally R., Panepinto Julie A., Weatherall David J., Costa Fernando F., and Vichinsky Elliott P., Sickle cell disease. Nature Reviews Disease Primers, 2018. 4(1): p. 18010. [DOI] [PubMed] [Google Scholar]
- 2.Belcher John D., Chen Chunsheng, Nguyen Julia, Milbauer Liming, Abdulla Fuad, Alayash Abdu I., Smith Ann, Nath Karl A., Hebbel Robert P., and Vercellotti Gregory M., Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood, 2014. 123(3): p. 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Camus Stephane M., De Moraes Joao A., Bonnin Philippe, Abbyad Paul, Sylvain Le Jeune Francois Lionnet, Loufrani Laurent, Grimaud Linda, Lambry Jean-Christophe, Charue Dominique, Kiger Laurent, Renard Jean-Marie, Larroque Claire, Herve Le Clesiau Alain Tedgui, Bruneval Patrick, Christina Barja-Fidalgo Antigoni Alexandrou, Tharaux Pierre-Louis, Boulanger Chantal M., and Blanc-Brude Olivier P. , Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood, 2015. 125(24): p. 3805–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Camus SM, Gausserès B, Bonnin P, Loufrani L, Grimaud L, Charue D, De Moraes JA, Renard JM, Tedgui A, Boulanger CM, Tharaux PL, and Blanc-Brude OP, Erythrocyte microparticles can induce kidney vaso-occlusions in a murine model of sickle cell disease. Blood, 2012. 120(25): p. 5050–8. [DOI] [PubMed] [Google Scholar]
- 5.Garnier Yohann, Ferdinand Séverine, Garnier Marie, Cita Kizzy-Clara, Hierso Régine, Claes Aurélie, Connes Philippe, Marie-Dominique Hardy-Dessources Claudine Lapouméroulie, Lemonne Nathalie, Maryse Etienne-Julan Wassim El Nemer, and Romana Marc, Plasma microparticles of sickle patients during crisis or taking hydroxyurea modify endothelium inflammatory properties. Blood, 2020. 136(2): p. 247–256. [DOI] [PubMed] [Google Scholar]
- 6.Noomuna P, Risinger M, Zhou S, Seu K, Man Y, An R, Sheik DA, Wan J, Little JA, Gurkan UA, Turrini FM, Kalfa T, and Low PS, Inhibition of Band 3 tyrosine phosphorylation: a new mechanism for treatment of sickle cell disease. Br J Haematol, 2020. 190(4): p. 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piccin A, Murphy C, Eakins E, Rondinelli MB, Daves M, Vecchiato C, Wolf D, Mc Mahon C, and Smith OP, Insight into the complex pathophysiology of sickle cell anaemia and possible treatment. Eur J Haematol, 2019. 102(4): p. 319–330. [DOI] [PubMed] [Google Scholar]
- 8.Piccin A, Murphy C, Eakins E, Kunde J, Corvetta D, Di Pierro A, Negri G, Guido M, Sainati L, Mc Mahon C, Smith OP, and Murphy W, Circulating microparticles, protein C, free protein S and endothelial vascular markers in children with sickle cell anaemia. J Extracell Vesicles, 2015. 4: p. 28414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piccin A, Murphy C, Eakins E, Kinsella A, McMahon C, Smith OP, and Murphy WG, Protein C and free protein S in children with sickle cell anemia. Ann Hematol, 2012. 91(10): p. 1669–71. [DOI] [PubMed] [Google Scholar]
- 10.Nader E, Romana M, Guillot N, Fort R, Stauffer E, Lemonne N, Garnier Y, Skinner SC, Etienne-Julan M, Robert M, Gauthier A, Cannas G, Antoine-Jonville S, Tressières B, Hardy-Dessources MD, Bertrand Y, Martin C, Renoux C, Joly P, Grau M, and Connes P, Association Between Nitric Oxide, Oxidative Stress, Eryptosis, Red Blood Cell Microparticles, and Vascular Function in Sickle Cell Anemia. Front Immunol, 2020. 11: p. 551441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaul DK, Nagel RL, Chen D, and Tsai HM, Sickle erythrocyte-endothelial interactions in microcirculation: the role of von Willebrand factor and implications for vasoocclusion. Blood, 1993. 81(9): p. 2429–38. [PubMed] [Google Scholar]
- 12.Ataga Kenneth I. and Key Nigel S., Hypercoagulability in Sickle Cell Disease: New Approaches to an Old Problem. Hematology, 2007. 2007(1): p. 91–96. [DOI] [PubMed] [Google Scholar]
- 13.Noubouossie D, Key NS, and Ataga KI, Coagulation abnormalities of sickle cell disease: Relationship with clinical outcomes and the effect of disease modifying therapies. Blood Rev, 2016. 30(4): p. 245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kato Gregory J., Steinberg Martin H., and Gladwin Mark T., Intravascular hemolysis and the pathophysiology of sickle cell disease. The Journal of clinical investigation, 2017. 127(3): p. 750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guillaume van Niel Gisela D Angelo, and Raposo Graça, Shedding light on the cell biology of extracellular vesicles. Nature Reviews Molecular Cell Biology, 2018. 19: p. 213. [DOI] [PubMed] [Google Scholar]
- 16.Thangaraju K, Neerukonda SN, Katneni U, and Buehler PW, Extracellular Vesicles from Red Blood Cells and Their Evolving Roles in Health, Coagulopathy and Therapy. Int J Mol Sci, 2020. 22(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puhm Florian, Afonyushkin Taras, Resch Ulrike, Obermayer Georg, Rohde Manfred, Penz Thomas, Schuster Michael, Wagner Gabriel, Rendeiro Andre F., Melki Imene, Kaun Christoph, Wojta Johann, Bock Christoph, Jilma Bernd, Mackman Nigel, Boilard Eric, and Binder Christoph J., Mitochondria Are a Subset of Extracellular Vesicles Released by Activated Monocytes and Induce Type I IFN and TNF Responses in Endothelial Cells. Circulation Research, 2019. 125(1): p. 43–52. [DOI] [PubMed] [Google Scholar]
- 18.Awojoodu Anthony O., Keegan Philip M., Lane Alicia R., Zhang Yuying, Lynch Kevin R., Platt Manu O., and Botchwey Edward A., Acid sphingomyelinase is activated in sickle cell erythrocytes and contributes to inflammatory microparticle generation in SCD. Blood, 2014. 124(12): p. 1941–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Westerman M, Pizzey A, Hirschman J, Cerino M, Weil-Weiner Y, Ramotar P, Eze A, Lawrie A, Purdy G, Mackie I, and Porter J, Microvesicles in haemoglobinopathies offer insights into mechanisms of hypercoagulability, haemolysis and the effects of therapy. Br J Haematol, 2008. 142(1): p. 126–35. [DOI] [PubMed] [Google Scholar]
- 20.Mahfoudhi E, Lecluse Y, Driss F, Abbes S, Flaujac C, and Garcon L, Red cells exchanges in sickle cells disease lead to a selective reduction of erythrocytes-derived blood microparticles. Br J Haematol, 2012. 156(4): p. 545–7. [DOI] [PubMed] [Google Scholar]
- 21.Nouraie Mehdi, Lee Janet S., Zhang Yingze, Kanias Tamir, Zhao Xuejun, Xiong Zeyu, Oriss Timothy B., Zeng Qilu, Kato Gregory J., J. Gibbs Simon R., Hildesheim Mariana E., Sachdev Vandana, Barst Robyn J., Machado Roberto F., Hassell Kathryn L., Little Jane A., Schraufnagel Dean E., Krishnamurti Lakshmanan, Novelli Enrico, Girgis Reda E., Morris Claudia R., Erika Berman Rosenzweig David B. Badesch, Lanzkron Sophie, Castro Oswaldo L., Goldsmith Jonathan C., Gordeuk Victor R., Gladwin Mark T., Phasst Investigators Walk, and Patients, The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica, 2013. 98(3): p. 464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bevers Edouard M., Wiedmer Therese, Comfurius Paul, Shattil Sanford J., Weiss Harvey J., Zwaal Robert F. A., and Sims Peter J., Defective Ca2+-Induced Microvesiculation and Deficient Expression of Procoagulant Activity in Erythrocytes From a Patient With a Bleeding Disorder: A Study of the Red Blood Cells of Scott Syndrome. Blood, 1992. 79(2): p. 380–388. [PubMed] [Google Scholar]
- 23.Wang Qinhong and Zennadi Rahima, The Role of RBC Oxidative Stress in Sickle Cell Disease: From the Molecular Basis to Pathologic Implications. Antioxidants, 2021. 10(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jagadeeswaran Ramasamy, Lenny Hong, Vazquez Benjamin, Muniz Joshua, Schad Aimee, Jain Shivi, Gowhari Michel, Lavelle Donald, Diamond Alan, Molokie Robert E., and Rivers Angela, The Abnormal Presence of Mitochondria in Circulating Red Blood Cells Cause an Increased Oxygen Consumption Rate, ROS Generation and Hemolysis in Patients with Sickle Cell Disease. Blood, 2017. 130(Supplement 1): p. 2237–2237.29170191 [Google Scholar]
- 25.Thi Bich Thuy Ly and Ingolf Bernhardt Duc Bach Nguyen, Microvesicles Released from Human Red Blood Cells: Properties and Potential Applications. Novel Implications of Exosomes in Diagnosis and Treatment of Cancer and Infectious Diseases. [Google Scholar]
- 26.Chen Shao-Yin, Wang Yulei, Telen Marilyn J., and Chi Jen-Tsan, The Genomic Analysis of Erythrocyte microRNA Expression in Sickle Cell Diseases. PLOS ONE, 2008. 3(6): p. e2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Beers EJ, Schaap MC, Berckmans RJ, Nieuwland R, Sturk A, van Doormaal FF, Meijers JC, and Biemond BJ, Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica, 2009. 94(11): p. 1513–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jana Sirsendu, Michael Brad Strader Fantao Meng, Hicks Wayne, Kassa Tigist, Tarandovskiy Ivan, Silvia De Paoli Jan Simak, Heaven Michael R., Belcher John D., Vercellotti Gregory M., and Alayash Abdu I., Hemoglobin oxidation–dependent reactions promote interactions with band 3 and oxidative changes in sickle cell–derived microparticles. JCI Insight, 2018. 3(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gemel Joanna, Mao Yifan, Lapping-Carr Gabrielle, and Beyer Eric C., Gap Junctions between Endothelial Cells Are Disrupted by Circulating Extracellular Vesicles from Sickle Cell Patients with Acute Chest Syndrome. International Journal of Molecular Sciences, 2020. 21(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gabrielle Lapping-Carr Joanna Gemel, Mao Yifan, Sparks Gianna, Harrington Margaret, Peddinti Radhika, and Beyer Eric C., Circulating extracellular vesicles from patients with acute chest syndrome disrupt adherens junctions between endothelial cells. Pediatric Research, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kucukal Erdem, Ilich Anton, Key Nigel S., Little Jane A., and Gurkan Umut A., Red blood cell adhesion to heme-activated endothelial cells reflects clinical phenotype in sickle cell disease. American Journal of Hematology, 2018. 93(8): p. 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smeets Michel W. J., Bierings Ruben, Meems Henriet, Mul Frederik P. J., Geerts Dirk, Vlaar Alexander P. J., Voorberg Jan, and Hordijk Peter L., Platelet-independent adhesion of calcium-loaded erythrocytes to von Willebrand factor. PLOS ONE, 2017. 12(3): p. e0173077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicolay Jan P., Thorn Verena, Daniel Christoph, Amann Kerstin, Siraskar Balasaheb, Lang Florian, Hillgruber Carina, Goerge Tobias, Hoffmann Stefan, Gorzelanny Christian, Huck Volker, Mess Christian, Obser Tobias, Schneppenheim Reinhard, Fleming Ingrid, Schneider Matthias F., and Schneider Stefan W., Cellular stress induces erythrocyte assembly on intravascular von Willebrand factor strings and promotes microangiopathy. Scientific Reports, 2018. 8(1): p. 10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J, and Townes TM, Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood, 2006. 108(4): p. 1183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Man Y, Kucukal E, An R, Watson QD, Bosch J, Zimmerman PA, Little JA, and Gurkan UA, Microfluidic assessment of red blood cell mediated microvascular occlusion. Lab Chip, 2020. 20(12): p. 2086–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Man Y, Kucukal E, An R, Bode A, Little JA, and Gurkan UA, Standardized microfluidic assessment of red blood cell-mediated microcapillary occlusion: Association with clinical phenotype and hydroxyurea responsiveness in sickle cell disease. Microcirculation, 2020: p. e12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kato Gregory J., Martyr Sabrina, Blackwelder William C., Nichols James S., Coles Wynona A., Hunter Lori A., Brennan Marie-Luise, Hazen Stanley L., and Gladwin Mark T., Levels of soluble endothelium-derived adhesion molecules in patients with sickle cell disease are associated with pulmonary hypertension, organ dysfunction, and mortality. British Journal of Haematology, 2005. 130(6): p. 943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El Nemer W, Wautier MP, Rahuel C, Gane P, Hermand P, Galacteros F, Wautier JL, Cartron JP, Colin Y, and Le Van Kim C, Endothelial Lu/BCAM glycoproteins are novel ligands for red blood cell alpha4beta1 integrin: role in adhesion of sickle red blood cells to endothelial cells. Blood, 2007. 109(8): p. 3544–51. [DOI] [PubMed] [Google Scholar]
- 39.Johnson C and Telen MJ, Adhesion molecules and hydroxyurea in the pathophysiology of sickle cell disease. Haematologica, 2008. 93(4): p. 481–5. [DOI] [PubMed] [Google Scholar]
- 40.Wick TM and Eckman JR, Molecular basis of sickle cell-endothelial cell interactions. Curr Opin Hematol, 1996. 3(2): p. 118–24. [DOI] [PubMed] [Google Scholar]
- 41.Swerlick RA, Eckman JR, Kumar A, Jeitler M, and Wick TM, Alpha 4 beta 1-integrin expression on sickle reticulocytes: vascular cell adhesion molecule-1-dependent binding to endothelium. Blood, 1993. 82(6): p. 1891–9. [PubMed] [Google Scholar]
- 42.Joneckis Christopher C., Ackley Rhonda L., Orringer Eugene P., Wayner Elizabeth A., and Parise Leslie V., Integrin a4b1 and glycoprotein IV (CD36) are expressed on circulating reticulocytes in sickle cell anemia. Blood, 1993. 82(12): p. 3548–3555. [PubMed] [Google Scholar]
- 43.Zennadi R, Moeller BJ, Whalen EJ, Batchvarova M, Xu K, Shan S, Delahunty M, Dewhirst MW, and Telen MJ, Epinephrine-induced activation of LW-mediated sickle cell adhesion and vaso-occlusion in vivo. Blood, 2007. 110(7): p. 2708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Jong K, Larkin SK, Styles LA, Bookchin RM, and Kuypers FA, Characterization of the phosphatidylserine-exposing subpopulation of sickle cells. Blood, 2001. 98(3): p. 860–7. [DOI] [PubMed] [Google Scholar]
- 45.Kuypers FA, Lewis RA, Hua M, Schott MA, Discher D, Ernst JD, and Lubin BH, Detection of altered membrane phospholipid asymmetry in subpopulations of human red blood cells using fluorescently labeled annexin V. Blood, 1996. 87(3): p. 1179–87. [PubMed] [Google Scholar]
- 46.Matsui NM, Borsig L, Rosen SD, Yaghmai M, Varki A, and Embury SH, P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood, 2001. 98(6): p. 1955–62. [DOI] [PubMed] [Google Scholar]
- 47.Kucukal Erdem, Man Yuncheng, Quinn Erina, Tewari Neil, An Ran, Ilich Anton, Key Nigel S., Little Jane A., and Gurkan Umut A., Red blood cell adhesion to ICAM-1 is mediated by fibrinogen and is associated with right-to-left shunts in sickle cell disease. Blood advances, 2020. 4(15): p. 3688–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Setty BN and Stuart MJ, Vascular cell adhesion molecule-1 is involved in mediating hypoxia-induced sickle red blood cell adherence to endothelium: potential role in sickle cell disease. Blood, 1996. 88(6): p. 2311–20. [PubMed] [Google Scholar]
- 49.Man Yuncheng, Kucukal Erdem, Liu Shichen, Manwani Deepa, Little Jane, and Gurkan Umut A., Heterogeneous Hypoxia-Mediated Neutrophil and Red Blood Cell Adhesion to E-Selectin in Microscale Flow. Blood, 2018. 132(Supplement 1): p. 3671–3671. [Google Scholar]
- 50.Wick TM, Moake JL, Udden MM, Eskin SG, Sears DA, and McIntire LV, Unusually large von Willebrand factor multimers increase adhesion of sickle erythrocytes to human endothelial cells under controlled flow. The Journal of Clinical Investigation, 1987. 80(3): p. 905–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Camus SM, De Moraes JA, Bonnin P, Abbyad P, Le Jeune S, Lionnet F, Loufrani L, Grimaud L, Lambry JC, Charue D, Kiger L, Renard JM, Larroque C, Le Clesiau H, Tedgui A, Bruneval P, Barja-Fidalgo C, Alexandrou A, Tharaux PL, Boulanger CM, and Blanc-Brude OP, Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood, 2015. 125(24): p. 3805–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Merle Nicolas S., Grunenwald Anne, Rajaratnam Helena, Gnemmi Viviane, Frimat Marie, Figueres Marie-Lucile, Knockaert Samantha, Bouzekri Sanah, Charue Dominique, Noe Remi, Tania Robe-Rybkine Marie Le-Hoang, Brinkman Nathan, Gentinetta Thomas, Edler Monika, Petrillo Sara, Tolosano Emanuela, Miescher Sylvia, Sylvain Le Jeune Pascal Houillier, Chauvet Sophie, Rabant Marion, Dimitrov Jordan D., Veronique Fremeaux-Bacchi Olivier P. Blanc-Brude, and Roumenina Lubka T., Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI insight, 2018. 3(12): p. e96910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Belcher JD, Bryant CJ, Nguyen J, Bowlin PR, Kielbik MC, Bischof JC, Hebbel RP, and Vercellotti GM, Transgenic sickle mice have vascular inflammation. Blood, 2003. 101(10): p. 3953–9. [DOI] [PubMed] [Google Scholar]
- 54.Wick Timothy M., Moake Joel L., Udden Mark M., and McLntire Larry V., Unusually large von willebrand factor multimers preferentially promote young sickle and nonsickle erythrocyte adhesion to endothelial cells. American Journal of Hematology, 1993. 42(3): p. 284–292. [DOI] [PubMed] [Google Scholar]
- 55.Chen Junmei, Hobbs William E., Le Jennie, Lenting Peter J., de Groot Philip G., and López José A., The rate of hemolysis in sickle cell disease correlates with the quantity of active von Willebrand factor in the plasma. Blood, 2011. 117(13): p. 3680–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Camus Stéphane M., Blandine Gausserès Philippe Bonnin, Loufrani Laurent, Grimaud Linda, Charue Dominique, De Moraes Joao A., Renard Jean-Marie, Tedgui Alain, Boulanger Chantal M., Tharaux Pierre-Louis, and Blanc-Brude Olivier P., Erythrocyte microparticles can induce kidney vaso-occlusions in a murine model of sickle cell disease. Blood, 2012. 120(25): p. 5050. [DOI] [PubMed] [Google Scholar]
- 57.Hebbel RP, Reconstructing sickle cell disease: a data-based analysis of the “hyperhemolysis paradigm” for pulmonary hypertension from the perspective of evidence-based medicine. Am J Hematol, 2011. 86(2): p. 123–54. [DOI] [PubMed] [Google Scholar]
- 58.Kato Gregory J., Vicki McGowan Roberto F. Machado, Little Jane A., Taylor James V. I., Morris Claudia R., Nichols James S., Wang Xunde, Poljakovic Mirjana, Morris Sidney M. Jr., and Gladwin Mark T., Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood, 2006. 107(6): p. 2279–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Faes C, Ilich A, Sotiaux A, Sparkenbaugh EM, Henderson MW, Buczek L, Beckman JD, Ellsworth P, Noubouossie DF, Bhoopat L, Piegore M, Renoux C, Bergmeier W, Park Y, Ataga KI, Cooley B, Wolberg AS, Key NS, and Pawlinski R, Red blood cells modulate structure and dynamics of venous clot formation in sickle cell disease. Blood, 2019. 133(23): p. 2529–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.