

Abstract
Diacylglycerol is a potent element of intracellular secondary signaling cascades whose production is enhanced by cell surface receptor agonism and function is regulated by enzymatic degradation by diacylglycerol kinases. In T cells, stringent regulation of the activity of this second messenger maintains an appropriate balance between effector function and anergy. Here we demonstrate that the DGKα is an indispensable regulator of T cell receptor-mediated activation of CD8 T cells in LCMV Clone 13 viral infection. In the absence of DGKα, Clone 13 infection in a murine model results in a pathologic, proinflammatory state and a multicellular immunopathologic host death that is predominantly driven by CD8 effector T cells.
Introduction
Diacylglycerol (DAG) is a ubiquitous intracellular signaling molecule that accumulates at the cell membrane following receipt of an extracellular stimulus, typically by a G protein-coupled receptor (GPCR)(1) or by a T cell receptor (TCR)(2) or B cell receptor (BCR)(3) in lymphocytes to promote secondary signaling mechanisms. Following cell surface receptor agonism, phospholipase C (PLC) is activated, hydrolyzing phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol triphosphate (IP3). The accumulation of intracellular DAG recruits and activates Ras-GTPases and protein kinase C (PKC), triggering secondary signaling pathways associated with cell proliferation and survival.(4)
Conversely, cell surface receptor agonism also recruits members of the DAG neutralizing family of enzymes, diacylglycerol kinases (DGKs), to the immunological synapse.(5) Mechanistically, DGKs cleave DAG into phosphatidic acid (PA) to deactivate DAG-mediated secondary signaling pathways. Ten known isoforms of DGK have been identified in mammals,(6) and are classified into five subgroups based on their regulatory motifs. Isoforms are incompletely redundant(6) in their specific functional control and most DGK isoforms are expressed in multiple tissues. Multiple isoforms can also be present within the same tissue or cell type.(2) Mutations and polymorphisms of DGK isoforms are implicated in diseases including hypospadias (κ)(7), psychiatric disorders (η)(8), diabetes mellitus (ε, ζ), atypical hemolytic uremic syndrome (ε), and cancer progression (α).(9)
In lymphocytes, tight regulation of DAG activity and metabolism manages a balance in activated T cells between effector function and anergy. Olenchock et al. showed that combined T cell receptor (TCR) stimulation with CD28 costimulation significantly increases production of DAG and leads to downregulation of two DGK isoforms (α, ζ) intrinsic to T cells.(10) In contrast, isolated TCR stimulation leads to lowered production of DAG and increased DGKα.(10)
In models of forced overexpression of DGKα, there is significantly impaired T cell activation. This occurs as a result of impaired transcription of activator protein-1 (AP-1) family transcription factors (TFs),(11) which normally heterodimerize with Nuclear Factor of Activated T-cells (NFAT) to upregulate the transcription of key molecules involved in T cell activation, including IL-2, IL-3, IL-4 and GM-CSF.(12) Consequently, the lack of AP-1 members results in unpartnered NFAT, inadequate transcription, and an anergic T cell state.(13)
As the absence of DGKα in an acute infection model has been shown to enhance effector T cell response and promote viral clearance when compared to wildtype controls,(14) we hypothesized that a similar effect would be observed in a chronic infection model. A prototypic model of chronic infection is the lymphocytic choriomeningitis virus (LCMV) Clone 13 strain. LCMV Clone 13 is an advantageous model as the virus is noncytopathic, ensuring that phenotypic observations within the host are a direct result of the host immune response and not viral cytotoxicity. Clone 13 infection in an immunocompetent host leads to a phenotype of persistent viral infection and T cell exhaustion. As a result of exhaustion, CD8 T cells develop dysfunction in their ability to proliferate and in their ability to produce IL-2, TNFα, and IFNγ. Further, exhausted T cells demonstrate high levels of inhibitory receptors, including PD-1 and LAG-3. PD-1 signaling is also shown to limit signaling through the mammalian target of rapamycin (mTOR) pathways, and inhibit glycolysis.(15)
We report our findings of unexpected mortality from an overwhelming immune response following LCMV Clone 13 infection in mice with germline DGKα deficiency. We further identified the same outcome occurs when DGKα deficiency is isolated to CD8 T cells, that DGKα deficiency in CD8 T cells is necessary for the observed mortality and contributes to an enhanced effector phenotype in virus specific CD8 T cells. Here, we demonstrate that DGKα limits pathologic immune responses to Clone 13 infection and characterize the effect of DGKα in innate and adaptive immune responses in this model.
Methods
Mice:
Mice deficient in DGKα were described previously.(10) C57Bl/6 mice were purchased from NCI. P14 mice, with transgenic expression of a TCR cognate to the LCMV epitope glycoprotein 33 (gp33) were maintained in-house. P14 DGKα−/− mice were generated through crossing DGKα−/− with P14 mice in-house. All experiments were performed in mice 6–12 weeks old. Animal housing and experimentation was done in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) at the Medical College of Wisconsin.
Generation of CD8 T cell restricted DGKα−/− mice by bone marrow chimera:
Mice were generated with DGKα deficiency restricted to CD8 T cells by bone marrow chimera. Irradiation was delivered to CD45.1+/+ recipient mice, followed by retroorbital injection of 2 × 106 total nucleated cells, comprised of single cell suspension of bone marrow cells derived from a CD8−/− mouse (70%) and a CD45.2+/+ DGKα-deficient mouse (30%). Control mice were also generated by the same protocol but were C57Bl/6, CD45.2+/+ hosts and received bone marrow cells derived from a CD8−/− mouse (70%) and a C57Bl/6 CD45.1+/+ mouse (30%). Recipient mice were CD8 T cell depleted by treatment with InVivoPlus anti-mouse CD8a mAb (clone YTS 169.4, BioXCell) and CD8 T cell reconstitution was confirmed by flow cytometry.
Statistics:
Statistical analysis was performed using GraphPad Prism software (La Jolla, CA, USA). Survival curve comparisons were performed using the Log-Rank (Mantel-Cox) test. Weight curve comparisons were performed by mixed effect analysis with the Geisser-Greenhouse correction for variability and Bonferroni correction for multiple comparisons with an alpha value of 0.05. Comparisons between frequency of cell subsets among experimental phenotypes were performed using an unpaired Welch’s t-test for cohorts with samples ≥ 5, or an unpaired Mann-Whitney Test for cohorts with sizes < 5. Serum analytes were compared using an unpaired Mann-Whitney test, using the Bonferroni correction for multiple comparisons, with an alpha value of 0.05. Paired t-test was used for competitive assay / co-transfer experiments. A p-value < 0.05 was considered to indicate statistical significance (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001).
LCMV infection:
Infection with LCMV Clone 13 was achieved by inoculation of at least 2.5 × 106 PFU or 5 × 106 focus forming units (FFU), injected in 500 μL by retroorbital intravenous injection.
Peripheral blood sampling:
Peripheral blood sampling was obtained by submandibular facial bleed. Following vein puncture, 3–4 drops of fresh blood were collected directly into a heparinized capillary tube. Blood was then emptied from the capillary tube into an empty 1.5 mL mini-Eppendorf tube. Immediately, 20 μL of fresh blood was transferred into 120 μL of CellPak (Sysmex) diluent augmented with 5 mM EDTA (Fisher Scientific) and 0.5 μg/mL PGE1 (Sigma-Aldrich). Samples were stored on ice until analysis on Sysmex XN-1000 Hematology Analyzer (Sysmex).
Serum viral titers:
Immediately prior to mouse euthanasia, 100 μL of peripheral blood were obtained by submandibular facial bleed. Blood was stored at room temperature for approximately 2 hours until coagulated, and then centrifuged at 1500 × g for 15 minutes at 4°C. Following centrifugation, serum comprised the supernatant, which was collected and stored at −20°C or −80°C until use in a focus forming assay.
Serum cytokine analysis:
Mouse Cytokine Array / Chemokine Array 31-Plex (MD31) was performed by Eve Technologies (Alberta, Canada). Serum was isolated as above. Reported results included serum levels of eotaxin, G-CSF, GM-CSF, IFNγ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-17A, IP-10, KC, LIF, LIX, MCP-1, M-CSF, MIG, MIP-1α, MIP-1b, MIP-2, RANTES, TNFα, and VEGF. For replicates with results that were either above or below the detectable limit of the assay, these samples were designated with the highest or lowest value obtained for that analyte, respectively, as recommended by the reference laboratory.
Focus forming assay:
Mouse serum containing LCMV Clone 13 was incubated in three serial dilutions with 3×104 Vero cells at 37°C, 5% CO2 for 20 hours, then fixed with 5% paraformaldehyde. After blocking (50 mM Tris, 0.14 M NaCl, 1% BSA) for 60 minutes at room temperature, wells were washed with PBS and rat anti-LCMV nucleoprotein mAb (BioXCell, clone VL-4) was added and incubated overnight at 4°C. After unbound antibody was washed from wells with PBS, secondary staining was performed using goat anti-rat IgG2a-FITC (Bethyl, clone A110–109F) and imaged using the Incucyte Live Cell Analysis System (Essen Bioscience). Fluorescent foci were counted manually (DotDotGoose open-source software, American Museum of Natural History Center for Biodiversity and Conservation) and titers were calculated as focus forming units (FFU) per milliliter.
Cellular phenotyping:
Cell types were classified as follows: CD4 Treg: CD4+, CD25+/−, FOXP3+; CD4 Th1: CD4+, CD44+, CXCR6+; CD4 Tfh: CD4+, CD44+, CXCR5+; macrophage: CD11b+, F4/80+; monocyte: CD11b+, F4/80−, Ly6Chigh, Ly6G−; granulocyte: CD11b+, F4/80−, Ly6Cint, Ly6G+; virus-specific CD8 T cell: CD8+, CD44+, GP33+; germinal center B cell: B220+, Fas+, GL7+. Single cell suspensions of splenocytes were stained as follows. For myeloid subsets, surface stains included: Ly6C-FITC (clone HK1.4), F4/80-PE/Cy7 (BM8), Ly6G-APC/Fire750 (1A8), CD11b-Pacific Blue (M1–70). For Treg cells, surface stains included CD4-PE (GK1.5) and CD25-APC (PC61) and transcription factor staining was performed using FOXP3-eFluor450 (Invitrogen, FJK-16s). CD4+ T cells were stained using CXCR6-PE/Cy7 (SA051D1), CXCR5-APC (L138D7), PD1-APC/Cy7 (29F.1A12), CD44-Pacific Blue (IM7) and CD4-BV510 (RM4–5). CD8 T cells were stained using CD8-FITC (53–6.7), GP33 tetramer-PE (NIH Tetramer Core Facility), PD1-PE/Cy7 (RMP1–30), LAG3-PerCP/Cy5.5 (C9B7W), and CD44-APC/Cy7 (IM7). B220-PE-Cy7 (RA3–6B2), Fas-PE(SA367H8), GL7-FITC (GL7). Intracellular stains include: granzyme B-PE/CF594 (QA16A02) and IFNγ-FITC (XMG1.2) All antibodies were purchased from BioLegend, unless otherwise noted. Data were collected using a FACSLSRII or a FACSCelesta flow cytometer (BD Biosciences) with FACSDiva acquisition software. Data analysis was performed using FlowJo software (Tree Star, Inc.).
RNA sequencing:
Peripheral blood was obtained from a CD45.1+/+ congenic marked P14 TCR transgenic C57Bl/6 mouse and a CD45.1+/+ DGKα−/− P14 mouse by submandibular bleed. Buffy coat was isolated by density centrifugation (Histopaque-1077, Sigma-Aldrich), and CD8 T cells were enriched by immunomagnetic negative selection (EasySep Mouse CD8+ T cell Isolation Kit, StemCell Technologies). On microscopy, 100% viability was grossly observed post-enrichment by trypan blue staining. CD8 T cell suspension was diluted to 6 × 103 cells/mL. C57Bl/6 recipient mice were inoculated with 2.5 × 106 PFU of LCMV Clone 13 in 500 μL DMEM and either 3 × 103 CD45.1+/+ DGKα−/− P14 CD8 T cells or 3 × 103 CD45.1+/+ P14 CD8 T cells. Four biological replicates were included in each experimental condition. On day 8 post-infection (p.i.), mice were sacrificed, spleens were harvested, and single-cell suspension of splenocytes was obtained by mashing tissue against a cell strainer followed by erythrocyte lysis (ACK Lysis Buffer, Lonza). CD8 T cells were enriched by cell sorting (FACSMelody, BD) by gating on CD8+ Ly5.1+ 7AAD– cells. The recovered cell count of DGKα−/− P14 cells and wildtype P14 T cells were not significantly different. Following sorting, complementary DNA (cDNA) was prepared using established protocols.(16) One nanogram of amplified cDNA from each mouse was used to generate a DNA library using the Nextera XT DNA Library Preparation Kit (Illumina). Four replicates from each group were subsequently sequenced. Uniquely barcoded libraries were pooled and sequenced with a NextSeq 500 sequencer using a NextSeq 500/550 high output V2 75 cycle kit (Illumina) with the following configuration: read 1, 38 cycles; read 2, 38 cycles; index read 1, 8 cycles; index read 2, 8 cycles. Bulk RNA-seq data were aligned to the Mus musculus mm10 genome and gene expression was quantified using Salmon.(17) A single biological replicate of the wildtype cohort was excluded from subsequent analyses based on quality control. RNA-seq libraries were then normalized and genes were tested for differential expression between DGKα-deficient and wild-type CD8 T cells with DESeq2 v1.24.0.(18) DESeq2 Wald tests were used to determine whether fold changes were significantly different from zero. For visualization, data were transformed using the regularized logarithmic transformation.(18) Pre-ranked gene set enrichment analyses were conducted using shrunken fold-changes and clusterProfiler v3.12.0.(19) KEGG,(20) Reactome,(21) and GO(22) databases were used for gene set enrichment analysis (GSEA). The Benjamini-Hochberg method was used to adjust p-values for false-discovery in both differential expression and GSEA analyses.
Competitive adoptive transfer:
Peripheral blood was collected by facial bleed from age-matched, female P14 and P14 DGKα−/− and buffy coat was isolated by density centrifugation (Lymphoprep, STEMCELL Technologies). Each recipient mouse (C57Bl/6) received congenic marked 2×103 P14 CD8 T cells and 2×103 P14 DGKα−/− CD8 T cells, along with concomitant LCMV Clone 13 infection. On day 8 post-infection, spleens were harvested and processed into single cell suspension. One million cells per mouse were taken for staining and analysis by flow cytometry. An in vitro stimulation assay was also performed by taking 1×106 splenocytes per mouse, which were cultured with 0.1μg of gp33 monomer, Brefeldin A (Biolegend) and PE-CD107a (1D4B, Biolegend) for 6 hours at 37°C, 5% CO2, and then were subsequently processed for flow cytometry. Cellular fixation and intracellular staining were performed using the Cytofast Fix/Perm Buffer Set (BioLegend).
CD8 T cell depletion:
DGKα−/− mice were pretreated (day −1) with either 400 μg of CD8 depleting Ab (clone 2.43, InVivoMab) in 500 μL PBS or PBS alone by IP injection. Mice were subsequently infected with LCMV Clone 13 by IV injection on day 0. Mice were weighed thrice weekly, and analysis for CD8 T cell depletion was performed by flow cytometric analysis of peripheral blood on day 7. For the duration of the study, mice were redosed weekly with either 250ug of CD8 depleting antibody in PBS or equivalent volume of PBS.
Data and code availability
Bulk RNA sequencing data from this paper are available in the GEO database with accession code GSE186256 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE186256). All other raw data and scripts are available from the corresponding author upon request.
Results
LCMV Clone 13 infection leads to mortality and severe weight loss in DGKα−/− hosts
We first used a systemic loss-of-function model to determine the role of DGKα in Clone 13 infection. Following infection with LCMV Clone 13, DGKα−/− mice suffered mortality (Figure 1A), with a median survival time of 12 days. When combining all experimental replicates, 27% of the DGKα−/− cohort survived to the completion of the study, compared to 94% of the wildtype cohort (p < 0.0001). Mice deficient in DGKα also suffered more severe weight loss following Clone 13 infection (Figure 1B). This cohort demonstrated persistent progression of weight loss during the observed period, with a mean of 26% loss of baseline weight on day 12 p.i. versus 14% weight loss in WT mice on day 12 p.i. (p = 0.014).
Figure 1:
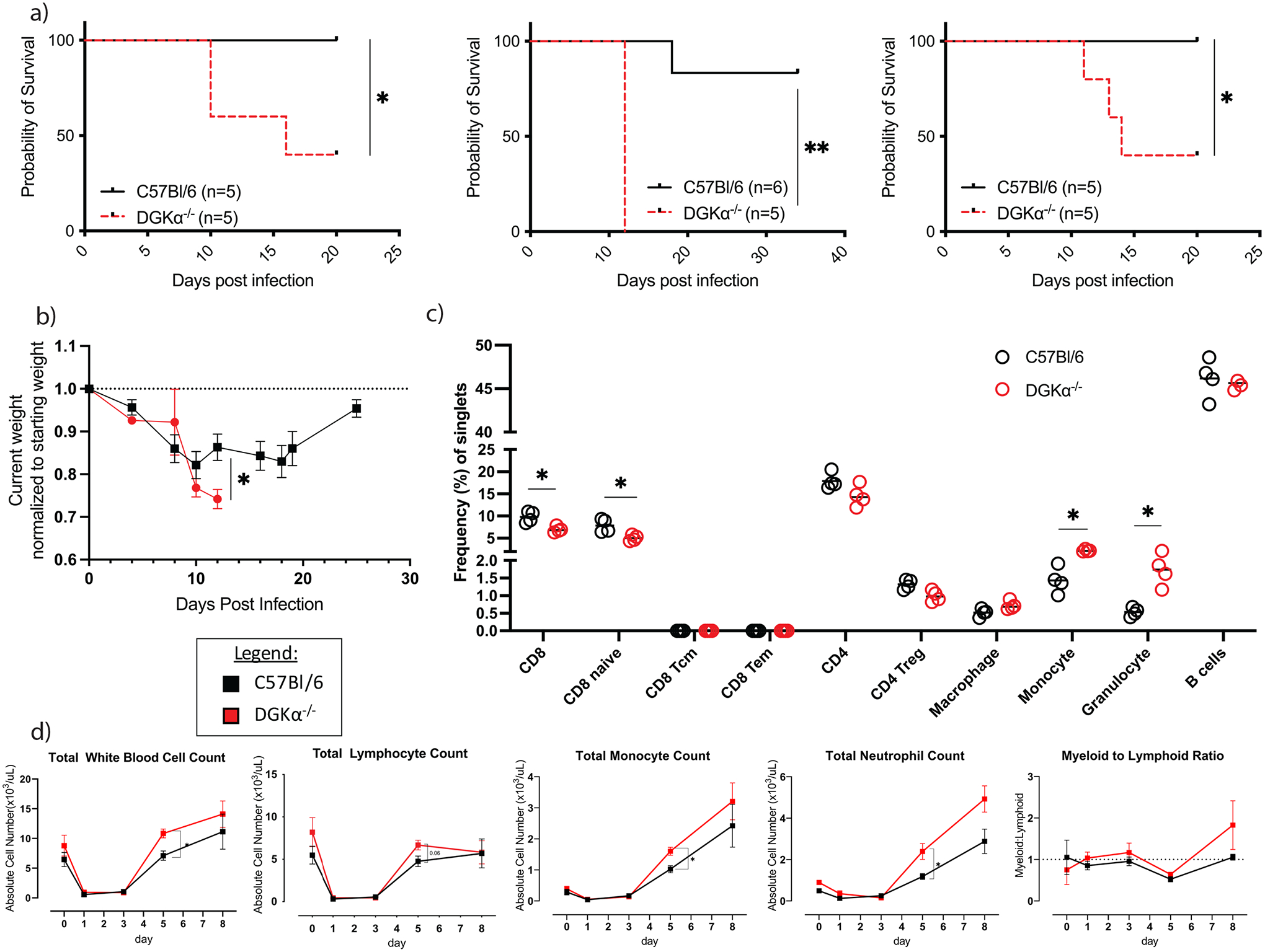
Characteristics of treatment cohorts following infection with LCMV Clone 13. a) Survival outcomes stratified by infection type in wildtype (C57Bl/6) or DGKα−/− host. Three replicate experiments with their subsequent survival outcomes are shown. b) Average change in host weight of treatment cohorts as a proportion of starting weight (n=5 per cohort, single experiment). c) Baseline composition of splenocytes from C57Bl/6 and DGKα−/− prior to infection with LCMV Cl 13. d) Total peripheral blood leukocyte, lymphocyte, monocyte, and neutrophil counts, and myeloid:lymphoid ratio prior to (day 0) and following LCMV Clone 13 infection. Peripheral blood counts are representative of n=6 mice per cohort from two experiments. Error bars, when present, represent standard error of the mean (SEM).
Clone 13 infection leads to enhanced peripheral immune cell expansion in DGKα−/− hosts
In the setting of this profound morbidity and mortality with LCMV Clone 13 infection, we hypothesized that the cause of death was an overwhelming immune response. To test this, we compared immune cell subsets for observable differences between the two groups at baseline. An immunophenotype of splenocytes at baseline (pre-infection) revealed small, but significant decreases in total CD8 T cells and naïve CD8 T cells in DGKα−/− mice and increases in monocyte and granulocyte subsets of myeloid cells (Figure 1C). Peripheral blood was sampled at pre-infection (day 0) and days 1,3,5, and 8 following LCMV Clone 13 infection (Figure 1D). On day 5 p.i., the DGKα−/− mice had a significantly higher average total white blood count, with significant increases in absolute monocyte and neutrophil (granulocyte) counts. There was also a nonsignificant trend towards the same in lymphocytes. Similar trends persisted through day 8 p.i., though they were not statistically significant. Post-infection myeloid to lymphoid cell ratios trended higher in the DGKα−/− group after infection compared to control (Figure 1D). As most changes in peripheral white cell counts of DGKα−/− persisted through the day 8 timepoint, shortly before mice first began to die around day 10 (Figure 1A), this suggested that a systemic proinflammatory state persisted until mortality.
Circulating analytes were subsequently compared in serum samples obtained from DGKα deficient mice and WT mice in an uninfected state (day 0) and day 8 p.i. with LCMV Clone 13 (Figure 2). There were no statistically significant differences between groups in assayed analytes at either time point when correcting for multiple hypotheses. An increase in IL-17 serum levels was observed in day 8 p.i. DGKα−/− mice as compared to day 8 p.i. wildtype mice (2.9 vs 0.43 pg/mL, respectively) with a trend towards significance (p=0.15). IL-17 is produced by activated T cells, and may mediate inflammation through recruitment of myeloid cells.(23)
Figure 2:

Serum measurements of selected cytokines at baseline, prior to infection (top, n=4 per group all collected at a single timepoint), and at day 8 post LCMV Clone 13 infection (bottom, n=3 per group all collected following a single infection). VEGF (day 0) and GM-CSF (day 8) serum levels were below the assayable range in all samples.
DGKα−/− increases myeloid derived cells in early Clone 13 infection and a blunted Tfh response
With the observed increases in white blood cell types in the peripheral blood, we next analyzed the role of DGKα in splenic immune cell subsets following Clone 13 infection. For this we performed flow cytometry on splenocytes harvested on day 8 p.i.. We elected to use day 8 p.i. as our timepoint to harvest splenocytes since host mortality began occurring as early as day 10 p.i., in DGKα−/− hosts (Figure 1A). Significant increases in frequency of myeloid cells were observed in DGKα−/− mice (Figure 3A–B). There was no difference observed in the frequency of splenic macrophages, monocytes, or granulocytes, suggesting that the increase in total myeloid cells was not due to a skewed response from a particular myeloid subset. (Figure 3C–G). Others have previously reported that Clone 13 infection leads to enhanced expansion of splenic monocytic and neutrophilic cells by day 7 p.i., when compared to Armstrong infection.(24) Increased hematopoiesis with myeloid differentiation is associated with elevated levels of cytokines including TNFα and IL-6.(25) We observed nonsignificant trends towards increase in these two cytokines in DGKα deficient mouse serum at day 8 p.i. compared to wildtype. Others have shown that splenic myeloid cells remain fully capable of stimulating T cell production in early (day 7 p.i.) Clone 13 infection,(24) which may contribute to our observed proinflammatory phenotype in our assessments at day 8 p.i.
Figure 3:
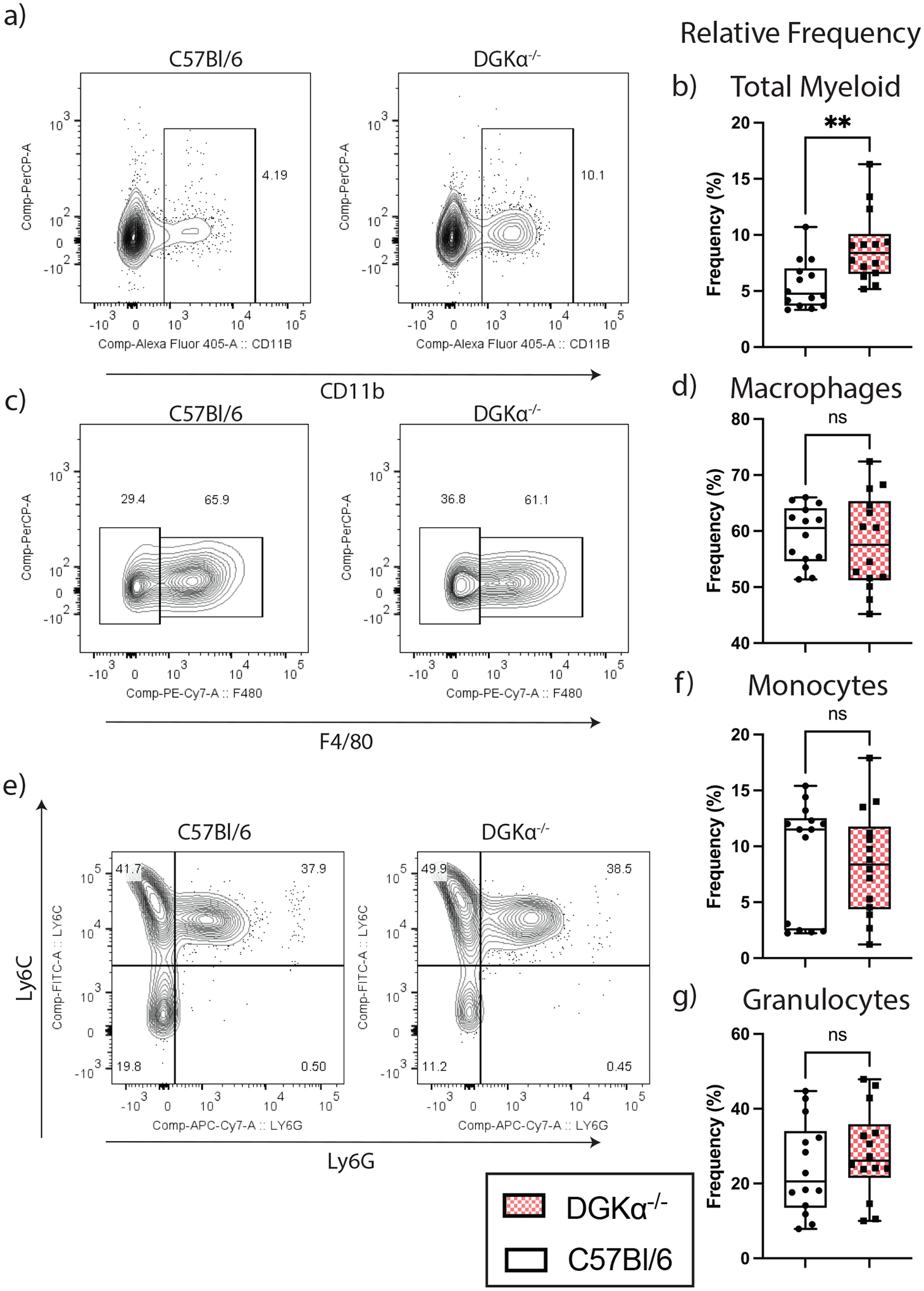
Relative frequencies of myeloid cell subsets among splenocytes on day 8 following LCMV Clone 13 infection. Representative flow plot and box and whisker plot for total splenic myelocytes (a-b), macrophages (c-d), monocytes, and granulocytes (e-g). Box plots demonstrate interquartile range and median; whisker error bars span from minimum to maximum values. Graphed relative frequency percentages are based on a ratio of the cell type listed to the following denominator listed in parenthesis: total myelocyte (singlet splenocytes); macrophages, monocytes and granulocytes (CD11b+ splenocytes). Data shown in the box and whisker plots are from fourteen mice total, aggregated from three independent infections.
We also compared the prevalence of Tregs between the wildtype and DGKα deficient states. As expected in an early Clone 13 infection, there was no significant difference in the frequency of Tregs between experimental states (Figure 4A–B). We further observed a decrease in the relative frequency of Tfh cells in DGKα−/− mice (Figure 4C–D), which is in line with previous research showing that virus-induced MDSCs can suppress Tfh responses.(26) Further, there was no difference in effector Th1 cells between genotypes (Figure 4E), which suggests that DGKα is not essential for limiting the Th1 cell activation in early chronic infection. Paradoxically, there was a significant increase in the frequency of germinal center B cells (GCB) within the spleen (Figure 4F–G). Previous studies have supported the role of DAG signaling in early plasma cell formation and enhanced antibody production when DGKζ is absent.(3) Our data suggests that DGKα may also be critical to attenuating GCB cell proliferation mediated by DAG signaling following Clone 13 infection, and this may be independent of Tfh interaction.
Figure 4:

Relative frequencies of CD4 lymphoid and germinal center (GCB) cell subsets among splenocytes on day 8 following LCMV Clone 13 infection. Representative flow plot and box and whisker plot for regulatory T cells (Treg) (a-b), follicular helper (Tfh) and type 1 helper (Th1) (c-e), and GCB (f-g). Box plots demonstrate interquartile range and median; whisker error bars from minimum to maximum values. Relative frequency percentages are based on a ratio of the cell type listed to the following denominator listed in parenthesis: Treg, Tfh, Th1 (CD4 splenocytes); GCB (B220 splenocytes). Data presented in a-e represent a single experiment and infection, whereas data in f-g are aggregated from three experiments with independent infections.
DGKα deficiency leads to enhanced CD8 T cell activation to early chronic viral infection and directly contributes to host mortality
Despite knowledge that DGKα functions to limit TCR signaling,(27) we did not identify a role for DGKα in limiting Th1 T cell expansion as a cause of a mortal immune response in early Clone 13 infection. We therefore hypothesized that DGKα was crucial to balancing the CD8 T cell response in a chronic viral infection to prevent a pathologic immune response. To test this, we examined splenic LCMV-specific CD8 T cells by flow cytometry. In contrast to our findings in cytotoxic CD4 T cells on day 8 p.i., we identified a significant increase in the relative frequency of activated, virus-specific CD8 T cells in the DGKα−/− host (Figure 5A–B). A significant reduction in serum viral titers in DGKα deficient mice occurred as compared to wildtype controls. (Figure 5C). Despite the enhanced cellular activation, we did not detect evidence of an increased exhaustion, as there was no difference in the mean fluorescent intensity (MFI) of the inhibitory receptors PD-1 and LAG-3 between knockout and wildtype CD44+ GP33 tetramer+ CD8 T cells (Figure 5D–E). These data suggest that a function of DGKα is to limit CD8 T cell activation, which is also a function of the zeta isoform.(28) As wildtype DGKζ is not inactivated in our system, this suggests an incomplete redundancy between these two isoforms in CD8 T cells. Finally, in the absence of DGKα, the increased frequency of activated, virus specific CD8 T cells and reduced serum viral titers suggests that in viral infection, DGKα function normally dampens viral clearance. This may exist to limit self-detrimental inflammation in this state.
Figure 5:

Relative and absolute frequencies of activated virus specific (gp33 tetramer bound) CD8 T cells on day 8 p.i. following LCMV Clone 13 infection. Representative flow plot and box and whisker plot for CD44+, gp33+ CD8 T cells (a-b), serum viral load (c), and mean fluorescent intensity (MFI) of PD-1 and LAG-3 checkpoint proteins. Box plots demonstrate interquartile range and median; whisker error bars from minimum to maximum values. Relative frequency percentages in Panel B are based on a ratio of the cell type listed to the following denominator listed in parenthesis: antigen specific CD8 T cells (CD8 splenocytes). Data presented are in a-d are aggregated from three experiments with independent infections. Data presented in e represents a single infection.
To determine the impact of an excessive CD8 T cell response to the mortality previously observed in early Clone 13 infection, we infected mice with LCMV Clone 13 that had previously received a chimeric bone marrow transplant with 7:3 admixture of marrow from a CD8−/− donor and either a WT or DGKα-deficient donor, respectively. Following reconstitution, this model provides selective ablation of DGKα in the CD8 T cell compartment, while the remainder of hemopoietic elements remain wild type (Figure 6A). In our system, following reconstitution of the chimera and prior to infection, peripheral blood CD8 T cells were observed at similar frequencies between the recipients of wildtype and DGKα−/− CD8 T cells. Further, the CD8 T cell compartment demonstrated a chimera of donor and recipient cells, where at least 50% of the circulating CD8 T cells were derived from the donor (Supplemental Figure 1). After infection with LCMV Clone 13, mice with DGKα−/− CD8 T cells suffered increased mortality as compared to controls (Figure 6B). The median survival time following Clone 13 infection among mice with DGKα deficiency restricted to CD8 T cells was 23 d.p.i. The findings in this chimeric system suggest that DGKα intrinsically limits CD8 T cell mediated immunopathology, and its absence in CD8 T cells directly contributes to host death following LCMV Clone 13 infection.
Figure 6:
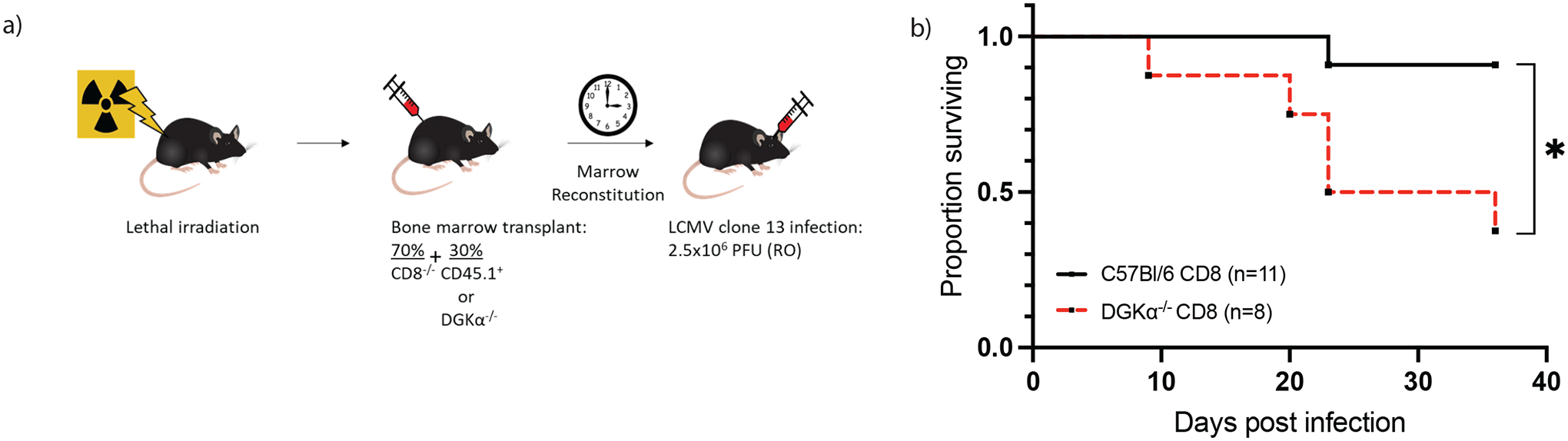
Characteristics of treatment cohorts following infection with LCMV Clone 13. a) Generation of bone marrow chimera. Following irradiation, and CD8 T cell depletion, mice were delivered a mixed bone marrow suspension by intravenous injection to generate hosts with DGKα deficiency restricted to CD8 T cells (n=8) or control (n=11). b) Survival outcomes in bone marrow chimeric mice stratified by presence or absence of DGKα in CD8 T cells. Data presented were collected from two independent experiments and infections.
Selective ablation of DGKα−/− CD8 T cells rescues mice from immune dysregulated mortality following LCMV Clone 13 infection.
To further characterize the contribution of DGKα in mitigating CD8 T cell mediated host mortality, we evaluated how selective, sustained depletion of CD8 T cells from mice globally deficient in DGKα would impact survival after Clone 13 infection (Figure 7A). One day prior to LCMV Clone 13 infection and every 7 days thereafter, mice were treated with CD8 depleting antibody. Mice dosed with antibody were confirmed to have complete loss of CD8 T cells (Figure 7B–C) one week after initial antibody dosing. As expected, mice that were depleted of CD8 T cells were rescued from mortality following Clone 13 infection, while those who received PBS control all died (Figure 7D). CD8 depleted mice also suffered significantly less weight loss than the CD8 replete group (Figure 7E) and appeared more alert, active, and groomed than the CD8 replete group. These results suggest that DGKα−/− CD8 T cells are necessary for the demise and mortality observed following LCMV Cl 13 infection in the global DGKα−/− system.
Figure 7:

CD8 T cell depletion rescues mice globally deficient in DGKα from lethality following LCMV Clone 13 infection. Figure is representative of two experiments with independent infections and a total of n=7 CD8 depleted and n=8 CD8 replete (control) mice. (a) Experimental schema. (b) Representative flow plot of CD8 analysis by peripheral blood on day 8 post infection demonstrates absence of CD8 T cells in depleted host. (c) Comparison of frequency of CD8 and CD4 T cells recovered from peripheral blood on day 7 post CD8 T cell depletion. Relative frequency percentage is based on a ratio of the cell type listed to the total singlet splenocytes collected by flow cytometry as the denominator. (d) Kaplan-Meier survival curve stratified by CD8 depleted or replete hosts. (e) Change in weight following infection stratified by CD8 depleted or replete hosts. Each data point is the mean weight; error bars represent standard error of measurement (SEM).
DGKα limits signaling pathways associated with cell replication and effector function
To correlate the DGKα−/− phenotype of CD8 T cells in early Clone 13 infection to an associated gene signature, we analyzed CD8 T cells with transgenic TCR specificity for an LCMV epitope, gp33, restricted to the murine MHC class I H-2Db haplotype, known as P14 CD8 T cells.(29) Eight days following Clone 13 infection and adoptive transfer of either wildtype P14 or DGKα−/− P14 CD8 T cells into wildtype recipients, P14 T cells were enriched by flow cytometry-based sorting of splenocyte single cell suspension by congenic marker for bulk RNA sequencing.
Principal component analysis (PCA) of wildtype P14 and DGKα−/− P14 biologic replicates demonstrated separation of the two phenotypes along PC1 (Figure 8A). Gene set enrichment analysis (GSEA) was performed to identify differences in cellular signaling pathways observed among DGKα−/− CD8 T cells compared to their wildtype counterparts. Of note, pathways enriched in DGKα deficient CD8 T cells included multiple unique cell cycle signaling pathways, including gene sets related to cellular entry into prometaphase, assembly of mitotic spindles, and progression of cells through the S, G2, and M phases (Figure 8B). A Venn diagram demonstrated the uniqueness of the enriched identified gene sets, with a 0–52% pairwise overlap between gene sets (Figure 8C). Additionally, genes associated with signaling through the mammalian target of rapamycin complex 1 (mTORC1) were also significantly enriched in the experimental cohort (NES = 1.53, padj=0.028) (Supplemental Figure 2A–B). Virus-specific DGKα−/− P14 CD8 T cells also demonstrated enhanced transcription of genes associated with glycolytic metabolism, effector function, and TCR signaling (Figure 8D). These findings correlate with our phenotypic observation that DGKα functions to limit the expansion of virus specific CD8 T cells (Figure 5B) and suggests that this occurs through restriction of TCR signaling pathways and shunting cellular metabolism away from glycolytic processes.
Figure 8:

Bulk RNA sequencing analysis of DGKα−/− and wildtype, virus specific CD8 T cells. On day 8 following infection with LCMV Clone 13 and adoptive transfer of either congenic marked P14+ (n=3) or P14+, DGKα−/− (n=4) CD8 T cells, congenic marked CD8 splenocytes from each mouse were harvested and processed to generate a transcriptome library for bulk RNA sequencing. (a) Principal component analysis of biologic replicates shows separation between experimental groups by first principal component (PC1). (b) Gene sets enriched among P14+DGKα−/− CD8 T cells relative to wildtype P14 CD8 T cells. (c) Venn diagram demonstrating frequency of distinct genes among gene sets. (d) Heatmap of selected genes sorted by associated function.
To corroborate the observed transcriptional profiles with the phenotypic function of DGKα, we compared functional characteristics of wildtype- and DGKα deficient-P14 CD8 T cells in a competitive, in vivo setting. In an unstimulated state, wildtype P14 and DGKα P14 CD8 T cell subsets of naïve (CD62L+, CD44−), effector memory (CD62L−, CD44+), and central memory (CD62L−, CD44+) were assessed and found to be in similar proportions to each other (Supplemental Figure 3). Following isolation of peripheral blood mononuclear cells from a P14 and P14-DGKα−/− donor, PBMC counts from each were normalized so that 2×103 of each of P14 and P14DGKα−/− CD8 T cells were co-adoptively transferred to recipient C57Bl/6 hosts, and inoculation of LCMV Cl 13 was performed (Figure 9A). Splenocytes collected on day 8 p.i. were analyzed by flow cytometry (Figure 9B), revealing significantly elevated numbers of tetramer positive, DGKα deficient P14 T cells compared to P14 wildtype CD8 T cells (Figure 9C). Further, a significant increase in granzyme B was observed on a per cell basis among DGKα−/− CD8 T cells (Figure 9D), and CD8 T cells lacking DGKα were the primary contributors of IFNγ (Figure 9E). DGKα deficient P14 CD8 T cells also had enhanced per-cell production of IFNγ as evidenced by MFI (Figure 9F). These data confirm an enhanced effector phenotype of DGKα−/− P14 CD8 T cells as compared to P14 CD8 T cells following LCMV Clone 13 infection and support the observed changes in enhanced transcription of effector molecules in CD8 T cells in the absence of DGKα.
Figure 9:

In vivo co-adoptive transfer of congenic marked P14+ and P14+DGKα−/− enables direct comparison of virus-specific CD8 T cell expansion following LCMV Clone 13 infection. (a) Experimental schema. Splenocytes harvested and analyzed on day 8 post infection and adoptive cell transfer. (b) Representative flow plots depicting gating strategy to identify P14+ and P14+DGKα−/− cell subsets among gp33 tetramer positive (top row), mean fluorescent index (middle) and interferon gamma (IFNg) producing CD8 T cells (bottom). (c) Paired analysis of composition of tetramer positive CD8 T cells by origin. (d) Mean fluorescent index (MFI) of granzyme B among activated CD8 T cells. (e) Paired analysis of the composition of IFNγ producing CD8 T cells by origin reveals P14+DGKα−/− cells are principally responsible for IFNγ production in this system. Presented results are aggregated data of two separate experiments comprising a total of 8 recipient mice.
In summary, our data demonstrate that the deficiency of DGKα in a murine host leads to mortality following LCMV Clone 13 infection. While the overwhelming immune response is likely multicellular in nature, CD8 T cells are a dominant effector of immune pathology in this system. Further DGKα−/− CD8 T cells are necessary for host mortality to occur following LCMV Clone 13 infection.
Discussion
We have demonstrated that DGKα is an essential regulator of CD8 T cell antiviral responses and plays an important role in balancing antiviral immunity with immunopathology in Clone 13 viral infection. Following Clone 13 infection, we observed profound host mortality beginning in the second week of infection. This mortality occurred following Clone 13 infection in both systemic and CD8 T cell restricted DGKα knockouts. The absence of DGKα regulation of TCR signaling enhances viral clearance, and leads to increased splenic CD44+, virus specific CD8 T cells. This profound activation did not appear to promote T cell exhaustion, as we did not observe increased expression of checkpoint proteins PD-1 and LAG-3. Finally, our data suggests that DGKα deficiency enhances mTOR cell signaling in virus specific CD8 T cells, and enhances transcription of genes associated with glycolytic metabolism, TCR signaling, and cytotoxic effector functions.
Previous studies have analyzed the role of DGKα in acute viral infection.(14) Like this study, we observed increases in virus specific CD8 T cells among splenocytes, and enhanced viral clearance in DGKα knockout. However, a key distinction between our study and studies evaluating the role of DGKα in acute viral infection is the profound mortality that occurred with Clone 13 infection. Oldstone, et al., recently demonstrated that Clone 13 infection also resulted in acute death in mouse models of immune dysregulation, including FVB, NZB and PL/J strains.(30) In these studies, mice had high numbers of antiviral CD8 T cells, increased levels of type I IFN (IFN-I), with death occurring as a result of enhanced pulmonary vascular permeability. Interestingly, blockade of IFN-I signaling CD8 prevented host death, as did CD8 T cell depletion. We similarly observed that in global DGKα knockout, depletion of CD8 T cells prevented host death after LCMV Clone 13 infection.
While studies commonly focus on the role of T cells in immune pathology, they are likely not the exclusive cause of immunopathology and mortality. In the absence of DGKα, effector CD8 T cells are a dominant driver of immune pathology but are just one component of a dysregulated multicellular immune process that results in a lethal phenotype, as changes in the frequencies of myeloid, CD4 T cell and B cell subtypes were also observed in our model.
A key limitation of our study is that we did not identify an organ-based cause of death in our model. Future studies incorporating tissue histology of vital organs including lung, liver and kidney may identify an end-organ dysfunction that may contribute to the cause of death. In our model, though we did not analyze serum IFN-I levels, we observed nonsignificant elevations in serum TNFα, which is a known proinflammatory cytokine and is associated with the pathogenesis of several autoimmune disease, including rheumatoid arthritis, inflammatory bowel disease and psoriatic arthritis.(31) We also observed a significant increase in the circulating levels of IL-17 following LCMV Clone 13 infection in mice with DGKα deficiency as compared to wildtype. Others have shown that the combined absence of both α and ζ isoforms of DGK results in enhanced differentiation of Th17 CD4 T cells, but does not occur if there is an isolated absence of either isoform.(32) As IL-17 is produced by a number of different immune cell subsets, including CD8 T cells,(33) future studies of selective deficiency of DGKα in CD8 T cells can explore how IL-17 producing CD8 T cells may specifically contribute to this phenotype.
A potential mechanism of how DGKα deficient CD8 T cells directly contribute to host mortality in Clone 13 infection may occur through resistance of DGKα−/− T cells to regulated TCR signaling. Following TCR mediated activation of CD8 T cells, protein tyrosine phosphatases (PTPs) SHP-1 and PTPN22 are recruited to the cell membrane to inactivate TCR-mediated phosphorylated Lck signaling. However PTP dephosphorylation of Lck can be avoided, as strong TCR-mediated activation of CD8 T cells also triggers rapid and sustained ERK activity, which enables resistance of Lck protein to PTP-mediated dephosphorylation by altering the conformational structure of Lck.(34) While our transcriptomic data demonstrate upregulation of SHP-1 and PTPN22 PTPs in DGKα−/− CD8 T cells, our data also demonstrate transcriptional upregulation of ERK (encoded by Mapk1), suggesting that a potential mechanism of sustained TCR signaling occurs as a result of DAG-mediated rapid ERK production. In the absence of DAG inactivation from cleavage of DAG into PA by DGKα, sustained ERK production may inhibit PTPs from performing their usual function. Future studies characterizing temporal changes in intracellular DAG, ERK and phosphorylated Lck levels following TCR activation of DGKα deficient CD8 T cells would provide data to support this hypothesis.
In summary, DGKα is an indispensable regulator of TCR-mediated cellular activation in LCMV Clone 13 viral infection. We have demonstrated that DGKα functions to modulate cellular components of the adaptive immune response to early Clone 13 viral infection, most notably in CD8 T cells. When DGKα is absent, Clone 13 infection results in excessive cell cycling and proinflammatory processes, likely through upregulated mTORC1, AP-1, and NF-κB mediated pathways. Subsequently, infected mice succumb to a pathologic proinflammatory state and demise.
Supplementary Material
Key Points.
DGKα is required to prevent immune mediated mortality in LCMV Clone 13 infection.
Immunopathologic host death in DGKα knockout is predominantly CD8 T cell mediated.
Depleting DGKα−/− CD8 T cells rescues mice from mortality in LCMV Clone 13 infection.
Acknowledgements
This research was completed in part with computational resources and technical support provided by the Research Computing Center at MCW. Some aspects of figures were created with BioRender.com.
Grant Support:
M.R.K. is supported by a St. Baldrick’s Foundation Fellowship (812679). M.Y.K. is a member of the Medical Scientist Training Program at the Medical College of Wisconsin (MCW), which is partially supported by a training grant from NIGMS (T32-GM080202). W.C. is supported by NIH grants AI125741 and AI148403, by an American Cancer Society Research Scholar Grant, and by an Advancing a Healthier Wisconsin Endowment Grant.
Footnotes
The authors declare no potential conflicts of interest.
Disclosure of Potential Conflicts of Interest
The authors have no financial conflicts of interest to disclose.
References
- 1.Wright DB, Tripathi S, Sikarwar A, Santosh KT, Perez-Zoghbi J, Ojo OO, Irechukwu N, Ward JPT, and Schaafsma D. 2013. Regulation of GPCR-mediated smooth muscle contraction: Implications for asthma and pulmonary hypertension. Pulm. Pharmacol. Ther 26: 121–131. [DOI] [PubMed] [Google Scholar]
- 2.Joshi RP, and Koretzky GA. 2013. Diacylglycerol kinases: Regulated controllers of T cell activation, function, and development. Int. J. Mol. Sci 14: 6649–6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wheeler ML, Dong MB, Brink R, Zhong XP, and De Franco AL. 2013. Diacylglycerol kinase ζ limits B cell antigen receptor-dependent activation of ERK Signaling to inhibit early antibody responses. Sci. Signal 6: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen SS, Hu Z, and Zhong XP. 2016. Diacylglycerol kinases in T cell tolerance and effector function. Front. Cell Dev. Biol 4: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chauveau A, Le Floc’h A, Bantilan NS, Koretzky GA, and Huse M. 2014. Diacylglycerol kinase α establishes T cell polarity by shaping diacylglycerol accumulation at the immunological synapse. Sci. Signal 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mérida I, Andrada E, Gharbi SI, and Ávila-Flores A. 2015. Redundant and specialized roles for diacylglycerol kinases α and ζ in the control of T cell functions. Sci. Signal 8: 1–12. [DOI] [PubMed] [Google Scholar]
- 7.Sakane F, Mizuno S, and Komenoi S. 2016. Diacylglycerol kinases as emerging potential drug targets for a variety of diseases: An update. Front. Cell Dev. Biol 4: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weber H, Kittel-Schneider S, Gessner A, Domschke K, Neuner M, Jacob CP, Buttenschon HN, Boreatti-Hümmer A, Volkert J, Herterich S, Baune BT, Gross-Lesch S, Kopf J, Kreiker S, Nguyen TT, Weissflog L, Arolt V, Mors O, Deckert J, Lesch KP, and Reif A. 2011. Cross-disorder analysis of bipolar risk genes: Further evidence of dGKH as a risk gene for bipolar disorder, but also unipolar depression and adult ADHD. Neuropsychopharmacology 36: 2076–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sim JA, Kim J, and Yang D. 2020. Beyond lipid signaling: Pleiotropic effects of diacylglycerol kinases in cellular signaling. Int. J. Mol. Sci 21: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olenchock BA, Guo R, Carpenter JH, Jordan M, Topham MK, Koretzky GA, and Zhong XP. 2006. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat. Immunol 7: 1174–1181. [DOI] [PubMed] [Google Scholar]
- 11.Arranz-Nicolás J, Ogando J, Soutar D, Arcos-Pérez R, Meraviglia-Crivelli D, Mañes S, Mérida I, and Ávila-Flores A. 2018. Diacylglycerol kinase α inactivation is an integral component of the costimulatory pathway that amplifies TCR signals. Cancer Immunol. Immunother 67: 965–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Macián F, García-Rodríguez C, and Rao A. 2000. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. EMBO J 19: 4783–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atsaves V, Leventaki V, Rassidakis GZ, and Claret FX. 2019. AP-1 transcription factors as regulators of immune responses in cancer. Cancers (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shin J, O’Brien TF, Grayson JM, and Zhong X-P. 2012. Differential Regulation of Primary and Memory CD8 T Cell Immune Responses by Diacylglycerol Kinases. J. Immunol 188: 2111–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kahan SM, and Zajac AJ. 2019. Immune exhaustion: Past lessons and new insights from lymphocytic choriomeningitis virus. Viruses 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, and Sandberg R. 2014. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9: 171–181. [DOI] [PubMed] [Google Scholar]
- 17.Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14: 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Love MI, Huber W, and Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu G, Wang LG, Han Y, and He QY. 2012. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol 16: 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanehisa M, and Gogo S. 2000. Kegg: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, Milacic M, Roca CD, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Viteri G, Weiser J, Wu G, Stein L, Hermjakob H, and D’Eustachio P. 2018. The Reactome Pathway Knowledgebase. Nucleic Acids Res 46: D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The Gene Ontology Consortium. 2000. Gene Ontology : tool for the unification of biology. Gene Expr 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mills KHG 2008. Induction, function and regulation of IL-17-producing T cells. Eur. J. Immunol 38: 2636–2649. [DOI] [PubMed] [Google Scholar]
- 24.Norris BA, Uebelhoer LS, Nakaya HI, Price AA, Grakoui A, and Pulendran B. 2013. Chronic but Not Acute Virus Infection Induces Sustained Expansion of Myeloid Suppressor Cell Numbers that Inhibit Viral-Specific T Cell Immunity. Immunity 38: 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maltby S, Hansbro NG, Tay HL, Stewart J, Plank M, Donges B, Rosenberg HF, and Foster PS. 2014. Production and Differentiation of Myeloid Cells Driven by Proinflammatory Cytokines in Response to Acute Pneumovirus Infection in Mice. J. Immunol 193: 4072–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C, Zhang N, Qi L, Yuan J, Wang K, Wang K, Ma S, Wang H, Lou W, Hu P, Awais M, Cao S, Fu ZF, and Cui M. 2017. Myeloid-Derived Suppressor Cells Inhibit T Follicular Helper Cell Immune Response in Japanese Encephalitis Virus Infection. J. Immunol 199: 3094–3105. [DOI] [PubMed] [Google Scholar]
- 27.Sanjuán MA, Jones DR, Izquierdo M, and Mérida I. 2001. Role of diacylglycerol kinase α in the attenuation of receptor signaling. J. Cell Biol 153: 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong XP, Hainey EA, Olenchock BA, Jordan MS, Maltzman JS, Nichols KE, Shen H, and Koretzky GA. 2003. Enhanced T cell responses due to diacylglycerol kinase ζ deficiency. Nat. Immunol 4: 882–890. [DOI] [PubMed] [Google Scholar]
- 29.Pirchner H, Moskophidis D, Rohrer U, Burki J, Hengartner H, and Zinkernagel RM. 1990. Viral escape by selection of cytotoxic T cell-resistant variants in vivo. Nature 346: 629–633. [DOI] [PubMed] [Google Scholar]
- 30.Oldstone MBA, Ware BC, Horton LE, Welch MJ, Aiolfi R, Zarpellon A, Ruggeri ZM, and Sullivan BM. 2018. Lymphocytic choriomeningitis virus Clone 13 infection causes either persistence or acute death dependent on IFN-1, cytotoxic T lymphocytes (CTLs), and host genetics. Proc. Natl. Acad. Sci. U. S. A 115: E7814–E7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jang DI, Lee AH, Shin HY, Song HR, Park JH, Kang TB, Lee SR, and Yang SH. 2021. The role of tumor necrosis factor alpha (Tnf-α) in autoimmune disease and current tnf-α inhibitors in therapeutics. Int. J. Mol. Sci 22: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang J, Wang HX, Xie J, Li L, Wang J, Wan ECK, and Zhong XP. 2020. DGK α and ζ Activities Control TH1 and TH17 Cell Differentiation. Front. Immunol 10: 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srenathan U, Steel K, and Taams LS. 2016. IL-17+ CD8+ T cells: Differentiation, phenotype and role in inflammatory disease. Immunol. Lett 178: 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fowler CC, Pao LI, Blattman JN, and Greenberg PD. 2010. SHP-1 in T Cells Limits the Production of CD8 Effector Cells without Impacting the Formation of Long-Lived Central Memory Cells. J. Immunol 185: 3256–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNA sequencing data from this paper are available in the GEO database with accession code GSE186256 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE186256). All other raw data and scripts are available from the corresponding author upon request.