

Abstract
Delivering the virus genome into the host nucleus through the nuclear pore complex (NPC) is pivotal in human immunodeficiency virus 1 (HIV-1) infection. The mechanism of this process remains mysterious owing to the NPC complexity and the labyrinth of molecular interactions involved. Here we built a suite of NPC mimics—DNA-origami-corralled nucleoporins with programmable arrangements—to model HIV-1 nuclear entry. Using this system, we determined that multiple cytoplasm-facing Nup358 molecules provide avid binding for capsid docking to the NPC. The nucleoplasm-facing Nup153 preferentially attaches to high-curvature regions of the capsid, positioning it for tip-leading NPC insertion. Differential capsid binding strengths of Nup358 and Nup153 constitute an affinity gradient that drives capsid penetration. Nup62 in the NPC central channel forms a barrier that viruses must overcome during nuclear import. Our study thus provides a wealth of mechanistic insight and a transformative toolset for elucidating how viruses like HIV-1 enter the nucleus.
To infect a host cell during interphase, human immunodeficiency virus 1 (HIV-1) must deliver its genome into the nucleus through the nuclear pore complex (NPC)1. NPCs form massive protein channels spanning the nuclear membranes and control the molecular exchange between cytoplasm and the nucleus2,3. The HIV-1 genome is housed inside the viral capsid, a cone-shaped protein shell that is ~120 nm long and ~60 nm wide. The capsid is assembled from ~250 copies of capsid protein (CA) hexamers and 12 CA pentamers4 and has a multitude of functions, such as protecting the viral genome, assisting in reverse transcription and facilitating the transport of virus from the cell periphery to the nucleus5–7. Although the HIV-1 capsid was previously thought to disassemble before entering the nucleus6,8,9, recent live-cell imaging, cryogenic electron tomography (cryo-ET) and biochemical studies have shown that HIV-1 capsids remain largely intact during the passage through the NPC, and even after nuclear entry10–16. These striking observations have raised fundamental questions about the mechanism of HIV nuclear import, including how the virus core could interact with and penetrate the NPC channel. The molecular interactions responsible for moving the virus capsid across the NPC, a conduit gated by multiple nucleoporin (nup) proteins, have not been elucidated. Delineating the molecular determinants and mechanisms of this vital process is essential to understanding HIV infection and developing new antiviral therapies.
Previous studies on the nuclear entry mechanisms of the HIV-1 capsid have primarily relied on two approaches. The first one is cell-based experiments, wherein capsid-NPC interactions are genetically or pharmacologically perturbed and virus trafficking and infectivity are monitored, to determine the role of certain nups or transport cofactors12,14,17–24. However, the complexity of the NPC has limited the capability of cell-based approaches to dissect the vast diversity of molecular interactions at the entry point into the nucleus. The human NPC contains ~30 types of nups, each with 8–48 copies distributed in eightfold symmetry2,25. To enter the nucleus, the capsid must pass through a 40- to 65-nm-wide channel at the center of the NPC11,26–28, breaching a diffusion barrier formed by hundreds of nups3,25. Although several CA-binding nups, including Nup358, Nup214, Nup88, Nup62 and Nup153, have been identified17,29–34, delineating their functional interactions with CA by conventional genetic methods has proven difficult owing to functional nup redundancy and off-target reductions that can occur through RNA interference17. Another approach to study HIV-1 nuclear entry leverages biochemical and structural-biology techniques to investigate CA-nup interactions14,17,29–32,35 in a cell-free environment. Although they are capable of determining binding specificity and high-resolution depictions of the binding events29,30,32,35,36, such approaches fall unacceptably short of reconstituting the multiple binding events that must occur during the interaction of the multivalent viral core with the NPC within the cell. Therefore, unlocking the mechanistic details of HIV-1 nuclear entry calls for a cell-free model system that robustly recapitulates the native environment of nuclear pores.
To bridge this gap, here we build DNA-origami-based NPC mimics to study the mechanism of HIV-1 nuclear transport. DNA-origami nanostructures feature well-defined shapes and chemically addressable surfaces, enabling the precise organization of biomolecules on self-assembled DNA scaffolds with nanoscale precision37–40. Recently, we and others have assembled selected nups in DNA-origami channels, which we term NuPODs (nups organized by DNA), to study the collective morphology, dynamics and function of nups in an NPC-like nano-environment41,42. Importantly, we showed that even a minimalist NPC mimic was able to recapitulate the essential function of the NPC as a selective barrier43. In this work, we expanded our NuPOD platform by building NPC mimics with greater structural and biochemical complexity. Specifically, we decorated DNA-origami channels (40–60 nm) with representative CA-binding nups at the NPC cytoplasmic ring (Nup358), central channel (Nup62) and nuclear basket (Nup153)3,44 and systematically varied the type, density and positioning of these nups to study their individual and collective contributions to HIV-1 capsid transport under NPC-like geometrical constraints. By analyzing the multivalent capsid-NuPOD interactions using a co-pelleting assay and electron microscopy (EM), our study delineates differential nup properties, including their capsid-binding strengths and curvature preferences. Uniquely enabled by the NuPOD system, we reveal a minimum diameter requirement for capsid penetration, as well as an intrinsic affinity gradient within the NPC that may drive capsid nuclear import. Our study suggests that the conical shape of the HIV-1 capsid and the nup organization within the NPC conspire to facilitate viral nuclear entry.
Results
Building DNA-origami channels as the scaffold for NPC mimics
Although we have previously established a minimalist NuPOD platform41,43, we sought to build more realistic NPC mimics to experimentally model the nuclear entry of the HIV-1 capsid. Our goal was to build an extended library of NuPODs to house a combination of capsid-interacting nups in various well-controlled configurations that either resemble the native NPC or purposely deviate from their natural arrangement to delineate critical functional elements. We continued to use our tried-and-tested cylindrical DNA channel41 (Fig. 1a and Extended Data Fig. 1; inner diameter, 45 nm; depth, 14 nm) to build NuPODs with single nup species, but closer mimics of the NPC necessitated deeper channels with more nup-anchoring positions. To this end, we designed two octagonal shaped DNA-origami channels that are ~30 nm deep and 40 nm or 60 nm wide (Fig. 1b,c and Extended Data Fig. 1). The 40-nm and 60-nm channels display up to 48 or 96 inward-facing single-stranded DNA (ssDNA) extensions (termed ‘handles’), respectively, with customizable handle sequences. These DNA-origami channels can accommodate up to two (40-nm channel, 24 copies/layer) or three (60-nm channel, 32 copies/layer) layers of nups. The 40-nm and 60-nm DNA-origami channels recapitulate the different reported transport channel widths of the human NPC and were accordingly designed to probe the influence of pore size on capsid penetration11,26–28. Both channels feature an octagonal cross-section to mimic the NPC’s eightfold rotational symmetry25 and carry protrusions on the exterior to serve as indexes for identifying the orientation of NuPODs under EM. Following our typical DNA-origami assembly, purification and characterization workflow (see Methods)41,45, we produced highly homogeneous DNA-origami channels. Gel electrophoresis (Extended Data Fig. 2) and negative-stain EM (Fig. 1, right column) confirmed the purity and the expected geometry of the DNA-origami channels.
Fig. 1 |. Design and assembly of DNA-origami channels as mimics of the NPC scaffold.
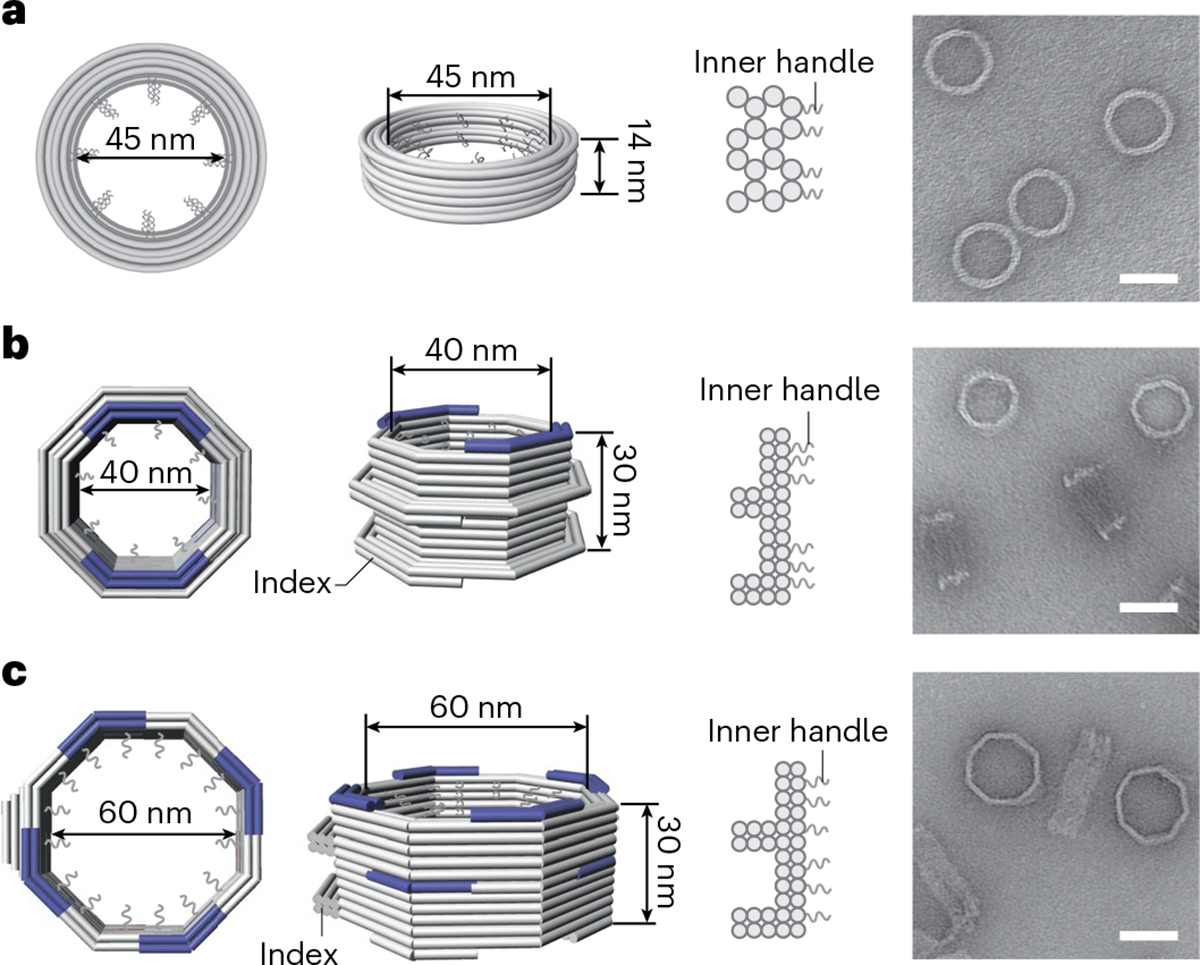
a, A DNA cylinder with an inner diameter of 45 nm, dubbed 45-nm channel. b, An open-ended DNA octagonal prism with an inner diameter of 40 nm, dubbed 40-nm channel. c, An open-ended DNA octagonal prism with an inner diameter of 60 nm, dubbed 60-nm channel. The 40-nm (b) and 60-nm (c) channels are designed with protruded regions on the side, or indexes, to mark the polarity of the channel, as well as shape-complementary interfaces (blue) at the top and bottom to facilitate vertical dimerization. Shown from left to right are cartoon models of the DNA-origami channels (in right, perspective and cross-section views) and representative negative-stain electron micrographs. All negative-stain EM experiments were repeated three times with similar results. The gray curls represent ssDNA extensions (handles) for organizing desired nups. Scale bar, 50 nm.
DNA-origami-guided assembly of nups in an NPC-like channel
To interrogate capsid-nup interactions within nanopore confinement, we first generated NuPODs with nups attached to the inner surface of the 45-nm DNA-origami channel41. We designed 32 handles inside the channel, with which complementary DNA oligonucleotides (anti-handles) conjugated to nups can hybridize, on the basis of previous findings that 32 copies of Nup358, Nup62 and Nup153, the three CA-binding nups under investigation (Fig. 2a) are present at the cytoplasmic face, the central channel and the nuclear face of the NPC, respectively25,46. We expressed recombinant proteins containing the known capsid-binding domains (Nup358RBD4-CycH, aa 2911–3225 and Nup153CTD, aa 896–1475, Fig. 2a) or the full-length protein (Nup62FL, aa 1–522, Fig. 2a), as its capsid-binding domain has not yet been characterized14,17,18,29,32,35 (Fig. 2a and Extended Data Fig. 3). For Nup153 and Nup62, an amino-terminal fusion of maltose-binding protein (MBP) was used to enhance solubility and stability. In addition to the three human nups, we used the phenylalanine-glycine rich domain (FG domain) of Nsp1 (amino acids (aa) 2–603), the yeast ortholog of Nup6247, as a negative control. Each nup was fused to a SNAP-tag at the carboxy terminus, which reacts with a benzylguanine-labeled DNA anti-handle to efficiently (>95% yield) form 1:1 nup–anti-handle conjugates43. After incubating anti-handle-conjugated nups with handle-displaying 45-nm channels, we used gel electrophoresis (Fig. 2b and Extended Data Fig. 4) and negative-stain EM to confirm NuPOD assembly. Notably, the DNA-encircled nups exhibited different morphologies, likely reflecting their inherent molecular properties (Fig. 2c). The folded Nup358 domains appeared as small puncta along the DNA channel’s inner rim; the other nups, containing unstructured FG domains, appeared to be more diffuse. Furthermore, cohesive Nup153 and Nup62 formed a dense condensate inside the DNA channel, whereas the less cohesive Nsp1 showed less contrast under negative-stain EM, in accordance with previous findings41,42.
Fig. 2 |. Interactions between HIV-1 capsids and NuPODs containing copies of a single nup species.
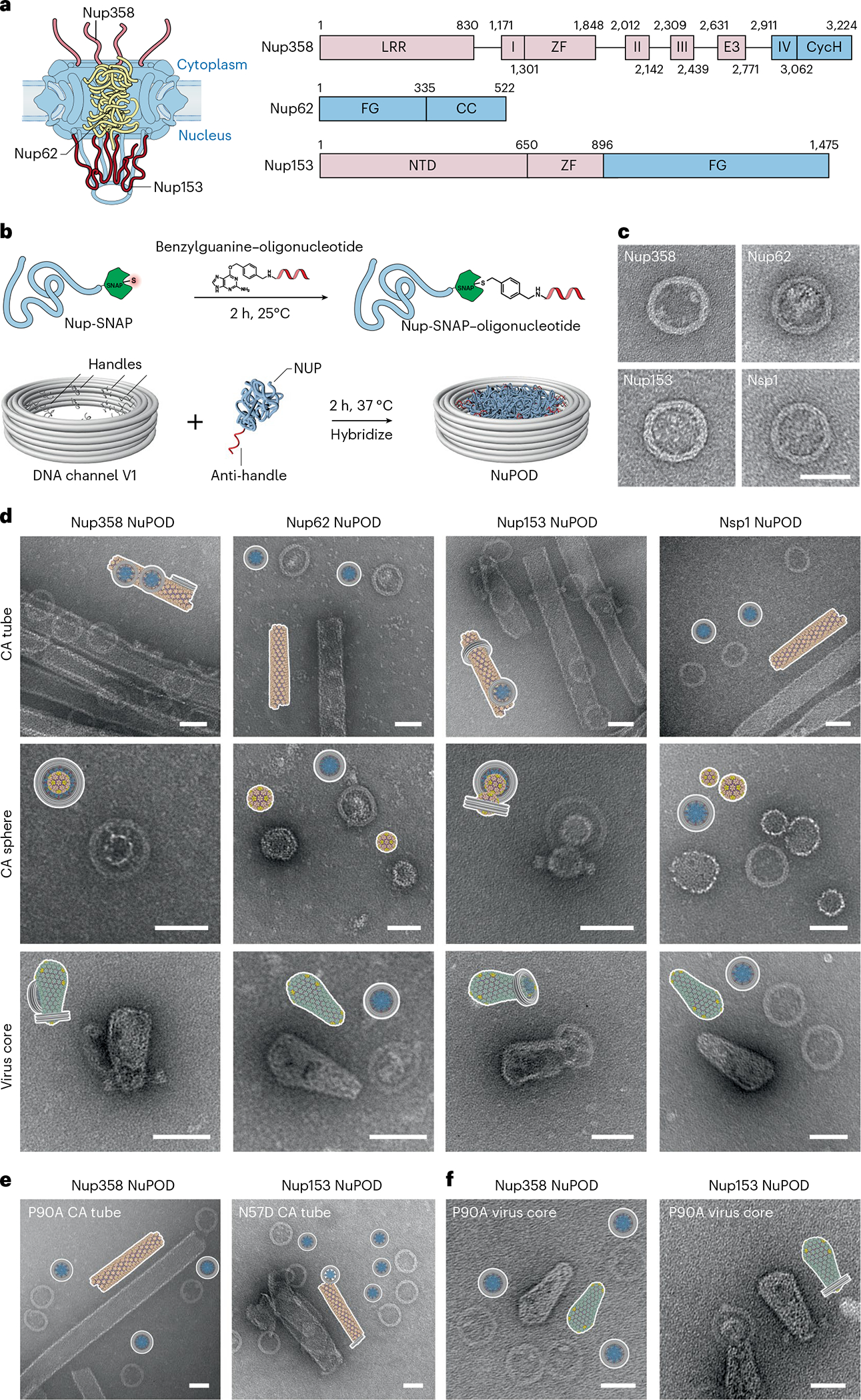
a, Left, a schematic diagram of the organization of capsid-interacting nups in the NPC: Nup358 on the cytoplasmic face, Nup62 in the central channel and Nup153 on the nuclear face. Right, schematic of the protein domains of Nup358, Nup62 and Nup153. LRR (leucine-rich region); Roman numbers I–IV (Ran binding domains I–IV); ZF (zinc finger); E3 (E3 ligase domain); CycH (cyclophilin homology domain); FG (phenylalanine-glycine rich domain); CC (coiled coil domain); NTD (N-terminal domain). The CA-binding regions used in this study are shown in blue. b, Schematic of the conjugation of benzylguanine-labeled anti-handle to SNAP-tag-labeled nup by click chemistry (top) and of attachment of nup–anti-handle conjugates to a DNA-origami channel by DNA hybridization (bottom). c, Negative-stain electron micrographs of various NuPODs. Scale bar, 50 nm. d, Schematics and negative-stain electron micrographs of the Nup358, Nup62, Nup153, and Nsp1 NuPODs (from left to right), mixed with CA tubes (top), CA spheres (middle) and purified virus cores (bottom) that were assembled in vitro. Scale bar, 50 nm. e, Schematics and negative-stain electron micrographs of the Nup358 NuPODs mixed with CA-P90A tubes that were assembled in vitro (left), and Nup153 NuPODs mixed with CA-N57D tubes (right). Scale bar, 50 nm. f, Schematics and negative-stain electron micrographs of the Nup358 (left) and the Nup153 (right) NuPODs mixed with purified virus cores with the P90A mutation. Scale bar, 50 nm. All negative-stain EM experiments were repeated three times with similar results.
Nup358 or Nup153 NuPODs bind to HIV-1 capsids
Although Nup358, Nup62 and Nup153 have been shown to bind with the capsid17,29–34, studying their multivalent interactions with the virus core in an in vitro system resembling the cellular NPC milieu is critical to understand the molecular mechanisms that govern the nuclear entry of HIV-1. Toward this goal, we first tested the binding of these nups to reconstituted nanotubes constructed from recombinant CAs using a co-pelleting assay. The cross-linked CA tubes (diameter 40–50 nm) with the A14C and E45C mutations were used owing to their stability under low salt conditions48 (Extended Data Fig. 4). The co-pelleting assay showed that, among the four nups tested, Nup153CTD had the strongest affinity to the CA tubes, followed by the modest affinities from Nup358RBD4-CycH and Nup62FL, whereas Nsp1 showed negligible binding (Extended Data Fig. 3). To analyze NuPOD interactions with CA lattices in greater detail, we performed EM studies on NuPODs (2 nM, containing 32 copies of nup) mixed with one of three CA structures: tubes as described above, spheres assembled from CA protein with the N21C and A22C mutations49,50 (1–2 μM CA), or bona fide virus cores (0.5 μM CA) purified from HIV-1-infected human T cells51. In accordance with the co-pelleting assay, Nup153 and Nup358 NuPODs showed robust binding to all three CA assemblies, whereas Nup62 and Nsp1 NuPODs failed to associate with any of them (Fig. 2d, see quantifications in Extended Data Fig. 4).
We also tested the effects of well-characterized CA mutants on capsid-NuPOD binding. Previous studies have shown that the mutation P90A in the CypA-binding loop of CA directly affected its binding to Nup35832,36, and that the N57D mutation in the FG-binding pocket of CA affected Nup153 binding29–31. In accordance with these findings29–32,36, our results showed that the CA-P90A tubes almost completely lost their affinity to Nup358 NuPODs, and that CA-N57D tubes displayed greatly reduced binding to Nup153 NuPODs (Fig. 2e and Extended Data Fig. 5). As for the purified virus cores with the P90A mutation, the P90A mutation in CA abolished capsid binding with the Nup358 NuPOD, but not with the Nup153 NuPOD (Fig. 2f). Furthermore, we performed a co-pelleting assay on CA tubes and cyanine-3 (Cy3)-labeled NuPODs and used fluorescence intensities in the supernatant and pellet to measure their binding strengths (see Methods and Extended Data Fig. 6 for details). These semi-quantitative ensemble assays reported capsid-NuPOD interactions that are highly consistent with the single-particle observations by negative-stain transmission EM (Extended Data Figs. 4 and 6). Therefore, the interactions between NuPODs and CA assemblies were robust and specific to the biologically relevant determinants in the process of HIV-1 infection.
In terms of binding geometry, the vast majority of Nup153 and Nup358 NuPODs attached to the side of the CA tubes or fully encircled a CA sphere (30–45 nm in diameter). We also observed CA tubes completely penetrating the entire depth of (that is, threading through) a small fraction of Nup153 NuPODs (Fig. 2d and Extended Data Fig. 4), indicating that multiple nups can collectively diffuse along the CA lattice. Interestingly, different from the Nup358 NuPOD, the Nup153 NuPODs almost exclusively bound to the curved regions of native HIV-1 cores, in most cases with the narrow end or ‘tip’ of the conical capsid penetrating the DNA channel (Fig. 2d,f). These findings suggest that Nup153 may have a curvature preference, specifically targeting the tip of the HIV-1 capsid.
Nup153 binds preferentially to curved CA lattices
The dominant tip-binding mode of HIV-1 cores to the Nup153 NuPOD prompted us to further investigate the preference of Nup153 and Nup358 to CA lattices with different curvatures. The cone-shaped HIV-1 capsid has a ‘flat’ surface on its sides and highly curved tip regions52 (Fig. 3a). It is well-established that the CA tube mimics the relatively flat region of the capsid, and the sphere mimics the highly curved regions49,50 (Fig. 3a and Extended Data Fig. 4), capturing two different surface geometries that are present in native capsids. We accordingly used these CA assemblies in binding-competition assays to test the curvature preference of Nup153 and Nup358 (see Methods for details). Nup153 NuPODs were challenged with equimolar mixtures of CA spheres and tubes and were analyzed by negative-stain EM (Fig. 3b). When CA assemblies were in excess of the NuPODs, almost all Nup153 NuPODs bound to CA spheres, and some bridged two spheres. We determined that ~85% of the NuPODs bound solely to CA spheres, which, compared with the ~1% of tube-bound NuPODs, showed a difference of nearly two orders of magnitude (Fig. 3b, top). When Nup153 NuPODs were added in excess of the mixture of CA spheres and tubes, we again found a ~60-fold preference for binding to spheres over tubes (Fig. 3b, bottom). The excess NuPODs did not lead to increased binding to CA tubes, but rather to greater NuPOD occupancy (up to three) on the same CA sphere. As the spheres present highly curved CA lattices, this reinforces the notion that Nup153 favors curved regions of the HIV-1 capsid, as was observed on capsid cones assembled in vitro (Extended Data Fig. 5) and native virus cores (Fig. 2d,f). In contrast, the percentages of sphere-binding and tube-binding Nup358 NuPODs were consistently almost the same, regardless of the NuPOD/CA ratio. Additionally, about 20–50% of Nup358 NuPODs bound CA spheres and CA tubes at the same time (Fig. 3c), suggesting that Nup358 lacks such a curvature preference.
Fig. 3 |. Nup153 prefers highly curved tip regions of the HIV-1 capsid for avid NuPOD association.
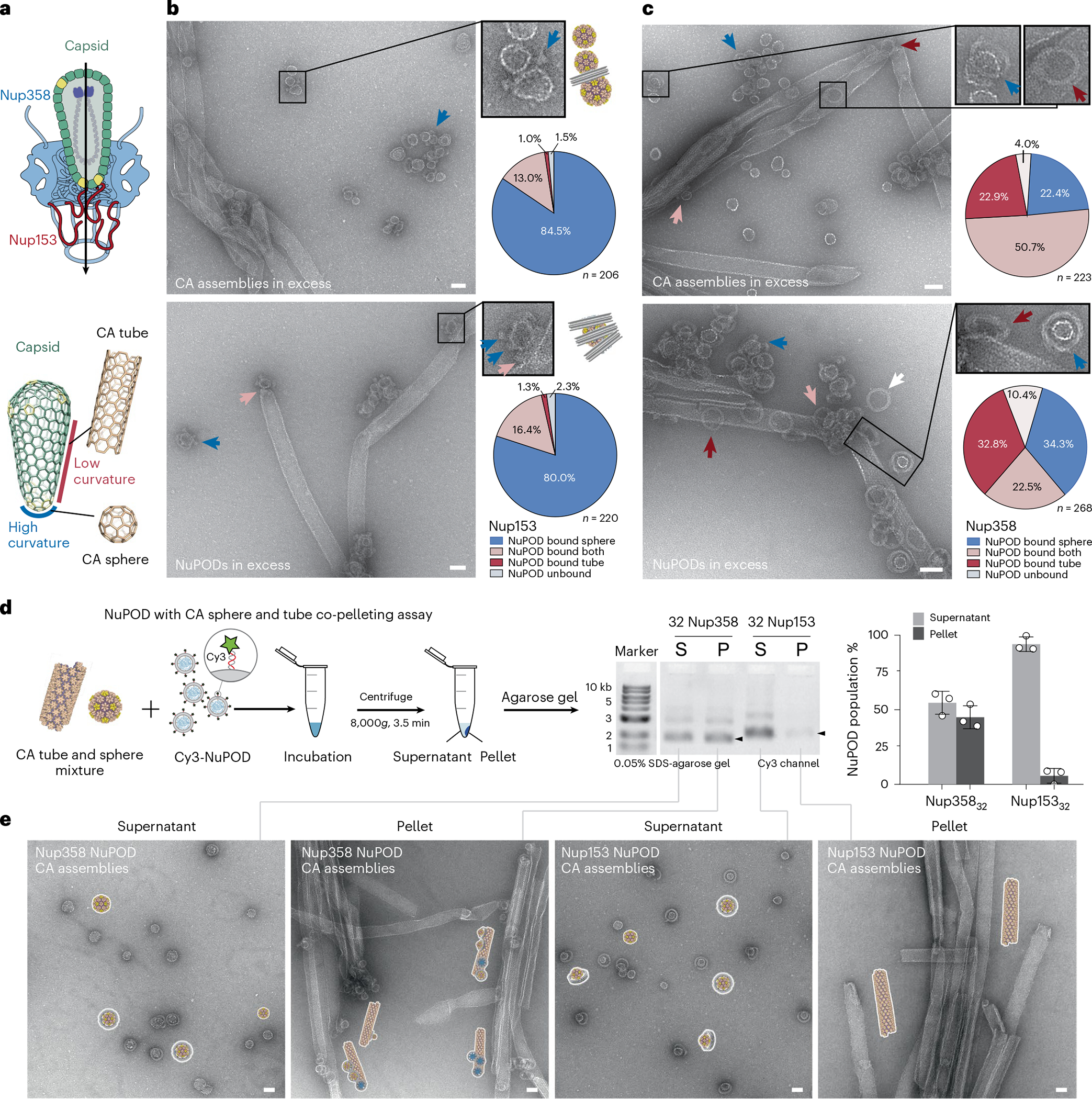
a, Top, a schematic of tip-first capsid insertion during HIV-1 nuclear transport. Bottom, illustrations showing that the capsid cone has a relatively flat surface on its sides, mimicked by the CA tube, and high curvature at the tips, mimicked by the CA sphere. b, Left, negative-stain electron micrographs showing the Nup153 NuPOD binding to a mixture of CA spheres and tubes. The CA assemblies were in excess in the top image, and the NuPOD was in excess in the bottom image. Blue arrows denote the CA-sphere-bound NuPODs, and pink arrows denote those simultaneously bound to CA tubes and spheres (insets for enlarged views). Right, schematics of the representative binding geometry and quantification of all binding events. n = total numbers of counted NuPODs. Scale bar, 50 nm. c, Left, negative-stain electron micrographs showing the Nup358 NuPOD binding to a mixture of CA spheres and tubes. The CA assemblies were in excess in the top image, and the NuPOD was in excess in the bottom. Blue, red, pink and white arrows denote the NuPODs bound to CA spheres only, CA tubes only, simultaneously to CA tubes and spheres, and free NuPODs, respectively. Right, quantification of all binding events. n = total numbers of counted NuPODs. Scale bar, 50 nm. d, Left, schematic of a co-pelleting assay using Cy3-NuPODs co-incubated with a mixture of CA tubes and spheres. Middle, the result of co-pelleting assays using the CA assemblies mixed with 32 copies of Nup358 or Nup153 NuPODs, analyzed by SDS–agarose gel electrophoresis. Fluorescence (Cy3) signals of the NuPOD bands (marked by black arrows) were used to quantify NuPODs in the supernatant (S) and pellet (P) fractions, as shown in the bar graph on the right. Data are plotted as mean ± s.d. of three independent experiments (n = 3), with individual data points shown as black circles. e, Schematics and negative-stain electron micrographs of the supernatant (S) and pellet (P) fractions collected after co-pelleting CA tubes, CA spheres and NuPODs. All negative-stain EM experiments were repeated three times with similar results. Scale bar, 50 nm.
To further analyze the preference for curvature, we again used the NuPOD–CA assembly co-pelleting assay. We first confirmed that centrifugation at 8,000g for 3.5 min completely separated CA spheres and tubes into the supernatant and the pellet fractions, respectively (Extended Data Fig. 7). We then incubated a mixture of CA spheres and CA tubes with Nup358 or Nup153 NuPODs. After centrifugation, we used gel electrophoresis to measure the distribution of fluorescently labeled NuPODs in the two fractions (Fig. 3d) and used negative-stain EM to confirm the NuPOD–CA lattice association (Fig. 3e). Strikingly, ~95% of the Nup153 NuPODs remained in the supernatant, demonstrating their strong preference for the highly curved, supernatant-residing CA spheres. In contrast, the supernatant and the pellet contained nearly equal amounts of the Nup358 NuPODs. The co-pelleting assay therefore corroborated the negative-stain EM analyses to confirm that the curvature-seeking interaction with the HIV-1 capsid is a property that is unique to Nup153.
We note that, as the nups were positioned inside the NuPOD, the observed CA sphere preference can be potentially owing to geometric constraints leading to reduced accessibility of CA tubes, although no preference was observed for the DNA-encircled Nup358. To further address this possibility, we repositioned the DNA handles to the exterior of the DNA-origami channel to create a NuPOD with fully exposed Nup153. This exterior-grafted Nup153 NuPOD (Nup153out NuPOD) efficiently bound to CA tubes (on the flat side) and spheres (six to eight spheres per NuPOD) in tube-only or sphere-only binding experiments, respectively (Extended Data Fig. 7). In accordance with the earlier competition results, the majority of Nup153out NuPODs clustered around the CA spheres in the mixture of spheres and tubes (Extended Data Fig. 7), confirming Nup153’s intrinsic preference for the curved CA lattice. It is conceivable that this preference facilitates the directional insertion of the capsid through its tip into the NPC.
Capturing HIV-1 capsid requires multiple Nup153 or Nup358
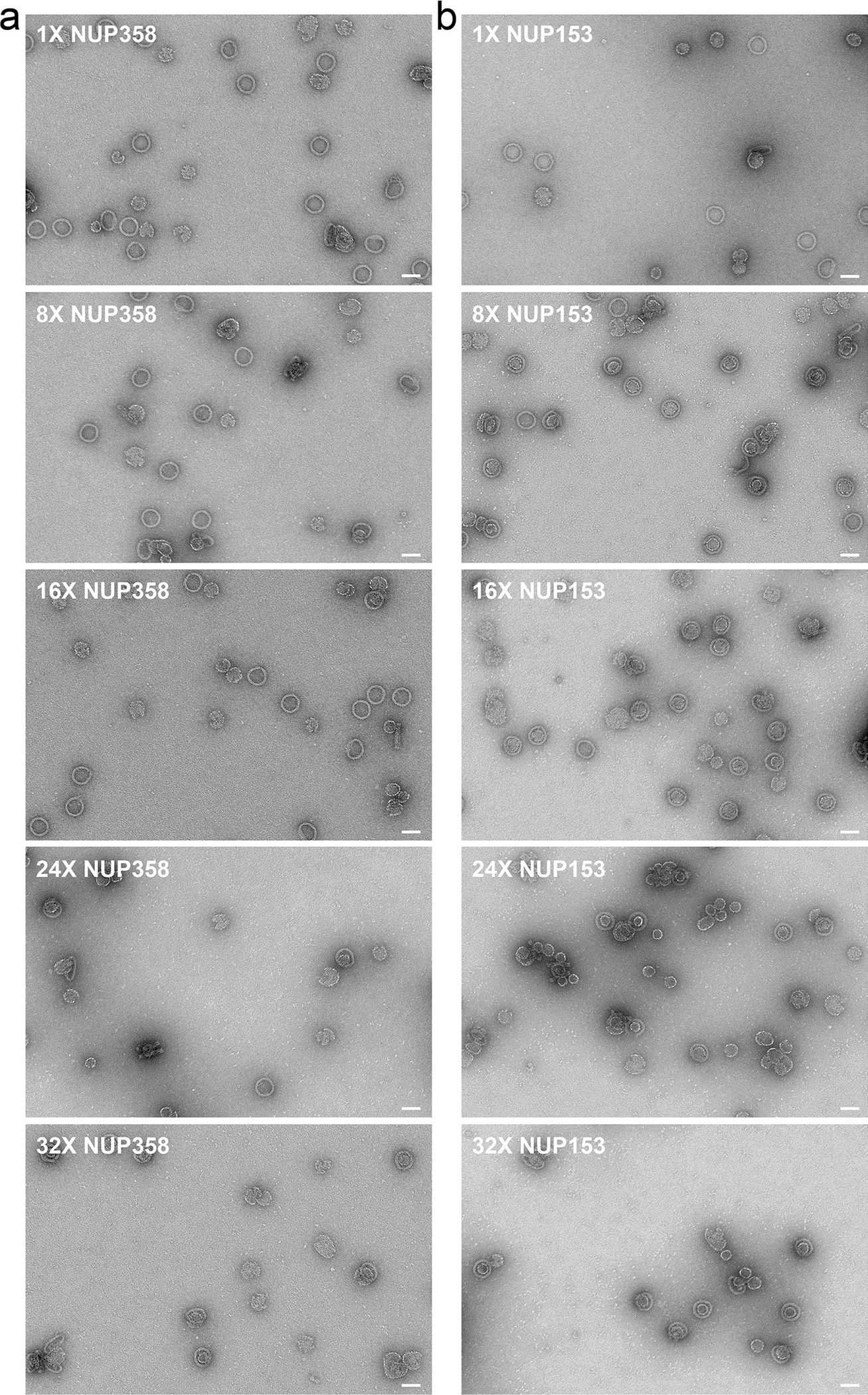
We observed that very low concentrations (2 nM) of NuPODs containing 32 copies of Nup358 or Nup153 efficiently bound to HIV-1 capsids (1–2 μM CA) (Fig. 2d). We hypothesized that densely grafted nups in a confined space enhanced their collective affinity to capsids. Taking advantage of the programmability of DNA origami, we tested this hypothesis by building DNA channels with varying numbers of nup anchoring points (1, 8, 16, 24 or 32) and evaluating the association between the resulting Nup358 or Nup153 NuPODs with CA spheres (Fig. 4a and Extended Data Fig. 8). After incubating NuPODs (2 nM) with spheres (1 μM CA), we imaged the mixture under negative-stain EM and sorted the NuPODs into three categories: those that fully encircled a sphere (stably bound), those with spheres attached to the rim (loosely bound) and those devoid of spheres (unbound). Plotting the binding-categorized NuPOD populations against the nominal nup stoichiometry showed a positive correlation between the number of nups per NuPOD and their capsid-binding ability. For example, ~75% of NuPODs with ≥24 Nup358 copies stably captured a CA sphere, whereas at least ~58% of NuPODs with ≤16 copies did not bind to CA spheres; similarly, more than 80% of the NuPODs carrying ≥8 Nup153 copies captured CA spheres. By contrast, NuPODs containing one Nup153 or Nup358 showed little to no binding to CA spheres, with only ~10% or <5% of NuPODs displaying loose capsid association, respectively (Fig. 4b). These results suggest that a single Nup153 or Nup358 is insufficient for firm attachment of the capsid to the NPC, and that physiologically relevant NPC-capsid associations need avidity that is provided by multiple nups. This observation is consistent with the dynamic Nup153–CA lattice binding with rapidly transitioning bound and unbound states found in molecular-dynamics simulations53. Such fast on/off kinetics are potentially important for the translocation of capsid within the NPC, as suggested by the CA tubes threading through the Nup153 NuPODs (Fig. 2d and Extended Data Fig. 4). Furthermore, our data suggest that nucleus-facing Nup153 is more capable of sequestering HIV-1 capsids than is cytosol-facing Nup358, implying that there could be a built-in binding-affinity gradient in the NPC that HIV exploits for nuclear import.
Fig. 4 |. Stable HIV-1 capsid binding requires multiple copies of Nup358 or Nup153 in a NuPOD.
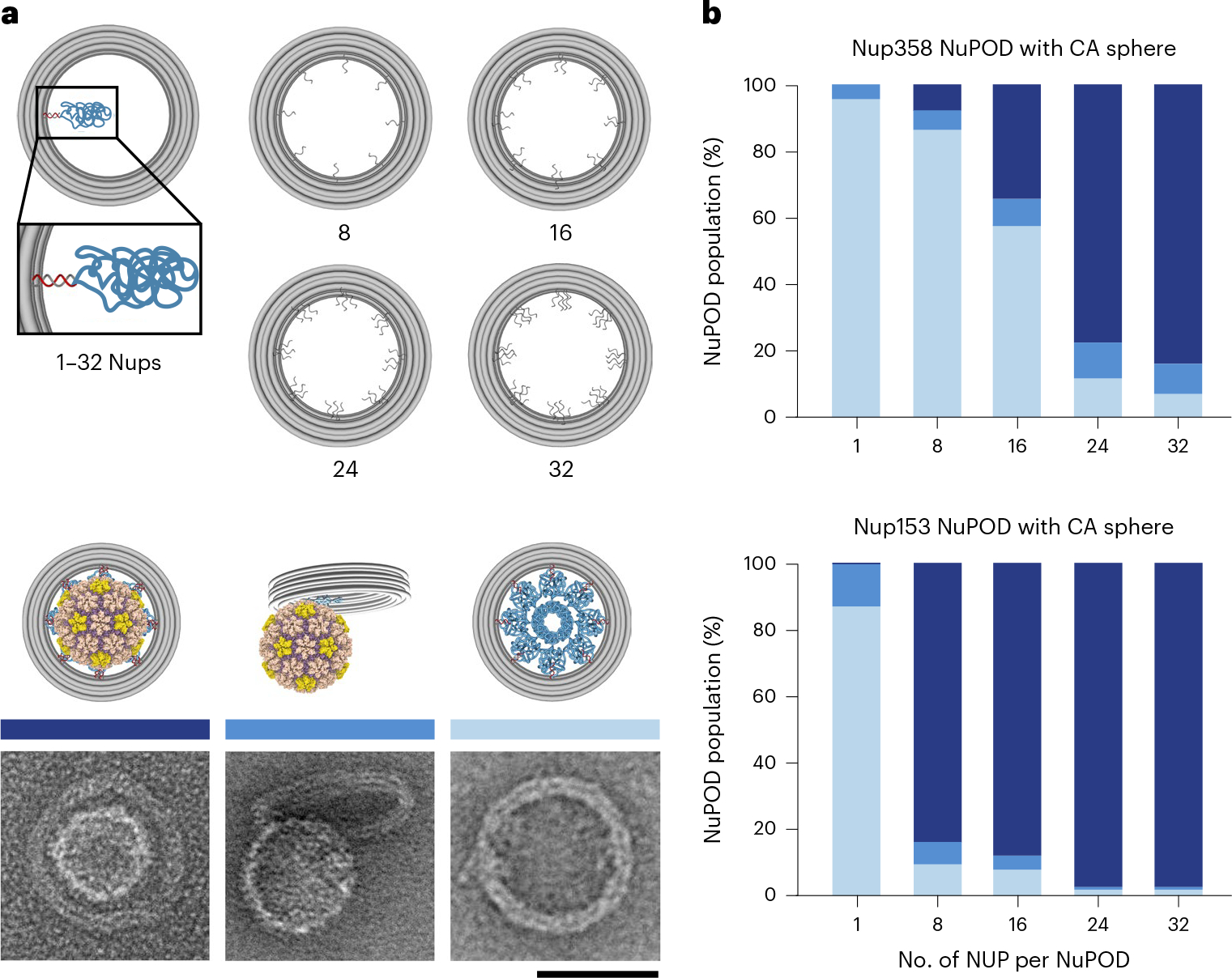
a, Top, schematic showing that the maximum copy number of nups in a NuPOD is dictated by the designed number of inner handles per channel. Bottom, cartoon models and representative EM images of NuPODs that stably bind to (left), loosely attach to (middle) or are free of (right) CA spheres. The negative-stain EM experiments were repeated three times with similar results. b, The capsid-binding ability of a NuPOD positively correlates with the number of Nup358 (top) and Nup153 (bottom) copies in the NuPOD. About 24 copies of Nup358 or 8 copies of Nup153 are needed to secure a CA sphere. n = 109–226, total numbers of counted NuPODs.
Nup arrangement and channel width modulate capsid insertion
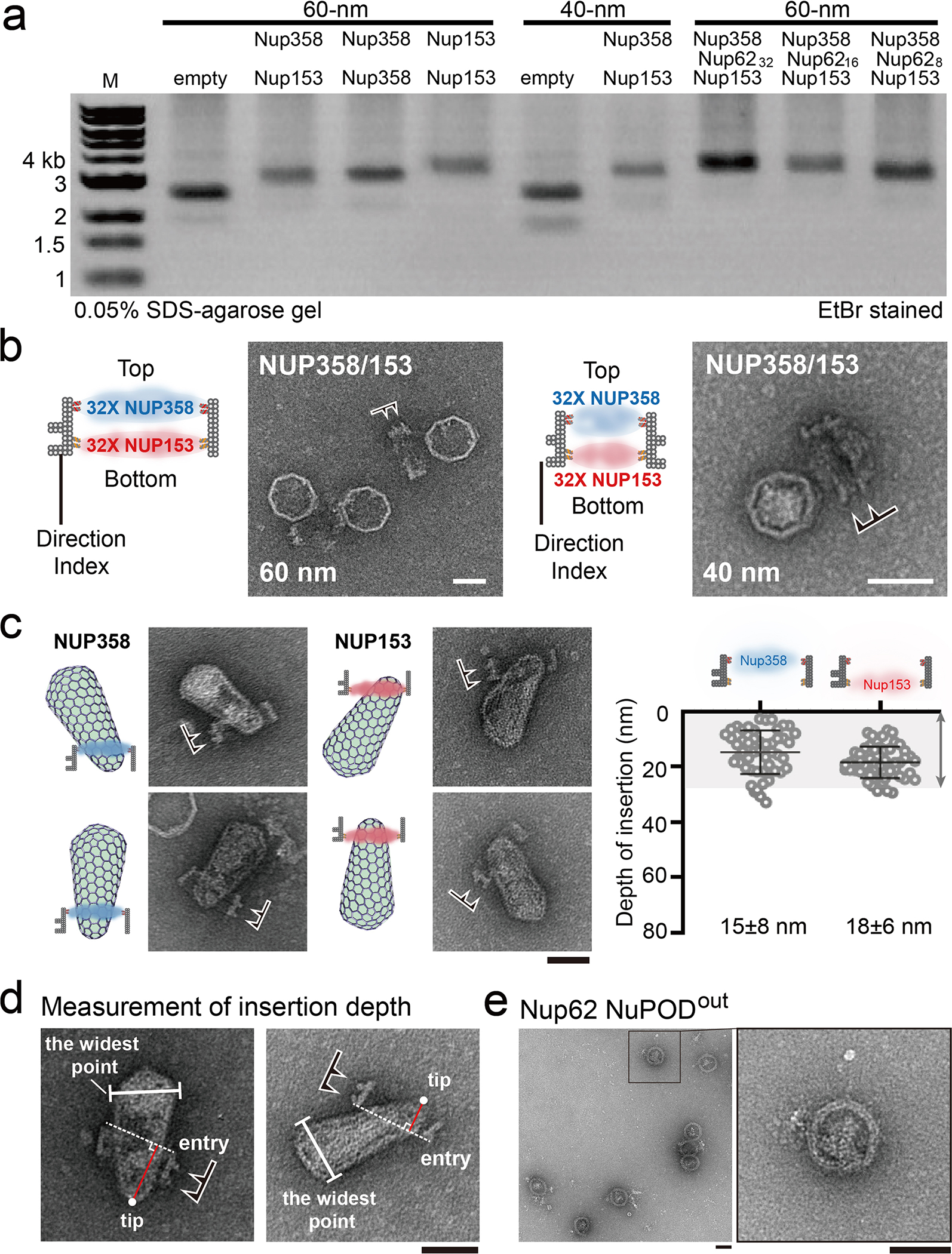
To test whether a binding-affinity gradient generated by Nup358 and Nup153 facilitates the translocation of the capsid through the nuclear pore, we built 30-nm-deep NuPODs containing two layers of nups, with up to 32 nups per layer (Extended Data Fig. 9a). To mimic the nup organization within the NPC, we created a two-layer NuPOD with the 60-nm-wide channel containing Nup358 in one layer and Nup153 in the other, thus forming a capsid-affinity gradient along the axial direction of the channel (Extended Data Fig. 9b). We also constructed a 40-nm-wide NuPOD with a similar nup arrangement to study the effect of channel width on capsid insertion. In both Nup358-Nup153 NuPODs, the Nup153 layer was placed near the indexed end of the channel (Fig. 5a). Negative-stain EM showed that ~75% of purified HIV-1 cores bound to these two-layer NuPODs. However, although a substantial number of capsids penetrated the 60-nm NuPODs, the 40-nm NuPODs did not permit such insertions, demonstrating that a largely intact virus core can penetrate only a sufficiently wide NuPOD that accommodates the width of the capsid.
Fig. 5 |. Penetration into multi-nup NuPODs is maximized when capsids travel along a built-in affinity gradient in the NPC.
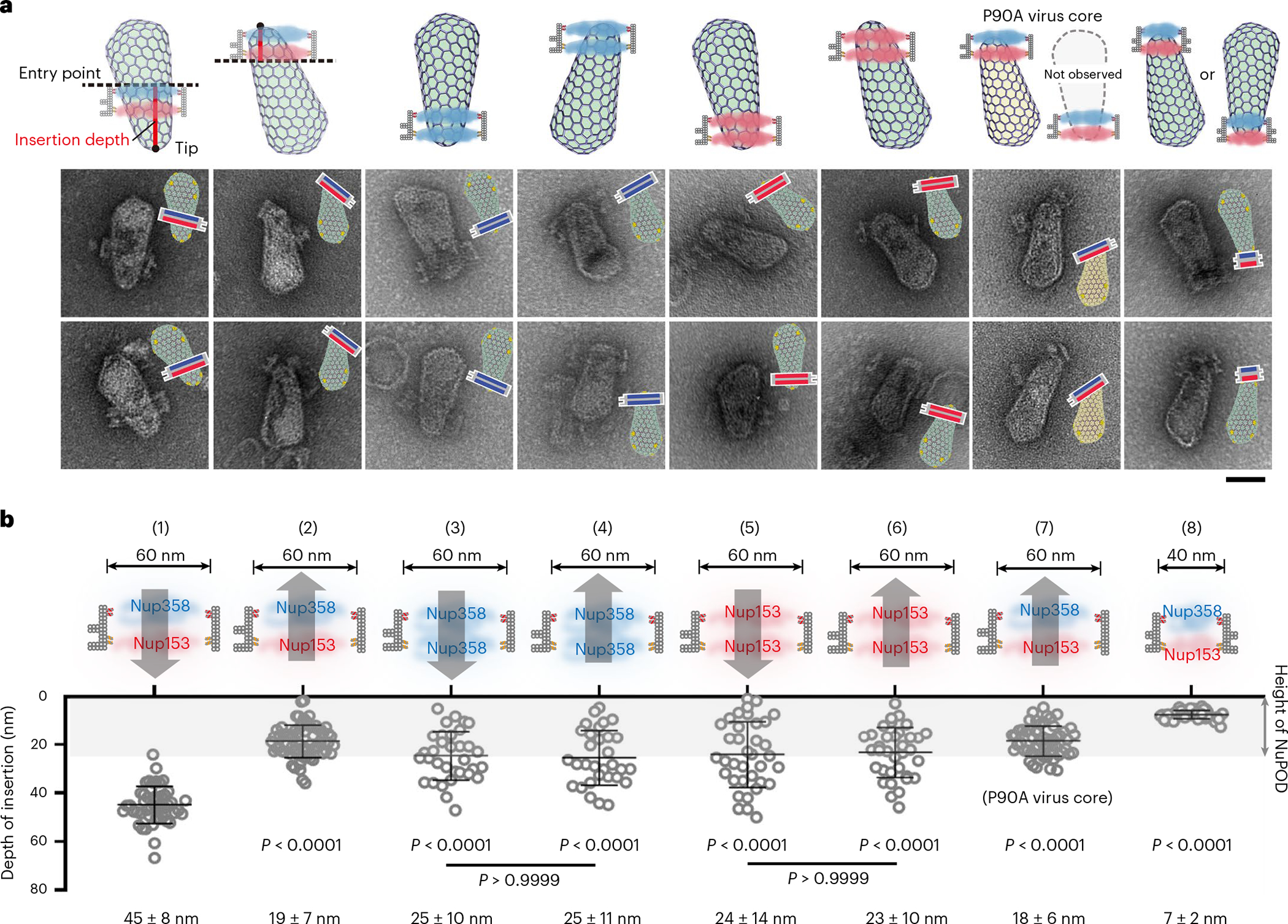
a, Cartoon models (top) and representative electron micrographs images (bottom) of purified virus cores inserted into two-layer NuPODs. Nup358 and Nup153 (32 copies each in a layer) are depicted in blue and red in the schematics, respectively. In the EM micrographs, schematics of the binding orientations of the DNA-origami channels are depicted. The negative-stain EM experiments were repeated three times with similar results. Scale bar, 50 nm. b, Top, schematics of NuPODs in cross-section views (32 nups/layer). Arrows denote the direction of HIV-1 core insertion. Bottom, depth of capsid insertion in various NuPODs, measured from the tip of the capsid to the point that it enters the DNA channel (entry point). Data are plotted as means ± s.d. The capsid–NuPOD binding events (n) were captured over three independent experiments: group 1 (n = 48), 2 (n = 68), 3 (n = 33), 4 (n = 31), 5 (n = 36), 6 (n = 35), 7 (n = 56), 8 (n = 52). Differences were determined by Kruskal–Wallis test and Dunn’s multiple-comparisons test. Unless marked on the graph, comparisons were made with insertion events following a nup capsid-affinity gradient (Nup358→Nup153). Full statistical results are provided in source data. Notably, capsids penetrate deeper into the NuPODs when following a nup capsid-affinity gradient (Nup358→Nup153) than going against (Nup153→Nup358) or in the absence of such a gradient (two layers of identical nups).
We subsequently focused on the 60-nm-wide NuPODs and measured the depth of capsid insertion from the tip of the virus core to its entry point on the NuPOD (Fig. 5a and Extended Data Fig. 9d, see Methods for details). Our data revealed a remarkable correlation between the depth and direction of capsid insertion (Fig. 5b). Namely, when the capsids entered NuPODs from the Nup358 end, similar to the natural nuclear import process, they traveled significantly deeper (45 ± 8 nm) than did those going in the opposite direction, which rarely went past the Nup153 layer they initially encountered (insertion depth, 19 ± 7 nm, Fig. 5b and Extended Data Fig. 10). In other words, because Nup153 has a higher capsid-binding affinity than does Nup358, the passage of capsids inside nup-gated channels was more efficient along, rather than against, the nup capsid-affinity gradient. To ascertain the effect of the affinity gradient on capsid insertion, we performed the penetration assay using NuPODs with two identical layers of nups (Fig. 5b and Extended Data Fig. 10) as controls. In the absence of an affinity gradient, the average penetration depth was less than 25 nm, without the directional bias observed for the Nup358-Nup153 NuPOD, thus showing inefficient diffusion-driven insertions under these conditions. We also tested the penetration of CA-P90A mutant viral cores with the Nup358-Nup153 NuPODs. These mutant cores entered the NuPODs almost exclusively from the Nup153 side, as P90A effectively eliminated the CA’s ability to bind Nup358, and the penetration depth was significantly reduced to ~18 nm (Fig. 5b and Extended Data Fig. 10). This was similar to the interaction of wild-type virus cores with the single-layer Nup153 NuPOD (Extended Data Figs. 9c and 10), confirming the necessity of both Nup358 and Nup153 for deep capsid insertion. These results suggest a mechanism whereby the capsid may first engage Nup358 and dwell at the entrance of the NPC before encountering Nup153, which helps it to migrate deeper into the nuclear pore.
Densely grafted Nup62 forms a barrier against HIV-1 capsid
In cells, the HIV-1 core must make its way through the central channel of the NPC, a conduit lined with mostly unstructured nups that collectively form a diffusion barrier54,55. Forming a trimeric complex with Nup54 and Nup58, Nup62 occupies a large volume of the NPC’s central channel and plays an important role in modulating nuclear transport56,57. Nup62 has been found to interact with CA tubes17, confirmed by our co-pelleting assay (Extended Data Fig. 3). However, NuPODs containing 32 copies of Nup62 (32 Nup62), in contrast to those containing 32 Nup358 or 32 Nup153, did not associate with CA assemblies or purified virus cores (Fig. 2d and Extended Data Fig. 6). Thus, it appears that capsid binding by Nup62 occurs differently when the capsid is grafted densely in a confined space.
To test the role of Nup62 in HIV-1 nuclear import, we built a 60-nm-wide, three-layer NuPOD with a Nup62 layer between the Nup358 and Nup153 layers (Fig. 6). Top-view electron micrographs showed that the three-layer NuPODs were more likely to bear channel-occluding, high-contrast protein masses than were the two-layer NuPODs (Fig. 6b and Extended Data Fig. 9b), suggesting that the additional layer of Nup62 may have formed a penetration-impeding barrier. Strikingly, even when grafted on the exterior of the 60-nm DNA channel, 32 copies of Nup62 managed to condense in the center (Extended Data Fig. 9e), while Nup153 stayed outside of the channel in similar setups (Extended Data Fig. 7b). In accordance with its strong tendency to self-interact, or its high cohesiveness54, Nup62 greatly hampered HIV-1 capsid penetration into NuPODs. Although ~75% of capsids bound to the three-layer NuPODs, their insertion was shallow (~11 nm), indicating that most virus cores stopped at the Nup62 layer in the middle of the channel, regardless of entry direction (Fig. 6a and Extended Data Fig. 10). Thus, not only did spatially confined 32 Nup62 lack affinity to HIV-1 capsids, they also formed an insulation layer between capsid-binding Nup358 and Nup153, effectively nullifying the affinity gradient that otherwise promoted capsid penetration. Systematically changing Nup62 copy number within the three-layer NuPODs (Extended Data Fig. 9a) revealed that such barrier-forming activity is dependent on protein density—although 8 copies of Nup62 permitted deeper penetration following the affinity gradient than 32 copies did, NuPODs lacking Nup62 supported the deepest penetration distance (Fig. 6c and Extended Data Fig. 10). These data suggest that the nuclear entry of capsid likely requires involvement of other host factors, such as Nup54 and Nup58, which normally form a complex with Nup62, and/or soluble nuclear transport receptors (NTRs), such as β-karyopherins, to overcome the barrier formed by condensed Nup62 proteins. Alternatively, Nup62 concentration may be reduced to weaken the barrier, either through nuclear pore expansion27 or dislodging a substantial portion of Nup62 from the NPC during HIV-1 infection14.
Fig. 6 |. Nup62 acts as a barrier to block capsid entry.
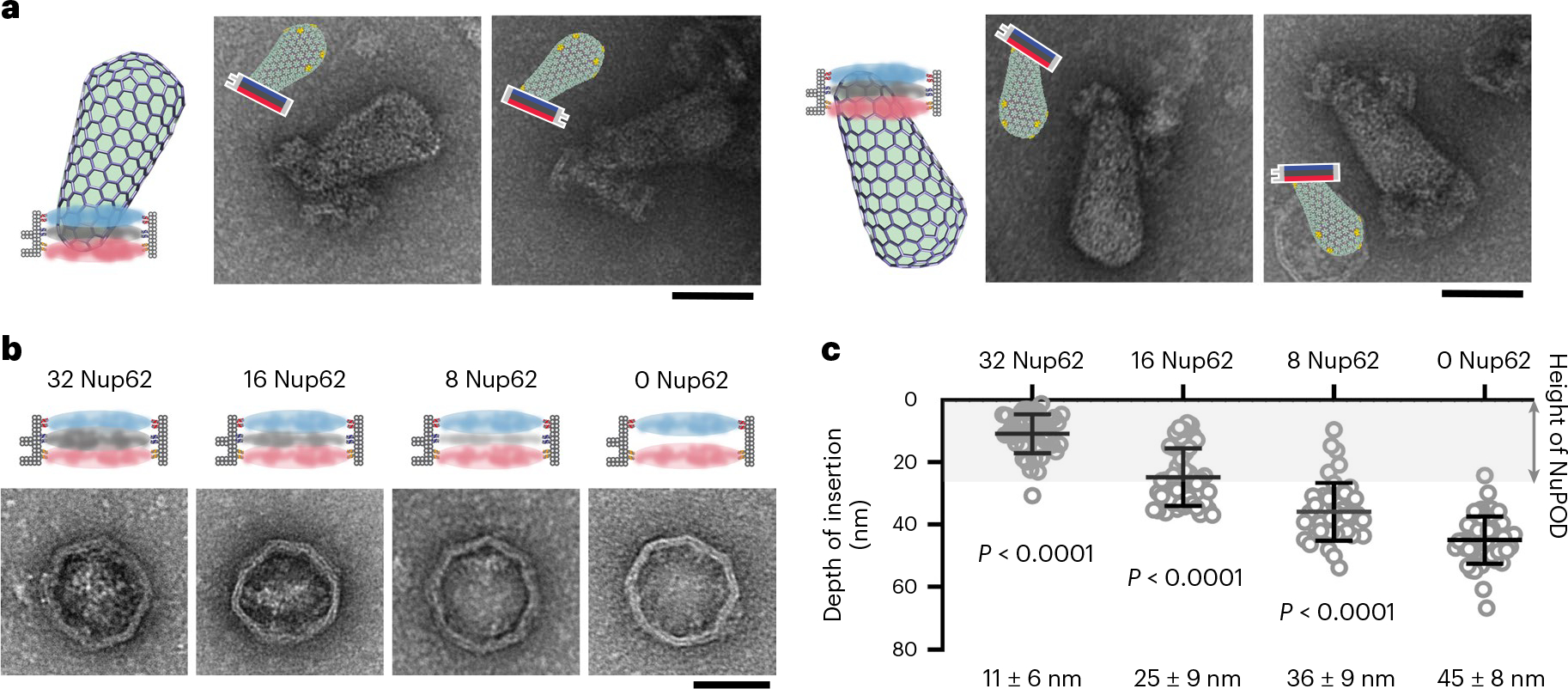
a, Schematics and representative EM images of virus cores inserted into Nup358-Nup62-Nup153 NuPODs. Nup358, Nup62 and Nup153 are depicted in blue, gray and red, respectively. The negative-stain EM experiments were repeated three times with similar results. Scale bar, 50 nm. b, Left, schematics and representative negative-stain EM images of 60-nm-wide NuPODs housing 32 Nup358, 0–32 copies of Nup62 and 32 Nup153 in three layers. The negative-stain EM experiments were repeated three times with similar results. Scale bar, 50 nm. c, Depth of virus core insertion into the three-layer NuPODs as a function of Nup62 copy number. Insertion depths were measured from the tip of the capsid to the point at which it enters the DNA channel from the Nup358 end. Data are plotted as mean ± s.d. The HIV-1 capsid-NuPOD binding events (n) were captured over three independent experiments: 32 Nup62 group (n = 46), 16 Nup62 group (n = 47), 8 Nup62 group (n = 42), 0 Nup62 group (n = 48). Differences were determined by one-way analysis of variance (ANOVA) and Tukey’s multiple-comparisons test, compared with the NuPOD without Nup62. Full statistical results are provided in source data. There is growing protein density in EM micrographs (Fig. 6b) and increasing impedance to capsid insertion (Fig. 6c) with increasing number of Nup62 in the DNA-origami channel.
Discussion
The nuclear import of the HIV-1 core is one of the least understood processes in the life cycle of this virus. Although recent studies have shown that the largely intact capsid can enter the nucleus through the NPC10–13, experimental data that elucidate the mechanisms of this process remain scarce. In this study, we model HIV-1 nuclear entry using NPC-mimicking DNA-origami channels and provide in-depth mechanistic insights into how the virus core interacts with critical NPC components. Our results depict a model (Fig. 7) in which the HIV-1 capsid docks onto the NPC through binding to multiple Nup358 molecules. Nup62 in the central channel forms a barrier, which is potentially overcome by the virus with the help of cellular transport factors or by dislodging Nup62 from the NPC. Nup153, which prefers to bind highly curved capsid regions, then positions the conical capsid for a tip-leading insertion. Subsequently, an intrinsic capsid-binding affinity gradient (Nup358→Nup153) may provide the driving force for the capsid to penetrate the nuclear pore.
Fig. 7 |. Model of HIV-1 nuclear entry.
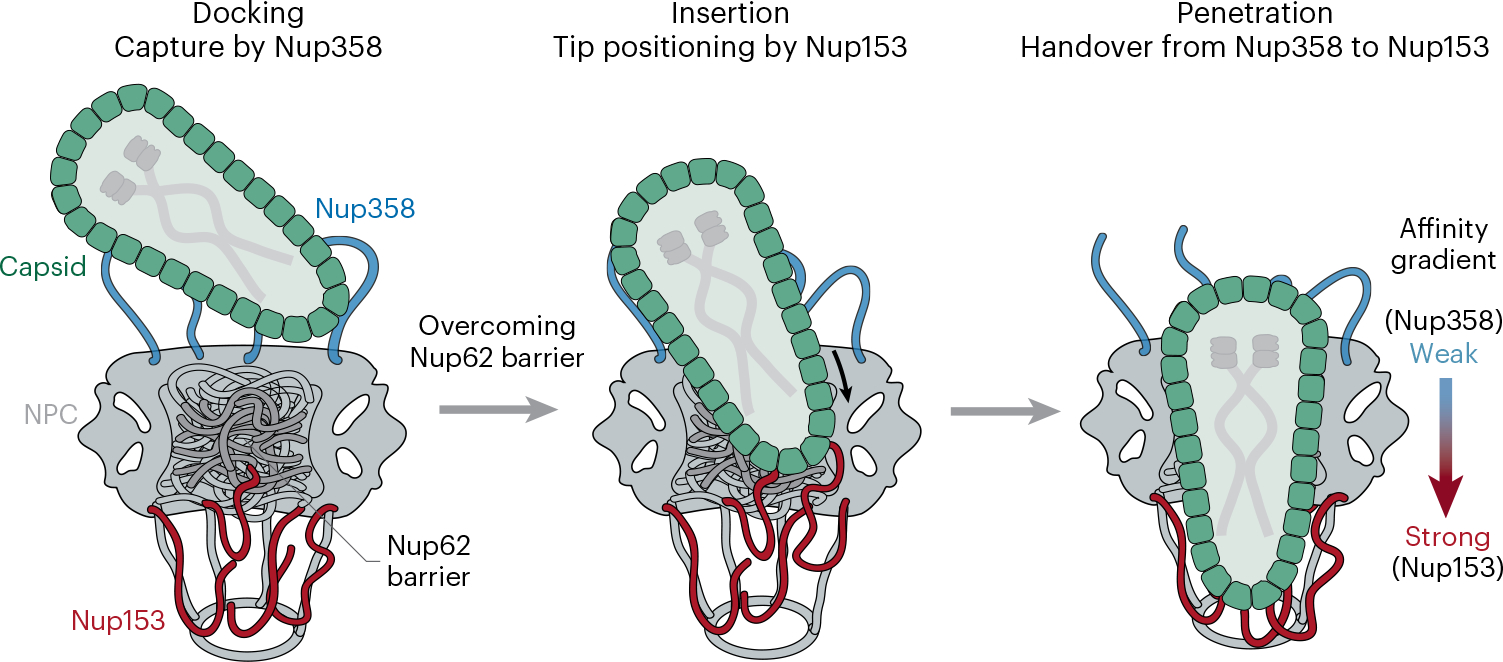
Schematic illustration of the Nup358-facilitated docking of HIV-1 capsid onto the NPC (left), Nup153-assisted tip positioning for insertion (after overcoming the Nup62 barrier) (middle) and a built-in capsid-binding affinity gradient from Nup358 to Nup153 driving capsid penetration (right).
Our NuPOD platform has allowed us to uncover mechanisms of HIV-1 nuclear import at the molecular level, unattainable by conventional methods. We found that multiple copies of nups were required to secure the virus core to the NPC mimics, indicating that avidity plays an important role in docking the capsid at and moving it through the nuclear pore28. These multivalent interactions require multiple binding sites in the CA lattice, supporting the notion that an at least somewhat intact capsid is needed for nuclear import10,11,13. Moreover, we showed that the Nup153 NuPOD had a higher affinity to the capsid than did the Nup358 NuPOD, suggesting an intrinsic capsid-binding affinity gradient from the cytoplasmic face (Nup358 localization site) to the nuclear face (Nup153 localization site) of the NPC. Although the RBD4-CycH protein confers the brunt of Nup358 binding affinity to CA, we note that a C-terminal-truncation construct lacking this region showed marginal CA-binding affinity in vitro18. Thus, other regions of Nup358 might also contribute to its overall binding affinity to capsids during HIV-1 infection. Nevertheless, our observations highlight a potential answer to the tantalizing question of what provides the driving force for the nuclear import of the virus core. Indeed, our experiments have demonstrated that the capsid penetration of nup-gated NPC mimics was vastly more efficient along, rather than against or in the absence of, the affinity gradient. Additionally, the newly discovered preference of Nup153 for the highly curved tip region of the HIV-1 core is intriguing. Although Nup153 as part of the NPC nuclear basket is localized toward the nuclear interior, its disordered C-terminal FG domain can extend into the cytoplasm58. In this way, its high-affinity capsid-binding domain can stabilize the virus core inside the NPC and position the capsid for tip-leading entry. This proposed mechanism is consistent with the in situ cryo-ET observation of the tip-first nuclear import of the HIV-1 core11. Our data further support that capsid transport, as depicted in this model, occurs only when the transport channel is sufficiently large to accommodate the width of the capsid, a state that has been observed in native NPCs in cells11,12,27,28.
Somewhat surprisingly, although we and others17,34 have found that Nup62 can interact weakly with CA tubes, multiple copies of Nup62 inside the NPC-like channel of the NuPODs formed a barrier preventing capsid binding and penetration. This phenomenon could be caused by the strong cohesiveness of Nup6254, which may lead to a condensate shielding the interaction interfaces from the capsid. Revealing this collective behavior of multiple spatially confined Nup62 molecules again highlights the unique strengths of the NuPOD platform. Capsids may enlist the assistance of NTRs to penetrate the Nup62 barrier, in a manner similar to the NTRs helping large cellular cargos pass through the NPC59. Alternatively, it has been reported that Nup62 relocates from the NPC to the cytoplasm during HIV-1 infection, suggesting that infection may cause the disassembly of this central barrier of the NPC14. Our in vitro tests found that reducing the copy number of Nup62 was indeed conducive to capsid passage, but this proposed mechanism requires further validation. It should be noted that, in native nuclear pores, Nup62, Nup58 and Nup54 form trimeric complexes56,57, and whether this heterotrimer affects the barrier formation and nuclear import of capsid remains to be examined.
In summary, our results demonstrate that the NuPOD platform bridges a technology gap in the study of the complex process of HIV nuclear entry that existed between powerful but over-simplified biochemical approaches and native but convoluted intact cells. Herein, we leveraged these precisely engineered nuclear-pore mimics to dissect the nuclear-entry mechanism of the HIV-1 core. Our current NuPODs resemble the channel-forming region of human nuclear pores in the overall architecture and the stoichiometry and spatial distribution of selected nup species. As it captures the collective properties of different nups with biologically relevant spatial arrangement, our NuPOD platform is especially useful for examining the interactions between the NPC and complex cargos, such as viral particles. It provides an effective approach to analyze the effects of mutations, small-molecule inhibitors60,61 or other cellular co-factors, including CypA5,36, MxB17,62, CPSF663,64 and various NTRs65,66, on the nuclear import of the virus core67. However, even with up to ~100 copies of the three major nups reported to interact with the HIV-1 capsid, our current NuPODs contain only a subset of the close to 1,000 nups in the natural NPC68, and lack their nuclear basket architecture. The complexity and functional redundancy of NPC components and transport regulation demand approaches that combine more advanced architectural designs of NPC-mimicking NuPODs with high-resolution imaging and functional studies. Furthermore, the results obtained from in vitro systems need to be validated in cell-based studies. Nonetheless, we envision that the NuPOD platform, with its many unique strengths, will continue to be an invaluable tool in the investigation of the nuclear import of HIV-1 and other viruses that rely on capsids for nuclear entry69,70.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41594-023-00925-9.
Methods
Cloning and expression
Nup358RBD4-CycH, Nup62FL, and Nup153CTD from Homo sapiens, as well as yeast nup Nsp1 from Saccharomyces cerevisiae, were cloned as 10 × His-MBP-SUMO-nup-SNAP constructs into a pET-28a-derived vector (Novagen) and expressed in BL21-Gold (DE3). Cells were grown in Terrific Broth at 37 °C in a shaking incubator until they reached an optical density at 600 nm (OD600) of 1.0. Protein expression was then induced with 1 mM IPTG for 4 h at 20 °C before collection by centrifugation. Cell pellets were refrigerated at −80 °C until use. All CA constructs were cloned into pET-11a (EMD Millipore). CA proteins were overexpressed in BL21-Gold (DE3) cells at 25 °C for 12 h by induction with 0.5 mM IPTG at an OD600 of 0.8. Cell pellets were collected by centrifugation and refrigerated at −80 °C until use.
Protein purification
Nup-expressing cell pellets were resuspended in lysis buffer (1 × phosphate-buffered saline (PBS), 0.1 mM PMSF, 1 × Roche cOmplete protease inhibitors, pH 7.4), and lysed in a homogenizer (EmulsiFlex-C3, Avestin). Whole-cell lysates were then spun at 35,000 r.p.m. for 45 min with a Type 45 Ti rotor (Beckman Coulter). Subsequently, the supernatant was decanted and filtered through a 0.45-μm-pore-size Millex syringe filter. The resulting filtered lysate was applied to a 5 mL HisTrap column (Cytiva) on an ÄKTA system (Cytiva) at a flow rate of 1 mL/min. The filtrate loaded to the column was rinsed with a wash buffer (1 × PBS, 25 mM imidazole, pH 7.4) and eluted with a gradient buffer (1 × PBS, 0–300 mM imidazole, pH 7.4). For Nup358, the MBP-sumo tag was removed by overnight digestion with TEV protease at 4 °C. The TEV-cleaved Nup358RBD4-CycH was further purified by anion-exchange chromatography. Concentrations of purified protein were determined by Nanodrop (Thermo Fisher). Samples were flash-frozen in liquid nitrogen and stored at −80 °C until use. Untagged CA proteins were purified by 25% wt/vol ammonium sulfate precipitation, dialyzed into a low-salt buffer (25 mM HEPES, 0.1 mM TCEP, pH 7.0) and further purified by cation-exchange chromatography. All CA constructs were dialyzed in CA storage buffer (50 mM Tris, 75 mM NaCl, 40 mM 2-mercaptoethanol, pH 8.0) before being frozen or used for experiments. Purified proteins at all steps were validated using SDS–PAGE.
CA tube co-pelleting assays
All nups (2 μM) were incubated in 21-μL reactions with disulfide-crosslinked CA-A14C E45C tubes (100 μM CA) in 1 × PBS buffer (150 mM NaCl), pH 7.4, for 30 min at room temperature, and then spun at 20,000g for 20 min at 4 °C. Supernatant and pellet fractions were collected and analyzed using SDS–PAGE.
DNA-origami assembly and purification
DNA-origami structures were designed in caDNAno71. ssDNA handles were extended from the 3′ end of staple strands at the positions indicated in Figure 1 and Extended Data Figure 1. The handle sequences are 5′-CTGATGATATTGATTGAAATG-3′ (inner), 5′-CTACCATCTCTCCTAAACTCA-3′ (inner) and 5′-AAATTATCTACCACAACTCAC-3′ (inner and outer). One of the three handle sequences or a combination thereof was used in a DNA channel to anchor one or more types of nups. These DNA-origami channels were assembled from an M13mp18 bacteriophage-derived circular ssDNA strand (8064 nt) and oligonucleotides (Integrated DNA Technologies) in a 1 × TE (5 mM Tris-HCl, 1 mM EDTA, pH 8.0), 14 mM MgCl2 buffer using an 85–25 °C annealing gradient over 36 h. The 45-nm DNA channel was purified as reported previously41. The 40-nm and 60-nm DNA channels were assembled as homodimers. The folded monomeric channels (see Extended Data Fig. 1 for design diagrams) were first purified away from staple strands by PEG fractionation. The DNA structures were incubated in a solution containing 4% wt/vol polyethylene glycol 8000 (VWR), 500 mM NaCl, 1 × TE and 14 mM MgCl2, pH 8.0 for 15 min and centrifuged at 15,000g for 30 min at 4 °C. The pellets were then resuspended in 1 × TE, 14 mM MgCl2, pH 8.0 buffer. For dimerization, monomers in equimolar quantities were mixed and incubated overnight at 37 °C in either 1 × TE, 14 mM MgCl2 (60-nm channel), pH 8.0 or 1 × TE, 30 mM MgCl2 (40-nm channel), pH 8.0. The dimerized channels were purified using rate-zonal centrifugation45. The dimerization reaction mixture was placed on top of a quasi-linear glycerol gradient (15–45%) in a 1 × TE, 15 mM MgCl2, pH 8.0 buffer and spun in an SW 55 rotor (Beckman Coulter) at 48,000 r.p.m. at 4 °C for 1 h, followed by fraction collection. Fractions containing dimerized DNA channels were buffer exchanged into a 1 × TE, 15 mM MgCl2, pH 8.0 buffer and stored at −20 °C.
Benzylguanine-DNA preparation
5′-N-terminally labeled DNA oligonucleotides (anti-handles; Integrated DNA Technologies) were resuspended in Milli-Q ultrapure water at 2 mM concentration. The crosslinker benzylguanine (BG)-GLA-NHS (New England Biolabs) was dissolved in DMSO at 20 mM. N-terminally-labeled anti-handles and BG-GLA-NHS were mixed in a 1:3 ratio in a 67 mM HEPES, pH 8.5, buffer, and incubated at room temperature for 1 h. BG-DNA was then purified from excess crosslinkers by ethanol precipitation. The BG-DNA pellets were dried and stored at −20 °C until use.
Protein–DNA conjugation
BG-DNA pellets were resuspended in Milli-Q ultrapure water and mixed with purified SNAP-tagged nups in 1 × PBS buffer (pH 7.4) at a final concentration of 20 μM BG-DNA and 10 μM nup. This reaction was incubated at 25 °C for 2 h. Excess DNA was removed from the reaction mixture by size exclusion chromatography using a Superdex 200 10/300 GL column (Cytiva) and an aqueous mobile phase (1 × PBS buffer, pH 7.4). Conjugation efficiency was verified through SDS-polyacrylamide gel electrophoresis (SDS–PAGE) followed by Coomassie-blue staining (Thermo Fisher).
Anchoring nups to DNA-origami channel
DNA-anti-handle-conjugated nups were added to handle-displaying DNA-origami channels at 1.5:1 anti-handle:handle ratio (for example, 2 nM channel × 32 handles/channel × 1.5 = 96 nM nup-anti-handle) in a hybridization buffer (1 × TE, 15 mM MgCl2, 150 mM NaCl, pH 8.0) and incubated at 37 °C for 2 h. Optionally, the resulting NuPODs were purified by rate-zonal centrifugation through a 15–45% glycerol gradient in the hybridization buffer.
CA assembly
Crosslinked CA tubes.
Disulfide crosslinked CA-A14C E45C tubes were assembled by dialyzing CA-A14C E45C into a 50 mM Tris, 1 M NaCl, pH 8.0 buffer at 10 mg/mL overnight, followed by dialysis into a 50 mM Tris, pH 8.0 buffer without salt for another night. The crosslinked tube was stored in a 50 mM Tris, 150 mM NaCl, pH 8.0 buffer and could remain stable for months.
CA spheres.
Following a previously established protocol50, purified CA-N21C A22C proteins (1 mg/mL) were dialyzed into a 50 mM Tris-HCl, 1 M NaCl, pH 8.0 buffer overnight at 4 °C to form the spheres, which were verified by negative-stain EM.
CA cones.
CA-A14C E45C protein was diluted in a 25 mM MES, 100 mM NaCl, pH 6.0 buffer to 200 μM and then mixed with IP6 (in 20- to 40-fold molar excess). The assembly reaction was maintained at room temperature for 5 min until the appearance of cloudy CA assemblies. Then, 200 uM CypA was added to reduce the aggregation of the cones. The CA assemblies were diluted 20 times for negative-stain EM sample preparation.
Isolation of HIV-1 cores
Native HIV-1 cores were prepared from concentrated HIV-1 particles by centrifugation through a layer of 1% Triton X-100 on the top of a linear sucrose density gradient as described51. For this purpose, HIV-1 particles were collected from 200 mL cultures of MT-4 cells72, infected with undiluted virus collected from 10-cm dishes transfected with the wild type HIV-1 molecular clone R973, which is nearly identical to NL4–3 in its protein-coding sequences. To produce the virus, cells were cultured in RPMI1640 medium supplemented with fetal bovine serum (10% vol/vol), penicillin (100 units/ml) and streptomycin (0.1 mg/ml) in a humidified incubator at 37 °C and 5% CO2. For virus production in MT-4 cells, 100 million cells were pelleted, resuspended in 50 mL of fresh medium. and inoculated with a quantity of HIV-1 corresponding to 5,000 ng of p24 in the presence of DEAE-dextran (20 μg/mL) to promote infection. Cells were subsequently cultured for 24 h, followed by removal of the inoculum by pelleting and aspiration. The cell pellet was resuspended in 200 mL of fresh medium and cultured for 4 to 5 days. The extent of infection was assessed daily by monitoring the culture visually for cytopathic effects. At approximately 5 days after inoculation, the cells were pelleted by centrifugation and the supernatants were collected and clarified by passing them through a 0.45-μm-pore-size vacuum filter. The HIV-1 particles were concentrated by ultracentrifugation, resuspended, and HIV-1 cores were isolated. The quantity of viral CA protein in the dense gradient fractions corresponding to HIV-1 cores was determined by p24 ELISA (Anti-HIV-1 CA, 183-H12–5C, 1 μg/mL)74,75. The above reagent was obtained through the US National Institutes of Health (NIH) HIV Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Disease, NIH: Anti-Human Immunodeficiency Virus 1 (HIV-1) p24 Hybridoma (183-H12–5C), ARP-1513 (cell lines), contributed by B. Chesebro and H. Chen. The presence of intact HIV-1 cores in these fractions was ascertained by negative-stain EM. Fractions containing HIV-1 cores were aliquoted and flash-frozen in liquid nitrogen and stored at −80 °C until use.
HIV-1 cores with the P90A mutation were prepared from concentrated virus particles produced by transfection of 293T cells with the envelope-defective plasmid HIV-GFP encoding the P90A codon substitution in CA. Cells were transfected in 10-cm dishes using PEI and cultured for 2 days, and the virions were collected, clarified by filtration and concentrated by ultracentrifugation. Cores were purified from the concentrated virus as described above.
NuPOD-capsid interaction assay
Unless noted otherwise, 2 nM NuPOD was mixed with reconstituted CA assemblies (1–2 μM CA) or virus core (0.5 μM CA) purified from HIV-1-infected human T cells in a hybridization buffer and incubated for 10 min at room temperature. NuPOD interactions with the capsid were then analyzed using negative-stain EM.
CA tube and sphere binding competition assay
When CA assemblies were in excess, 0.5 nM NuPOD was added to a mixture of CA tubes (1 μM CA) and CA spheres (1 μM CA) in a hybridization buffer and incubated at room temperature for 10 min. When NuPODs were in excess, 3 nM NuPOD was added to a mixture of CA tubes (0.25 μM CA) and CA spheres (0.25 μM CA) in a hybridization buffer and incubated at room temperature for 10 min. The mixtures were imaged using negative-stain EM.
NuPOD–CA assembly co-pelleting assay
Anchoring Cy3 to DNA-origami channel.
Cy3-modified DNA anti-handles were added to eight outer-handle-displaying DNA-origami channels at 1.1:1 anti-handle:handle ratio (for example, 24 nM channel × 8 outer handles/channel × 1.1 = 211 nM Cy3-anti-handle) in a hybridization buffer and incubated at 37 °C for 1 h. Optionally, the resulting NuPODs were purified by rate-zonal centrifugation through a 15–45% glycerol gradient in the hybridization buffer.
Sample preparation.
Unless noted otherwise, 4 nM Cy3-labeled NuPODs were mixed with disulfide crosslinked CA-A14C E45C nanotubes (5 μM CA) in the hybridization buffer and incubated for 15 min at room temperature and then spun at 12,000g for 5 min at 4 °C. Samples of the supernatant and the pellet fractions were collected and analyzed using SDS–agarose gel electrophoresis (to detect Cy3-labeled NuPOD), SDS–PAGE gel electrophoresis (to detect nup protein and CA) and negative-stain EM. Empty DNA-origami channels served as the negative control, which were incubated with the CA nanotubes under identical conditions.
For experiments testing the capsid curvature preference of nups, 4 nM Cy3-labeled NuPODs were incubated with a mixture of disulfide-crosslinked CA-A14C E45C nanotubes (5 μM CA) and disulfide crosslinked CA-N21C A22C spheres (2.5 μM CA) in the hybridization buffer and incubated for 15 min at room temperature. The mixture was then spun at 8,000g for 35 min at 4 °C. CA spheres were pre-centrifuged at 5,000g for 2 min if they had visible sediment before being mixed with CA tubes. Samples of the supernatant and the pellet fractions were collected and analyzed using SDS–agarose gel electrophoresis and negative-stain EM.
SDS–agarose gel electrophoresis.
Samples were loaded in an SDS–agarose gel (1.5% agarose in 0.5 × TBE, 10 mM MgCl2 and 0.05% SDS, pH 8.0). Electrophoresis was carried out at 5.8 V/cm for 90 min in 0.5 × TBE buffer containing 10 mM MgCl2 and 0.05% SDS, pH 8.0. Gels were imaged on a Typhoon FLA 9500 scanner (Cytiva) for the in-gel fluorescence (Cy3) first, stained with ethidium bromide and then imaged again for the ethidium bromide fluorescence. For ethidium bromide staining, the gel was first soaked in deionized H2O and shaken for 1 h to remove SDS, and then was submerged in an ethidium bromide solution (Sigma-Aldrich, 20,000 × dilution in H2O to 0.5 μg/mL) for 1 h. Gels were destained for 1 h in deionized H2O before imaging (acquired using software Typhoon FLA 9500 v1.0).
SDS–polyacrylamide gel electrophoresis.
All SDS–PAGE experiments were carried out using 4–12% acrylamide NuPAGE Bis-Tris gel (Thermo Fisher Scientific). Samples were boiled in 1 × Laemmli sample buffer (Thermo Fisher Scientific) at 90 °C for 5 min before loading to the gels. The gels were run for 40 min at 25 V/cm in MES-SDS buffer (Thermo Fisher Scientific). Gels were stained with Coomassie Blue. Gel images were acquired using software ImageQuant LAS4000 v1.2 and analyzed using software ImageJ v1.48.
Negative-stain EM
Capsid assemblies with NuPOD samples were deposited onto glow-discharged 400 mesh formvar/carbon-coated copper grids (Electron Microscopy Sciences). Grids were then stained with 2% uranyl formate. Imaging was performed on a JEOL JEM-1400 Plus microscope operated at 80 kV with a bottom-mount 4k × 3k CCD camera (Advanced Microscopy Technologies). TEM images were collected using software AMT Image Capture Engine v602.
Measurement of capsid insertion depth
The sizes of the DNA-origami channels used in this study do not allow the broad end of the virus core to penetrate the NuPOD. Therefore, when a long edge of a NuPOD was observed in between the widest point and the tip of an intact capsid, the capsid was considered to have been inserted into the DNA channel from the tip (Fig. 5a). We subsequently selected the NuPOD binding events that have inserted virus cores and observable direction indexes for quantitative analysis (Extended Data Fig. 10 and Supplementary Figs. 1–7). The insertion depth was defined as the distance measured from the tip of the virus core to the entry point of the NuPOD, in the direction normal to the NuPOD edge (Extended Data Fig. 9d). TEM images were analyzed using software ImageJ v1.48.
Statistical analysis
The data analysis was performed using the software Prism 8.0.0 (Graph-Pad). All data were presented as mean ± s.d. unless noted otherwise. The Kruskal–Wallis test and Dunn’s multiple-comparisons test were used to evaluate differences in the depth of capsid insertion in various NuPODs. The data met the assumption of normality, and equal variances (nups bound to CA tubes in the co-pelleting assays and virus core insertions into three-layer NuPODs with different copies of Nup62) were tested with one-way ANOVA and Tukey’s multiple-comparisons test.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. DNA-origami designs rendered in caDNAno.

a, Cross-sections (left) and strand diagrams (right) of the 45-nm channel design: (1) 0–32 inner handle version; (2) 48 outer handle version. The scaffold strand is in gray. Staples carrying inner handles and outer handles are shown in red and orange, respectively. Handles (see Methods for sequences, not shown here for clarity) were extended from the 3′ end (marked by a triangle) of staple strands. The rest of the staple strands are in black. b, Cross-sections (left) and strand diagrams (right) of the 40-nm channel design: (P1) top monomer; (P2) bottom monomer. The scaffold strand is in gray. Inner handles and sticky ends are shown in red and blue, respectively. Handles (see Methods for sequences, not shown here for clarity) were extended from the 3′ end of staple strands. The rest of the staple strands are in black. c, Same as (b) but showing the 60-nm channel design.
Extended Data Fig. 2 |. DNA-origami assembly and purification.

a, Agarose gel electrophoreses showing the dimerization yields of the 40-nm and 60-nm wide DNA channels at different MgCl2 concentrations. Channel (dimer) bands are denoted by asterisks. The experiment was repeated three times with similar results. b, Different sizes of DNA channels purified by rate-zonal centrifugation. Agarose gel (1.5%) electrophoreses show the enrichment of DNA channel in fractions 11–13 (40 nm), 9–10 (60 nm), and 10–12 (45 nm), respectively. Well-folded nanostructure bands are denoted by asterisks. This experiment was repeated three times with similar results.
Extended Data Fig. 3 |. Nup purification and CA nanotube co-pelleting assays.
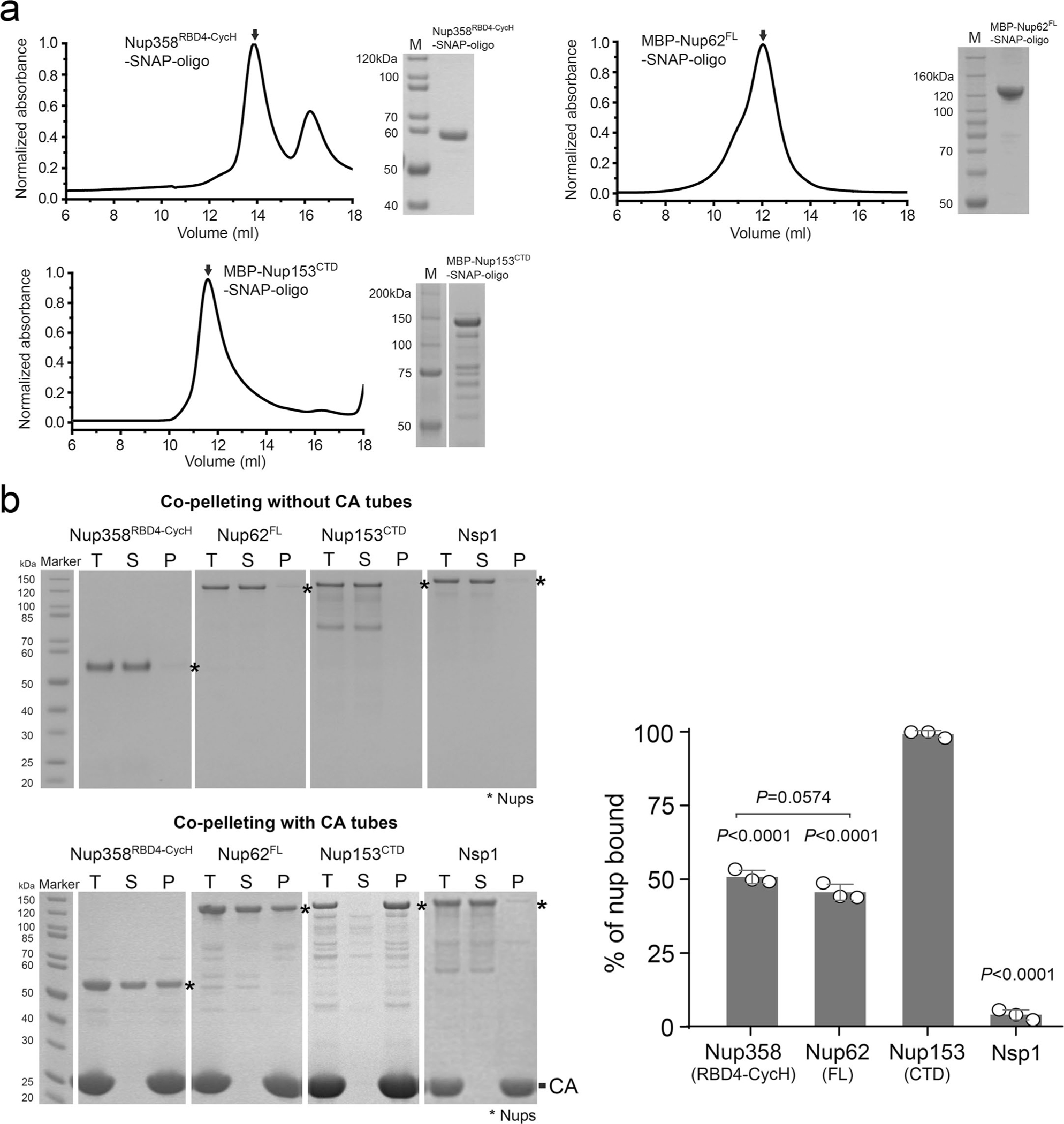
a, Size exclusion chromatography analysis showing that the DNA-conjugated of Nup358RBD4-CycH, MBP-Nup62FL, and MBP-Nup153CTD are mainly monodispersed monomers in solution. Right panels: SDS-PAGE analysis of protein purity. The experiment was repeated three times with similar results. b, Left: co-pelleting assay of purified nups using A14C/E45C disulfide crosslinked CA nanotubes. Soluble (S) and pellet (P) denote fractions of co-pelleting assays. Total (T) denotes the nup-CA tube mixture before fractionation. Right: percentages of various nups bound to CA tubes in the co-pelleting assays in PBS buffer. Data are plotted as mean±SD of three independent experiments (n = 3), with individual data points shown as black circles. Differences were determined by one-way ANOVA and Tukey’s multiple comparisons test. Unless marked in the graph, comparisons were made with MBP-Nup153CTD. Full statistical results are provided in source data.
Extended Data Fig. 4 |. Characterizing 45-nm NuPODs and their interactions with CA assemblies and virus cores.
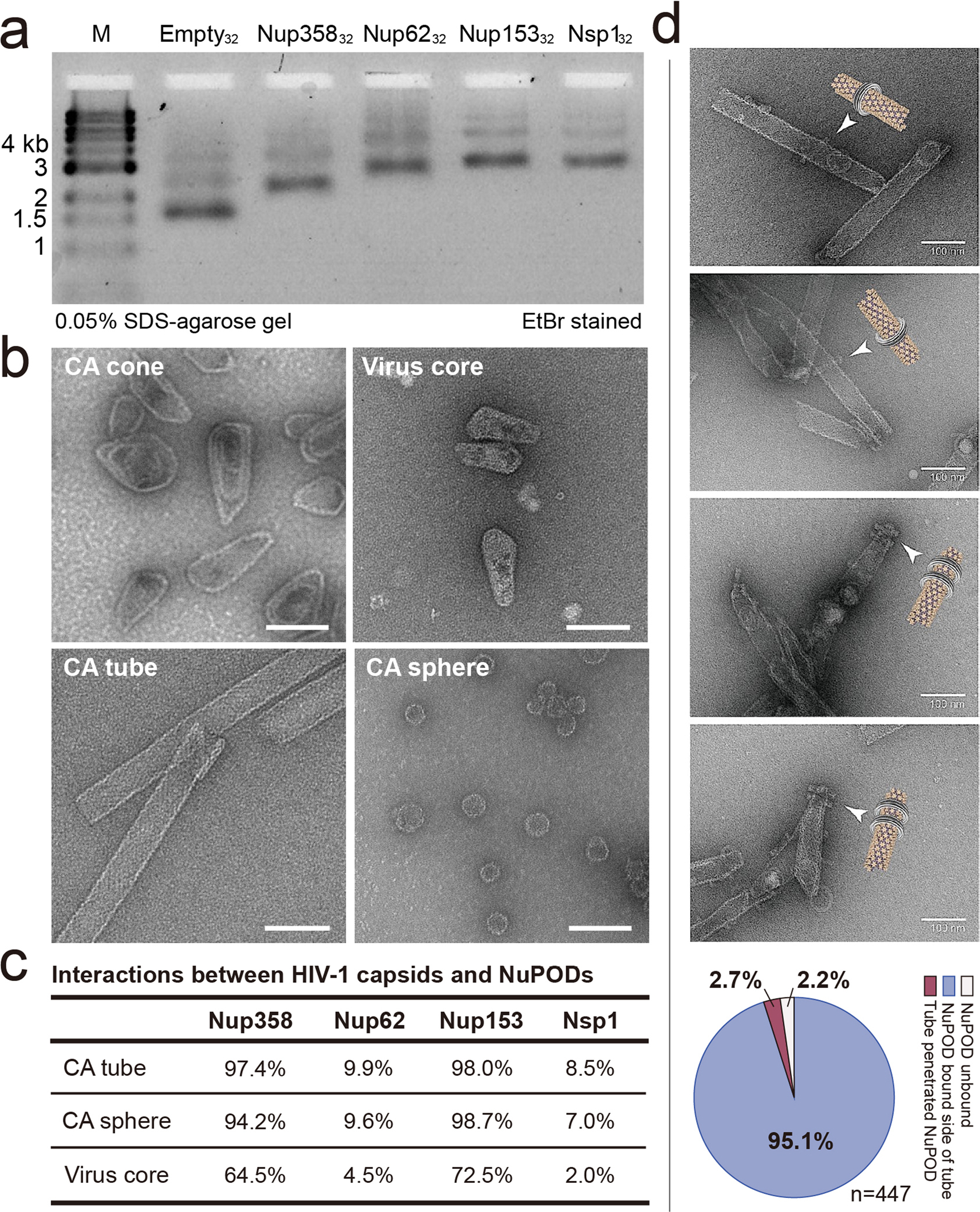
a, The NuPODs assembled with 32 copies of nups are characterized by SDS–agarose gel electrophoresis. The experiment was repeated three times with similar results. b, Negative-stain electron micrographs of CA assemblies and purified virus cores. The negative-stain EM experiments were repeated three times with similar results. Scale bar, 100 nm. c, Quantification of the interactions between HIV-1 capsid assemblies and NuPODs with various nups. Percentage values in the “CA tube” and “CA sphere” rows describe the population of NuPODs bound to CA assemblies. As the concentration of purified natural HIV-1 cores is low, percentage values in the “Virus core” row describe the population of cores occupied by NuPODs. n = 121–223. d, Schematics and negative-stain electron micrographs of CA nanotubes threading through the Nup153 NuPOD. The negative-stain EM results were repeated three times with similar results. Scale bar: 100 nm. Bottom: quantification of all binding events between Nup153 NuPODs and CA tubes. n = 447.
Extended Data Fig. 5 |. Nup358 or Nup153 NuPODs interacting with CA assemblies.
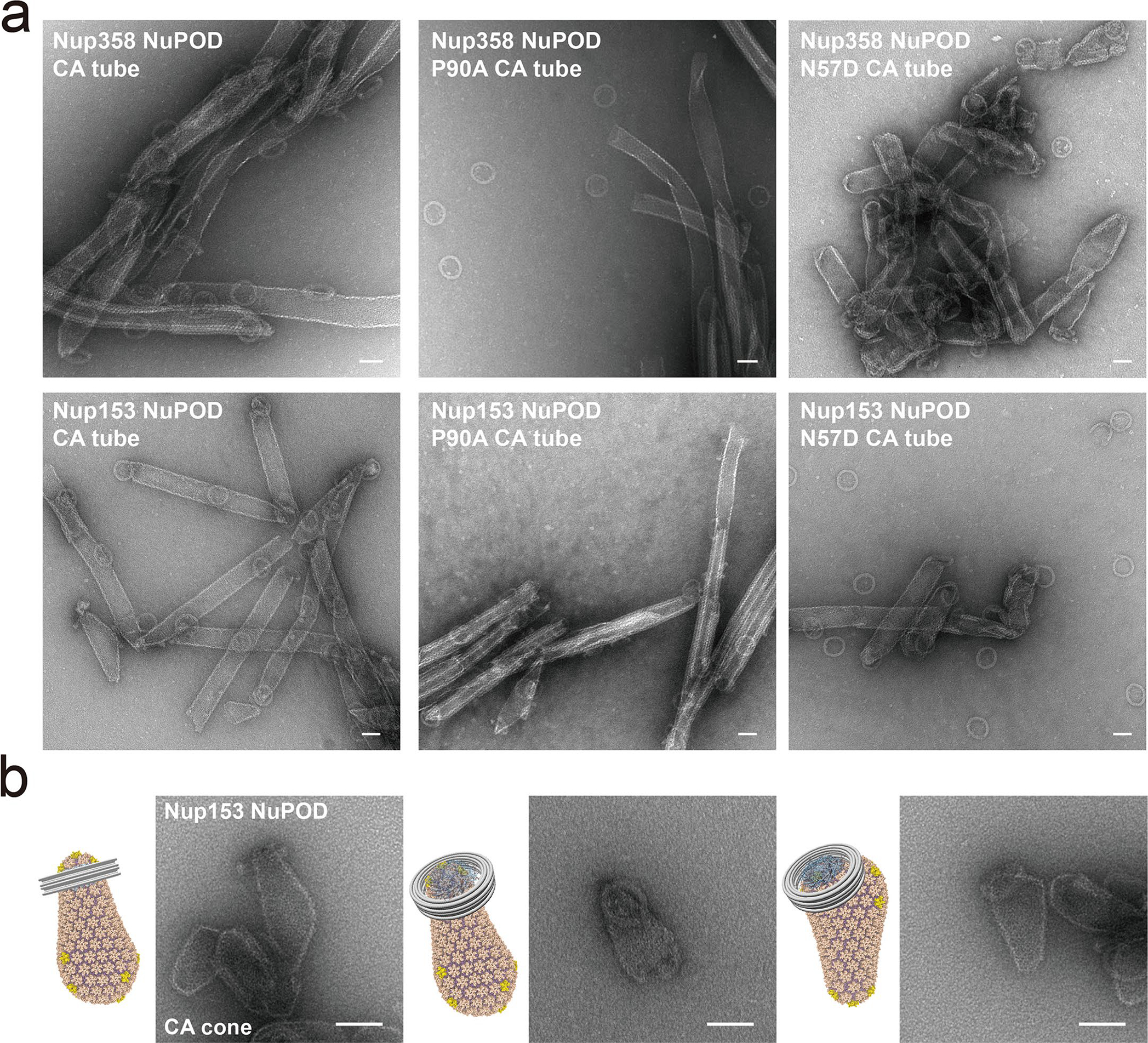
a, Negative-stain electron micrographs of Nup358 NuPODs (top) or Nup153 NuPODs (bottom) mixed with in vitro assembled WT (left), P90A (middle), or N57D (right) CA tubes. All negative-stain EM experiments were repeated three times with similar results. Scale bar, 50 nm. b, Schematics and negative-stain electron micrographs of the Nup153 NuPOD bound to in vitro assembled capsid cones. The experiment was repeated three times with similar results. Scale bar, 50 nm.
Extended Data Fig. 6 |. Co-pelleting assays of NuPODs with variants of CA tubes.
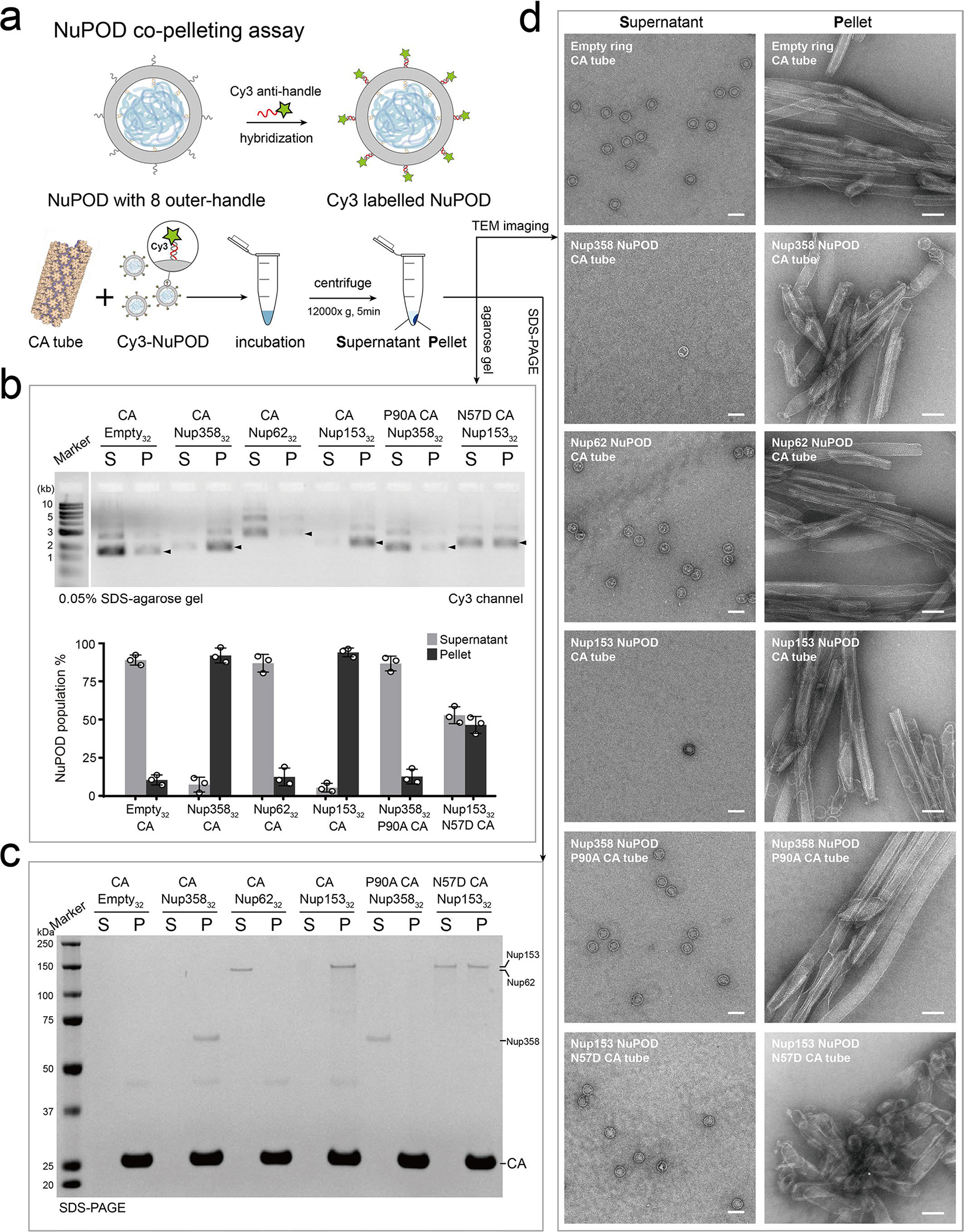
a, Schematic of the NuPOD-CA nanotube co-pelleting assay. b, Top: Supernatant (S) and pellet (P) fractions collected after co-pelleting Cy3-labelled NuPODs and A14C/E45C disulfide crosslinked CA tubes were analyzed on a 0.05% SDS–agarose gel. Fluorescence signals were used to detect the Cy3-labelled NuPODs. Black arrowheads indicate the NuPOD bands. Bottom: a plot showing the populations of Cy3-labelled NuPODs in the supernatant and pellet fractions. Data are plotted as mean±SD of three independent experiments (n = 3), with individual data points shown as black circles. c, Supernatant (S) and pellet (P) fractions from the co-pelleting assays analyzed by SDS-PAGE to detect the protein component of the NuPODs. The experiment was repeated three times with similar results. d, Negative-stain electron micrographs of supernatant (S) and pellet (P) samples from the co-pelleting assays. The experiment was repeated three times with similar results. Scale bar, 100 nm.
Extended Data Fig. 7 |. Nup153 prefers highly-curved regions of HIV-1 capsid.
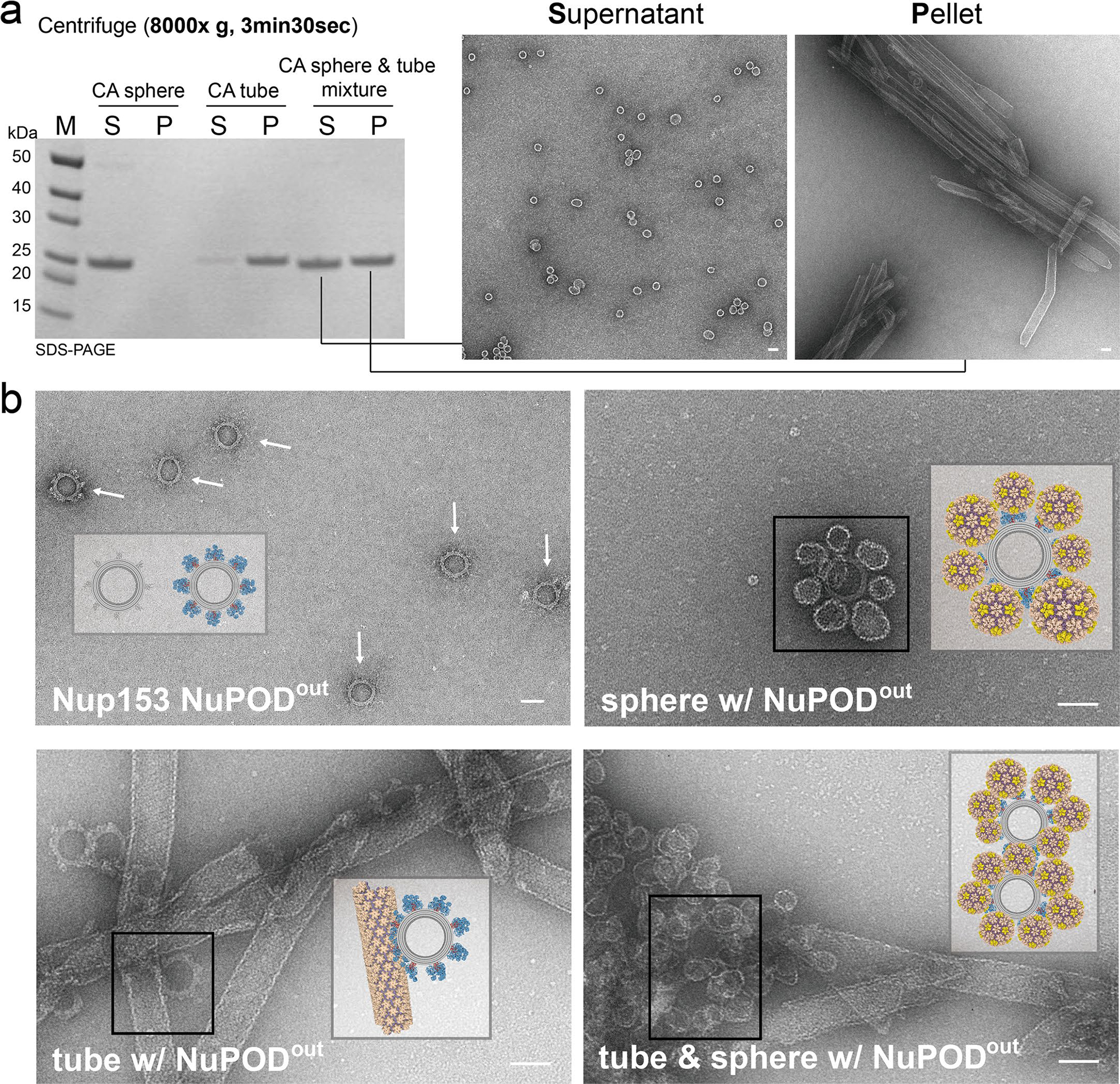
a, Left: supernatant (S) and pellet (P) fractions of CA spheres, nanotubes, or their mixture after centrifugation at 8,000 × g, analyzed by SDS-PAGE. Right: negative-stain electron micrographs of supernatant (S) and pellet (P) fractions of the CA sphere/nanotube mixture. The CA spheres remained in the supernatant, while the CA tubes were completely pelleted. Scale bar, 50 nm. This experiment was repeated three times with similar results. b, Schematics and negative-stain electron micrographs of Nup153 NuPODout (top left) binding to CA nanotubes (top right), CA spheres (bottom left), and a 1:1 mixture of CA spheres and nanotubes (bottom right). Note the lack of NuPOD binding to the flat tube surface in the competition binding experiment. All negative-stain EM experiments were repeated three times with similar results. Scale bar, 50 nm.
Extended Data Fig. 8 |. Negative-stain electron micrographs of CA spheres mixed with NuPODs.
a, 1 to 32 copies of Nup358RBD4-CycH. b, 1 to 32 copies of Nup153CTD. All negative-stain EM experiments were repeated three times with similar results. Scale bar, 50 nm.
Extended Data Fig. 9 |. Penetration assay for multilayer NuPODs and controls.
a, The 40-nm and 60-nm wide NuPODs containing multiple layers of nups analyzed by SDS–agarose gel electrophoresis. The experiment was repeated three times with similar results. b, Schematics and negative-stain electron micrographs of the 40-nm (right) and 60-nm (left) wide two-layer Nup358-Nup153 NuPODs (32 nups per layer). Direction indexes on NuPODs are marked by black arrowheads. The experiment was repeated three times with similar results. Scale bar, 50 nm. c, Left: cartoon models and representative negative-stain electron micrographs of purified virus cores inserted into single-layer Nup358 or Nup153 NuPODs. Scale bar, 50 nm. Right: Quantification of capsid insertion depths. Data are plotted as mean±SD, The HIV-1 capsid-NuPOD binding events (n) were captured over three independent experiments: Nup358 group (n = 50), Nup153 group (n = 52). d, Capsid insertion depths into NuPODs, measured from the tip of the capsid to the point it enters the DNA channel (entry point). Direction indexes on NuPODs are marked by black arrowheads. The experiment was repeated three times with similar results. Scale bar, 50 nm. e, Negative-stain electron micrographs of 32 copies of Nup62 grafted on the exterior of the 45-nm DNA-origami channel. This experiment was repeated three times with similar results. Scale bar, 50 nm.
Extended Data Fig. 10 |. Galleries of representative negative-stain electron micrographs of HIV-1 core-NuPOD interactions.

The purified virus cores bound to NuPODs loaded with different nups, with schematics on the left. All negative-stain EM experiments were repeated three times with similar results. Scale bar, 50 nm. More high-magnification images of NuPOD-virus core interactions are included in Supplementary Fig. 1–7.
Supplementary Material
Acknowledgements
We thank the Xiong and Lin labs for discussion. This work was supported by NIH grants P50AI150481 (Y. X., C. A. and A. N. E.), U54AI170791 (Y. X., C. A. and A. N. E.), R01AI052014 (A. N. E.), T32AI007386 (G. J. B.), R21GM109466 (C. L. and C. P. L.), R01GM105672 (C. P. L.), R01AI162260 (Y. X. and C. L.), a Collaboration Development Award Program from the Pittsburgh Center for HIV Protein Interactions (C. L.) and a Singapore Agency for Science, Technology and Research Graduate Scholarship (Q. X.)
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41594-023-00925-9.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41594-023-00925-9.
Peer review information Nature Structural & Molecular Biology thanks Grigory Tikhomirov and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Editor recognition statement (if applicable to your journal): Florian Ullrich, Carolina Perdigoto, Beth Moorefield and Dimitris Typas were the primary editors on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Data availability
All the data are available within this paper and its Supplementary Information. Source data are provided with this paper.
References
- 1.Yamashita M & Emerman M Retroviral infection of non-dividing cells: old and new perspectives. Virology 344, 88–93 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Strambio-De-Castillia C, Niepel M & Rout MP The nuclear pore complex: bridging nuclear transport and gene regulation. Nat. Rev. Mol. Cell Biol. 11, 490–501 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Beck M & Hurt E The nuclear pore complex: understanding its function through structural insight. Nat. Rev. Mol. Cell Biol. 18, 73–89 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Ganser BK, Li S, Klishko VY, Finch JT & Sundquist WI Assembly and analysis of conical models for the HIV-1 core. Science 283, 80–83 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Yamashita M & Engelman AN Capsid-dependent host factors in HIV-1 infection. Trends Microbiol. 25, 741–755 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hulme AE, Kelley Z, Okocha EA & Hope TJ Identification of capsid mutations that alter the rate of HIV-1 uncoating in infected cells. J. Virol. 89, 643–651 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Temple J, Tripler TN, Shen Q & Xiong Y A snapshot of HIV-1 capsid-host interactions. Curr. Res Struct. Biol. 2, 222–228 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Francis AC & Melikyan GB Single HIV-1 imaging reveals progression of infection through CA-dependent steps of docking at the nuclear pore, uncoating, and nuclear transport. Cell Host Microbe 23, 536–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Francis AC, Marin M, Shi J, Aiken C & Melikyan GB Time-resolved imaging of single HIV-1 Uncoating in vitro and in living cells. PLoS Pathog. 12, e1005709 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burdick RC et al. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl Acad. Sci. USA 117, 5486–5493 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zila V et al. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. Cell 184, 1032–1046 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muller TG et al. HIV-1 uncoating by release of viral cDNA from capsid-like structures in the nucleus of infected cells. eLife 10, e64776 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li C, Burdick RC, Nagashima K, Hu WS & Pathak VK HIV-1 cores retain their integrity until minutes before uncoating in the nucleus. Proc. Natl Acad. Sci. USA 118, e2019467118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dharan A, Bachmann N, Talley S, Zwikelmaier V & Campbell EM Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 5, 1088–1095 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selyutina A, Persaud M, Lee K, KewalRamani V & Diaz-Griffero F Nuclear import of the HIV-1 core precedes reverse transcription and uncoating. Cell Rep. 32, 108201 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guedan A et al. HIV-1 requires capsid remodelling at the nuclear pore for nuclear entry and integration. PLoS Pathog. 17, e1009484 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kane M et al. Nuclear pore heterogeneity influences HIV-1 infection and the antiviral activity of MX2. eLife 7, e35738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Nunzio F et al. Human nucleoporins promote HIV-1 docking at the nuclear pore, nuclear import and integration. PLoS ONE 7, e46037 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebina H, Aoki J, Hatta S, Yoshida T & Koyanagi Y Role of Nup98 in nuclear entry of human immunodeficiency virus type 1 cDNA. Microbes Infect. 6, 715–724 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Lee K et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7, 221–233 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang RN, Mehla R & Chauhan A Perturbation of host nuclear membrane component RanBP2 impairs the nuclear import of human immunodeficiency virus-1 preintegration complex (DNA). PLoS ONE 5, e15620 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matreyek KA & Engelman A The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J. Virol. 85, 7818–7827 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dharan A et al. KIF5B and Nup358 coperatively mediate the nuclear import of HIV-1 during infection. PLoS Pathog. 12, e1005700 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo J et al. The transmembrane nucleoporin Pom121 ensures efficient HIV-1 pre-integration complex nuclear import. Virology 521, 169–174 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin DH & Hoelz A The structure of the nuclear pore complex (an update). Annu. Rev. Biochem. 88, 725–783 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Appen A et al. In situ structural analysis of the human nuclear pore complex. Nature 526, 140–143 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zimmerli CE et al. Nuclear pores dilate and constrict in cellulo. Science 374, eabd9776 (2021). [DOI] [PubMed] [Google Scholar]
- 28.Schuller AP et al. The cellular environment shapes the nuclear pore complex architecture. Nature 598, 667–671 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matreyek KA, Yucel SS, Li X & Engelman A Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog. 9, e1003693 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price AJ et al. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 10, e1004459 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhattacharya A et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl Acad. Sci. USA 111, 18625–18630 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bichel K et al. HIV-1 capsid undergoes coupled binding and isomerization by the nuclear pore protein NUP358. Retrovirology 10, 81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Nunzio F et al. Nup153 and Nup98 bind the HIV-1 core and contribute to the early steps of HIV-1 replication. Virology 440, 8–18 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buffone C et al. Nup153 unlocks the nuclear pore complex for HIV-1 nuclear translocation in nondividing cells. J. Virol. 92, e00648–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin DH, Zimmermann S, Stuwe T, Stuwe E & Hoelz A Structural and functional analysis of the C-terminal domain of Nup358/RanBP2. J. Mol. Biol. 425, 1318–1329 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaller T et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 7, e1002439 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rothemund PWK Folding DNA to create nanoscale shapes and patterns. Nature 440, 297–302 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Seeman NC & Sleiman HF DNA nanotechnology. Nat. Rev. Mater. 3, 17068 (2018). [Google Scholar]
- 39.Dietz H, Douglas SM & Shih WM Folding DNA into twisted and curved nanoscale shapes. Science 325, 725–730 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dey S et al. DNA origami. Nat. Rev. Methods Prim. 1, 13 (2021). [Google Scholar]
- 41.Fisher PDE et al. A programmable DNA origami platform for organizing intrinsically disordered nucleoporins within nanopore confinement. ACS Nano. 12, 1508–1518 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ketterer P et al. DNA origami scaffold for studying intrinsically disordered proteins of the nuclear pore complex. Nat. Commun. 9, 902 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen Q et al. DNA-origami nanotrap for studying the selective barriers formed by phenylalanine-glycine-rich nucleoporins. J. Am. Chem. Soc. 143, 12294–12303 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang GXY et al. Structure of the cytoplasmic ring of the Xenopus laevis nuclear pore complex by cryo-electron microscopy single particle analysis. Cell Res. 30, 520–531 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin C, Perrault SD, Kwak M, Graf F & Shih WM Purification of DNA-origami nanostructures by rate-zonal centrifugation. Nucleic Acids Res. 41, e40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ori A et al. Cell type-specific nuclear pores: a case in point for context-dependent stoichiometry of molecular machines. Mol. Syst. Biol. 9, 648 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carmofonseca M, Kern H & Hurt EC Human nucleoporin P62 and the essential yeast nuclear-pore protein Nsp1 show sequence homology and a similar domain organization. Eur. J. Cell Biol. 55, 17–30 (1991). [PubMed] [Google Scholar]
- 48.Selyutina A, Bulnes-Ramos A & Diaz-Griffero F Binding of host factors to stabilized HIV-1 capsid tubes. Virology 523, 1–5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao G et al. Rhesus TRIM5α disrupts the HIV-1 capsid at the inter-hexamer interfaces. PLoS Pathog. 7, e1002009 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Z et al. T = 4 icosahedral HIV-1 capsid as an immunogenic vector for HIV-1 V3 loop epitope display. Viruses 10, 667 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shah VB & Aiken C In vitro uncoating of HIV-1 cores. 10.3791/3384 (2011). [DOI] [PMC free article] [PubMed]
- 52.Liu J, Sadre-Marandi F, Tavener S & Chen C Curvature concentrations on the HIV-1 capsid. 10.1515/mlbmb-2015-0003 (2015). [DOI]
- 53.Shen Q, et al. A DNA-origami nuclear pore mimic reveals nuclear entry mechanisms of HIV-1 capsid. Preprint at bioRxiv 10.1101/2020.08.10.245522 (2020). [DOI] [Google Scholar]
- 54.Labokha AA et al. Systematic analysis of barrier-forming FG hydrogels from Xenopus nuclear pore complexes. EMBO J. 32, 204–218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patel SS, Belmont BJ, Sante JM & Rexach MF Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell 129, 83–96 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Chug H, Trakhanov S, Hulsmann BB, Pleiner T & Gorlich D Crystal structure of the metazoan Nup62*Nup58*Nup54 nucleoporin complex. Science 350, 106–110 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Hu TH, Guan TL & Gerace L Molecular and functional characterization of the p62 complex, an assembly of nuclear pore complex glycoproteins. J. Cell Biol. 134, 589–601 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fahrenkrog B et al. Domain-specific antibodies reveal multiple-site topology of Nup153 within the nuclear pore complex. J. Struct. Biol. 140, 254–267 (2002). [DOI] [PubMed] [Google Scholar]
- 59.Wente SR & Rout MP The nuclear pore complex and nuclear transport. Cold Spring Harb. Perspect. Biol. 2, a000562 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Link JO et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 584, 614–618 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bester SM et al. Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science 370, 360–364 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Busnadiego I et al. Host and viral determinants of Mx2 antiretroviral activity. J. Virol. 88, 7738–7752 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Price AJ et al. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 8, e1002896 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bejarano DA et al. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the nuclear pore complex. eLife 8, e41800 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fernandez J et al. Transportin-1 binds to the HIV-1 capsid via a nuclear localization signal and triggers uncoating. Nat. Microbiol. 4, 1840–1850 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Song Y et al. Importin KPNA2 confers HIV-1 pre-integration complex nuclear import by interacting with the capsid protein. Antivir. Res. 200, 105289 (2022). [DOI] [PubMed] [Google Scholar]
- 67.Shen Q, Wu C, Freniere C, Tripler TN & Xiong Y Nuclear import of HIV-1. Viruses 13, 2242 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoelz A, Debler EW & Blobel G The structure of the nuclear pore complex. Annu. Rev. Biochem. 80, 613–643 (2011). [DOI] [PubMed] [Google Scholar]
- 69.Fay N & Pante N Nuclear entry of DNA viruses. Front. Microbiol. 6, 467 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gallucci L & Kann M Nuclear import of hepatitis B virus capsids and genome. Viruses 9, 21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Douglas SM et al. Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. 37, 5001–5006 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pauwels R et al. Sensitive and rapid assay on Mt-4 cells for detection of antiviral compounds against the AIDS virus. J. Virol. Methods 16, 171–185 (1987). [DOI] [PubMed] [Google Scholar]
- 73.Gallay P, Hope T, Chin D & Trono D HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc. Natl Acad. Sci. USA 94, 9825–9830 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wehrly K & Chesebro B p24 antigen capture assay for quantification of human immunodeficiency virus using readily available inexpensive reagents. Methods 12, 288–293 (1997). [DOI] [PubMed] [Google Scholar]
- 75.Chesebro B, Wehrly K, Nishio J & Perryman S Macrophage-tropic human-immunodeficiency-virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates—definition of critical amino-acids involved in cell tropism. J. Virol. 66, 6547–6554 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data are available within this paper and its Supplementary Information. Source data are provided with this paper.