

Abstract
Huntington’s disease (HD) is a dominantly inherited neurodegenerative disorder caused by a trinucleotide repeat expansion in the huntingtin gene resulting in long stretches of polyglutamine repeats in the huntingtin protein. The disease involves progressive degeneration of neurons in the striatum and cerebral cortex resulting in loss of control of motor function, psychiatric problems, and cognitive deficits. There are as of yet no treatments that slow disease progression in HD. Recent advances in gene editing using CRISPR-Cas systems and demonstrations of their ability to correct gene mutations in animal models of a range of diseases suggest that gene editing may prove effective in preventing or ameliorating HD. Here we describe potential CRISPR-Cas designs and cellular delivery methods for the correction of mutant genes that cause inherited diseases, and recent preclinical findings demonstrating efficacy of such gene editing approaches in animal models with a focus on HD.
Keywords: base editing, CRISPR, gene therapy, polyglutamine, prime editing, striatal neurons
The genetics and manifestation of HD
Huntington’s disease (HD) is caused by trinucleotide (CAG) expansions in the Huntingtin (HTT) gene resulting in long stretches of the amino acid glutamine in the huntingtin protein. The presence of forty or more CAG repeats leads invariably to HD, while shorter expansions may or may not lead to HD. The symptoms typically manifest between the ages of 30 and 50 but can appear before the age of 20 when there are more than 60 CAG repeats. Mutant huntingtin self-aggregates inside neurons and is associated with mitochondrial dysfunction and impaired autophagy which likely contribute to neuronal death [1]. Fast-spiking GABAergic medium spiny neurons with a high energy demand are the first to die in HD and their demise is thought to involve an excitotoxic process [2]. There are no treatments that delay the onset or slow disease progression. Reproductive choices for individuals harboring mutant genes that cause fatal inherited neurological disorders have been described previously [3].
Gene therapy for persons with disease causing mutation, along with the scientific and ethical challenges associated with them, were discussed as early as the 1970s [4]. Since then, several methods for gene therapy have been developed including those that deploy zinc finger proteins (ZFPs), transcription activator-like effector nucleases (TALENs), antisense DNA, or small interfering RNA. The recent development of gene editing methods based on CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR-associated protein 9) has enabled efficient correction of missense and other gene mutations and has resulted an explosion of translational research studies in animal models of a range of genetic disorders [4]. Here we describe CRISPR-based gene editing technologies, approaches for targeted cellular delivery of the gene editing cargos, and translational studies in experimental models of HD.
CRISPR-base Gene Editing Technologies
CRISPR-Cas9 systems utilize a single guide RNA (sgRNA) that recognizes the target sequence via complementary base pairing (Figure 1A). Cas9 endonuclease uses the sgRNA to form base pairs with DNA target sequences enabling a site-specific double-stranded break in the DNA. The portion of the sgRNA sequence that is complementary to the target sequence is called the ‘spacer’. An additional requirement for Cas9 to function is a specific protospacer adjacent motif (PAM) located directly downstream of the target sequence in the non-target strand. Through RNA-directed Cas9 nucleases this system can modify DNA with greater precision than ZFPs and TALENs. [5]. The double strand break introduced by Cas9 is repaired by either non-homologous end joining (NHEJ) or homology-directed repair (HDR). NHEJ is more efficient than HDR but tends to be error-prone and can produce insertions, deletions, translocations, or other DNA rearrangements [6], making the outcome of the repair process unpredictable. However, progress has been made in predicting the outcome of some types of insertions and/or deletions (InDels) [7]. The end products of the NHEJ repair process often harbor larger DNA alterations that cause disruption of the target gene [8]. In contrast, HDR exhibits high repair precision but low efficiency [7]. Thus, without additional features CRISPR-Cas9 is not suitable for the precise sequence manipulations of genomic DNA required in gene therapy. Recent efforts have therefore focused on developing technologies that enable the precise replacement of individual bases in the DNA without generating double strand breaks and without the need for exogenous repair templates [9, 10].
Figure 1.
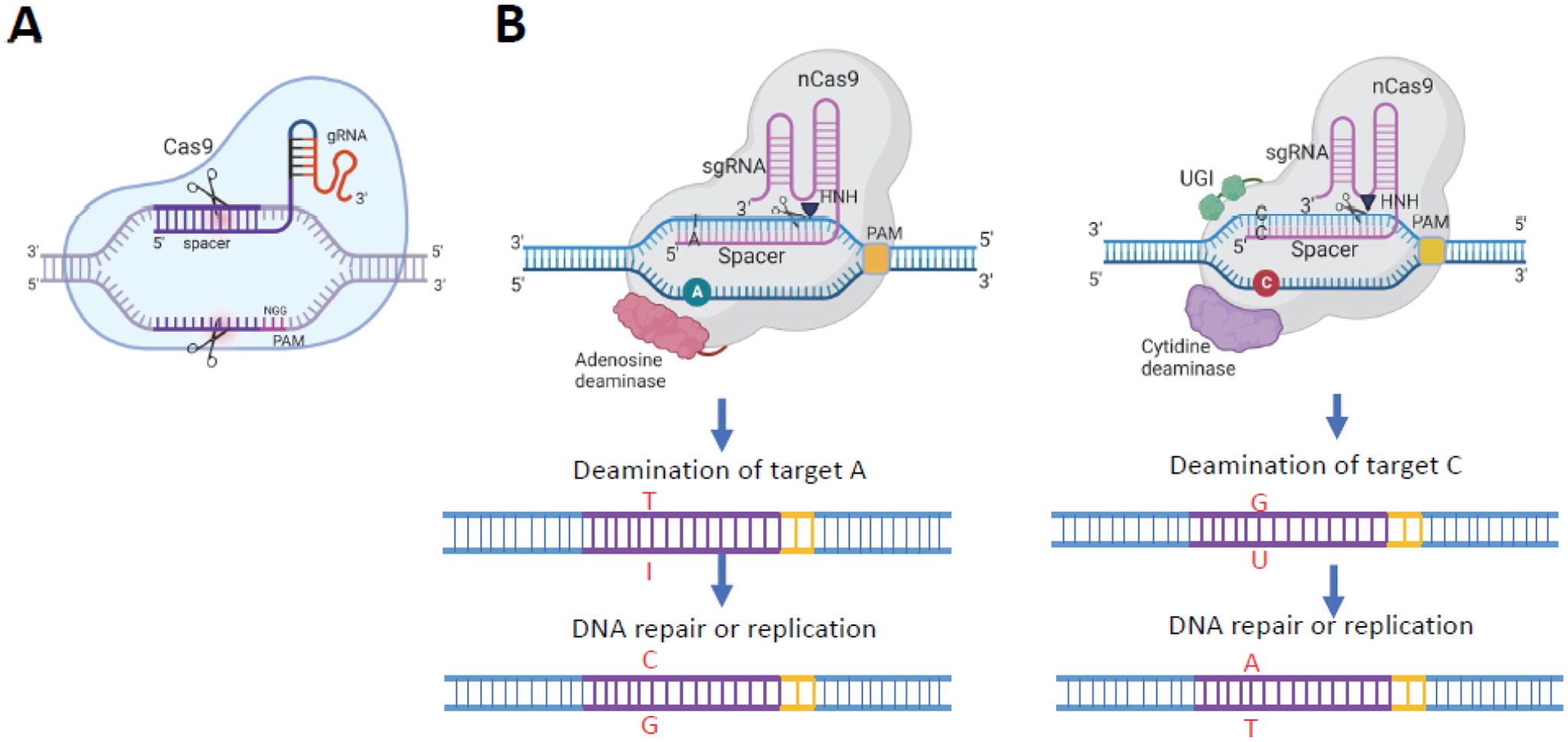
Illustration of CRISPR-Cas9 and base editing. A. CRISPR-Cas9-mediated gene editing. B. Adenine base editing (left) and cytosine base editing (right) are illustrated. Adenine base editors consist of Cas9 nickase (nCas9) and an adenosine deaminase that mediates deamination of adenosine (A) to inosine (I). Inosine is recognized as guanosine (G) upon DNA replication, thus resulting in fixation of an A-to-G mutation. Cytosine base editors consist of a nCas9, a cytidine deaminase, and a uracil DNA glycosylase inhibitor (UGI). The UGI protects the uracils arising from cytidine deamination by preventing their excision by uracil DNA glycosylase. The nick in the unedited strand is recognized by the cellular mismatch repair pathway that then can use the edited strand as a repair template to form a U:A base pair, which can become fixed as a T:A base pair by DNA repair and/or DNA replication. Figure created using BioRender.com
Base editing
Base editing is a novel technology that can generate gene disruptions or correct DNA mutations. Base editors consist of a Cas nickase (nCas9) fused to a nucleoside deaminase [11,12]. This feature enables construction of fusion proteins that provide CRISPR-Cas systems with new enzymatic properties, including nucleotide-modifying activities. nCas9 can introduce single strand breaks in a manner that largely prevents the introduction of InDels, while fully retaining the DNA recognition properties conferred by the sgRNA [13].
Two different nucleoside deaminases are used for base editing (Figure 1B). Adenine base editing (ABE) uses adenosine deaminase fused to nCas9 and cytosine base editing (CBE) uses cytidine deaminase fused to nCas9 [10,12]. In ABE, the adenosine deaminase catalyzes the oxidative deamination of deoxyadenosine to deoxyinosine. Because deoxyinosine mimics deoxyguanosine, this conversion causes A-to-G transitions. CBEs cause C-to-T transitions by deaminating deoxycytidine to deoxyuridine. ABEs and CBEs enable all four base transitions (A to G, T to C, C to T, and G to A). These base editors do not require provision of a template or additional protein factors for nucleotide modification to occur [9,14]. However, because uracil and hypoxanthine are both alien bases in DNA they are recognized and removed by the base excision repair pathway. This makes the correction of the mutation introduced by base editing dependent on improper repair or lack of repair prior to DNA replication and poses a significant limitation on the efficiency of the editing. Additional strategies for improving the efficiency of deaminase-based base editing are therefore being developed [15].
During complex formation between the base editing fusion protein and sgRNA with the target DNA sequence an R-loop is formed by annealing of the sgRNA with the complementary DNA strand. The displaced single-stranded DNA provides the substrate for the deaminase. The sequence stretch of the displaced strand that is accessible to the deaminase domain of the fusion protein is referred to as the ‘base editing window’. Since base editing depends on direct interaction between the catalytic domain of the deaminase and the substrate nucleotide, Cas protein variants differing in their PAM sequence requirements provide different editing windows. The base editing window can also be modified by the orientation of the domains within the base editor fusion protein. The linkers that connect the different domains of base editing fusion proteins can also influence the location and width of the editing window as well as editing activity. However, ABE and CBE may induce RNA editing in an sgRNA-independent manner because of their affinity to RNA which in the case of ABEs is the natural substrate of the deaminase.
The editing activities of all currently available base editors is not strictly limited to the intended target site, and off-target DNA editing can occur in either an sgRNA-dependent or sgRNA-independent manner. Guide RNA-dependent off-target editing can be largely avoided by using high fidelity Cas variants [16–18], careful sgRNA design, use of truncated sgRNAs [19, 20], and/or delivery of the base editor as a preassembled ribonucleoprotein particle [21, 22]. Guide RNA-independent off-target mutations can lead to deamination of accessible nucleotides preferentially in coding regions. This phenomenon is predominantly observed in CBEs and less so in ABEs [22, 23]. Base editing can also cause unwanted bystander mutations within the editing window. Bystander mutations occur when a C or A nucleotide other than the targeted one is deaminated, or multiple Cs or As within the editing window undergo editing [24].
A major potential application of base editing is the reversion of gain of function disease-causing point mutations [25]. However, a major limitation of base editing is the ability to generate precise base-edits beyond the four transition mutations. Recently, prime editing has been developed to overcome this shortcoming [26].
Prime editing
Prime editing can mediate targeted insertions, deletions, and all possible base-to-base conversions without double strand breaks or donor DNA templates (Figure 2).An engineered prime editing guide RNA (pegRNA) that specifies the target site and contains the desired edit(s) engages the prime editor protein consisting of a nCas9 fused to a reverse transcriptase (RT). The nCas9 is guided to the DNA target site by the pegRNA. After nicking by nCas9, the RT domain uses the pegRNA as a template for reverse transcription of the desired edit directly polymerizing DNA onto the nicked target DNA strand. The edited DNA strand replaces the original DNA strand creating a heteroduplex containing one edited strand and one unedited strand. The prime editor then guides the heteroduplex to copy the edit onto the unedited strand.
Figure 2.
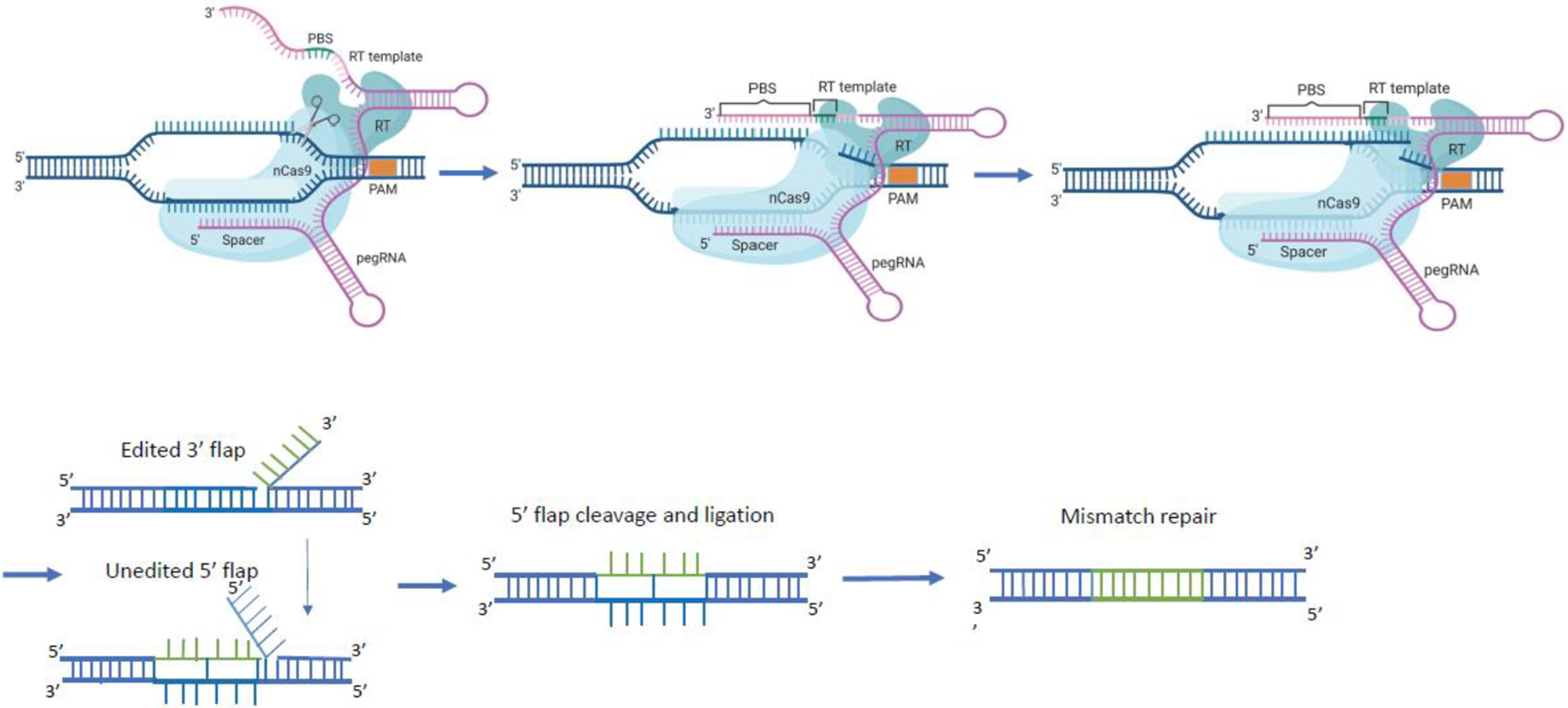
Prime editing components are comprised of a fusion protein of nCas9 (neon blue) with a reverse transcriptase (RT; turquoise) domain and an engineered prime editing single guide RNA (pegRNA; purple). The pegRNA harbors a primer binding site (PBS) and a reverse transcription template (RT) as 3′ extension of the sgRNA scaffold. The pegRNA guides the nCas9 domain to the target site to cut the protospacer adjacent motif (PAM)-containing strand and directs the synthesis of an edited DNA strand starting from the 3′ end of the nicked strand and using the RT sequence (green) as template. The 3′ newly synthesized edited DNA strand can displace the 5′ unmodified DNA strand, resulting in a hybridization of the uncut strand with two DNA flaps. The 3’ flap contains the newly synthesized (edited) sequence and the 5’ flap contains the dispensable, unedited DNA sequence. The 5’ flap is then cleaved by structure-specific endonucleases or 5’ exonucleases. The edited 3’ flap proceeds to ligation and forms a heteroduplex DNA composed of one edited strand and one unedited strand. The reannealed double stranded DNA contains nucleotide mismatches at the location where editing took place. The mismatch repair mechanism is initiated and the information in the edited strand is copied into the complementary strand. Figure created using BioRender.com
The pegRNA is a guide RNA with a 3′ end that contains two additional functional domains: a primer-binding sequence (PBS) complementary to the 3′ end of the nicked DNA strand, and an upstream RNA sequence that serves as a template for reverse transcription (RT) (Figure 2). Thus, peg RNA not only guides the prime editor to its target site but also facilitates the annealing of the primer-binding sequence with the nicked DNA strand. The 3′ end of the DNA is extended by reverse transcription of the RNA template containing the sequence to be introduced by prime editing. cDNA synthesis leads to the formation of a branched intermediate at the nicked site consisting of two DNA flaps, the extended and edited DNA (3′ flap), and the original DNA strand (5′ flap). Hybridization of the two flaps to the uncut DNA strand occurs in an equilibrium that is then resolved by the DNA repair machinery. Removal of the unedited 5′ flap results in formation of a DNA heteroduplex consisting of one edited and one unedited strand. The cellular DNA mismatch repair system subsequently replaces the original sequence with the edited sequence.
Prime editing is less constrained by PAM sequence and more versatile and precise than base editing, and is more efficient than HDR [26]. Prime editing can incorporate point mutations into relatively large sequence stretches and can enable multiple mutations up to 30 nucleotides away from the cut site in the DNA strand. Prime editors can target about 90% of the pathogenic gene variants that are currently listed in the ClinVar databasei [27–29]. The pegRNA is believed to be the main component limiting prime editing efficiency [26] but other factors such as the orientation of the RT relative to the Cas9 nickase and the lengths of both the primer-binding sequence and RT sequences of the pegRNA may also affect editing efficiency [18,26]. Additionally, the cellular DNA mismatch repair system lowers the efficiency of prime editing, but this can be alleviated by co-expression of an engineered mismatch repair-inhibiting protein [30].
While prime editing is currently the most versatile method for DNA editing several challenges remain. The efficiency of prime editing still varies greatly depending on the type of edits to be made, the surrounding sequence context, and the target cell type. The large size of the prime editor complex also poses a challenge for packaging and cell delivery.
Delivery and Cell Targeting
Gene editing cargos can be delivered as DNA, RNA, or ribonucleoproteins (RNP). Delivering DNA encoding Cas9 (or related endonucleases) and a sgRNA results in stable expression of these gene editing cargos which can be advantageous for some applications but also increases the probability of off-target effects. Delivering mRNAs encoding Cas9 proteins in combination with sgRNA results in transient expression of the gene editing machinery. Because RNA is rapidly degraded by RNAses, methods of chemical modification of the RNA to increase stability have been developed [31]. For RNP, recombinant Cas protein is combined with sgRNA.
There are several features of a vehicle for gene editing cargos that are required for effective delivery to the desired cells [32, 33]. First, the vehicle must contain the cargoes and protect them from degradation prior to their delivery to the target cells. This is typically accomplished by enclosing the cargos in a lipid or protein shell. Second, the delivery vehicle must bind to the surface of the target cells. Typically this is mediated by an antibody or glycoprotein on the surface of the delivery vehicle that recognizes a molecule on the surface of the target cell. Third, the delivery vehicle must enter the cell, for example by receptor-mediated endocytosis. Fourth, the gene editing cargos must be released into the cytosol and trafficked to the cell nucleus; to accomplish this, vehicles typically include features that exploit the acidic pH of endosomes resulting in release of cargos into the cytosol.
The technologies used for delivery of the gene editing cargos can be categorized as physical, chemical, or biological [32,33]. Physical methods include microinjection, electroporation, and ultrasonication. Physical methods are useful for delivery of cargos into cultured cells but are generally impractical for in vivo applications. The most commonly used chemical delivery methods are incorporation of the gene editing machineries into nanoparticles comprised of lipids or gold. Lipid nanoparticles (LNP) consist of a combination of cationic lipids, polyethylene glycol, ‘helper’ phospholipid, and cholesterol (Figure 3a). This combination enhances cell entry and intracellular release of the gene editing cargos. LNP are useful for delivery of gene editing cargos to cultured cells and have also been used to correct genetic defects in mouse models of neurological disorders [34]. For example, in a mouse model of hereditary deafness caused by a missense mutation in the Tmc1 gene, injection into the cochlea of LNP with Cas9-sgRNA complexes targeting the mutant allele reduced hearing loss and prevented degeneration of auditory hair cells [35]. LNP delivery has several advantages over viral delivery of gene editing cargos. LNPs have a much larger cargo capacity, result in transient expression of the gene editing cargos which reduces the probability of off-target editing.
Figure 3.
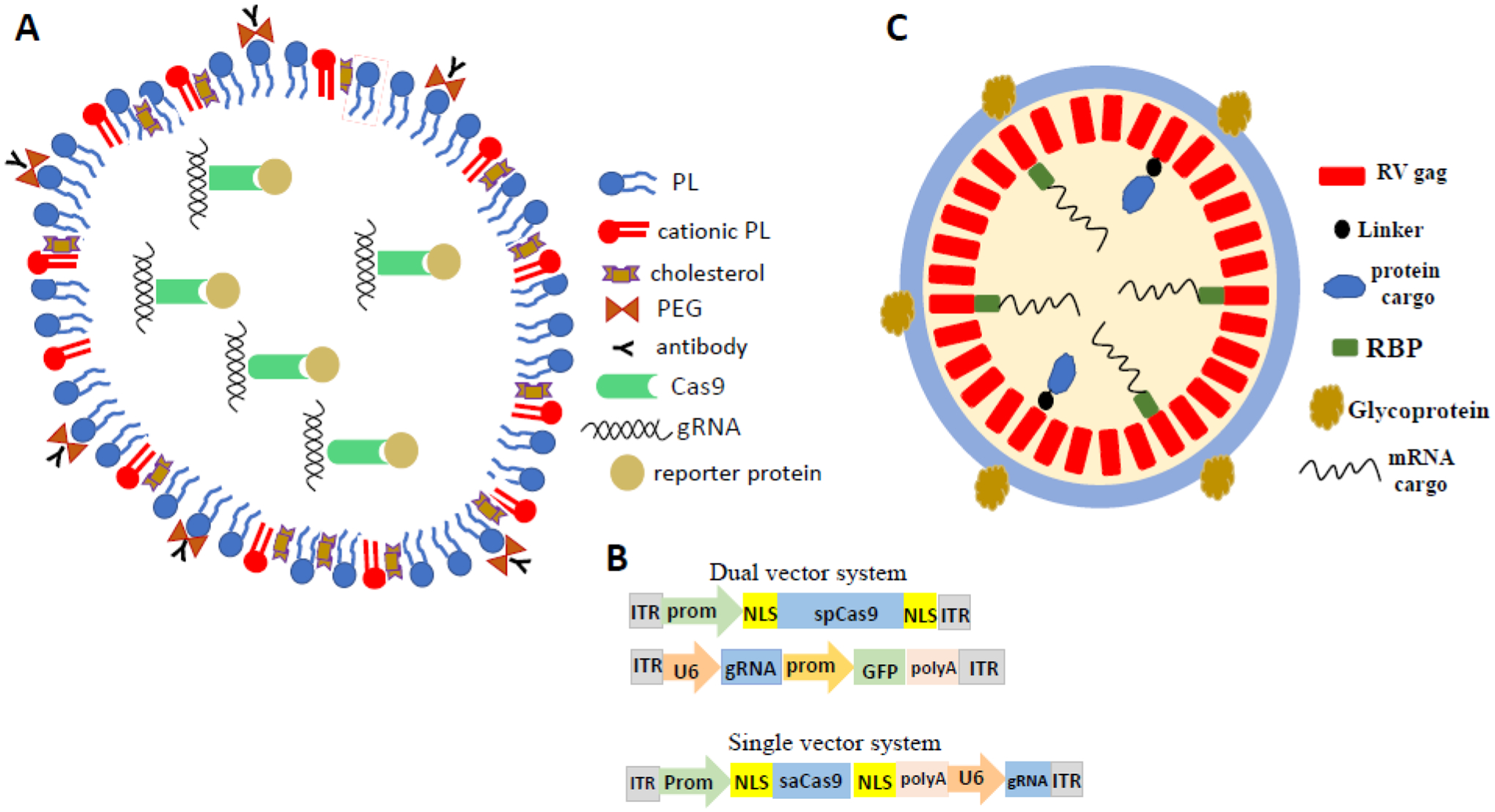
Lipid nanoparticles (LNP), adeno-associated virus (AAV), and virus-like particles (VLP) as systems for delivery of CRISPR-Cas9 gene editing cargos. A. Molecular composition of a LNP for delivery of CRISPR-Cas9 gene editing cargos (see [34]). gRNA, guide RNA; PEG, polyethyleneglycol; PL, phospholipid. B. Examples of AAV constructs for gene editing. GFP, green fluorescent protein; ITR, inverted terminal repeat; NLS, nuclear localization sequence; prom, promoter; saCas9, Cas9 from staphylococcus aureus; spCas9, Cas9 from streptococcus pyrogenes; U6, an RNA polymerase II promoter. C. An example of a VLP containing CRISPR-Cas9 cargos. The viral gag protein contains the protease required for cleaving polyproteins into subunits and also provides structural stability to VLP. RBP, RNA-binding protein; RV, retroviral.
CRISPR-gold nanoparticles consist of 15 nanometer diameter gold particles conjugated to oligonucleotides with Cas9 RNP incorporated into the oligonucleotides. The nanoparticles are coated with polyaspartic acid which is degraded upon entry of the nanoparticles into endosomes thereby releasing the CRISPR cargos to the cell cytoplasm. Gold nanoparticle delivery of CRISPR-Cas9 RNP cargos targeting the metabotropic glutamate receptor 5 gene ameliorated behavioral abnormalities in a mouse model of fragile X syndrome [36].
Viral vector-based delivery of gene editing tools has been widely used in translational neuroscience research. More recently, this approach has been incorporated into various clinical trials as well. It is important to note that viral delivery methods carry several limitations that require careful consideration in any potential clinical application, including: (i) limited cargo capacity; (ii) potential viral-mediated toxicity; (iii) immune responses, which may limit the number of viral delivery administrations and/or the unity of virus-like particles; (iv) decreased efficiency and/or off-target effects due to in-cell generation of active Cas9 enzyme from nucleic acids; (v) side effects of long term expression of endonucleases following viral delivery; (vi) risk of oncogenesis, particularly for lentiviruses, due to integration. We refer the readers to prior articles for more detailed discussion of the advantages and limitations of potential clinical applications of viral vectors [37]. Adeno-associated viruses (AAVs) have been widely employed as vectors for the delivery of genes and small interfering RNAs to neurons [38]. AAVs are small (~4.8 kilobase) linear single-stranded DNA viruses that are replication-defective. Although AAV remains episomal in infected target cells it is stabilized by circularization and concatemer formation which enables long-term transgene expression in non-mitotic cells such as neurons. Importantly, in contrast to other viruses, AAV elicits limited immune responses compared to other viral vectors[39].
In applications where a single engineered AAV is insufficient to deliver all of the desired cargos, two AAV constructs have been employed (Figure 3b). Alternatively, AAV systems with smaller Cas9 orthologues and minimal promoters can be used. For example, Cas9 dual AAV gene editing approach incorporating a protein auto-processing domain (intein) was used to introduce a nonsense coding sequence into transgenic mice expressing a mutant SOD1 that causes familial ALS [40]. Expression of CRISPR components using such AAVs has also proven effective in generating transgenic mouse models of several different neurological disorders and for the rapid generation of brain cell type-specific gene knockout mice [41–44].
Lentivirus readily infects neurons, provides sustained expression of cargos, and has a carrying capacity (8.5 kilobases) that is greater than AAV (4.7 kilobases). For example, inactivation of the synaptotagmin gene with a lentivirus CRISPR interference method resulted in a decrease in excitation of glutamatergic neurons in the hippocampus [45]. In another study, lentivirus was used for CRISPR activation and interference systems engineered with intron-containing Cre recombinase to drive gRNAs targeting several genes including Fos, GRM2, and Gadd45b in neurons [46]. However, among the major drawbacks of lentiviruses is that they can induce unwanted immune responses.
Another vehicle for delivery of gene editing cargos that is being developed is virus-like particles (VLP) [32]. VLP are assemblies of viral proteins in which desired gene editing cargos are packaged (Figure 3c). VLP have several advantages including: transient expression of gene editing cargos which reduces the chance of off-target editing. VLP have been used extensively for the preparation of vaccines including SARS-CoV2 vaccines [47] but neuroscientists have yet to exploit their advantages for gene editing.
The most common routes of delivery of gene editing cargos into the central nervous system in animal studies are stereotaxic injection into desired locations, intrathecal injection or intraventricular injection into the cerebrospinal fluid. In addition, AAV variants that efficiently infect axons and are retrogradely transported to neuronal cell bodies have been developed and shown to express cargos in projection neurons in the mouse brain [48]. Intravenous delivery suffers from several problems including dilution of the CRISPR cargo vehicles, limited access to the central nervous system, and increased chance of adverse effects on peripheral tissues.
Preclinical studies of gene therapy for HD
Gene editing using CRISPR-Cas9 techniques have been used to correct gene mutations in cell culture and animal models of several neurological disorders (Table 1). One common approach is to first establish the efficacy of a CRISPR-Cas9 construct designed to target a gene mutation in cultured neurons derived from pluripotent stem cells (iPSC) generated from fibroblasts taken from a human carrier of that mutation. Once the efficacy and specificity of a gene editing system is established in cultured cells, the system can be tested in transgenic mouse models.
Table 1.
Preclinical gene-editing studies in iPSC and animal models of neurological disorders.
| Disease | Method | Model | Major findings | Reference |
|---|---|---|---|---|
| ALS | CRISPR/Cas9 | SOD1 G93A mice | Reduction of mutant SOD1 protein in the lumbar and thoracic spinal cord improves motor function and reduces muscle atrophy | [64] |
| ALS | CRISPR/Cas9 | SOD1 G93A mice | A nonsense-coding insertion into a mutant SOD1 gene prolongs survival and markedly slows disease progression | [40] |
| SCA3 | CRISPR/Cas9 | Human iPSC | iPSC derived from an SCA3 patient retain pluripotency and neural differentiation following deletion of the expanded polyQ-encoding region of ataxin 3 | [65] |
| Fragile-X syndrome | CRISPR/Cas9 | Human iPSC | After removal of the CGG repeats, the upstream CpG island of the FMR1 promoter shows extensive demethylation, an open chromatin state, and transcription initiation | [66] |
| ALS | CRISPR/Cas9 | Human iPSC | Excision of the large C9orf72 repeat expansion mutation rescues RNA foci formation and promoter hypermethylation without altering C9orf72 transcript and protein expression | [67] |
| HD | CRISPR/Cas9 | Human iPSC | Excision of mHtt on the disease chromosome prevents the generation of mutant huntingtin mRNA and protein | [55] |
| HD | CRISPR/Cas9 | HD140Q-KI mice | Permanent suppression of mutant Htt expression in the striatum depletes huntingtin aggregates and attenuates early neuropathology | [49] |
| HD | ZFN | R6/2 mice | One-time striatal AAV-ZFP infusion can correct histopathological, electrophysiological and biomarker deficits that are characteristic of human HD pathology | [68] |
| AD | CRISPR/Cas9 | C57Bl/6J mice | Genome editing of intron −1/exon 1 of Mapt in C57Bl/6J mice generates a strain that shows a significantly reduced susceptibility to excitotoxic seizures, with normal learning and memory | [69] |
| PD | CRISPR/Cas9 | Synuclein A53T rats | Gene deletion of A53T-SNCA significantly ameliorates α-synuclein pathology, dopaminergic neurodegeneration, and parkinsonian motor symptoms | [70] |
| HD | CRISPR/Cas9 | Human iPSC | CAG repeat deletion ameliorates mitochondrial phenotypes in motor neurons | [71] |
| ALS | CRISPR/Cas9 | Human iPSC | Normal cellular phenotypes are restored following focused deletion of the G4C2 hexanucleotide repeat expansion in C9orf72 | [72] |
| ALS | CRISPR/Cas9 | G93ASOD1 mice | The deletion of mutant SOD1 prolongs survival | [73] |
| ALS/FTD | CRISPR/Cas9 | Human iPSC | The excision of the repeat expansion in CRISPR/Cas9 MNs reverses several physiological defects in Ca2+ homeostasis | [74] |
| ALS | CRISPR/Cas9 | SOD1 G93A mice | hSOD1-G93A transgene editing in SOD1-linked ALS mice prevents the development of ALS-like disease | [75] |
| AD | CRISPR/Cas9 | 5XFAD mice | The deposition of amyloid-beta, as well as microgliosis, neurite dystrophy and cognitive performance impairments are ameliorated using brain-wide selective disruption of a mutant APP allele | [76] |
| Fragile-X Syndrome | CRISPR/Cas9 | FMR1KO mice | Reduction of local mGluR5 levels in the striatum can rescue mice from the exaggerated repetitive behaviours | [77] |
| HD | CRISPR/Cas9 | Huntingtinz Q175 mice | Non-allele-specific HTT silencing in striatal neurons restores altered arteriolar cerebral blood volumes in premanifest mice, delays onset of striatal atrophy, and slows the progression of motor phenotype and brain pathology | [50] |
The therapeutic potential of CRISPR-Cas9 based gene therapy for HD has been tested in several preclinical models. In HD140Q knock-in mice expressing human HTT exon-1 with 140 CAG repeats, a nonallele-specific targeting strategy repressed mutant human HTT expression, and was associated with reduced mutant huntingtin protein aggregates and improved motor function [49]. In a study from our group using the same Cas9 system, altered arteriolar cerebral blood volumes were restored and striatal atrophy and the progression of motor phenotype were slowed by CRISPR-Cas9-mediated mutant HTT silencing in striatal neurons of zQ175 knock-in HD mice [50].
Because the difference between normal and mutant HTT alleles is the size of CAG expansions, classical CRISPR-Cas9 systems that target CAG will not achieve allele-specific silencing or excision. Discrimination between the two CAG tracts therefore requires specificity for repeat expansions greater than ~35 CAGs, corresponding to about 105 base pairs, which is not achievable using current methods. In addition, other genes containing repetitive CAG tracts would likely be affected by targeting the CAG expansions.
One study deployed CRISPR-Cas9 designed to selectively target human mutant HTT in a transgenic mouse model expressing exon-1 of the human HD gene, the R6/2 model. This study demonstrated reduction of mutant huntingtin protein and improved lifespan and motor symptoms in mice receiving the CRISPR-Cas9 therapy [51]. But this selectivity was possible only because the sgRNA selectively targeted a non-CAG sequence specific for the human HTT allele thereby leaving the endogenous mouse Htt allele unaffected.
Wild type huntingtin is critical for normal brain development and adult neuroplasticity. Because people with HD have one normal and one mutant allele, it is critical to develop gene editing systems that target only the mutant allele. Several such approaches have been evaluated in cultured cells derived from HD patients. For example, a paired nCas9 strategy selectively excised expanded CAG repeats in exon 1 of the HTT gene in three different HD patient fibroblast lines, inactivating only mutant HTT [52]. A strategy using CRISPR-Cas9 and piggyBac transposon was also successfully used in HD human iPSCs, correcting the mutant HTT allele and amending HD mediated phenotypic abnormalities [53]. So far, to our knowledge, these allele-selective approaches have only been applied to cultured cells.
One promising method for selectively targeting mutant HTT is to take advantage of allele-specific single nucleotide polymorphisms (SNP) which allow for allele discrimination based on the creation or destruction of a PAM sequence by a particular SNP, leaving a PAM solely on the mutant HTT allele at a given location [54]. The CRISPR-Cas9 system has been used to achieve silencing of the mutant HTT gene through heterozygous PAM sites in patient-derived fibroblast cells significantly reducing expression of the mutant huntingtin protein [55]. In this approach, two SNPs and two cut sites were required to excise large portions of the gene that included the promoter and exon 1. A similar CRISPR-Cas9 system requiring a single SNP and a shared target site in an intronic region selectively targeted the disease allele and reduced mutant HTT expression in neural cells of the BACHD mouse model [56]. These approaches require at least two target sites in the genome, which may increase the risk of off-target side effects.
But not all people with a mutant HTT gene contain SNPs. Therefore, haplotypes should be examined to identify gRNAs which lead to allele-specific silencing of the mutant HTT gene. A potential challenge is that screening patients for SNPs and determining on which allele the SNP is present requires specific sequencing techniques [57]. Additionally, the mutation that causes HD involves expanded CAG repeats that contain a larger number of nucleotides than the gRNA construct for Cas9 can recognize. In those instances, indirect methods have to be used to specifically identify a target sequence outside of the mutation. The success of these approaches relies on SNPs within both the promoter region and just beyond exon 1, which significantly limits the targetable SNPs because there are 67 exons in the HTT gene. A more robust approach would allow for the use of SNPs outside of the promoter and first intron.
Prime editing may overcome the difficulties in targeting mutant HTT because prime editing has the potential to insert or delete short sequences to disrupt the promoter and alter splicing sites which could lead to a selective reduction in expression of the mutant HTT gene. Indeed, prime editing technology was applied to target progerin, a dominant negative mutant protein that accumulates in Hutchinson-Gilford Progeria, a rare form of accelerated aging. Encouragingly, in a mouse progeria model, there was a correction of the disease-causing mutation and median lifespan was extended [58].
Another CRISPR-based approach for HD is to target the mutant huntingtin mRNA rather than the mutant HTT gene. The efficacy of this approach preclinically was recently reported in a study that employed a CRISPR-Cas13d system designed with a gRNA-Cas13d that selectively targets and cleaves CAG-expanded huntingtin mRNA (CRISPR-Cas13d-HTTEX) [59]. When packaged in a constitutive lentiviral vector and delivered into cultured HD patient fibroblasts and neurons derived from HD iPSC, CRISPR-Cas13d-HTTEX eliminated the accumulation of mutant huntingtin mRNA and protein. AAV-mediated delivery of CRISPR-Cas13d-HTTEX into the striatum of mutant HTT transgenic mice reduced the accumulation of mutant huntingtin aggregates, reduced neuronal degeneration, and improved motor function. These phenotypic improvements lasted for at least eight months without noticeable adverse effects and with minimal off-target transcriptomic effects.
Concluding Remarks
Base and prime editing are currently the most promising approaches for correcting mutations that cause disease. Methods for delivery of gene editing cargos to desired cellular targets in vivo have been developed, and a rapidly growing number of preclinical studies are testing the efficacy of these gene editing approaches for correction of gene mutations in animal models of a wide range of inherited diseases including neurological disorders.
In the instances of gain of function missense mutations, correcting a single-base on one allele is sufficient. However, because HD results from CAG expansions in the HTT gene, the CAG repeat region cannot be directly targeted without also altering the wildtype HTT allele. To overcome this problem HD requires a different gene editing approach (see Outstanding Questions). One promising approach to selectively disrupt the mutant HTT gene is to target a SNP that is specific for the mutant allele. While the results of recent preclinical tests of such gene editing systems for HD are promising, clinical trials in HD patients will require considerable further refinement of the methods in animal models.
Outstanding questions.
How can gene-editing technologies be further refined in ways that eliminate or greatly reduce off-target side effects?
At what time in relation to the onset of HD symptoms should gene-editing therapy be administered?
Can cell delivery platforms be developed that selectively target the striatal and cortical neurons that are most severely affected in HD?
Recent clinical trials have deployed CRISPR-Cas9 systems for delivery of gene therapy cargos in cells from a few patients with sickle cell anemia, beta thalassemia, lymphocyte cancers, inherited childhood blindness, and transthyretin amyloidosis [60–63]. These trials have provided evidence that gene editing is feasible and can be efficacious in cells in human patients. However, so far clinical trials have only targeted cells located in readily accessible compartments (blood and eye) and it will likely prove more difficult to deliver gene editing cargos into the brain cells affected in HD.
Acknowledgements:
This work was supported by grants to W. D. from the National Institutes of Health (R01NS124084 and R21NS118079), the CHDI Foundation, and the Bev Hartig Huntington’s Disease Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: MPM serves on the Advisory Board of Trends in Neurosciences. The authors have no other competing interests to declare.
References
- 1.Soares TR et al. (2019) Targeting the proteostasis network in Huntington’s disease. Ageing Res Rev 49, 92–103. 10.1016/j.arr.2018.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fan MM and Raymond LA (2007) N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington’s disease. Prog Neurobiol 81, 272–293. 10.1016/j.pneurobio.2006.11.003 [DOI] [PubMed] [Google Scholar]
- 3.Mattson MP (2021) Applying available knowledge and resources to alleviate familial and sporadic neurodegenerative disorders. Prog Mol Biol Transl Sci 177, 91–107. 10.1016/bs.pmbts.2020.09.001 [DOI] [PubMed] [Google Scholar]
- 4.Saha K et al. (2021) The NIH Somatic Cell Genome Editing program. Nature 592, 195–204. 10.1038/s41586-021-03191-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilbert LA et al. (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. 10.1016/j.cell.2013.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang HHY et al. (2017) Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18, 495–506. 10.1038/nrm.2017.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molla KA et al. (2022) Predictable NHEJ Insertion and Assessment of HDR Editing Strategies in Plants. Front Genome Ed 4, 825236. 10.3389/fgeed.2022.825236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jinek M et al. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rees HA and Liu DR (2018) Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet 19, 770–788. 10.1038/s41576-018-0059-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaudelli NM et al. (2017) Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471. 10.1038/nature24644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jinek M et al. (2014) Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343, 1247997. 10.1126/science.1247997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komor AC et al. (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. 10.1038/nature17946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hess GT et al. (2017) Methods and Applications of CRISPR-Mediated Base Editing in Eukaryotic Genomes. Mol Cell 68, 26–43. 10.1016/j.molcel.2017.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Komor AC et al. (2017) CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 168, 20–36. 10.1016/j.cell.2016.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zafra MP et al. (2018) Optimized base editors enable efficient editing in cells, organoids and mice. Nat Biotechnol 36, 888–893. 10.1038/nbt.4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu JH et al. (2018) Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63. 10.1038/nature26155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim D et al. (2019) Genome-wide target specificity of CRISPR RNA-guided adenine base editors. Nat Biotechnol 37, 430–435. 10.1038/s41587-019-0050-1 [DOI] [PubMed] [Google Scholar]
- 18.Xu W et al. (2020) Discriminated sgRNAs-Based SurroGate System Greatly Enhances the Screening Efficiency of Plant Base-Edited Cells. Mol Plant 13, 169–180. 10.1016/j.molp.2019.10.007 [DOI] [PubMed] [Google Scholar]
- 19.Fu Y et al. (2014) Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32, 279–284. 10.1038/nbt.2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rose JC et al. (2020) Suppression of unwanted CRISPR-Cas9 editing by co-administration of catalytically inactivating truncated guide RNAs. Nat Commun 11, 2697. 10.1038/s41467-020-16542-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rees HA et al. (2017) Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat Commun 8, 15790. 10.1038/ncomms15790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuo E et al. (2019) Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 364, 289–292. 10.1126/science.aav9973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin S et al. (2019) Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 364, 292–295. 10.1126/science.aaw7166 [DOI] [PubMed] [Google Scholar]
- 24.Anzalone AV et al. (2020) Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38, 824–844. 10.1038/s41587-020-0561-9 [DOI] [PubMed] [Google Scholar]
- 25.Landrum MJ et al. (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44, D862–868. 10.1093/nar/gkv1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anzalone AV et al. (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157. 10.1038/s41586-019-1711-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hampton T (2020) DNA Prime Editing: A New CRISPR-Based Method to Correct Most Disease-Causing Mutations. JAMA 323, 405–406. 10.1001/jama.2019.21827 [DOI] [PubMed] [Google Scholar]
- 28.Matsoukas IG (2020) Prime Editing: Genome Editing for Rare Genetic Diseases Without Double-Strand Breaks or Donor DNA. Front Genet 11, 528. 10.3389/fgene.2020.00528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newby GA and Liu DR (2021) In vivo somatic cell base editing and prime editing. Mol Ther 29, 3107–3124. 10.1016/j.ymthe.2021.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen PJ et al. (2021) Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 184, 5635–5652 e5629. 10.1016/j.cell.2021.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rozners E (2022) Chemical Modifications of CRISPR RNAs to Improve Gene-Editing Activity and Specificity. J Am Chem Soc 144, 12584–12594. 10.1021/jacs.2c02633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raguram A et al. (2022) Therapeutic in vivo delivery of gene editing agents. Cell 185, 2806–2827. 10.1016/j.cell.2022.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taha EA et al. (2022) Delivery of CRISPR-Cas tools for in vivo genome editing therapy: Trends and challenges. J Control Release 342, 345–361. 10.1016/j.jconrel.2022.01.013 [DOI] [PubMed] [Google Scholar]
- 34.Mirjalili Mohanna SZ et al. (2022) LNP-mediated delivery of CRISPR RNP for widespread in vivo genome editing in mouse cornea. Control Release. 350, 401–413. doi: 10.1016/j.jconrel.2022.08.042. [DOI] [PubMed] [Google Scholar]
- 35.Gao X et al. (2018) Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553, 217–221. 10.1038/nature25164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee B et al. (2018) Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile X syndrome from exaggerated repetitive behaviours. Nat Biomed Eng 2, 497–507. 10.1038/s41551-018-0252-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uddin F, Rudin CM and Sen T. (2020) CRISPR gene therapy: applications, limitations, and implications for the future. Front. Oncol 10:1387. doi: 10.3389/fonc.2020.01387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang D et al. (2020) CRISPR-Based Therapeutic Genome Editing: Strategies and In Vivo Delivery by AAV Vectors. Cell 181, 136–150. 10.1016/j.cell.2020.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bravo-Hernandez M et al. (2020) Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat Med 26, 118–130. 10.1038/s41591-019-0674-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim CKW et al. (2020) Treatment of a mouse model of ALS by in vivo base editing. Mol Ther 28, 1177–1189. 10.1016/j.ymthe.2020.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hunker AC et al. (2020) Conditional single vector CRISPR/SaCas9 viruses for efficient mutagenesis in the adult mouse nervous system. Cell Rep 30, 4303–4316 e4306. 10.1016/j.celrep.2020.02.092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun H et al. (2020) Development of a CRISPR-SaCas9 system for projection- and function-specific gene editing in the rat brain. Sci Adv 6, eaay6687. 10.1126/sciadv.aay6687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hana S et al. (2021) Highly efficient neuronal gene knockout in vivo by CRISPR-Cas9 via neonatal intracerebroventricular injection of AAV in mice. Gene Ther 28, 646–658. 10.1038/s41434-021-00224-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiao D et al. (2021) CRISPR-mediated rapid generation of neural cell-specific knockout mice facilitates research in neurophysiology and pathology. Mol Ther Methods Clin Dev 20, 755–764. 10.1016/j.omtm.2021.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng Y et al. (2018) CRISPR interference-based specific and efficient gene inactivation in the brain. Nat Neurosci 21, 447–454. 10.1038/s41593-018-0077-5 [DOI] [PubMed] [Google Scholar]
- 46.Carullo NVN et al. (2021) A Cre-dependent CRISPR/dCas9 system for gene expression regulation in neurons. eNeuro 8. 10.1523/ENEURO.0188-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohsen MO and Bachmann MF (2022) Virus-like particle vaccinology, from bench to bedside. Cell Mol Immunol 19, 993–1011. 10.1038/s41423-022-00897-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tervo DG et al. (2016) A designer AAV variant permits efficient retrograde access to projection neurons. Neuron 92, 372–382. 10.1016/j.neuron.2016.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang S et al. (2017) CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J Clin Invest 127, 2719–2724. 10.1172/JCI92087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu H et al. (2021) Huntingtin silencing delays onset and slows progression of Huntington’s disease: a biomarker study. Brain 144, 3101–3113. 10.1093/brain/awab190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ekman FK et al. (2019) CRISPR-Cas9-mediated genome editing increases lifespan and improves motor deficits in a Huntington’s disease mouse model. Mol Ther Nucleic Acids 17, 829–839. 10.1016/j.omtn.2019.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dabrowska M et al. (2018) Precise Excision of the CAG Tract from the Huntingtin Gene by Cas9 Nickases. Front Neurosci 12, 75. 10.3389/fnins.2018.00075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu X et al. (2017) Reversal of Phenotypic Abnormalities by CRISPR/Cas9-Mediated Gene Correction in Huntington Disease Patient-Derived Induced Pluripotent Stem Cells. Stem Cell Reports 8, 619–633. 10.1016/j.stemcr.2017.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wild EJ and Tabrizi SJ (2017) Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol 16, 837–847. 10.1016/S1474-4422(17)30280-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin JW et al. (2016) Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum Mol Genet 25, 4566–4576. 10.1093/hmg/ddw286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Monteys AM et al. (2017) CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo. Mol Ther 25, 12–23. 10.1016/j.ymthe.2016.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi Y et al. (2018) Comparison of phasing strategies for whole human genomes. PLoS Genet 14, e1007308. 10.1371/journal.pgen.1007308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koblan LW et al. (2021) In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature 589, 608–614. 10.1038/s41586-020-03086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morelli KH et al. (2023) An RNA-targeting CIRSPR-Cas13d system alleviates disease-related phenotypes in Huntington’s disease models. Nat. Neurosci 26, 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frangoul H et al. (2021) CRISPR-Cas9 gene editing for sickle cell disease and betathalassemia. N Engl J Med 384, 252–260. 10.1056/NEJMoa2031054 [DOI] [PubMed] [Google Scholar]
- 61.Chiu W et al. (2021) An update on gene therapy for inherited retinal dystrophy: experience in Leber congenital amaurosis clinical trials. Int J Mol Sci 22. 10.3390/ijms22094534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hu Y et al. (2021) CRISPR/Cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia. Clin Cancer Res 27, 2764–2772. 10.1158/1078-0432.CCR-20-3863. [DOI] [PubMed] [Google Scholar]
- 63.Gillmore JD et al. (2021) CRISPR-Cas9 in Vivo gene editing for transthyretin amyloidosis. N Engl J Med 385, 493–502. [DOI] [PubMed] [Google Scholar]
- 64.Gaj T et al. (2017) In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci Adv 3 (12), eaar3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ouyang S, Xie Y, Xiong Z, Yang Y, Xian Y, Ou Z, Song B, Chen Y, Xie Y, Li H, & Sun X (2018). CRISPR/Cas9-targeted deletion of polyglutamine in spinocerebellar ataxia type 3-derived induced pluripotent stem cells. Stem Cells and Dev. 27, 756–770. [DOI] [PubMed] [Google Scholar]
- 66.Park C-Y, Halevy T, Lee DR, Sung JJ, Lee JS, Yanuka O, Benvenisty N, & Kim D-W (2015). Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile x IPSC-derived neurons. Cell Rep. 13, 234–241. [DOI] [PubMed] [Google Scholar]
- 67.Pribadi M, Yang Z, Kim TS, Swartz EW, Huang AY, Chen JA, Dokuru D, Baek J, Gao F, Fua AT, Wojta K, Wang Q, Karydas A, Fong J, Lezcano E, Ng S, Chehab FF, Vinters HV, Miller BL, & Coppola G (2016). CRISPR-Cas9 targeted deletion of the c9orf72 repeat expansion mutation corrects cellular phenotypes in patient-derived IPS cells. BioRxiv. 10.1101/051193. [DOI] [Google Scholar]
- 68.Zeitler B, Froelich S, Marlen K, Shivak DA, Yu Q, Li D, Pearl JR, Miller JC, Zhang L, Paschon DE, Hinkley SJ, Ankoudinova I, Lam S, Guschin D, Kopan L, Cherone JM, Nguyen H-OB, Qiao G, Ataei Y, … Zhang HS (2019). Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nature Med. 25, 1131–1142 [DOI] [PubMed] [Google Scholar]
- 69.Tan DCS, Yao S, Ittner A, Bertz J, Ke YD, Ittner LM, & Delerue F (2018). Generation of a new tau knockout (tauδex1) line using CRISPR/cas9 genome editing in mice. Journal of Alzheimer’s Disease, 62, 571–578. [DOI] [PubMed] [Google Scholar]
- 70.Yoon HH, Ye S, Lim S, Jo A, Lee H, Hong F, Lee SE, Oh S-J, Kim N-R, Kim K, Kim B-J, Kim H, Lee CJ, Nam M-H, Hur JW, & Jeon SR (2022). CRISPR-Cas9 gene editing protects from the A53T-SNCA overexpression-induced pathology of parkinson’s disease in vivo. The CRISPR Journal, 5, 95–108. [DOI] [PubMed] [Google Scholar]
- 71.Lopes C, Tang Y, Anjo SI, Manadas B, Onofre I, de Almeida LP, Daley GQ, Schlaeger TM, & Rego AC (2020). Mitochondrial and redox modifications in Huntington disease induced pluripotent stem cells rescued by CRISPR/Cas9 cags targeting. Frontiers in Cell and Developmental Biology, 8, 576592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ababneh NA et al. (2020) Correction of amyotrophic lateral sclerosis related phenotypes in induced pluripotent stem cell-derived motor neurons carrying a hexanucleotide expansion mutation in C9orf72 by CRISPR/Cas9 genome editing using homology-directed repair. Hum Mol Genet 29 (13), 2200–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duan W, Guo M, Yi L, Liu Y, Li Z, Ma Y, Zhang G, Liu Y, Bu H, Song X, & Li C (2020). The deletion of mutant SOD1 via CRISPR/Cas9/sgrna prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Therapy, 27, 157–169. [DOI] [PubMed] [Google Scholar]
- 74.Dafinca R, Barbagallo P, Farrimond L, Candalija A, Scaber J, Ababneh NA, Sathyaprakash C, Vowles J, Cowley SA, & Talbot K (2020). Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in IPS-derived motor neurons from patients with ALS/FTD. Stem Cell Rep, 14, 892–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Deng HX et al. (2021) Efficacy and long-term safety of CRISPR/Cas9 genome editing in the SOD1-linked mouse models of ALS. Commun Biol 4 (1), 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Duan Y et al. (2022) Brain-wide Cas9-mediated cleavage of a gene causing familial Alzheimer’s disease alleviates amyloid-related pathologies in mice. Nat Biomed Eng 6 (2), 168–180. [DOI] [PubMed] [Google Scholar]
- 77.Lee B et al. (2018) Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile X syndrome from exaggerated repetitive behaviours. Nat Biomed Eng. 2: 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]