

Abstract
Left ventricular hypertrophy is a leading risk factor for cardiovascular morbidity and mortality. Although reverse ventricular remodelling was long thought to be irreversible, evidence from the past three decades indicates that this process is possible with many existing heart disease therapies. The regression of pathological hypertrophy is associated with improved cardiac function, quality of life and long-term health outcomes. However, less than 50% of patients respond favourably to most therapies, and the reversibility of remodelling is influenced by many factors, including age, sex, BMI and disease aetiology. Cardiac hypertrophy also occurs in physiological settings, including pregnancy and exercise, although in these cases, hypertrophy is associated with normal or improved ventricular function and is completely reversible postpartum or with cessation of training. Studies over the past decade have identified the molecular features of hypertrophy regression in health and disease settings, which include modulation of protein synthesis, microRNAs, metabolism and protein degradation pathways. In this Review, we summarize the evidence for hypertrophy regression in patients with current first-line pharmacological and surgical interventions. We further discuss the molecular features of reverse remodelling identified in cell and animal models, highlighting remaining knowledge gaps and the essential questions for future investigation towards the goal of designing specific therapies to promote regression of pathological hypertrophy.
Introduction
Despite advances in treatment over the past 50 years, heart disease remains the leading cause of morbidity and mortality in the world1. Heart failure onset is typically preceded by cardiac hypertrophy, a compensatory thickening of the muscle in response to elevated left ventricular (LV) wall stress. However, the response becomes maladaptive with time, and this pathological hypertrophy is an independent predictor of adverse cardiovascular outcomes including myocardial infarction, arrhythmia and heart failure2,3. Although pathological hypertrophy was long thought to be irreversible, partial regression is observed with many existing therapies in a subset of patients and is associated with improved long-term health outcomes4,5. Importantly, in almost all cases, the reversal of cardiac hypertrophy leads to improved ventricular function6. A similar adaptive response is observed with exercise, in which increased workload precipitates increased cardiac muscle mass7. However, this physiological hypertrophy is associated with normal or increased ventricular function and is completely reversible with cessation of training8. Physiological hypertrophy also occurs in pregnancy but is rapidly reversible in most cases postpartum9. These observations lend strong support for the development of therapies directed at hypertrophy reversal as a treatment for heart disease. Molecular studies over the past decade have begun to shed light on the complexity of hypertrophy and its regression at the level of the cardiomyocyte, and to identify new therapeutic targets for reversal of pathological remodelling.
In this Review, we present an integration of clinical studies and basic science discoveries on cardiac hypertrophy regression. We discuss evidence for the benefits of reverse remodelling from clinical studies with current heart failure therapies. We also examine the molecular features of pathological hypertrophy and regression identified in cell and animal models and human tissue studies, comparing where possible with physiological models. The objective of this Review is to highlight therapies and mechanisms involved in the regression of existing cardiac hypertrophy and, therefore, studies on the prevention of hypertrophy development, although important, are not the focus.
Overview of cardiac hypertrophy
Cardiac hypertrophy is defined as a LV mass increase arising from thickening of the LV wall and/or enlargement of chamber diameter10. The structural manifestation of hypertrophy is determined by the type of cardiac insult, broadly categorized as either pressure or volume overload. Examples of pressure overload are aortic stenosis and hypertension, in which increased afterload on the heart triggers ventricular muscle growth. Volume overload underlies hypertrophy in the case of mitral regurgitation and also in physiological growth settings, including postnatal life, exercise and pregnancy11. The organ-level structural changes are mediated by distinct responses to increased pressure or volume at the cellular level by cardiomyocytes, the predominant cardiac cell type12. Postnatal cardiomyocytes have limited proliferative capacity and consequently, to increase organ size, the individual cells grow larger13. An exception in which cardiomyocyte proliferation does have a minor role is in early postnatal development; however, in this setting, cardiomyocyte hypertrophy is also the predominant factor14. With sustained pressure, cardiomyocytes undergo concentric hypertrophy, becoming thicker by the parallel addition of sarcomeres — the contractile units of striated muscle cells. Serial addition of sarcomeres occurs with volume overload, extending cardiomyocyte length to drive eccentric growth and leading to increased chamber diameter.
Molecular mechanisms of cardiac hypertrophy
The cellular and molecular mechanisms of physiological and pathological hypertrophy are well established. Although both manifestations of hypertrophy are initially adaptive, pathological hypertrophy becomes maladaptive and can lead to heart failure. The stimuli and mechanisms that regulate these two types of growth are distinct and, in some cases, antagonistic15 (Fig. 1). In this section, we discuss the factors known to initiate each type of hypertrophy and the downstream cellular signalling cascades.
Fig. 1 |. Cellular signalling pathways of physiological and pathological hypertrophy.
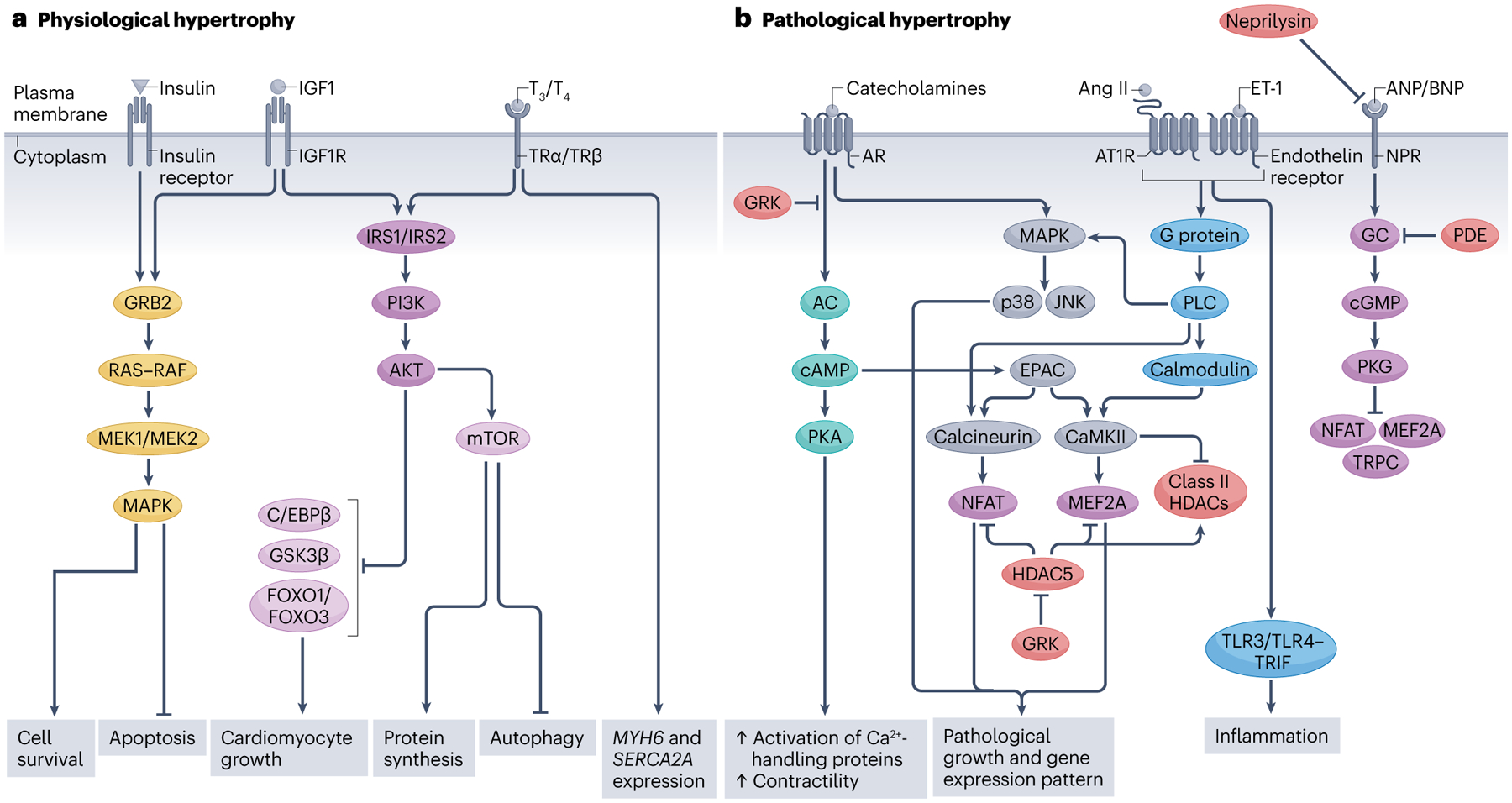
a, Cellular pathways of physiological hypertrophy. Insulin or insulin-like growth factor 1 (IGF1) bind to the insulin receptor or IGF1 receptor (IGF1R), respectively, and activate RAS–mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)–AKT pathways, which promote cell survival and growth and protein synthesis. The thyroid hormones triiodothyronine (T3) or thyroxine (T4) also trigger the PI3K–AKT pathway and cause increased transcription of MHY6 and SERCA2A. The PI3K–AKT pathway inhibits CCAAT/enhancer-binding protein-β (C/EBPβ), glycogen synthase kinase 3β (GSK3β), forkhead box protein O1 (FOXO1) and FOXO3, all of which are negative regulators of hypertrophy. b, Cellular pathways of pathological hypertrophy. Catecholamines, angiotensin II (Ang II) and endothelin 1 (ET-1) trigger pathways that promote maladaptive gene expression and growth. Catecholamines and Ang II–ET-1 activate the downstream effectors calcineurin and calcium–calmodulin-dependent protein kinase II (CaMKII), which stimulate the transcription factors nuclear factor of activated T cells (NFAT) and myocyte-specific enhancer factor 2A (MEF2A), responsible for pathological growth. G protein-coupled receptor kinases (GRKs) mediate adrenergic receptor (AR) desensitization and promote pathological hypertrophy by inhibiting histone deacetylases (HDACs). Atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP) typically inhibit NFAT, MEF2A and transient receptor potential canonical channel (TRPC) but in heart failure, their receptors become desensitized. Increased neprilysin activity in heart failure triggers degradation of circulating BNP levels, further repressing natriuresis. Additionally, phosphodiesterases (PDEs) inhibit cGMP production resulting in maladaptive gene expression. AC, adenylyl cyclase; AT1R, angiotensin II type 1 receptor; EPAC, exchange protein directly activated by cAMP; GC, guanylate cyclase; GRB2, growth factor receptor-bound protein 2; IRS, insulin receptor substrate; JNK, JUN N-terminal kinase; MEK, MAP/ERK kinase; mTOR, mechanistic target of rapamycin; NPR, natriuretic peptide receptor; PKA, protein kinase A; PKG, protein kinase G; PLC, phospholipase C; TLR, Toll-like receptor; TRIF, TIR domain-containing adapter molecule 1; TR, thyroid hormone receptor.
Physiological hypertrophy
During development, pregnancy or long-term exercise, cardiomyocytes undergo hypertrophy to meet increased volume demands. This adaptation leads to unchanged or improved cardiac function15,16. At the cellular level, circulating factors trigger signalling cascades that mediate survival and growth and thereby permit a fully adaptive response15. Several factors contribute to physiological hypertrophy and herein we focus primarily on hypertrophy-promoting circulating factors and the organ-level changes and cellular signalling cascades they initiate.
Insulin and insulin-like growth factor 1 (IGF1) regulate a broad range of cellular functions, including growth, survival and metabolism. Their receptors are highly expressed in cardiomyocytes and have a prominent role in regulating cardiac function and mediating physiological growth17,18. Both hormones and their respective tyrosine kinase receptors are structurally similar and stimulate shared pathways, including the phosphoinositide 3-kinase (PI3K)–AKT and RAS–MAPK pathways15,19. Binding of insulin or IGF1 to their receptors activates the PI3K–AKT pathway by triggering the recruitment of insulin receptor substrate 1 (IRS1) and IRS2 (refs.20,21). The p100α isoform of PI3K is the master regulator of exercise-induced hypertrophy and has a crucial protective role for cardiomyocytes in the context of pathological stimuli21,22. AKT activation downstream of PI3K leads to the inhibition of several negative regulators of hypertrophy, including glycogen synthase kinase 3β (GSK3β), forkhead box protein O1 (FOXO1), FOXO3 and CCAAT/enhancer-binding protein-β (C/EBPβ)23–25. AKT also indirectly activates mechanistic target of rapamycin (mTOR), which contributes to hypertrophy by increasing protein synthesis and inhibiting autophagy26. Insulin and IGF1 also activate the RAS–MEK–MAPK pathway by recruiting the growth factor receptor-bound protein 2 (GRB2)18,21. When recruited, GRB2 activates RAS–RAF, which in turn activates MEK1 or MEK2 to trigger the MAPK cascade18. Stimulation of MEK1 or MEK2 is linked to physiological hypertrophy and cell survival through inhibition of apoptosis27.
The thyroid hormones triiodothyronine (T3) and thyroxine (T4) also mediate physiological growth28. These hormones are released after birth and inhibit the expression of fetal-related genes while activating the expression of genes that promote cardiomyocyte maturation15,28. T3 activates the PI3K–AKT pathway after binding to either of its receptors, thereby leading to physiological growth28,29. Additionally, in rat and mouse hearts, T3 and T4 stimulate the expression of Myh6 and Serca2a30,31, which encode proteins that are important for maintaining cardiomyocyte contractility. Several studies in animal models have shown that low thyroid hormone levels are associated with heart failure and impaired cardiac function, but pathological hypertrophy is reversed with increased T3 levels, resulting in improved cardiac function32–34. Other circulating factors, including fatty acids, have also been shown to promote physiological hypertrophy. A study of hypertrophy in the infrequently feeding Burmese python identified three circulating fatty acids (myristic acid, palmitic acid and palmitoleic acid) that regulate hypertrophy in response to a meal35. Injection of these fatty acids into fasted pythons and mice led to the recapitulation of cardiac hypertrophy35.
Several non-coding RNAs (ncRNAs) also regulate physiological hypertrophy. MicroRNA-222 (miR-222) and miR-17–3p are each upregulated in exercise-induced hypertrophy in mice36,37. Both microRNAs activate pathways that promote cell proliferation, growth and survival36,37. The long ncRNA (lncRNA) cardiac physiological hypertrophy-associated regulator (CPhar) was shown in mice to be cardioprotective as well as necessary for exercise-induced hypertrophy in a C/EBPβ-dependent manner38. Conversely, the lncRNA lncExACT1 is downregulated in exercise-induced physiological hypertrophy and upregulated in pathological hypertrophy39. Furthermore, inhibition of lncExACT1 led to physiological hypertrophy and increased cardiomyocyte proliferation through regulation of miRNA-222 and the Hippo–Yap signalling pathway39. Additional involvement of ncRNAs in reverse remodelling has been extensively reviewed previously6.
One factor that distinguishes physiological hypertrophy from pathological hypertrophy is the degree of angiogenesis. The capacity of the heart to maintain vascularization and perfusion largely depends on increased capillary density mediated by vascular endothelial growth factor40. Increased cardiac angiogenesis allows proper supply of nutrients and oxygen, which contributes to the fully adaptive physiological growth41. Angiogenesis decreases in pathological hypertrophy, thereby resulting in hypoxia and insufficient nutrients40,42. Therefore, angiogenesis is a key process not only for promoting adaptive growth but also for preventing maladaptive responses.
Pathological hypertrophy
Maladaptive cardiac hypertrophy develops in pathological settings, including hypertension, obesity and certain genetic cardiomyopathies. Compared with physiological growth, the stimuli and signalling mechanisms that trigger this maladaptive response promote fibrosis, apoptosis, and other cellular and structural dysfunction, which can ultimately lead to heart failure15,43. Circulating factors including catecholamines, natriuretic peptides and peptide hormones contribute to pathological remodelling. In this section, we discuss these factors and the downstream signalling mechanisms (Fig. 1).
Catecholamines are neuroendocrine hormones that are released after activation of the adrenergic nervous system in response to stress44. These hormones bind to cardiomyocyte adrenergic receptors, G protein-coupled receptors that subsequently activate adenylyl cyclase44. After activation of adenylyl cyclase, cAMP levels increase, which in turn activates protein kinase A (PKA) and exchange protein directly activated by cAMP (EPAC). Activation of PKA stimulates the upregulation of the activity of calcium-handling proteins and increases basal cardiac contractility44. EPAC, which is independently activated by cAMP, stimulates the phosphatase calcineurin and its downstream effector nuclear factor of activated T cells (NFAT), which regulates pathological growth and gene expression45. EPAC also activates calcium/calmodulin-dependent protein kinase II (CaMKII), which stimulates the transcription factor myocyte-specific enhancer factor 2A (MEF2A) to induce pathological hypertrophic gene expression46,47. The continual release of catecholamines eventually leads to adrenergic receptor desensitization and heart failure44,48. This process is mediated by G protein-coupled receptor kinase 2 (GRK2), GRK5 and β-arrestins49. GRK5 also positively regulates MEF2A in a histone deacetylase 5 (HDAC5)-dependent manner, as well as NFAT50–52. Additionally, catecholamines activate MAPK proteins and the downstream p38 and JNK pathways, which trigger pathological hypertrophic gene expression53,54.
The circulating levels of atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP) are elevated in heart failure and are a common indicator of pathological hypertrophy55. In healthy physiology, ANP and BNP are antihypertrophic by preventing increases in intracellular calcium levels in cardiomyocytes56,57. Circulating ANP and BNP bind to natriuretic peptide receptors, which activate guanylyl cyclase and lead to increases in intracellular cGMP concentration57,58. cGMP activates PKG, which inhibits the prohypertrophic molecules calcineurin–NFAT, MEF2A and transient receptor potential canonical channel (TRPC)55,59. However, chronically elevated levels of circulating ANP and BNP in heart disease cause natriuretic peptide receptor desensitization, increased intracellular calcium levels in cardiomyocytes and pathological cardiac remodelling60,61. Additionally, in pathological hypertrophy, the expression and activity of phosphodiesterases (PDEs), which regulate cGMP homeostasis, are increased59,62. Inhibition of PDEs blocks prohypertrophic signalling pathways59,62,63.
The circulating levels of the peptide hormones angiotensin II and endothelin 1 are upregulated in heart failure64,65. These humoral factors are released in response to mechanical stress in the vascular system and promote cardiac structural and functional changes66–68. Binding of these hormones to their receptors on cardiomyocytes stimulates G proteins, which activate the phospholipase C–calcineurin–NFAT, MAPK and calmodulin–CaMKII pathways, leading to maladaptive gene expression69–71. CaMKII phosphorylates class II HDACs, which repress cardiomyocyte hypertrophy by inhibiting MEF2A expression72. After phosphorylation, HDACs are exported from the nucleus, enabling the upregulated expression of prohypertrophic genes73. Angiotensin II also regulates hypertrophy through the Toll-like receptor 3 (TLR3)–TIR domain-containing adapter molecule 1 (TRIF) and TLR4–TRIF immune pathways, which are activated by inflammation caused by hypertension74.
Clinical relevance of hypertrophy regression
Before 1990, pathological hypertrophy was considered to be an irreversible condition. However, it has become clear that modest regression is possible in a subset of patients (Table 1), and this reverse remodelling is associated with improved cardiac function and long-term health75,76. In this section, we discuss the data supporting reverse remodelling with existing pharmacological and surgical therapies, as well as with implementation of exercise and dietary changes.
Table 1 |.
Reverse left ventricular structural remodelling with heart failure therapies
| Therapy | Regression (%) | Patients responding (%) | Refs. | ||
|---|---|---|---|---|---|
| ≤3 months | 6–8 months | ≥12 months | |||
| ACE inhibitors | 4–12 | 1–21 | 10–26 | 36–46 | 75,80–84,102,273–276 |
| Angiotensin receptor blockers | 2–11 | 12–20 | 9–22 | 21–47 | 75,80,85,87,277–279 |
| ARNIs | 8–12 | 15 | 26–27 | 52–58 | 92,94,241,275,280,281 |
| Aortic valve replacement | 10 | 17 | 15–44 | 90–100 | 128–130,132,133,135,282 |
| Bariatric surgery | 7–22 | 9–18 | 19–32 | 70 | 137,139–142,283,284 |
| β-Blockers | 0–4 | 0–7 | 6–10 | 30 | 75,106,278,285–287 |
| Calcium channel blockers | 10–14 | 2–18 | 3–14 | 40–60 | 75,78,110–113,273,285,288,289 |
| CRT | 3 | 4–5 | 10–16 | 46–72 | 146,149,290–294 |
| LVAD | 20–40 | 20–40 | 40–50 | 70–90 | 153–155,295,296 |
| Mineralocorticoid receptor antagonists | 10–16 | 10–20 | 8–18 | 40 | 89,90,297,298 |
| SGLT2 inhibitors | Not reported | 4 | 5–9 | Not reported | 120,121,299 |
ACE, angiotensin-converting enzyme; ARNI, angiotensin receptor–neprilysin inhibitor; CRT, cardiac resynchronization therapy; LVAD, left ventricular assist device; SGLT2, sodium–glucose cotransporter 2.
Pharmacological therapies
RAAS blockade.
In heart disease, compromised haemodynamic function triggers sustained renin–angiotensin–aldosterone system (RAAS) activation, leading to fluid retention, high blood pressure and elevated LV preload and afterload77. Therefore, several compounds have been developed to antagonize the RAAS. Angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) were among the first identified and remain the most effective monotherapies for reversing cardiac hypertrophy78–80. Patients receiving ACE inhibitors show progressive reductions in LV mass index (LVMI; where LVMI = LV mass (grams)/body surface area (square metres)) beginning 3 months after treatment81–83. ACE inhibition also prevents the development of LV dilatation, a feature of end-stage heart failure84. ARBs have similar effects to those of ACE inhibitors on regression of hypertrophy and the concurrent improvement in LV function and are effective in reversing both eccentric and concentric hypertrophy80,85–87.
Another approach targeting the RAAS is the use of mineralocorticoid receptor antagonists. Mineralocorticoid receptor antagonists reduce LVMI, restore ventricular compliance and reduce all-cause mortality88,89. Mineralocorticoid receptor antagonists are also effective in reducing cardiac hypertrophy and diastolic dysfunction in patients with heart failure with preserved ejection fraction90. A newer class of compounds targeting both the angiotensin receptor and the membrane metallopeptidase neprilysin (angiotensin receptor–neprilysin inhibitors (ARNIs)) is the most effective for reversing cardiac remodelling91,92 (Box 1). More pronounced regression of cardiac hypertrophy with ARNIs is associated with reduction of plasma amino-terminal pro-BNP (NT-proBNP) levels93–96. In patients with heart failure with reduced ejection fraction, ARNI therapy increases LV ejection fraction by 5% compared with ACE inhibitors or ARBs alone, suggesting that increased regression of hypertrophy is associated with further functional improvements97. Various factors affect the efficacy of RAAS blockade in inducing cardiac hypertrophy regression, and less regression is observed in patients with ischaemic heart disease, older age, longer duration of hypertension, greater degree of pathological remodelling or higher BMI87,98–100. Interestingly, reverse remodelling with RAAS blockade can occur even when hypertension is not rectified89,101,102. The mechanisms underlying pressure-independent hypertrophy regression are an important topic for future study.
Box 1. Mechanism of action of ARNI therapy.
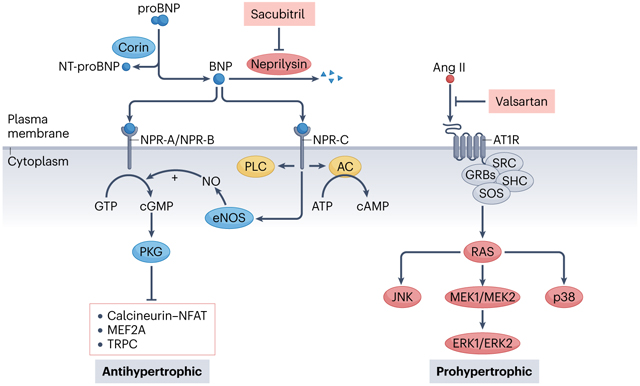
Angiotensin receptor–neprilysin inhibitors (ARNIs) are a promising class of heart failure therapies that target both the renin–angiotensin–aldosterone system (RAAS) and natriuretic peptide system. Data suggest that ARNI therapy with sacubitril–valsartan (Entresto) is the most effective first-line therapy for patients with systolic heart failure92. Sacubitril promotes the activation of the antihypertrophic natriuretic peptide system, and valsartan antagonizes the prohypertrophic RAAS. The benefits of sacubitril–valsartan include improved ventricular function, reduced risk of arrhythmia, mitigation of hypertension and stimulation of cardiac hypertrophy regression.
B-type natriuretic peptide (BNP) is synthesized in cardiomyocytes as an inactive pro-hormone (proBNP), which is released into the bloodstream in response to high pressure or stretch300. In the circulation, the biologically active BNP is released from its precursor proBNP by the enzyme corin, which generates the inactive amino-terminal pro-BNP (NT-proBNP) as a by-product301 (see the figure). In healthy physiology, BNP acts on the natriuretic peptide receptors (NPR-A, NPR-B and NPR-C) expressed on cardiomyocytes and triggers antihypertrophic signalling through endothelial nitric oxide synthase (eNOS) and protein kinase G (PKG)55. However, increased activity of neprilysin in heart failure leads to reduced circulating levels of biologically active BNP, mitigating its antihypertrophic effects. Neprilysin is a membrane-spanning, proteolytic enzyme expressed predominantly in kidney cells that actively degrades BNP302. Neprilysin does not degrade NT-proBNP, which is why circulating levels of this inactive factor are used as a gold-standard biomarker in heart failure303. Sacubitril was the first-developed neprilysin inhibitor and successfully restores natriuresis and vasodilatation in patients with heart failure304. However, inhibiting neprilysin alone triggers a compensatory increase in RAAS activity, thereby necessitating dual treatment with an angiotensin receptor blocker304. Valsartan is an angiotensin II type 1 receptor (AT1R) antagonist and prevents binding of the receptor to circulating angiotensin II (Ang II), thereby blocking downstream pathways associated with pathological hypertrophy, including JUN N-terminal kinase (JNK), p38, ERK1 and ERK2 signalling. In the PARADIGM-HF trial94, improved outcomes with sacubitril–valsartan were associated with reduced circulating NT-proBNP levels.
AC, adenylyl cyclase; GRBs, growth factor receptor-bound proteins; MEF2A, myocyte-specific enhancer factor 2A; MEK, MAP/ERK kinase; NFAT, nuclear factor of activated T cells; NO, nitric oxide; PLC, phospholipase C; SHC, SHC-transforming protein; SOS, son-of-sevenless homologue; SRC, proto-oncogene tyrosine protein kinase SRC; TRPC, transient receptor potential canonical channel.
β-Adrenergic receptor blockade.
Chronic sympathetic activity in heart disease leads to downregulation of cardiomyocyte β-adrenergic receptors and reduced contractility103. β-Adrenergic receptor antagonists increase receptor expression and membrane density, leading to restoration of cardiomyocyte contractility104. β-Blockers lower all-cause mortality and mildly reduce existing hypertrophy in patients with heart failure, while increasing LV ejection fraction in a dose-dependent manner105. Compared with other therapies, β-blockers are less effective in reversing hypertrophy, and several studies found no effect on reverse remodelling (Table 1). However, combination therapy with RAAS blockade has additive benefits for ventricular geometry and function106–108.
Calcium channel blockade.
Calcium channel blockers are antihypertensive medications that act on L-type calcium channels, inhibiting calcium uptake in the vasculature and stimulating vasodilatation109. Several studies suggest that calcium channel blockers are the most effective pharmacological therapy for stimulating reverse remodelling in the short-term, consistently reducing LVMI by ≥10% after 3 months110–113. However, unlike RAAS-targeting therapies, calcium channel blockers reverse only concentric hypertrophy and have limited additional benefit on regression with longer duration of treatment112 (Table 1). As with other treatments, the benefits of calcium channel blockers for hypertrophy regression are limited to non-ischaemic heart disease114. Calcium channel blockade increases hypertrophy regression when used as an add-on treatment with RAAS antagonists or thiazide diuretics115,116. Calcium channel blockers also induce hypertrophy regression, without affecting blood pressure levels, in patients with LV hypertrophy and normal blood pressure117.
Other drug therapies.
Sodium–glucose cotransporter 2 (SGLT2) inhibitors, which prevent the reabsorption of glucose in the proximal renal tubules, were developed as a treatment for patients with type 2 diabetes mellitus118. However, SGLT2 inhibitors have since proven to be effective for improving outcomes in patients with heart failure regardless of diabetes presence119. The mechanisms underlying heart failure reduction with SGLT2 inhibitors are unclear; however, studies have found that SGLT2 inhibition mildly reduces LV mass and ambulatory blood pressure120,121. Improved prognosis with SGLT2 inhibitors in patients with heart failure is associated with reverse remodelling and might be blood-pressure independent120. In the MET-REMODEL trial122, another diabetes drug, metformin, reduced LVMI by 5% after 12 months compared with placebo in patients with coronary artery disease without diabetes. Another study found that adding the PDE5 inhibitor sildenafil to a traditional treatment regimen with RAAS inhibition and β-blockade increases hypertrophy regression123. Modest reverse remodelling has also been observed with cardiac myosin inhibitors and mTOR inhibitors124–126.
Surgery and device therapies
Aortic valve replacement.
Pressure overload from aortic valve stenosis or insufficiency leads to pathological hypertrophy. Aortic valve replacement (AVR) reduces ventricular outflow gradient pressure and triggers reverse remodelling127. The effects of AVR are immediate, beginning 7 days after surgery with a reduction in LV mass of 10% by 1 month128,129. This reduction in LV mass is driven by a decrease in cardiomyocyte volume in the early remodelling phase and later (6–12 months) by fibrosis reduction130,131. Greater regression with AVR is associated with lower serum NT-proBNP levels and rehospitalization rates128,130. Despite most patients experiencing structural improvements, <20% of patients show a regression to a normal LVMI in the first 18 months132. However, longer-term studies identified LVMI normalization in most patients 8–10 years after AVR4,129. Stented bioprostheses (compared with stentless bioprostheses), higher BMI and systemic hypertension are associated with less hypertrophy regression after AVR128,133–135.
Bariatric surgery.
Obesity causes diastolic dysfunction and is a leading risk factor for heart disease136. Bariatric surgery reduces long-term cardiovascular morbidity and concentric cardiac hypertrophy137. Beneficial cardiac structural remodelling and improved diastolic function are observed within 3 months after surgery, and normal LV geometry is achieved in ~70% of patients by 3 years138–140. LV mass reduction after bariatric surgery occurs regardless of cardiovascular comorbidities or medications used141. However, patients receiving β-blockers have the most pronounced hypertrophy regression, compared with patients receiving other compounds141. The role of blood pressure reduction in hypertrophy regression after bariatric surgery is unclear139,141,142.
Cardiac resynchronization therapy.
Optimal ventricular function requires tightly coordinated electrical activation. In many patients with heart disease, dyssynchrony owing to ventricular pacing or left bundle branch block contributes to the pathology. Cardiac resynchronization therapy (CRT) was developed to resynchronize contraction, and multiple clinical trials have indicated its benefits for both long-term health and reverse remodelling143–145. Reduced LV dimensions and volume with CRT are associated with a small but significant increase in LV ejection fraction by 3 months that is independent of concurrent pharmacological therapy146. The structural effects of CRT peak at 2 years and are sustained until at least 5 years147–149. Early intervention is important given that patients with NYHA class IV heart failure have the least reverse remodelling with CRT150.
Left ventricular assist device therapy.
The number of patients with heart failure who require heart transplantation far exceeds donor heart availability. LV assist devices (LVADs) mechanically pump blood from the left ventricle to the aortic root and have been used as a bridge-to-transplantation for almost three decades in patients with end-stage heart disease151. Numerous studies indicate that LVADs restore cardiac output and reverse hypertrophy in these patients152. The extent of hypertrophy regression is positively correlated with the duration of LVAD support, although the majority of remodelling occurs in the first 40 days153,154. Importantly, the effects of LVAD on hypertrophy are similar in ischaemic and non-ischaemic heart disease155. LVADs induce the most pronounced hypertrophy reduction of any existing therapy for heart failure (Table 1), highlighting the pressure-dependent aspect of hypertrophy regression. Interestingly, LVAD therapy does not reduce right ventricular hypertrophy, suggesting that reduced LV preload drives the hypertrophy regression156.
Lifestyle modifications
Aerobic exercise.
Regular exercise in healthy individuals is associated with physiological hypertrophy. Interestingly, reduced LV volumes and increased LV ejection fraction have been observed when patients with heart disease adopt an aerobic exercise routine157. Exercise-induced hypertrophy regression requires months of training, and strength training has no added effects158,159. The regression is most pronounced in patients with non-ischaemic heart failure, whereas for patients after myocardial infarction the benefits of exercise are greatest for those who start early and continue for more than 3 months160,161. Exercise leads to a reduction in circulating angiotensin II, noradrenaline, BNP and aldosterone levels158, which mitigates hypertension and might underlie hypertrophy regression in this patient population159. Notably, one study found that exercise-induced weight loss did not contribute to the reverse remodelling because BMI was unchanged whereas LV mass decreased by 12% after 4 months of low-intensity exercise159. These studies suggest that the effect of exercise on reverse remodelling is similar to that of most drug therapies.
Calorie restriction.
Preclinical models suggest that calorie restriction and intermittent fasting paradigms might be effective for preventing and reversing adverse cardiac remodelling162,163. In rodent models of metabolic syndrome-induced and obesity-induced cardiomyopathy, calorie restriction attenuated cardiomyocyte hypertrophy, oxidative stress and fibrosis162,164. Starting alternate-day intermittent fasting in rats before or directly after a myocardial infarction reduced cardiomyocyte hypertrophy and LV dilatation 12 weeks after the myocardial infarction163. In another study, starting calorie restriction 4 weeks after myocardial infarction also ameliorated cardiac dysfunction, reduced heart mass and restored adrenergic sensitivity165. Using calorie restriction and naturally occurring compounds that mimic calorie restriction (such as curcumin and resveratrol) to treat heart failure is an appealing therapeutic strategy that requires further investigation166.
Determinants of reversibility in pathological hypertrophy
One feature that distinguishes pathological from physiological hypertrophy is that pathological hypertrophy is typically only partially reversible. The reasons for the often-incomplete regression remain unclear. However, one possible explanation is the presence of extensive fibrotic remodelling driven by proliferation and activation of cardiac fibroblasts in pathological settings167. Indeed, for most therapies, patients with the lowest baseline levels of fibrosis experience greater regression of hypertrophy168. Interestingly, one of the factors thought to underlie the more rapid regression observed in female patients after AVR is that they develop less pressure-overload-associated fibrosis than male patients169. This observation might also explain why less regression is observed in ischaemic heart disease, which is associated with greater fibrosis than non-ischaemic heart failure98. However, longer-term studies of AVR found that fibrosis only slows the rate of reverse remodelling131,170, and long-term LVAD therapy has also been shown to reduce fibrosis and expression of collagen-encoding genes153. Inhibiting the progression of fibrosis with antifibrotic drugs has received interest in the past decade, and a clinical trial of the antifibrotic drug pirfenidone in patients with heart failure with preserved ejection fraction indicated that fibrosis reduction is possible in this patient population171. Future research on combination treatments of pirfenidone with existing first-line therapies for heart failure are needed to explore the potential for rapid and complete reversal of pathological hypertrophy.
Like fibroblasts, immune cells also uniquely contribute to the development of pathological cardiac hypertrophy and might have a role in the incomplete reversibility172. Although the role of immune cells in reverse remodelling remains to be fully investigated, preclinical studies indicate that modulation of these cells might be efficacious173,174. Treating mice with pressure-overload-induced cardiac hypertrophy with an antibody against CD20, which suppresses B cells, reversed hypertrophy and inhibited pathological transforming growth factor-β (TGFβ)–SMAD2/SMAD3 and ERK1/ERK2 signalling173. In other studies using the same mouse model, treatment with granulocyte colony-stimulating factor, which caused neutrophil infiltration and increased IL-1β expression, led to regression of fibrosis174. Further study is required to clarify the involvement of the immune system in reverse remodelling.
Mechanisms of hypertrophy regression
Regression of physiological hypertrophy
Pregnancy.
Chronic volume overload in pregnancy leads to eccentric cardiac hypertrophy. This physiological growth, which is mediated by the IGF1–PI3K–AKT and ERK pathways175,176, is associated with normal cardiac function and is completely reversible postpartum. In humans, the timing of cardiac hypertrophy development and regression is temporally coupled to haemodynamic load177. Pregnancy-associated changes in contractility, valve area and LV mass are reversed over several months postpartum178,179. Similar results have been observed in rodents, but hypertrophy regression is evident after 1–3 days postpartum and is complete at 2–3 weeks180–182. In addition to the reduction in heart weight, changes in LV pressure, contractility and angiogenesis are notable at 1 week postpartum180,182. The regression might be mediated by the ubiquitin–proteasome system, given that proteasome activity and expression of Fbxo32, encoding an E3 ligase, are upregulated immediately postpartum in mice176. These findings are further supported by the observation in mice that protein polyubiquitination is reduced in late pregnancy and subsequently increases 1 day postpartum182. Micro-array analysis immediately postpartum also identified changes in the expression of genes related to chemokine, glucocorticoid receptor and cytochrome P450 pathways176. Non-fibrotic extracellular matrix remodelling is an important feature of hypertrophy regression after pregnancy, and increased expression of genes encoding extracellular matrix-related proteins (Adam15, Mmp2 and Timp1) has been identified in multiple studies176,180,183,184. Notably, hypertrophy regression does not occur in lactating rats because increased circulating blood volume sustains volume overload on the heart9,185. However, stopping lactation by separating newborn pups from the mother induces rapid hypertrophy regression9,186. Lactation is associated with increased phosphorylation of ERK1, ERK2, FOXO1 and FOXO3 at 1 week postpartum in mice186, which might contribute to the extended hypertrophy.
Exercise.
Exercise-induced cardiac hypertrophy is associated with normal or improved cardiac function and reverses rapidly when training is stopped. In one study, rats with 9 weeks of swim training had substantial cardiac hypertrophy that had completely reversed 2 weeks after training cessation187. In these rats, reduced heart weight was associated with diminished mRNA content, suggesting that reduced protein synthesis contributes to hypertrophy regression187. Given that the serum IGF1 level is known to increase with endurance exercise188, reduced protein synthesis in sedentary animals might be due to decreased growth pathway activity downstream of IGF1. However, this represents an important area for future research. In humans, complete hypertrophy regression is observed within 1 month of stopping activity and is mediated by a decrease in the intracellular compartment, suggesting reduction of myofibrils8. Interestingly, exercise preconditioning ingrains antihypertrophic memory to future pathological insults. In mice that underwent 3 weeks of swim training, complete hypertrophy regression had occurred 1 week after training cessation and was associated with upregulation of lncRNA Mhrt779 expression189. When these mice were later subjected to pathological stimuli, Mhrt779 conveyed anti-hypertrophic effects through regulation of HDAC2 and the AKT–GSK3β pathway189. The calcium-binding proteins S100A8 and S100A9 are also upregulated with exercise and suppress calcineurin–NFAT signalling, which mitigates pressure-overload-induced cardiac hypertrophy190. Other mechanisms by which exercise is thought to reverse pathological cardiac hypertrophy include reduction of circulating catecholamines and blood pressure levels157,191.
Regression of pathological hypertrophy
LVAD mechanical unloading.
Owing to the number of studies using tissue samples obtained before and after LVAD therapy, the molecular mechanisms of cardiac hypertrophy regression after mechanical unloading are the best characterized (Fig. 2). The effects of LVADs are numerous and include changes to myocardial metabolism, contractility, protein synthesis, protein degradation and immune response152. The detrimental reversion to fetal-like metabolism, in which glucose is preferentially used over fatty acids, in heart failure is well established192. Mechanical unloading induces partial restoration of oxidative phosphorylation, upregulation of fatty acid metabolism, increased expression of genes encoding components of the mitochondrial respiratory chain complex and repression of fetal-like gene expression193–197. Additionally, the pentose phosphate pathway and one-carbon metabolism have been implicated in recovery after LVAD therapy, given that NADH — a key by-product of these pathways — is involved in the biosynthesis of other metabolites and helps protect against oxidative damage198. Several studies have identified an upregulation of calcium-handling proteins, including sarcoplasmic–endoplasmic reticulum calcium ATPase 2A (SERCA2A), ryanodine receptor 2 and the sodium–calcium exchanger, after LVAD therapy, which is associated with increased cardiomyocyte developed force199,200. Furthermore, the β-adrenergic responsiveness that is lost in heart failure is restored after LVAD therapy, permitting increased contractility in response to inotropic stimulation201. LVADs are one of few therapies that substantially reduce existing fibrosis, which is reflected at the molecular level by reduced expression of genes encoding collagen and matrix metalloproteinases (MMPs)194,196,202. Individuals who respond favourably to LVAD therapy show a reduction in the expression of Il1B, NPPB and EPAC2 at the time of LVAD explantation compared with non-responders203.
Fig. 2 |. Molecular features of hypertrophy regression with mechanical unloading.
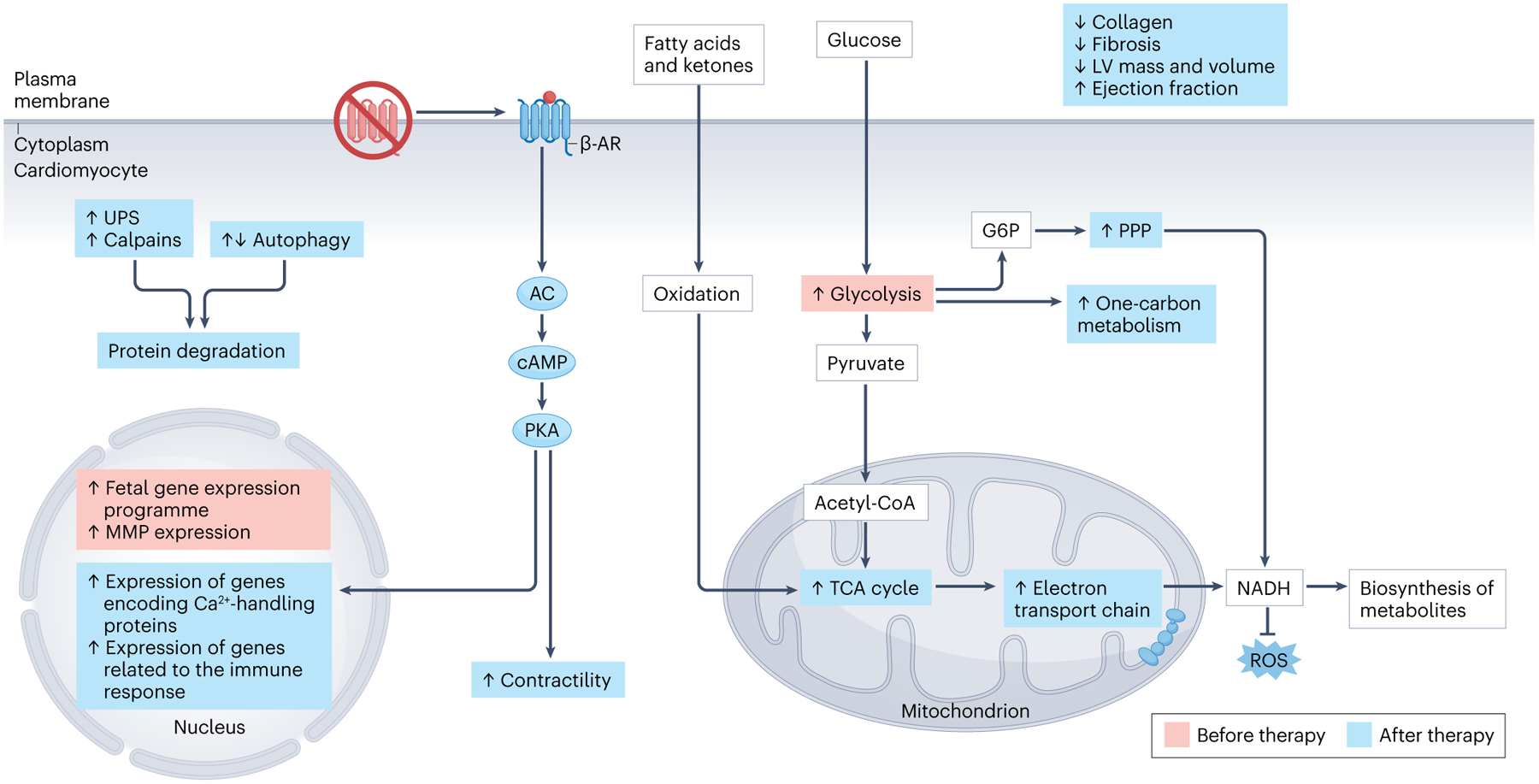
Mechanical unloading induced by left ventricular (LV) assist device (LVAD) therapy alters cardiomyocyte metabolism, protein turnover, gene expression and contractility as well as immune responses. Before LVAD therapy (red boxes), hypertrophic cardiomyocytes have immature phenotypes, such as increased glucose metabolism and a fetal gene expression pattern. Heart failure also causes desensitization of β-adrenergic receptors (β-AR) in cardiomyocytes. After LVAD therapy (blue boxes), β-AR responsiveness is restored, resulting in increased contractility and transcription of genes encoding calcium-handling proteins. The pentose phosphate pathway (PPP), oxidative phosphorylation through the electron transport chain, and one-carbon metabolism increase with LVAD treatment, resulting in the generation of nicotinamide adenine dinucleotide (NADH), a by-product that participates in other metabolic pathways and protects against oxidative damage. The ubiquitin–proteasome system (UPS) as well as the calpain protein degradation pathways become upregulated after LVAD treatment; however, autophagy activation and gene expression have been observed either to increase or to decrease. Altogether, these molecular changes result in decreased fibrosis and LV mass and volume as well as increased ejection fraction and contractility. AC, adenylyl cyclase; G6P, glucose-6-phosphate; MMP, matrix metalloproteinase; PKA, protein kinase A; ROS, reactive oxygen species; TCA, tricarboxylic acid.
Protein degradation pathways have been well characterized after mechanical unloading, with some seemingly conflicting results. The ubiquitin–proteasome system is upregulated after LVAD placement, as indicated by increased proteasome activity and upregulation of the expression of FBXO32 (encoding F-box only protein 32; also known as atrogin 1) and TRIM63 (encoding E3 ubiquitin protein ligase TRIM63; also known as MURF1)204–206. The involvement of atrogin 1 and TRIM63 in mechanical unloading-induced regression was corroborated in mouse studies showing that these proteins are required for regression of heart mass206,207. Expression and activity of the calcium-activated calpain proteases also increase following mechanical unloading in humans and rats208.
Data on the role of autophagy after mechanical unloading are less clear. One study of human tissue samples obtained after LVAD therapy found significant decreases in beclin 1, autophagy-related gene 5 (ATG5), ATG12 and microtubule-associated protein light chain 3II (LC3II), suggesting reduced autophagy, whereas levels of BNIP3 (an adaptor protein for mitophagy) increased205. However, in mouse and cell models of mechanical unloading, increased autophagic activity was required for cardiac hypertrophy regression downstream of FOXO1, which upregulated the expression of the autophagy genes Bnip3, Gabarapl1 and Ulk2 (ref.209). FOXO1 and FOXO3A gene expression was also shown to increase after LVAD therapy in a study of human heart samples210. The apparent incongruence between the findings of these studies might be due to timing. In the mouse study, autophagy activity was assessed 1 week after mechanical unloading, whereas the mean duration of unloading in the initial LVAD study was 214 days. Therefore, autophagy might be upregulated in the acute phase of mechanical unloading and then deactivated later to prevent further hypertrophy regression. Another study of mechanical unloading in rats found that the transcriptional factor eyes absent homologue 2 (EYA2) was involved in cardiac hypertrophy regression211, indicating the importance of its downstream gene targets in this process.
Other surgical and device therapies.
Compared with LVAD, the molecular changes and potential mechanisms underlying cardiac hypertrophy regression with other surgical therapies are poorly characterized. One study in patients receiving CRT suggests that miR-30d is an important determinant of a positive patient response to CRT212. Elevated plasma miR-30d levels were associated with reduced myocardial necrosis, and an increase in cardiomyocyte miR-30d levels was protective against detrimental tumour necrosis factor (TNF) signalling212. In a dog model, CRT was found to increase mitochondrial ATPase activity through reduction of cysteine disulfide bonds and to upregulate the expression of redox enzymes, leading to improved cardiomyocyte metabolism213,214. CRT is also associated with reduced expression and activity of CaMKII, p38 and TNF, and reversal of calcium channel and potassium channel remodelling215. Patients who respond favourably to CRT have increased SERCA2A and MYH6 expression and reduced NPPB expression216.
Modest molecular-level data are available for AVR. In a study in patients, multiple microRNAs were dysregulated 1 week after AVR and increased circulating levels of miR-122–5p were associated with LV dysfunction at 7 days and 6 months after AVR217. This finding was recapitulated in a mouse model, in which miR-122–5p reduced the expression of BCL2 (encoding an anti-apoptotic protein) and decreased cardiomyocyte viability217. Therefore, in patients who respond poorly to AVR, targeting miR-122–5p might be of therapeutic value.
Pharmacological therapies.
Molecular-level studies with other models of pathological hypertrophy regression are scarce; however, similar pathways to those involved in mechanical unloading are implicated. In rats with hyperthyroidism-induced cardiac hypertrophy, ARNI therapy reduced the levels of cardiac inflammatory markers, increased autophagy and suppressed TGFβ–SMAD signalling compared with the control group218. Similar findings were noted for therapy with ARB alone; however, the effects were milder than for treatment with an ARNI218. One study used mice treated with angiotensin II or the β-agonist isoprenaline to induce cardiac hypertrophy followed by cessation of treatment (mimicking ARB and β-blocker therapy, respectively) and cardiac hypertrophy regression was assessed over 1 week219. Regression in both cases was associated with increased autophagy, increased or no change in proteasome activity and reduced TGFβ–SMAD signalling, with evident sex-related differences219, as discussed below. In another study, hypertensive rats treated with a thiazide diuretic had reverse remodelling and reduced fibrosis, which was associated with reduced expression of genes encoding collagens and TGFβ, reduced reactive oxygen species production and suppression of RHO kinase activity220. The mechanisms regulating hypertrophy regression with drug therapies remain very poorly defined and further investigation is warranted.
Hypertrophy regression in genetic cardiomyopathy
Monogenic variants are estimated to account for 25–50% of cases of dilated cardiomyopathy (DCM) and 30–60% of hypertrophic cardiomyopathy (HCM)221. In a cohort of patients with DCM, hypertrophy regression potential with standard drug therapies varied according to the causative gene variants, with pathogenic variants in genes encoding the sarcomere Z-disc components (DES, DMD and FLNC) inversely correlated with reverse remodelling222. Another study in patients with DCM found that reverse remodelling was less frequent in genotype-positive patients with DCM than in patients with idiopathic DCM (39.6% versus 46.2%)223. TTN pathogenic variants (the leading genetic cause of DCM) are linked to improved reverse remodelling with therapy223–225, whereas regression is limited in patients with LMNA variants, the second leading genetic cause of DCM224,225.
HCM is predominantly caused by variants in MYH7 and MYBPC3 (ref.221). Exercise can help promote reverse remodelling in this patient population, as shown by a study in which 4 months of modest exercise in a patient with genetic HCM led to a reduction in LV mass of 12% and improved quality of life226. However, the type of exercise is an important consideration, and it is generally agreed that patients with HCM should avoid intense exercise to reduce the risk of arrhythmia227. The benefits of exercise in patients with HCM are supported by findings from studies in rodents. Voluntary cage-wheel running in mice with an HCM-causing MYH variant reversed cardiac hypertrophy, improved cardiomyocyte array and reduced apoptosis228. Hypertrophy reversibility has also been observed in non-sarcomeric genetic HCM, a less common aetiology of HCM. Cardiac remodelling associated with variants in PTPN11 — which cause Leopard syndrome, an autosomal dominant RASopathy — is completely reversible in animal models by treatment with rapamycin229.
These studies indicate an important contribution of genetics to reverse remodelling. However, the influence of the genetic background on hypertrophy regression remains poorly understood. Studies of hypertrophy development using the Hybrid Mouse Diversity Panel, a collection of 107 inbred mouse strains, identified key loci for heritability of cardiac mass230,231. Hypertrophy regression potential is likely to be similarly affected by genetic background, which represents a fertile area for future research.
Biological sex and hypertrophy regression
Sex has a role in regression of cardiac hypertrophy, with female individuals generally experiencing more favourable outcomes. This sex-related difference might be due to sex hormones and/or genes encoded on the X and Y chromosomes. Oestrogens, for instance, have been linked to upregulation of genes encoding mitochondrial proteins and lipases, whereas testosterone controls glucose tolerance and reduces fatty acid oxidation232. Furthermore, hearts from female individuals have higher levels of AKT signalling, better function after pressure overload and less fibrosis development in response to pathological stimuli than hearts from male individuals, suggesting that these features elicit cardioprotective mechanisms and might aid in the more favourable regression outcomes observed in female patients233. Among patients receiving CRT, LVAD therapy or AVR, female patients had more pronounced LV volume reduction, improved LV ejection fraction and lower serum NT-proBNP levels compared with male patients169,234–237. However, in female patients undergoing AVR, hypertrophy regression is inversely correlated with serum levels of miR-29b, a microRNA that regulates pathological hypertrophy238.
Sex-related differences are also observed in hypertrophy regression mediated by pharmacological therapies. ARNI therapy is effective in both male and female patients; however, female sex is associated with more reverse remodelling, higher LV ejection fraction and lower serum NT-proBNP levels239–241. Other studies have yielded conflicting results. One study of antihypertensive medications found that the extent of cardiac hypertrophy regression was lower in women than in men99. Conversely, another study found that female patients treated with ARBs or β-blockers had more regression of cardiac hypertrophy than male patients242. A mechanistic study found that regression of pathological hypertrophy shows stimulus-specific and sex-specific regulation219. In mice treated with angiotensin II or isoprenaline, female mice had no cardiac hypertrophy regression 1 week after angiotensin II removal, whereas isoprenaline removal was associated with rapid regression219. Hearts from male mice showed hypertrophy regression with removal of either stimulus. Regression from isoprenaline-induced hypertrophy was associated with increased autophagy in both sexes219. However, only hearts from male mice had increased proteasome activity after stimulus removal. The sex-related difference in regression of angiotensin II-induced hypertrophy might be due to TGFβ–SMAD signalling, which increased during pathological cardiac hypertrophy in both sexes but was sustained after angiotensin II removal in female mice219. In a rat model of mechanical unloading, female rats had a greater reduction in fibrosis and fetal-like gene expression than male rats243. These studies highlight the importance of considering sex when choosing a treatment strategy for pathological hypertrophy regression.
Regression versus atrophy
The distinction between hypertrophy regression and cardiac atrophy — the latter of which occurs with cancer, starvation and spaceflight — remains poorly defined244–246. The simplest discrimination might be that atrophy is a progressive, active, muscle-wasting condition, whereas regression occurs only to a certain baseline level without continued cardiac mass loss. Additionally, hypertrophy regression is associated with improved structure and function. However, in cardiac atrophy, fibrosis, sarcomere structural disarray and reduced LV performance are observed245,247. At the molecular level, several features of cardiac atrophy overlap with hypertrophy regression (Fig. 3). Cancer-associated cachexia, a severe form of muscle wasting that occurs in 30–80% of patients with cancer, is associated with cardiac dysfunction248,249. Mice with colon cancer-induced cachexia have a reduction in heart mass of ~20% compared with healthy controls, which was more pronounced in male mice and was associated with upregulation of gene expression and activity of autophagy proteins250. The involvement of FBXO32, TRIM63 and the proteasome in cancer-associated cachexia is unclear, with studies showing either increased activity or no change250,251. Other studies in mice with cancer-induced cachexia found inactivation of protein synthesis pathways downstream of AKT and mTOR252,253. Chemotherapies are also independent drivers of cardiac atrophy, and their use is associated with exacerbated muscle wasting in cancer-associated cachexia254. In mice, cardiac atrophy induced by the chemotherapy drug doxorubicin is dependent on Trim63 upregulation255. Less is known about the mechanisms of starvation-induced cardiac atrophy. However, a study in rabbits found reduced protein synthesis and myofibrillar protein half-life during starvation-induced cardiac atrophy, suggesting that similar mechanisms to those underlying cancer-associated cachexia are involved246.
Fig. 3 |. Molecular features of cardiac atrophy.
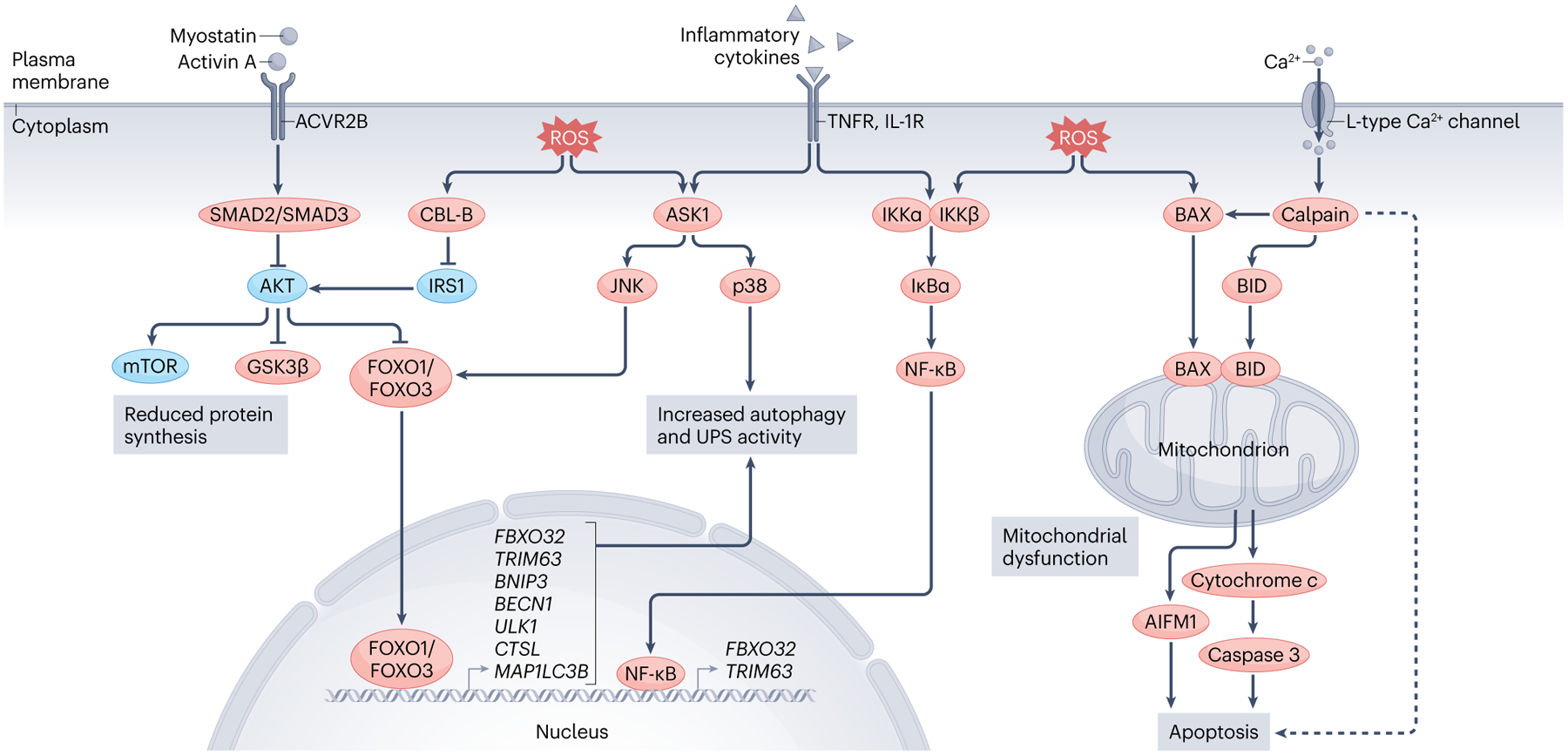
Atrophy can be initiated by an increase in the circulating levels of certain factors (such as myostatin, activin A and inflammatory cytokines) and/or increased cellular oxidative stress. Myostatin and activin A bind to the activin receptor type 2B (ACVR2B), which stimulates downstream SMAD2 and SMAD3 signalling. SMAD2 and SMAD3 inhibit pro-growth pathways (AKT and mechanistic target of rapamycin (mTOR) pathways) and activate FOXO transcription factors, which translocate to the nucleus and increase the expression of genes related to autophagy and the ubiquitin–proteasome system (UPS). Inflammatory cytokines (such as tumour necrosis factor) activate the p38 and JUN N-terminal kinase (JNK) MAP kinase pathways and nuclear factor-κB (NF-κB), which stimulate atrophic-related gene expression patterns and protein degradation. Increased intracellular levels of reactive oxygen species (ROS) act on similar pathways, while also causing the translocation of apoptosis regulator BAX and BH3-interacting domain death agonist (BID) to the mitochondria, leading to mitochondrial dysfunction and triggering apoptosis through apoptosis-inducing factor mitochondria associated 1 (AIFM1; also known as AIF) and cytochrome c–caspase 3 signalling. ROS also inhibit growth pathways and promote FOXO-dependent degradation by activating the E3 ubiquitin protein ligase CBL-B, which mediates insulin receptor substrate 1 (IRS1) degradation and thus represses downstream AKT signalling. Calpain activation owing to increased intracellular calcium levels in cardiac atrophy also triggers proteolysis, mitochondrial dysfunction and (in some instances; dashed line) apoptosis. ASK1, apoptosis signal-regulating kinase 1; GSK3β, glycogen synthase kinase 3β; IL-1R, IL-1 receptor; IκBα, NF-κB inhibitor-α; IKK, inhibitor of nuclear factor-κB kinase; TNFR, tumour necrosis factor receptor.
Spaceflight causes cardiac atrophy owing to microgravity-induced mechanical unloading of the heart256. LVAD therapy also induces mechanical unloading but the acute effect on cardiac mass is more pronounced with spaceflight than with LVAD therapy, with a reduction in LV mass of 12% in astronauts after just 10 days in space257. An in vitro study of cardiomyocytes in microgravity found a bias towards mitochondrial protein synthesis at the expense of whole-cell protein production and no change in apoptosis or protein degradation258. In a mouse microgravity model, the levels of the proteolytic proteins calpain 1 and calpain 2 were upregulated compared with normal-weight-bearing controls, whereas cardiomyocyte-specific knockout of Capns1 prevented microgravity-induced cardiac atrophy259. Increased expression and activity of ubiquitin–proteasome system components were observed in Drosophila flies reared on the International Space Station (microgravity) compared with flies reared on Earth (normal gravity)260. In mice, 15 days of spaceflight altered the expression in the heart of genes related to oxidative stress, cell cycle and senescence, suggestive of rapid cardiac ageing261,262. Spaceflight is associated with increased cardiac stiffness and risk of arrhythmia245. By comparison, with LVAD-induced mechanical unloading, reduced protein synthesis, increased calpain and proteasome activity, and expression changes in genes related to mitochondria and metabolism are observed193,205,208,263. In this case, the mechanical unloading is generally beneficial and is associated with improved cardiomyocyte developed force and LV ejection fraction, albeit with an increased risk of arrhythmia199,264,265.
One commonality of all forms of cardiac atrophy is an increase in the levels of circulating factors that stimulate downstream atrophic remodelling, including myostatin, activin A and inflammatory cytokines266–269. Myostatin and activin A trigger signalling cascades that inhibit protein synthesis and activate atrophy-related gene expression270. Whereas atrophy is widely agreed to be an active process driven by these factors, whether regression of hypertrophy is an active process or a passive response to the removal or inhibition of a stimulus remains unclear. Further investigation is needed to address this question.
Conclusions
Hundreds of clinical studies in the past almost 40 years indicate that regression of pathological cardiac hypertrophy is unequivocally linked to improved health outcomes. However, many factors have a role in determining whether and to what extent reverse remodelling occurs with existing therapies. These factors include hypertension duration, type of hypertrophy (concentric versus eccentric), age, BMI, sex, physical activity, genetic background, disease aetiology and concurrent medications. The result is that <50% of patients receiving heart failure therapies currently undergo hypertrophy regression, and those without hypertrophy regression have poor quality of life and increased mortality compared with patients who have reverse remodelling271,272. However, if regression of cardiac hypertrophy were considered a primary end point in the treatment of heart disease, as many studies suggest it should be, data-informed choice of therapy is possible thanks to the wealth of clinical literature. The use of computational approaches towards this goal might make it possible to predict whether a patient will respond to a given therapy with reverse remodelling and thereby inform clinician decision-making.
In the subset of patients who undergo regression of cardiac hypertrophy with existing therapies, full normalization of LV mass in most patients does not occur even after years of treatment87. Therefore, the development of new therapies informed by molecular studies to target LV hypertrophy specifically might prove more effective than current medications in restoring heart health in this patient population and in non-responding patients. However, despite the long-established functional and quality-of-life benefits of reverse remodelling, the molecular characterization of this process remains in its infancy. What is apparent from molecular studies is the involvement of protein degradation pathways (calpains, ubiquitin–proteasome system and autophagy) in hypertrophy regression. This finding might not be surprising, given that regulation of cardiac mass can be fundamentally distilled to a balance between protein synthesis and degradation, in which inhibiting protein synthesis and/or activating protein degradation results in net protein loss and thus tissue mass regression. However, targeting these crucial pathways with broadly acting compounds will have off-target effects on other tissues, and without careful titration might be cardiotoxic. Development of therapies that target specific molecules in these pathways with tissue specificity should permit a wider dose range and limit adverse effects. However, the lead molecular candidates for drug development remain to be elucidated, and future study in this area is paramount to advancing the field. One particularly under-studied area in which such targets might be identified is in the regression from physiological cardiac hypertrophy, in which reverse remodelling is rapid and complete. We hope this Review stimulates discussion and investigation in these areas towards the goal of reducing morbidity and mortality in the millions of people living with heart disease worldwide.
Key points.
Pathological cardiac hypertrophy is a leading risk factor for cardiovascular morbidity and mortality and is associated with increased fibrosis and apoptosis that lead to ventricular stiffness, risk of arrhythmia and impaired cardiac function.
Partial reversal of cardiac hypertrophy occurs with many existing heart failure therapies, including renin–angiotensin–aldosterone system, β-adrenergic receptor, calcium-channel and SGLT2 antagonists, but is achieved in only a subset of patients.
The potential for reverse remodelling in heart disease is influenced by many factors, including biological sex, genetics, duration of hypertension, BMI, age and disease aetiology.
Exercise-induced and pregnancy-induced physiological cardiac hypertrophy is driven by cellular mechanisms distinct from those in pathological hypertrophy, and is completely reversible, whereas moderate exercise in heart failure antagonizes pathological cellular pathways and promotes hypertrophy regression.
At the molecular level, hypertrophy regression is associated with metabolic shifts, reduced protein synthesis, extracellular matrix remodelling and altered activity of proteolytic pathways.
Acknowledgements
The authors acknowledge support from the National Institutes of Health (T32 HL007822 to T.G.M., T32 GM142607 to M.A.J., and R01HL117138 and R01GM029090 to L.A.L.).
Footnotes
Competing interests
L.A.L. is a co-founder of MyoKardia, acquired by Bristol Myers Squib, and is a paid member of their Scientific Advisory Board. The other authors declare no competing interests.
References
- 1.Ahmad FB & Anderson RN The leading causes of death in the US for 2020. JAMA 325, 1829–1830 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bluemke DA et al. The relationship of left ventricular mass and geometry to incident cardiovascular events. The MESA (Multi-Ethnic Study of Atherosclerosis) study. J. Am. Coll. Cardiol 52, 2148–2155 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levy D, Garrison RJ, Savage DD, Kannel WB & Castelli WP Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N. Engl. J. Med 322, 1561–1566 (1990). [DOI] [PubMed] [Google Scholar]
- 4.Izumi C et al. Effect of left ventricular reverse remodeling on long-term outcomes after aortic valve replacement. Am. J. Cardiol 124, 105–112 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Daubert MA et al. NT-proBNP goal achievement is associated with significant reverse remodeling and improved clinical outcomes in HFrEF. JACC Heart Fail. 7, 158–168 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Kim GH, Uriel N & Burkhoff D Reverse remodelling and myocardial recovery in heart failure. Nat. Rev. Cardiol 15, 83–96 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Rawlins J, Bhan A & Sharma S Left ventricular hypertrophy in athletes. Eur. J. Echocardiogr 10, 350–356 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Swoboda PP et al. Regression of left ventricular mass in athletes undergoing complete detraining is mediated by decrease in intracellular but not extracellular compartments. Circ. Cardiovasc. Imaging 12, e009417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung E & Leinwand LA Pregnancy as a cardiac stress model. Cardiovascular Res. 101, 561–570 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nauta JF et al. Concentric vs. eccentric remodelling in heart failure with reduced ejection fraction: clinical characteristics, pathophysiology and response to treatment. Eur. J. Heart Fail 22, 1147–1155 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melenovsky V Cardiac adaptation to volume overload. Card. Adaptations: Mol. Mechanisms 4, 167–199 (2013). [Google Scholar]
- 12.Litviňuková M et al. Cells of the adult human heart. Nature 588, 466–472 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carreño JE, Apablaza F, Ocaranza MP & Jalil JE Cardiac hypertrophy: molecular and cellular events. Rev. Esp. Cardiol 59, 473–486 (2006). [PubMed] [Google Scholar]
- 14.Bergmann O et al. Dynamics of cell generation and turnover in the human heart. Cell 161, 1566–1575 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Nakamura M & Sadoshima J Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol 15, 387–407 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Weeks KL & McMullen JR The athlete’s heart vs. the failing heart: Can signaling explain the two distinct outcomes? Physiology 26, 97–105 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Troncoso R, Ibarra C, Vicencio JM, Jaimovich E & Lavandero S New insights into IGF-1 signaling in the heart. Trends Endocrinol. Metab 25, 128–137 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Dale Abel E Insulin signaling in the heart. Am. J. Physiol. Endocrinol. Metab 321, 130–145 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagao H et al. Distinct signaling by insulin and IGF-1 receptors and their extra- and intracellular domains. Proc. Natl Acad. Sci. USA 118, e2019474118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riehle C et al. Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Mol. Cell. Biol 34, 3450–3460 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McMullen JR et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110α) pathway. J. Biol. Chem 279, 4782–4793 (2004). [DOI] [PubMed] [Google Scholar]
- 22.McMullen JR et al. Phosphoinositide 3-kinase(p110α) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc. Natl Acad. Sci. USA 100, 12355–12360 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skurk C et al. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J. Biol. Chem 280, 20814–20823 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boström P et al. C/EBPβ controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell 143, 1072–1083 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haq S et al. Glycogen synthase kinase-3β is a negative regulator of cardiomyocyte hypertrophy. J. Cell Biol 151, 117–129 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sciarretta S, Forte M, Frati G & Sadoshima J New insights into the role of mtor signaling in the cardiovascular system. Circ. Res 122, 489–505 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bueno OF et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 19, 6341–6350 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ojamaa K Signaling mechanisms in thyroid hormone-induced cardiac hypertrophy. Vasc. Pharmacol 52, 113–119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simoncini T et al. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 407, 538–541 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang KC et al. Thyroid hormone improves function and Ca2+ handling in pressure overload hypertrophy. Association with increased sarcoplasmic reticulum Ca2+-ATPase and α-myosin heavy chain in rat hearts. J. Clin. Invest 100, 1742–1749 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trivieri MG et al. Cardiac-specific elevations in thyroid hormone enhance contractility and prevent pressure overload-induced cardiac dysfunction. Proc. Natl Acad. Sci. USA 103, 6043–6048 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iliopoulou I et al. Time-dependent and independent effects of thyroid hormone administration following myocardial infarction in rats. Mol. Med. Rep 18, 864–876 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pantos C et al. Thyroid hormone at supra-physiological dose optimizes cardiac geometry and improves cardiac function in rats with old myocardial infarction. J. Physiol. Pharmacol 60, 49–56 (2009). [PubMed] [Google Scholar]
- 34.Ojamaa K, Kenessey A, Shenoy R & Klein I Thyroid hormone metabolism and cardiac gene expression after acute myocardial infarction in the rat. Am. J. Physiol. Endocrinol. Metab 279, E1319–E1324 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Riquelme CA et al. Fatty acids identified in the Burmese python promote beneficial cardiac growth. Science 334, 528–531 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X et al. MiR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. 21, 584–595 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi J et al. miR-17–3p contributes to exercise-induced cardiac growth and protects against myocardial ischemia-reperfusion injury. Theranostics 7, 664–676 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao R et al. Long noncoding RNA cardiac physiological hypertrophy-associated regulator induces cardiac physiological hypertrophy and promotes functional recovery after myocardial ischemia-reperfusion injury. Circulation 144, 303–317 (2021). [DOI] [PubMed] [Google Scholar]
- 39.Li H et al. lncExACT1 and DCHS2 regulate physiological and pathological cardiac growth. Circulation 145, 1218–1233 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gogiraju R, Bochenek ML & Schäfer K Angiogenic endothelial cell signaling in cardiac hypertrophy and heart failure. Front. Cardiovasc. Med 6, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oka T, Akazawa H, Naito AT & Komuro I Angiogenesis and cardiac hypertrophy: Maintenance of cardiac function and causative roles in heart failure. Circ. Res 114, 565–571 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Shiojima I et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Invest 115, 2108–2118 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oldfield CJ, Duhamel TA & Dhalla NS Mechanisms for the transition from physiological to pathological cardiac hypertrophy. Can. J. Physiol. Pharmacol 98, 74–84 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Lymperopoulos A, Rengo G & Koch WJ Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ. Res 113, 739–753 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morel E et al. cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ. Res 97, 1296–1304 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Métrich M et al. Epac activation induces histone deacetylase nuclear export via a Ras-dependent signalling pathway. Cell Signal. 22, 1459–1468 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Métrich M et al. Epac mediates β-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res 102, 959–965 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Osadchii OE Cardiac hypertrophy induced by sustained β-adrenoreceptor activation: pathophysiological aspects. Heart Fail. Rev 12, 66–86 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Sato PY, Chuprun JK, Schwartz M & Koch WJ The evolving impact of G protein-coupled receptor kinases in cardiac health and disease. Physiol. Rev 95, 377–404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hullmann JE et al. GRK5-mediated exacerbation of pathological cardiac hypertrophy involves facilitation of nuclear NFAT activity. Circ. Res 115, 976–985 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martini JS et al. Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc. Natl Acad. Sci. USA 105, 12457–12462 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gold JI, Gao E, Shang X, Premont RT & Koch WJ Determining the absolute requirement of G protein-coupled receptor kinase 5 for pathological cardiac hypertrophy: short communication. Circ. Res 111, 1048–1053 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rapacciuolo A et al. Important role of endogenous norepinephrine and epinephrine in the development of in vivo pressure-overload cardiac hypertrophy. J. Am. Coll. Cardiol 38, 876–882 (2001). [DOI] [PubMed] [Google Scholar]
- 54.Dash R et al. Differential regulation of p38 mitogen-activated protein kinase mediates gender-dependent catecholamine-induced hypertrophy. Cardiovasc. Res 57, 704–714 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Calvieri C, Rubattu S & Volpe M Molecular mechanisms underlying cardiac antihypertrophic and antifibrotic effects of natriuretic peptides. J. Mol. Med 90, 5–13 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Hall EJ et al. Cardiac natriuretic peptide deficiency sensitizes the heart to stress-induced ventricular arrhythmias via impaired CREB signalling. Cardiovasc. Res 118, 2124–2138 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holtwick R et al. Pressure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J. Clin. Invest 111, 1399–1407 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Potter LR, Yoder AR, Flora DR, Antos LK & Dickey DM Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb. Exp. Pharmacol 191, 341–366 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rainer PP & Kass DA Old dog, new tricks: novel cardiac targets and stress regulation by protein kinase G. Cardiovasc. Res 111, 154–162 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klaiber M et al. A cardiac pathway of cyclic GMP-independent signaling of guanylyl cyclase A, the receptor for atrial natriuretic peptide. Proc. Natl Acad. Sci. USA 108, 18500–18505 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vinnakota S & Chen HH The importance of natriuretic peptides in cardiometabolic diseases. J. Endocr. Soc 4, bvaa052 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsai EJ & Kass DA Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther 122, 216–238 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takimoto E et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med 11, 214–222 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Orsborne C, Chaggar PS, Shaw SM & Williams SG The renin-angiotensin-aldosterone system in heart failure for the non-specialist: the past, the present and the future. Postgrad. Med. J 93, 29–37 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Zhang CL et al. Plasma endothelin-1-related peptides as the prognostic biomarkers for heart failure: a PRISMA-compliant meta-analysis. Medicine 96, e9342 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamazaki T et al. Angiotensin II partly mediates mechanical stress-induced cardiac hypertrophy. Circ. Res 77, 258–265 (1995). [DOI] [PubMed] [Google Scholar]
- 67.Zablocki D & Sadoshima J Solving the cardiac hypertrophy riddle: the angiotensin II–mechanical stress connection. Circ. Res 113, 1192–1195 (2013). [DOI] [PubMed] [Google Scholar]
- 68.Lyon RC, Zanella F, Omens JH & Sheikh F Mechanotransduction in cardiac hypertrophy and failure. Circ. Res 116, 1462–1476 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Molkentin JD Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res 63, 467–475 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Wilkins BJ et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res 94, 110–118 (2004). [DOI] [PubMed] [Google Scholar]
- 71.Mehta PK & Griendling KK Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol 292, C82–C97 (2007). [DOI] [PubMed] [Google Scholar]
- 72.Zhang CL et al. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110, 479–488 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Z, Zhao YT & Zhao TC Histone deacetylases in modulating cardiac disease and their clinical translational and therapeutic implications. Exp. Biol. Med 246, 213–225 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Singh MV et al. Angiotensin II-induced hypertension and cardiac hypertrophy are differentially mediated by TLR3- and TLR4-dependent pathways. Am. J. Physiol. Heart Circ. Physiol 316, H1027–H1038 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pierdomenico SD, Lapenna D & Cuccurullo F Regression of echocardiographic left ventricular hypertrophy after 2 years of therapy reduces cardiovascular risk in patients with essential hypertension. Am. J. Hypertens 21, 464–470 (2008). [DOI] [PubMed] [Google Scholar]
- 76.Moon MG et al. Reverse remodelling by sacubitril/valsartan predicts the prognosis in heart failure with reduced ejection fraction. ESC Heart Fail. 8, 2058–2069 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sayer G & Bhat G The renin-angiotensin-aldosterone system and heart failure. Cardiol. Clin 32, 21–32 (2014). [DOI] [PubMed] [Google Scholar]
- 78.Yasunari K et al. Comparative effects of valsartan versus amlodipine on left ventricular mass and reactive oxygen species formation by monocytes in hypertensive patients with left ventricular hypertrophy. J. Am. Coll. Cardiol 43, 2116–2123 (2004). [DOI] [PubMed] [Google Scholar]
- 79.Misra KH, Das MC & Ramani YJ Effect of telmisartan on the regression of the left ventricular hypertrophy in the patients of essential hypertension. J. Clin. Diagn. Res 7, 1352–1355 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nalbantgil S et al. Effects of valsartan and enalapril on regression of left ventricular hypertrophy in patients with mild to moderate hypertension: a randomized, double-blind study. Curr. Ther. Res. Clin. Exp 61, 331–338 (2000). [Google Scholar]
- 81.Dyadyk AI et al. ACE inhibitors captopril and enalapril induce regression of left ventricular hypertrophy in hypertensive patients with chronic renal failure. Nephrol. Dial. Transplant 12, 945–951 (1997). [DOI] [PubMed] [Google Scholar]
- 82.Hernandez D et al. Regression of left ventricular hypertrophy by lisinopril after renal transplantation: role of ACE gene polymorphism. Kidney Int. 58, 889–897 (2000). [DOI] [PubMed] [Google Scholar]
- 83.Nakashima Y, Fouad FM & Tarazi RC Regression of left ventricular hypertrophy from systemic hypertension by enalapril. Am. J. Cardiol 53, 1044–1049 (1984). [DOI] [PubMed] [Google Scholar]
- 84.Konstam MA et al. Effects of the angiotensin converting enzyme inhibitor enalapril on the long-term progression of left ventricular dysfunction in patients with heart failure. Circulation 86, 431–438 (1992). [DOI] [PubMed] [Google Scholar]
- 85.Wong M et al. Valsartan benefits left ventricular structure and function in heart failure: Val-HeFT echocardiographic study. J. Am. Coll. Cardiol 40, 970–975 (2002). [DOI] [PubMed] [Google Scholar]
- 86.Wong M et al. Severity of left ventricular remodeling defines outcomes and response to therapy in heart failure: valsartan heart failure trial (Val-HeFT) echocardiographic data. J. Am. Coll. Cardiol 43, 2022–2027 (2004). [DOI] [PubMed] [Google Scholar]
- 87.Cuspidi C et al. Effects of angiotensin II receptor blockade-based therapy with losartan on left ventricular hypertrophy and geometry in previously treated hypertensive patients. Blood Press. 15, 107–115 (2006). [DOI] [PubMed] [Google Scholar]
- 88.Edwards NC, Steeds RP, Stewart PM, Ferro CJ & Townend JN Effect of spironolactone on left ventricular mass and aortic stiffness in early-stage chronic kidney disease. a randomized controlled trial. J. Am. Coll. Cardiol 54, 505–512 (2009). [DOI] [PubMed] [Google Scholar]
- 89.Feniman Stefano GMM et al. Spironolactone is secure and reduces left ventricular hypertrophy in hemodialysis patients. Ther. Adv. Cardiovasc. Dis 9, 158–167 (2015). [DOI] [PubMed] [Google Scholar]
- 90.Edelmann F et al. Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: the Aldo-DHF randomized controlled trial. JAMA 309, 781–791 (2013). [DOI] [PubMed] [Google Scholar]
- 91.Packer M et al. Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation 131, 54–61 (2015). [DOI] [PubMed] [Google Scholar]
- 92.Martens P, Beliën H, Dupont M, Vandervoort P & Mullens W The reverse remodeling response to sacubitril/valsartan therapy in heart failure with reduced ejection fraction. Cardiovasc. Ther 36, e12435 (2018). [DOI] [PubMed] [Google Scholar]
- 93.Piña IL et al. Improvement of health status following initiation of sacubitril/valsartan in heart failure and reduced ejection fraction. JACC Heart Fail. 9, 42–51 (2021). [DOI] [PubMed] [Google Scholar]
- 94.Januzzi JL et al. Association of change in N-terminal pro-B-type natriuretic peptide following initiation of sacubitril-valsartan treatment with cardiac structure and function in patients with heart failure with reduced ejection fraction. JAMA 322, 1085–1095 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ibrahim NE et al. Racial and ethnic differences in biomarkers, health status, and cardiac remodeling in patients with heart failure with reduced ejection fraction treated with sacubitril/valsartan. Circ. Heart Fail 13, E007829 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ibrahim NE et al. Sex-based differences in biomarkers, health status, and reverse cardiac remodelling in patients with heart failure with reduced ejection fraction treated with sacubitril/valsartan. Eur. J. Heart Fail 22, 2018–2025 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang Y et al. Effects of the angiotensin-receptor neprilysin inhibitor on cardiac reverse remodeling: meta-analysis. J. Am. Heart Assoc 8, e012272 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Safdar O et al. Impact of sacubitril/valsartan in cardiac reverse remodeling in ischemic vs. nonischemic cardiomyopathy. J. Heart Lung Transplant 39, S239–S240 (2020). [Google Scholar]
- 99.Lønnebakken MT et al. Left ventricular hypertrophy regression during antihypertensive treatment in an outpatient clinic (the Campania salute network). J. Am. Heart Assoc 6, e004152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gerdts E et al. Impact of age on left ventricular hypertrophy regression during antihypertensive treatment with losartan or atenolol (the LIFE study). J. Hum. Hypertens 18, 417–422 (2004). [DOI] [PubMed] [Google Scholar]
- 101.Xie X et al. Early prediction of left ventricular reverse remodeling in first-diagnosed idiopathic dilated cardiomyopathy: a comparison of linear model, random forest, and extreme gradient boosting. Front. Cardiovasc. Med 8, 684004 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cannella G et al. Prolonged therapy with ACE inhibitors induces a regression of left ventricular hypertrophy of dialyzed uremic patients independently from hypotensive effects. Am. J. Kidney Dis 30, 659–664 (1997). [DOI] [PubMed] [Google Scholar]
- 103.Bristow MR et al. β1- and β2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective β1-receptor down-regulation in heart failure. Circ. Res 59, 297–309 (1986). [DOI] [PubMed] [Google Scholar]
- 104.de Lucia C, Eguchi A & Koch WJ New insights in cardiac β-adrenergic signaling during heart failure and aging. Front. Pharmacol 9, 904 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bristow MR et al. Carvedilol produces dose-related improvements in left ventricular function and survival in subjects with chronic heart failure. Circulation 94, 2807–2816 (1996). [DOI] [PubMed] [Google Scholar]
- 106.Colucci WS et al. Metoprolol reverses left ventricular remodeling in patients with asymptomatic systolic dysfunction: the REversal of VEntricular Remodeling with Toprol-XL (REVERT) trial. Circulation 116, 49–56 (2007). [DOI] [PubMed] [Google Scholar]
- 107.Waagstein F et al. Beneficial effects of metoprolol in idiopathic dilated cardiomyopathy. Lancet 342, 1441–1446 (1993). [DOI] [PubMed] [Google Scholar]
- 108.Packer M et al. Double-blind, placebo-controlled study of the effects of carvedilol in patients with moderate to severe heart failure: the PRECISE trial. Circulation 94, 2793–2799 (1996). [DOI] [PubMed] [Google Scholar]
- 109.Godfraind T Discovery and development of calcium channel blockers. Front. Pharmacol 8, 286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Singh NK, Gupta SK & Agrawal BV Left ventricular mass regression with amlodipine in elderly hypertensives. Clin. Exp. Hypertens 21, 113–119 (1999). [DOI] [PubMed] [Google Scholar]
- 111.Adalet K et al. The effect of amlodipine on the mass and functions of the left ventricle in patients with primary hypertension and left ventricular hypertrophy. Curr. Ther. Res 56, 607–616 (1995). [Google Scholar]
- 112.Fak AS, Okucu M, Tezcan H, Bodur G & Oktay A The effects of amlodipine on left ventricular mass and diastolic function in concentric and eccentric left ventricular hypertrophy. J. Cardiovasc. Pharmacol. Ther 1, 95–100 (1996). [DOI] [PubMed] [Google Scholar]
- 113.Szlachcic J, Tubau JF, Vollmer C & Massie BM Effect of diltiazem on left ventricular mass and diastolic filling in mild to moderate hypertension. Am. J. Cardiol 63, 198–201 (1989). [DOI] [PubMed] [Google Scholar]
- 114.Natale E et al. The effect of verapamil on left ventricular remodelling and diastolic function after acute myocardial infarction (the Verapamil Infarction Study on Remodelling and Relaxation – VISOR). Cardiovasc. Drugs Ther 13, 315–324 (1999). [DOI] [PubMed] [Google Scholar]
- 115.Ahmed SN, Jhaj R, Sadasivam B & Joshi R Regression of the left ventricular hypertrophy in patients with essential hypertension on standard drug therapy. Discoveries 8, e115 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sun S, Lu F, Zhao Y, Liu Z & Wang S Effects and reversal of left ventricular hypertrophy of amlodipine plus amiloride/hydrochlorothiazide versus amlodipine plus telmisartan in patients with mild to moderate hypertension. Int. J. Cardiol 152, S24 (2011). [Google Scholar]
- 117.Polonia J et al. Lisinopril and diltiazem reduce left ventricular mass without changing blood pressure in normotensive subjects with exaggerated blood pressure response to exercise. Rev. Port. Cardiol 15, 185–193 (1996). [PubMed] [Google Scholar]
- 118.Wiviott SD et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med 380, 347–357 (2019). [DOI] [PubMed] [Google Scholar]
- 119.Anker SD et al. Empagliflozin in heart failure with a preserved ejection fraction. N. Engl. J. Med 385, 1451–1461 (2021). [DOI] [PubMed] [Google Scholar]
- 120.Verma S et al. Effect of empagliflozin on left ventricular mass in patients with type 2 diabetes mellitus and coronary artery disease: the EMPA-HEART CardioLink-6 randomized clinical trial. Circulation 140, 1693–1702 (2019). [DOI] [PubMed] [Google Scholar]
- 121.Brown AJM et al. A randomized controlled trial of dapagliflozin on left ventricular hypertrophy in people with type two diabetes: the DAPA-LVH trial. Eur. Heart J 41, 3421–3432 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mohan M et al. A randomized controlled trial of metformin on left ventricular hypertrophy in patients with coronary artery disease without diabetes: the MET-REMODEL trial. Eur. Heart J 40, 3409–3417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Guazzi M, Vicenzi M, Arena R & Guazzi MD PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: result of a 1-year, prospective, randomized, placebo-controlled study. Circ. Heart Fail 4, 8–17 (2011). [DOI] [PubMed] [Google Scholar]
- 124.Paoletti E et al. Everolimus for regression of left ventricular hypertrophy of renal transplant recipients: a randomized controlled trial [abstract 16]. Am. J. Transplant 12 (Suppl. 3), 31 (2012). [Google Scholar]
- 125.Paoletti E et al. Effect of sirolimus on left ventricular hypertrophy in kidney transplant recipients: a 1-year nonrandomized controlled trial. Am. J. Kidney Dis 52, 324–330 (2008). [DOI] [PubMed] [Google Scholar]
- 126.Saberi S et al. Mavacamten favorably impacts cardiac structure in obstructive hypertrophic cardiomyopathy: EXPLORER-HCM cardiac magnetic resonance substudy analysis. Circulation 143, 606–608 (2021). [DOI] [PubMed] [Google Scholar]
- 127.Koga-Ikuta A et al. Reverse remodelling after aortic valve replacement for chronic aortic regurgitation. Interact. Cardiovasc. Thorac. Surg 33, 10–18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lindman BR et al. Early regression of severe left ventricular hypertrophy after transcatheter aortic valve replacement is associated with decreased hospitalizations. JACC Cardiovasc. Interv 7, 662–673 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Monrad ES et al. Time course of regression of left ventricular hypertrophy after aortic valve replacement. Circulation 77, 1345–1355 (1988). [DOI] [PubMed] [Google Scholar]
- 130.Treibel TA et al. Reverse myocardial remodeling following valve replacement in patients with aortic stenosis. J. Am. Coll. Cardiol 71, 860–871 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Flett AS et al. Diffuse myocardial fibrosis in severe aortic stenosis: an equilibrium contrast cardiovascular magnetic resonance study. Eur. Heart J. Cardiovasc. Imaging 13, 819–826 (2012). [DOI] [PubMed] [Google Scholar]
- 132.Lund O & Erlandsen M Changes in left ventricular function and mass during serial investigations after valve replacement for aortic stenosis. J. Heart Valve Dis 9, 583–593 (2000). [PubMed] [Google Scholar]
- 133.Lund O, Emmertsen K, Dørup I, Jensen FT & Flø C Regression of left ventricular hypertrophy during 10 years after valve replacement for aortic stenosis is related to the preoperative risk profile. Eur. Heart J 24, 1437–1446 (2003). [DOI] [PubMed] [Google Scholar]
- 134.Pibarot P, Dumesnil JG, Leblanc MH, Cartier P & Metras J Changes in left ventricular mass and function after aortic valve replacement: a comparison between stentless and stented bioprosthetic valves. J. Am. Soc. Echocardiogr 12, 981–987 (1999). [DOI] [PubMed] [Google Scholar]
- 135.Walther T et al. Prospectively randomized evaluation of stentless versus conventional biological aortic valves: impact on early regression of left ventricular hypertrophy. Circulation 100(Suppl. 2), Ii-6–Ii-10 (1999). [DOI] [PubMed] [Google Scholar]
- 136.Powell-Wiley TM et al. Obesity and cardiovascular disease a scientific statement from the American Heart Association. Circulation 143, E984–E1010 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ippisch HM et al. Reversibility of cardiac abnormalities in morbidly obese adolescents. J. Am. Coll. Cardiol 51, 1342–1348 (2008). [DOI] [PubMed] [Google Scholar]
- 138.Cunha L et al. Evolutive echocardiographic study of the structural and functional heart alterations in obese individuals after bariatric surgery. Arq. Bras. Cardiol 87, 615–622 (2006). [DOI] [PubMed] [Google Scholar]
- 139.Hsuan CF et al. The effect of surgical weight reduction on left ventricular structure and function in severe obesity. Obesity 18, 1188–1193 (2010). [DOI] [PubMed] [Google Scholar]
- 140.Kanoupakis E et al. Left ventricular function and cardiopulmonary performance following surgical treatment of morbid obesity. Obes. Surg 11, 552–558 (2001). [DOI] [PubMed] [Google Scholar]
- 141.Syed M, Torosoff M, Rosati C, Alger S & Fein S Effect of comorbidities and medications on left ventricular mass regression after bariatric surgery. J. Clin. Hypertens 12, 223–227 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jhaveri RR et al. Cardiac remodeling after substantial weight loss: a prospective cardiac magnetic resonance study after bariatric surgery. Surg. Obes. Relat. Dis 5, 648–652 (2009). [DOI] [PubMed] [Google Scholar]
- 143.Bristow MR et al. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N. Engl. J. Med 350, 2140–2150 (2004). [DOI] [PubMed] [Google Scholar]
- 144.Linde C et al. Randomized trial of cardiac resynchronization in mildly symptomatic heart failure patients and in asymptomatic patients with left ventricular dysfunction and previous heart failure symptoms. J. Am. Coll. Cardiol 52, 1834–1843 (2008). [DOI] [PubMed] [Google Scholar]
- 145.Moss AJ et al. Cardiac-resynchronization therapy for the prevention of heart-failure events. N. Engl. J. Med 361, 1329–1338 (2009). [DOI] [PubMed] [Google Scholar]
- 146.St John Sutton M et al. Effect of cardiac resynchronization therapy on left ventricular size and function in chronic heart failure. Circulation 107, 1985–1990 (2003). [DOI] [PubMed] [Google Scholar]
- 147.Linde C et al. Long-term impact of cardiac resynchronization therapy in mild heart failure: 5-year results from the resynchronization reverses remodeling in systolic left ventricular dysfunction (REVERSE) study. Eur. Heart J 34, 2592–2599 (2013). [DOI] [PubMed] [Google Scholar]
- 148.Naqvi SY et al. Left ventricular reverse remodeling in cardiac resynchronization therapy and long-term outcomes. JACC Clin. Electrophysiol 5, 1001–1010 (2019). [DOI] [PubMed] [Google Scholar]
- 149.Matsumoto K et al. Reverse remodelling induces progressive ventricular resynchronization after cardiac resynchronization therapy ‘from vicious to virtuous cycle’. Eur. J. Echocardiogr 12, 782–789 (2011). [DOI] [PubMed] [Google Scholar]
- 150.Alvarez-Alvarez B et al. Long-term cardiac reverse remodeling after cardiac resynchronization therapy. J. Arrhythmia 37, 653–659 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jefferson HL et al. Left ventricular assist devices: a comprehensive review of major clinical trials, devices, and future directions. J. Card. Surg 36, 1480–1491 (2021). [DOI] [PubMed] [Google Scholar]
- 152.Burkhoff D, Topkara VK, Sayer G & Uriel N Reverse remodeling with left ventricular assist devices. Circ. Res 128, 1594–1612 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bruckner BA et al. Regression of fibrosis and hypertrophy in failing myocardium following mechanical circulatory support. J. Heart Lung Transplant 20, 457–464 (2001). [DOI] [PubMed] [Google Scholar]
- 154.Madigan JD et al. Time course of reverse remodeling of the left ventricle during support with a left ventricular assist device. J. Thorac. Cardiovasc. Surg 121, 902–908 (2001). [DOI] [PubMed] [Google Scholar]
- 155.Zafeiridis A, Jeevanandam V, Houser SR & Margulies KB Regression of cellular hypertrophy after left ventricular assist device support. Circulation 98, 656–662 (1998). [DOI] [PubMed] [Google Scholar]
- 156.Barbone A et al. Comparison of right and left ventricular responses to left ventricular assist device support in patients with severe heart failure: a primary role of mechanical unloading underlying reverse remodeling. Circulation 104, 670–675 (2001). [DOI] [PubMed] [Google Scholar]
- 157.Johnson EJ, Dieter BP & Marsh SA Evidence for distinct effects of exercise in different cardiac hypertrophic disorders. Life Sci. 123, 100–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chen YM, Li ZB, Zhu M & Cao YM Effects of exercise training on left ventricular remodelling in heart failure patients: an updated meta-analysis of randomised controlled trials. Int. J. Clin. Pract 66, 782–791 (2012). [DOI] [PubMed] [Google Scholar]
- 159.Kokkinos PF et al. Effects of regular exercise on blood pressure and left ventricular hypertrophy in African-American men with severe hypertension. N. Engl. J. Med 333, 1462–1467 (1995). [DOI] [PubMed] [Google Scholar]
- 160.Haykowsky M et al. A meta-analysis of the effects of exercise training on left ventricular remodeling following myocardial infarction: start early and go longer for greatest exercise benefits on remodeling. Trials 12, 92 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Delagardelle C et al. Reverse remodelling through exercise training is more pronounced in non-ischemic heart failure. Clin. Res. Cardiol 97, 865–871 (2008). [DOI] [PubMed] [Google Scholar]
- 162.Takatsu M et al. Calorie restriction attenuates cardiac remodeling and diastolic dysfunction in a rat model of metabolic syndrome. Hypertension 62, 957–965 (2013). [DOI] [PubMed] [Google Scholar]
- 163.Okoshi K et al. Influence of intermittent fasting on myocardial infarction-induced cardiac remodeling. BMC Cardiovasc. Disord 19, 126 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.An HS et al. Caloric restriction reverses left ventricular hypertrophy through the regulation of cardiac iron homeostasis in impaired leptin signaling mice. Sci. Rep 10, 7176 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.De Lucia C et al. Long-term caloric restriction improves cardiac function, remodeling, adrenergic responsiveness, and sympathetic innervation in a model of postischemic heart failure. Circ. Heart Fail 11, e004153 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sciarretta S et al. Caloric restriction mimetics for the treatment of cardiovascular diseases. Cardiovasc. Res 117, 1434–1449 (2021). [DOI] [PubMed] [Google Scholar]
- 167.Hall C, Gehmlich K, Denning C & Pavlovic D Complex relationship between cardiac fibroblasts and cardiomyocytes in health and disease. J. Am. Heart Assoc 10, e019338 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dobson LE et al. Acute reverse remodelling after transcatheter aortic valve implantation: a link between myocardial fibrosis and left ventricular mass regression. Can. J. Cardiol 32, 1411–1418 (2016). [DOI] [PubMed] [Google Scholar]
- 169.Petrov G et al. Regression of myocardial hypertrophy after aortic valve replacement: faster in women? Circulation 122, S23–S28 (2010). [DOI] [PubMed] [Google Scholar]
- 170.Puls M et al. Impact of myocardial fibrosis on left ventricular remodelling, recovery, and outcome after transcatheter aortic valve implantation in different haemodynamic subtypes of severe aortic stenosis. Eur. Heart J 41, 1903–1914 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lewis GA et al. Pirfenidone in heart failure with preserved ejection fraction: a randomized phase 2 trial. Nat. Med 27, 1477–1482 (2021). [DOI] [PubMed] [Google Scholar]
- 172.Frieler RA & Mortensen RM Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation 131, 1019–1030 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ma XL et al. Rituximab prevents and reverses cardiac remodeling by depressing B cell function in mice. Biomed. Pharmacother 114, 108804 (2019). [DOI] [PubMed] [Google Scholar]
- 174.Szardien S et al. Regression of cardiac hypertrophy by granulocyte colony-stimulating factor-stimulated interleukin-1β synthesis. Eur. Heart J 33, 595–605 (2012). [DOI] [PubMed] [Google Scholar]
- 175.Chung E, Yeung F & Leinwand LA Akt and MAPK signaling mediate pregnancy-induced cardiac adaptation. J. Appl. Physiol 112, 1564–1575 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chung E, Heimiller J & Leinwand LA Distinct cardiac transcriptional profiles defining pregnancy and exercise. PLoS One 7, e42297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mone SM, Sanders SP & Colan SD Control mechanisms for physiological hypertrophy of pregnancy. Circulation 94, 667–672 (1996). [DOI] [PubMed] [Google Scholar]
- 178.Robson SC, Dunlop W, Moore M & Hunter S Haemodynamic changes during the puerperium: a Doppler and M‐mode echocardiographic study. BJOG. Int. J. Obstet. Gynaecol 94, 1028–1039 (1987). [DOI] [PubMed] [Google Scholar]
- 179.Clapp JF & Capeless E Cardiovascular function before, during, and after the first and subsequent pregnancies. Am. J. Cardiol 80, 1469–1473 (1997). [DOI] [PubMed] [Google Scholar]
- 180.Umar S et al. Cardiac structural and hemodynamic changes associated with physiological heart hypertrophy of pregnancy are reversed postpartum. J. Appl. Physiol 113, 1253–1259 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gonzalez AMD et al. Hypertrophy signaling during peripartum cardiac remodeling. Am. J. Physiol. Heart Circ. Physiol 293, H3008–H3013 (2007). [DOI] [PubMed] [Google Scholar]
- 182.Iorga A, Dewey S, Partow-Navid R, Gomes AV & Eghbali M Pregnancy is associated with decreased cardiac proteasome activity and oxidative stress in mice. PLoS One 7, e48601 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Limon-Miranda S et al. Pregnancy differentially regulates the collagens types I and III in left ventricle from rat heart. Biomed. Res. Int 2014, 984785 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Parrott ME et al. Feature article: Maternal cardiac messenger RNA expression of extracellular matrix proteins in mice during pregnancy and the postpartum period. Exp. Biol. Med 243, 1220–1232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bond CF Blood volume changes in the lactating rat. Endocrinology 63, 285–289 (1958). [DOI] [PubMed] [Google Scholar]
- 186.Lunsford T Cardiac Hypertrophy and Regresion during Postpartum in C57B1/6 Mice. Thesis, Texas Tech Univ. Honors College; (2014). [Google Scholar]
- 187.Frenzel H et al. Regression of cardiac hypertrophy: morphometric and biochemical studies in rat heart after swimming training. J. Mol. Cell. Cardiol 20, 737–751 (1988). [DOI] [PubMed] [Google Scholar]
- 188.Rubin DA et al. Endocrine response to acute resistance exercise in obese versus lean physically active men. Eur. J. Appl. Physiol 115, 1359–1366 (2015). [DOI] [PubMed] [Google Scholar]
- 189.Lin H et al. Antihypertrophic memory after regression of exercise-induced physiological myocardial hypertrophy is mediated by the long noncoding RNA Mhrt779. Circulation 143, 2277–2292 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wei X et al. Myocardial hypertrophic preconditioning attenuates cardiomyocyte hypertrophy and slows progression to heart failure through upregulation of S100A8/A9. Circulation 131, 1506–1517 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kinney LaPier TL & Rodnick KJ Effects of aerobic exercise on energy metabolism in the hypertensive rat heart. Phys. Ther 81, 1006–1017 (2001). [PubMed] [Google Scholar]
- 192.Chen L, Song J & Hu S Metabolic remodeling of substrate utilization during heart failure progression. Heart Fail. Rev 24, 143–154 (2019). [DOI] [PubMed] [Google Scholar]
- 193.Gupte AA et al. Mechanical unloading promotes myocardial energy recovery in human heart failure. Circ. Cardiovasc. Genet 7, 266–276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Blaxall BC, Tschannen-Moran BM, Milano CA & Koch WJ Differential gene expression and genomic patient stratification following left ventricular assist device support. J. Am. Coll. Cardiol 41, 1096–1106 (2003). [DOI] [PubMed] [Google Scholar]
- 195.Schaefer A et al. Analysis of fibrosis in control or pressure overloaded rat hearts after mechanical unloading by heterotopic heart transplantation. Sci. Rep 9, 5710 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gao XM et al. Regression of pressure overload-induced left ventricular hypertrophy in mice. Am. J. Physiol. Heart Circ. Physiol 288, H2702–H2707 (2005). [DOI] [PubMed] [Google Scholar]
- 197.Diakos NA et al. Evidence of glycolysis up-regulation and pyruvate mitochondrial oxidation mismatch during mechanical unloading of the failing human heart: implications for cardiac reloading and conditioning. JACC Basic. Transl. Sci 1, 432–444 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Badolia R et al. The role of nonglycolytic glucose metabolism in myocardial recovery upon mechanical unloading and circulatory support in chronic heart failure. Circulation 142, 259–274 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Heerdt PM et al. Chronic unloading by left ventricular assist device reverses contractile dysfunction and alters gene expression in end-stage heart failure. Circulation 102, 2713–2719 (2000). [DOI] [PubMed] [Google Scholar]
- 200.Birks EJ Molecular changes after left ventricular assist device support for heart failure. Circ. Res 113, 777–791 (2013). [DOI] [PubMed] [Google Scholar]
- 201.Ogletree-Hughes ML et al. Mechanical unloading restores β-adrenergic responsiveness and reverses receptor downregulation in the failing human heart. Circulation 104, 881–886 (2001). [DOI] [PubMed] [Google Scholar]
- 202.Li YY et al. Downregulation of matrix metalloproteinases and reduction in collagen damage in the failing human heart after support with left ventricular assist devices. Circulation 104, 1147–1152 (2001). [DOI] [PubMed] [Google Scholar]
- 203.Felkin LE, Lara-Pezzi EA, Hall JL, Birks EJ & Barton PJR Reverse remodelling and recovery from heart failure are associated with complex patterns of gene expression. J. Cardiovasc. Transl. Res 4, 321–331 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wohlschlaeger J et al. Ventricular unloading is associated with increased 20S proteasome protein expression in the myocardium. J. Heart Lung Transplant 29, 125–132 (2010). [DOI] [PubMed] [Google Scholar]
- 205.Kassiotis C et al. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation 120, S191–S197 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Willis MS et al. Muscle ring finger 1 mediates cardiac atrophy in vivo. Am. J. Physiol. Heart Circ. Physiol 296, H997–H1006 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Baskin KK et al. MAFbx/Atrogin-1 is required for atrophic remodeling of the unloaded heart. J. Mol. Cell. Cardiol 72, 168–176 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Razeghi P et al. Mechanical unloading of the heart activates the calpain system. J. Mol. Cell. Cardiol 42, 449–452 (2007). [DOI] [PubMed] [Google Scholar]
- 209.Hariharan N et al. Autophagy plays an essential role in mediating regression of hypertrophy during unloading of the heart. PLoS One 8, e51632 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hall JL et al. Genomic profiling of the human heart before and after mechanical support with a ventricular assist device reveals alterations in vascular signaling networks. Physiol. Genomics 17, 283–291 (2004). [DOI] [PubMed] [Google Scholar]
- 211.Yang DK et al. Gene profiling during regression of pressure overload-induced cardiac hypertrophy. Physiol. Genomics 30, 1–7 (2007). [DOI] [PubMed] [Google Scholar]
- 212.Melman YF et al. Circulating microRNA-30d is associated with response to cardiac resynchronization therapy in heart failure and regulates cardiomyocyte apoptosis: a translational pilot study. Circulation 131, 2202–2216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wang SB, Murray CI, Chung HS & Van Eyk JE Redox regulation of mitochondrial ATP synthase. Trends Cardiovasc. Med 23, 14–18 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Agnetti G et al. Modulation of mitochondrial proteome and improved mitochondrial function by biventricular pacing of dyssynchronous failing hearts. Circ. Cardiovasc. Genet 3, 78–87 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Cho H, Barth AS & Tomaselli GF Basic science of cardiac resynchronization therapy: molecular and electrophysiological mechanisms. Circ. Arrhythm. Electrophysiol 5, 594–603 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Vanderheyden M et al. Myocardial gene expression in heart failure patients treated with cardiac resynchronization therapy. Responders versus nonresponders. J. Am. Coll. Cardiol 51, 129–136 (2008). [DOI] [PubMed] [Google Scholar]
- 217.Hosen MR et al. Circulating microRNA-122–5p is associated with a lack of improvement in left ventricular function after transcatheter aortic valve replacement and regulates viability of cardiomyocytes through extracellular vesicles. Circulation 10.1161/CIRCULATIONAHA.122.060258 (2022). [DOI] [PubMed] [Google Scholar]
- 218.Khamis T, Alsemeh AE & Abdullah DM Sacubitril/valsartan (LCZ696) ameliorates hyperthyroid-induced cardiac hypertrophy in male rats through modulation of miR-377, let-7b, autophagy, and fibrotic signaling pathways. Sci. Rep 12, 14654 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Muehleman DL, Crocini C, Swearingen AR, Ozeroff CD & Leinwand LA Regression from pathological hypertrophy in mice is sexually dimorphic and stimulus specific. Am. J. Physiol. Heart Circ. Physiol 322, H785–H797 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Mondaca-Ruff D et al. Hydrochlorothiazide reduces cardiac hypertrophy, fibrosis and rho-kinase activation in DOCA-salt induced hypertension. J. Cardiovasc. Pharmacol. Ther 26, 724–735 (2021). [DOI] [PubMed] [Google Scholar]
- 221.Garfinkel AC, Seidman JG & Seidman CE Genetic pathogenesis of hypertrophic and dilated cardiomyopathy. Heart Fail. Clin 14, 139–146 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Dal Ferro M et al. Association between mutation status and left ventricular reverse remodelling in dilated cardiomyopathy. Heart 103, 1704–1710 (2017). [DOI] [PubMed] [Google Scholar]
- 223.Escobar-Lopez L et al. Association of genetic variants with outcomes in patients with nonischemic dilated cardiomyopathy. J. Am. Coll. Cardiol 78, 1682–1699 (2021). [DOI] [PubMed] [Google Scholar]
- 224.Verdonschot JAJ et al. Clinical phenotype and genotype associations with improvement in left ventricular function in dilated cardiomyopathy. Circ. Heart Fail 11, e005220 (2018). [DOI] [PubMed] [Google Scholar]
- 225.Tobita T et al. Genetic basis of cardiomyopathy and the genotypes involved in prognosis and left ventricular reverse remodeling. Sci. Rep 8, 1998 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Petto J et al. Reverse myocardial remodeling in hypertrophic cardiomyopathy: little explored benefit of exercise. Int. J. Exerc. Sci 14, 1018–1026 (2021). [PMC free article] [PubMed] [Google Scholar]
- 227.Snir AW, Connelly KA, Goodman JM, Dorian D & Dorian P Exercise in hypertrophic cardiomyopathy: restrict or rethink. Am. J. Physiol. Heart Circ. Physiol 320, H2101–H2111 (2021). [DOI] [PubMed] [Google Scholar]
- 228.Konhilas JP et al. Exercise can prevent and reverse the severity of hypertrophic cardiomyopathy. Circ. Res 98, 540–548 (2006). [DOI] [PubMed] [Google Scholar]
- 229.Marin TM et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J. Clin. Invest 121, 1026–1043 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Rau CD et al. Mapping genetic contributions to cardiac pathology induced by beta-adrenergic stimulation in mice. Circ. Cardiovasc. Genet 8, 40–49 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Wang JJC et al. Genetic dissection of cardiac remodeling in an isoproterenol-induced heart failure mouse model. PLoS Genet. 12, e1006038 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Regitz-Zagrosek V, Oertelt-Prigione S, Seeland U & Hetzer R Sex and gender differences in myocardial hypertrophy and heart failure. Circ. J 74, 1265–1273 (2010). [DOI] [PubMed] [Google Scholar]
- 233.Beale AL, Meyer P, Marwick TH, Lam CSP & Kaye DM Sex differences in cardiovascular pathophysiology: why women are overrepresented in heart failure with preserved ejection fraction. Circulation 138, 198–205 (2018). [DOI] [PubMed] [Google Scholar]
- 234.Lilli A et al. Cardiac resynchronization therapy: gender related differences in left ventricular reverse remodeling. Pacing Clin. Electrophysiol 30, 1349–1355 (2007). [DOI] [PubMed] [Google Scholar]
- 235.Kenigsberg BB et al. Sex-associated differences in cardiac reverse remodeling in patients supported by contemporary left ventricular assist devices. J. Card. Fail 26, 494–504 (2020). [DOI] [PubMed] [Google Scholar]
- 236.Aimo A et al. Effect of sex on reverse remodeling in chronic systolic heart failure. JACC Heart Fail. 5, 735–742 (2017). [DOI] [PubMed] [Google Scholar]
- 237.Stangl V et al. Impact of gender on three-month outcome and left ventricular remodeling after transfemoral transcatheter aortic valve implantation. Am. J. Cardiol 110, 884–890 (2012). [DOI] [PubMed] [Google Scholar]
- 238.García R et al. Sex-specific regulation of miR-29b in the myocardium under pressure overload is associated with differential molecular, structural and functional remodeling patterns in mice and patients with aortic stenosis. Cells 9, 833 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Savage PS, Campbell PC & Adams SA Gender differences in reverse cardiac remodeling following initiation of sacubitril/valsartan in heart failure with reduced ejection fraction: real world experience [abstract]. Eur. Heart J 42, (Suppl. 1) ehab724.0900 (2021). [Google Scholar]
- 240.Ibrahim NE et al. Sex-based differences in biomarkers, quality of life, and reverse cardiac remodeling in patients with heart failure with reduced ejection fraction treated with sacubitril/valsartan. J. Card. Fail 26, S9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Paolini C et al. Effects and clinical implications of sacubitril/valsartan on left ventricular reverse remodeling in patients affected by chronic heart failure: a 24-month follow-up. IJC Heart Vasc. 35, 100821 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Bella JN et al. Sex-related difference in regression of left ventricular hypertrophy with antihypertensive treatment: the LIFE study. J. Hum. Hypertens 18, 411–416 (2004). [DOI] [PubMed] [Google Scholar]
- 243.Ruppert M et al. Sex similarities and differences in the reverse and anti-remodeling effect of pressure unloading therapy in a rat model of aortic banding and debanding. Am. J. Physiol. Heart Circ. Physiol 323, H204–H222 (2022). [DOI] [PubMed] [Google Scholar]
- 244.Barkhudaryan A, Scherbakov N, Springer J & Doehner W Cardiac muscle wasting in individuals with cancer cachexia. Esc. Heart Fail 4, 458–467 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Vernice NA, Meydan C, Afshinnekoo E & Mason CE Long-term spaceflight and the cardiovascular system. Precis. Clin. Med 3, 284–291 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Samarel AM, Parmacek MS, Magid NM, Decker RS & Lesch M Protein synthesis and degradation during starvation-induced cardiac atrophy in rabbits. Circ. Res 60, 933–941 (1987). [DOI] [PubMed] [Google Scholar]
- 247.Belloum Y, Rannou-Bekono F & Favier FB Cancer-induced cardiac cachexia: pathogenesis and impact of physical activity (review). Oncol. Rep 37, 2543–2552 (2017). [DOI] [PubMed] [Google Scholar]
- 248.Inui A Cancer anorexia-cachexia syndrome: current issues in research and management. CA Cancer J. Clin 52, 72–91 (2002). [DOI] [PubMed] [Google Scholar]
- 249.Fearon K et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 12, 489–495 (2011). [DOI] [PubMed] [Google Scholar]
- 250.Cosper PF & Leinwand LA Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res. 71, 1710–1720 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Tian M, Asp ML, Nishijima Y & Belury MA Evidence for cardiac atrophic remodeling in cancer-induced cachexia in mice. Int. J. Oncol 39, 1321–1326 (2011). [DOI] [PubMed] [Google Scholar]
- 252.Springer J et al. Prevention of liver cancer cachexia-induced cardiac wasting and heart failure. Eur. Heart J 35, 932–941 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Manne NDPK et al. Altered cardiac muscle mTOR regulation during the progression of cancer cachexia in the ApcMin/+ mouse. Int. J. Oncol 42, 2134–2140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Murphy KT The pathogenesis and treatment of cardiac atrophy in cancer cachexia. Am. J. Physiol. Heart Circ. Physiol 310, H466–H477 (2016). [DOI] [PubMed] [Google Scholar]
- 255.Willis MS et al. Doxorubicin exposure causes subacute cardiac atrophy dependent on the striated muscle-specific ubiquitin ligase MuRF1. Circ. Heart Fail 12, e005234 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.MacNamara JP et al. Cardiac effects of repeated weightlessness during extreme duration swimming compared with spaceflight. Circulation 143, 1533–1535 (2021). [DOI] [PubMed] [Google Scholar]
- 257.Perhonen MA et al. Cardiac atrophy after bed rest and spaceflight. J. Appl. Physiol 91, 645–653 (2001). [DOI] [PubMed] [Google Scholar]
- 258.Feger BJ et al. Microgravity induces proteomics changes involved in endoplasmic reticulum stress and mitochondrial protection. Sci. Rep 6, 34091 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Liang L et al. Calpain activation mediates microgravity-induced myocardial abnormalities in mice via p38 and ERK1/2 MAPK pathways. J. Biol. Chem 295, 16840–16851 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Walls S et al. Prolonged exposure to microgravity reduces cardiac contractility and initiates remodeling in Drosophila. Cell Rep. 33, 108445 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Kumar A, Tahimic CGT, Almeida EAC & Globus RK Spaceflight modulates the expression of key oxidative stress and cell cycle related genes in heart. Int. J. Mol. Sci 22, 9088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Hu C, Zhang X, Teng T, Ma ZG & Tang QZ Cellular senescence in cardiovascular diseases: a systematic review. Aging Dis. 13, 103–128 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Baba HA et al. Dynamic regulation of MEK/Erks and Akt/GSK-3β in human end-stage heart failure after left ventricular mechanical support: myocardial mechanotransduction-sensitivity as a possible molecular mechanism. Cardiovasc. Res 59, 390–399 (2003). [DOI] [PubMed] [Google Scholar]
- 264.Gopinathannair R et al. Device therapy and arrhythmia management in left ventricular assist device recipients: a scientific statement from the American Heart Association. Circulation 139, E967–E989 (2019). [DOI] [PubMed] [Google Scholar]
- 265.Jakovljevic DG et al. Left ventricular assist device as a bridge to recovery for patients with advanced heart failure. J. Am. Coll. Cardiol 69, 1924–1933 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Rodgers BD & Ward CW Myostatin/activin receptor ligands in muscle and the development status of attenuating drugs. Endocr. Rev 43, 329–365 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Smith RC et al. Inhibition of myostatin prevents microgravity-induced loss of skeletal muscle mass and strength. PLoS One 15, e0230818 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Loumaye A et al. Role of activin A and myostatin in human cancer cachexia. J. Clin. Endocrinol. Metab 100, 2030–2038 (2015). [DOI] [PubMed] [Google Scholar]
- 269.Allen DL, Cleary AS, Lindsay SF, Loh AS & Reed JM Myostatin expression is increased by food deprivation in a muscle-specific manner and contributes to muscle atrophy during prolonged food deprivation in mice. J. Appl. Physiol 109, 692–701 (2010). [DOI] [PubMed] [Google Scholar]
- 270.Han HQ, Zhou X, Mitch WE & Goldberg AL Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int. J. Biochem. Cell Biol 45, 2333–2347 (2013). [DOI] [PubMed] [Google Scholar]
- 271.Reis Filho JRAR, Cardoso JN, Cardoso CMR & Pereira-Barretto AC Reverse cardiac remodeling: a marker of better prognosis in heart failure. Arq. Bras. Cardiol 104, 502–506 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Boulet J & Mehra MR Left ventricular reverse remodeling in heart failure: remission to recovery. Struct. Heart 5, 466–481 (2021). [Google Scholar]
- 273.Picca M, Bisceglia J, Zocca A & Pelosi G Effects of enalapril and amlodipine on left ventricular hypertrophy and function in essential hypertension. Clin. Drug. Investig 13, 29–35 (1997). [Google Scholar]
- 274.Greenberg B et al. Effects of long-term enalapril therapy on cardiac structure and function in patients with left ventricular dysfunction results of the SOLVD echocardiography substudy. Circulation 91, 2573–2581 (1995). [DOI] [PubMed] [Google Scholar]
- 275.Desai AS et al. Effect of sacubitril-valsartan vs enalapril on aortic stiffness in patients with heart failure and reduced ejection fraction: a randomized clinical trial. JAMA 322, 1077–1084 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Seeman T et al. Regression of left-ventricular hypertrophy in children and adolescents with hypertension during ramipril monotherapy. Am. J. Hypertens 20, 990–996 (2007). [DOI] [PubMed] [Google Scholar]
- 277.Park K et al. The impact of a dose of the angiotensin receptor blocker valsartan on post-myocardial infarction ventricular remodelling. Esc. Heart Fail 5, 354–363 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Degirmenci H et al. Comparison of effects of nebivolol, carvedilol and irbesartan on left ventricular hypertrophy associated with hypertension. Eur. Rev. Med. Pharmacol. Sci 18, 630–637 (2014). [PubMed] [Google Scholar]
- 279.Barrios V et al. Regression of left ventricular hypertrophy by a candesartan-based regimen in clinical practice. the VIPE study. J. Renin Angiotensin Aldosterone Syst 7, 236–242 (2006). [DOI] [PubMed] [Google Scholar]
- 280.Khan MS et al. Reverse cardiac remodeling following initiation of sacubitril/valsartan in patients with heart failure with and without diabetes. JACC Heart Fail. 9, 137–145 (2021). [DOI] [PubMed] [Google Scholar]
- 281.Maizels L et al. Characterization of heart failure patients with reverse left ventricular remodelling post-angiotensin receptor blockers/neprilysin inhibitors therapy. Esc. Heart Fail 9, 1682–1688 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Chau KH et al. Regression of left ventricular mass after transcatheter aortic valve replacement: the PARTNER trials and registries. J. Am. Coll. Cardiol 75, 2446–2458 (2020). [DOI] [PubMed] [Google Scholar]
- 283.Algahim MF et al. Progressive regression of left ventricular hypertrophy two years after bariatric surgery. Am. J. Med 123, 549–555 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Ikonomidis I et al. Weight loss after bariatric surgery improves aortic elastic properties and left ventricular function in individuals with morbid obesity: a 3-year follow-up study. J. Hypertens 25, 439–447 (2007). [DOI] [PubMed] [Google Scholar]
- 285.Weiss RJ & Bent B Diltiazem-induced left ventricular mass regression in hypertensive patients. J. Clin. Hypertens 3, 135–143 (1987). [PubMed] [Google Scholar]
- 286.Awata N et al. Regression of left ventricular hypertrophy in hypertensive patients treated with metoprolol. Curr. Ther. Res. Clin. Exp 50, 175–182 (1991). [Google Scholar]
- 287.Cherchi A, Sau F & Seguro C Possible regression of left ventricular hypertrophy during antihypertensive treatment with diuretics and/or beta blockers. J. Clin. Hypertens 3, 216–225 (1987). [PubMed] [Google Scholar]
- 288.Islim IF, Watson RD, Ihenacho HNC, Ebanks M & Singh SP Amlodipine: effective for treatment of mild to moderate essential hypertension and left ventricular hypertrophy. Cardiology 96, 10–18 (2001). [DOI] [PubMed] [Google Scholar]
- 289.Rosendorff C, Dubiel R, Xu J & Chavanu KJ Comparison of olmesartan medoxomil versus amlodipine besylate on regression of ventricular and vascular hypertrophy. Am. J. Cardiol 104, 359–365 (2009). [DOI] [PubMed] [Google Scholar]
- 290.Gu M et al. Clinical outcome of cardiac resynchronization therapy in dilated-phase hypertrophic cardiomyopathy. J. Geriatr. Cardiol 14, 238–244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Sugano A et al. Optimal cut-off value of reverse remodeling to predict long-term outcome after cardiac resynchronization therapy in patients with ischemic cardiomyopathy. J. Cardiol 69, 456–461 (2017). [DOI] [PubMed] [Google Scholar]
- 292.Ocaranza MP et al. Reverse remodeling in human heart failure after cardiac resynchronization therapy is associated with reduced RHO-kinase activation. Front. Pharmacol 12, 565724 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Solomon SD et al. Effect of cardiac resynchronization therapy on reverse remodeling and relation to outcome: multicenter automatic defibrillator implantation trial: cardiac resynchronization therapy. Circulation 122, 985–992 (2010). [DOI] [PubMed] [Google Scholar]
- 294.St John Sutton MG et al. Sustained reverse left ventricular structural remodeling with cardiac resynchronization at one year is a function of etiology: quantitative Doppler echocardiographic evidence from the Multicenter InSync Randomized Clinical Evaluation (MIRACLE). Circulation 113, 266–272 (2006). [DOI] [PubMed] [Google Scholar]
- 295.Drakos SG et al. Reverse electrophysiologic remodeling after cardiac mechanical unloading for end-stage nonischemic cardiomyopathy. Ann. Thorac. Surg 91, 764–769 (2011). [DOI] [PubMed] [Google Scholar]
- 296.Muthiah K et al. Longitudinal structural, functional, and cellular myocardial alterations with chronic centrifugal continuous-flow left ventricular assist device support. J. Heart Lung Transplant 36, 722–731 (2017). [DOI] [PubMed] [Google Scholar]
- 297.Gupta A et al. Effect of spironolactone on diastolic function in hypertensive left ventricular hypertrophy. J. Hum. Hypertens 29, 241–246 (2015). [DOI] [PubMed] [Google Scholar]
- 298.Ori Y et al. Regression of left ventricular hypertrophy in patients with primary aldosteronism/low-renin hypertension on low-dose spironolactone. Nephrol. Dial. Transplant 28, 1787–1793 (2013). [DOI] [PubMed] [Google Scholar]
- 299.Kosugi D et al. Beneficial effects of sodium glucose cotransporter 2 inhibitors on left ventricular mass in patients with diabetes mellitus. J. Diabetes 13, 847–856 (2021). [DOI] [PubMed] [Google Scholar]
- 300.Rørth R et al. Comparison of BNP and NT-proBNP in patients with heart failure and reduced ejection fraction. Circ. Heart Fail 2, e006541 (2020). [DOI] [PubMed] [Google Scholar]
- 301.Ichiki T, Huntley BK & Burnett JC BNP molecular forms and processing by the cardiac serine protease corin. Adv. Clin. Chem 61, 1–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Sutanto H, Dobrev D & Heijman J Angiotensin receptor-neprilysin inhibitor (ARNI) and cardiac arrhythmias. Int. J. Mol. Sci 22, 8994 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.McKie PM & Burnett JC NT-proBNP: the gold standard biomarker in heart failure. J. Am. Coll. Cardiol 68, 2437–2439 (2016). [DOI] [PubMed] [Google Scholar]
- 304.Pascual-Figal D et al. Sacubitril-valsartan, clinical benefits and related mechanisms of action in heart failure with reduced ejection fraction. A review. Front. Cardiovasc. Med 8, 754499 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]